

Abstract
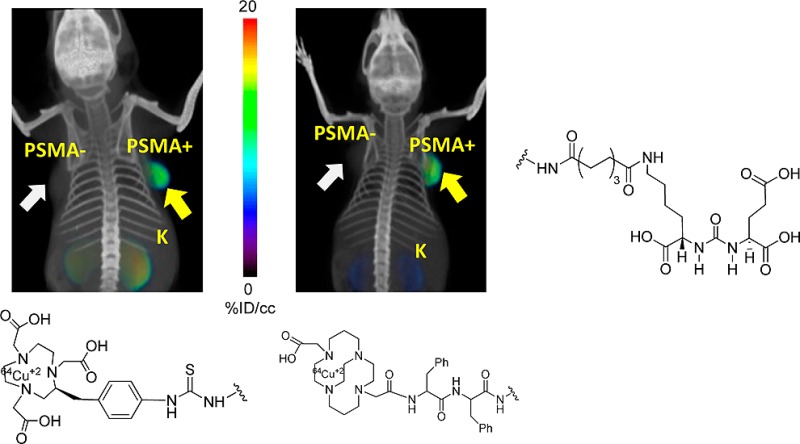
Prostate-specific membrane antigen (PSMA) is a well-recognized target for identification and therapy of a variety of cancers. Here we report five 64Cu-labeled inhibitors of PSMA, [64Cu]3–7, which are based on the lysine–glutamate urea scaffold and utilize a variety of macrocyclic chelators, namely NOTA(3), PCTA(4), Oxo-DO3A(5), CB-TE2A(6), and DOTA(7), in an effort to determine which provides the most suitable pharmacokinetics for in vivo PET imaging. [64Cu]3–7 were prepared in high radiochemical yield (60–90%) and purity (>95%). Positron emission tomography (PET) imaging studies of [64Cu]3–7 revealed specific accumulation in PSMA-expressing xenografts (PSMA+ PC3 PIP) relative to isogenic control tumor (PSMA– PC3 flu) and background tissue. The favorable kinetics and high image contrast provided by CB-TE2A chelated [64Cu]6 suggest it as the most promising among the candidates tested. That could be due to the higher stability of [64Cu]CB-TE2A as compared with [64Cu]NOTA, [64Cu]PCTA, [64Cu]Oxo-DO3A, and [64Cu]DOTA chelates in vivo.
Introduction
The prostate-specific membrane antigen (PSMA) is emerging as an attractive target for addressing cancer, whether for diagnosis or therapy, due to its restricted expression within normal tissue,1 its elevated expression in the epithelium of prostate tumors, and within the neovasculature of most solid tumors tested.2 With respect to prostate cancer, elevated expression of PSMA is associated with metastasis,3 castrate resistance,4,5 and progression.6 PSMA has also been used to guide antibody–drug conjugates and nanoparticles to PSMA-expressing tissues, including for human studies, some of which do not involve prostate cancer.7−11 Radiohalogenated, urea-based, low-molecular-weight inhibitors of PSMA have recently been explored to image expression of PSMA in prostate tumor xenografts12,13 as well as in clinical studies.14−16 Radiometals, including 99mTc,17−23111In,27−2964Cu,3086Y,31 and 89Zr,32,33 have also recently been implemented for imaging PSMA, in part to leverage the longer physical half-life of these nuclides, which will be necessary for tracking large peptides, aptamers, minibodies, antibodies, and nanoparticles. To enable targeting agents to bind with high affinity to PSMA, a spacer of approximately 20 Å is generally employed between the PSMA-targeting group and the metal chelator.21 Moreover, we have shown that the chelating moiety has a significant effect on the pharmacokinetics of this class of low-molecular-weight PSMA-based imaging agents when radiolabeled with 99mTc.34 The search for small-molecule, functionalized affinity agents for PSMA that have longer retention and superior pharmacokinetics properties for imaging and therapeutic applications is ongoing.
64Cu-Labeled molecules are promising imaging agents for positron emission tomography (PET) due to the favorable nuclear characteristics of the isotope (t1/2 = 12.7 h, β+ 17.4%, Emax = 0.656 MeV, β– 39%, Emax = 0.573 MeV) and its availability in high specific activity.35 The longer physical half-life of 64Cu compared to 11C (t1/2 = 20 min) and 18F (t1/2 = 110 min) enables imaging at delayed time points, which allows sufficient time for clearance from background tissues, resulting in increased image contrast, particularly for targeting agents that demonstrate long circulation times such as antibodies and nanoparticles. Moreover, 64Cu-based PET radiotracers have demonstrated efficacy for radioimmunotherapy36 comparable to that for the strictly therapeutic radionuclide, 67Cu (t1/2 = 61.5 h, β– 100%, Emax = 0.121 MeV).37 Accordingly, 64Cu could be used for imaging and therapy concurrently.38 In vivo stability of 64Cu-labeled targeted biomolecules depends on the stability of the corresponding Cu(II)-chelated coordination complex employed.39−41 Numerous bifunctional chelating agents have been developed to form stable 64Cu(II) complexes based on well-developed copper coordination chemistry42 and have been used to functionalize small molecules,43,44 peptides,45−48 aptamers,49 and antibodies.50,51 Acyclic and macrocyclic polyamines tend to have limited stability in vivo with respect to chelation of copper(II).52,53 Complexes of copper based on macrocyclic polyamino carboxylates, such as copper chelates of 1,4,8,11-tetraazacyclotetradecane-N,N′,N″,N‴-tetraacetic acid (TETA) and 1,4,7,10-tetraazacyclodoadecane-N,N′,N″,N‴-tetraacetic acid (DOTA), have greater kinetic inertness than acyclic analogues, although this has not eliminated transchelation.53,54 The cross-bridged tetraamine bicyclic polyaminocarboxylates, specifically 1,4,8,11-tetraazabicyclo[6.6.2]hexadecane-4,11-diacetic acid (CB-TE2A), provide an improvement over the monocyclic counterparts. The superior kinetic stability of the copper(II) cross-bridged complexes relative to DOTA and TETA analogues55−57 is due to the rigidity of the cross-bridge system. Derivatives of the polyaminocarboxylate NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid) are encouraging both because of the convenience of radiolabeling with 64Cu(II) and the lack of subsequent transmetalation in vivo.46,47,58
Here we study the effect of various chelators for labeling 64Cu on PSMA-targeted ureas with respect to pharmacokinetics for imaging prostate tumor xenografts in vivo. Although these low-molecular-weight compounds may clear relatively rapidly from nontarget sites, that is not always the case, and increasingly, larger PSMA-specific probes with slower kinetics are sometimes used as diagnostic and/or therapeutic probes for PSMA, requiring the longer physical half-life of 64Cu. Optimization of the chelator for radiolabeling with 64Cu is an important initial step in its implementation for imaging species that target PSMA. We investigated the in vitro binding affinity, mouse biodistribution, and prostate tumor xenograft uptake of five 64Cu-labeled PSMA inhibitors. To do that, we prepared urea-based PSMA inhibitors that utilize the Lys-Glu urea motif and conjugated them with five different macrocyclic chelating agents, NOTA, CB-TE2A, 3,6,9,15-tetraazabicyclo[9.3.1]-pentadeca-1(15),11,13-triene)-3,6,9-triacetic acid (PCTA), oxa-4,7,1-tetraazacyclododecane-4,7,10-triacetic acid (oxo-DO3A),59 and DOTA. Each conjugate was labeled with 63/65Cu/64Cu. Imaging and biodistribution studies in NOD/SCID mice harboring prostate tumor xenografts demonstrated that both 64Cu-labeled NOTA- and CB-TE2A-conjugated radiotracers exhibited favorable pharmacokinetics over the PCTA, oxo-DO3A, and DOTA-conjugated compounds. Between the NOTA- and CB-TE2A-chelated compounds, the 64Cu-labeled CB-TE2A conjugated [64Cu]6B exhibited superior tumor-to-background ratios and is the most promising agent from the series.
Results
Chemical and Radiochemical Syntheses
Macrocyclic Chelator-Conjugated PSMA Inhibitors
We have synthesized two different types of ligands as shown in Figure 1 using Lys-Glu urea as the targeting moiety. For ligands 3–5 we used a Lys-suberate linker21 and conjugated the linker with commercially available chelating agents, NOTA-Bn-SCN, PCTA-Bn-SCN, and oxo-DOTA-Bn-SCN via thiourea bond formation. For those three bifunctional chelators, the macrocyclic backbone was functionalized to carry a benzyl isocyanate for conjugation with an amine function. Copper(II) compounds for 3–5 are predicted to demonstrate distorted octahedron geometry.41 Because NOTA is a hexadentate N3O3 chelator, [64Cu(II)]3 is expected to carry a negative charge. On the other hand, both PCTA (N4O3) and oxo-DOTA (N3O4) are heptadentate macrocycles and likely form neutral compounds with one pendant acetate arm after complexation.35,41 The Lys-suberate linker was extended using two phenylalanine residues to prepare 6 and 7 containing CB-TE2A and DOTA, respectively, as chelating agents. The purpose of attaching those two phenylalanine residues to the linker of 6 and 7 is mainly to compensate for the lipophilicity of the phenylthiourea moiety of compounds 3–5.22,24−26 Note that one of the pendant acetate arms of the CB-TE2A and DOTA chelators has been modified to attach the PSMA targeting moiety via amide bond formation to produce 6 and 7, respectively. [64Cu]CB-TE2A forms a positively charged radiotracer when conjugated to a biomolecule, whereas DOTA-conjugated [64Cu]7 is expected to carry one pendant acetate, providing an overall neutral compound.
Figure 1.

Proposed structures of 64Cu-labeled inhibitors of PSMA.
Syntheses of PSMA targeting ligands 3, 4, and 5 are shown in Scheme 1. Compounds 1 and 2 were prepared as previously reported.28 Compound 2 was conjugated with commercially available NOTA-Bn-SCN, PCTA-Bn-SCN, and oxo-DOTA-Bn-SCN in DMSO in the presence of diisopropylethylamine (DIEA) at 40 °C for 4 h, to give 3, 4, and 5 in ∼30–40% yield, respectively, after purification by high-performance liquid chromatography (HPLC). Synthesis of CB-TE2A-conjugated ligand 6 was performed using standard fluorenylmethoxycarbonyl (Fmoc) solid phase peptide synthesis (SPPS), starting from Fmoc-Lys(Boc)-Wang resin according to Scheme 2. After two phenylalanine residues were coupled with the resin bound lysine, CB-TE2A was conjugated at the N-terminal of the second phenylalanine residue, after which the compound was cleaved from the resin by a 1:1 mixture of TFA/CH2Cl2 to produce 8 in moderate yield (∼20%). The free ε-amine of lysine of 8 was then conjugated with 1(28) to produce 6. DOTA-conjugated 7 was prepared according to our previous report.26 NMR and mass spectrometry (MS) were used to confirm the identity of newly synthesize compounds. Copper (63/65Cu) complexes were prepared by incubating conjugates 3–7 with an aqueous solution of Cu(NO3)2 at 95 °C as shown in Schemes 1–2. The mass spectra of the copper-labeled compounds showed the expected isotope distribution pattern for natural copper, which is a mixture of 63Cu (69%) and 65Cu (31%). The stable copper-labeled conjugates were used as authentic reference material for chromatography to identify the corresponding 64Cu-labeled compounds.
