

Abstract

NMDA receptors are tetrameric complexes composed of GluN1 and GluN2A–D subunits that mediate a slow Ca2+-permeable component of excitatory synaptic transmission. NMDA receptors have been implicated in a wide range of neurological diseases and thus represent an important therapeutic target. We herein describe a novel series of pyrrolidinones that selectively potentiate only NMDA receptors that contain the GluN2C subunit. The most active analogues tested were over 100-fold selective for recombinant GluN2C-containing receptors over GluN2A/B/D-containing NMDA receptors as well as AMPA and kainate receptors. This series represents the first class of allosteric potentiators that are selective for diheteromeric GluN2C-containing NMDA receptors.
Introduction
N-Methyl-d-aspartate (NMDA) receptors are members of the family of ionotropic glutamate receptors that mediate excitatory neurotransmission. NMDA receptors are tetrameric assemblies of two GluN1 subunits, which bind the coagonist glycine, and two GluN2 subunits, which bind glutamate.1 Both GluN1 and GluN2 subunits share a similar architecture, comprised of an extracellular amino-terminal domain (ATD), an extracellular ligand-binding domain (LBD), a transmembrane domain (TMD), and an intracellular carboxyl-terminal domain (CTD).2 The GluN2 subunit is encoded by four distinct gene products (GluN2A-D), which have temporally and spatially distinct expression patterns in the brain.3 The GluN2 subunit controls pharmacological characteristics such as agonist sensitivity, deactivation time course, mean open time, and open probability.2,3b,4
The distinct anatomical locations of the GluN2 subunits could allow subunit-selective modulators (either potentiators or inhibitors) to target specific brain regions for therapeutic gain. NMDA receptors are thought to play a role in neuronal development, learning, and memory formation,5 as well as being implicated in ischemia,6 dementia,7 schizophrenia,8 treatment resistant depression,9 and Parkinson’s disease.10 Recently discovered modulators have demonstrated selectivity for GluN2A, 3-chloro-4-fluoro-N-[4-[[2-(phenylcarbonyl)hydrazino]carbonyl]benzyl]benzenesulfonamide (TCN201); GluN2A/GluN2B, 9-cyclopropylphenanthrene-3-carboxylic acid (UBP710); and GluN2C/GluN2D, (3-chlorophenyl) [3,4-dihydro-6,7-dimethoxy-1-[(4-methoxyphenoxy)methyl]-2(1H)-isoquinolinyl]methanone (CIQ), 4-[6-methoxy-2-[(1E)-2-(3-nitrophen yl)ethenyl]-4-oxo-3(4H)quinazolinyl]benzoic acid (QNZ46), 5-(4-bromophenyl)-3-(1,2-dihydro-6- methyl-2-oxo-4-phenyl-3-quinolinyl)-4,5-dihydro-g-oxo-1H-pyrazole-1-butanoic acid (DQP1105), and (2R,3S)-1-(phenanthrenyl-3-carbonyl)piperazine-2,3-dicarboxylic acid (UBP141).11 Here, we describe the development of the first class of positive allosteric modulators that are selective for GluN2C-containing NMDA receptors over GluN2A-, GluN2B-, and GluN2D-containing receptors.
To identify this class of ligands, a GluN1/GluN2C cell line and multiwell fluorescence-based assay were developed to enable screening of compound libraries for NMDA receptor modulators. We screened two commercial diversity libraries to identify several compounds that modulate GluN2C-containing NMDA receptors. One of these screening hits established a novel class of subunit-selective potentiators for recombinant GluN1/GluN2C NMDA receptors, exemplified by compound 1 (Figure 1). Optimization of the initial lead pyrrolidinone scaffold involved the development of a structure–activity relationship, which led to the identification of a novel series of compounds with potency in the low micromolar range and high selectivity for recombinant GluN2C-containing receptors over GluN2A/B/D-containing NMDA receptors. In addition, no detectable potentiation was observed at recombinant AMPA, kainate, GABA, glycine, serotonin, nicotinic, or purinergic receptors (data not shown). These analogues represent a novel class of NMDA receptor modulators that are highly selective for diheteromeric GluN1/GluN2C receptor subtypes and provide a useful tool with which to evaluate the physiological role of GluN2C in normal and neuropathological conditions.
Figure 1.

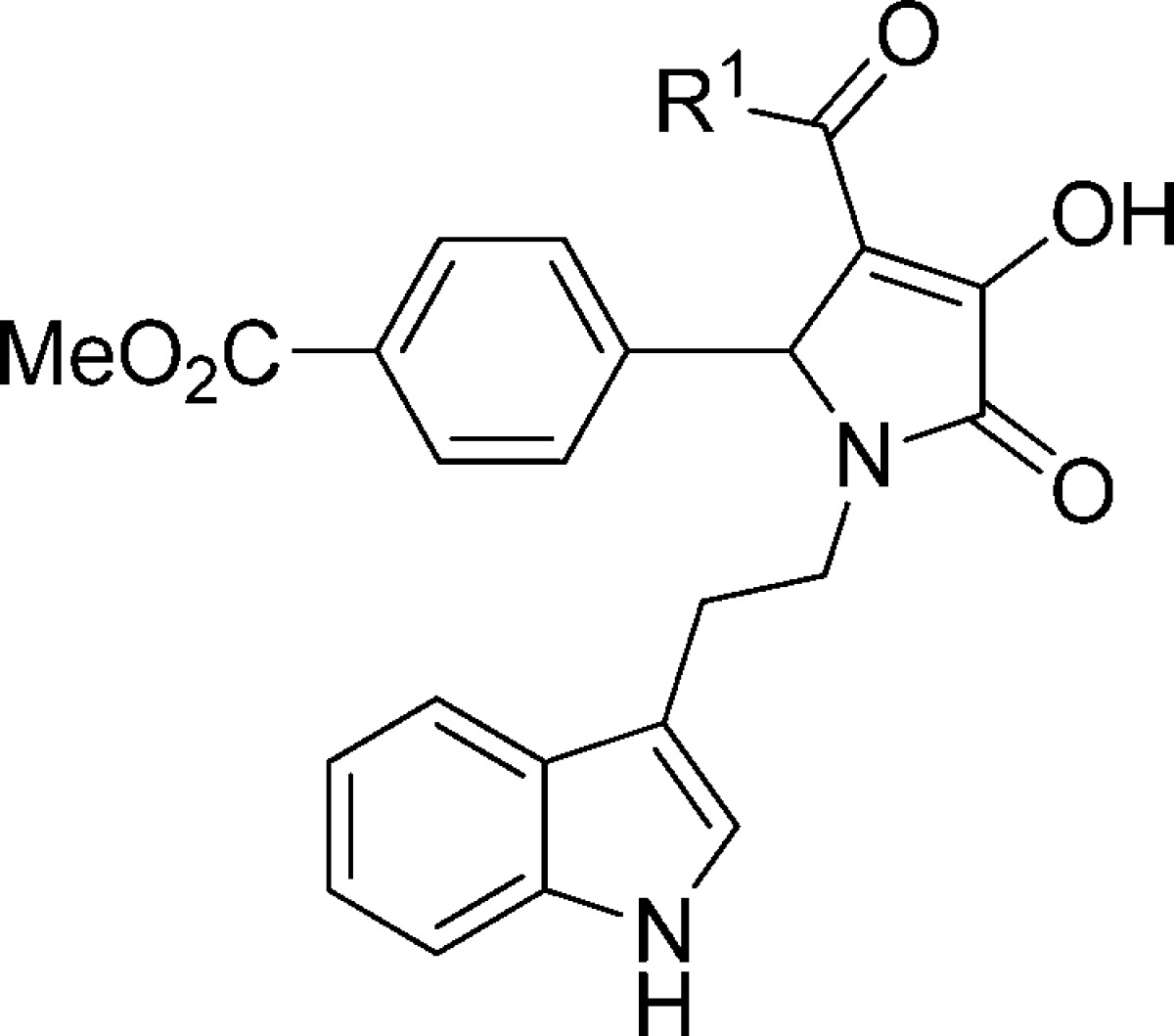
Structure of screening hit. Chemical structure of methyl 4-(1-(2-(1H-indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (compound 1) that was identified as a positive modulator using a fluorescence-based screen of compound libraries in a cell line expressing diheteromeric GluN1/GluN2C NMDA receptors.
