

Abstract

Carbohydrate moieties were strategically transported from the indolocarbazole topoisomerase I (Top1) inhibitor class to the indenoisoquinoline system in search of structurally novel and potent Top1 inhibitors. The syntheses and biological evaluation of 20 new indenoisoquinolines glycosylated with linear and cyclic sugar moieties are reported. Aromatic ring substitution with 2,3-dimethoxy-8,9-methylenedioxy or 3-nitro groups exerted strong effects on antiproliferative and Top1 inhibitory activities. While the length of the carbohydrate side chain clearly correlated with antiproliferative activity, the relationship between stereochemistry and biological activity was less clearly defined. Twelve of the new indenoisoquinolines exhibit Top1 inhibitory activity equal to or better than that of camptothecin. An advanced synthetic intermediate from this study was also used to efficiently prepare indotecan (LMP400) and indimitecan (LMP776), two anticancer agents currently under investigation in a Phase I clinical trial at the National Institutes of Health.
Introduction
Human topoisomerase type I (Top1) relieves the torsional strain that develops in DNA as a natural consequence of transcription, replication, and chromatin remodeling.1 X-ray crystallographic studies support a Top1-mediated DNA relaxation mechanism where one strand of the DNA is reversibly cleaved to allow for relaxation of DNA supercoiling. The proposed mechanism includes a “cleavage complex” intermediate in which Top1 catalytic residue Tyr723 becomes covalently bound to the 3′ end of the scissile strand of DNA, allowing the cleaved DNA strand to rotate around the uncleaved strand in a “controlled rotation” mechanism.2−4 Top1 poisons are Top1 inhibitors that bind at the DNA cleavage site and stabilize the “cleavage complex” as “interfacial poisons”,5,6 thereby inhibiting the religation of the scissile strand, a process that normally occurs rapidly enough that the complexes are undetectable in cells.7 Collision of the advancing replication fork with the stabilized single-strand DNA break in the cleavage complex causes irreversible double-strand DNA breaks that lead to cell death.1 Top1 inhibitors are effective antitumor agents because cancer cells express higher levels of Top1 and replicate DNA more often than normal cells, enhancing their susceptibility to the effects of these drugs.1,8
Examples of Top1 poisons include the camptothecins 1–3, the indenoisoquinolines 4–6, the indolocarbazole 7, and the dibenzonaphthyridinone 8 (Figure 1).9−11 At present, there are only two FDA-approved Top1 inhibitors, the camptothecin analogues topotecan (2) and irinotecan (3). Out of hundreds of biologically evaluated indenoisoquinolines, indotecan (LMP400, 4)12 and indimitecan (LMP776, 5)12 were promoted to a Phase I clinical trial at the National Institutes of Health, which began in 2010.13 Likewise, the indolocarbazole class is a potential source of therapeutic Top1 inhibitors with advantages similar to the indenoisoquinoline class.14 One exemplary indolocarbazole, the Top1 inhibitor edotecarin (7), advanced to a Phase III clinical trial, where it unfortunately failed.15
Figure 1.

Representative Top1 inhibitors.
Previous studies of indenoisoquinoline glycosides were inspired by the possibility that “platform hopping” carbohydrates from indolocarbazoles to indenoisoquinolines might yield potent, novel Top1 poisons.16 Alignment of the X-ray crystal structures of Top1–DNA cleavage complexes bound with the indenoisoquinoline 9 (MJ238) or the indolocarbazole 10 (SA315F) shows similar positions and orientations for the two small molecules within the ternary complex (Figure 2).17 The fused polycyclic cores of each molecule intercalate between consecutive DNA base pairs, allowing their side chains (butanoic acid in 9, glucopyranosyl in 10) to project into the major groove. The lactam nitrogen of the indenoisoquinoline appears to be an excellent candidate for attachment of a carbohydrate moiety because it is so near the glycosylated indole nitrogen of the indolocarbazole.
Figure 2.

Overlay of the X-ray crystal structures of the ternary Top1–DNA–drug cleavage complexes containing 9 (PDB code 1SC7, purple) or the indolocarbazole 10 (PDB code 1SEU, green). Top1 and DNA from 1SC7 are shown. The stereoview is programmed for wall-eyed (relaxed) viewing.
Within the indolocarbazole class, glycosylation is a critical feature for Top1 inhibitory activity, and its absence severely compromises potency.18,19 Attachment of a carbohydrate moiety to the indenoisoquinoline system has the potential to enhance binding affinity for the Top1–DNA cleavage complex because of improved hydrogen bonding interactions between the molecule and its binding site. Substitution of indenoisoquinolines at the lactam nitrogen with side chains containing hydrogen bond donors and acceptors is established as a necessary feature for potent Top1 inhibitory and antiproliferative activity,12,20 and carbohydrates fulfill this requirement. Glycosylation could also result in more selective uptake of these compounds by cancer cells overexpressing glucose transporters.21
Only three previously studied indenoisoquinoline glycosides have aromatic ring substitution, and these three exhibit the most promising biological activities. The other glycosides do not feature aromatic ring substitution, and most have only micromolar mean graph midpoints (MGM, similar to GI50) and Top1 inhibitory activity that is 50–75% that of 1 μM camptothecin (1).16 The 2,3-dimethoxy-8,9-methylenedioxy pattern was previously found to improve Top1 and growth inhibitory potencies when added to unsubstituted analogues (see Figure 1 for indenoisoquinoline numbering and lettering systems).22 Prior studies have also shown a substantial contribution to the same biological activities by a 3-nitro substituent.23−27 The present investigation was therefore undertaken to determine whether or not the combination of cyclic and noncyclic N-carbohydrate substituents with the 2,3-dimethoxy-8,9-methylenedioxy or 3-nitro substitution pattern could yield new indenoisoquinolines with greatly enhanced activities. Maximum synthetic efficiency and versatility might be achieved if the carbohydrates were installed at a late stage using a divergent pathway from common intermediate 20, and a synthesis of 20 was therefore undertaken (Scheme 1). If it could be prepared in an efficient and economical way, intermediate 20 might also provide the phase I experimental anticancer drugs indotecan (4) and indimitecan (5) at lower cost. A similar synthetic strategy involving late-stage common intermediate 25 was adopted and executed for the preparation of 3-nitroindenoisoquinoline glycosides (Scheme 5). These efforts have resulted in extremely potent, new indenoisoquinolines substituted with carbohydrates on the lactam nitrogen. The present series of compounds was also examined as part of a program to determine factors that selectively modulate tyrosyl DNA phosphodiesterase I (TDP1) inhibitory activity relative to Top1 activity, since recent work has revealed that certain indenoisoquinolines can express both types of activities.28,29 There is increasing interest in drugs that rationally exploit multiple targets to achieve more advantageous therapeutic effects.30,31
Scheme 1.

Scheme 5.

Chemistry
2,3-Dimethoxy-8,9-methylenedioxyindenobenzopyran 20 was prepared via the condensation of hydroxyphthalide 14 and phthalide 18 (Scheme 1). The synthesis of 14 began with veratric acid (11), which was alkylated with formaldehyde under acidic conditions to give phthalide 12.32 Lactone 12 was brominated by free radical halogenation to provide intermediate 13, which was hydrolyzed with water to provide hydroxyphthalide 14.33 The synthesis of 18 started with piperonal (15), which was brominated to give benzaldehyde 16.34 Reduction of intermediate 16 with NaBH4 afforded the benzylic alcohol 17.35 Compound 17 was treated with n-BuLi at −78 °C to effect a halogen–lithium exchange, and the resulting intermediate was quenched with CO2 and treated with aqueous HCl to provide 18, according to the procedure reported by Noire and Franck.36 The two phthalides 14 and 18 were condensed in the presence of NaOMe in refluxing EtOAc–MeOH to give indenedione intermediate 19, which was then cyclized in refluxing Ac2O to yield indenobenzopyran 20. The known 3-nitroindenobenzopyran 25 was produced via similar chemistry, as shown in Scheme 2.26 Beginning with phthalide 21, sequential aromatic nitration,37 radical bromination, and alkaline hydrolysis provided hydroxyphthalide 24, which was condensed with phthalide 21 and cyclized as before to produce indenobenzopyran 25. The synthetic pathways leading to 20 and 25 are highly convergent, thus increasing the overall efficiencies of these routes.
Scheme 2.

Eleven different primary amines derived from aldose sugars were either purchased or prepared according to published procedures (Scheme 3). Glyceraldehyde-derived primary amines 26–28 are commercially available. Five- and six-carbon d-aldose sugars were converted to primary amines (31, 34, 37, 40, 43, 46, and 49) by condensation with hydroxylamine under basic conditions to provide mixtures of E- and Z-oximes, which were then subjected to hydrogenation over PtO2.16,38 Commercially available β-glucopyranose nitromethane (50) was hydrogenated over PtO2 to provide cyclic β-glucose derivative 51. The aminosugars were condensed with indenobenzopyran 20 (Scheme 4) or indenobenzopyran 25 (Scheme 5) to provide carbohydrate-substituted indenoisoquinolines.
Scheme 3.

Scheme 4.

The synthesis of 20 conveniently facilitated the investigation of new syntheses of the Phase I anticancer agents indotecan (4) and indimitecan (5) (Schemes 1 and 6). Indenobenzopyran 20 was condensed with commercially available 3-morpholinopropylamine or 3-imidazolylpropylamine to provide 4 or 5, respectively. The alternative route involving the primary bromide 72 as the last intermediate has previously been reported (Schemes 6 and 7).12
Scheme 6.

Scheme 7.

