

Abstract
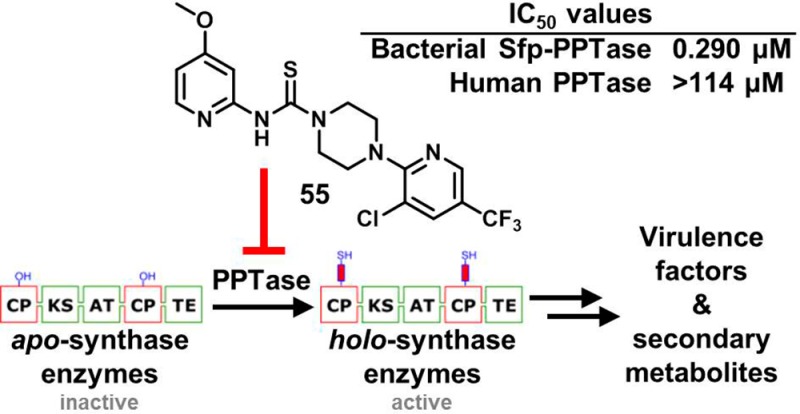
4′-Phosphopantetheinyl transferases (PPTases) catalyze a post-translational modification essential to bacterial cell viability and virulence. We present the discovery and medicinal chemistry optimization of 2-pyridinyl-N-(4-aryl)piperazine-1-carbothioamides, which exhibit submicromolar inhibition of bacterial Sfp-PPTase with no activity toward the human orthologue. Moreover, compounds within this class possess antibacterial activity in the absence of a rapid cytotoxic response in human cells. An advanced analogue of this series, ML267 (55), was found to attenuate production of an Sfp-PPTase-dependent metabolite when applied to Bacillus subtilis at sublethal doses. Additional testing revealed antibacterial activity against methicillin-resistant Staphylococcus aureus, and chemical genetic studies implicated efflux as a mechanism for resistance in Escherichia coli. Additionally, we highlight the in vitro absorption, distribution, metabolism, and excretion and in vivo pharmacokinetic profiles of compound 55 to further demonstrate the potential utility of this small-molecule inhibitor.
Introduction
Despite the rapidity with which genomic science has enabled the identification of genes essential to bacterial homeostasis, the translation of these targets into pharmacological agents has proven to be much more difficult than anticipated.1−3 Post-translational modification (PTM) pathways represent a unique subset of these targets since such events are scarce in prokaryotes relative to their frequency of occurrence in eukaryotes. In metabolism, bacterial PTMs involve the attachment of prostheses that impart a chemical functionality distinct from that of the 20 proteogenic amino acids. Thus, effector enzyme function is wholly dependent upon the installation of these groups. One such PTM is performed by 4′-phosphopantetheinyl transferase (PPTase) enzymes (Figure 1A), which install 4′-phosphopantetheinyl (4′-PP) arms to carrier protein domains of synthase enzymes (Figure 1B).4 This functionality serves to store and channel reactive intermediates that would otherwise be lost to the cellular milieu by diffusion.
Figure 1.
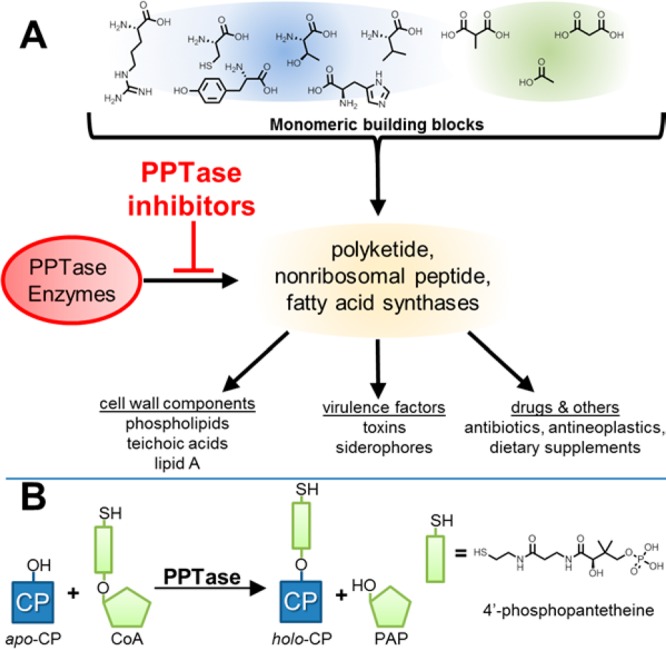
PPTase enzymes in bacterial metabolism. (A) Bacteria utilize synthase enzymatic pathways to assemble small carboxylic and amino acid monomers into complex polymeric molecules, including membrane components, virulence factors, and medicinally important compounds. A key feature of these synthases is the presence of a post-translational appendage to which the nascent polymer is tethered during extension and elaboration; the attachment of this appendage is catalyzed by PPTase enzymes. (B) PPTase enzymes utilize CoA as a 4′-phosphopantetheinyl group donor and install the functionality to conserved serine residues within apo-CP domains of synthase enzymes, generating biochemically active holo-CP-containing synthases and the nucleotide PAP as products. PPTase = phosphopantetheinyl transferase, CP = carrier protein, and PAP = 3′-phosphoadenosine 5′-monophosphate.
Pathways activated by PPTases include microbial fatty acid synthase (FAS), a multidomain enzyme complex that has received considerable attention for both its orthogonal structural characteristics relative to the mammalian FAS system and its novelty as a drug target.5 Reduction in FAS anabolic capacity is anticipated to exert pleotropic effects on cell viability by inhibiting the production of the primary membrane component palmitate and halting the assembly of virulence-determining components of the Gram-negative, Gram-positive, and mycobacterial cell walls (lipid A, lipoteichoic acid, and mycolic acids, respectively). Furthermore, FAS as a drug target has been validated in vivo with chemical probes that act upon β-ketoacyl-ACP synthase.6
Additionally, bacterial secondary metabolism contains polyketide and nonribosomal synthases which require 4′-PP groups from PPTases to produce metabolites needed for bacteria to thrive in environmental and infection settings.7 Representative compounds from these pathways include siderophores that are secreted to scavenge iron8 and the phenolic glycolipids of Mycobacterium spp. that dampen host defense mechanisms.9 In many cases, the capacity to manufacture these compounds has been linked to virulence, and disruption of synthase genes precludes the establishment of infection.10,11 These avirulent phenotypes have been recapitulated in vitro with chemical probes that target synthase enzymes.7,12,13
Since PPTase enzymes represent the gatekeeper of these pathways, their inhibition stands to attenuate bacterial cell viability through direct inactivation of FAS. At the same time, the concomitant disruption of secondary metabolism offers a path to mitigate numerous aspects of pathogenicity. These hypotheses have been confirmed at the genetic level, where disruption of PPTase genes has been observed as either lethal or severely compromising to the fitness of both Escherichia coli(14)and Mycobacterium tuberculosis.11 Thus, PPTase represents an important enzyme in bacterial metabolism and is well-deserving of further study.
It is noteworthy that, in most cases, bacterial genomes contain two structurally distinct PPTase enzymes, the AcpS- and Sfp-PPTases. Enzymes of the former class are typically responsible for activating FAS, while enzymes of the latter type modify secondary metabolism synthases. Given these associations and the above-mentioned therapeutic potential of targeting PPTases, there have been several reports by academic and industrial researchers describing the development of small-molecule AcpS-PPTase inhibitors, yet no reports of agents targeting Sfp-PPTase (see Figure 2). Chu and co-workers isolated SCH-538415 (Figure 2), a symmetrical and highly planar compound, from an unidentified bacterial extract and reported it to have moderate inhibitory activity toward AcpS.15 Prior to the initiation of our current program, we had found that analogues of anthranilic acid-based leads originating from a Wyeth Research program16 targeting AcpS-PPTase (e.g., UCSD-18ae, Figure 2) possessed no activity toward Sfp-PPTase and had overall weak activity.17 An additional publication from Wyeth Research came out shortly thereafter detailing further medicinal chemistry efforts leading to Wyeth-16 (Figure 2), which was optimized for potency toward AcpS-PPTase (IC50 = 1.4 μM) but had limited antimicrobial activity.18 On the basis of the limited antibacterial activity observed for these AcpS-PPTase-specific compounds, we have put forth a hypothesis that the presence of an Sfp-PPTase in the bacterial genome provides an inborn mechanism of resistance.17 Therefore, it is our presumption that a viable antibacterial agent must simultaneously target both classes of PPTase enzymes to effectively halt bacterial proliferation.
Figure 2.

Previously reported inhibitors of Sfp- and AcpS-PPTase.
Toward this end, we developed a strategy to search for small-molecule inhibitors of Sfp-PPTase.19 These efforts identified SCH-202676 as the first compound reported to have activity against Sfp-PPTase (Figure 2). However, compounds belonging to this class are known covalent modifiers20 and possess promiscuous activity (∼20% hit rate in PubChem assays), which ultimately limits their utility as probe compounds.
Therefore, we conducted a high-throughput screen (HTS) in search of additional small-molecule inhibitors of Sfp-PPTase. Herein we describe the discovery and SAR investigation of the 2-pyridinyl-N-(4-aryl)piperazine-1-carbothioamides as best in class small-molecule inhibitors of Sfp-PPTase. This work led to the development of ML267 (55), a new chemical probe and potent inhibitor of this bacterial enzyme. We demonstrate that 55 possesses the required selectivity, mechanism of action, physicochemical properties, and cellular activities to interrogate the role of PPTases in bacterial metabolism, and we utilize this compound to test hypotheses about the importance of PPTase to the function of whole bacterial cells.
Chemistry
As shown in Scheme 1, compound 1 was readily synthesized by 1,1′-thiocarbonyldiimidazole-assisted coupling of commercially available 4-methylpyridin-2-amine and 1-(3-(trifluoromethyl)phenyl)piperazine at 40 °C. The same procedure was utilized to generate a small library of compounds (Table 1) around the 4-methylpyridine region using differentially substituted pyridylamines, 3-toluidine, and other heterocycles (in Table 1, analogues 1–11 and 19–21).
Scheme 1. General Procedure for the Synthesis of Analogues 1–11 and 19–21.
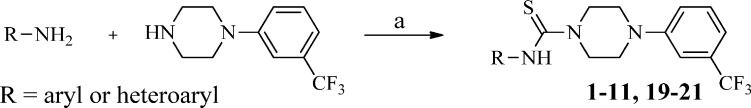
Reagents and conditions: (a) 1,1′-thiocarbonyldiimidazole (TCDI), CH2Cl2, 40 °C, 1 h.
Table 1. Inhibition of B. subtilis Sfp-PPTase by Analogues 1–21a.