Results
Chemistry
We used bioinformatic searches and medicinal chemistry to obtain analogues for our initial screening hit, compound 1 (see below). Both commercially available analogues and compounds synthesized via a mi-component Biginelli-like reaction (Scheme 1) were assessed at 30 μM. We determined the EC50 and maximal potentiation from concentration–effect curves for compounds that showed potentiation of more than 120% of control at 30 μM. No compounds in this class potentiated GluN2A-, GluN2B-, or GluN2D-containing receptors, suggesting remarkable selectivity for this class (see below). Modifications were made at either R1, the A-ring, or the B-ring using alternative methodologies to access the appropriate precursor.
Scheme 1. Synthesis of 1H-Pyrrol-2(5H)-ones.

Reaction conditions: PPTS, rt, 1–24 h, 2% to >99% (procedure I). Final compounds 161–180, in which either the A or B ring is replaced, were also prepared using these conditions.
Addition of diethyl oxalate and sodium ethoxide to a methyl ketone generated a series of pyruvate analogues (3–20) containing modifications at R1 (Scheme 2). Only when R1 was a phenol was it necessary to first protect the hydroxyl group with triisopropyl chloride (TIPSCl) before the addition of diethyl oxalate. Standard deprotection afforded the target pyruvate (21).
Scheme 2. Route for the Synthesis of Pyruvate Derivatives.

Reaction conditions: (a) diethyl oxalate, NaOEt, EtOH, 0 °C to rt, 4 h, 15% to >99% (procedure II); (b) TIPSCl, imidazole, rt, 6 h, >99%; (c) diethyl oxalate, NaOEt, EtOH, 0 °C to rt, 4 h, 28% (procedure II); (d) TBAF, 0 °C to rt, 1 h, 43%.
Analogues containing disubstituted A-rings were synthesized using several procedures based on the commercially available precursors (Scheme 3). Benzaldehydes 32–41 were prepared from methyl esters 145–154. Dibromination and hydrolysis afforded analogues 32 and 33.12 Suzuki coupling between dibutyl vinylboronate and the appropriately substituted methyl 4-iodobenzoate, followed by ozonolysis, gave phenols 34 and 35. Alternatively, addition of a Grignard reagent and N,N-dimethylformamide (DMF) led to isolation of benzaldehyde 36. A palladium-catalyzed formylation was used to access benzaldehydes 37–40.13 Finally, anisole 41 was prepared via a dialkylation of both the hydroxyl and carboxylic acid functional groups.
Scheme 3. Synthetic Routes to Access Substituted Benzaldehydes 32–41.

Reaction conditions: (a) 2.0 equiv NBS, (PhCOO)2, reflux, 4 h, then AgNO3, rt, 3 h, 38–63%; (b) dibutyl vinylboronate, 5 mol % (PPh3)2PdCl2, NaCO3, reflux, 2 h, 68–80%; (c) O3; then (CH3)2S, −78 °C to rt, 12 h, 60–87%; (d) i-PrMgCl, DMF, −15 °C to rt, 3 h, 70%; (e) CO(g), (PPh3)2PdCl2, NaCO3, 110 °C, 8–24% (procedure IV); (f) CH3I, K2CO3, rt, 3 h, 58%.
Benzaldehydes containing a para-amide (42–44) or para-ester (45–47) substituent were synthesized as illustrated in Scheme 4. Primary amide 42 was synthesized from carboxylic acid 155 by generating the acid chloride in situ. Standard amide coupling conditions were employed for the preparation of amides 43 and 44. Alkylation of carboxylic acid 155 with the appropriate alkyl iodide afforded esters 45 and 46, while t-butyl ester 47 was prepared using a method previously described.14
Scheme 4. Routes for the Synthesis of Amides 42–44 and Esters 45–47.

Reaction conditions: (a) Vilsmeier reagent, aq NH3, 0 °C, 16 h, 30%; (b) R2aR2bN where R2a = H and R2b = Me or where R2a = R2b = Me, DMAP, EDCI, 0 °C to rt, 24 h, 14–51%; (c) R2I where R2 = Et or R2 = i-Pr, K2CO3, rt, 4 h, 24–87%; (d) (CH3)2NCH(Ot-Bu)2, reflux, 11/2 h, 81%.
An alternative strategy was used to synthesize analogues containing a modification at R11 starting from pyrrolidinones 1 and 106 (Scheme 5). Protection of analogue 1 with trimethylsilyl diazomethane afforded methoxy 156. Amine 157 was generated by reaction with ammonium formate. Esterification of 1 with acetic anhydride gave acetate 158. Alternatively, esters 159 and 160 were synthesized from enol 106 using the appropriate acyl chloride and triethylamine.
Scheme 5. Route to Modifications at R11.

Reaction conditions: (a) TMSCH2N2, rt, 5 h, 46%; (b) NH4HCO2, reflux, 3 h, 14%; (c) Ac2O, pyridine, rt, 61/2 h, 7%; (d) R11C(O)Cl where R11 = CH2CH2CH3 or R11 = CH=CH2, TEA, −30 °C, 2 h, 20–35%.
Pyrrolidinones Selectively Potentiate GluN2C-Containing Receptors
A fluorescence-based screen of 57504 compounds obtained from Asinex and ChemDiv libraries was performed in BHK cells with inducible expression of GluN1/GluN2C receptors. Hits were defined as compounds that produced changes that were 2.5 standard deviations away from the average response to maximally effective agonist (i.e., glutamate and glycine) application. In this primary screen, 1% of the compounds met these criteria. Compounds that showed potentiation were further evaluated for their ability to produce responses in cells with no NMDA receptor expression (in uninduced cells) in order to identify false positive hits. False positive results can occur when the compounds directly release Ca2+ from intracellular stores, enhance Ca2+ channel function, possess fluorescent properties in the excitation/emission range of Fluo-4, or otherwise produce an increase in intracellular Ca2+ signal independent of NMDA receptor activation. Compounds that showed potentiation of glutamate responses in induced cells and did not produce responses in uninduced cells were subsequently studied by two-electrode voltage-clamp recording of NMDA receptor responses.
A single compound was found to selectively potentiate the GluN1/GluN2C receptors and did not show any activity at GluN2A/B/D-containing NMDA receptors expressed in Xenopus laevis oocytes (Figure 2A). Compound 1, which contains a pyrrolidinone core motif, potentiated GluN1/GluN2C responses to 238 ± 8.2% of control at 100 μM with an EC50 of 24 ± 2.4 μM (n = 12) (Figure 2B). Compound 1 had no agonist activity on its own in that it did not induce current responses in oocytes expressing GluN1/GluN2C in the absence of glutamate and glycine (n = 4). In addition, 30 μM of compound 1 did not potentiate homomeric recombinant GluA1 AMPA receptor responses (97 ± 1.1% control, n = 16). In addition, 120 μM of compound 1 did not potentiate homomeric GluK2 recombinant kainate receptors (95 ± 2.3% of control, n = 5).
Figure 2.