Biological Results and Discussion
All of the new carbohydrate-substituted indenoisoquinolines were evaluated for antiproliferative activity in cultured human cancer cells in the National Cancer Institute’s NCI-60 screen, so named because it commonly employs a panel of 60 different human cancer cell lines.39,40 The NCI-60 screen first evaluates new compounds at 10–5 M in a one-dose assay to measure average growth percent of treated versus untreated cells. For each of the new compounds prepared in this study, the average growth percent is reported in Table 1. Compounds displaying significant growth inhibition at 10–5 M are promoted to a five-dose assay where testing concentrations range from 10–8 to 10–4 M. A repeat of the latter assay may be carried out if the first five-dose assay shows potent growth inhibition. Mean graph midpoint (MGM) values from the five-dose assays are reported in Table 1. The MGM value is similar to an average GI50 except that in situations in which the GI50 falls below or above the testing concentration range of 10–8 or 10–4 M it is recorded as 10–8 or 10–4 M. The Top1 inhibitory activity of each compound was evaluated in a Top1-mediated DNA cleavage assay,41 and the resulting data are presented in Table 1 and Figure 3. The Top1 inhibition potency scoring rubric is defined in the Table 1 legend.
Table 1. Antiproliferative and Top1 and TDP1 Inhibitory Activities of Carbohydrate-Substituted Indenoisoquinolines.
| cytotoxicity
GI50, μM |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| side chain | compd | Top1 cleavagea | TDP1 inhibitionb | mean growth, %d | lung, HOP-62 | colon, HCT-116 | CNS, SF-539 | melanoma, UACC-62 | ovarian, OVCAR-3 | renal, SN12C | prostate, DU-145 | breast, MCF7 | MGMc |
| 1 | ++++ | 0.01 | 0.03 | 0.01 | 0.01 | 0.22 | 0.02 | 0.01 | 0.01 | 0.0405 ± 0.0187 | |||
| 4 | ++++ | 1.78 | 1.15 | 0.04 | 0.03 | 74.1 | 0.813 | 0.155 | 0.37 | 4.64 ± 1.25 | |||
| 5 | ++++ | <0.01 | <0.01 | 0.037 | <0.01 | 0.085 | <0.01 | <0.01 | 0.01 | 0.079 ± 0.023 | |||
| 2,3-Dimethoxy-8,9-methylenedioxy | |||||||||||||
| racemic propanediol | 52 | ++++ | 0 | 27.11 | 0.031 | 0.026 | 0.023 | 0.023 | 0.052 | 0.035 | 0.036 | 0.021 | 0.065 ± 0.011 |
| S-propanediol | 53 | ++++ | 0 | 7.10 | 0.028 | 0.021 | 0.025 | 0.017 | 0.048 | 0.030 | 0.034 | 0.010 | 0.049 ± 0.003 |
| R-propanediol | 54 | ++++ | 0 | 31.56 | 0.036 | 0.036 | 0.032 | 0.025 | 0.037 | 0.040 | 0.029 | 0.166 ± 0.005 | |
| arabityl | 55 | +++ | 0 | 52.80 | 50.1 | 0.355 | 0.174 | 0.155 | 50.1 | 0.234 | 50.1 | 0.148 | 5.62 ± 0.18 |
| ribityl | 56 | +++ | + | 46.90 | 0.251 | 0.148 | 0.028 | 0.062 | 0.022 | 0.229 | 0.479 | 0.059 | 1.43 ± 0.44 |
| lyxityl | 57 | ++++ | 0 | 34.68 | 0.676 | 0.891 | 0.214 | 0.195 | 0.794 | 0.457 | 2.51 | 0.079 | 2.94 ± 0.52 |
| xylityl | 58 | ++++ | 0 | 5.32 | 0.027 | 0.059 | 0.021 | 0.052 | 0.096 | 0.085 | 0.138 | 0.034 | 0.150 ± 0.012 |
| galactityl | 59 | ++ | + | 82.29 | |||||||||
| glucosyl | 60 | +++ | 0 | 87.53 | |||||||||
| mannityl | 61 | ++++ | 0 | 79.47 | |||||||||
| glucopyranosylmethyl | 62 | ++ | + | 97.65 | |||||||||
| 3-Nitro | |||||||||||||
| racemic propanediol | 63 | +++ | 0 | –17.51 | 0.324 | 0.138 | 0.339 | 0.479 | 0.355 | 0.363 | 0.060 | 0.389 ± 0.013 | |
| R-propanediol | 64 | +++ | 0 | –26.99 | 0.347 | 0.138 | 0.309 | 0.398 | 0.380 | 0.427 | 0.056 | 0.332 ± 0.022 | |
| arabityl | 65 | ++++ | + | –9.21 | 1.91 | 1.35 | 1.45 | 1.70 | 2.69 | 3.16 | 0.912 | 1.58 | |
| ribityl | 66 | ++++(+) | 0 | –27.46 | 0.407 | 0.219 | 0.234 | 0.525 | 0.380 | 0.550 | 0.145 | 0.508 ± 0.041 | |
| lyxityl | 67 | ++++ | ++ | –16.07 | 1.74 | 1.20 | 1.74 | 1.10 | 2.14 | 2.69 | 3.47 | 0.501 | 1.66 |
| xylityl | 68 | ++++(+) | + | –14.59 | 0.363 | 0.141 | 0.214 | 0.407 | 0.295 | 0.457 | 0.096 | 0.372 ± 0.024 | |
| galactityl | 69 | ++++ | 0 | 51.53 | 5.13 | 3.39 | 2.57 | 6.03 | 4.27 | 9.55 | 2.95 | 7.94 | |
| mannityl | 70 | ++++ | + | 73.17 | >100 | 3.39 | 3.39 | 3.47 | 5.25 | >100 | 0.724 | 16.6 | |
| glucopyranosylmethyl | 71 | ++ | ++ | 77.36 | |||||||||
| S-propanediol | 79(16) | ++++ | 0.089 | 0.025 | 0.157 | 0.098 | 0.309 | 0.241 | 0.040 | 0.016 | 0.156 ± 0.061 | ||
| glucosyl | 80(16) | ++++ | 58.05 | 7.35 | 5.08 | 2.61 | 2.98 | 4.26 | 17.7 | 4.76 | 0.88 | 4.60 ± 0.53 | |
Compound-induced DNA cleavage due to Top1 inhibition, with scores given according to the following system based on the activity of 1 μM camptothecin: 0, no inhibition; +, between 20% and 50% activity; ++, between 50% and 75% activity; +++, between 75% and 95% activity; ++++, equal activity; ++++(+), greater activity.
TDP1 inhibition scores are given according to IC50: 0, >111 μM; +, 37–111 μM; ++, 12–37 μM; +++, 1–12 μM; ++++, < 1 μM.
Mean growth midpoint of growth inhibition from five dose assay, ranging from 10–8 to 10–4 M.
Growth percent of cultured cells of ∼60 cancer cell lines treated with 10 μM compound relative to vehicle-treated control.
Figure 3.

Top1-mediated DNA cleavage assay gel: lane 1, DNA alone; lane 2, DNA and Top1; lane 3, DNA and Top1 and 1 μM camptothecin (1); lane 4, DNA and Top1 and 1 μM MJ-III-65 (6); lanes 5–28, DNA and Top1 and indicated concentration (μM) of test compound. The numbers and arrows at the left indicate cleavage site positions (see Experimental Section). Gel-based assays are commonly acquired twice for each compound, and they are always run with positive controls, including camptothecin and an indenoisoquinoline.
Combination chemotherapy using both Top1 and TDP1 inhibitors may be a promising approach to the treatment of cancer. Top1–DNA cleavage complexes are converted into irreversible cleavage complexes following collision with replication or transcription forks. TDP1 assists in the removal of irreversible cleavage complexes by catalyzing the hydrolysis of the phosphodiester linkage between Top1 catalytic residue Tyr723 and the 3′-end of DNA.42 TDP1 has been implicated both in the repair of transcription- and replication-associated irreversible Top1-DNA cleavage complexes.43−45 Inhibition of TDP1 denies the cell the ability to repair such adducts by this mechanism and may therefore sensitize cells to the effects of Top1 poisons.46 Since the discovery of TDP1 in 1996, a variety of inorganic and organic inhibitors have appeared in the literature, many of which exhibit only micromolar IC50 concentrations. The structure–activity relationships of indenoisoquinoline TDP1 inhibitors are being developed by our laboratory, and already several promising dual inhibitors of Top1 and TDP1 have been identified.28,29 The structural features that contribute to TDP1 inhibitory activity vs Top1 inhibitory activity are of current interest because they might allow selective modulation of each type of activity. Inhibitory activity against recombinant TDP1 was measured in a gel-based assay,47 and these data are presented in Table 1 and Figure 4.
Figure 4.

TDP1 inhibition assay gel. The concentrations of test compounds are indicated above each lane. N14Y is 5′-end labeled DNA oligonucleotide with 3′ phosphotyrosyl, and N14P is 5′-end labeled DNA oligonucleotide (see Experimental Section). Gel-based assays are commonly acquired twice for each compound, and they are always run with positive controls, including camptothecin and an indenoisoquinoline.
In the 2,3-dimethoxy-8,9-methylenedioxy linear sugar side chain series (Scheme 4), 6 of the 10 compounds (52–54, 57, 58, and 61) scored ++++ for Top1 inhibition, three (55, 56, and 60) earned +++, and the remaining one (59) received ++. Two compounds, 56 and 59, secured a TDP1 inhibition score of +, while the remaining compounds were inactive vs TDP1. The number of carbons in the side chain does not appear to correlate with Top1 or TDP1 inhibition. Interestingly, the only compounds in this series not promoted to the NCI-60 five-dose assay because of low growth inhibition were the three featuring six-carbon side chains. Cells treated with 61, 59, and 60 in the one-dose assay displayed growth percentages 79.47%, 82.29%, or 87.53%, indicating weak growth inhibition, especially in comparison with the most potent growth inhibitors in this series, 58, 53, and 52, whose growth percentages were 5.32%, 7.10%, and 27.11%, respectively, at 10 μM.
In general, the 3-nitro-substituted compounds (Scheme 5 and Figure 5) appended with linear sugar side chains outperformed the 2,3-dimethoxy-8,9-methylenedioxy-substituted series in both Top1 inhibition and growth inhibition, and a higher percentage of its members inhibit TDP1, although very weakly, at concentrations below 111 μM (Table 1). Two molecules (66 and 68) in the series outscored camptothecin in Top1 inhibition at ++++(+), six (65, 67, 69, 70, 79, and 80) were equipotent to camptothecin at ++++, and two others (63 and 64) scored +++. Four compounds (65, 67, 68, and 70) scored at least + in TDP1 inhibition, with 67 securing a ++ score. The most potent growth inhibitor (66) in the series at 10 μM is one of the two most potent Top1 poisons reported in this study. The molecules in this series fall neatly into two separate groups based on their growth inhibitory activity. Those with six-carbon side chains (69, 70, and 80) delivered positive growth percentages between 51.53% and 73.17%, and the six compounds with three- or five-carbon side chains (63–68) have negative growth percentages ranging from −27.46% to −9.21%. The weak growth inhibitory activities displayed by 3-nitro compounds with six-carbon carbohydrate side chains mirror the low activities of 59, 60, and 61 in the 2,3-dimethoxy-8,9-methylenedioxy series. This trend occurs even though the six-carbon carbohydrate series contains potent Top1 poisons.
Figure 5.