Scheme 1. Synthesis of [64Cu]3–5.

Scheme 2. Synthesis of [64Cu]6.

Radiochemistry
The radiolabeling of new PSMA ligands with 64Cu is shown in Schemes 1–2. Briefly, for ligands 3–5 and 7, radiolabeling was performed at pH ∼5.5–6 in acetate buffer at ∼65 °C for 30 min, whereas for ligand 6, radiolabeling was achieved in a boiling water bath (95 °C) for 1 h at pH ∼7.5–8 in acetate buffer. Radiolabeled products were obtained in high yield (>60%, without decay correction) and radiochemical purity (>95%) as determined by HPLC and radio thin-layer chromatography (radio-TLC). Two radiolabeled peaks were observed for [64Cu]4, likely due to the separation of diastereomers as previously reported for this class of bifunctional chelators.61 On the other hand, free ligand 6 contained two nearly inseparable isomeric peaks by HPLC. Therefore, radiolabeling of 6 was performed using the isomeric mixture to give two HPLC separable products designated [64Cu]6A and [64Cu]6B, isolated in 45% and 35% yield, respectively. We assumed that the presence of an asymmetric center at the Phe residue of the linker moiety adjacent to CB-TE2A was responsible for producing those two diastereomeric compounds. All radioligands were obtained in specific activities ranging from 2.9 to 9.1 GBq/μmol (78–347 mCi/μmol). The Log Poct/w values for the radioligands were determined by octanol–water partition and are shown in Table 1. Surprisingly, [64Cu]6A and [64Cu]6B were found to be more hydrophilic than [64Cu]3 in spite of possessing two Phe residues on the linker and only one carboxylate groups on the chelator backbone.
Table 1. Radiolabeling Yield, Log Po/w, and PSMA Inhibitory Activity.
| yield (%) | Log Poct/w | Ki (nM) | ||
|---|---|---|---|---|
| 3 | 2.84 | |||
| [64Cu]3 | ∼65–70 | –1.17 | [63/65Cu]3 | 6.23 |
| 4 | 2.03 | |||
| [64Cu]4 | ∼70–90 | –1.42 | [63/65Cu]4 | 10.76 |
| 5 | 3.10 | |||
| [64Cu]5 | ∼75–85 | –1.56 | [63/65Cu]5 | 5.47 |
| 6 | 0.19 | |||
| [64Cu]6A | ∼40–45 | –2.68 | [63/65Cu]6A | 3.98 |
| [64Cu]6B | ∼30–35 | –2.31 | [63/65Cu]6B | 4.65 |
| 7 | 11.07 | |||
| [64Cu]7 | ∼65–70 | ND | [63/65Cu ]7 | 13.26 |
In Vitro Binding
Ligands and the corresponding stable metal-labeled compounds demonstrated high binding affinity to PSMA, with Ki values ranging from 0.19 to 13.26 nM (Table 1). The known, high-affinity PSMA inhibitor ZJ43 was used as a reference ligand (Supporting Information Figure S1) and exhibits a Ki of 4.3 nM.62 The low nanomolar affinities displayed by these five new copper compounds are similar to the affinities of the majority PSMA-targeted imaging agents we have synthesized to date.21,28
Small Animal PET-CT Imaging
Whole body PET-CT images were obtained for [64Cu]3–7 in intact male severe combined immunodeficient (SCID) mice (Figures 2–4) bearing PSMA+ PC3 PIP and PSMA– PC3 flu xenografts in opposite, upper flanks. Pharmacokinetics derived from the images were used to determine which compound(s) would be further evaluated through ex vivo biodistribution. Irrespective of charge and lipophilicity, all radiotracers, [64Cu]3–7, enabled visualization of PSMA+ PC3 PIP tumor and kidneys (Figure 2). Renal uptake of the radiotracers is partially due to the route of excretion of these agents as well as to specific uptake from the expression of PSMA in mouse proximal renal tubules.63 Although [64Cu]4, [64Cu]5, and [64Cu]7 demonstrated moderate to high PSMA+ PC3 PIP tumor uptake, they also showed significant accumulation within liver at 2.5 h postinjection (Figure 2), which remained high until 6 h (data not shown). Radioligands [64Cu]5 and [64Cu]7 exhibited significantly higher background uptake, probably related to lower stability of these 64Cu complexes,58 resulting in trans-chelation of copper to other, endogenous proteins.56
Figure 2.
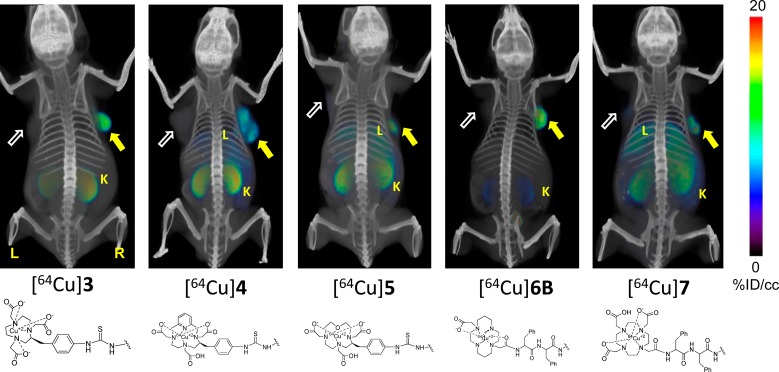
Whole body PET-CT imaging of PSMA+ PC3 PIP and PSMA – PC3 flu tumor-bearing mice with compounds [64Cu]3, [64Cu]4, [64Cu]5, [64Cu]6B, and [64Cu]7 at 2.5 h postinjection, respectively. Mice were injected with ∼11.1 MBq (∼300 μCi) of radiotracer IV. PIP = PSMA+ PC3 PIP (solid arrow); flu = PSMA– PC3 flu (unfilled arrow); K= kidney; B = bladder; L = left; R = right. All images are decay-corrected and adjusted to the same maximum value.
Figure 4.

Whole body PET-CT imaging of PC3 PIP and PC3 flu tumor bearing mice with [64Cu]6A (top row) and [64Cu]6B (bottom row) at 20 min, 2.5 h, 12 and 22 h postinjection. Abdominal radioactivity is primarily due to uptake within kidneys and bladder. PIP = PC3 PSMA+ PIP (solid arrow); flu = PC3 PSMA− flu (unfilled arrow); K= kidney; L = left; R = right, B = bladder. All images are decay-corrected and adjusted to the same maximum value.
PET-CT images acquired at 20 min, 6 and 28 h postinjection of [64Cu]3 (Figure 3) (n = 2) showed clear uptake in PSMA+ PC3 PIP tumor. At 20 min and 6 h postinjection, the most visible tissues were PSMA+ PC3 PIP tumor and kidneys, with some accumulation of radioactivity observed in liver and urinary bladder. Radioactivity in liver and kidneys cleared significantly by 28 h.
Figure 3.
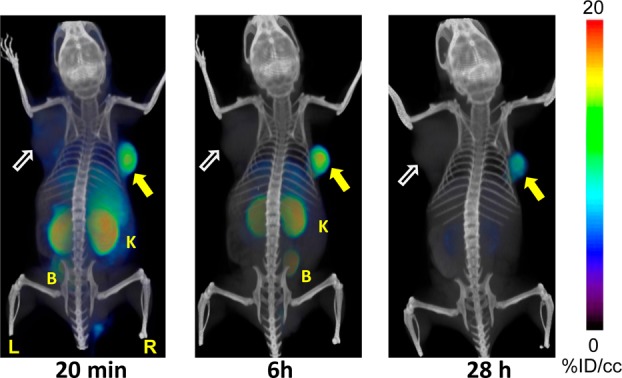
Whole body PET-CT imaging of PC3 PIP and PC3 flu tumor bearing mice with [64Cu]3 at 20 min (left), 6 h (middle), 28 h (right). Abdominal radioactivity is primarily due to uptake within kidneys and bladder. PIP = PC3 PSMA+ PIP (solid arrow); flu = PC3 PSMA– flu (unfilled arrow); K= kidney; L = left; R = right, B = bladder. All images are decay-corrected and adjusted to the same maximum value.
Compounds [64Cu]6A and [64Cu]6B exhibited high radiotracer concentration both within PSMA+ PC3 PIP tumor and kidneys, similar to the distribution profile observed with [64Cu]3. Significantly, both CB-TE2A conjugated diastereomers [64Cu]6A and [64Cu]6B exhibited similar PET imaging profiles as shown in Figure 4. Both compounds showed low liver uptake as early as 20 min after the injection. Consequently, clear delineation of tumor was achieved even at early time points. By 2.5 h postinjection, radioactivity was largely cleared from kidneys for both isomers, producing clear target-to-background contrast for these radiotracers. As a further test of binding specificity, we imaged animals administered [64Cu]6B after pretreating them with 50 mg/kg of ZJ43 30 min prior to radiotracer.62 ZJ43 proved capable of blocking binding of [64Cu]6B (Supporting Information Figure S2), not only within the tumor but also within the renal cortex, confirming that uptake observed in these tissues is PSMA-mediated.63
Biodistribution
On the basis of PET-CT imaging results, [64Cu]3, [64Cu]6A, and [64Cu]6B were further assessed in a biodistribution assays using the same isogenic human prostate cancer PSMA+ PC3 PIP and PSMA– PC3 flu tumor models (n = 4). Tables 2 and 3 show the pharmacokinetics in percentage of injected dose per gram of tissue (% ID/g) in selected organs for [64Cu]3 and [64Cu]6A at 30 min, 1 h, 2 h, and at 4 or 5 h postinjection, while Table 4 shows the % ID/g at the optimized time point of 2 h for [64Cu]3, [64Cu]4, [64Cu]6A, and[64Cu]6B. All compounds exhibited clear PSMA-dependent binding in PSMA+ PC3 PIP tumor xenografts, with [64Cu]3 demonstrating high tumor uptake as early as 30 min postinjection (33.78 ± 9.68% ID/g), peaking at 2 h (38.51 ± 5.68% ID/g) (Table 2), with extended retention of radioactivity up to 4 h postinjection (20.64 ± 5.06% ID/g). PSMA+ PC3 PIP to PSMA– PC3 flu tumor uptake ratios were 14.03 ± 3.71 and 23.50 ± 4.65 at 30 min at 4 h, respectively. The distribution within normal organs and tissues was also favorable, with low blood and normal tissue uptake and rapid clearance. The highest nonspecific uptake observed was in the liver (7.37 ± 0.40% ID/g) and spleen (28.02 ± 5.82% ID/g) at 30 min postinjection. However, those values decreased to 4.18 ± 1.61% ID/g and 4.72 ± 1.49% ID/g, respectively, by 4 h. Kidney uptake was expectedly high and peaked at 195.44 ± 29.82% ID/g at 30 min and decreased to 108.05 ± 20.50% ID/g by 4 h.