Compound 1 selectively potentiates the GluN1/GluN2C response. (A) Current traces for 1 at the GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, and the GluN1/GluN2D receptors. (B) Compound 1 selectively potentiates the GluN1/GluN2C receptor to a fitted maximum of 275 ± 10% with an EC50 of 24 ± 2.4, n = 12. (C) The EC50 for glycine in the absence and presence of 1 is 0.20 ± 0.01 μM (n = 6) and 0.16 ± 0.02 μM (n = 4), respectively. The EC50 for glutamate in the absence and presence of 1 is 0.8 ± 0.07 μM (n = 8) and 1.2 ± 0.04 μM (n = 6), respectively. The presence of 1 did not shift the glycine or glutamate EC50 values significantly. (D) The reversal potential is −5.1 ± 0.8 mV when activated by coagonists (100 μM glutamate and 30 μM glycine) and is −5.0 ± 1.2 mV (n = 6) when the GluN1/GluN2C receptor is potentiated by 1. The reversal potential was not significantly shifted in the presence of 1, suggesting that potentiation is independent of membrane potential.
Compound 1 (68 μM) did not detectably alter the EC50 of glycine or glutamate (n = 4–6; Figure 2C). Additionally, the reversal potential of glutamate and glycine induced current responses was unchanged in the presence (−5.0 + 1.2 mV, n = 6) or absence (−5.1 + 0.8 mV, n = 6) of compound 1. Potentiation was not significantly different at −40 mV (202 ± 11%) compared to +30 mV (180 ± 12%; p = 0.2679; paired t test), indicating that potentiation of GluN2C-containing receptors by compound 1 at 20 μM was voltage-independent (n = 6; Figure 2D).
Effect of Modifications to R1 on Potency at GluN2C-Containing Receptors
We subsequently evaluated the response to 30 μM of all pyrrolidinone analogues at GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, and GluN1/GluN2D and proceeded to determine the concentration–effect curve when potentiation exceeded 120% of control. Exploration of the effects of keto-linked R1 (Scheme 1; see Chemistry section) substitutions on potentiation of GluN2C-containing receptors in oocytes revealed that additional steric bulk was tolerated, with only minimal improvements in potency (Table 1, 62–65). For example, replacement of R1 with a phenyl group, as in 65, produced a small increase in potency (EC50 = 17 ± 2.3) accompanied by a modest decrease in maximal potentiation compared to compound 1 (Table 1). Analogues containing m-substituted phenyl rings (66–70) offered variable potentiation, while analogues with o- and p-substituted phenyl rings were inactive (data not shown). Notably, 66, with a meta-hydroxyl group, displayed a considerably higher potency at GluN2C-containing receptors (7.0 ± 0.9 μM) but caused significant inhibition of GluN2A-, GluN2B-, and GluN2D-containing receptors at 100 μM (responses were 76 ± 2.0%, 42 ± 1.6%, and 48 ± 2.4% of control, respectively, normalized to agonist activated current). Such mixed-action modulators that potentiate one subunit while inhibiting another are intriguing but of little utility as pharmacological probes. Two compounds containing a pyridine ring at R1 potentiated responses up to ∼200% with EC50 values of 12 ± 1.9 μM (72) and 8.9 ± 1.3 μM (73). Interestingly, 71, which contains a 2-substituted pyridine ring, was inactive at all receptor subunits. These initial experiments confirmed the ability of derivatives within this class to selectively potentiate GluN2C-containing receptors compared to other NMDA receptor subtypes.
Table 1. Optimization of Potency through Evaluation of Keto-Linked Substituents.
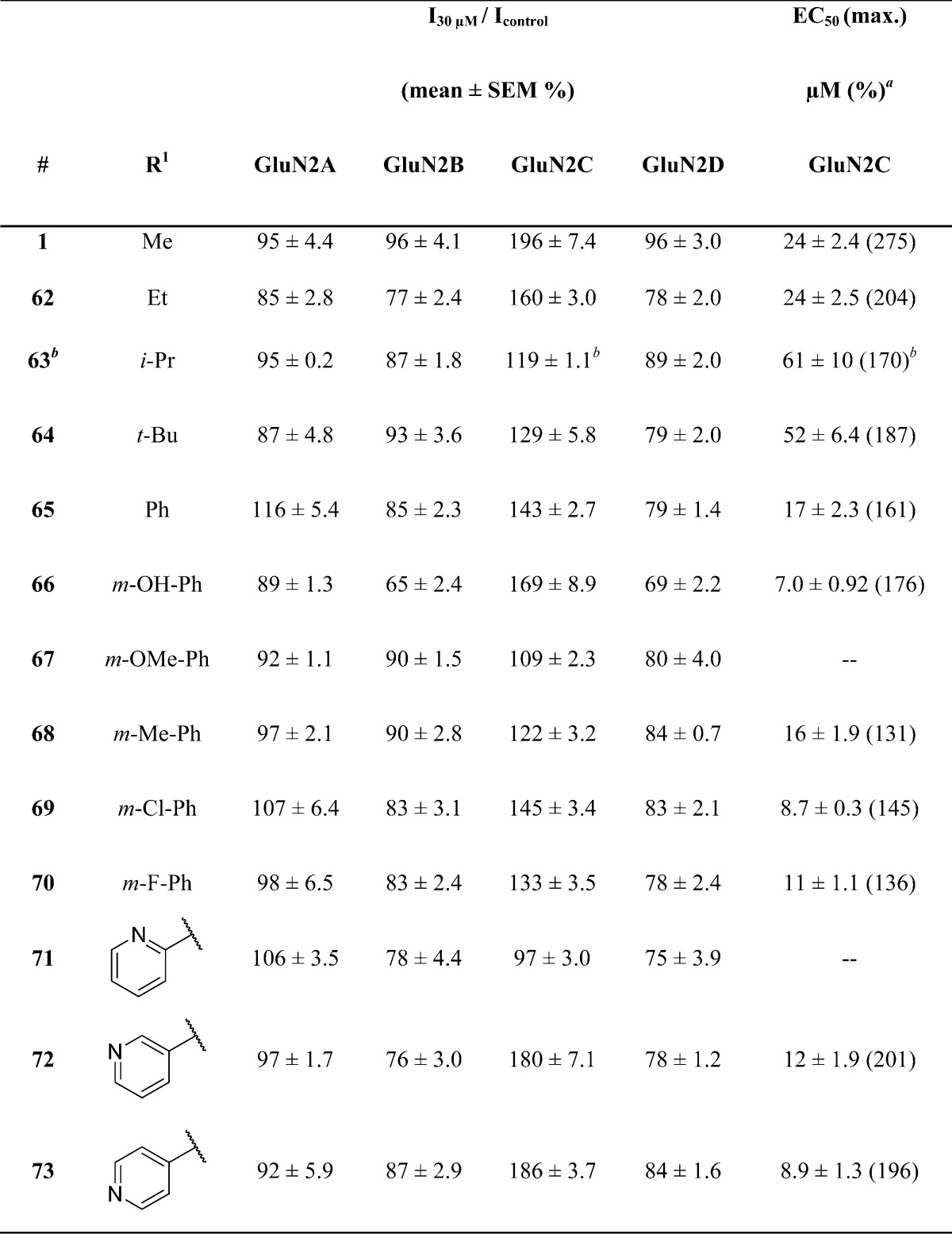
Fitted EC50 values are shown for GluN1/GluN2C to two significant figures when potentiation at 30 μM of the test compound exceeded 120% of control; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) response. Hill slopes varied between 1.2 and 2.0. Data for active compounds at GluN1/GluN2C are from between 6 and 12 oocytes from 2–3 frogs for each compound. When no effect was found (n = 3–15 oocytes), the lack of effect was confirmed by testing at 100 μM (data not shown, n ≥ 3 oocytes all compounds). For all tables, GluN2 subunits were coexpressed with GluN1 in Xenopus oocytes and evaluated using two-electrode voltage-clamp recordings.
The response to 100 μM of test compound was greater than 140% of control.