Previously reported 3-nitro-substituted indenoisoquinoline glycosides.16
The nitrated indenoisoquinoline glycoside 68 containing a xylityl side chain is among the most potent Top1 poisons reported to date, and it was therefore selected for a docking study in an attempt to rationalize its exceptional activity (Figure 6). Morrell et al. previously proposed a dual role for 3-nitro substitution that attempts to explain its striking effect on the Top1 inhibitory activity of the indenoisoquinolines.23,24 First, the nitro group may engage in hydrogen bonding interactions with nearby Top1 enzyme residues, including Asn722. In the present case, the closer of the two nitro group oxygens is 2.6 Å away from the side chain nitrogen of Asn722. A second role for the nitro group is to withdraw electron density from the “A” aromatic ring of the indenoisoquinoline, which may facilitate the formation of stabilizing π-stacking interactions between the flanking base pairs and the indenoisoquinoline through increased charge transfer interactions and electrostatic charge complementarity.48
Figure 6.
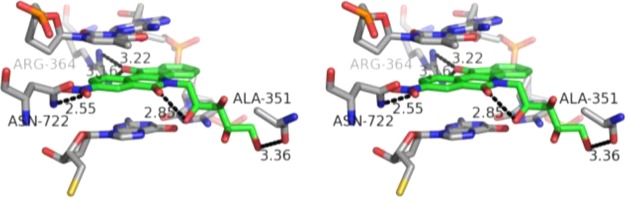
Energy-minimized hypothetical binding pose of 68 (green) within the X-ray crystal structure of a stalled Top1–DNA cleavage complex cocrystallized with 9 (PDB code 1SC7). Potential hydrogen bonding interactions are indicated by dashed lines. Heavy atom distances appear next to the dashed lines. The stereoview is programmed for wall-eyed (relaxed) viewing.
The β-glucopyranosylmethyl moiety present in 62 and 71 is structurally and stereochemically identical to the sugar appended to edotecarin (7) except for the addition of a single methylene spacer between the glycosyl group and the nitrogen in 62 and 71. In a previous study seeking to optimize the carbohydrate moiety of indolocarbazole NB-506, β-glucopyranosyl was found to outperform most other furanose and pyranose sugars in terms of in vitro cytotoxicity, Top1 inhibition, and safety margin in a tumor xenograft experiment.49 On the basis of these observations, potent Top1 inhibition and growth inhibition activities were anticipated with the most accurate β-glucopyranosyl translation. However, Top1, TDP1, and growth inhibitory activity data for the two molecules were disappointing. Both compounds scored ++ for Top1 inhibition, and neither inhibited TDP1 at an IC50 under 12 μM. Growth percentage at 10 μM was 97.65% and 77.36% for 62 and 71, respectively, and therefore, neither met the threshold for MGM evaluation in a five-dose assay. Each of the two compounds was the least active in their respective series for growth inhibition and Top1 inhibition (Tables 2 and 3).
Table 2. Rank-Order for Top1-Mediated DNA Cleavage Assay Score vs Side Chain for Three Carbohydrate-Substituted Indenoisoquinoline Seriesa.
| unsubstituted | Top1 cleavage | 2,3-dimethoxy-8,9-methylenedioxy | Top1 cleavage | 3-nitro | Top1 cleavage | |||
|---|---|---|---|---|---|---|---|---|
| 86 | lyxityl | +++ | 53 | S-propanediol | ++++ | 68 | xylityl | ++++(+) |
| 82 | S-propanediol | +++ | 52 | racemic propanediol | ++++ | 66 | ribityl | ++++(+) |
| 84 | arabityl | ++ | 54 | R-propanediol | ++++ | 69 | galactityl | ++++ |
| 81 | racemic propanediol | ++ | 57 | lyxityl | ++++ | 65 | arabityl | ++++ |
| 89 | glucosyl | ++ | 61 | mannityl | ++++ | 67 | lyxityl | ++++ |
| 87 | xylityl | ++ | 58 | xylityl | ++++ | 70 | mannityl | ++++ |
| 90 | mannityl | + | 60 | glucosyl | +++ | 80 | glucosyl | ++++ |
| 88 | galactityl | + | 56 | ribityl | +++ | 79 | S-propanediol | ++++ |
| 83 | R-propanediol | 0/+ | 55 | arabityl | +++ | 63 | racemic propanediol | +++ |
| 85 | ribityl | 0 | 59 | galactityl | ++ | 64 | R-propanediol | +++ |
| 62 | glucopyranosylmethyl | ++ | 71 | glucopyranosylmethyl | ++ |
Data for compounds 79–90 from Peterson et al.16
Table 3. Rank-Order for Mean Growth Percent vs Side Chain for Three Carbohydrate-Substituted Indenoisoquinoline Seriesa.
| unsubstituted | mean growth, % | 2,3-dimethoxy-8,9-methylenedioxy | mean growth, % | 3-nitro | mean growth, % | |||
|---|---|---|---|---|---|---|---|---|
| 85 | ribityl | 17.28 | 58 | xylityl | 5.32 | 66 | ribityl | –27.46 |
| 87 | xylityl | 34.91 | 53 | S-propanediol | 7.10 | 64 | R-propanediol | –26.99 |
| 86 | lyxityl | 50.62 | 52 | racemic propanediol | 27.11 | 63 | racemic propanediol | –17.51 |
| 84 | arabityl | 58.44 | 54 | R-propanediol | 31.56 | 67 | lyxityl | –16.07 |
| 88 | galactityl | 67.99 | 57 | lyxityl | 34.68 | 68 | xylityl | –14.59 |
| 90 | mannityl | 69.35 | 56 | ribityl | 46.90 | 65 | arabityl | –9.21 |
| 89 | glucosyl | 97.16 | 55 | arabityl | 52.80 | 69 | galactityl | 51.53 |
| 61 | mannityl | 79.47 | 80 | glucosyl | 58.05 | |||
| 59 | galactityl | 82.29 | 70 | mannityl | 73.17 | |||
| 60 | glucosyl | 87.53 | 71 | glucopyranosylmethyl | 77.36 | |||
| 62 | glucopyranosylmethyl | 97.65 |
Data for compounds 80 and 84–90 from Peterson et al.16
A docking study was carried out to investigate whether indenoisoquinoline 62 is predicted to assume a binding pose similar to indolocarbazole 10 in the Top1–DNA cleavage complex. The highest-ranked docking pose of 62 is similar to the experimentally observed binding pose of indenoisoquinoline 9 in the PDB code 1SC7 X-ray crystal structure.17 The polycyclic cores of 62 and 10 overlap, and their carbohydrate moieties project into the major groove (Figure 7). However, a significant difference is that the β-glucopyranosylmethyl group of 62 projects toward Asn352, while the β-glucopyransoyl group of 10 points in the opposite direction, away from Asn352. It is possible that the glucopyransoyl moiety is not suitably accommodated in the binding site when it is attached to the indenoisoquinoline scaffold. It is unclear why the cyclic carbohydrate moiety confers less Top1 inhibitory activity than linear carbohydrates. The entropy penalty that must be paid upon ligand binding would be less for the rigid β-glucopyranose versus a linear chain. On the other hand, conformational freedom of five- and six-carbon linear sugar side chains may allow them to reach and interact with far-away residues in the binding site better than a rigid β-glucopyranose; although three-carbon side chains also conferred excellent Top1 inhibitory activity. The possibilities that alternative linkage lengths between the carbohydrate and the indenoisoquinoline lactam nitrogen, and other cyclic sugar moieties besides β-glucopyranose, may confer greater Top1 inhibitory activity and cytotoxicity should not be discounted at this point.
Figure 7.
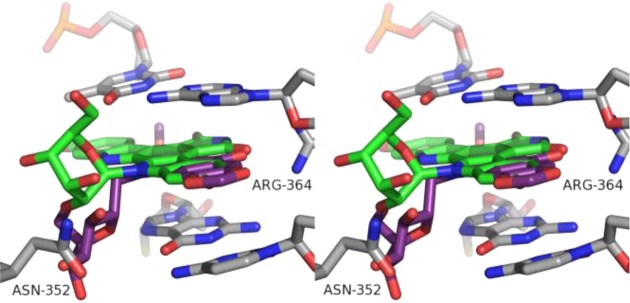
Overlay of 10 (green) and 62 (purple) within the X-ray crystal structure of a stalled Top1–DNA cleavage complex cocrystallized with 9 (PDB code 1SC7). The stereoview is programmed for wall-eyed (relaxed) viewing.
A rank-order comparison of three series of indenoisoquinoline glycosides (ring-unsubstituted (Figure 8), 3-nitro, and 2,3-dimethoxy-8,9-methylenedioxy) was carried out to elucidate structure–activity relationships, using Top1 inhibition (Table 2) or the mean growth percent (Table 3) as criteria. In terms of Top1 inhibition, however, no clear trends emerge. Three-, five-, and six-carbon linear sugars are all among the best Top1 inhibitors within each series. One clear conclusion from the rank-order comparison is that 3-nitro substitution enhances or maintains Top1 inhibitory activity relative to analogous compounds in the 2,3-dimethoxy-8,9-methylenedioxy or unsubstituted series. This is true in nearly all cases, with propanediols 63 and 64 being the sole exceptions. The ribityl side chain was enigmatic. While ring-unsubstituted 85 received a 0 score for Top1 inhibition, the 3-nitro analogue 66 scored an exceptional ++++(+) score (Table 2). Meanwhile, the 2,3-dimethoxy-8,9-methylenedioxy analogue 56 earned +++ (Table 2). For some reason, the effect of ribityl substitution is particularly sensitive to its context.
Figure 8.

Previously reported ring-unsubstituted indenoisoquinoline glycosides.16
On the other hand, rank-ordering the compounds within each series using mean growth percent as the criterion reveals a clear dependence of biological activity on the number of carbons in the side chain (Table 3). Consistently, the worst growth inhibitors in each series bear a linear, six-carbon or β-glucopyranosylmethyl side chain, and none of such compounds expressed a mean growth percent above 50% when tested at 10 μM. Standout side chains in this regard include ribityl, xylityl, and S-, R-, and racemic propanediols, and they were distinguished by their consistent presence near the top of the rank-order within each series. As seen with the rank-order comparison by Top1 inhibitory activity, 3-nitro substitution is present on the best growth inhibitors. With only one exception, 3-nitro substitution conferred a better inhibition of mean growth percent to a compound with a given side chain than 2,3-dimethoxy-8,9-methylenedioxy substitution or nonsubstitution. No such trend is detected when comparing the unsubstituted series compounds with a given side chain to their analogue in the 2,3-dimethoxy-8,9-methylenedioxy series. In that case, aromatic substitution conferred better activity to compounds with xylityl, lyxityl, arabityl, and glucosyl side chains, while worse activity resulted when ribityl, galactityl, and mannityl chains were present.
Comparisons between side chains on the basis of stereochemistry are considerably more complicated than when using length or shape as the basis. Only in cases in which the side chain length was held constant and a single stereocenter inverted have comparisons been made. Several interesting stereochemical effects were observed in the 2,3-dimethoxy-8,9-methylenedioxy series. While all three propanediol side chains conferred ++++ Top1 activity, wide variance was seen between R- (54) and S-propanediols (53) for growth inhibition, which achieved mean growth percentages of 31.56% and 7.10%, respectively (Table 3). Among the five-carbon side chains, 2′S stereochemistry appeared optimal for growth inhibition (e.g., 2′S58, 5.32% vs 2′R57, 34.68%), although it did not appear to influence Top1 inhibition scores (Tables 2 and 3). 3′R stereochemistry appeared to confer improved growth inhibition and Top1 inhibition (e.g., 3′R58, 5.32%, ++++ vs 3′S56, 46.90%, +++). Finally, two separate comparisons were made within the six-carbon side chain trio. While 2′ and 4′ stereochemistry had a slight effect on growth inhibition, 2′R61 (++++) outperformed 2′S60 (+++), and 4′R60 (+++) bested 4′S59 (++) in Top1 inhibition scores (Table 3).
Similar comparisons within the 3-nitro series show that its stereochemical preferences do not necessarily follow those seen above, complicating the development of a comprehensive SAR for the carbohydrate-substituted indenoisoquinolines. While 2′R stereochemistry confers better growth inhibition than 2′R,S to the propanediol side chain (64, −26.99%; 63, −17.51%), Top1 inhibition is best with 2′S vs 2′R (79, Figure 5, ++++; 64, Scheme 5 +++). A preference for S vs R stereochemistry on the propanediol side chain was also found for the ring-unsubstituted series (82, +++; 83, 0/+; Figure 8). When five-carbon side chains are being considered, 2′S is best for Top1 inhibition [2′R65, ++++ vs 2′S66, ++++(+)]. 2′S stereochemistry on six-carbon side chains appears to hold a slight edge over 2′R for growth inhibition (2′S80, 58.05% vs 2′R70, 73.17%).
In an effort to explain the apparent preference for 2′S over R stereochemistry observed in compounds 64 and 79, we constructed molecular models of the two compounds within the Top1–DNA cleavage complex (Figure 9). The calculated binding poses revealed the potential for a network of intra- and intermolecular hydrogen bonding interactions that could stabilize 79 in the Top1–DNA cleavage complex to a greater extent than 64. The isoquinoline carbonyl oxygen of 79 is predicted to engage in an intramolecular hydrogen bond with the side chain 2′S hydroxyl. Such an interaction is not possible in the case of 64, where the 2′R hydroxyl is oriented away and at a further distance from the carbonyl oxygen.
Figure 9.

Energy-minimized hypothetical top-ranked binding pose of 64 (top, green) and 79 (bottom, green) within the X-ray crystal structure of a stalled Top1–DNA cleavage complex cocrystallized with 9 (PDB code 1SC7). Potential hydrogen bonding interactions are indicated by dashed lines. Heavy atom distances appear next to the dashed lines. The stereoview is programmed for wall-eyed (relaxed) viewing.
Molecular modeling also exposed an interesting relationship between intramolecular hydrogen bonding and stereochemistry of the side chain 2′ hydroxyl for the other indenoisoquinoline glycosides prepared in this study. It was consistently observed that the top-ranked docking poses could form an intramolecular hydrogen bond between the isoquinoline carbonyl oxygen and, if present, the side chain 2′S hydroxyl (figures not shown). The presence of the intramolecular hydrogen bond did not translate to increased docking scores for compounds with a side chain 2′S hydroxyl vs a 2′R hydroxyl. It is noted that molecular modeling studies with highly flexible ligands, such as the compounds bearing five- and six-carbon linear carbohydrate side chains, are less reliable than those with more rigid ligands.50
To evaluate whether carbohydrate side chains are viable for consideration in future structure optimization efforts of indenoisoquinolines, exceptionally potent antiproliferative Top1 poisons from the present work were compared to analogues bearing 3-morpholinopropyl or 3-imidazolylpropyl side chains (Figure 10). 3-Nitro-substituted indenoisoquinoline 68, bearing a xylityl side chain, is as potent or more potent in the antiproliferative (MGM 0.372 ± 0.024 μM) and the Top1 inhibition [++++(+)] assays than its analogues 91 and 92(24) in either respect. 2,3-Dimethoxy-8,9-methylenedioxy-substituted indenoisoquinoline 53, bearing an S-propanediol side chain, displays more potent antiproliferative activity (MGM 0.049 ± 0.003 μM) than 4 or 5(12) while maintaining equivalent Top1 inhibitory activity. This finding contradicts the “conventional wisdom” that nitrogen heterocycles and amine moieties ought to be attached to indenoisoquinolines and structurally related platforms for maximal antiproliferative and Top1 inhibitory activity potency. The gel data presented in Figure 3 reveal that in contrast to many aminoalkyl-substituted indenoisoquinolines, the carbohydrate-substituted compounds do not suppress Top1-mediated DNA cleavage at high drug concentrations.
Figure 10.