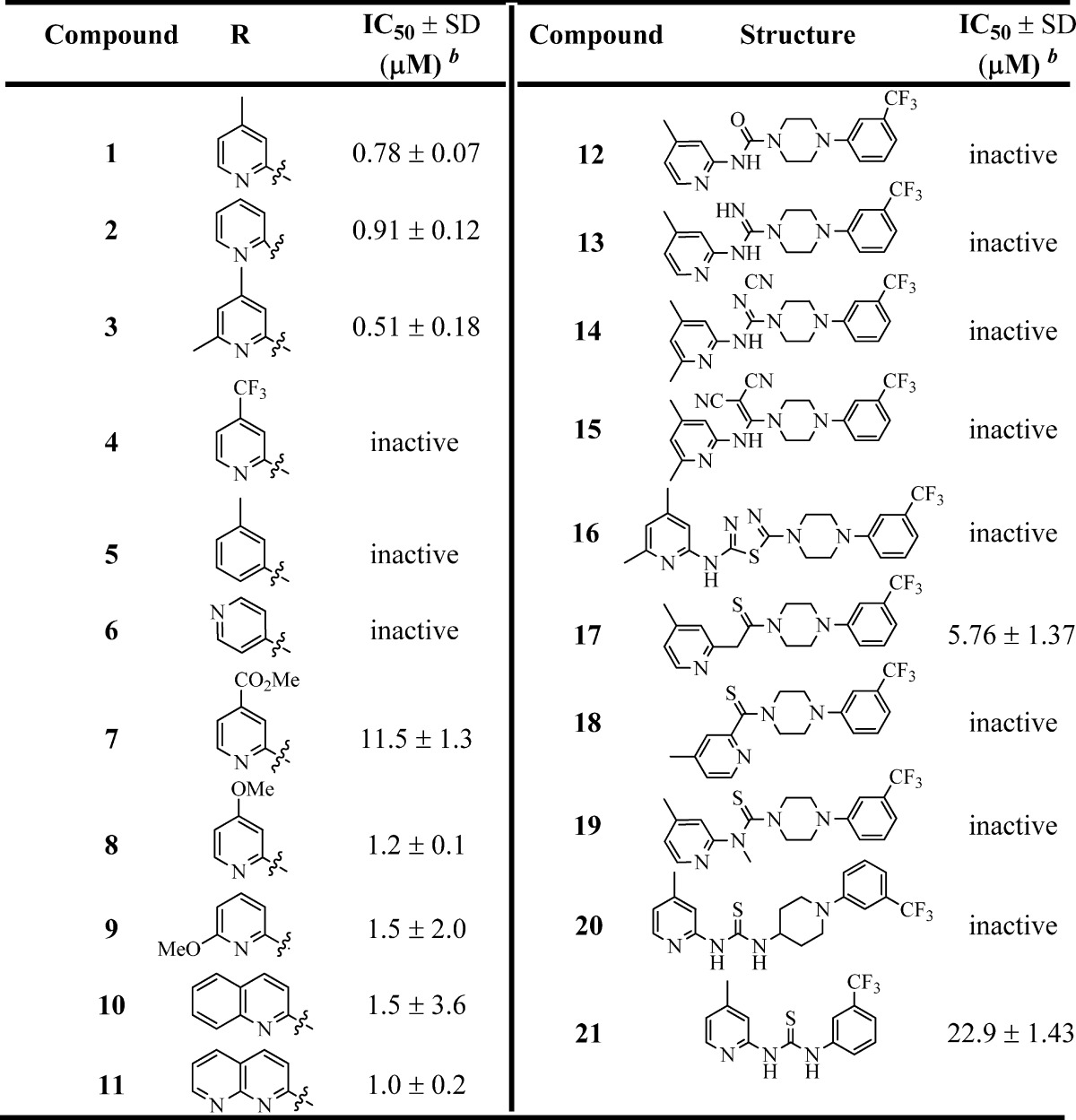
IC50 values represent the half-maximal (50%) inhibitory concentration as determined in the HTS assay, and the experiment was performed in triplicate.
The term “inactive” refers to compounds with IC50 ≥ 114 μM.
Through bioisosteric replacement of the thiourea, analogues 12–17 were synthesized utilizing known protocols reported for similar compounds in the literature as shown in Scheme 2 (see the Supporting Information for details).21−24 Phenoxycarbonyl chloride-assisted coupling of 1-(3-(trifluoromethyl)phenyl)piperazine with 4-picolin-2-amine afforded the urea analogue 12. Analogue 13 was prepared by stirring the analogue 1 with ammonium hydroxide and sodium periodate at 80 °C in a DMF–water solvent mixture. Compounds 14 and 15, which represent the bioisosteric replacement of the thiourea functionality, were prepared by refluxing 1-(3-(trifluoromethyl)phenyl)piperazine and 4-picolin-2-amine with diphenyl N-cyanocarbonimidate (for 14) or 2-[bis(methylthio)methylene]malononitrile (for 15) in acetonitrile. The thiadiazole derivative 16 was prepared utilizing xanthphos-catalyzed amination of the 2-bromo-5-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)-1,3,4-thiadiazole in the presence of Pd2(dba)3 and cesium carbonate. The Kindler reaction of 2-acetyl-4-picoline and 2-formyl-4-picoline with 1-(3-(trifluoromethyl)phenyl)piperazine under microwave (MW) conditions furnished analogues 17 and 18, respectively.
Scheme 2. General Procedure for the Synthesis of Analogues 12–18.

Reagents and conditions: (a) PhOCOCl, DIPEA, DMAP, 0 °C to rt, 3 h; (b) NH4OH, NaIO4, DMF–H2O, 80 °C, 1 h; (c) acetonitrile, reflux, 16 h; (d) acetonitrile, 70 °C, 24 h; (e) xantphos, Pd2(dba)3t-BuONa, toluene, 110 °C, 3 h; (f) S, DMF, MW, 130 °C, 0.5 h.
To explore the SAR around the 3-trifluorophenyl region as shown in Table 2, a robust synthetic method was required to generate a library of arylpiperazine precursors, since many of the targeted compounds were not commercially available. Given the large number of available boronic acids, we found the Chan-Lam reaction25,26 to be an ideal method to generate several mono- and disubstituted arylpiperazine precursors for preparation of analogues 27, 28, 36, and 40. However, the use of a large excess of copper or a constant supply of oxygen gas is necessary to regenerate the copper(II) catalyst, making this method less compatible with parallel library synthesis. To overcome this drawback, we used a modified version of the Chan-Lam reaction reported by Quach et al.27 which requires only 10 mol % copper(II) acetate but still uses an oxygen atmosphere. To facilitate analogue synthesis, we further modified this method by carrying out this reaction in a sealable microwave vial. In this case, all the reactants were mixed together, the vessel was charged with oxygen gas, and then the vessel contents were stirred at 45 °C for 12–24 h as shown in Scheme 3A. Several mono- and disubstituted arylpiperazines were prepared using this method followed by Boc deprotection with trifluoroacetic acid in dichloromethane.
Table 2. Inhibition of B. subtilis Sfp-PPTase by Analogues 22–56a.
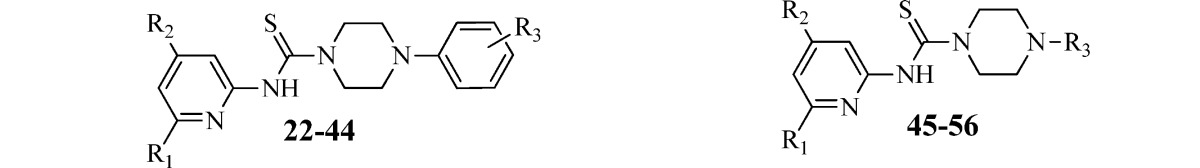
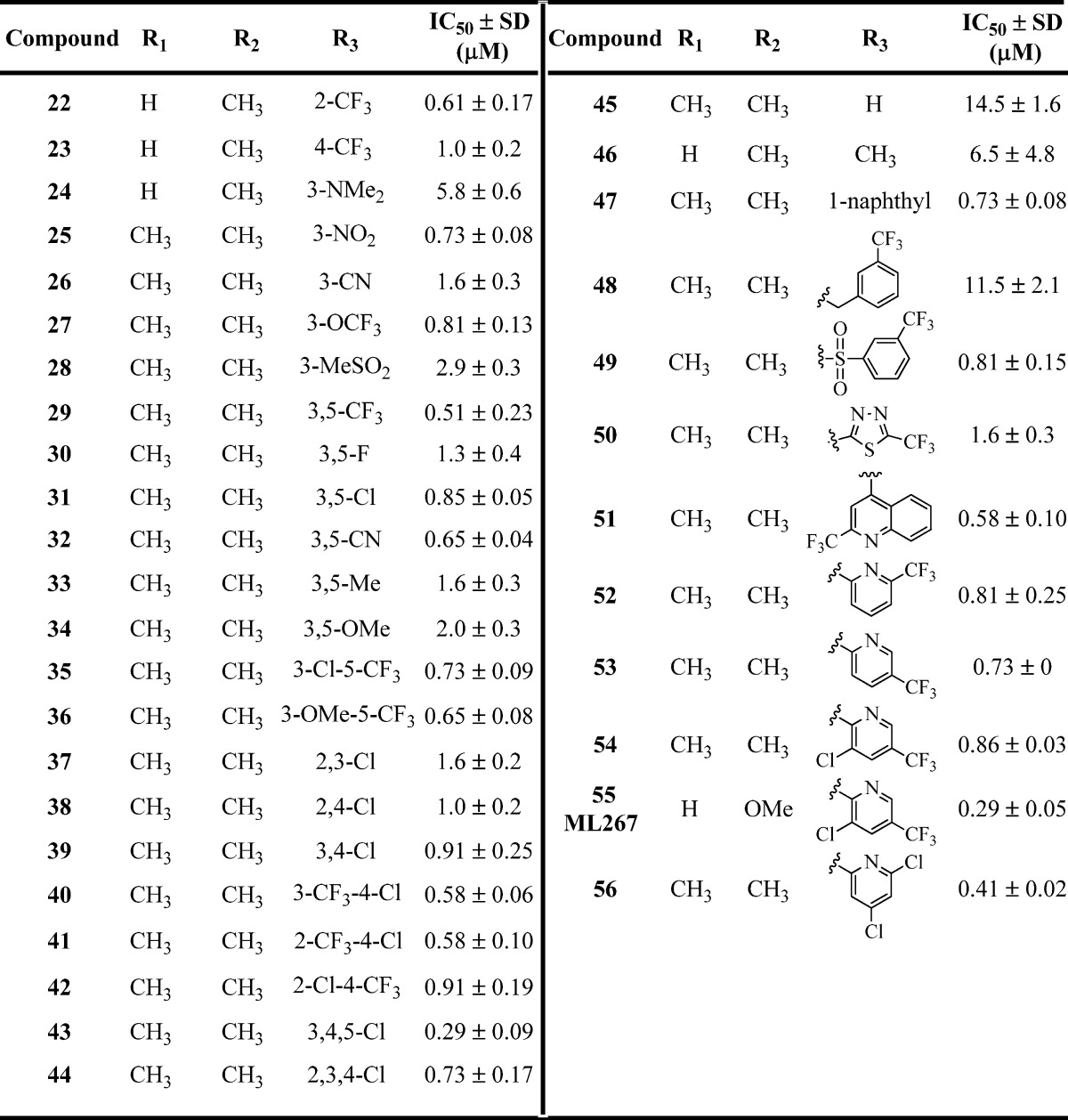
IC50 values represent the half-maximal (50%) inhibitory concentration as determined in the HTS assay, and the experiment was performed in triplicate.
Scheme 3. Synthesis of Requisite Aryl/Heteroarylpiperazines and Analogues 22–58.

Reagents and conditions: (a) Cu(OAc)2 (10 mol %), 4 Å molecular sieves, O2, CH2Cl2, 45 °C, 12–24 h; (b) TFA/CH2Cl2, rt, 1 h; (c) BINAP (10 mol %), Pd(OAc)2 (5 mol %), Cs2CO3 (1.5 equiv), toluene, 110 °C, 12–24 h; (d) JohnPhos (10 mol %), Pd2(dba)3 (5 mol %), t-BuONa (1.5 equiv), toluene, 110 °C, 2–8 h; (e) BINOL (20 mol %), CuBr (20 mol %), K3PO4 (2 equiv), DMF, rt, 12–24 h; (f) 1,1′-thiocarbonyldiimidazole (TCDI), CH2Cl2, 40 °C; (g) DMF, 90 °C, 1 h.
Despite these encouraging results, this method was insufficient for use with several di- and trisubstituted aryl and heterocyclic boronic acids. In these cases, Buchwald–Hartwig amination conditions were utilized. Amination of the requisite aryl or heteroaryl bromides was achieved using BINAP or JohnPhos ligands in combination with Pd(OAc)2 or Pd2(dba)3 with sodium tert-butoxide or cesium carbonate in toluene. The arylation of Boc-piperazine with aryl iodides was accomplished using a BINOL/CuBr catalytic system in the presence of potassium phosphate in DMF at room temperature (Scheme 3B). Subsequent Boc deprotection with trifluoroacetic acid (TFA) in dichloromethane gave the desired free piperazinamines (see the Supporting Information for details). As shown in Scheme 3C, the commercially available arylpiperazines and compounds obtained from the above methods were treated with various 4,6-substituted pyridin-2-amines in the presence of 1,1′-thiocarbonyldiimidazole to furnish analogues 22–56 (Table 2). To explore the SAR of the piperazine core itself, analogues 59–67 were synthesized using 1,1′-thiocarbonyldiimidazole-assisted thiourea synthesis (Table 3). The acyclic analogues 57 and 58 were prepared by condensation of the 4-picoline-2-isothiocyanate with corresponding acyclic amines as shown in Scheme 3D.
Table 3. Inhibition of B. subtilis Sfp-PPTase by Analogues 57–67a.