Table 2. Tissue Biodistribution for [64Cu]3 in Tumor-Bearing Micea.
| 0.5 h | 1 h | 2 h | 4 h | |
|---|---|---|---|---|
| blood | 2.37 ± 0.90 | 1.12 ± 0.21 | 0.82 ± 0.20 | 0.45 ± 0.20 |
| heart | 1.78 ± 0.41 | 1.39 ± 0.24 | 1.11 ± 0.13 | 0.71 ± 0.26 |
| lung | 6.40 ± 0.59 | 5.99 ± 0.70 | 4.51 ± 0.97 | 2.49 ± 0.99 |
| liver | 7.37 ± 0.40 | 7.50 ± 1.43 | 6.44 ± 0.83 | 4.18 ± 1.61 |
| spleen | 28.02 ± 5.82 | 27.58 ± 10.05 | 12.33 ± 6.32 | 4.72 ± 1.49 |
| fat | 1.78 ± 0.31 | 1.43 ± 0.38 | 0.76 ± 0.34 | 0.65 ± 0.75 |
| kidney | 195.44 ± 29.82 | 233.39 ± 22.76 | 199.69 ± 48.44 | 108.05 ± 20.50 |
| muscle | 0.57 ± 0.32 | 0.60 ± 0.14 | 0.34 ± 0.12 | 0.21 ± 0.10 |
| small intestine | 4.60 ± 1.42 | 5.58 ± 1.43 | 3.68 ± 0.41 | 2.44 ± 1.30 |
| large intestine | 4.51 ± 1.64 | 6.01 ± 2.54 | 3.80 ± 1.67 | 2.42 ± 1.19 |
| bladder | 5.65 ± 2.77 | 2.48 ± 0.42 | 13.33 ± 8.35 | 1.70 ± 0.97 |
| PC-3 PIP | 33.79 ± 9.68 | 29.11 ± 3.02 | 38.51 ± 5.68 | 20.64 ± 5.06 |
| PC-3 flu | 2.40 ± 0.17 | 2.02 ± 0.18 | 1.92 ± 0.23 | 1.04 ± 0.37 |
| PIP:flu | 14.03 ± 3.71 | 14.37 ± 0.43 | 19.96 ± 0.81 | 23.50 ± 4.65 |
| PIP:blood | 15.11 ± 5.38 | 26.64 ± 5.12 | 48.75 ± 10.27 | 50.01 ± 15.54 |
Values expressed are in % ID/g ± standard deviation; n = 4 for all tissues.
Table 3. Tissue Biodistribution for [64Cu]6B in Tumor-Bearing Micea.
| 0.5 h | 1 h | 2 h | 5 h | |
|---|---|---|---|---|
| blood | 2.50 ± 1.08 | 0.64 ± 0.24 | 0.26 ± 0.13 | 0.06 ± 0.05 |
| heart | 0.92 ± 0.27 | 0.32 ± 0.08 | 0.13 ± 0.05 | 0.06 ± 0.01 |
| lung | 2.70 ± 0.73 | 0.83 ± 0.11 | 0.45 ± 0.21 | 0.15 ± 0.01 |
| liver | 0.73 ± 0.17 | 0.42 ± 0.04 | 0.30 ± 0.06 | 0.21 ± 0.03 |
| stomach | 1.02 ± 0.47 | 0.48 ± 0.28 | 0.25 ± 0.06 | 0.10 ± 0.03 |
| pancreas | 0.90 ± 0.23 | 0.31 ± 0.19 | 0.13 ± 0.05 | 0.05 ± 0.04 |
| spleen | 5.80 ± 2.61 | 2.14 ± 0.58 | 0.65 ± 0.15 | 0.25 ± 0.07 |
| fat | 0.87 ± 0.15 | 0.87 ± 1.32 | 0.19 ± 0.10 | 0.05 ± 0.05 |
| kidney | 256.11 ± 75.72 | 148.60 ± 48.41 | 65.39 ± 21.16 | 3.54 ± 0.04 |
| muscle | 0.33 ± 0.01 | 1.71 ± 2.29 | 0.17 ± 0.05 | 0.05 ± 0.02 |
| small intestine | 0.85 ± 0.41 | 1.38 ± 1.89 | 0.26 ± 0.07 | 0.11 ± 0.10 |
| large intestine | 0.88 ± 0.42 | 0.77 ± 0.65 | 0.30 ± 0.08 | 0.14 ± 0.05 |
| bladder | 3.99 ± 0.26 | 7.06 ± 3.75 | 2.21 ± 0.57 | 1.43 ± 1.41 |
| PC-3 PIP | 23.14 ± 2.20 | 29.50 ± 8.10 | 20.46 ± 2.90 | 18.69 ± 2.88 |
| PC-3 flu | 1.29 ± 0.12 | 0.66 ± 0.26 | 0.26 ± 0.04 | 0.13 ± 0.03 |
| PIP:flu | 19.36 ± 3.98 | 48.41 ± 14.25 | 79.40 ± 15.83 | 146.79 ± 32.30 |
| PIP:blood | 9.13 ± 5.23 | 47.47 ± 7.24 | 101.17 ± 6.70 | 1123.96 ± 25.83 |
| PIP:muscle | 69.35 ± 4.29 | 91.30 ± 93.17 | 132.99 ± 43.97 | 436.14 ± 202.71 |
Values expressed are in % ID/g ± standard deviation; n = 4 for all tissues.
Table 4. Tissue Biodistibistion for [64Cu]3, [64Cu]4, [64Cu]6A, and [64Cu]6B at 2 h Post-Injection in Tumor-Bearing Micea.
| [64Cu]3 | [64Cu]4 | [64Cu]6A | [64Cu]6B | |
|---|---|---|---|---|
| blood | 1.06 ± 0.29 | 1.87 ± 0.63 | 0.20 ± 0.03 | 0.20 ± 0.07 |
| heart | 1.48 ± 0.14 | 2.71 ± 0.13 | 0.26 ± 0.10 | 0.35 ± 0.21 |
| lung | 5.31 ± 0.74 | 10.16 ± 2.29 | 1.17 ± 0.61 | 0.89 ± 0.15 |
| liver | 8.63 ± 0.92 | 17.04 ± 1.44 | 1.68 ± 0.38 | 1.63 ± 0.72 |
| stomach | 3.88 ± 0.82 | 6.37 ± 0.78 | 0.62 ± 0.42 | 0.62 ± 0.17 |
| pancreas | 2.22 ± 0.34 | 3.51 ± 0.67 | 0.46 ± 0.44 | 0.80 ± 0.92 |
| spleen | 13.42 ± 1.18 | 9.27 ± 2.28 | 0.39 ± 0.30 | 0.90 ± 0.23 |
| fat | 0.76 ± 0.77 | 1.18 ± 0.59 | 0.27 ± 0.26 | 0.05 ± 0.02 |
| kidney | 125.40 ± 42.47 | 166.57 ± 42.39 | 23.55 ± 8.96 | 24.87 ± 11.26 |
| muscle | 0.33 ± 0.08 | 0.71 ± 0.32 | 0.11 ± 0.05 | 0.12 ± 0.05 |
| small intestine | 6.24 ± 1.46 | 10.95 ± 3.70 | 0.74 ± 0.32 | 0.68 ± 0.13 |
| large intestine | 10.13 ± 6.83 | 10.10 ± 4.76 | 1.49 ± 0.75 | 1.28 ± 0.70 |
| bladder | 12.47 ± 10.30 | 10.56 ± 6.20 | 7.98 ± 8.32 | 5.92 ± 4.94 |
| PC-3 PIP | 16.58 ± 3.15 | 24.13 ± 10.06 | 11.10 ± 5.42 | 16.89 ± 5.73 |
| PC-3 flu | 2.12 ± 0.25 | 3.88 ± 0.62 | 0.36 ± 0.09 | 0.40 ± 0.12 |
| PIP:flu | 11.00 ± 0.73 | 6.22 ± 1.86 | 30.90 ± 7.92 | 41.75 ± 5.54 |
| PIP:blood | 16.52 ± 5.14 | 12.87 ± 1.87 | 52.41 ± 17.34 | 86.97 ± 15.22 |
| PIP:muscle | 46.98 ± 19.05 | 38.34 ± 21.69 | 97.31 ± 42.46 | 138.88 ± 30.86 |
Values expressed are in % ID/g ± standard deviation; n = 4 for all tissues.
Table 3 shows the organ-related % ID/g of uptake for [64Cu]6B. Similar to [64Cu]3, [64Cu]6B showed the highest PSMA-dependent tumor uptake with 29.50 ± 8.10% ID/g at 1 h postinjection. Tumor uptake remained high, decreasing to 18.69 ± 2.88% ID/g at 5 h. The PSMA+ PC3 PIP to PSMA– PC3 flu ratios were 19.36 ± 3.98 at 30 min, 79.40 ± 15.83 at 2 h, and 146.79 ± 32.20 at 5 h. The PSMA+ PC3 PIP tumor-to-muscle ratio reached a maximum value of 436.14 ± 202.71 at 5 h, nearly 3-fold higher than those observed with [64Cu]3. Renal uptake for [64Cu]6B was highest at 30 min at 256.11 ± 75.72% ID/g, much higher than that seen for [64Cu]3, however, it cleared much more rapidly, decreasing to 3.54 ± 0.04% ID/g by 5h. Nontarget organs, such as blood, heart, liver, spleen, stomach, and pancreas, showed lower uptake (∼2% ID/g at 1 h) and much faster clearance than for [64Cu]3 and decreased to below 1% ID/g by 5 h.