Indenoisoquinolines bearing 3-morpholino- and 3-imidazolylpropyl side chains for comparison to indenoisoquinoline glycosides.
Finally, the synthetic routes pioneered here open up the possibility for new syntheses of the Phase I clinical drugs indotecan (4) and indimitecan (5). Overall yields of each route were calculated from the longest linear sequence, which was six steps in each case (Table 4). The yields given in Schemes 6 and 7 have previously been reported.12,51,52 The new routes produce 4 and 5 in 30.8% and 26.3% overall yields, respectively, from starting material 11 (Schemes 1 and 6). The previously developed routes afforded 4 and 5 in 16.5% and 18.1% overall yields, respectively, starting from 73 (Schemes 6 and 7).12,51,52 Advantages of the old routes are that they do not require the use of a pyrophoric material (n-BuLi), nor cryogenic conditions, at any step. Advantages that the new routes hold over the previous ones include significantly improved overall yields and scale-up-friendly purification procedures, which eliminate the need to perform column chromatography at any step. All purifications in the new routes were performed by solvent washes, recrystallization, trituration, or precipitation.
Table 4. Comparison of Yields of 4 and 5 by Synthetic Routes Depicted in Schemes 1, 6, and 7.
Conclusion
Although both topotecan (2) and irinotecan (3) are effective antitumor agents, the camptothecin class is not without serious flaws, including (1) dose-limiting side effects such as leucopenia and severe diarrhea, (2) chemical instability of the α-hydroxylactone and its hydrolysis to the ring-open carboxylate form, which binds to plasma proteins, (3) removal from cells by drug efflux transporters, and (4) long infusion times for patients due to the rapid reversibility of the camptothecin-bound cleavage complex. In general, the indenoisoquinolines can overcome several of these drawbacks because of their (1) chemical stability, (2) cleavage site specificity that is different from camptothecin, (3) less rapidly reversible binding to cleavage complexes, (4) greater activity vs camptothecin-resistant enzyme mutants, and (5) the fact that some of them are not substrates for drug efflux pumps or are ejected from cells less efficiently than the camptothecins.1,11,47
Two series of carbohydrate-substituted indenoisoquinolines bearing 3-nitro or 2,3-dimethoxy-8,9-methylenedioxy substituents on the aromatic rings were produced through advantageous synthetic routes, diverging at a late stage from intermediate 20 or 25 and thus avoiding costly linear routes to each compound. Fourteen of the 20 compounds inhibited the growth of cultured cancer cells by 52.80% or better when tested at a concentration of 10 μM. Twelve new compounds achieved Top1 inhibition assay scores of ++++ or greater. These highly active carbohydrate-substituted indenoisoquinolines provide a set of new lead compounds for further optimization studies. TDP1 inhibition assay scores were very weak, with 12 compounds scoring 0, six compounds scoring +, and only two compounds scoring ++. Incorporation of a carbohydrate therefore offers a way to ensure low TDP1 inhibitory activity while maintaining Top1 inhibition. Although the structural features that confer TDP1 vs Top1 inhibitory activity to the indenoisoquinolines is an open question that is presently under investigation, previous reports have documented structural characteristics that contribute to TDP1 inhibition, including the presence of primary amines connected to the lactam nitrogen through polymethylene chains, and bis(indenoisoquinoline) dimers having polyamine linkers.28,29
One significant difference between the 3-nitro and 2,3-dimethoxy-8,9-methylenedioxy series is that only those in the former series showed negative growth percent, indicative of greater cytotoxicity. Carbohydrate side chain length appears to have no correlation to Top1 inhibitory activity in each series, but it does appear to influence growth inhibitory activity, with three- and five-carbon sugar side chains appearing exclusively on the list of the most potent growth inhibitors. Stereochemical effects of the 2′, 3′, and 4′ positions of the sugar side chain were found to be important for 2,3-dimethoxy-8,9-methylenedioxy-substituted indenoisoquinoline glycosides, while the 2′ and 3′ positions were found to be of import for 3-nitro-substituted indenoisoquinoline glycosides. Through rank-order comparisons, it was revealed that the aromatic ring substitution pattern plays a large role in both growth inhibition and Top1 inhibitory activity for indenoisoquinolines bearing identical sugar side chains. Indenobenzopyran 20 was investigated as a common intermediate in new synthetic routes to the anticancer agents indotecan (4) and indimitecan (5). The new routes were found to be higher-yielding than previous ones, and are therefore of interest for the more efficient production of these Phase I clinical anticancer drugs.
This investigation has established a framework for future studies of indenoisoquinoline topoisomerase I poisons and their glycosides, which may include (1) incorporation of other indenoisoquinoline aromatic ring substitution patterns that have previously been found to confer high Top1 inhibitory and antiproliferative activities, such as 3-nitro-9-methoxy24,25,27 and 2,3,8-trimethoxy-9-hydroxy;53 (2) analysis of alternative linkages of cyclic carbohydrate moieties to the indenoisoquinoline lactam nitrogen; and (3) consideration of aminoglycoside moieties.18
Experimental Section
General Methods
Reactions were monitored by silica gel analytical thin-layer chromatography, and 254 nm UV light was used for visualization. Melting points were determined using capillary tubes and are uncorrected. 1H nuclear magnetic resonance spectrometry was performed using a 300 MHz spectrometer, and 13C NMR was performed using a 500 MHz spectrometer. Infrared spectra were obtained from KBr pellets using an FTIR spectrometer. High-resolution mass spectra were recorded on a double-focusing sector mass spectrometer with magnetic and electrostatic mass analyzers. Compound purities were estimated by reversed phase C18 HPLC, with UV detection at 254 nm, and the major peak area of each tested compound was >95% of the combined total peak area.
2,3-Dimethoxy-6-(3-morpholinopropyl)-5,6-dihydro-8,9-methylenedioxy-5,12-dioxoindeno[1,2-c]isoquinoline (4)
3-Morpholinopropylamine (0.081 g, 0.56 mmol) was added to a stirring solution of indenobenzopyran 20 (0.069 g, 0.20 mmol) in CHCl3 (21 mL). The solution was stirred and heated at reflux under Ar for 16 h. The cooled reaction mixture was washed with water (2 × 15 mL) and brine (1 × 15 mL), dried over Na2SO4, and concentrated to provide 4 (0.092 g, 96%) as a dark purple solid: mp 294–296 °C (lit.12 mp 290–292 °C). 1H NMR (300 MHz, CDCl3) δ 8.04 (s, 1 H), 7.64 (s, 1 H), 7.43 (s, 1 H), 7.08 (s, 1 H), 6.10 (s, 2 H), 4.51 (m, 2 H), 4.05 (s, 3 H), 3.99 (s, 3 H), 3.75 (m, 4 H), 2.58–2.43 (m, 6 H), 2.02, (m, 2 H).
6-(3-(1H-Imidazol-1-yl)propyl)-2,3-dimethoxy-5,6-dihydro-8,9-methylendioxy-5,12-dioxoindeno[1,2-c]isoquinoline (5)
3-Imidazolylpropylamine (0.078 g, 0.62 mmol) was added to a stirring solution of indenobenzopyran 20 (0.079 g, 0.22 mmol) in CHCl3 (22 mL). The solution was stirred and heated at reflux under Ar for 16 h. Additional amine was added (0.071 g, 0.57 mmol), and stirring and heating at reflux were continued for 24 h. The cooled reaction mixture was washed with H2O (20 mL), and the aqueous layer containing emulsified product was extracted with 95:5 CHCl3–MeOH (4 × 100 mL). The combined organic layers were washed with H2O (4 × 50 mL) and brine (1 × 50 mL), dried over Na2SO4, and concentrated to provide 5 (0.083, 82%) as a dull purple solid: mp 320–322 °C (lit.12 mp 270–272 °C). 1H NMR (300 MHz, CDCl3) δ 8.02 (s, 1 H), 7.64 (s, 1 H), 7.62 (s, 1 H), 7.16 (s, 1 H), 7.06 (s, 1 H), 7.05 (s, 1 H), 6.41 (s, 1 H), 6.09 (s, 2 H), 4.46 (t, J = 7.2 Hz, 2 H), 4.22 (t, J = 6.7 Hz, 2 H), 4.05 (s, 3 H), 4.00 (s, 3 H), 2.46–2.27 (m, 2 H).
2,3-Dimethoxy-8,9-methylenedioxyindeno[1,2-e]benzopyran-6,11-dione (20)
Hydroxyphthalide 14(33) (3.44 g, 16.4 mmol) and phthalide 18(35) (2.94 g, 16.5 mmol) were suspended with stirring in EtOAc (40 mL). A NaOMe solution was prepared by carefully dissolving Na (3.24 g, 141 mmol) in MeOH (anhydrous, 80 mL), and this solution was added to the suspension. The reaction mixture was stirred and heated at reflux under an Ar atmosphere for 24 h. The solution was cooled to room temperature, acidified to pH 1 with 37% HCl, and concentrated in vacuo to dryness. Ac2O (40 mL) was added to the residue and the mixture was heated at reflux with stirring for 72 h under an Ar atmosphere. After the mixture was cooled to room temperature, hexanes (50 mL) were added, and the mixture was filtered. The residue was washed with hexanes (100 mL), suspended in H2O (75 mL), and extracted with CHCl3 (8 × 250 mL). The combined organic layers were concentrated in vacuo and azeotroped once with PhMe (20 mL). The residue was purified by two precipitations from CHCl3–hexanes (30 mL:25 mL) and one from CHCl3–Et2O (30 mL:25 mL) to yield 20 as a dark purple solid (2.92 g, 51%): mp 311–314 °C. IR (film) 1732, 1707, 1520, 1476, 1364, 1315, 1243 cm–1; 1H NMR (300 MHz, CDCl3) δ 7.69 (s, 1 H), 7.64 (s, 1 H), 7.08 (s, 1 H), 6.99 (s, 1 H), 6.09 (s, 2 H), 4.05 (s, 3 H), 3.98 (s, 3 H); ESIMS m/z (rel intensity) 353 (MH+, 100); HRESIMS m/z calcd for C19H13O7 353.0661 (MH+), found 353.0664 (MH+); HPLC purity, 100% (90% MeOH, 10% H2O).
3-Hydroxy-6-nitrophthalide (24)
Phthalide 22(37) (7.0 g, 39 mmol) and NBS (7.0 g, 39 mmol) were suspended in CCl4 (140 mL) with stirring, and then benzoyl peroxide (68 mg, catalytic) was added. The mixture was heated at reflux with stirring under argon, and then it was exposed to the light of a 250 W, 125 V sunlamp located 4” from the flask. After 2 h the reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated to yield a yellowish oil. Water (40 mL) was added to the oil, and the mixture was heated at reflux with stirring for 2 h, at which time the solution was nearly colorless. The solution was then kept at 5 °C for 16 h and then gently shaken one time, which appeared to initiate crystallization. The mixture was kept at 5 °C for 2 h more and then filtered to provide 24 as a white crystalline solid intermixed with some yellowish solid (2.3 g, 30%): mp 160–164 °C (lit.54 mp 156–160 °C). 1H NMR (300 MHz, DMSO-d6) δ 8.60 (dd, J = 8.3 Hz, J = 2.1 Hz, 1 H), 8.49 (d, J = 2.1 Hz, 1 H), 7.96 (d, J = 8.3 Hz, 1 H), 6.82 (s, 1 H).