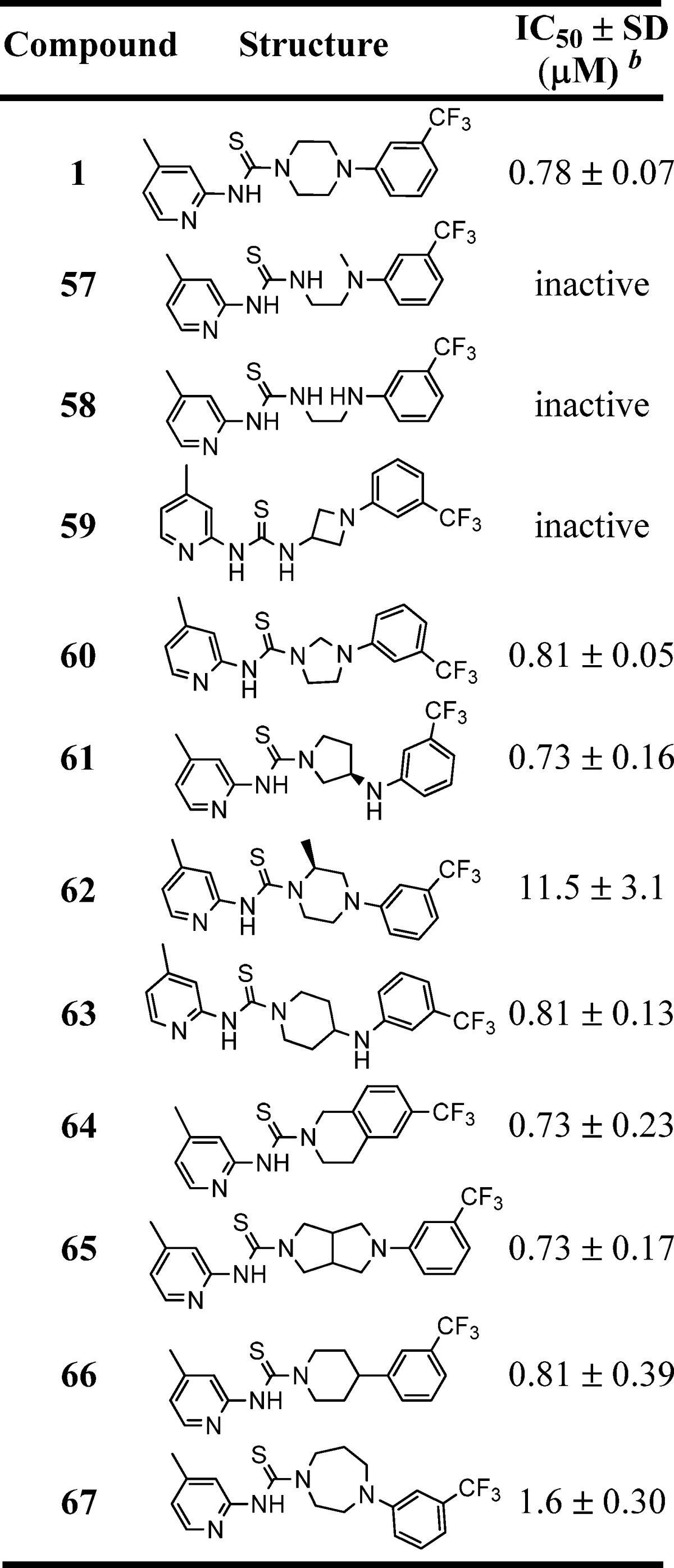
IC50 values represent the half-maximal (50%) inhibitory concentration as determined in the HTS assay, and the experiment was performed in triplicate.
The term “inactive” refers to compounds with IC50 ≥ 114 μM.
Specifically, the synthesis of analogues 59–63, 65, and 67 was accomplished by arylation28 of the requisite Boc-protected amine cores with 3-trifluorophenyl iodide utilizing the 2-isobutyrylcyclohexanone/CuI system (Scheme 4), followed by 1,1′-thiocarbonyldiimidazole-assisted coupling with 4-methylpyridin-2-amine. Analogues 64 and 66 were synthesized utilizing the general procedure outlined in Scheme 3 using commercially available precursors 6-(trifluoromethyl)-1,2,3,4-tetrahydroisoquinoline and 4-(3-(trifluoromethyl)phenyl)piperidine, respectively.
Scheme 4. Synthesis of Requisite Aryl/Heteroarylpiperazines and Analogues 59–63, 65, and 67.

Reagents and conditions: (a) 2-isobutyrylcyclohexanone, Cs2CO3, CuI, DMF, 70 °C, 2–10 h; (b) TFA, DCM, rt, 1 h; (c) 1,1′-thiocarbonyldiimidazole (TCDI), CH2Cl2, 40 °C.
Results and Discussion
Discovery of N-(4-Methylpyridin-2-yl)-4-(3-(trifluoromethyl)phenyl)piperazine-1-carbothioamide (1) as an Inhibitor of Sfp-PPTase
We profiled the Molecular Libraries Small Molecule Repository (at the time totaling 311 260 compounds) for PPTase inhibitors with a previously described activity assay for Sfp-PPTase.19 The highly miniaturized format enabled the experiment to be conducted at eight concentrations ranging from 3.6 nM to 114 μM following the quantitative HTS methodology.29 Data generated by this experiment are deposited in the public database PubChem under assay identifier number (AID) 1490. Data analysis and chemoinformatic filtering identified 388 structurally diverse compounds that were selected for follow-up in a battery of assays. This characterization began with retest of freshly prepared serial dilutions of hit compounds in the Sfp-PPTase screening assay (AID 2701) and included further activity profiling with AcpS-PPTase (AID 602360). In addition, this set of compounds was evaluated with Sfp-PPTase in a gel-based phosphopantetheinylation assay to confirm biochemical inhibition with an orthogonal detection format (AID 2707).19 Finally, since prior work has established the essential nature of PPTases to bacterial viability,30−32 we profiled these compounds for antibacterial activity with Bacillus subtilis HM489, whose viability depends solely on Sfp (AID 602366).32 The sum of these experiments identified 1 (Figure S1A, Supporting Information) as a confirmed screening hit with inhibitory activity against both AcpS- and Sfp-PPTases (Figure S1B) and modest antibacterial activity against B. subtilis. We subsequently confirmed the identity and integrity of the chemical matter by resynthesis and reaffirmed the above findings for the resynthesized sample.
Compound 1 is a Reversible, Noncompetitive Inhibitor of Sfp-PPTase
To further characterize the biochemical activity of 1, we first addressed the use of fluorescent substrates in our primary and confirmatory assays. As recently highlighted, the potential exists that the observed inhibition could be a result of compound–label interaction that leads to an artifactual mechanism of inhibition.33−35 To this end, we developed and implemented a label-free biochemical assay that assessed PPTase activity on the basis of differences in the electrophoretic mobility of the apo and holo states of the carrier protein under native PAGE conditions. These experiments revealed that 1 elicited inhibition through a mechanism independent of the fluorescent label, with a dose-dependent inhibition being observed in the presence of the natural CoA and acyl carrier protein (ACP) substrates (Figure S1E,F, Supporting Information).
To better understand the mechanism through which 1 inhibits the target, we evaluated the effects that a varying substrate concentration had on potency. In these experiments, which utilized an HPLC assay and operated under an initial-rates regimen, 1 exhibited a near constant IC50 of 300 nM when CoA or apo-ACP acceptor substrates were increased to concentrations 50- and 100-fold above their Km values, respectively (Figure S1C,D, Supporting Information). This trend is consistent with a noncompetitive mechanism of inhibition36 and indicates that the compound binds to the free enzyme, enzyme–substrate complex, or ternary complex with similar affinities at a location other than the active site: a form of allosteric modulation.
Finally, we probed the reversibility of inhibition with a rescue-by-dilution experiment that assessed the recovery of enzymatic activity after a large (100-fold) dilution from conditions producing 90% inhibition of enzymatic activity.37 These experiments included control compounds PAP and SCH-202676, the latter of which was discovered during assay optimization as an active compound from LOPAC1280.19 Inhibition by the former proved to be reversible, as anticipated by the fact that it is one of the reaction products (Figure S2A, Supporting Information), while the latter was found be an irreversible inhibitor, a result that is consistent with other reports of the compound’s reactivity (Figure S2B).20 Parallel evaluation of our primary hit 1 (Figure S1G, Supporting Information) in the same manner revealed completely reversible inhibition of Sfp, with enzymatic activity restored upon dilution of the enzyme–1 solution in a compound-free buffer (orange data symbols, Figure S1G), relative to a buffer containing a constant concentration of the compound (green data symbols, Figure S1G).
Structure–Activity Relationship Studies of 1
After confirming the activity of 1, we planned systematic structural modifications to the various regions of the molecule in an effort to establish tractable SAR. Our first area of exploration was the western 2-aminopyridine moiety as shown in Table 1. Removal of the 4-Me group (2) resulted in only a slight drop in potency, whereas addition of electron-withdrawing groups such as 4-CF3 (4) and 4-C(O)OMe (7) resulted in a significant loss of activity. Moreover, removal of the pyridine nitrogen (5) or changing the position of the pyridine (6) also led to inactive compounds. Thus, it appears that the pyridine nitrogen needs to be adjacent to the thiourea motif to maintain Sfp-PPTase inhibitory activity. Conversely, modification with electron-donating groups, such as addition of an extra methyl group adjacent to the nitrogen (3), 4-OMe (8), and other heterocycles (10 and 11), led to compounds with activity comparable to that of 1.
With initial SAR explorations of the western region of the molecule complete, we then focused our attention on the thiourea moiety. As shown in Table 1, replacement of the thiourea was not well tolerated with the urea (12), guanidine (13), and thiadiazole (16) all being inactive. Attempts to replace the thiourea structural motif with bioisosteres such as cyanoguanidine (14) or a 1,1-dicyanoketene group (15) also led to complete loss of activity. Methylation of the nitrogen (19) or changing the nitrogen from being a part of a ring to being exocyclic (20) resulted in loss of potency. Interestingly, replacement of the 2-amino group with a methylene spacer (17) retained some activity toward Sfp-PPTase albeit much reduced, with an IC50 value of 5.8 μM. As such, these data suggested that the thiourea moiety was clearly the most optimal group for the desired activity. However, the thiourea functionality is often considered an undesirable structural feature, primarily due to its potential in forming toxic metabolites via oxidation to the S-oxide or sulfinic acid (typically P450 or FMO-catalyzed), followed by GSH-trapping. These covalent GSH adducts could ultimately lead to oxidative-stress-induced cell death.38 Therefore, upon completion of the SAR efforts, we planned to investigate the propensity of this undesired reaction to occur with our lead compound(s) (vide infra).
Given that the 4,6-dimethyl substitution in 3 displayed potency comparable to that of the 4-methylpyridine moiety of 1, we decided to further investigate both pyridine derivatives when exploring the SAR of the eastern phenyl region. In these efforts, a large number of analogues were synthesized, and representative compounds are shown in Table 2. Overall, substitution of the pendant phenyl group was very well tolerated, with most of the analogues 22–44 having comparable activity. The general trend suggested that electron-withdrawing groups are preferred, with most of these analogues exhibiting submicromolar potency. The analogues which showed the most potent activity included 29 (R = 3,5-CF3), 40 (R = 3-CF3-4-Cl), 41 (R = 2-CF3-4-Cl), and 43 (R = 3,4,5-Cl). In contrast, compounds in which electron-donating substituents were incorporated (e.g., 24 (R = 3-NMe2) and 34 (R = 3,5-OMe)) had weaker potency with IC50 values of 5.8 and 2.0 μM, respectively. These data suggest that this region of the molecule may provide a handle for modulating the ADME properties (e.g., solubility) in future SAR efforts given the general tolerance for structural modifications. With analogues 45–56 (Table 2), we aimed to investigate more significant structural changes to this region, including removal of the aryl group (45) or replacing it with smaller groups such as a methyl group (46), and both changes significantly diminished the potency (IC50 = 14.5 and 6.5 μM respectively). Addition of a methylene spacer between the aryl group and piperazine (48) was not well tolerated (IC50 = 11.5 μM). However, incorporation of a sulfonamide moiety (49) resulted in potency comparable to that of 1. Replacement of the aryl group with a thiadiazole (50) resulted in a ∼2-fold drop in potency.
In an effort to improve the overall solubility of the molecule, various nitrogen-containing heterocyles were synthesized (compounds 51–56). Gratifyingly, this change was tolerated and led to the discovery of analogue 55, which was ultimately determined to be our probe molecule (ML267) after all compound attributes such as potency, in vitro ADME properties, and antibacterial activity were taken into consideration (vide infra). Notably, compound 55 possesses a 4-OMe group in contrast to the 4,6-dimethyl derivative, which led to a 2–3-fold improvement in potency (IC50 = 0.81 μM (54) vs IC50 = 0.29 μM (55)) but also contributed to improvement in other attributes mentioned above. We had synthesized and tested a small set of analogues containing the 4-methoxypyridine combined with the best arylpiperazines in the right-hand side of the molecule. These analogues showed similar or less potency compared to compound 55 and exhibited a consistent SAR trend (data not shown).