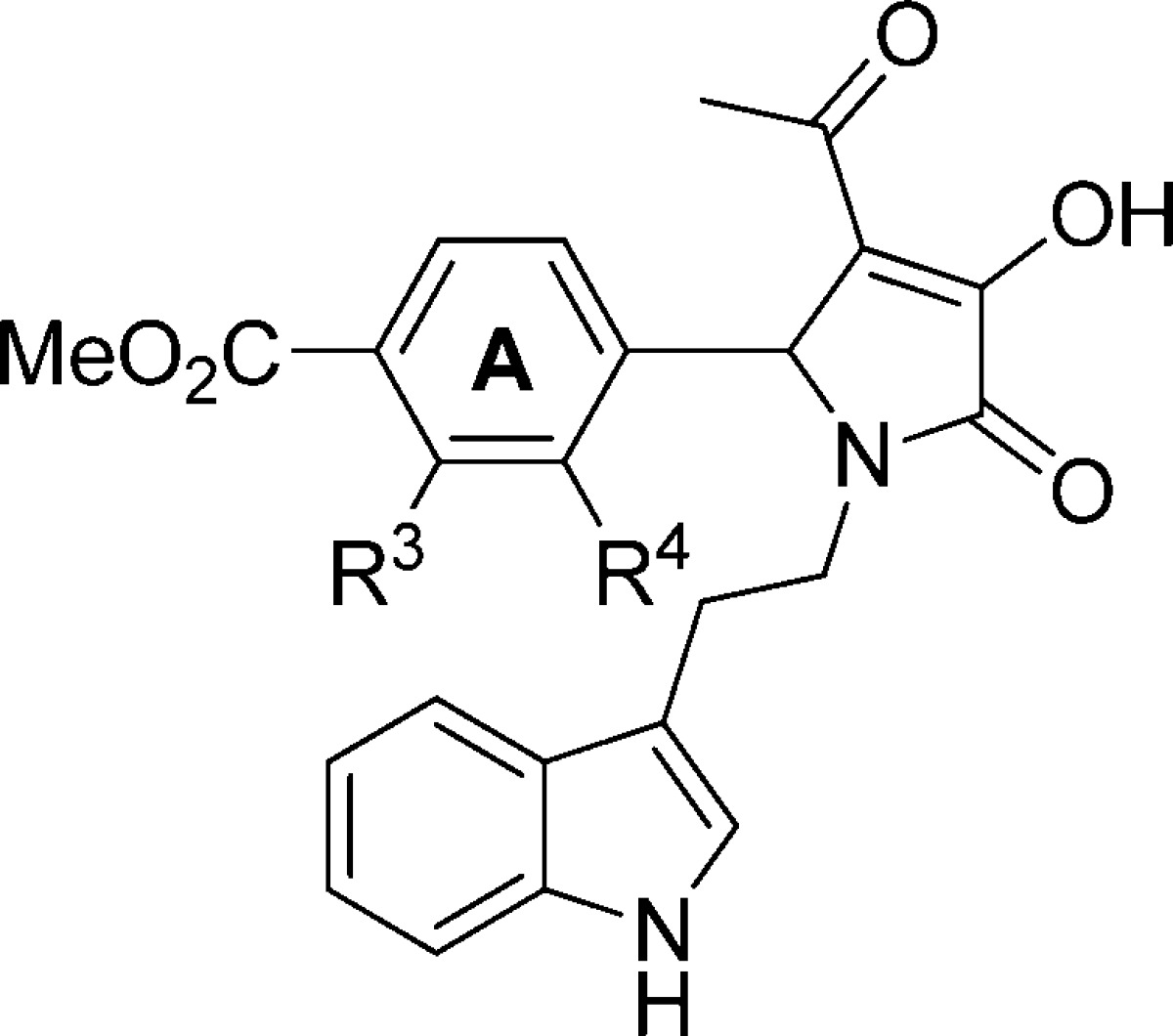
Effect of A-Ring Modifications
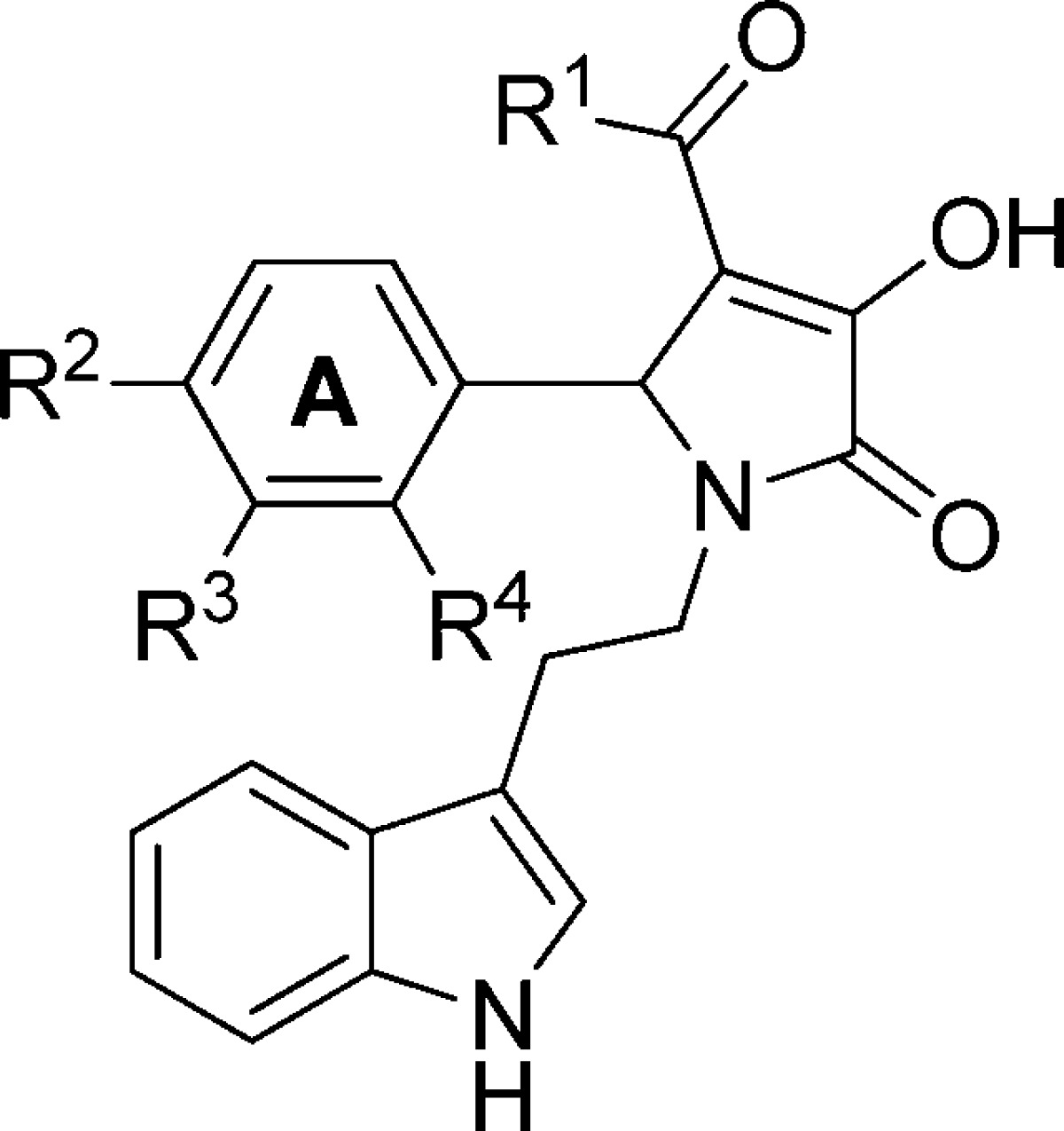
Next, we evaluated the effects of various A-ring substituents (Table 2) utilizing R1 substitutions shown to offer the desired activity. Positional isomer analogues 81 and 82 were inactive at GluN1/GluN2C. One compound, 84, which contains an ethyl ester at ring position R2, displayed comparable potency compared to screening hit 1. Analogues containing bulkier ester substituents (e.g., 85 with an iso-propyl ester and 86 with a tert-butyl ester) led to inactivity. A series of compounds containing ester isosteres including a nitrile (87), nitro (88), amide (89–91), and sulfonamide (92 and 93) were also evaluated for their ability to potentiate GluN2C-containing NMDA receptors. Unfortunately, none of these analogues exhibited any activity.