3-Nitroindeno[1,2-c]isochromene-5,11-dione (25).26
Phthalide (21, 0.9 g, 6.7 mmol) and hydroxyphthalide 24 (1.3 g, 6.7 mmol) were stirred as a suspension in EtOAc (25 mL), and a NaOMe in MeOH solution (0.8 g Na dissolved in 50 mL of MeOH) was added. The mixture was heated at reflux with stirring, and after 19 h an additional portion of phthalide (0.3 g, 2.2 mmol) was added. After 39 h the reaction mixture was cooled to room temperature and acidified with 37% HCl (∼2 mL) to pH 1. The mixture was concentrated in vacuo and azeotroped once with PhMe (30 mL). Acetic anhydride (60 mL) was added to the residue, and the mixture was heated at reflux for 29 h. The mixture was filtered and concentrated, and the residue was dissolved in CHCl3 (80 mL) and washed with saturated NaHCO3 solution (1 × 40 mL) and H2O (2 × 40 mL). The organic solution was concentrated to ∼5 mL, loaded onto a silica gel column, and purified by flash chromatography (100% CHCl3, 13 cm × 2 cm, 25 mL fractions). The impure, product-containing fractions were combined and concentrated to ∼5 mL. Hexanes (1 mL) were added, and the mixture was filtered. The residue was resuspended in EtOAc (20 mL) and filtered to provide 25 as a bright orange solid (224 mg, 11%): mp 271–274 °C (lit.26 mp 271–272 °C). 1H NMR (300 MHz, DMSO-d6) δ 8.81 (d, J = 2.4, 1 H), 8.69 (dd, J = 8.8 Hz, 2.5 Hz, 1 H), 8.40 (d, J = 2.4 Hz, 1 H), 7.73–7.55 (m, 4 H); EIMS m/z (rel intensity) 293 (MH+, 100).
β-d-Glucopyranosylmethylamine (51)
PtO2·H2O (36 mg) was added to a stirring solution of β-d-glucopyranosylnitromethane (50, 309 mg, 1.38 mmol) in MeOH (20 mL) and H2O (5 mL) in a 50 mL oven-dried flask, which was then stoppered by a rubber septum. The system was flushed with argon and evacuated using a high-vacuum pump, and this cycle was repeated twice more. A balloon containing H2 gas was affixed, and the mixture was allowed to stir at room temperature under the H2 atmosphere for 18 h. The mixture was filtered and the residue washed with MeOH (5 mL). The filtrate was concentrated in vacuo to provide 51 as a colorless oil that solidified upon standing as a white solid (225 mg, 84%): mp 157–160 °C (lit.55 mp 168 °C). 1H NMR (300 MHz, CD3OD) δ 3.86 (dd, J = 11.9 Hz, 2.0 Hz, 1 H), 3.60 (dd, J = 11.9 Hz, 6.1 Hz, 1 H), 3.38–2.96 (m, 7 H), 2.66 (dd, J = 13.3 Hz, 7.4 Hz, 1 H); ESIMS m/z (rel intensity) 192 [(M – H+)−, 92].
(2′RS)-5,6-Dihydro-6-(2′,3′-dihydroxypropyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (52)
Racemic 3-aminopropane-1,2-diol (26, 43 mg, 0.47 mmol) was dissolved in MeOH (1 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (66 mg, 0.19 mmol) in CHCl3 (25 mL). The reaction mixture was heated at reflux with stirring for 18 h. The product precipitated and was collected by filtration of the cooled reaction mixture. The residue was dried by evaporation of solvents in vacuo to provide compound 52 as a purple solid (56 mg, 69%): mp 323 °C (dec). IR 3588, 2980, 1657, 1647, 1530, 1501, 1421, 1053 cm–1; 1H NMR (DMSO-d6) δ 7.89 (s, 1 H), 7.63 (s, 1 H), 7.46 (s, 1 H), 7.03 (s, 1 H), 6.14 (d, J = 1.5 Hz, 2 H), 5.11 (d, J = 5.0 Hz, 1 H), 5.00 (t, J = 5.3 Hz, 1 H), 4.54–4.27 (m, 2 H), 3.97–3.88 (m, 1 H), 3.88 (s, 3 H), 3.83 (s, 3 H), 3.53 (t, J = 5.1 Hz, 2 H); MALDI m/z (rel intensity) 426 (MH+, 27); HREIMS m/z calcd for C22H19NO8 425.1111 (M+), found 425.1108 (M+); HPLC purity, 99.16% (90% MeOH, 10% H2O).
(2′S)-5,6-Dihydro-6-(2′,3′-dihydroxypropyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (53)
(S)-3-Aminopropane-1,2-diol (27, 35 mg, 0.38 mmol) was dissolved in MeOH (1 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (55 mg, 0.16 mmol) in CHCl3 (25 mL). The reaction mixture was heated at reflux with stirring for 18 h. The product precipitated and was collected by filtration of the cooled reaction mixture. The residue was dried by evaporation of solvents in vacuo to yield compound 53 as a purple solid (50 mg, 73%): mp 315–318 °C (dec). IR 3369, 2926, 1696, 1655, 1628, 1560, 1497, 1396, 1305 cm–1; 1H NMR (DMSO-d6) δ 7.90 (s, 1 H), 7.65 (s, 1 H), 7.48 (s, 1 H), 7.04 (s, 1 H), 6.16 (d, J = 1.8 Hz, 2 H), 5.12 (d, J = 5.1 Hz, 1 H), 5.00 (t, J = 5.2 Hz, 1 H), 4.56–4.29 (m, 2 H), 3.99–3.90 (m, 1 H), 3.90 (s, 3 H), 3.85 (s, 3 H), 3.55 (t, J = 5.1 Hz, 2 H); MALDI m/z (rel intensity) 426 (MH+, 100); HRESIMS m/z calcd for C22H20NO8 426.1189 (MH+), found 426.1202 (MH+); HPLC purity, 98.51% (90% MeOH, 10% H2O).
(2′R)-5,6-Dihydro-6-(2′,3′-dihydroxypropyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (54)
(R)-3-Aminopropane-1,2-diol (28, 54 mg, 0.59 mmol) was dissolved in MeOH (1 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (64 mg, 0.18 mmol) in CHCl3 (25 mL). The reaction mixture was heated at reflux with stirring for 26 h. The product precipitated and was collected by filtration of the cooled reaction mixture. The residue was dried by evaporation of solvents in vacuo to furnish compound 54 as a purple solid (58 mg, 76%): mp 321 °C (dec). IR 3463, 2925, 1694, 1629, 1555, 1509, 1389, 1205, 1046 cm–1; 1H NMR (DMSO-d6) δ 7.88 (s, 1 H), 7.62 (s, 1 H), 7.46 (s, 1 H), 7.02 (s, 1 H), 6.15 (d, J = 1.7 Hz, 2 H), 5.11 (d, J = 5.1 Hz, 1 H), 5.00 (t, J = 5.4 Hz, 1 H), 4.53–4.27 (m, 2 H), 3.96–3.89 (m, 1 H), 3.89 (s, 3 H), 3.84 (s, 3 H), 3.54 (t, J = 5.1 Hz, 2 H); MALDI m/z (rel intensity) 426 (MH+, 71); HREIMS m/z calcd for C22H19NO8 425.1111 (M+), found 425.1119 (M+); HPLC purity, 99.39% (90% MeOH, 10% H2O).
(2′R,3′S,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (55)
d-Arabitylamine (31, 69 mg, 0.46 mmol) was dissolved in MeOH (15 mL) and H2O (0.5 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (51 mg, 0.14 mmol) in CHCl3 (20 mL). The reaction mixture was heated at reflux with stirring for 48 h. The product precipitated and was collected by filtration of the cooled reaction mixture. The residue was washed with MeOH (10 mL) and CHCl3 (10 mL) and dried in vacuo. Compound 55 was obtained as a purple solid (54 mg, 79%): mp 318–319 °C (dec). IR 3365, 2912, 1697, 1656, 1646, 1575, 1554, 1462, 1050 cm–1; 1H NMR (DMSO-d6) δ 7.93 (s, 1 H), 7.80 (s, 1 H), 7.50 (s, 1 H), 7.05 (s, 1 H), 6.16 (d, J = 3.5 Hz, 2 H), 4.99 (d, J = 6.7 Hz, 1 H), 4.83 (d, J = 5.8 Hz, 1 H), 4.50 (d, J = 5.7 Hz, 1 H), 4.47–4.36 (m, 3 H), 3.91 (s, 3 H), 3.85 (s, 3 H); APCIMS m/z (rel intensity) 486 (MH+, 64); HRESIMS m/z calcd for C24H24NO10 486.1400 (MH+), found 486.1396 (MH+); HPLC purity, 98.43% (90% MeOH, 10% H2O).
(2′S,3′S,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (56)
d-Ribitylamine (34, 74 mg, 0.49 mmol) was dissolved in MeOH (15 mL) and H2O (0.5 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (76 mg, 0.22 mmol) in CHCl3 (20 mL). The reaction mixture was heated at reflux with stirring for 18 h, at which time additional d-ribitylamine (39 mg, 0.26 mmol) dissolved in MeOH (1 mL) and H2O (3 drops) was added. After 48 h, the product precipitated and was collected by filtration of the cooled reaction mixture. The residue was washed with MeOH (10 mL) and CHCl3 (10 mL) and dried in vacuo. Compound 56 was obtained as a purple solid (74 mg, 69%): mp 307–310 °C (dec). IR 3312, 1746, 1676, 1581, 1500, 1409, 1221, 969 cm–1; 1H NMR (DMSO-d6) δ 7.93 (s, 1 H), 7.80 (s, 1 H), 7.50 (s, 1 H), 7.05 (s, 1 H), 6.16 (d, J = 3.5 Hz, 2 H), 4.99 (d, J = 6.7 Hz, 1 H), 4.83 (d, J = 5.8 Hz, 1 H), 4.50 (d, J = 5.7 Hz, 1 H), 4.47–4.36 (m, 3 H), 3.91 (s, 3 H), 3.85 (s, 3 H); MALDI m/z (rel intensity) 486 (MH+, 100); HREIMS m/z calcd for C24H23NO10 485.1322 (M+), found 485.1330 (M+); HPLC purity, 97.39% (90% MeOH, 10% H2O).
(2′R,3′R,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (57)
d-Lyxitylamine (37, 72 mg, 0.48 mmol) was dissolved in MeOH (15 mL) and H2O (1 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (68 mg, 0.19 mmol) in CHCl3 (20 mL). The reaction mixture was heated at reflux with stirring for 40 h. The product precipitated and was collected by filtration of the cooled reaction mixture. The residue was washed with MeOH (10 mL) and CHCl3 (10 mL) and dried in vacuo. Compound 57 was obtained as a purple solid (64 mg, 69%): mp 305–309 °C (dec). IR 3212, 2899, 1701, 1632, 1552, 1530, 1499, 1261, 1087 cm–1; 1H NMR (DMSO-d6) δ 7.91 (s, 1 H), 7.65 (s, 1 H), 7.48 (s, 1 H), 7.04 (s, 1 H), 6.16 (d, J = 6.1 Hz, 2 H), 5.05 (d, J = 5.5 Hz, 1 H), 4.85 (d, J = 6.1 Hz, 1 H), 4.74–4.35 (m, 2 H), 4.33 (d, J = 6.4 Hz, 1 H), 4.00 (s, 1 H), 3.90 (s, 3 H), 3.85 (s, 3 H), 3.71–3.57 (m, 2 H), 3.54–3.42 (m, 1 H); MALDI m/z (rel intensity) 486 (MH+, 100); HRESIMS m/z calcd for C24H24NO10 486.1400 (MH+), found 486.1401 (MH+); HPLC purity, 96.04% (90% MeOH, 10% H2O).
(2′S,3′R,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxyhexyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinolone (58)
d-Xylitylamine (40, 65 mg, 0.43 mmol) was dissolved in MeOH (15 mL) and H2O (3 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (70 mg, 0.20 mmol) in CHCl3 (20 mL). The reaction mixture was heated at reflux with stirring for 16 h, at which time additional d-xylitylamine (40, 93 mg, 0.62 mmol) was added as a solution in MeOH (2 mL) and H2O (5 drops). After the mixture was stirred and heated at reflux for 16 h, the product precipitated and was collected by filtration of the cooled reaction mixture. The residue was washed with MeOH (10 mL) and CHCl3 (10 mL) and dried in vacuo. Compound 58 was obtained as a dark purple solid (68 mg, 70%): mp 310 °C (dec). IR 3272, 2967, 1700, 1651, 1608, 1476, 1430, 1030 cm–1; 1H NMR (DMSO-d6) δ 7.87 (s, 1 H), 7.76 (s, 1 H), 7.45 (s, 1 H), 7.02 (s, 1 H), 6.14 (s, 2 H), 5.00 (d, J = 5.0 Hz, 1 H), 4.90 (d, J = 5.4 Hz, 1 H), 4.64–4.46 (m, 4 H), 3.89 (s, 3 H), 3.83 (s, 3 H), 3.66–3.51 (m, 5 H); MALDI m/z (rel intensity) 486 (MH+, 100); HRESIMS m/z calcd for C24H24NO10 486.1400 (MH+), found 486.1409 (MH+); HPLC purity, 98.33% (90% MeOH, 10% H2O).
(2′S,3′R,4′S,5′R)-5,6-Dihydro-6-(2′,3′,4′,5′,6′-pentahydroxyhexyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (59)