Our final area of SAR exploration was modification of the piperazine linker, as shown in Table 3. A literature search uncovered Troviridine [1-(5-bromopyridin-2-yl)-3-(2-(pyridin-2-yl)ethyl)thiourea] (also known as LY300045-HCl), a thiourea-containing small molecule that was originally developed by Eli Lilly as a non-nucleoside reverse transcriptase inhibitor but later was found to have antimicrobial properties.39 Similar to our lead scaffold, Troviridine contains a 2-aminopyridine attached to the thiourea moiety; however, instead of a piperazine, it contains an acyclic linker. Therefore, we replaced the piperazine core with two acyclic linkers (57 and 58); however, these compounds lost all activity. Despite this, we were encouraged that a compound with a structure comparable to that of our lead had advanced into human trials. Moreover, the inactivity of “Troviridine-like” analogues 57 and 58 assured us that our compound has a divergent mechanisms of action, particularly in reference to the antimicrobial activity. Additional SAR explorations of the piperazine motif included varying the ring size to smaller, larger, and fused ring systems. The smaller rings showed similar activity (60), whereas larger rings (67) or the substituted piperazine (62) had reduced potency. The fused pyrrolidine motif (65) had comparable potency (IC50 = 0.73 μM), but the solubility was markedly reduced. Interestingly, when the nitrogen linked to thiourea is exocyclic (e.g., 59), all activity is lost, whereas the piperazine nitrogen attached to an aryl ring is not essential (e.g., 66, IC50 = 0.81 μM).
Initial SAR explorations around compound 1 led to a thorough understanding of the structural features that are required for activity. Thus, we then sought to more thoroughly characterize the best Sfp-PPTase inhibitors for their activity against AcpS-PPTase and antibacterial activity. Additionally, we profiled this panel for activity with the human PPTase, an important antitarget, and cytotoxicity with human HepG2 cells (Table 4). As mentioned above, our primary target of interest was Sfp-PPTase, yet activity against AcpS-PPTase is desirable for maximal antimicrobial activity. As shown in Table 4, most compounds display activity toward AcpS-PPTase, but the IC50 values were generally 5–10-fold less potent. To assess acute cytotoxicity, we chose to profile the top compounds using HepG2 (hepatocellular carcinoma) cells since hepatotoxicity has been reported for some thiourea-containing compounds, and this profiling not only reveals potential toxicity of the parent compound, but also its metabolites. While this method is certainly not to be considered a thorough investigation of toxicity, it provided us a means to quickly profile numerous compounds. Notably, despite the generally favorable activity of the original compound 1, it demonstrated moderate toxicity toward this cell line, whereas most other compounds were inactive. Finally, as part of our initial biological activity profile, we sought to further characterize the compounds for antibacterial activity using the B. subtilis HM489 strain.32 This strain contains sfp as the only locus encoding a functional PPTase gene product, making the allele essential to viability of this organism. These experiments revealed that we had modest inhibitors of bacterial growth that generally tracked with SAR in the biochemical assay. This important finding is exemplified by urea derivative 12, which was inactive against Sfp/AcpS-PPTase and lacked activity in the antibacterial assays (Table 4). While the antibacterial activity of these compounds was modest in these high-throughput assays, in subsequent antibacterial studies using more traditional methods of minimum inhibitory concentration (MIC) determination, we found the compounds to be generally more potent (vide infra). After careful analysis of the data in Table 4, and profiling of select compounds for their in vitro ADME properties, 55 emerged as the compound with the best balance of properties. In these profiling studies, 55 demonstrated dual activity toward the bacterial Sfp- and AcpS-PPTase targets (Figure S4, Supporting Information), presenting IC50 values of 290 nM and 8.1 μM, respectively. Furthermore, we profiled top compounds for activity with the human PPTase, an important antitarget. While we observed inhibition of this enzyme with PAP and SCH202676, 55 exhibited no inhibition at concentrations up to 125 μM (Figure S4B, lower panel). Comparison of these data to the inhibition observed in the same biochemical assay for Sfp-PPTase (Figure S4B, upper panel) indicated the selectivity index, the ratio of inhibition observed for the bacterial target relative to the human enzyme, to be greater than 500-fold.
Table 4. Biological Activity Profile of Select Compoundsa.
| compd | Sfp-PPTase IC50 (μM) | AcpS-PPTase IC50 (μM) | cytotoxicity (HepG2), IC50 (μM) | B. subtilis (HM489) IC50 (μM) |
|---|---|---|---|---|
| 1 | 0.78 | 4.6 | 14 | 9.8 |
| 55 | 0.29 | 8.1 | inactive | 9.3 |
| 12 | inactive | inactive | 7.0 | inactive |
| 22 | 0.41 | 10.2 | inactive | 9.4 |
| 41 | 0.58 | 10.2 | inactive | 18.6 |
| 53 | 0.73 | 16.2 | 15.8 | 13.2 |
| 54 | 0.86 | 18.6 | 30 | 36.2 |
| 63 | 0.81 | 12.9 | inactive | 13.2 |
Sfp-PPTase and AcpS-PPTase IC50 values were determined using the HTS assay. Compound toxicity toward HepG2 cells was assessed by measuring the cellular ATP content using a luciferase-coupled ATP quantitation assay (CellTiter-Glo, Promega Corp., Madison, WI). Antibacterial activity against B. subtilis (HM489) was accessed by measuring the cellular ATP content using a luciferase-coupled ATP quantitation assay (Bac-Titer Glo, Promega Corp.).
Compound 55 Possesses Specific Gram-Positive-Targeted Bactericidal Activity
To evaluate the antibacterial spectrum of activity possessed by 55, we assembled a panel of microorganisms which included B. subtilis strains HM489, 168, and OKB105, which possess all possible PPTase genotype combinations sfp+/acpS–, sfp–/acpS+, and sfp+/acpS+, respectively, in a strain 168 isogenic background, as well as E. coli K12 and the laboratory strain BW25113. With respect to human pathogens, the panel contained Pseudomonas aeruginosa ATCC9028, a Gram-negative organism of clinical importance, as well as three strains of methicillin-sensitive and community-acquired methicillin-resistant Staphylococcus aureus (ATCC6538, USA300, and USA500). Finally, the panel also contained fluconazole-sensitive and -resistant strains of Candida albicans as archetypical fungal pathogens (ATCC90028 and ATCC96901, respectively). Compound 55 displayed a spectrum of activity that targets Gram-positive organisms, where, in addition to inhibiting the growth of B. subtilis at a concentration of 1.7 μg/mL, it also thwarted the growth of both methicillin-sensitive and -resistant strains of S. aureus in a similar concentration range [Figure 3, where the growth indicator resazurin (purple) is reduced to resorufin (pink) by metabolically active microorganisms].’ No growth inhibition was observed for the Gram-negative organisms E. coli and P. aeruginosa, a result that was investigated further (vide infra), or for fungal organisms. The finding of null activity with eukaryotic microbes was not discouraging, as total coverage of the panel would have been taken as nonspecific inhibition of growth and could be considered suspicious for an antibacterial candidate.2
Figure 3.
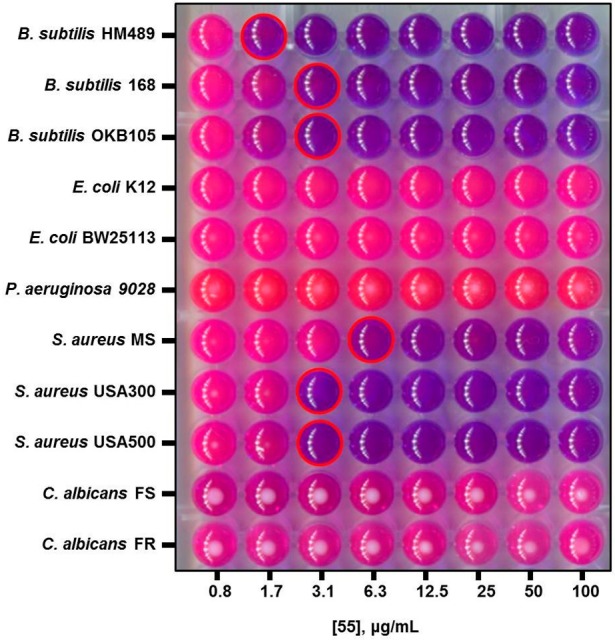
Spectrum of antibacterial activity possessed by 55. Broth dilution experiments were performed to assess the microbicidal activity of 55 with different organisms. In these experiments, growth is detected with the indicator resazurin, a purple dye which is reduced to the pink compound resorufin by metabolically active organisms. Wells corresponding to the MIC are outlined in red. The antibacterial activity of 55 was restricted to Gram-positive organisms, represented here by strains of B. subtilis and S. aureus, with no growth inhibition observed for Gram-negative bacteria or fungi.
We followed up on this finding with a study of bacterial cell membrane integrity, as Gram-positive whole-cell experiments can be convoluted by nonspecific damage to the lipid bilayer. We employed the differential nucleic acid staining of B. subtilis 168 bacterial cells by Syto9 and propidium iodide in the BacLight LIVE/DEAD microscopy assay, which utilizes green and red staining to differentiate healthy and compromised cells, respectively.40 Chloramphenicol (CAM) and cetyltrimethylammonium bromide (CTAB), two compounds that possess minimum inhibitory concentration (MIC) values similar to that of 55 (∼2 μg/mL), were included as controls to serve as known specific and nonspecific agents, respectively. Testing of these agents indicated a rapid compromise of bacterial cell integrity for CTAB after 15 min of incubation, where similar testing of chloramphenicol and 55 revealed continued cellular viability over the short incubation period at concentrations that exceeded MIC by 10-fold (Figure 4A). Therefore, we concluded that compound 55 does not elicit antibacterial activity through the nonspecific disruption of the Gram-positive membrane and likely acts on an intracellular target.
Figure 4.

Compound 55 does not disturb bacterial membranes and is expelled by efflux pumps in Gram-negative bacteria. (A) Microscopy experiments using the exclusion of propidium iodide (PI) were conducted to evaluate the effects of 55 on the integrity of the membrane in B. subtilis. In these, Syto9 stains DNA of all bacteria green, while PI can only penetrate compromised bacterial cells. CAM, CTAB, and 55 were evaluated at 1×, 4×, and 10× their MIC values, and the number of observed cells was counted. Chloramphenicol (CAM), a non-membrane-active antibiotic, and cetyltrimethylammonium bromide (CTAB), a known membrane-disrupting agent, were included as controls for the experiment. (B) Disruption of tolC, the primary efflux pump of E. coli, induced a 55-sensitive phenotype in broth dilution experiments, where concentrations as low as 12 μM inhibited bacterial growth (red-outlined well). (C) Building upon the results of (B), checkerboard synergy experiments were conducted with the AcrAB-TolC inhibitor phenylarginine-β-napthamide (PAβN) and demonstrate an effective chemical knockout strategy to induce a 55-sensitive phenotype in wild-type E. coli. Wells containing no metabolically active bacteria are observed as purple in this experiment, and wells corresponding to the lowest concentration of PAβN capable of synergizing with 55 to halt bacterial proliferation at each concentration of 55 are outlined in red.
With this information, we further characterized the antibacterial activity of 55 to discern whether the compound merely halted proliferation of bacteria or possessed bactericidal activity. The results of these experiments, summarized in Table S1 (Supporting Information), demonstrated that the minimum bactericidal concentration (MBC) of 55 fell within a range of 1×–4× the MIC in S. aureus and B. subtilis. The generally accepted criteria of defining antibacterial activity as cidal is a ratio of MBC to MIC less than or equal to 4.41 By this definition, 55 exhibited bactericidal activity in all cases tested.
Efflux of Compound 55 by the AcrAB–TolC Complex Induces a Resistant Phenotype in E. coli
The limited spectrum of activity possessed by 55 was surprising, since our biochemical testing indicated that the compound was active with AcpS in the direct enzymatic assay. This led us to hypothesize that the Gram-negative outer membrane was the source of this complication, as it presents a formidable barrier possessing both low penetrability and an active efflux mechanism.42 In E. coli, a tripartite complex of the TolC–AcrA–AcrB gene products constitutes the primary efflux pump and source of resistance through this mechanism.43 Thus, we tested E. coli BW25113 and JW0451, the wild-type strain and an efflux-defective mutant that contains a lesion in the acrB locus, respectively,44 for susceptibility to 55. We anticipated that active transport out of the cell by the AcrAB–TolC system would manifest as an observed hypersusceptibility of the ΔacrB strain, while insensitivity would indicate cellular penetration or another mechanism as the source of resistance. For these experiments, we observed a clear differential susceptibility between the two strains (Figure 4B), where disruption of the acrB locus rendered the organism susceptible to 55 at concentrations as low as 12.5 μg/mL. These findings indicated that efflux is the primary mechanism of resistance in this organism and that 55 is capable of penetrating the Gram-negative bacterial cell.