Table 2. Optimization of A-Ring Substituents.
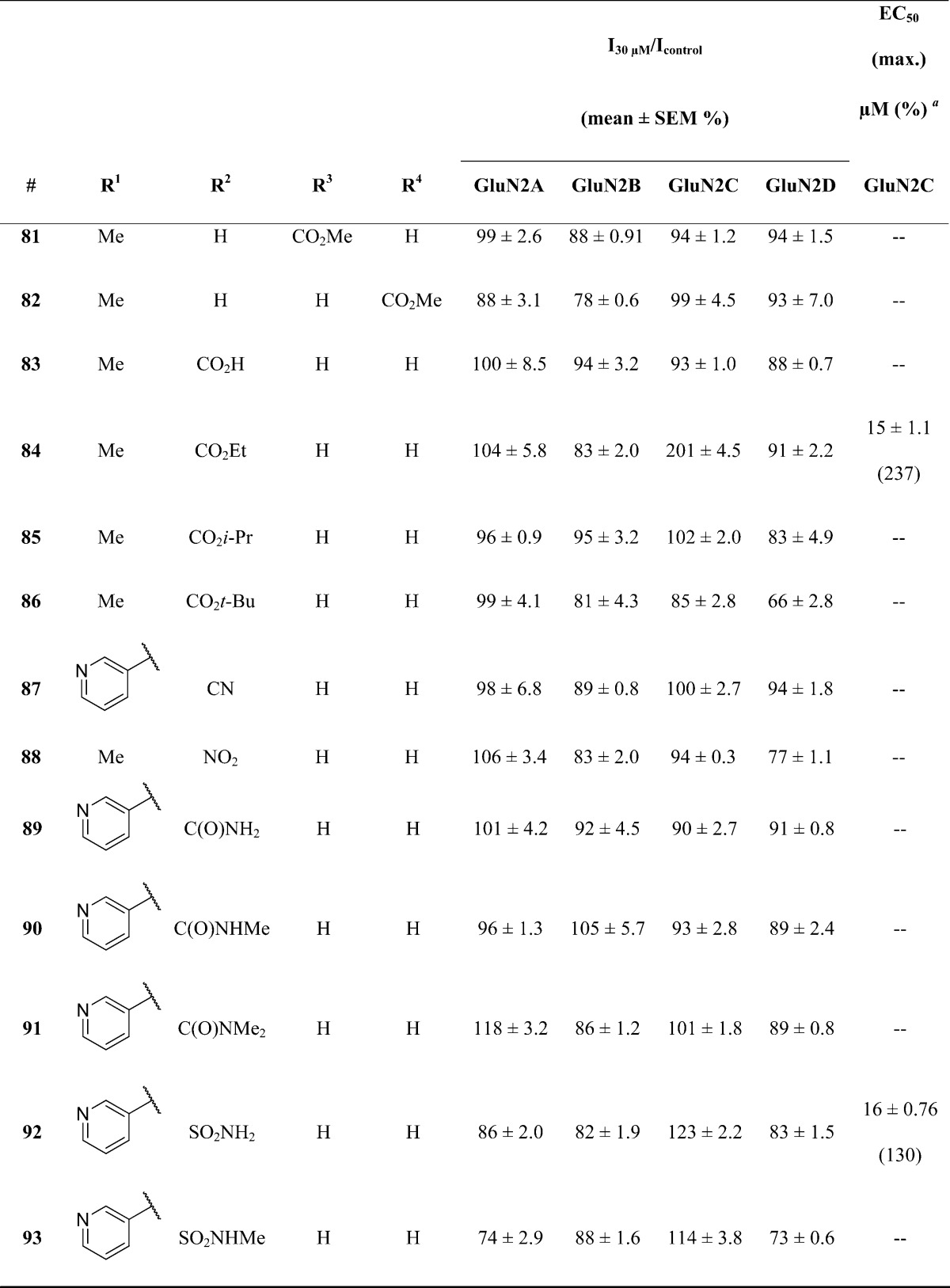
Fitted EC50 values are shown for GluN1/GluN2C to two significant figures when potentiation at 30 μM of the test compound exceeded 120% of control; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) response. Hill slopes varied between 1.3 and 1.7. Data for active compounds at GluN1/GluN2C are from between 8 and 12 oocytes from 2–3 frogs for each compound. When no effect was found (n = 3–11 oocytes), the lack of effect was confirmed by testing at 100 μM (data not shown, n ≥ 3 oocytes all compounds).
A variety of substituents at A-ring positions R3 and R4 were systematically tested while holding the para-methyl ester constant at R2 (Table 3). Substitution at the meta position (R3) revealed either a reduction in potency (95) or complete inactivity (96–99). Evaluation of a series of ortho (R4) ring substituents demonstrated a preference for electron donating groups. For example, analogues containing an ortho-hydroxyl (100) exhibited potentiation with a modest increase in potency, whereas ortho-chloro (103) or -fluoro (104) substituents were slightly less active.
Table 3. Evaluation of Combinations of A-Ring Substituents.
| I30 μM/Icontrol (mean ± SEM %) |
EC50 (max) μM (%)a | ||||||
|---|---|---|---|---|---|---|---|
| # | R3 | R4 | GluN2A | GluN2B | GluN2C | GluN2D | GluN2C |
| 95 | OH | H | 81 ± 1.7 | 79 ± 1.2 | 123 ± 2.5 | 90 ± 3.0 | 29 ± 2.8 (151) |
| 96 | OMe | H | 98 ± 3.3 | 80 ± 1.8 | 92 ± 2.4 | 86 ± 1.4 | |
| 97 | Me | H | 108 ± 3.4 | 91 ± 2.5 | 88 ± 2.1 | 86 ± 0.4 | |
| 98 | Cl | H | 88 ± 4.3 | 95 ± 5.1 | 113 ± 4.2 | 83 ± 1.5 | |
| 99 | F | H | 99 ± 4.1 | 83 ± 2.2 | 114 ± 3.3 | 93 ± 5.3 | |
| 100 | H | OH | 107 ± 3.9 | 86 ± 3.8 | 173 ± 3.0 | 88 ± 1.9 | 15 ± 0.6 (202) |
| 101 | H | OMe | 102 ± 6.1 | 83 ± 0.3 | 132 ± 3.3 | 100 ± 3.1 | 46 ± 19 (183) |
| 102 | H | Me | 101 ± 1.8 | 95 ± 4.2 | 129 ± 3.6 | 85 ± 1.9 | 35 ± 1.4 (165) |
| 103 | H | Cl | 103 ± 3.9 | 87 ± 1.7 | 139 ± 2.8 | 90 ± 0.6 | 36 ± 3.0 (191) |
| 104 | H | F | 93 ± 2.5 | 96 ± 2.0 | 123 ± 3.4 | 91 ± 1.1 | 37 ± 2.6 (155) |
Fitted EC50 values are shown for GluN1/GluN2C to two significant digits when potentiation at 30 μM of the test compound exceeded 120% of control; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) response; Hill slopes ranged between 1.3 and 1.8. Data for active compounds at GluN1/GluN2C are from between 3 and 12 oocytes from 2–3 frogs for each compound. When no effect was found (n = 3–15 oocytes), the lack of effect was confirmed by testing at 100 μM (data not shown, n ≥ 5 oocytes for all compounds).