d-Galactitylamine (43, 84 mg, 0.46 mmol) was dissolved in MeOH (15 mL) and H2O (0.25 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (74 mg, 0.21 mmol) in CHCl3 (20 mL). The reaction mixture was heated at reflux with stirring for 72 h. The product precipitated and was collected by filtration of the cooled reaction mixture. The residue was washed with MeOH (10 mL) and CHCl3 (10 mL) and dried in vacuo. Compound 59 was obtained as a purple solid (84 mg, 78%): mp 300 °C (dec). IR 3315, 2938, 1605, 1506, 1551, 1535, 1511, 1386, 1084 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 7.91 (s, 1 H), 7.80 (s, 1 H), 7.48 (s, 1 H), 7.03 (s, 1 H), 6.15 (d, J = 4.0 Hz, 2 H), 4.95 (d, J = 4.0 Hz, 1 H), 4.75 (d, J = 5.8 Hz, 1 H), 4.54–4.24 (m, 4 H), 4.20 (d, J = 6.6 Hz, 1 H), 4.14–4.06 (m, 1 H), 3.90 (s, 3 H), 3.84 (s, 3 H), 3.77 (d, J = 6.6 Hz, 1 H), 3.59–3.37 (m, 2 H); MALDI m/z (rel intensity) 516 (MH+, 100); HRCIMS m/z calcd for C25H26NO11 516.1506 (MH+), found 516.1501 (MH+); HPLC purity, 100% (90% MeOH, 10% H2O).
(2′S,3′R,4′R,5′R)-5,6-Dihydro-6-(2′,3′,4′,5′,6′-pentahydroxyhexyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinolone (60)
d-Glucamine (46, 130 mg, 0.72 mmol) was dissolved in MeOH (15 mL) and H2O (0.5 mL), and the obtained solution was added to a stirring solution of indenobenzopyran 20 (87 mg, 0.25 mmol) in CHCl3 (20 mL). The reaction mixture was heated at reflux with stirring for 72 h. The product precipitated and was collected by filtration of the cooled reaction mixture. The residue was washed with MeOH (10 mL) and CHCl3 (10 mL) and dried in vacuo. Compound 60 was obtained as a dark purple solid (135 mg, 100%): mp 289 °C (dec). IR 3274, 2903, 1706, 1618, 1514, 1468, 1390, 1350, 1262, 1098 cm–1; 1H NMR (DMSO-d6) δ 7.89 (s, 1 H), 7.75 (s, 1 H), 7.46 (s, 1 H), 7.02 (s, 1 H), 6.15 (s, 2 H), 5.11 (m, 1 H), 4.91 (m, 1 H), 4.59–4.47 (m, 6 H), 4.10 (m, 2 H), 3.89 (s, 3 H), 3.84 (s, 3 H), 3.57 (m, 4 H); APCIMS m/z (rel intensity) 516 (MH+, 100); HRESIMS m/z calcd for C25H26NO11 516.1506 (MH+), found 516.1508 (MH+); HPLC purity, 99.33% (90% MeOH, 10% H2O).
(2′R,3′R,4′R,5′R)-5,6-Dihydro-6-(2′,3′,4′,5′,6′-pentahydroxyhexyl)-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (61)
d-Mannitylamine (49, 92 mg, 0.51 mmol) was added as a solution in MeOH (5 mL) to a stirring solution of indenobenzopyran 20 (112 mg, 0.318 mmol) in CHCl3 (20 mL) and MeOH (10 mL). The reaction mixture was heated at reflux with stirring for 48 h. The product precipitated and was collected by filtration of the cooled reaction mixture. The residue was washed with MeOH (10 mL) and CHCl3 (10 mL) and dried in vacuo. Compound 61 was obtained as a dark purple solid (159 mg, 97%): mp 285 °C (dec). IR 3525, 2975, 1703, 1570, 1512, 1465, 1398, 1346, 1250, 1066 cm–1; 1H NMR (DMSO-d6) δ 7.88 (s, 1 H), 7.80 (s, 1 H), 7.46 (s, 1 H), 7.00 (s, 1 H), 6.15 (d, J = 2.8 Hz, 2 H), 5.23 (d, J = 4.2 Hz, 1 H), 4.94 (d, J = 5.2 Hz, 1 H), 4.67 (d, J = 4.6, 1 H), 4.57–4.44 (m, 3 H), 4.13 (m, 1 H), 3.89 (s, 3 H), 3.84 (s, 3 H), 3.60 (m, 3 H), 3.47 (m, 1 H); MALDI m/z (rel intensity) 516 (MH+, 100); HRESIMS m/z calcd for C25H26NO11 516.1506 (MH+), found 516.1508 (MH+); HPLC purity, 99.28% (90% MeOH, 10% H2O).
(2′S,3′R,4′R,5′S,6’R)-5,6-Dihydro-6-[(3′,4′,5′-trihydroxy-6′-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)methyl)]-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (62)
β-d-Glucopyranosylmethylamine (51, 59 mg, 0.31 mmol) was added to a stirring solution of indenobenzopyran 20 (75 mg, 0.21 mmol) in CHCl3 (20 mL) and MeOH (10 mL). The reaction mixture was stirred and heated at reflux for 20 h, cooled to room temperature, and concentrated in vacuo. The residue was suspended in CHCl3 (20 mL) and filtered and then suspended in MeOH (20 mL), filtered, and dried by evaporation of solvents in vacuo to provide 62 as a purple solid (70 mg, 63%): mp 324 °C (dec). IR 3374, 2913, 1686, 1527, 1495, 1420, 1384, 1245, 1109 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 7.88 (s, 1 H), 7.58 (s, 1 H), 7.46 (s, 1 H), 7.01 (s, 1 H), 6.15 (s, 2 H), 5.44–5.37 (m, 1 H), 5.05 (d, J = 4.2 Hz, 1 H), 4.89 (d, J = 5.1 Hz, 1 H), 4.78–4.67 (m, 1 H), 4.53 (s, 1 H), 4.00–3.93 (m, 1 H), 3.89 (s, 3 H), 3.84 (s, 3 H); MALDI m/z (rel intensity) 528 (MH+, 100); HRCIMS m/z calcd for C26H26NO11 528.1506 (MH+), found 528.1484 (MH+); HPLC purity, 98.80% (90% MeOH, 10% H2O).
(2′RS)-5,6-Dihydro-6-(2′,3′-dihydroxypropyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (63)
Indenobenzopyran 25 (71 mg, 0.24 mmol) was suspended with stirring in MeOH (20 mL) at room temperature. A solution of racemic 3-aminopropane-1,2-diol (26, 56 mg, 0.62 mmol) in MeOH (2 mL) was added to the stirring suspension, which was then heated at reflux with stirring for 23 h. The suspension was cooled to room temperature, filtered, and dried in vacuo to provide 63 as a red solid (55 mg, 63%): mp 272–273 °C. IR 3419, 2923, 1751, 1701, 1648, 1609, 1541, 1420, 1041 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.87 (d, J = 2.5 Hz, 1 H), 8.73 (d, J = 9.0 Hz, 1 H), 8.56 (dd, J = 9.0 Hz, 2.5 Hz, 1 H), 8.15 (d, J = 7.3 Hz, 1 H), 7.66–7.49 (m, 4 H), 5.16 (d, J = 5.0 Hz, 1 H), 5.02 (t, J = 5.5 Hz, 1 H), 4.63–4.53 (m, 2 H), 4.05–3.96 (m, 1 H), 3.61 (t, J = 5.3 Hz, 2 H); negative ion ESIMS m/z (rel intensity) 401/403 (M + Cl¯, 100/30); negative ion HRESIMS m/z calcd for C19H13N2O6 365.0774 [(M – H+)¯], found 365.0777 [(M – H+)¯]; HPLC purity, 99.71% (MeOH–H2O, 90:10).
(2′R)-5,6-Dihydro-6-(2′,3′-dihydroxypropyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (64)
Indenobenzopyran 25 (71 mg, 0.24 mmol) was suspended with stirring in MeOH (20 mL) at room temperature. (R)-3-Aminopropane-1,2-diol (28, 52 mg, 0.57 mmol) was added to the stirring suspension, which was then heated at reflux with stirring for 23 h. The product mixture was absorbed onto silica gel (2 g) and purified by flash chromatography (SiO2, 100% CHCl3 to 95:5 CHCl3–MeOH, 11 cm × 2 cm, 10 mL fractions). The impure, product-containing fractions were pooled and concentrated. The residue was suspended in CHCl3 (15 mL), filtered, and dried in vacuo to provide 64 as a yellow-orange solid (31 mg, 35%): mp 226–229 °C (dec). IR 3303, 2929, 1707, 1657, 1624, 1583, 1542, 1497, 1456, 1328, 1034 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.89 (d, J = 2.5 Hz, 1 H), 8.76 (d, J = 9.0 Hz, 1 H), 8.58 (dd, J = 8.9, 2.5 Hz, 1 H), 8.17 (d, J = 7.3 Hz, 1 H), 7.67–7.52 (m, 3 H), 5.17 (d, J = 5.0 Hz, 1 H), 5.02 (t, J = 5.5 Hz, 1H), 4.62–4.56 (m, 2 H), 4.05–3.99 (m, 1 H), 3.61 (t, J = 5.3 Hz, 2 H); negative ion ESIMS m/z (rel intensity) 401/403 (M + Cl¯, 100/33); negative ion HRESIMS m/z calcd for C19H13N2O6 365.0774 [(M – H+)¯], found 365.0777 [(M – H+)¯]; HPLC purity, 99.71% (MeOH–H2O, 90:10).
(2′R,3′S,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (65)
Indenobenzopyran 25 (77 mg, 0.26 mmol) was suspended with stirring in CHCl3 (20 mL) and MeOH (15 mL) at room temperature. A solution of d-arabitylamine (31, 97 mg, 0.64 mmol) in H2O (0.5 mL) was added to the stirring suspension, which was then heated at reflux with stirring for 22 h. The suspension was cooled to room temperature, filtered, and the residue was washed with CHCl3 (10 mL) and dried in vacuo to provide 65 as a yellow-brown solid (47 mg, 42%): mp 252–254 °C (dec). IR 3282, 2928, 1710, 1671, 1648, 1547, 1322, 1203 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.85 (d, J = 2.5 Hz, 1 H), 8.71 (d, J = 8.9 Hz, 1 H), 8.54 (dd, J = 9.0, 2.5 Hz, 1 H), 8.28 (d, J = 7.1 Hz, 1 H), 7.63–7.47 (m, 3 H), 4.99 (d, J = 7.4 Hz, 1 H), 4.89 (d, J = 5.8 Hz, 1 H), 4.72–4.59 (m, 1 H), 4.55 (d, J = 5.4 Hz, 1 H), 4.50–4.38 (m, 2 H), 4.38–4.27 (m, 1 H), 3.71–3.62 (m, 1 H), 3.60–3.41 (m, 2 H); negative ion ESIMS m/z (rel intensity) 461/463 (M + Cl¯, 69, 31); negative ion HRESIMS m/z calcd for C21H17N2O8 425.0985 [(M – H+)¯], found 425.0991 [(M – H+)¯]; HPLC purity, 98.85% (MeOH–H2O, 90:10).
(2′S,3′S,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (66)
Indenobenzopyran 25 (38 mg, 0.13 mmol) was suspended with stirring in MeOH (25 mL) at room temperature. A solution of d-ribitylamine (34, 56 mg, 0.37 mmol) in H2O (0.25 mL) was added to the stirring suspension, which was then heated at reflux with stirring for 72 h. The suspension was cooled to room temperature, filtered, and the residue was washed with MeOH (2 mL) and dried in vacuo to provide 66 as a yellow-orange solid (16 mg, 29%): mp 260–262 °C (dec). IR 3395, 2928, 1669, 1553, 1622, 1507, 1432, 1353, 1262 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.93–8.86 (m, 2 H), 8.76 (d, J = 9.5 Hz, 1 H), 8.63–8.52 (m, 1 H), 8.93–8.81 (m, 1 H), 8.34 (d, J = 7.0 Hz, 1 H), 7.70–7.49 (m, 2 H), 5.25 (d, J = 4.8 Hz, 1 H), 5.05 (d, J = 5.2 Hz, 1 H), 4.80 (d, J = 5.1 Hz, 1 H), 4.74–4.58 (m, 2 H), 4.56–4.41 (m, 1 H), 4.29–4.15 (m, 1 H), 3.74–3.54 (m, 3 H); negative ion ESIMS m/z (rel intensity) 425 [(M – H+)¯, 100]; negative ion HRESIMS m/z calcd for C21H17N2O8 425.0985 [(M – H+)¯], found 425.0991 [(M – H+)¯]; HPLC purity, 99.83% (MeOH–H2O, 90:10).
(2′R,3′R,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (67)
Indenobenzopyran 25 (70 mg, 0.24 mmol) was suspended with stirring in MeOH (30 mL) at room temperature. A solution of d-lyxitylamine (37, 75 mg, 0.50 mmol) in H2O (1 mL) was added to the stirring suspension, which was then heated at reflux with stirring for 21 h. The suspension was cooled to room temperature, filtered, and dried in vacuo to provide 67 as an orange solid (38 mg, 37%): mp 245 °C (dec). IR 3282, 2929, 1697, 1612, 1561, 1507, 1344, 1121 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.90 (d, J = 2.5 Hz, 1 H), 8.76 (d, J = 9.0 Hz, 1 H), 8.58 (dd, J = 9.0, 2.5 Hz, 1 H), 8.17 (d, J = 7.3 Hz, 1 H), 7.68–7.48 (m, 3 H), 4.84–4.55 (m, 2 H), 4.09 (s, 1 H), 3.67 (t, J = 6.3 Hz, 2 H); negative ion ESIMS m/z (rel intensity) 461/463 (M + Cl¯, 100, 50); negative ion HRESIMS m/z calcd for C21H18N2O8Cl 461.0751 (M + Cl¯), found 461.0761 (M + Cl¯); HPLC purity, 99.65% (MeOH–H2O, 90:10).
(2′S,3′R,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (68)