To further validate the above findings using a chemical genetic model, we tested 55 for synergistic activity with the broad-spectrum efflux pump inhibitor phenylarginine-β-naphthamide (PAβN).45 In these experiments, neither compound alone was found to possess antibacterial activity against the wild-type strain BW25113 at the top concentrations tested, but offered pronounced growth inhibition in combination (Figure 4C). The fractional inhibition concentration index, a ratio of the potency of the combined test articles relative to that of the singular components,46 was estimated to be less than 0.25, a finding strongly indicative of synergism, although we were not able to precisely determine this parameter since neither compound elicited growth inhibition at the top concentrations tested.
Compound 55 Attenuates Surfactin Production in B. subtilis
To further understand the on-target engagement of PPTase enzymes within the bacterial cell, we exploited the association of Sfp-PPTase with secondary metabolism in B. subtilis. The lipopeptide surfactin 68 is produced by a nonribosomal peptide synthetase pathway, diagrammed in Figure 5A. This canonical megasynthase complex contains 24 functional domains, 7 of which are 4′-PP-accepting carrier protein domains. A singly missed 4′-PP transfer event will disrupt processivity in the megasynthase and render the complex incapable of producing the metabolite. As such, this system represents a strategy to study target engagement by 55 in the bacterial cell through monitoring of the fermentative yield of 68. Toward this end, we conducted time course experiments for cultures of B. subtilis in the presence of increasing concentrations of 55, where we periodically measured both bacterial growth and surfactin titer over 28 h (Figure 5B,C). Given the potent antibacterial activity of 55 against this organism, delicate selection of test conditions was required to supply high enough concentrations of compound to elicit a response but also provide for normal bacterial growth. In these experiments, cultures treated with 55 at concentrations of 0.53 or 1.1 μg/mL resulted in 33% and 41% reductions in surfactin titer relative to a vehicle control after 28 h of culture, respectively (Figure 5B). These reductions in titer were accompanied by modest extensions in the culture lag phase, although continued culture revealed that 55 had a null effect on both the doubling time in the log phase and final cellular densities (Figure 5C). This dose-dependent attenuation of surfactin titer in cultures which accumulated similar quantities of biomass is consistent with engagement of Sfp-PPTase by 55 inside living bacteria.
Figure 5.

Inhibition of surfactin production by 55. (A) Surfactin 68 is produced by a nonribosomal peptide synthetase pathway in B. subtilis that contains 24 distinct functional domains, represented as cubes. Seven of these are CP domains that require post-translational activation by Sfp-PPTase, and the monitoring of the fermentative yield provides a means to assess the blockade of PPTase-dependent processes in the bacterial cell. (B) Culture time course experiments demonstrate that sublethal concentrations of 55 attenuate surfactin production by B. subtilis OKB105. (C) Culture density data are plotted for the culture time course experiment in (B) and demonstrate a modest effect of 55 on the growth of the cultures, indicating that the compound did not impede their ability to reach a terminal density.
While our preliminary studies involved some characterization of the in vitro ADME properties, after selection of 55 as a lead compound, a more detailed analysis was obtained. As shown in Table 5, 55 has a modest solubility in PBS buffer (pH 7.4) of 20 μM, which is approximately 60 times higher than the recorded IC50 value (290 nM). Moreover, 55 displayed stability to mouse and rat liver microsomes with T1/2 values of 49.5 and >30 min, respectively. The compound was completely stable (measured for 1 h) in the absence of the NADPH cofactor, suggesting a CYP-mediated oxidation event. Passive permeability was assessed using the PAMPA assay, and very favorable permeability was observed (1122 × 10–6 cm/s). Next, we wanted to examine whether the probe compound inhibits specific cytochrome P450 enzymes 2D6 and 3A4 as these two isoforms account for the metabolism of approximately 80% of drugs.47 To assess the potential for CYP inhibition and drug–drug interactions (DDIs), we looked at the effect of coincubation of our compound(s) at 3 μM with known CYP substrates (2D6 with dextromethorphan and 3A4 with 6β-hydroxytestosterone). We found that 55 exhibits modest CYP inhibition of 25% and 20% for 2D6 and 3A4, respectively. To further elucidate the potential for CYP liabilities, follow-up IC50 values and testing of a more expansive representation of CYP isoforms will be required. However, we also investigated CYP inhibition for other analogues, and inhibition was inconsistent across the series, so we are confident that these liabilities could be addressed through targeted structural modifications. Given the potential instability of the thiourea moiety in aqueous and biological media,38 we were encouraged that 55 showed no signs of degradation in aqueous media at a wide pH range (2–9), buffers (PBS and HEPES) (Figure S7, Supporting Information), and mouse plasma (Table 5). In addition, the thiourea functionality is a known structural alert that can cause toxicity due to formation of reactive metabolites under physiological conditions (vide supra). However, our probe compound 55 and analogue 41 are not susceptible to bioactivation and did not show any GSH adducts in follow-up studies (Table S3, Supporting Information).
Table 5. In Vitro ADME Profile of 55a.
| compd | aq solubility (pH 7.4) | cLogP | microsomal stability (T1/2, min) |
CYP inh (3 μM) (%) | PAMPA | plasma stability (mouse) (%) | ||
|---|---|---|---|---|---|---|---|---|
| 55 | 20 μM | 3.35 | 40 (mouse) | >30 (rat) | 25 (2D6) | 20 (3A4) | 1122 | 100 (at 2 h) |
Aqueous solubility (PBS buffer), mouse liver microsome (MLM) stability, CYP2D6/3A4 inhibition, and plasma stability were determined at Pharmaron Inc. All other studies were conducted at NCATS. The microsomal stability data represent the stability in the presence of NADPH. Compound 55 showed no degradation without NADPH present over a 1 h period. Dextromethorphan and 6β-hydroxytestosterone were the substrates used for the CYP2D6 and CYP3A4 inhibition studies, respectively. PAMPA represents passive permeability measured as 1 × 10–6 cm/s.
Having demonstrated a favorable in vitro ADME profile of 55, we next sought to investigate the in vivo PK profile in support of testing this compound in proof of concept (POC) antibacterial animal models (vide infra). As shown in Table 6, 55 was administered to CD1 mice via both iv and ip routes at 3 and 30 mg/kg doses, respectively, using a solution formation consisting of 5/10/85 (w/w/w) DMSO, Solutol, and water. Compound 55 exhibits favorable systemic exposure levels (AUCinf = 68860 h·ng/mL) that are 40–50-fold higher than the MIC values against S. aureus strains at 30 mg/kg dosing. Moreover, the compound has a reasonable T1/2 (2.0 h), and low clearance (7.2 mL/min/kg) results in an exposure (total drug concentration, not free drug concentration) that exceeds the in vitro antibacterial MIC value for over 8 h. The compound also efficiently crosses the BBB and thus could be potentially used for bacterial infections which reside in the brain. Furthermore, dosing of 55 at these concentrations did not result in any adverse clinical observations over the 24 h period, which seems to indicate that no acute toxicity is to be expected at the doses used for in vivo POC studies.
Table 6. In Vivo PK Profile of 55a.
| compd | route | T1/2 (h) | Cmax (ng/mL) | AUCinf (h·ng/mL) | Vd (L/kg) | MRT (h) | clearance (mL/min/kg) | P/B |
|---|---|---|---|---|---|---|---|---|
| 55 | iv | 2.4 | 4114 | 6983 | 1.5 | 3.4 | 7.2 | 2.5 |
| ip | 2.0 | 17633 | 68860 | 2.1 |
All experiments were conducted at Pharmaron Inc. using male CD1 mice (6–8 weeks of age). Data were collected in triplicate at eight time points over a 24 h period. 55 was formulated as a solution (5% DMSO and 10% Solutol in H2O). For iv, dosed at 3 mg/kg. For ip, dosed at 30 mg/kg. MRT = mean residence time (the time for elimination of 63.2% of the iv dose). P/B = plasma to brain ratio [AUClast(plasma)/AUClast(brain)].
As discussed above, 55 exhibits antibacterial activity against MRSA and has a desirable ADME/PK profile, and thus, it was poised to be tested in vivo. For these studies, we profiled a panel of clinical isolates to assess the susceptibility of various genotypes (Table S2, Supporting Information), and this revealed similar potencies (within 3-fold) for all strains tested. With this information, we pursued an in vivo POC study that utilized the MRSA-USA500 strain (BK2395) since our compound had demonstrated favorable in vitro activity against it and it has demonstrated suitable virulence in mouse models of sepsis.48 Unfortunately, in this experiment, 55 did not increase survival compared to the vehicle control, while vancomycin treatment completely protected animals from morbidity (Figure S6, Supporting Information). These findings led us to investigate the source of discrepancy between our in vitro and in vivo results. To rule out the rapid development of resistance in vivo, bacteria were harvested from the kidneys of mice treated with the vehicle or 55. For each crude isolate, five representative strains were isolated. We then determined MIC values for these strains in parallel along with the parental MRSA-USA500 strain and observed identical susceptibility of all strains to 55. These results indicated that neither passage of the organism in mice nor challenge with 55 in vivo led to reduced susceptibility to the compound. Realizing that the antibacterial activity of compounds can be attenuated in the presence of whole blood and/or serum, we repeated the MIC determinations of 55 in the presence of calf serum (20%, v/v) in cation-adjusted Mueller Hinton II broth and observed a significant shift in potency (>57 μM vs 3.4 μM). These data, along with subsequent analysis via equilibrium dialysis, revealed that 55 displays a high degree (98.6%) of plasma protein binding (PPB), and thus, the lack of activity is likely a consequence of the lipophilic nature of this compound. While disappointing, other groups have shown the ability to successfully modulate the lipophilicity of the lead compound through the strategic placement of polar moieties to improve the antibacterial activity in vivo. In fact, a group of researchers at Pfizer recently reported improved in vivo efficacy of their lead antibacterial candidate by lowering PPB via lowering cLogP.49 As described above, the eastern region of the molecule is highly tolerant to structural modifications (Table 2), which could be exploited to modulate the cLogP and thus hopefully improve activity in vivo. In addition, we plan on conducting more extensive PK studies to look at tissue distribution and free drug concentration, which may guide not only a future medicinal chemistry effort, but also aid the design of our in vivo proof of concept models.
Over the course of this work, a heated debate has emerged as to the suitability of the FAS as a drug target.50−54 It appears that, in some Gram-positive pathogens, FAS is dispensable when bacteria are propagated in the presence of serum, which is a nutrient source rich in fatty acids. Further study has found that the results may not be accurately predictive for even closely related bacteria, and further investigation of this phenomenon is warranted to tease apart the details with finer resolution. Nonetheless, this discrepancy provides another mechanism through which bacterial tolerance to 55 may be occurring in vivo, and such a mechanism may confound the advancement of in vitro PPTase-targeting inhibitors through in vivo POC studies of staphylococcal septicemia. However, this does not speak to the role that PPTase plays in the production of virulence factors and elaborate cell wall components, especially in the case of Mycobacterium spp., where recent findings have confirmed the essential nature of both AcpS- and Sfp-PPTases.55 These organisms depend on the concerted effort of FAS and PKS systems for the assembly of mycolic acids, and these PPTase dependencies have been substantiated in rodent models of infection.56 Thus, PPTase inhibitors remain a target worthy of pursuit and may find a more immediate applicability as tools to pharmacologically probe the role of PPTases in mycobacterial pathogenesis.
Conclusion
PPTase enzymes catalyze an essential PTM that activates the assembly of fatty acid, polyketide, and nonribosomal peptides. Metabolites from all three of these classes are necessary for bacterial cell viability and virulence. As a result, inhibition of these pathways has received attention as an attractive approach to the development of new antimicrobial compounds. Rather than targeting the machinery involved in the production of a single metabolite, we have chosen to investigate the inhibition of this PTM as it represents a unifying feature between these pathways. PPTase inhibitors may leverage this overlap to downregulate the production of key cellular components and virulence-determining factors.
The critical role of this PTM in bacterial metabolism has been studied by others, but to date these efforts have not produced compounds with appreciable antibacterial activity. This limited success can likely be attributed to the inability of these programs to consider the presence of an Sfp-PPTase in addition to their primary target, AcpS-PPTase. This secondary enzyme is capable of compensating for loss of the AcpS locus32,57,58 and thus provides a direct mechanism through which bacterial organisms may develop resistance through simple transcriptional/translational upregulation.17 To address this issue, we have pursued the development of a chemical probe with dual PPTase inhibitory activity.