Effect of B-Ring Modifications
Replacement of the B-ring with an assortment of acyclic, cyclic, and heterocyclic systems generated a series of compounds that were evaluated for potency and subunit selectivity while retaining optimal R1 and A-ring substitutions (Table 4). Interestingly, substitution with a napthyl derivative, as in 162, led to strong inhibition at all four subunits. Replacement of the indole NH with an oxygen atom led only to weak activity (163), suggesting the presence of a hydrogen bond in the binding pocket. In all other instances, removal of the indole led to complete inactivity (i.e., 164 and 165). These data suggest that the indole functionality is preferred for activity.
Table 4. Effect of Replacing the B-Ring.
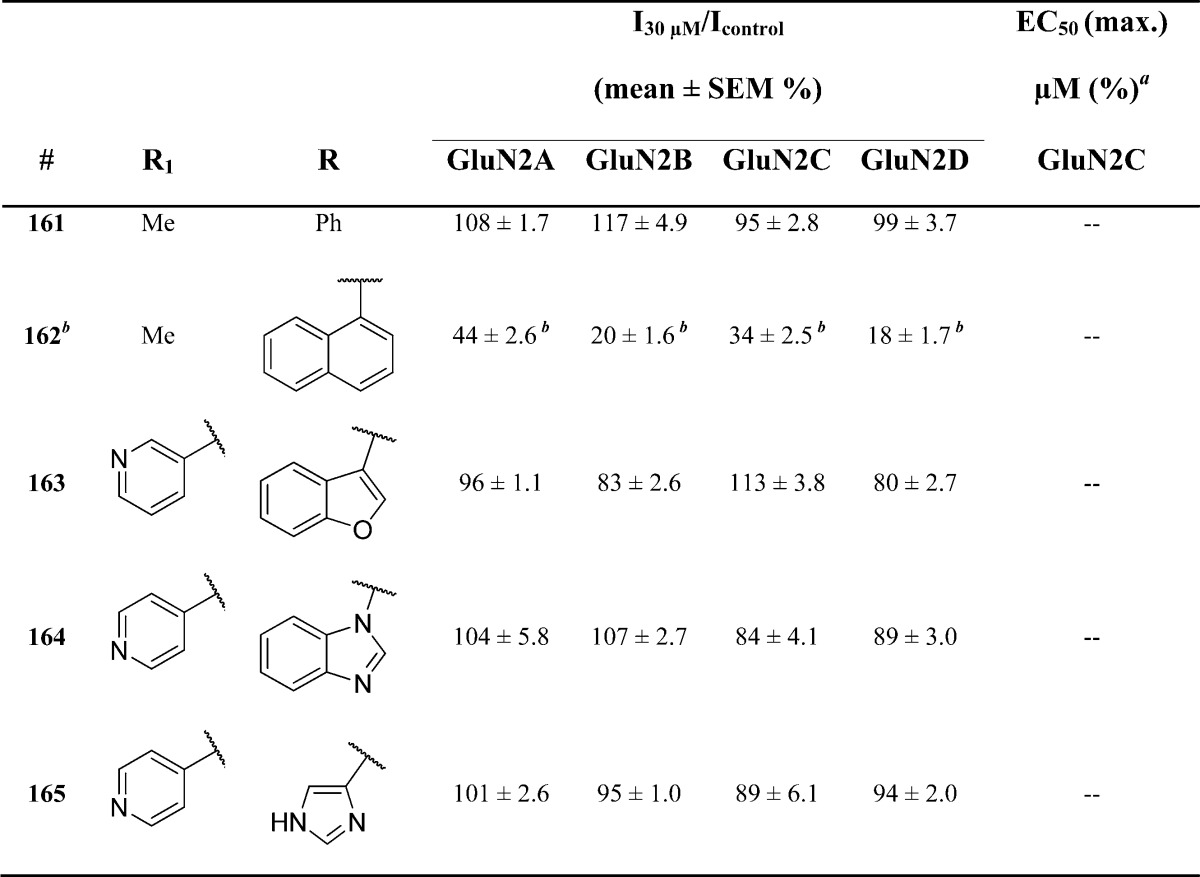
Fitted EC50 values are shown for GluN1/GluN2C to two significant digits when potentiation at 30 μM of the test compound exceeded 120% of control; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) response. All data are from 3–14 oocytes from 2–3 frogs. When no effect was found, the lack of effect was confirmed by testing at 100 μM (data not shown, n ≥ 3 oocytes for all compounds).
Inhibited at GluN1/GluN2A with an IC50 of 18 μM, at GluN1/GluN2B with an IC50 of 7.2 μM, at GluN1/GluN2C with an IC50 of 11 μM, and GluN1/GluN2D with an IC50 of 5.7 μM.
This led us to examine B-ring substituents as an alternative strategy to access increased potency. The data describing these compounds is summarized in Table 5. Methylation of the indole nitrogen led to inactivity (105), further suggesting the importance of a hydrogen atom at this position in the binding pocket. The best potency was obtained for analogues with substitutions at B-ring position R9. Compound 111 demonstrated an ability to selectively potentiate GluN2C-containing NMDA receptors up to 218% with an EC50 value of 4.3 ± 0.3 μM. It is unclear whether the increase in potency observed for 111 can be ascribed to a steric effect or, alternatively, to a mildly electropositive effect. Consistent with a steric effect, analogues which contain larger R9 substituents such as R9 = OMe (112) revealed a loss of potency compared to 111. Analogues containing strongly electron withdrawing R9 substituents such as R9 = F (109) also decreased the observed activity.
Table 5. Optimization of B-Ring Substituents.