Indenobenzopyran 25 (71 mg, 0.24 mmol) was suspended with stirring in CHCl3 (20 mL) and MeOH (15 mL) at room temperature. A solution of d-xylitylamine (40, 116 mg, 0.77 mmol) in H2O (0.5 mL) was added to the stirring suspension, which was then heated at reflux with stirring for 22 h. The suspension was cooled to room temperature, filtered, and the residue was washed with CHCl3 (15 mL) until the filtrate that drained was nearly colorless. The residue was dried in vacuo to provide 68 as a red-orange solid (48 mg, 47%): mp 205 °C (dec). IR 3467, 2942, 1717, 1655, 1638, 1541, 1329, 1106 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.90 (d, J = 2.5 Hz, 1 H), 8.77 (d, J = 9.0 Hz, 1 H), 8.59 (dd, J = 9.0, 2.5 Hz, 1 H), 8.32 (d, J = 7.4 Hz, 1 H), 7.68–7.52 (m, 3 H), 5.08 (d, J = 5.1 Hz, 1 H), 4.91 (d, J = 6.2 Hz, 1 H), 4.68 (d, J = 5.3 Hz, 1 H), 4.59 (s, 2 H), 4.14 (s, 1 H), 3.67 (s, 2 H); negative ion ESIMS m/z (rel intensity) 461/463 (M + Cl¯, 100/30); negative ion HRESIMS m/z calcd for C21H18N2O8Cl 461.0751 (M + Cl¯), found 461.0760 (M + Cl¯); HPLC purity, 95.09% (MeOH–H2O, 90:10).
(2′S,3′R,4′S,5′R)-5,6-Dihydro-6-(2′,3′,4′,5′,6′-pentahydroxyhexyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (69)
Indenobenzopyran 25 (70 mg, 0.24 mmol) was partially dissolved with stirring in CHCl3 (20 mL) and MeOH (15 mL) at room temperature. A solution of d-galactitylamine (43, 130 mg, 0.72 mmol) in H2O (0.5 mL) was added to the stirring suspension, which was then heated at reflux with stirring for 39 h. The suspension was cooled to room temperature, filtered, and the residue was washed with CHCl3, H2O, and MeOH (10 mL of each) and dried in vacuo to provide 69 as a dull yellow solid (51 mg, 51%): mp 284–287 °C (dec). IR 3536, 2931, 1624, 1561, 1524, 1332, 1206, 1090 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.84 (d, J = 2.5 Hz, 1 H), 8.71 (d, J = 9.0 Hz, 1 H), 8.54 (dd, J = 9.0, 2.5 Hz, 1 H), 8.30 (d, J = 6.6 Hz, 1 H), 7.70–7.38 (m, 4 H), 4.95 (d, J = 6.9 Hz, 1 H), 4.83 (d, J = 5.9 Hz, 1 H), 4.65 (d, J = 12.0 Hz, 1 H), 4.55–4.40 (m, 3 H), 4.40–4.29 (m, 8 H), 4.25 (d, J = 6.5 Hz, 1 H), 4.15 (d, J = 7.2 Hz, 1 H), 3.80 (d, J = 6.4 Hz, 1 H), 3.58 (t, J = 7.1 Hz, 2 H), 3.41 (d, J = 6.1 Hz, 3 H); negative ion ESIMS m/z (rel intensity) 491/493 (M + Cl¯, 100, 42); negative ion HRESIMS m/z calcd for C22H19N2O9 455.1090 [(M – H+)¯], found 455.1094 [(M – H+)¯]; HPLC purity, 98.99% (MeOH–H2O, 90:10).
(2′R,3′R,4′R,5′R)-5,6-Dihydro-6-(2′,3′,4′,5′,6′-pentahydroxyhexyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (70)
Indenobenzopyran 25 (78 mg, 0.27 mmol) was suspended with stirring in MeOH (30 mL) at room temperature. A solution of d-mannitylamine (49, 108 mg, 0.60 mmol) in H2O (0.5 mL) was added to the stirring suspension, which was then heated at reflux with stirring for 20 h. The suspension was cooled to room temperature, filtered, and the residue was washed with CHCl3 (5 mL) and dried in vacuo to provide 70 as a red-orange solid (66 mg, 54%): mp 279–282 °C (dec). IR 3210, 2970, 1712, 1638, 1519, 1389, 1348, 1245, 1009 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.90 (d, J = 2.5 Hz, 1 H), 8.76 (d, J = 9.0 Hz, 1 H), 8.58 (dd, J = 8.9, 2.5 Hz, 1 H), 8.15 (d, J = 7.3 Hz, 1 H), 7.71–7.50 (m, 3 H), 5.08 (d, J = 5.5 Hz, 1 H), 4.85 (d, J = 7.0 Hz, 1 H), 4.80–4.77 (m, 1 H), 4.52 (d, J = 4.9 Hz, 1 H), 4.42 (t, J = 5.6 Hz, 1 H), 4.27 (d, J = 7.5 Hz, 1 H), 4.09 (s, 1 H), 3.87 (d, J = 7.5 Hz, 1 H), 3.67–3.50 (m, 1 H); negative ion ESIMS m/z (rel intensity) 491/493 (M + Cl¯, 100/30); negative ion HRESIMS m/z calcd for C22H20N2O9Cl 491.0857 (M + Cl¯), found 491.0868 (M + Cl¯); HPLC purity, 99.64% (MeOH–H2O, 90:10).
(2′S,3′R,4′R,5′S,6′R)-5,6-Dihydro-6-[(3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)methyl)]-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (71)
β-d-Glucopyranosylmethylamine (51, 95 mg, 0.49 mmol) was added to a stirring suspension of indenobenzopyran 25 (93 mg, 0.32 mmol) in MeOH (30 mL). The reaction mixture was stirred and heated at reflux for 23 h, cooled to room temperature, and filtered. The residue was washed with CHCl3 and MeOH (5 mL of each) and dried in vacuo to provide 71 as a red-orange solid (91 mg, 61%): mp 297–300 °C (dec). IR (KBr) 3312, 2944, 1698, 1649, 1622, 1501, 1349, 1210, 683 cm–1; 1H NMR (300 MHz, DMSO-d6) δ 8.88 (d, J = 2.5 Hz, 1 H), 8.73 (d, J = 9.0 Hz, 1 H), 8.56 (dd, J = 9.0, 2.5 Hz, 1 H), 8.11 (d, J = 7.2 Hz, 1 H), 7.67–7.48 (m, 3 H), 5.38 (d, J = 5.4 Hz, 1 H), 5.06 (d, J = 4.5 Hz, 1 H), 4.98–4.85 (m, 2 H), 4.71–4.55 (m, 1 H), 3.94 (t, J = 5.4 Hz, 1 H), 3.65–3.56 (m, 1 H), 3.50–3.39 (m, 1 H), 3.20–2.91 (m, 3 H); negative ion ESIMS m/z (rel intensity) 503/505 [(M + Cl¯), 100/39]; HRESIMS m/z calcd for C23H20N2O9Na 491.1067 (M + Na+), found 491.1067 (M + Na+); HPLC purity, 99.83% (MeOH–H2O, 90:10).
Topoisomerase I Mediated DNA Cleavage Reactions
A brief description of the assay follows.41 A 3′-[32P]-labeled 117-bp DNA oligonucleotide was prepared as previously described. Approximately 2 nM radiolabeled DNA substrate was incubated with recombinant Top1 in 20 μL of reaction buffer [10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL BSA] at 25 °C for 20 min in the presence of various concentrations of test compounds. The reactions were terminated by adding SDS (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of each reaction mixture were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a phosphoimager and ImageQuant software (Molecular Dynamics). For simplicity, cleavage sites were numbered as previously described in the 161-bp fragment.
Gel-Based Assay Measuring the Inhibition of Recombinant TDP1
A brief description of the assay follows.47 A 5′-[32P]-labeled single-stranded DNA oligonucleotide containing a 3′-phosphotyrosine (N14Y) was generated as described by Dexheimer et al. The DNA substrate was then incubated with 5 pM recombinant TDP1 in the absence or presence of an indicated concentration of the test compound for 15 min at room temperature in a buffer containing 50 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween-20. Reactions were terminated by the addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to a 16% denaturing PAGE and gels were exposed after drying to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600 (GE Healthcare), and densitometry analyses were performed using ImageQuant software (GE Healthcare).
Docking and Modeling Studies
The 1SC7 X-ray crystal structure file was prepared as previously described.25 Carbohydrate-substituted indenoisoquinolines were constructed and optimized using the Tripos force field with default parameters in SYBYL.56 Ligands were docked into the prepared crystal structure using GOLD 3.2 software.57 The centroid of the binding site was defined by the crystallized ligand. Ten GOLD algorithm runs were executed per ligand. Default parameters were used. The top 10 docking poses per ligand were inspected visually following the docking runs. Energy minimizations were performed for selected ligands within SYBYL. Ligand SYBYL atom types were inspected and corrected as necessary prior to minimization. Each ligand was merged with the prepared crystal structure. Minimization was executed by allowing only the ligand to move while freezing the surrounding crystal structure, which was defined as a static set. The details of the minimization were set as follows: Powell method; MMFF94s force field; MMFF94 charges; and 0.05 kcal mol–1 Å–1 energy gradient convergence criterion. Overlays of two ligands were performed in SYBYL, using the “Align Structures by Homology” tool. The α-carbons were used as reference points.
Acknowledgments
This work was made possible by the National Institutes of Health (NIH) through support with Research Grant UO1 CA89566. In vitro cytotoxicity testing was performed by the Developmental Therapeutics Program at the National Cancer Institute, under Contract NO1-CO-56000. D.E.B. thanks Wei Lv, Martin Conda-Sheridan, Rubayat Khan, and Karl Wood for technical assistance.
Glossary
Abbreviations Used
- CPT
camptothecin
- Top1
human topoisomerase type I
- TDP1
human tyrosyl DNA phosphodiesterase I
- HPLC
high performance liquid chromatography
- Ac2O
acetic anhydride
- NaBH4
sodium borohydride
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Pommier Y. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat. Rev. Cancer 2006, 6, 789–802. [DOI] [PubMed] [Google Scholar]
- Stewart L.; Redinbo M. R.; Qiu X.; Hol W. G. J.; Champoux J. J. A Model for the Mechanism of Human Topoisomerase I. Science 1998, 279, 1534–1541. [DOI] [PubMed] [Google Scholar]
- Koster D. A.; Palle K.; Bot E. S. M.; Bjornsti M. A.; Dekker N. H. Antitumour Drugs Impede DNA Uncoiling by Topoisomerase I. Nature 2007, 448, 213–217. [DOI] [PubMed] [Google Scholar]
- Seol Y.; Zhang H. L.; Pommier Y.; Neuman K. C. A Kinetic Clutch Governs Religation by Type IB Topoisomerases and Determines Camptothecin Sensitivity. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 16125–16130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Marchand C. Interfacial Inhibitors: Targeting Macromolecular Complexes. Nat. Rev. Drug Discovery 2012, 11, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand C.; Antony S.; Kohn K. W.; Cushman M.; Ioanoviciu A.; Staker B. L.; Burgin A.; Stewart L.; Pommier Y. A Novel Norindenoisoquinoline Structure Reveals a Common Interfacial Inhibitor Paradigm for Ternary Trapping of the Topoisomerase I-DNA Complex. Mol. Cancer Ther. 2006, 5, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champoux J. J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- Thomas C. J.; Rahier N. J.; Hecht S. M. Camptothecin: Current Perspectives. Bioorg. Med. Chem. 2004, 12, 1585–1604. [DOI] [PubMed] [Google Scholar]
- Teicher B. Next Generation of Topoisomerase I Inhibitors: Rationale and Biomarker Strategies. Biochem. Pharmacol. 2008, 75, 1262–1271. [DOI] [PubMed] [Google Scholar]
- Khadka D. B.; Cho W.-J. Topoisomerase Inhibitors as Anticancer Agents: A Patent Update. Expert Opin. Ther. Pat. 2013, 23, 1033–1056. [DOI] [PubMed] [Google Scholar]
- Pommier Y.; Cushman M. The Indenoisoquinoline Noncamptothecin Topoisomerase I Inhibitors: Update and Perspectives. Mol. Cancer Ther. 2009, 8, 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan M.; Morrell A.; Ioanoviciu A.; Antony S.; Kohlhagen G.; Agama K.; Hollingshead M.; Pommier Y.; Cushman M. Synthesis and Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Substituted with Nitrogen Heterocycles. J. Med. Chem. 2006, 49, 6283–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A Phase I Study of Indenoisoquinolines LMP400 and LMP776 in Adults with Relapsed Solid Tumors and Lymphomas. http://clinicaltrials.gov/ct2/show/study/NCT01051635 (accessed November 17, 2013).