Here we have detailed the development and characterization of 55, a novel small molecule that possesses these characteristics. We have demonstrated the qualities of this chemical probe and evaluated it according to the benchmarks suggested by the community.59 The features of 55 can be summarized according to the five principles of a quality chemical probe as follows: (i) We have performed detailed molecular profiling with relevant molecular targets, with observed potencies of 290 nM and 8.1 μM against the primary development target Sfp-PPTase and bacterial orthologue AcpS, respectively, which is contrasted by null activity with the human enzyme. In the context of an antibacterial discovery campaign, where the end goal is the inhibition of bacterial growth in man, this indicates a selectivity index of >500-fold with respect to the human orthologue. (ii) With respect to the mechanism of action, we demonstrate that the chemotype inhibits Sfp-PPTase independent of a fluorescent label through an allosteric mechanism that is noncompetitive with substrates and is rapidly reversible. (iii) Regarding the identity of the active species, synthetic/medicinal chemistry efforts verified the structure, purity, stability, and chemical tractability of the chemotype. In addition, a structurally similar but inactive control was identified which was devoid of AcpS/Sfp-PPTase inhibitory activity as well as antibacterial activity. (iv) To demonstrate the utility of 55 as a probe, we confirmed the ability of a dual-specific PPTase inhibitor to thwart the growth of bacteria in the absence of a rapid cytotoxic response. Additionally, 55 demonstrated activity against clinically relevant microorganisms, where it stifled the growth of methicillin-resistant S. aureus. Expanding on these findings, we also identified the primary mechanism of resistance in E. coli to be efflux by the AcrAB–TolC system and demonstrated a chemical genetic approach to mitigate this issue and extend the spectrum of antibacterial activity to include Gram-negative organisms. We recognize that optimizing a chemotype to circumvent efflux is a formidable task and is exacerbated in clinically relevant Gram-negative pathogens where efflux pathways are more complex than in the model organism E. coli.60 Beyond these assessments of antimicrobial activity, the ability of 55 to attenuate the production of surfactin, a metabolite dependent on the biochemical functionality of Sfp-PPTase (Figure 5B), strongly indicates that 55 is acting on-target inside the bacterial cell. (v) Finally, in conjunction with the spirit and principles of the scientific community and the probe characteristic of availability, we will provide samples of 55 freely upon request.
Experimental Procedures
General Methods for Chemistry
All air- or moisture-sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Anhydrous solvents such as dichloromethane, N,N-dimethylformamide (DMF), acetonitrile, methanol, and triethylamine were purchased from Sigma-Aldrich. Preparative purification was performed on a Waters semipreparative HPLC system. The column used was a Phenomenex Luna C18 (5 μm, 30 × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10–50% acetonitrile over 8 min was used during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis was performed on an Agilent LC/MS system (Agilent Technologies, Santa Clara, CA). Method 1: A 7 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 min run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 μm, 3 × 75 mm) was used at a temperature of 50 °C. Method 2: A 3 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 min run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 μm, 3 × 100 mm) was used at a temperature of 50 °C. Purity determination was performed using an Agilent diode array detector for both method 1 and method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical shifts are reported in parts per million with undeuterated solvent (DMSO-d6 at 2.49 ppm) as the internal standard for DMSO-d6 solutions. All of the analogues tested in the biological assays have purity greater than 95% on the basis of both analytical methods. High-resolution mass spectrometry was recorded on an Agilent 6210 time-of-flight LC/MS system. Confirmation of the molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
General Procedure for the Synthesis of N-(Aryl/heteroaryl)-4-arylpiperazine-1-carbothioamides
A mixture of substituted pyridin-2-amine (1 equiv) and 1,1′-thiocarbonyldiimidazole (TCDI) (1.05 equiv) in dichloromethane (2 mL/mmol) was stirred for 15 min at room temperature. To the clear yellow solution was added arylpiperazine (1.1 equiv), and the reaction mixture was stirred at 40 °C for 1 h. The solvent was evaporated, and the crude product was taken up in 2 mL of DMSO and purified via reversed-phase chromatography to give the products as TFA salts (the details and characterization for all the compounds is depicted in the Supporting Information). Characterization data for representative compounds are given below.
N-(4-Methylpyridin-2-yl)-4-(3-(trifluoromethyl)phenyl)piperazine-1-carbothioamide Trifluoroacetate (1)
This compound was prepared following the above general procedure starting from commerically available 4-methylpyridin-2-amine and 1-(3-(trifluoromethyl)phenyl)piperazine: LC/MS retention time t1 (method 1, 7 min) = 4.91 min and t2 (method 2, 3 min) = 3.13 min; 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.22 (d, J = 5.4 Hz, 1H), 7.53–7.42 (m, 2H), 7.33–7.18 (m, 2H), 7.16–7.07 (m, 1H), 7.03 (d, J = 5.4 Hz, 1H), 4.08 (t, J = 5.2 Hz, 4H), 3.40 (dd, J = 6.5, 3.9 Hz, 4H), 2.36 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 181.16, 158.49, 153.14, 150.95, 130.53, 130.49, 130.22, 126.22, 120.70, 119.17, 118.96, 115.15, 111.29, 111.25, 48.49, 47.41, 21.38; HRMS (ESI) m/z (M + H)+ calcd for C18H20F3N4S 381.1355, found 381.1341.
N-(4-Methylpyridin-2-yl)-4-(2-(trifluoromethyl)phenyl)piperazine-1-carbothioamide Trifluoroacetate (22)
This compound was prepared following the above general procedure starting from commerically available 4-methylpyridin-2-amine and 1-(2-(trifluoromethyl)phenyl)piperazine: LC/MS retention time t1 (method 1, 7 min) = 4.993 min and t2 (method 2, 3 min) = 3.215 min; 1H NMR (400 MHz, DMSO-d6) δ 8.18 (d, J = 5.3 Hz, 1H), 7.76–7.57 (m, 3H), 7.46–7.34 (m, 2H), 6.96 (d, J = 5.1 Hz, 1H), 4.09–3.95 (m, 4H), 2.93 (m, 4H), 2.32 (s, 3H); HRMS (ESI) m/z (M + H)+ calcd for C18H20F3N4S 381.1355, found 381.1361.
4-(4-Chloro-2-(trifluoromethyl)phenyl)-N-(4,6-dimethylpyridin-2-yl)piperazine-1-carbothioamide Trifluoroacetate (41)
This compound was prepared following the above general procedure starting from intermediate 1-(4-chloro-2-(trifluoromethyl)phenyl)piperazine (see the Supporting Information for the synthesis) and commerically available 2,4-dimethylpyridin-2-amine: LC/MS retention time t1 (method 1, 7 min) = 5.384 min and t2 (method 2, 3 min) = 3.247 min; 1H NMR (400 MHz, DMSO-d6) δ 7.75 (dd, J = 7.1, 2.5 Hz, 2H), 7.68–7.59 (m, 1H), 7.23 (s, 1H), 6.85 (s, 1H), 4.01 (d, J = 5.6 Hz, 4H), 2.93 (q, J = 4.6, 3.7 Hz, 4H), 2.40 (s, 3H), 2.29 (s, 3H); HRMS (ESI) m/z (M + H)+ calcd for C19H21ClF3N4S 429.1122, found 429.1133.
N-(4,6-Dimethylpyridin-2-yl)-4-(5-(trifluoromethyl)pyridin-2-yl)piperazine-1-carbothioamide Trifluoroacetate (53)
This compound was prepared following the above general procedure starting from commerically available precursors 2,4-dimethylpyridin-2-amine and 1-(5-(trifluoromethyl)pyridin-2-yl)piperazine: LC/MS retention time t1 (method 1, 7 min) = 4.615 min and t2 (method 2, 3 min) = 3.114 min; 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 2.5 Hz, 1H), 7.85 (dd, J = 9.2, 2.6 Hz, 1H), 7.29 (s, 1H), 6.96 (d, J = 9.5 Hz, 2H), 4.10–3.99 (m, 4H), 3.79 (dd, J = 6.5, 4.0 Hz, 4H), 2.44 (s, 3H), 2.33 (s, 3H); HRMS (ESI) m/z (M + H)+ calcd for C18H21F3N5S 396.1464, found 396.1469.
4-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4,6-dimethylpyridin-2-yl)piperazine-1-carbothioamide Trifluoroacetate (54)
This compound was prepared following the above general procedure starting from commerically available precursors 2,4-dimethylpyridin-2-amine and 1-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)piperazine: LC/MS retention time t1 (method 1, 7 min) = 5.113 min and t2 (method 2, 3 min) = 3.287 min; 1H NMR (400 MHz, DMSO-d6) δ 8.58 (d, J = 2.1 Hz, 1H), 8.23 (d, J = 2.1 Hz, 1H), 7.25 (s, 1H), 6.93 (s, 1H), 4.06 (dd, J = 6.5, 3.6 Hz, 4H), 3.61 (dd, J = 6.6, 3.7 Hz, 4H), 2.43 (s, 3H), 2.31 (s, 3H); HRMS (ESI) m/z (M + H)+ calcd for C18H20ClF3N5S 430.1075, found 430.1089.
4-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-methoxypyridin-2-yl)piperazine-1-carbothioamide Trifluoroacetate (ML267) (55)
This compound was prepared following the above general procedure starting from commerically available precursors 4-methoxypyridin-2-amine and 1-(3-chloro-5-(trifluoromethyl)pyridin-2-yl)piperazine: LC/MS retention time t1 (method 1, 7 min) = 4.88 min and t2 (method 2, 3 min) = 3.04 min; 1H NMR (400 MHz, DMSO-d6) δ 8.56 – 8.61 (m, 1H), 8.16–8.26 (m, 2H), 7.15 (s, 1H), 6.83 (s, 1H), 4.05–4.13 (m, 4H), 3.88 (s, 3H), 3.58–3.67 (m, 4 H); 13C NMR (101 MHz, DMSO-d6) δ 180.45, 168.50, 159.01, 153.62, 143.08, 136.44, 136.41, 124.78, 122.09, 119.61, 118.77, 118.44, 107.22, 102.47, 102.37, 56.43, 56.29, 48.06, 47.56; HRMS (ESI) m/z (M + H)+ calcd for C17H18ClF3N5OS 432.0867, found 432.0856.
N-(4-Methylpyridin-2-yl)-4-((3-(trifluoromethyl)phenyl)amino)piperidine-1-carbothioamide Trifluoroacetate (63)
A mixture of 4-methylpyridin-2-amine (0.1 g, 0.925 mmol, 1 equiv) and TCDI (0.165 g, 0.925 mmol, 1.0 equiv) in dichloromethane (3 mL) was stirred for 15 min at room temperature. To the clear yellow solution was added commercially available N-(3-(trifluoromethyl)phenyl)piperidin-4-amine (0.226 g, 0.925 mmol, 1 equiv), and the resulting solution was stirred at 40 °C for 1 h. The solvent was evaporated, and the crude product was taken up in 2 mL of DMSO and purified via reversed-phase chromatography to give the product as a TFA salt: LC/MS retention time t1 (method 1, 7 min) = 4.954 min and t2 (method 2, 3 min) = 3.025 min; 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 5.4 Hz, 1H), 7.39 (d, J = 1.6 Hz, 1H), 7.31–7.22 (m, 1H), 7.05–7.00 (m, 1H), 6.91–6.85 (m, 2H), 6.83–6.77 (m, 1H), 5.50 (br s, 1H), 4.54 (d, J = 13.2 Hz, 2H), 3.68 (tt, J = 9.4, 3.9 Hz, 1H), 3.43 (ddd, J = 13.7, 11.1, 2.8 Hz, 2H), 2.35 (s, 3H), 2.07–1.89 (m, 2H), 1.43 (dtd, J = 13.6, 10.2, 3.8 Hz, 2H); HRMS (ESI) m/z (M + H)+ calcd for C19H22F3N4S 395.1512, found 395.1510.