Because [64Cu]3, [64Cu]4, and [64Cu]6 demonstrated superior pharmacokinetics to [64Cu]5 and [64Cu]7, a one-time point (2 h) biodistribution study (Table 4) was performed with [64Cu]3, [64Cu]4, and [64Cu]6B. Syntheses and biodistribution studies were performed on the same day, as close to one another as possible to minimize the changes in specific activity of the radiotracer. The biodistribution and imaging data reveal several important points regarding the in vivo properties of these compounds. The NOTA-chelated radiotracers [64Cu]3, PCTA-chelated [64Cu]4, and CB-TE2A-chelated [64Cu]6B showed high uptake in PSMA+ PC3 PIP tumor, highest for [64Cu]4 (24.13 ± 10.06% ID/g). However, [64Cu]4 also exhibited slightly higher nonspecific uptake in PSMA– PC3 flu tumor and other organs, including blood, muscle, heart, liver, and stomach, resulting in lower tumor-to-organ ratios than for [64Cu]3 and [64Cu]6B. In the imaging studies, [64Cu]4 exhibited considerable background radioactivity primarily due to high liver uptake of [64Cu]4. Diastereomers [64Cu]6A and [64Cu]6B, each containing the CB-TE2A chelating agent, possessed similar in vivo properties and exhibited significantly lower normal tissue uptake, including kidney, than either [64Cu]3 or [64Cu]4.
Discussion
We have evaluated five 64Cu-labeled PSMA-targeted, urea conjugates using different chelating agents with the aim of optimizing in vivo PET imaging properties of this class of compounds for clinical application. It is generally accepted that the overall biologic profile of radiolabeled ligands is determined not only by receptor-specific binding but also by nonspecific interactions. Such off-target effects rely on the overall physicochemical features of the radiolabeled compound, e.g., molecular weight, charge, lipophilicity, and metabolic stability. The chelator used to attach the radionuclide to the targeting agent plays a key role in establishing those physicochemical features, particularly for agents smaller than antibodies and nanoparticles, namely those of relatively low molecular weight (<1500 Da). Two key findings emerged from this study. First, both NOTA and CB2-TE2A-conjugated radiotracers [64Cu]3 and [64Cu]6 demonstrated significantly higher in vivo stability, as evidenced by their lower liver uptake than the other three radiotracers, [64Cu]4, [64Cu]5, and [64Cu]7, which utilized PCTA, oxo-DOTA, and DOTA, respectively. High liver uptake and slow blood clearance for those latter three radiotracers are indicative of free Cu(II), which is sequestered in liver.56,64 Second, liver uptake and blood concentration are much lower at all time points for [64Cu]6A–B compared to [64Cu]3, suggesting higher in vivo stability of CB-TE2A-conjugated [64Cu]6A–B compared to NOTA-conjugated [64Cu]3. That might also be related to the higher hydrophilicity of [64Cu]6A–B compared to [64Cu]3 (Log Po/w = −2.68 for [64Cu]6B compared to −1.17 for [64Cu]3). Low liver uptake of CB-TE2A-chelated [64Cu]6 ligands might also be related their higher stability in forming complexes with Cu(II) and Cu(I), assuming Cu(II) reduction is the primary reason for transchelation, as reported previously.56 It is worth mentioning that [64Cu]3, which possesses an overall negative charge, exhibited higher kidney and spleen uptake that was similar to that observed for 99mTc-oxo labeled PSMA-targeted compounds.34
Both kidney and tumor uptake of [64Cu]6B were specifically blocked by ZJ43, demonstrating PSMA-mediated renal uptake, however, much faster renal clearance of [64Cu]6B was observed compared to clearance from PSMA+ PIP tumor. We and others have seen a similar pattern of faster clearance of radioactivity from PSMA-expressing kidney than PSMA+ PIP tumor, presumably associated with more rapid flow through normally organized renal parenchymal vasculature compared to the relatively disorganized vasculature of the xenograft.26,65,66 As an extension of that particular pharmacokinetic feature, [64Cu]6 exhibited the highest tumor-to-normal tissue ratios in this series of agents and the ratios increased with time, thereby providing high image contrast. Moreover, no bone uptake was observed for [64Cu]6, indicating that this agent may find application for detection of prostate cancer metastases, which are preferentially found in bone.
The imaging and biodistribution results demonstrate higher in vivo stability and renal clearance for CB-TE2A-chelated [64Cu]6 compared to NOTA-chelated [64Cu]3, which appears to be in contradistinction to previous reports that involve similar comparison for agents that target somatostatin receptor and αvβ3 integrin. In those reports [64Cu]NOTA-conjugated radiopeptides provided faster renal clearance than those conjugated with [64Cu]CB-TE2A.46,47 Although unclear at this time, this difference between 64Cu-labeled species for targeting of PSMA vs somatostatin receptor or integrin may be associated with other physical properties unique to each system such as binding affinity or capacity for internalization of the cognate ligands. It nevertheless underscores the uniqueness of each system and indicates that one chelator cannot be assumed to outperform another in a new, untested system.
There are several classes of imaging agents available for PET-based molecular imaging of PSMA, including antibodies,4,5,32,67 antibody fragments,68 aptamers,49 and low-molecular-weight PSMA-binding affinity agents.14,65 They have widely varying pharmacokinetics, however, each class has shown PSMA-specific binding in preclinical studies. Rockey and colleagues have recently optimized conditions for conjugating 64Cu to a PSMA-targeting aptamer with different chelators, which has shown PSMA-mediated uptake in PSMA+ 22Rv1 prostate tumor cells relative to PSMA– PC3 cells.49 The radiolabeled monoclonal antibodies [64Cu]DOTA-3/A1214 and [89Zr]DFO-J59155 both showed values of approximately 3:1 at 48 h post-injection for PSMA+ to PSMA- tumor. Among the long-lived PET radionuclides, 64Cu and 89Zr, the stability of the [89Zr]DFO complex is limited as shown in several in vitro and in vivo in studies, which is reflected in a bone accumulation ranging from 3 to 15% and assumed to be associated with the nonlinear nature of the desferrioxamine (DFO) chelating agent.69 More recently, studies in biopsy tissue from patients with prostate cancer and cell lines have demonstrated a correlation between PSMA cell–surface expression and androgen activity using 64Cu-labeled anti-PSMA antibody ([64Cu]J591).20 ImmunoPET detection of PSMA could provide a path for quantitative monitoring of successful androgen blockade in patients. Low-molecular-weight urea-based agent [64Cu]6 could be an inexpensive alternative for those applications. Furthermore, results obtained with [64Cu]6 suggest that delayed imaging with 64Cu-labeled PSMA-targeted ligands using CB-TE2A can improve the visualization of PSMA+ tumors in vivo anywhere outside of central nervous system and may allow imaging of tumors with lower PSMA expression levels than possible with existing 18F- or 68Ga-labeled PSMA-targeted imaging agents.
Conclusion
We describe five new low-molecular-weight, urea-based, 64Cu-labeled, PSMA-targeted radiotracers that incorporated well-established chelating agents. All compounds demonstrated high tumor uptake and retention with the choice of chelator having a profound effect on pharmacokinetics. Our data revealed that [64Cu]6, which utilizes the CB-TE2A chelator, demonstrated improved biodistribution with rapid clearance from normal tissues, including kidney, resulting in significantly improved image contrast. Accordingly, [64Cu]6 provides a potentially clinically viable imaging agent for PSMA+ tissues, particularly if delayed imaging, obtainable with a 64Cu-labeled agent, is required.
Experimental Section
Solvents and chemicals purchased from commercial sources were of analytical grade or better and used without further purification. All 9-fluorenylmethyloxycarbonyl (Fmoc) protected amino acids including the Fmoc-Lys(Boc)-Wang resin, 1-hydroxybenzotriazole monohydrate, and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Chem Impex International Inc. (Wooddale, IL). [64Cu]CuCl2 was purchased from the University of Wisconsin. DOTA-tris(t-butyl ester)-monoacid, p-SCN-Bn-NOTA, p-SCN-Bn-PCTA, and p-SCN-Bn-oxo-DO3A were received from Macrocyclics Inc. (Dallas, TX). Copper(II) chloride, triethylsilane (Et3SiH), diisopropylethylamine (DIEA), and triethylamine (TEA) were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were purchased from Thermo Fisher Scientific (Pittsburgh, PA) unless otherwise specified. Analytical thin-layer chromatography (TLC) was performed using Aldrich aluminum-backed 0.2 mm silica gel Z19, 329-1 plates and visualized by ultraviolet light (254 nm), I2, and 1% ninhydrin in EtOH. Flash chromatography was performed using silica gel (MP SiliTech 32-63 D 60 Å) purchased from Bodman (Aston, PA). All in vitro PSMA binding studies and determination of partition coefficient experiments were performed in triplicate to ensure reproducibility. 1H NMR spectra were recorded on a Bruker UltrashieldTM 400 MHz spectrometer. Chemical shifts (δ) are reported in ppm downfield by reference to proton resonances resulting from incomplete deuteration of the NMR solvent. Low resolution ESI mass spectra were obtained on a Bruker Daltonics Esquire 3000 Plus spectrometer. High resolution mass spectra were obtained by the University of Notre Dame Mass Spectrometry and Proteomics Facility, Notre Dame, IN, using ESI either by direct infusion on a Bruker microTOF-II or by LC elution via an ultrahigh pressure Dionex RSLC with C18 column coupled with a Bruker microTOF-Q II.
HPLC purification of compounds 3–7 was performed using a Phenomenex C18 Luna 10 × 250 mm2 column on a Waters 600E Delta LC system with a Waters 486 variable wavelength UV/vis detector, both controlled by Empower software. HPLC was performed using the following methods. Method 1: solvent A (0.1% TFA in water) and solvent B (0.1% TFA in in acetonitrile), flow rate 8 mL/min. The elution gradient was 90% A and 10% B for 0–5 min and 90% A to 0% A and 10% B to 100% B over 6–30 min. Methods 2–5 were isocrati,c with a flow rate 4 mL/min. Method 2: 85% A and 15% B for 0–50 min. Method 3: 82% A and 18% B for 0–40 min. Method 4: 83% A and 17% B for 0–40 min. Method 5: 78% A and 22% B for 0–30 min. Method 6: 80% A and 20% B for 0–30 min, flow rate 1 mL/min, and a Waters Novapak C18 150 × 3.9 mm2 column was used. HPLC purification of [64Cu]3-7 were performed on a Varian Prostar System (Palo Alto, CA), equipped with a Varian ProStar 325 UV–vis variable wavelength detector and a Bioscan Flow-Count in-line radioactivity detector, all controlled by Galaxie software. The specific radioactivity was calculated as the radioactivity eluting at the retention time of product during the preparative HPLC purification divided by the mass corresponding to the area under the curve of the UV absorption. The purity of tested compounds as determined by analytical HPLC with absorbance at 220 nm were >95%.