- Teicher B. A.Topoisomerase I Inhibitors: Chemical Biology. In DNA Topoisomerases and Cancer; Pommier Y., Ed.; Springer: New York, 2012; pp 185–210. [Google Scholar]
- Neidle S.Design of CPT Derivatives: An Endless Series. Cancer Drug Design and Discovery; Academic Press: New York, 2007. [Google Scholar]
- Peterson K. E.; Cinelli M. A.; Morrell A. E.; Mehta A.; Dexheimer T. S.; Agama K.; Antony S.; Pommier Y.; Cushman M. Alcohol-, Diol-, and Carbohydrate-Substituted Indenoisoquinolines as Topoisomerase I Inhibitors: Investigating the Relationships Involving Stereochemistry, Hydrogen Bonding, and Biological Activity. J. Med. Chem. 2011, 54, 4937–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staker B. L.; Feese M. D.; Cushman M.; Pommier Y.; Zembower D.; Stewart L.; Burgin A. B. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J. Med. Chem. 2005, 48, 2336–2345. [DOI] [PubMed] [Google Scholar]
- Prudhomme M. Rebeccamycin Analogues as Anti-Cancer Agents. Eur. J. Med. Chem. 2003, 38, 123–140. [DOI] [PubMed] [Google Scholar]
- Pereira E. R.; Belin L.; Sancelme M.; Prudhomme M.; Ollier M.; Rapp M.; Severe D.; Riou J. F.; Fabbro D.; Meyer T. Structure–Activity Relationships in a Series of Substituted Indolocarbazoles: Topoisomerase I and Protein Kinase C Inhibition and Antitumoral and Antimicrobial Properties. J. Med. Chem. 1996, 39, 4471–4477. [DOI] [PubMed] [Google Scholar]
- Nagarajan M.; Xiao X.; Antony S.; Kohlhagen G.; Pommier Y.; Cushman M. Design, Synthesis, and Biological Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Featuring Polyamine Side Chains on the Lactam Nitrogen. J. Med. Chem. 2003, 46, 5712–5724. [DOI] [PubMed] [Google Scholar]
- Lin Y. S.; Tungpradit R.; Sinchaikul S.; An F. M.; Liu D. Z.; Phutrakul S.; Chen S. T. Targeting the Delivery of Glycan-Based Paclitaxel Prodrugs to Cancer Cells via Glucose Transporters. J. Med. Chem. 2008, 51, 7428–7441. [DOI] [PubMed] [Google Scholar]
- Nagarajan M.; Morrell A.; Fort B. C.; Meckley M. R.; Antony S.; Kohlhagen G.; Pommier Y.; Cushman M. Synthesis and Anticancer Activity of Simplified Indenoisoquinoline Topoisomerase I Inhibitors Lacking Substituents on the Aromatic Rings. J. Med. Chem. 2004, 47, 5651–5661. [DOI] [PubMed] [Google Scholar]
- Morrell A.; Antony S.; Kohlhagen G.; Pommier Y.; Cushman M. Synthesis of Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 3659–3663. [DOI] [PubMed] [Google Scholar]
- Morrell A.; Placzek M.; Parmley S.; Antony S.; Dexheimer T. S.; Pommier Y.; Cushman M. Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors: A Systematic Study and Optimization. J. Med. Chem. 2007, 50, 4419–4430. [DOI] [PubMed] [Google Scholar]
- Morrell A.; Antony S.; Kohlhagen G.; Pommier Y.; Cushman M. A Systematic Study of Nitrated Indenoisoquinolines Reveals a Potent Topoisomerase I Inhibitor. J. Med. Chem. 2006, 49, 7740–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell A.; Antony S.; Kohlhagen G.; Pommier Y.; Cushman M. Synthesis of Benz[d]indeno[1,2-b]pyran-5,11-diones: Versatile Intermediates for the Design and Synthesis of Topoisomerase I Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1846–1849. [DOI] [PubMed] [Google Scholar]
- Zhang X. Y.; Wang R. B.; Zhao L.; Lu N.; Wang J. B.; You Q. D.; Li Z. Y.; Guo Q. L. Synthesis and Biological Evaluations of Novel Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1276–1281. [DOI] [PubMed] [Google Scholar]
- Conda-Sheridan M.; Reddy P. V. N.; Morrell A.; Cobb B. T.; Marchand C.; Agama K.; Chergui A.; Renaud A.; Stephen A. G.; Bindu L. K.; Pommier Y.; Cushman M. Synthesis and Biological Evaluation of Indenoisoquinolines That Inhibit Both Tyrosyl-DNA Phosphodiesterase I (Tdp1) and Topoisomerase I (Top1). J. Med. Chem. 2013, 56, 182–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. X.; Morrell A.; Conda-Sheridan M.; Marchand C.; Agama K.; Bermingam A.; Stephen A. G.; Chergui A.; Naumova A.; Fisher R.; O’Keefe B. R.; Pommier Y.; Cushman M. Synthesis and Biological Evaluation of the First Dual Tyrosyl-DNA Phosphodiesterase I (Tdp1)-Topoisomerase I (Top1) Inhibitors. J. Med. Chem. 2012, 55, 4457–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [DOI] [PubMed] [Google Scholar]
- Lv W.; Liu J.; Lu D.; Flockhart D. A.; Cushman M. Synthesis of Mixed (E,Z)-, (E)-, and (Z)-Norendoxifen with Dual Aromatase Inhibitory and Estrogen Receptor Modulatory Activities. J. Med. Chem. 2013, 56, 4611–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlmark M.; Baeckstroem R.; Luiro A.; Psystynen J.; Tiainen E.. Preparation of Benzothiophenes, Benzothiazoles, Isobenzofurans, and Their Analogs as Inhibitors of Catechol-O-methyltransferase (COMT) for the Treatment of Parkinson’s Disease. Patent filed January 25, 2007.
- Che C.; Xiang J.; Wang G. X.; Fathi R.; Quan J. M.; Yang Z. One-Pot Synthesis of Quinoline-Based Tetracycles by a Tandem Three-Component Reaction. J. Comb. Chem. 2007, 9, 982–989. [DOI] [PubMed] [Google Scholar]
- Conrad P. C.; Kwiatkowski P. L.; Fuchs P. L. Synthesis via Vinyl Sulfones 18. Rapid Access to a Series of Highly Functionalized α,β-Unsaturated Cyclopentenones. A Caveat on Aminospirocyclization. J. Org. Chem. 1987, 52, 586–591. [Google Scholar]
- Khanapure S. P.; Biehl E. R. Preparation of Novel 4-Substituted 6-Methoxy-, 6,7-Dimethoxy, and 6,7-(Methylenedioxy)isochroman-3-ones. 2. J. Org. Chem. 1990, 55, 1471–1475. [Google Scholar]
- Noire P. D.; Franck R. W. A Facile Synthesis of 5,7-Dimethoxy-1(3H)-isobenzofuranone (5,7-Dimethoxyphthalide). Synthesis 1980, 882–883. [Google Scholar]
- Borsche W.; Diacont K.; Hanau H. Regarding 5-Carboxyhomophthalic Acid, 6-Nitrophthalide and 6-Nitroisoindolin-1-one. Chem. Ber. 1934, 67, 675–686. [Google Scholar]
- Weinstock C.; Plaut G. W. E. Synthesis and Properties of Certain Substituted Lumazines. J. Org. Chem. 1961, 26, 4456–4462. [Google Scholar]
- Shoemaker R. H. The NCI60 Human Tumour Cell Line Anticancer Drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
- Holbeck S. L.; Collins J. M.; Doroshow J. H. Analysis of Food and Drug Administration-Approved Anticancer Agents in the NCI60 Panel of Human Tumor Cell Lines. Mol. Cancer Ther. 2010, 9, 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexheimer T. S.; Pommier Y. DNA Cleavage Assay for the Identification of Topoisomerase I Inhibitors. Nat. Protoc. 2008, 3, 1736–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Leo E.; Zhang H. L.; Marchand C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z. H.; Agama K.; Sordet O.; Povirk L.; Kohn K. W.; Pommier Y. Hereditary Ataxia SCAN1 Cells Are Defective for the Repair of Transcription-Dependent Topoisomerase I Cleavage Complexes. DNA Repair 2006, 5, 1489–1494. [DOI] [PubMed] [Google Scholar]
- Hudson J. J. R.; Chiang S. C.; Wells O. S.; Rookyard C.; El-Khamisy S. F. SUMO Modification of the Neuroprotective Protein TDP1 Facilitates Chromosomal Single-Strand Break Repair. Nat. Commun. 2012, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B. B.; Sen N.; Ganguly A.; Majumder H. K. Reconstitution and Functional Characterization of the Unusual Bi-Subunit Type I DNA Topoisomerase from Leishmania donovani. FEBS Lett. 2004, 565, 81–88. [DOI] [PubMed] [Google Scholar]
- Dexheimer T. S.; Antony S.; Marchand C.; Pommier Y. Tyrosyl-DNA Phosphodiesterase as a Target for Anticancer Therapy. Anti-Cancer Agents Med. Chem. 2008, 8, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony S.; Marchand C.; Stephen A. G.; Thibaut L.; Agama K. K.; Fisher R. J.; Pommier Y. Novel High-Throughput Electrochemiluminescent Assay for Identification of Human Tyrosyl-DNA Phosphodiesterase (Tdp1) Inhibitors and Characterization of Furamidine (NSC 305831) as an Inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Antony S.; Pommier Y.; Cushman M. On the Binding of Indeno[1,2-c]isoquinolines in the DNA-Topoisomerase I Cleavage Complex. J. Med. Chem. 2005, 48, 3231–3238. [DOI] [PubMed] [Google Scholar]
- Ohkubo M.; Nishimura T.; Kawamoto H.; Nakano M.; Honma T.; Yoshinari T.; Arakawa H.; Suda H.; Morishima H.; Nishimura S. Synthesis and Biological Activities of NB-506 Analogues Modified at the Glucose Group. Bioorg. Med. Chem. Lett. 2000, 10, 419–422. [DOI] [PubMed] [Google Scholar]
- Erickson J. A.; Jalaie M.; Robertson D. H.; Lewis R. A.; Vieth M. Lessons in Molecular Recognition: The Effects of Ligand and Protein Flexibility on Molecular Docking Accuracy. J. Med. Chem. 2004, 47, 45–55. [DOI] [PubMed] [Google Scholar]
- Cushman M.; Jayaraman M.; Vroman J. A.; Fukunaga A. K.; Fox B. M.; Kohlhagen G.; Strumberg D.; Pommier Y. Synthesis of New Indeno[1,2-c]isoquinolines: Cytotoxic Non-Camptothecin Topoisomerase I Inhibitors. J. Med. Chem. 2000, 43, 3688–3698. [DOI] [PubMed] [Google Scholar]
- Rastogi S. N.; Bindra J. S.; Anand N. α-Methyldopa in a Rigid Framework: 3-Amino-3-methyl-6,7-dihydroxy-3,4-dihydroxycoumarins and 2-Amino-6,7-dihydroxytetralin-2-carboxylic Acids. Indian J. Chem. 1971, 9, 1175–1182. [Google Scholar]
- Cinelli M. A.; Reddy P. V. N.; Lv P. C.; Liang J. H.; Chen L.; Agama K.; Pommier Y.; van Breemen R. B.; Cushman M. Identification, Synthesis, and Biological Evaluation of Metabolites of the Experimental Cancer Treatment Drugs Indotecan (LMP400) and Indimitecan (LMP776) and Investigation of Isomerically Hydroxylated Indenoisoquinoline Analogues as Topoisomerase I Poisons. J. Med. Chem. 2012, 55, 10844–10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson C. M.; Brown C. W.; Simpson J. C. E. Some Reactions in the Phthalazine Series. J. Chem. Soc. 1956, 1081–1083. [Google Scholar]
- Bhat A. S.; Gervay-Hague J. Efficient Syntheses of β-Cyanosugars Using Glycosyl Iodides Derived from Per-O-silylated Mono- and Disaccharides. Org. Lett. 2001, 3, 2081–2084. [DOI] [PubMed] [Google Scholar]
- SYBYL, version 8.1.1; Tripos International: St. Louis, MO. [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved Protein–Ligand Docking Using GOLD. Proteins: Struct., Funct., Genet 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]