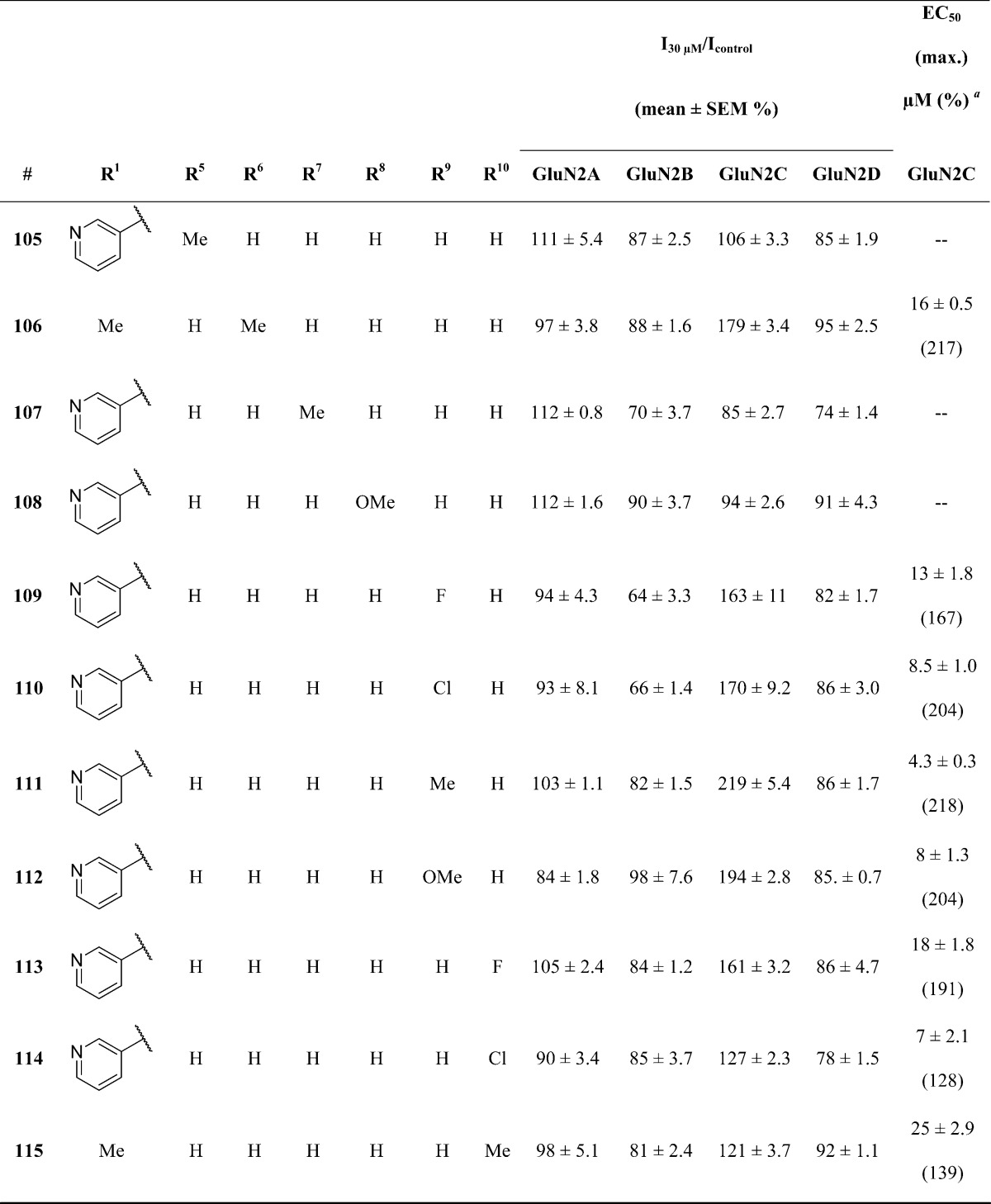
Fitted EC50 values are shown for GluN1/GluN2C to two significant digits when potentiation at 30 μM of the test compound exceeded 120% of control; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) response. Hill slopes were between 1.3 and 1.9. Data for active compounds at GluN1/GluN2C are from between 6 and 27 oocytes from 2–3 frogs for each compound. When no effect was found (n = 4–11 oocytes), the lack of effect was confirmed by testing at 100 μM (data not shown, n ≥ 4 oocytes for all compounds).
Effect of Combinatorial Modifications on Potency at GluN2C-Containing Receptors
We subsequently evaluated the effect of combining modifications at R1, the A-ring and the B-ring that had previously demonstrated an improvement in potency (Table 6). Substitution with either a meta- or para-substituted pyridine ring at R1 and a para-ethyl ester at R2 revealed potentiation of GluN1/GluN2C responses with EC50 values of 8.2 ± 0.9 μM (116) and 9.7 ± 0.6 μM (117), respectively. Modification of the B-ring and either R1 (R1 = m-pyridine) or R2 (R2 = p-CO2Et) exhibited a similar increase in on-target potency. For example, substitution with a methyl group at R6 and a para-ethyl ester at R2, as in analogue 119, resulted in a 2-fold potency enhancement.
Table 6. Optimization of Potency though Additional Modifications.
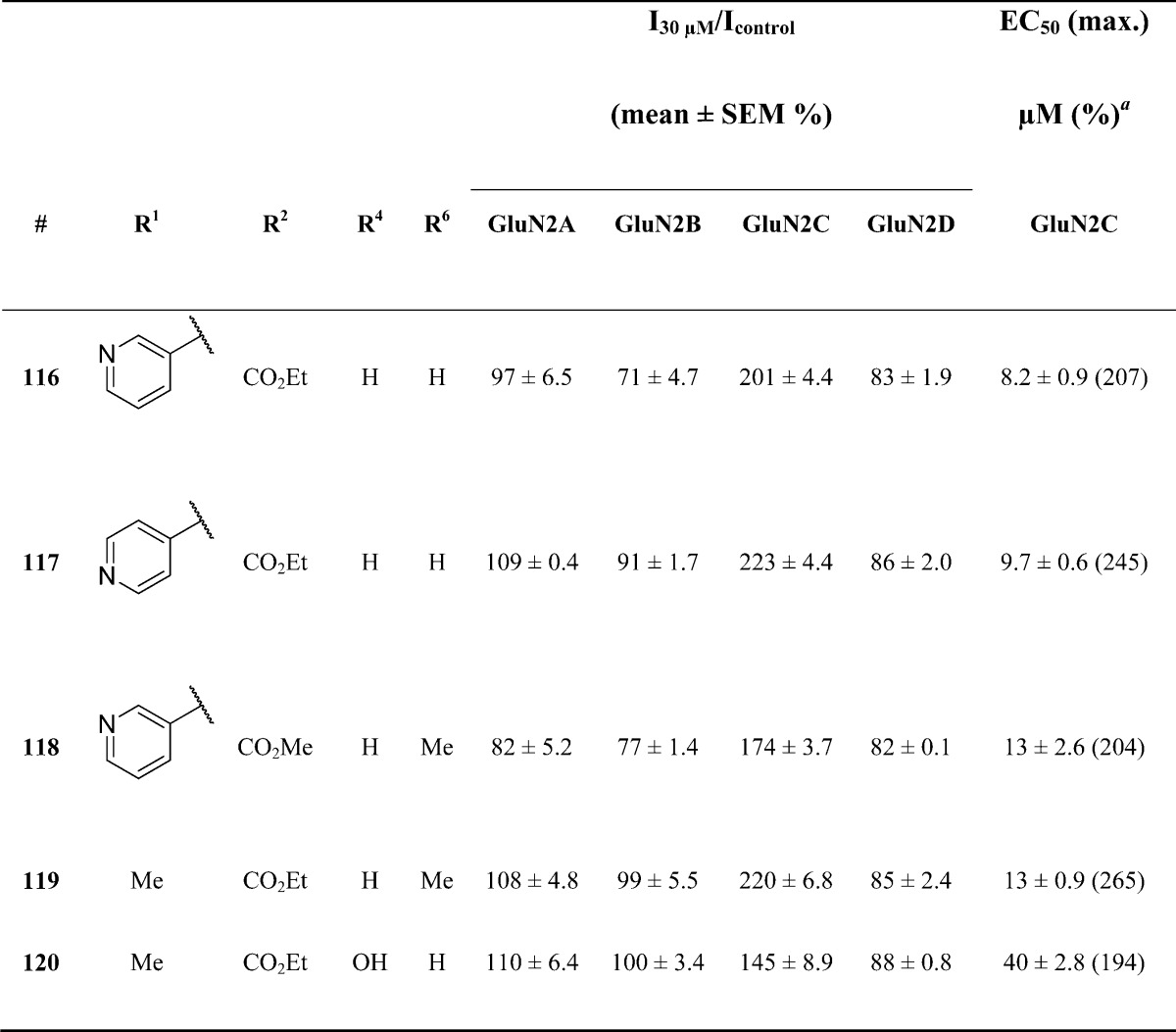
Fitted EC50 values are shown for GluN1/GluN2C to two significant digits when potentiation at 30 μM of the test compound exceeded 120% of control; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) response. Data for active compounds at GluN1/GluN2C are from between 8 and 14 oocytes from 2 frogs for each compound; the Hill slope varied between 1.2 and 1.5. When no effect was found at 30 μM (n = 3–6 oocytes), the lack of effect was confirmed by testing at 100 μM (data not shown, n ≥ 7 oocytes for all compounds).
Effect of Linker Modifications on Potency of 1616-Series
The original screening hit, 1, contains a two carbon region linking the B-ring with the core pyrrolidinone. The linker modifications explored are illustrated in Table 7. Both shortening (121) and extending (122) the linker eliminated all activity, suggesting that the potency of pyrrolidinone analogues is highly dependent on the length of the carbon linkage.
Table 7. Optimization of Potency though Linker Modifications.