Synthesis and Characterization of Compounds 1–7
Compounds 1,28 and 2,26 and 7(26) were prepared following our previous reports. Compounds 3–5 were prepared following a general procedure as described for compound 3.
9,16,24-Trioxo-1-thioxo-1-((4-(((S)-1,4,7-tris(carboxymethyl)-1,4,7-triazonan-2-yl)methyl)phenyl)amino)-2,8,17,23,25-pentaazaoctacosane-7,22,26,28-tetracarboxylic Acid, Compound 3
To a solution of 2 (20 mg, 0.33 μmol in 500 μL of DMSO) was added NOTA-Bn-SCN (18.5 mg, 0.33 μmol in 500 μL of DMSO) and DIEA (100 μL), and the solution was kept at 40 °C for 4 h. The reaction mixture was then evaporated to dryness, and the crude residue was dissolved in 2 mL of water and loaded on a prepacked C18 column (5.5 g, Agilent SF10). The product was eluted with 80/20–70/30 H2O (0.1%TFA)/CH3CN (0.1% TFA) solution. The fractions containing the products were combined together and evaporated to dryness to obtain a colorless solid. The solid thus obtained was further purified by HPLC (method 1, retention time, Rt 15.80 min). Yield ∼8 mg (0.076 μmol, ∼23%) after HPLC purification and lyphilization. 1H NMR (DMSO-d6) δ: 8.15 (m, 1H, HNCO(Lys-linker)), 7.75 (m, 1H, HNCO(Lys)), 7.14 (d, 2H, Bz), 6.78 (d, 2H, Bz), 6.34 (m, 2H, NH(CO)NH), 4.40–3.99 (m, 3H, HC(NHCO2(Glu), HC(NHCO2(Lys), HC(NHCO2(Lys)), 3.40–3.30 (m, 7H, NCH2, CHCH2, (NOTA)), 3.21–3.03(m, 16H, (CH2)2(NOTA), H2C-Bz, H2CNH(Lys-linker), H2CNH(Lys)), 2.40–2.09 (m, 6H, H2CCO2(Glu), H2CCO2(linker), H2CCO2(linker)),1.89–1.55 (m, 6H, H2CCH(Glu), H2CCH(Lys-linker), H2CCH(Lys)), 1.55–1.21 (m, 16H, (CH2)2(Lys), (CH2)2(Lys-Linker), (CH2)4(linker)). ESMS m/z: 1054 [M + H]+. HRESI+ MS: calcd for C46H72N9O17S, 1054.4767 [M + H]+; found, 1054.4771.
9,16,24-Trioxo-1-thioxo-1-((4-(((4S)-3,6,9-tris(carboxymethyl)-3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(14),12-dien-4-yl)methyl)phenyl)amino)-2,8,17,23,25-pentaazaoctacosane-7,22,26,28-tetracarboxylic Acid, Compound 4
The compound was purified by HPLC (method 1; Rt 15.60 min). Yield ∼12 mg (0.076 μmol, ∼23%) after HPLC purification and lyophilized. 1H NMR (DMSO-d6) δ: 8.14 (m, 1H, HNCO(Lys-linker)), 7.80–7.75 (m, 2H, HNCO(Lys), Py), 7.32 (d, 2H, Py), 7.15 (d, 2H, Bz), 6.78 (d, 2H, Bz), 6.35 (m, 2H, NH(CO)NH), 4.42–3.99 (m, 7H, HC(NHCO2(Glu), HC(NHCO2(Lys), HC(NHCO2(Lys), H2C-Py)), 3.42–3.31 (m, 7H, CHCH2, NCH2(PCTA)), 3.21–3.02 (m, 12H, (CH2)2(PCTA), H2C-Bz, H2CNH(Lys-linker), H2CNH(Lys)), 2.41–2.08 (m, 6H, H2CCO2(Glu), H2CCO2(linker), H2CCO2(linker)), 1.89–1.55 (m, 6H, H2CCH(Glu), H2CCH(Lys-linker), H2CCH(Lys)), 1.55–1.21 (m, 16H, (CH2)2 (Lys), (CH2)2(Lys-Linker), (CH2)4(linker)). ESMS m/z: 1132 [M + H]+. HRESI+ MS: calcd for C51H75N10O17S, 1131.5032 [M + H]+; found, 1131.5011.
9,16,24-Trioxo-1-thioxo-1-((4-((4,7,10-tris(carboxymethyl)-1-oxa-4,7,10-triazacyclododecan-2-yl)methyl)phenyl)amino)-2,8,17,23,25-pentaazaoctacosane-7,22,26,28-tetracarboxylic Acid, Compound 5
The compound was purified by HPLC (method 1; Rt 13.40 min). Yield ∼10 mg (0.076 μmol, ∼23%) after HPLC purification and lyophilization. 1H NMR (DMSO-d6) δ: 8.12 (m, 1H, HNCO(Lys-linker)), 7.80–7.75 (m, 1H, HNCO(Lys)), 7.14 (d, 2H, Bz), 6.79 (d, 2H, Bz), 6.32 (m, 2H, NH(CO)NH), 4.42–3.99 (m, 3H, HC(NHCO2(Glu), HC(NHCO2(Lys), HC(NHCO2(Lys)), 3.49–3.31 (m, 11H, NCH2(DO3A), CHCH2(DO3A), OCH2)), 3.21–3.02 (m, 16H, (CH2)2(DO3A), H2C-Bz, H2CNH(Lys-linker), H2CNH(Lys)), 2.41–2.08 (m, 6H, H2CCO2(Glu), H2CCO2(linker), H2CCO2(linker)), 1.91–1.50 (m, 6H, H2CCH(Glu), H2CCH(Lys-linker), H2CCH(Lys)), 1.50–1.22 (m, 16H, (CH2)2(Lys), (CH2)2(Lys-Linker), (CH2)4(linker). ESMS m/z: 1098 [M + 1]+, HRESI+ MS: calcd for C48H76N9O18S, 1098.5029 [M + H]+; found, 1098.5048.
(14S,29S,33S)-4,7-Dibenzyl-1-(11-(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecan-4-yl)-2,5,8,16,23,31-hexaoxo-3,6,9,15,24,30,32-heptaazapentatriacontane-14,29,33,35-tetracarboxylic Acid, Compound 6
Fmoc-Lys(Boc)-Wang resin (100 mg, 0.43 mM) was allowed to swell with CH2Cl2 (3 mL) followed by DMF (3 mL). A solution of 20% piperidine in DMF (3 × 3 mL) was added to the resin that was then shaken gently on a mechanical shaker for 30 min at ambient temperature. The resin was washed with DMF (3 × 3 mL) and CH2Cl2 (3 × 3 mL). Formation of free amine was assessed by the Kaiser test. After swelling the resin in DMF, a solution of Fmoc-Phe-OH (3 equiv), HBTU (3 equiv), HOBt (3 equiv), and DIPEA (4.0 equiv) in DMF was added and gently shaken for 2 h. The resin was then washed with DMF (3 × 3 mL) and CH2Cl2 (3 × 3 mL). The coupling efficiency was assessed by the Kaiser test. The aforementioned sequence was repeated for two more coupling steps with Fmoc-Phe-OH and CB-TE2A, respectively. The final compound was cleaved from the resin using TFA:CH2Cl2 (1:1) and concentrated under vacuum to produce 8. The concentrated product was purified by using a C18 SepPak Vac 2g column. The product was eluted with a solution 70/30 water/acetonitrile (0.1% TFA in each). ESI-MS: 635 [M]+. To a solution of 8 (20 mg, 31 μmol in 500 μL of DMSO) (27 mg, 47 μmol) and DIEA (50 μL) was added 1, and the reaction mixture was left at ambient temperature for 2 h. The solution was diluted in 10 mL of water and was purified by HPLC (method 1, time (Rt), 20 min). Yield ∼10 mg (0.076 μmol, ∼22%) after HPLC purification and lyophilization. 1H NMR (DMSO-d6): 9.02–8.07 (m, 12H, HNCO(Lys-linker), HNCO(Lys), PhCH2), 6.34 (m, 2H, NH(CO)NH), 4.62 (m, 2H, NHCO2(Phe)), 4.37–4.28 (m, 3H, (m, 3H, HC(NHCO2(Glu), HC(NHCO2(Lys), HC(NHCO2(Lys)), 3.65–3.01 (m, 14H, CH2 N(CB-TE2A), H2CPhe, H2CNH(Lys), H2CNH(Lys)), 2.74–2.41 (m, 20H, NCH2CH2(CB-TE2A), 2.41–2.08 (m, 6H, H2CCO2(Glu), H2CCO2(linker), H2CCO2(linker)), 1.91–1.50 (m, 10H, CH2(CB-TE2A), H2CCH(Glu), H2CCH(Lys-linker), H2CCH(Lys)), 1.56–1.22 (m, 16H, (CH2)2(Lys), (CH2)2(Lys-Linker), (CH2)4(linker). ESMS m/z: 1222 [M + H]+. HRESI+ MS: calcd for C60H91N11O16, 1221.6645; found, 1221.4951.
Copper Complexes of Ligand 3, 4, 5, 6, and 7
Copper compounds [63/65 Cu]3–5 and[63/65 Cu]7 were prepared by the same general procedure as presented for compound 3 below. [63/65Cu]6A–B were prepared by same conditions as described for their radiosynthesis.
[63/65Cu]3. To a solution of Cu(NO3)2 (1 mg, 20 μmol in 100 μL) in deionized water was added 3 (1 mg, 0.95 μmol) in 500 μL of 0.2 M NaOAc. The resulting solution was heated in boiling water for 0.5 h. The solution was purified by HPLC method 2. The retention time for the product was 34 min. Yield: ∼50%. ESIMS m/z: 1115 [M + H]+. HRMS: calcd for C46H70CuN9O17S, 1115.3906 [M + H]+; found, 1115.3828.
[63/65Cu]4. The compound was purified by HPLC method 3. Retention time for the product was at 22 min. Yield: ∼50%. ESIMS m/z: 1192 [M + H]+. HRMS: calcd for C51H73CuN10O17S, 1192.4172 [M + H]+; found, 1192.4364.
[63/65Cu]5. The compound was purified by HPLC method 4. Retention time for the product was at 14 min. Yield: ∼60%. ESIMS m/z: 1160 [M + H]+. HRMS: calcd for C48H76CuN9O18S, 1160.4247 [M + H]+; found, 1160.3828.