N-(4-Methylpyridin-2-yl)-4-(3-(trifluoromethyl)phenyl)piperazine-1-carboxamide Trifluoroacetate (12)
To a mixture of 4-methylpyridin-2-amine (0.5 g, 4.62 mmol, 1 equiv), (i-Pr)2NEt (0.970 mL, 5.55 mmol, 1.2 equiv), and DMAP (0.113 g, 0.925 mmol, 0.2 equiv) in dichloromethane (25 mL) was added phenyl carbonochloridate (0.664 mL, 5.09 mmol, 1.1 equiv) at 0 °C, and the reaction was allowed to stir at room temperature for 1 h. To the resulting clear solution was added 1-(3-(trifluoromethyl)phenyl)piperazine (1.13 mL, 6.01 mmol, 1.3 equiv), and the reaction mixture was stirred for an additional 2 h at room temperature. Volatiles were removed by forced air, and the residue was dissolved in DMF and purified via reversed-phase chromatography to give 12 as a TFA salt: LC/MS retention time t1 (method 1, 7 min) = 4.43 min and t2 (method 2, 3 min) = 3.095 min; 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.17 (d, J = 5.9 Hz, 1H), 7.54–7.39 (m, 2H), 7.29–7.18 (m, 2H), 7.17–7.05 (m, 2H), 3.71–3.63 (m, 4H), 3.31–3.29 (m, 4H), 2.40 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 158.27, 153.81, 150.90, 150.53, 140.63, 130.07, 130.04, 129.76, 119.59, 119.03, 115.02, 114.70, 111.33, 111.29, 47.41, 43.50, 21.45; HRMS (ESI) m/z (M + H)+ calcd for C18H20F3N4O 365.1584, found 365.1590.
Methods
Sfp- and AcpS-PPTase qHTS Assays
The assay was performed in 50 mM HEPES·Na (pH 7.6), 10 mM MgCl2, 0.01% Nonidet P-40, and 0.01% BSA. A 3 μL volume of each reagent, consisting of buffer (in columns 3 and 4 as a negative control) and Sfp- or AcpS-PPTase (in columns 1, 2, and 5–48, final concentration of 15 or 100 nM, respectively) were dispensed into a 1536-well Greiner black solid-bottom plate. Compounds (23 nL) were transferred via a Kalypsys pintool equipped with a 1536-pin array. The plate was incubated for 15 min at room temperature, followed by the addition of 1 μL of substrate (final concentrations for rhodamine–CoA and BHQ-2-YbbR were 5 and 12.5 μM, respectively) to start the reaction. The plate was then centrifuged at 1000 rpm for 15 s, and the fluorescence intensity was recorded on a ViewLux high-throughput charge-coupled device (CCD) imager (Perkin-Elmer) using standard BODIPY optics (525 nm excitation and 598 nm emission). The plate was then incubated for 30 or 60 min (Sfp or AcpS, respectively), and a second read on the ViewLux was performed. The fluorescence intensity difference over the 30 or 60 min period (Sfp or AcpS, respectively) was used to calculate the respective reaction rate for each well. All screening operations were performed on a fully integrated robotic system (Kalypsys Inc., San Diego, CA) as described elsewhere. Plates containing DMSO only (instead of compound solutions) were included approximately every 50 plates throughout the screen to monitor any systematic trend in the assay signal associated with reagent dispenser variation or a decrease in enzyme specific activity.
PPTase Gel Assay
A DMSO solution of confirmed hits (0.5 μL) was added to a 1.33× enzyme solution (15 μL, containing 26.6 nM Sfp, 66 mM HEPES·Na, 13.3 mM MgCl2, 0.0133% NP-40, and 0.133% BSA, pH 7.6). After a 10 min incubation, the enzymatic reaction was initiated by the addition of a 4× substrate solution (4 μL, containing 50 μM rhodamine–CoA and 50 μM apo-actinorhodin–ACP). The reactions were terminated after a 30 min incubation at room temperature by the addition of a 2× quench solution (20 μL, containing 4 M urea, 25 mM EDTA, and 0.004% phenol red, pH 8.0).
Samples were separated under native conditions on a 20% polyacrylamide gel using standard Laemmli conditions. Following the run, the gels were imaged with a Chemi-Doc Plus imager (Bio-Rad, Hercules, CA), and the band intensity was quantified using the ImageJ software package. Pixel density values were normalized to control wells and fit with the four-parameter Hill equation using in-house tools. PubChem AID 602362.
Minimum Inhibitory Concentration Determination
Methods for MIC determination were made in accordance with standards put forth by the National Clinical Laboratory Standards Institute detailed in documents M07-A861 and M27-A262 for bacterial and fungal species, respectively. Briefly, organisms were maintained on a solid medium. Innoculum was prepared from overnight liquid cultures of bacteria or by suspending 2 day old colonies in RPMI 1640 medium. Test articles dissolved in DMSO (1 μL) were added to sterile medium [50 μL, cation-adjusted Mueller Hinton II broth for bacteria (BD BBL, Franklin Lakes, NJ) or RMPI 1640 for fungi (Invitrogen Corp., Carlsbad, CA)] followed by innoculum prepared in the same medium (50 μL, containing ∼1 × 103 cfu) and incubated at 30–37 °C for 16–20 h (48 h for fungi). The plates were visually inspected for microbial growth, and the first well containing no visible microbial growth was scored as the MIC. Strains evaluated in this manner included B. subtilis 168, B. subtilis HM489,32E. coli K12, P. aeruginosa ATCC 9028, S. aureus ATCC 6538 (methicillin-sensitive), S. aureus ATCC BAA-1717 (community-acquired methicillin-resistant strain USA300-HOU-MR), C. albicans ATCC 90028 (fluconazole-sensitive), and C. albicans ATCC 96901 (fluconazole-resistant).
For the acquisition of images for Figures 3D and 4C, resazurin was employed as an indicator to assist in visualization in a method similar to that of Sarker et al.63 A 10 μL volume of sterile aqueous resazurin solution (0.7% w/v) was added to the wells of test plates resulting from the protocol above. After further incubation 4 h at 37 °C, the plates were then imaged with an Epson photoscanner.
Acknowledgments
T.L.F., G.R., A.Y., T.D., H.L.B., M.A.-R., W.L., A.J., A.S., and D.J.M. were supported by the intramural research program of the National Center for Advancing Translational Sciences and the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research (Grant U54MH084681). N.M.K. and M.D.B. were supported by NIH Grants R21AI090213 and R01GM094924. We thank Sam Michael, Michael Balcom (deceased), and Richard Jones for automation support, Paul Shinn and Danielle van Leer for the assistance with compound management, Edward H. Kerns and Kimloan Nguyen for determination of in vitro ADME properties, Elizabeth Fernandez, Christopher Leclair, and Heather Baker for assistance with compound purification and processing, Mohamed Marahiel (Philipps-Universität Marburg), the Bacillus Genomic Stock Center, and The National BioResource Project of the Japanese National Institute for Genetics for bacterial strains, Udo Oppermann (University of Oxford) for the human PPTase expression plasmid, and Victor Torres (Anti-infectives Screening Core, New York University) for MIC studies on clinical isolates of MRSA and conducting the mouse model of staphylococcal septicemia.
Glossary
Abbreviations Used
- PPTase
4′-phosphopantetheine transferase
- Sfp
PPTase involved in surfactin production
- AcpS
PPTase involved in FAS, acyl carrier protein synthase
- CoA
coenzyme A
- NRPS
nonribosomal peptide synthethase
- PKS
polyketide synthetase
- FAS
fatty acid synthetase
- CA-MRSA
community-acquired methicillin-resistant Staphylococcus aureus
- SAR
structure–activity relationship
- HTS
high-throughput screen
- MIC
minimum inhibitory concentration
- ADME
absorption, distribution, metabolism, and excretion
- ip
intraperitoneal
- iv
intravenous
- mpk
milligrams per kilogram
- RLMs
rat liver microsomes
- MLMs
mouse liver microsomes
- POC
proof of concept
- BBB
blood–brain barrier
- PAMPA
parallel artificial membrane permeability assay
- NADPH
nicotinamide adenine dinucleotide phosphate
- CYP
cytochrome P450
- PBS
phosphate-buffered saline
- PaβN
phenylarginine β-naphthylamide
- DDI
drug–drug interaction
- PPB
plasma protein binding
- PK
pharmacokinetics
- GSH
glutathione
- LOPAC
Library of Pharmacologically Active Compounds
Supporting Information Available
Additional supplemental figures, experimental procedures, and spectroscopic data (1H NMR, LC/MS, and HRMS) for representative compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Miller A. A.; Miller P. F.. Emerging Trends in Antibacterial Discovery: Answering the Call to Arms; Caister Academic Press: Norfolk, U.K., 2011. [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Silver L. L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambalot R. H.; Gehring A. M.; Flugel R. S.; Zuber P.; LaCelle M.; Marahiel M. A.; Reid R.; Khosla C.; Walsh C. T. A new enzyme superfamily—The phosphopantetheinyl transferases. Chem. Biol. 1996, 3, 923–936. [DOI] [PubMed] [Google Scholar]
- Zhang Y. M.; White S. W.; Rock C. O. Inhibiting bacterial fatty acid synthesis. J. Biol. Chem. 2006, 281, 17541–17544. [DOI] [PubMed] [Google Scholar]
- Wang J.; Soisson S. M.; Young K.; Shoop W.; Kodali S.; Galgoci A.; Painter R.; Parthasarathy G.; Tang Y. S.; Cummings R.; Ha S.; Dorso K.; Motyl M.; Jayasuriya H.; Ondeyka J.; Herath K.; Zhang C.; Hernandez L.; Allocco J.; Basilio A.; Tormo J. R.; Genilloud O.; Vicente F.; Pelaez F.; Colwell L.; Lee S. H.; Michael B.; Felcetto T.; Gill C.; Silver L. L.; Hermes J. D.; Bartizal K.; Barrett J.; Schmatz D.; Becker J. W.; Cully D.; Singh S. B. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 2006, 441, 358–361. [DOI] [PubMed] [Google Scholar]
- Torres A. G.; Redford P.; Welch R. A.; Payne S. M. TonB-dependent systems of uropathogenic Escherichia coli: Aerobactin and heme transport and TonB are required for virulence in the mouse. Infect. Immun. 2001, 69, 6179–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley T. L.; Simeonov A. Targeting iron assimilation to develop new antibacterials. Expert Opin. Drug Discovery 2012, 7, 831–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed M. B.; Domenech P.; Manca C.; Su H.; Barczak A. K.; Kreiswirth B. N.; Kaplan G.; Barry C. E. 3rd. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 2004, 431, 84–87. [DOI] [PubMed] [Google Scholar]
- Heinekamp T.; Thywissen A.; Macheleidt J.; Keller S.; Valiante V.; Brakhage A. A. Aspergillus fumigatus melanins: interference with the host endocytosis pathway and impact on virulence. Front. Microbiol. 2012, 3, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalut C.; Botella L.; de Sousa-D’Auria C.; Houssin C.; Guilhot C. The nonredundant roles of two 4′-phosphopantetheinyl transferases in vital processes of mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8511–8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreras J. A.; Stirrett K. L.; Lu X.; Ryu J.-S.; Soll C. E.; Tan D. S.; Quadri L. E. N. Mycobacterial phenolic glycolipid virulence factor biosynthesis: Mechanism and small-molecule inhibition of polyketide chain initiation. Chem. Biol. 2008, 15, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreras J. A.; Ryu J. S.; Di Lello F.; Tan D. S.; Quadri L. E. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005, 1, 29–32. [DOI] [PubMed] [Google Scholar]
- Flugel R. S.; Hwangbo Y.; Lambalot R. H.; Cronan J. E. Jr.; Walsh C. T. Holo-(acyl carrier protein) synthase and phosphopantetheinyl transfer in Escherichia coli. J. Biol. Chem. 2000, 275, 959–968. [DOI] [PubMed] [Google Scholar]
- Chu M.; Mierzwa R.; Xu L.; Yang S. W.; He L.; Patel M.; Stafford J.; Macinga D.; Black T.; Chan T. M.; Gullo V. Structure elucidation of Sch 538415, a novel acyl carrier protein synthase inhibitor from a microorganism. Bioorg. Med. Chem. Lett. 2003, 13, 3827–3829. [DOI] [PubMed] [Google Scholar]
- Gilbert A. M.; Kirisits M.; Toy P.; Nunn D. S.; Failli A.; Dushin E. G.; Novikova E.; Petersen P. J.; Joseph-McCarthy D.; McFadyen I.; Fritz C. C. Anthranilate 4H-oxazol-5-ones: Novel small molecule antibacterial acyl carrier protein synthase (AcpS) inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 37–41. [DOI] [PubMed] [Google Scholar]
- Foley T. L.; Young B. S.; Burkart M. D. Phosphopantetheinyl transferase inhibition and secondary metabolism. FEBS J. 2009, 276, 7134–7145. [DOI] [PubMed] [Google Scholar]
- Joseph-McCarthy D.; Parris K.; Huang A.; Failli A.; Quagliato D.; Dushin E. G.; Novikova E.; Severina E.; Tuckman M.; Petersen P. J.; Dean C.; Fritz C. C.; Meshulam T.; DeCenzo M.; Dick L.; McFadyen I. J.; Somers W. S.; Lovering F.; Gilbert A. M. Use of structure-based drug design approaches to obtain novel anthranilic acid acyl carrier protein synthase inhibitors. J. Med. Chem. 2005, 48, 7960–7969. [DOI] [PubMed] [Google Scholar]
- Yasgar A.; Foley T. L.; Jadhav A.; Inglese J.; Burkart M. D.; Simeonov A. A strategy to discover inhibitors of Bacillus subtilis surfactin-type phosphopantetheinyl transferase. Mol. BioSyst. 2010, 6, 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowicz A. M.; Vepsalainen J.; Laitinen J. T. The ‘allosteric modulator’ SCH-202676 disrupts G protein-coupled receptor function via sulphydryl-sensitive mechanisms. Br. J. Pharmacol. 2006, 147, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima T.; Takahashi K.; Funasaka S.; Obaishi H.. Novel pyridine derivative and pyrimidine derivative. U.S. Patent 12,558,982, Sept 14, 2009.