[63/65Cu]6A–B .The compound was purified by HPLC method 5. Retention time for the products were at 24 and 26 min. Yield: ∼50%. ESIMS m/z: 1285 [M + H]+. HRMS: calcd for C60H91CuN11O16, 1284.5930 [M + H]+; found, 1284.5786.
[63/65Cu]7. The compound was purified by HPLC method 6. Rt for the product was at 13 min. Yield: ∼55%. ESIMS m/z: 1349 [M + H]+. HRMS: calcd for C60H90CuN11O20, 1348.5650 [M + H]+; found, 1348.5671
64Cu Radiolabeling
Radioligands [64Cu]3–5 and 7 were prepared by the same general method as described for [64Cu]3. For each radiolabeling reaction, approximately 30–40 μg of ligand and in 200 mM NaOAc (purged under N2 for 2–3 min) was incubated with 3–4 mCi 64CuCl2 at pH 5.5–6 for 0.3 h in a water bath at 65 °C. Radiolabeling was monitored by injecting aliquots of 20–40 μL of the solution onto the HPLC. When the reaction reached completion, the reaction mixture was diluted with 1 mL of water then loaded onto a preparative HPLC for purification using method 2, Rt 41 min, for the desired product and Rt 38 min for the free ligand. The radiolabeled product [64Cu]3 was obtained in ∼60–70% radiochemical yield, and the radiochemical purity was >98% as measured by ITLC (Gelman ITLC strips, 10 mM EDTA). HPLC method 2 was used to purify the radiolabeled product [64Cu]3. Rt 34 min for the desired product and Rt 42 min for the free ligand. The specific activity of the probe was 2.9–9.1 GBq/μmol (n = 4). The acidic eluate was neutralized with 50 μL of 1 M Na2(CO3), and the volume of the eluate was reduced under vacuum to dryness. The solid residue was diluted with saline to the desired radioactivity concentration for biodistribution and imaging studies. HPLC method 3 was used for [64Cu]4, Rt = 20–26 min for [64Cu]4 and Rt = 15 min for the free ligand 4. [64Cu]4 was isolated in two peaks, Rt 20–22 min for the first fraction and 23–26 min for the second fraction. On the basis of the initial results (in PET imaging both fractions displayed similar in vivo distributions), only the second fraction was used for all studies. HPLC method 4 was used for [64Cu]5. Retention times for [64Cu]5 and free ligand were and 14.5 and 9.5 min, respectively. The radiotracer [64Cu]7 was purified by HPLC method 6. Rt for [64Cu]7 was at 13 min.
For [64Cu]6A–B, radiolabeling was done in a boiling water bath for 1 h by incubation of 64CuCl2 with the corresponding ligand using 0.2 NaOAc buffer at pH = 7.5–8. HPLC method 5, Rt = 24.9 min (A) and 26.8 min (B) for the desired products and Rt = 19.8 min (A) and 21.5 min (B) for the free ligands. The specific activity of the probe was 2.9 GBq–12.84 GBq/μmol.
NAALADase Assay
The PSMA inhibitory activities of 3–7 and the corresponding copper-labeled analogues were determined using a fluorescence-based assay according to a previously reported procedure. 28 Briefly, lysates of LNCaP cell extracts (25 μL) were incubated with the inhibitor (12.5 μL) in the presence of 4 μM N-acetylaspartylglutamate (NAAG) (12.5 μL) for 120 min. The amount of the glutamate released by NAAG hydrolysis was measured by incubating with a working solution (50 μL) of the Amplex Red glutamic acid kit (Life Technologies, Grand Island, NY) for 60 min. Fluorescence was measured with a VICTOR3 V multilabel plate reader (Perkin-Elmer Inc., Waltham, MA) with excitation at 490 nm and emission at 642 nm. Inhibition curves were determined using semilog plots, and IC50 values were determined at the concentration at which enzyme activity was inhibited by 50%. Enzyme inhibitory constants (Ki values) were generated using the Cheng–Prusoff conversion.70 Assays were performed in triplicate. Data analysis was performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, California).
Cell Lines
Sublines of the androgen-independent PC3 human prostate cancer cell line, originally derived from an advanced androgen independent bone metastasis, were used. These sublines have been modified to express high (PC3 PIP) or possess low (PC3 flu) levels of PSMA and were generously provided by Dr. Warren Heston (Cleveland Clinic). PSMA-expressing (PC3 PIP), nonexpressing (PC3 flu) PCa cell lines, were grown in RPMI 1640 medium (Corning Cellgro, Manassas, VA) containing 10% fetal bovine serum (FBS) (Sigma-Aldrich, St.Louis, MO) and 1% penicillin–streptomycin (Corning Cellgro, Manassas, VA) and PC-3 PIP cells were grown under 20 μg/mL of puromycin to maintain PSMA expression. All cell cultures were maintained in an atmosphere containing 5% carbon dioxide (CO2) at 37.0 °C in a humidified incubator.
Tumor Models
Animal studies were carried out in full compliance with the regulations of the Johns Hopkins Animal Care and Use Committee. Six-to-eight-week-old male, nonobese diabetic (NOD)/severe-combined immunodeficient (SCID) mice (Johns Hopkins Immune Compromised Core) were implanted subcutaneously (sc) with PC3 PIP (PSMA+) and PC3 flu (PSMA−) cells (1 × 106 in 100 μL of HBSS (Corning Cellgro, Manassas, VA) at the forward right and left flanks, respectively. Mice were imaged or used in ex vivo biodistribution assays when the xenografts reached 5– 7 mm in diameter.
Small-Animal PET Imaging and Analysis
Dynamic and whole-body PET and CT images were acquired on an eXplore VISTA small-animal PET (GE Healthcare) and an X-SPECT small SPECT/CT system (Gamma Medica Ideas), respectively. For imaging studies, mice were anesthetized with 3% and maintained under 1.5% isoflurane (v/v). PET-CT Imaging studies were performed on NOD/SCID mice bearing PSMA+ PC3 PIP and PSMA– PC3 flu tumors. Immediately after intravenous injection of [64Cu]3–7, changes in radiotracer accumulation were recorded over the whole body using an imaging sequence consisting of eight frames for a total of 30 min with variable dwell times as described previously.43,65 After dynamic imaging, whole-body PET images (two bed positions, 15 min emission per bed position) were acquired at the indicated time points after injection of radiotracer. For binding specificity studies, a mouse was subcutaneously administered with a blocking dose of the known PSMA inhibitor ZJ43 (50 mg/kg) at 30 min before the injection of [64Cu]6B, and another mouse was injected with [64Cu]6B alone. After each PET scan, a CT scan was acquired as 512 projections using a 50 keV beam for anatomic coregistration. PET emission data were corrected for decay and dead time and were reconstructed using the three-dimensional ordered-subsets expectation maximization algorithm. Data were displayed and analyzed using AMIDE software (http://sourceforge.net/amide), and volume-rendered images were generated using Amira 5.2.0 software (Visage Imaging Inc.; http://www.vsg3d.com/amira).
Ex Vivo Biodistribution
Mice bearing PSMA+ PC3 PIP and PSMA– PC3 flu xenografts were injected via the tail vein with 740 kBq (20 μCi) of 64Cu in 200 μL of saline. At 30, 60, 240, and 240 or 300 min postinjection, the mouse was sacrificed by cervical dislocation and the blood immediately collected by cardiac puncture. The heart, lungs, liver, stomach, pancreas, spleen, fat, kidney, muscle, small and large intestines, urinary bladder, PSMA+ PC3 PIP and PSMA– PC3 flu tumors were collected. Each organ was weighed, and the tissue radioactivity was measured with an automated gamma counter (1282 Compugamma CS, Pharmacia/LKBNuclear, Inc., Mt. Waverly, Victoria, Australia). The % ID/g was calculated by comparison with samples of a standard dilution of the initial dose. All measurements were corrected for decay.
Data Analysis
Data are expressed as mean ± standard deviation (SD). Prism software (GraphPAD, San Diego, California) was used to determine statistical significance. Statistical significance was calculated using a paired t test. A P-value <0.0001 was considered significant.
Acknowledgments
We thank the University of Wisconsin Cyclotron Research Group for providing [64Cu]CuCl2. We also thank Nordion and Macrocyclics, Inc., for providing PCTA-Bn-SCN and Oxo-DO3A-Bn-SCN chelating agent, Drs. Cara Ferreira and Russell Redshaw for helpful discussion, and James Fox, Gilbert Green, and Dr. Yuchuan Wang for assistance with imaging and image analysis. We are grateful for the following sources of support: K25 CA148901, NIH NCI U54CA151838, NCI CA134675, and NCI R01 CA093375.
Glossary
Abbreviations Used
- PSMA
prostate-specific membrane antigen
- GCPII
glutamate carboxypeptidase II
- NAALADase
N-acetylated-α-linked acidic dipeptidase
- PET
positron emission tomography
- SPECT
single photon emission computed tomography
- NOTA
1,4,7-triazacyclononane-1,4,7-triacetic acid
- CB-TE2A
4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane
- PCTA
3,6,9,15-tetraazabicyclo[9.3.1]-pentadeca-1(15),11,13-triene)-3,6,9-triacetic acid
- oxo-DO3A
oxa-4,7,1-tetraazacyclododecane-4,7,10-triacetic acid
- DOTA
1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid
Supporting Information Available
Detailed spectral data and supporting PET-CT blocking study image. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ghosh A.; Heston W. D. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J. Cell Biochem. 2004, 91, 528–539. [DOI] [PubMed] [Google Scholar]
- Rajasekaran A. K.; Anilkumar G.; Christiansen J. J. Is prostate-specific membrane antigen a multifunctional protein?. Am. J. Physiol., Cell Physiol. 2005, 288, C975–C981. [DOI] [PubMed] [Google Scholar]
- Chang S. S.; Reuter V. E.; Heston W. D.; Gaudin P. B. Comparison of anti-prostate-specific membrane antigen antibodies and other immunomarkers in metastatic prostate carcinoma. Urology 2001, 57, 1179–1183. [DOI] [PubMed] [Google Scholar]
- Wright G. L. Jr.; Grob B. M.; Haley C.; Grossman K.; Newhall K.; Petrylak D.; Troyer J.; Konchuba A.; Schellhammer P. F.; Moriarty R. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology 1996, 48, 326–334. [DOI] [PubMed] [Google Scholar]
- Evans M. J.; Smith-Jones P. M.; Wongvipat J.; Navarro V.; Kim S.; Bander N. H.; Larson S. M.; Sawyers C. L. Noninvasive measurement of androgen receptor signaling with a positron-emitting radiopharmaceutical that targets prostate-specific membrane antigen. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9578–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner S.; Hofer M. D.; Kim R.; Shah R. B.; Li H.; Moller P.; Hautmann R. E.; Gschwend J. E.; Kuefer R.; Rubin M. A. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Human Pathol. 2007, 38, 696–701. [DOI] [PubMed] [Google Scholar]
- Sanna V.; Pintus G.; Roggio A. M.; Punzoni S.; Posadino A. M.; Arca A.; Marceddu S.; Bandiera P.; Uzzau S.; Sechi M. Targeted biocompatible nanoparticles for the delivery of (−)-epigallocatechin 3-gallate to prostate cancer cells. J. Med. Chem. 2011, 54, 1321–1332. [DOI] [PubMed] [Google Scholar]
- DiPippo V. A.; Magargal W. W.; Moorji S. M.; Murga J. D.; Olson W. C. Antiandrogen modulation of prostate-specific membrane antigen (PSMA): dynamics and synergy with PSMA-targeted therapy. ASCO Meet. Abstr. 2013, 31, e16007. [Google Scholar]
- Petrylak D. P.; Kantoff P. W.; Mega A. E.; Vogelzang N. J.; Stephenson J.; Fleming M. T.; Stambler N.; Petrini M.; Blattman S.; Israel R. J. Prostate-specific membrane antigen antibody drug conjugate (PSMA ADC): a phase I trial in metastatic castration-resistant prostate cancer (mCRPC) previously treated with a taxane. ASCO Meet. Abstr. 2013, 31, 5018. [Google Scholar]
- Rotshteyn Y.; Mercier F.; Bruno R.; Stambler N.; Israel R. J.; Wong V. Correlation of PSMA ADC exposure with reduction in tumor growth rate determined using serial PSA measurements from a phase I clinical trial. ASCO Meet. Abstr. 2013, 31, e16047. [Google Scholar]
- Hrkach J.; Von Hoff D.; Mukkaram Ali M.; Andrianova E.; Auer J.; Campbell T.; De Witt D.; Figa M.; Figueiredo M.; Horhota A.; Low S.; McDonnell K.; Peeke E.; Retnarajan B.; Sabnis A.; Schnipper E.; Song J. J.; Song Y. H.; Summa J.; Tompsett D.; Troiano G.; Van Geen Hoven T.; Wright J.; LoRusso P.; Kantoff P. W.; Bander N. H.; Sweeney C.; Farokhzad O. C.; Langer R.; Zale S. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci. Transl. Med. 2012, 4, 128ra39. [DOI] [PubMed] [Google Scholar]
- Hillier S. M.; Maresca K. P.; Femia F. J.; Marquis J. C.; Foss C. A.; Nguyen N.; Zimmerman C. N.; Barrett J. A.; Eckelman W. C.; Pomper M. G.; Joyal J. L.; Babich J. W. Preclinical evaluation of novel glutamate–urea–lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009, 69, 6932–6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease R. C.; Foss C. A.; Pomper M. G. PET imaging in prostate cancer: focus on prostate-specific membrane antigen. Curr. Top. Med. Chem. 2013, 13, 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S. Y.; Gage K. L.; Mease R. C.; Senthamizhchelvan S.; Holt D. P.; Jeffrey-Kwanisai A.; Endres C. J.; Dannals R. F.; Sgouros G.; Lodge M.; Eisenberger M. A.; Rodriguez R.; Carducci M. A.; Rojas C.; Slusher B. S.; Kozikowski A. P.; Pomper M. G. Biodistribution, tumor detection, and radiation dosimetry of 18F–DCFBC, a low-molecular-weight inhibitor of prostate-specific membrane antigen, in patients with metastatic prostate cancer. J. Nucl. Med. 2012, 53, 1883–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshar-Oromieh A.; Malcher A.; Eder M.; Eisenhut M.; Linhart H. G.; Hadaschik B. A.; Holland-Letz T.; Giesel F. L.; Kratochwil C.; Haufe S.; Haberkorn U.; Zechmann C. M. PET imaging with a [(68)Ga]gallium-labelled PSMA ligand for the diagnosis of prostate cancer: biodistribution in humans and first evaluation of tumour lesions. Eur. J. Nucl. Med. Mol. Imaging 2012, 40, 486–495. [DOI] [PubMed] [Google Scholar]
- Barrett J. A.; Coleman R. E.; Goldsmith S. J.; Vallabhajosula S.; Petry N. A.; Cho S.; Armor T.; Stubbs J. B.; Maresca K. P.; Stabin M. G.; Joyal J. L.; Eckelman W. C.; Babich J. W. First-in-man evaluation of 2 high-affinity PSMA-avid small molecules for imaging prostate cancer. J. Nucl. Med. 2013, 54, 380–387. [DOI] [PubMed] [Google Scholar]
- Maresca K. P.; Hillier S. M.; Lu G.; Marquis J. C.; Zimmerman C. N.; Eckelman W. C.; Joyal J. L.; Babich J. W. Small molecule inhibitors of PSMA incorporating technetium-99m for imaging prostate cancer: effects of chelate design on pharmacokinetics. Inorg. Chim. Acta 2012, 389, 168–172. [Google Scholar]
- Nedrow-Byers J. R.; Moore A. L.; Ganguly T.; Hopkins M. R.; Fulton M. D.; Benny P. D.; Berkman C. E. PSMA-targeted SPECT agents: Mode of binding effect on in vitro performance. Prostate 2013, 73, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; Maresca K. P.; Hillier S. M.; Zimmerman C. N.; Eckelman W. C.; Joyal J. L.; Babich J. W. Synthesis and SAR of 99mTc/Re-Labeled Small Molecule Prostate Specific Membrane Antigen Inhibitors with Novel Polar Chelates. Biorg. Med. Chem. Lett. 2013, 23, 1557–1563. [DOI] [PubMed] [Google Scholar]
- Nedrow-Byers J. R.; Jabbes M.; Jewett C.; Ganguly T.; He H.; Liu T.; Benny P.; Bryan J. N.; Berkman C. E. A phosphoramidate-based prostate-specific membrane antigen-targeted SPECT agent. Prostate 2012, 72, 904–912. [DOI] [PubMed] [Google Scholar]
- Banerjee S. R.; Foss C. A.; Castanares M.; Mease R. C.; Byun Y.; Fox J. J.; Hilton J.; Lupold S. E.; Kozikowski A. P.; Pomper M. G. Synthesis and evaluation of technetium-99m- and rhenium-labeled inhibitors of the prostate-specific membrane antigen (PSMA). J. Med. Chem. 2008, 51, 4504–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kularatne S. A.; Zhou Z.; Yang J.; Post C. B.; Low P. S. Design, synthesis, and preclinical evaluation of prostate-specific membrane antigen targeted (99m)Tc-radioimaging agents. Mol. Pharmaceutics 2009, 6, 790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; DiFilipp F.; Doke A.; Huang J.; Heston W.; Huang S. Preliminary micro-SPECT and biodistribution study of a novel Tc99m-labeled PSMA tracer derived from RBI1033. J. Nucl. Med. Meet. Abstr. 2012, 53Suppl 11661. [Google Scholar]
- Schafer M.; Bauder-Wust U.; Leotta K.; Zoller F.; Mier W.; Haberkorn U.; Eisenhut M.; Eder M. A dimerized urea-based inhibitor of the prostate-specific membrane antigen for 68Ga-PET imaging of prostate cancer. EJNMMI Res. 2012, 2, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder M.; Schafer M.; Bauder-Wust U.; Hull W. E.; Wangler C.; Mier W.; Haberkorn U.; Eisenhut M. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjugate Chem. 2012, 23, 688–697. [DOI] [PubMed] [Google Scholar]
- Banerjee S. R.; Pullambhatla M.; Byun Y.; Nimmagadda S.; Green G.; Fox J. J.; Horti A.; Mease R. C.; Pomper M. G. 68Ga-labeled inhibitors of prostate-specific membrane antigen (PSMA) for imaging prostate cancer. J. Med. Chem. 2010, 53, 5333–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S. R.; Pullambhatla M.; Shallal H.; Lisok A.; Mease R. C.; Pomper M. G. A Modular Strategy to Prepare Multivalent Inhibitors of Prostate-Specific Membrane Antigen (PSMA). Oncotarget 2011, 2, 1244–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S. R.; Pullambhatla M.; Byun Y.; Nimmagadda S.; Foss C. A.; Green G.; Fox J. J.; Lupold S. E.; Mease R. C.; Pomper M. G. Sequential SPECT and optical imaging of experimental models of prostate cancer with a dual modality inhibitor of the prostate-specific membrane antigen. Angew. Chem. 2011, 50, 9167–9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Penet M. F.; Nimmagadda S.; Li C.; Banerjee S. R.; Winnard P. T. Jr.; Artemov D.; Glunde K.; Pomper M. G.; Bhujwalla Z. M. PSMA-targeted theranostic nanoplex for prostate cancer therapy. ACS Nano 2012, 6, 7752–7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao G.; Kumar A.; Dobin T.; Oz O. K.; Hsieh J. T.; Sun X. A multivalent approach of imaging probe design to overcome an endogenous anion binding competition for noninvasive assessment of prostate specific membrane antigen. Mol. Pharmacol. 2013, 10, 2975–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S. R.; Pullambhatla M.; Byun Y.; Nimmagadda S.; Baidoo K. E.; Brechbiel M. W.; Mease R. C.; Pomper M. G.. Preclinical Evaluation of 86Y-Labeled Inhibitors of Prostate Specific Membrane Antigen. J. Labelled Compd. Radiopharm. 2011, 54, Suppl S1, p S65.