- Sammond D. M.; Nallor K. E.; Veal J. M.; Nolte R. T.; Wang L. P.; Knick V. B.; Rudolph S. K.; Truesdale A. T.; Nartey E. N.; Stafford J. A.; Kumar R.; Cheung M. Discovery of a novel and potent, and urea isostere inhibitors series of dianilinopyrimidineurea of VEGFR2 tyrosine kinase. Bioorg. Med. Chem. Lett. 2005, 15, 3519–3523. [DOI] [PubMed] [Google Scholar]
- Trinka P.; Reiter J. A convenient large-scale synthesis of ethyl (2-cyanoimino-5,6-dichloro-1,2,3,4-tetrahydroquinazoline-3-yl)acetate. J. Prakt. Chem./Chem.-Ztg. 1997, 339, 750–753. [Google Scholar]
- Pohl M.; Bechstein U.; Patzel M.; Liebscher J.; Jones P. G. Synthesis of partially saturated condensed triazoles by reaction of ω-aminoalkyl-1,2,4-triazoles with electrophiles. J. Prakt. Chem./Chem.-Ztg. 1992, 334, 630–636. [Google Scholar]
- Chan D. M. T.; Monaco K. L.; Li R. H.; Bonne D.; Clark C. G.; Lam P. Y. S. Copper promoted C-N and C-O bond cross-coupling with phenyl and pyridylboronates. Tetrahedron Lett. 2003, 44, 3863–3865. [Google Scholar]
- Lam P. Y. S.; Clark C. G.; Saubern S.; Adams J.; Winters M. P.; Chan D. M. T.; Combs A. New aryl/heteroaryl C-N bond cross-coupling reactions via arylboronic acid cupric acetate arylation. Tetrahedron Lett. 1998, 39, 2941–2944. [Google Scholar]
- Quach T. D.; Batey R. A. Ligand- and base-free copper(II)-catalyzed C–N bond formation: Cross-coupling reactions of organoboron compounds with aliphatic amines and anilines. Org. Lett. 2003, 5, 4397–4400. [DOI] [PubMed] [Google Scholar]
- Shafir A.; Buchwald S. L. Highly selective room-temperature copper-catalyzed C–N coupling reactions. J. Am. Chem. Soc. 2006, 128, 8742–8743. [DOI] [PubMed] [Google Scholar]
- Inglese J.; Auld D. S.; Jadhav A.; Johnson R. L.; Simeonov A.; Yasgar A.; Zheng W.; Austin C. P. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 11473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiff H. E.; Baker T.; Copeland T.; Chen S. M.; Court D. L. Locating essential Escherichia coli genes by using mini-Tn10 transposons: The pdxJ operon. J. Bacteriol. 1992, 174, 1544–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambalot R. H.; Walsh C. T. Cloning, overproduction, and characterization of the Escherichia coli holo-acyl carrier protein synthase. J. Biol. Chem. 1995, 270, 24658–24661. [DOI] [PubMed] [Google Scholar]
- Mootz H. D.; Finking R.; Marahiel M. A. 4′-Phosphopantetheine transfer in primary and secondary metabolism of Bacillus subtilis. J. Biol. Chem. 2001, 276, 37289–37298. [DOI] [PubMed] [Google Scholar]
- Borra M. T.; Smith B. C.; Denu J. M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M.; McDonagh T.; Heltweg B.; Hixon J.; Westman E. A.; Caldwell S. D.; Napper A.; Curtis R.; DiStefano P. S.; Fields S.; Bedalov A.; Kennedy B. K. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [DOI] [PubMed] [Google Scholar]
- Pacholec M.; Bleasdale J. E.; Chrunyk B.; Cunningham D.; Flynn D.; Garofalo R. S.; Griffith D.; Griffor M.; Loulakis P.; Pabst B.; Qiu X. Y.; Stockman B.; Thanabal V.; Varghese A.; Ward J.; Withka J.; Ahn K. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Copeland R. A.; Lai Z. Defining balanced conditions for inhibitor screening assays that target bisubstrate enzymes. J. Biomol. Screening 2009, 14, 111–120. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Basavapathruni A.; Moyer M.; Scott M. P. Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal. Biochem. 2011, 416, 206–210. [DOI] [PubMed] [Google Scholar]
- Stevens G. J.; Hitchcock K.; Wang Y. K.; Coppola G. M.; Versace R. W.; Chin J. A.; Shapiro M.; Suwanrumpha S.; Mangold B. L. K. In vitro metabolism of N-(5-chloro-2-methylphenyl)-N′-(2-methylpropyl)thiourea: Species comparison and identification of a novel thiocarbamide–glutathione adduct. Chem. Res. Toxicol. 1997, 10, 733–741. [DOI] [PubMed] [Google Scholar]
- Ahgren C.; Backro K.; Bell F. W.; Cantrell A. S.; Clemens M.; Colacino J. M.; Deeter J. B.; Engelhardt J. A.; Hogberg M.; Jaskunas S. R.; Johansson N. G.; Jordan C. L.; Kasher J. S.; Kinnick M. D.; Lind P.; Lopez C.; Morin J. M.; Muesing M. A.; Noreen R.; Oberg B.; Paget C. J.; Palkowitz J. A.; Parrish C. A.; Pranc P.; Rippy M. K.; Rydergard C.; Sahlberg C.; Swanson S.; Ternansky R. J.; Unge T.; Vasileff R. T.; Vrang L.; West S. J.; Zhang H.; Xhou X. X. The PETT series, a new class of potent nonnucleoside inhibitors of human immunodeficiency-virus type-i reverse transcriptase. Antimicrob. Agents Chemother. 1995, 39, 1329–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M. P. Rapid test for distinguishing membrane-active antibacterial agents. J. Microbiol. Methods 2006, 67, 125–130. [DOI] [PubMed] [Google Scholar]
- Peterson L. R.; Shanholtzer C. J. Tests for bactericidal effects of antimicrobial agents: Technical performance and clinical relevance. Clin. Microbiol. Rev. 1992, 5, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. E. W. The bacterial outer membrane as a drug barrier. Trends Microbiol. 1997, 5, 37–42. [DOI] [PubMed] [Google Scholar]
- Pos K. M. Drug transport mechanism of the AcrB efflux pump. Biochim. Biophys. Acta 2009, 1794, 782–793. [DOI] [PubMed] [Google Scholar]
- Baba T.; Ara T.; Hasegawa M.; Takai Y.; Okumura Y.; Baba M.; Datsenko K. A.; Tomita M.; Wanner B. L.; Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomovskaya O.; Warren M. S.; Lee A.; Galazzo J.; Fronko R.; Lee M.; Blais J.; Cho D.; Chamberland S.; Renau T.; Leger R.; Hecker S.; Watkins W.; Hoshino K.; Ishida H.; Lee V. J. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. J.; Middleton R. F.; Westmacott D. The fractional inhibitory concentration (FIC) index as a measure of synergy. J. Antimicrob. Chemother. 1983, 11, 427–433. [DOI] [PubMed] [Google Scholar]
- Clarke S. E., Jones B. C.. Human cytochrome P450s and their role in metabolism-based drug-drug interactions. In Drug-Drug Interactions, 2nd ed.; Rodrigues A. D., Ed.; Marcel Dekker: New York, 2002; Vol. 179, pp 55–88. [Google Scholar]
- Alonzo F.; Benson M. A.; Chen J.; Novick R. P.; Shopsin B.; Torres V. J. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol. Microbiol. 2012, 83, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan M. E.; Brickner S. J.; Lall M.; Casavant J.; Deschenes L.; Finegan S. M.; George D. M.; Granskog K.; Hardink J. R.; Huband M. D.; Thuy H.; Lamb L.; Marra A.; Mitton-Fry M.; Mueller J. P.; Mullins L. M.; Noe M. C.; O’Donnell J. P.; Pattavina D.; Penzien J. B.; Schuff B. P.; Sun J.; Whipple D. A.; Young J.; Gootz T. D. Preparation, Gram-negative antibacterial activity, and hydrolytic stability of novel siderophore-conjugated monocarbam diols. ACS Med. Chem. Lett. 2011, 2, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster S.; Lamberet G.; Staels B.; Trieu-Cuot P.; Gruss A.; Poyart C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 2009, 458, 83–85. [DOI] [PubMed] [Google Scholar]
- Balemans W.; Lounis N.; Gilissen R.; Guillemont J.; Simmen K.; Andries K.; Koul A. Essentiality of FASII pathway for Staphylococcus aureus. Nature 2010, 463, E3–E4. [DOI] [PubMed] [Google Scholar]
- Brinster S.; Lamberet G.; Staels B.; Trieu-Cuot P.; Gruss A.; Poyart C. Essentiality of FASII pathway for Staphylococcus aureus reply. Nature 2010, 463, E4–E5. [Google Scholar]
- Parsons J. B.; Rock C. O. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery?. Curr. Opin. Microbiol. 2011, 14, 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. B.; Frank M. W.; Rosch J. W.; Rock C. O. Staphylococcus aureus fatty acid auxotrophs do not proliferate in mice. Antimicrob. Agents Chemother. 2013, 57, 5729–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalut C.; Botella L.; de Sousa-D’Auria C.; Houssin C.; Guilhot C. The nonredundant roles of two 4′-phosphopantetheinyl transferases in vital processes of mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8511–8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc C.; Prudhomme T.; Tabouret G.; Ray A.; Burbaud S.; Cabantous S.; Mourey L.; Guilhot C.; Chalut C. 4′-Phosphopantetheinyl transferase PptT, a new drug target required for Mycobacterium tuberculosis growth and persistence in vivo. PLoS Pathog. 2012, 8, e1003097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finking R.; Solsbacher J.; Konz D.; Schobert M.; Schafer A.; Jahn D.; Marahiel M. A. Characterization of a new type of phosphopantetheinyl transferase for fatty acid and siderophore synthesis in Pseudomonas aeruginosa. J. Biol. Chem. 2002, 277, 50293–50302. [DOI] [PubMed] [Google Scholar]
- Barekzi N.; Joshi S.; Irwin S.; Ontl T.; Schweizer H. P. Genetic characterization of pcpS, encoding the multifunctional phosphopantetheinyl transferase of Pseudomonas aeruginosa. Microbiology (Reading, U. K.) 2004, 150, 795–803. [DOI] [PubMed] [Google Scholar]
- Frye S. V. The art of the chemical probe. Nat. Chem. Biol. 2010, 6, 159–161. [DOI] [PubMed] [Google Scholar]
- Schweizer H. P. Understanding efflux in Gram-negative bacteria: Opportunities for drug discovery. Expert Opin. Drug Discovery 2012, 7, 633–642. [DOI] [PubMed] [Google Scholar]
- Wilkner M. A.; et al. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard, 8th ed.; CLSI Document M07-A8; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, 2009; Vol. 2. [Google Scholar]
- Pfaller M. A.; Chaturvedi V.; Espinel-Ingroff A.; Ghannoum M. A.; Gosey L. L.; Odds F. C.; Rex J. H.; Rinaldi M. G.; Sheehan D. J.; Walsh T. J.; Warnock D. W.. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Approved Standard, 2nd ed.; NCCLS Document M27-A2; National Committee for Clinical Laboratory Standards (NCCLS): Wayne, PA, 2002; Vol. 15. [Google Scholar]
- Sarker S. D.; Nahar L.; Kumarasamy Y. Microtitre plate-based antibacterial assay incorporating resazurin as an indicator of cell growth, and its application in the in vitro antibacterial screening of phytochemicals. Methods 2007, 42, 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.