- Holland J. P.; Divilov V.; Bander N. H.; Smith-Jones P. M.; Larson S. M.; Lewis J. S. 89Zr–DFO–J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J. Nucl. Med. 2010, 51, 1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola-Villegas N.; Evans H.; Bartlett D.; Wu A.; Lewis J. Preclinical development of Zr-89 labeled anti-PSMA minibody and cys-diabody. J Nucl. Med. Meet. Abstr. 2012, 53, 347. [Google Scholar]
- Ray Banerjee S.; Pullambhatla M.; Foss C. A.; Falk A.; Byun Y.; Nimmagadda S.; Mease R. C.; Pomper M. G. Effect of chelators on the pharmacokinetics of (99m)Tc-labeled imaging agents for the prostate-specific membrane antigen (PSMA). J. Med. Chem. 2013, 56, 6108–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadas T. J.; Wong E. H.; Weisman G. R.; Anderson C. J. Copper chelation chemistry and its role in copper radiopharmaceuticals. Curr. Pharm. Des. 2007, 13, 3–16. [DOI] [PubMed] [Google Scholar]
- Connett J. M.; Anderson C. J.; Guo L. W.; Schwarz S. W.; Zinn K. R.; Rogers B. E.; Siegel B. A.; Philpott G. W.; Welch M. J. Radioimmunotherapy with a 64Cu-labeled monoclonal antibody: a comparison with 67Cu. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 6814–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower P. J.; Lewis J. S.; Zweit J. Copper radionuclides and radiopharmaceuticals in nuclear medicine. Nucl. Med. Biol. 1996, 23, 957–980. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Parry J. J.; Laforest R.; Rogers B. E.; Anderson C. J. The role of p53 in combination radioimmunotherapy with 64Cu–DOTA–cetuximab and cisplatin in a mouse model of colorectal cancer. J. Nucl. Med. 2013, 54, 1621–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly P. S. The role of coordination chemistry in the development of copper and rhenium radiopharmaceuticals. Dalton Trans. 2011, 40, 999–1010. [DOI] [PubMed] [Google Scholar]
- Smith S. V. Molecular imaging with copper-64. J. Inorg. Biochem. 2004, 98, 1874–1901. [DOI] [PubMed] [Google Scholar]
- Wadas T. J.; Wong E. H.; Weisman G. R.; Anderson C. J. Molecular imaging of cancer with copper-64 radiopharmaceuticals and positron emission tomography (PET). Chem. Rev. 2010, 110, 2858–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokeen M.; Anderson C. J. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Acc. Chem. Res. 2009, 110, 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva R. A.; Peyre K.; Pullambhatla M.; Fox J. J.; Pomper M. G.; Nimmagadda S. Imaging CXCR4 expression in human cancer xenografts: evaluation of monocyclam 64Cu–AMD3465. J. Nucl. Med. 2011, 52, 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Schroeder T.; Bowsher J. E.; Hedlund L. W.; Wong T.; Dewhirst M. W. Intertumoral differences in hypoxia selectivity of the PET imaging agent 64Cu(II)-diacetyl-bis(N4-methylthiosemicarbazone). J. Nucl. Med. 2006, 47, 989–998. [PubMed] [Google Scholar]
- Liu Z.; Li Z. B.; Cao Q.; Liu S.; Wang F.; Chen X. Small-animal PET of tumors with (64)Cu-labeled RGD–bombesin heterodimer. J. Nucl. Med. 2009, 50, 1168–1177. [DOI] [PubMed] [Google Scholar]
- Dumont R. A.; Deininger F.; Haubner R.; Maecke H. R.; Weber W. A.; Fani M. Novel (64)Cu- and (68)Ga-labeled RGD conjugates show improved PET imaging of alpha(nu)beta(3) integrin expression and facile radiosynthesis. J. Nucl. Med. 2011, 52, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Fani M.; Del Pozzo L.; Abiraj K.; Mansi R.; Tamma M. L.; Cescato R.; Waser B.; Weber W. A.; Reubi J. C.; Maecke H. R. PET of somatostatin receptor-positive tumors using 64Cu– and 68Ga–somatostatin antagonists: the chelate makes the difference. J. Nucl. Med. 2011, 52, 1110–1118. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Li Z. B.; Cao Q.; Liu S.; Wang F.; Chen X.; Small-animal P. E. T. of tumors with (64)Cu-labeled RGD–bombesin heterodimer. J. Nucl. Med. 2009, 50, 1168–1177. [DOI] [PubMed] [Google Scholar]
- Rockey W. M.; Huang L.; Kloepping K. C.; Baumhover N. J.; Giangrande P. H.; Schultz M. K. Synthesis and radiolabeling of chelator–RNA aptamer bioconjugates with copper-64 for targeted molecular imaging. Bioorg. Med. Chem. 2011, 19, 4080–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou G. L.; Li Y. H.; Zhang Z. L.; Xiong Y. H.; Chen X. F.; Yao K.; Liu Z. W.; Han H.; Qin Z. K.; Zhou F. J. A modified technique for neourethral anastomosis in orthotopic neobladder reconstruction. Urology 2009, 74, 1145–1149. [DOI] [PubMed] [Google Scholar]
- Cooper M. S.; Ma M. T.; Sunassee K.; Shaw K. P.; Williams J. D.; Paul R. L.; Donnelly P. S.; Blower P. J. Comparison of (64)Cu-complexing bifunctional chelators for radioimmunoconjugation: labeling efficiency, specific activity, and in vitro/in vivo stability. Bioconjugate Chem. 2012, 23, 1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moi M. K.; Meares C. F.; McCall M. J.; Cole W. C.; DeNardo S. J. Copper chelates as probes of biological systems: stable copper complexes with a macrocyclic bifunctional chelating agent. Anal. Biochem. 1985, 148, 249–253. [DOI] [PubMed] [Google Scholar]
- Jones-Wilson T. M.; Deal K. A.; Anderson C. J.; McCarthy D. W.; Kovacs Z.; Motekaitis R. J.; Sherry A. D.; Martell A. E.; Welch M. J. The in vivo behavior of copper-64-labeled azamacrocyclic complexes. Nucl. Med. Biol. 1998, 25, 523–530. [DOI] [PubMed] [Google Scholar]
- Garrison J. C.; Rold T. L.; Sieckman G. L.; Figueroa S. D.; Volkert W. A.; Jurisson S. S.; Hoffman T. J. In vivo evaluation and small-animal PET/CT of a prostate cancer mouse model using 64Cu bombesin analogs: side-by-side comparison of the CB-TE2A and DOTA chelation systems. J. Nucl. Med. 2007, 48, 1327–1337. [DOI] [PubMed] [Google Scholar]
- Sun X.; Wuest M.; Weisman G. R.; Wong E. H.; Reed D. P.; Boswell C. A.; Motekaitis R.; Martell A. E.; Welch M. J.; Anderson C. J. Radiolabeling and in vivo behavior of copper-64-labeled cross-bridged cyclam ligands. J. Med. Chem. 2002, 45, 469–477. [DOI] [PubMed] [Google Scholar]
- Boswell C. A.; Sun X.; Niu W.; Weisman G. R.; Wong E. H.; Rheingold A. L.; Anderson C. J. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J. Med. Chem. 2004, 47, 1465–1474. [DOI] [PubMed] [Google Scholar]
- Sprague J. E.; Peng Y.; Sun X.; Weisman G. R.; Wong E. H.; Achilefu S.; Anderson C. J. Preparation and biological evaluation of copper-64-labeled tyr3-octreotate using a cross-bridged macrocyclic chelator. Clin. Cancer Res. 2004, 10, 8674–8682. [DOI] [PubMed] [Google Scholar]
- Ait-Mohand S.; Fournier P.; Dumulon-Perreault V.; Kiefer G. E.; Jurek P.; Ferreira C. L.; Benard F.; Guerin B. Evaluation of 64Cu-labeled bifunctional chelate–bombesin conjugates. Bioconjugate Chem. 2011, 22, 1729–1735. [DOI] [PubMed] [Google Scholar]
- Ferreira C. L.; Lamsa E.; Woods M.; Duan Y.; Fernando P.; Bensimon C.; Kordos M.; Guenther K.; Jurek P.; Kiefer G. E. Evaluation of Bifunctional Chelates for the Development of Gallium-Based Radiopharmaceuticals. Bioconjugate Chem. 2010, 21, 531–536. [DOI] [PubMed] [Google Scholar]
- Ferreira C. L.; Yapp D. T.; Lamsa E.; Gleave M.; Bensimon C.; Jurek P.; Kiefer G. E. Evaluation of novel bifunctional chelates for the development of Cu-64-based radiopharmaceuticals. Nucl. Med. Biol. 2008, 35, 875–882. [DOI] [PubMed] [Google Scholar]
- Olszewski R. T.; Bukhari N.; Zhou J.; Kozikowski A. P.; Wroblewski J. T.; Shamimi-Noori S.; Wroblewska B.; Bzdega T.; Vicini S.; Barton F. B.; Neale J. H. NAAG peptidase inhibition reduces locomotor activity and some stereotypes in the PCP model of schizophrenia via group II mGluR. J. Neurochem. 2004, 89, 876–885. [DOI] [PubMed] [Google Scholar]
- Silver D. A.; Pellicer I.; Fair W. R.; Heston W. D.; Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–95. [PubMed] [Google Scholar]
- Prasanphanich A. F.; Nanda P. K.; Rold T. L.; Ma L.; Lewis M. R.; Garrison J. C.; Hoffman T. J.; Sieckman G. L.; Figueroa S. D.; Smith C. J. [64Cu-NOTA-8-Aoc-BBN(7–14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12462–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Pullambhatla M.; Foss C. A.; Byun Y.; Nimmagadda S.; Senthamizhchelvan S.; Sgouros G.; Mease R. C.; Pomper M. G. 2-(3-{1-Carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pen tanedioic acid, [18F]DCFPyL, a PSMA-based PET imaging agent for prostate cancer. Clin. Cancer Res. 2011, 17, 7645–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier S. M.; Maresca K. P.; Lu G.; Merkin R. D.; Marquis J. C.; Zimmerman C. N.; Eckelman W. C.; Joyal J. L.; Babich J. W. 99mTc-Labeled Small-Molecule Inhibitors of Prostate-Specific Membrane Antigen for Molecular Imaging of Prostate Cancer. J. Nucl. Med. 2013, 54, 1369–1376. [DOI] [PubMed] [Google Scholar]
- Elsasser-Beile U.; Reischl G.; Wiehr S.; Buhler P.; Wolf P.; Alt K.; Shively J.; Judenhofer M. S.; Machulla H. J.; Pichler B. J. PET imaging of prostate cancer xenografts with a highly specific antibody against the prostate-specific membrane antigen. J. Nucl. Med. 2009, 50, 606–611. [DOI] [PubMed] [Google Scholar]
- Alt K.; Wiehr S.; Ehrlichmann W.; Reischl G.; Wolf P.; Pichler B. J.; Elsasser-Beile U.; Buhler P. High-resolution animal PET imaging of prostate cancer xenografts with three different 64Cu-labeled antibodies against native cell-adherent PSMA. Prostate 2010, 70, 1413–1421. [DOI] [PubMed] [Google Scholar]
- Fischer G.; Seibold U.; Schirrmacher R.; Wangler B.; Wangler C. (89)Zr, a radiometal nuclide with high potential for molecular imaging with PET: chemistry, applications and remaining challenges. Molecules 2013, 18, 6469–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.