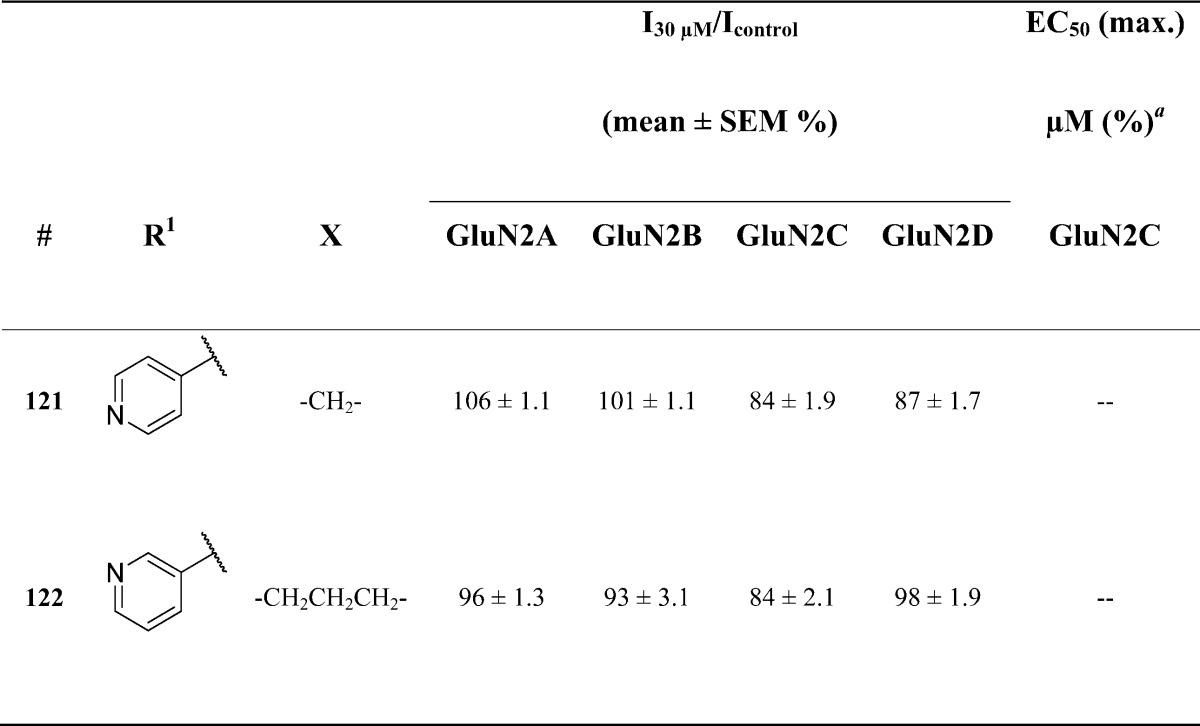
Fitted EC50 values are shown for GluN1/GluN2C to two significant digits when potentiation at 30 μM of the test compound exceeded 120%; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) response. Data for active compounds at GluN1/GluN2C are from between 7 and 8 oocytes from 2 frogs for each compound tested; the Hill slope varied between 1.3 and 1.4. When no effect was found at 30 μM (n = 3–11 oocytes), the lack of effect was confirmed by testing at 100 μM (data not shown, n ≥ 4 oocytes for all compounds).
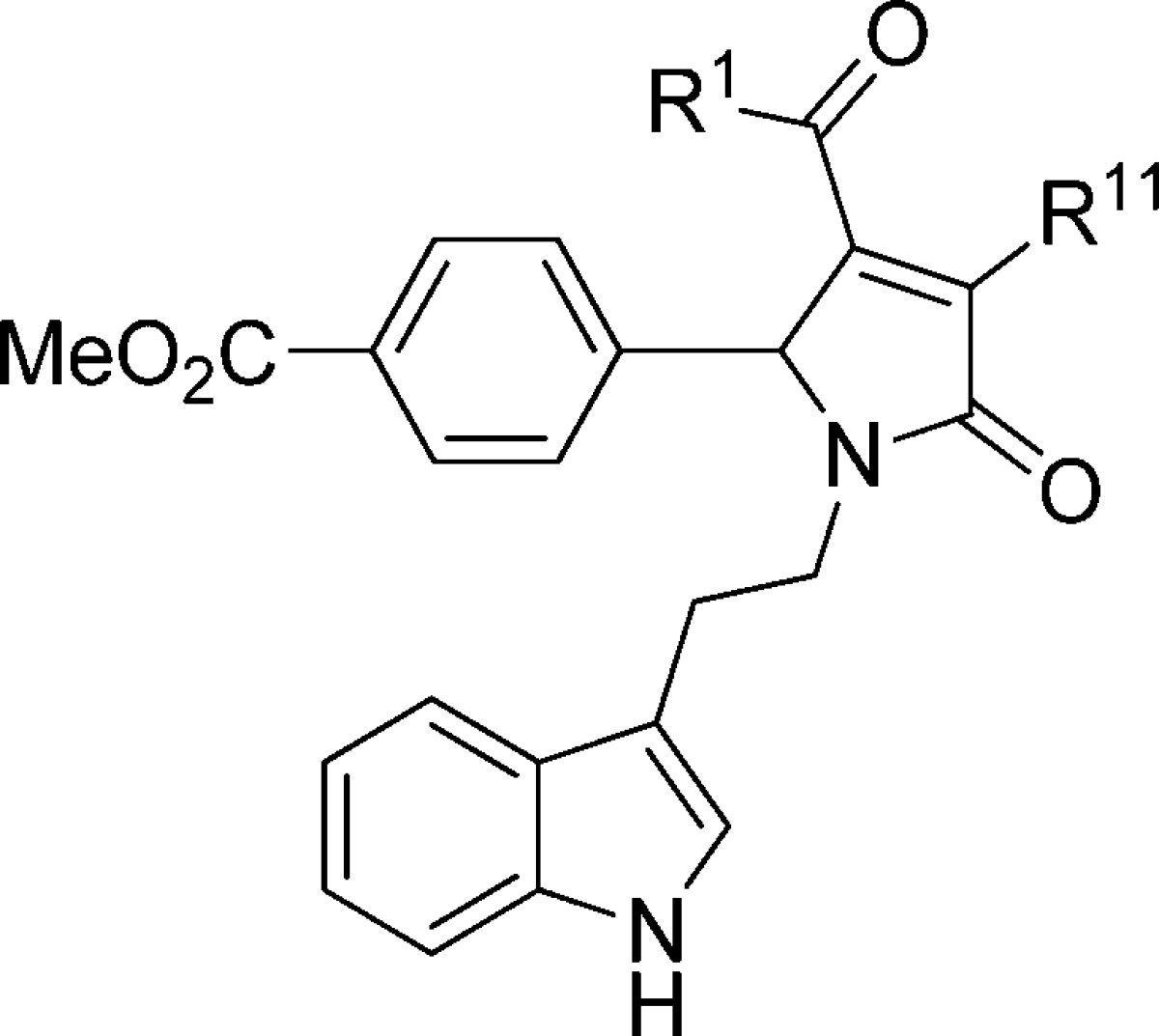
Effect of Modifications to R11 on Potency at GluN2C-Containing Receptors
Several modifications were made at R11 to determine the significance of the enol in controlling potency and selectivity (Table 8). Replacement with an amine, as in 157, led to a complete loss of potentiation at concentrations up to 100 μM. In most instances, compounds containing a protected alcohol led to less potent analogues. For example, a 2-fold decrease in potency was observed for acetate 158. In contrast, propyl ester 159 maintained activity comparable to lead analogue 1, with an EC50 of 17 ± 1.8 μM. These data suggest that enhancements in potency cannot be gained though modifications of the enol.
Table 8. Optimization of Potency though Evaluation of Vinyl Substituents.
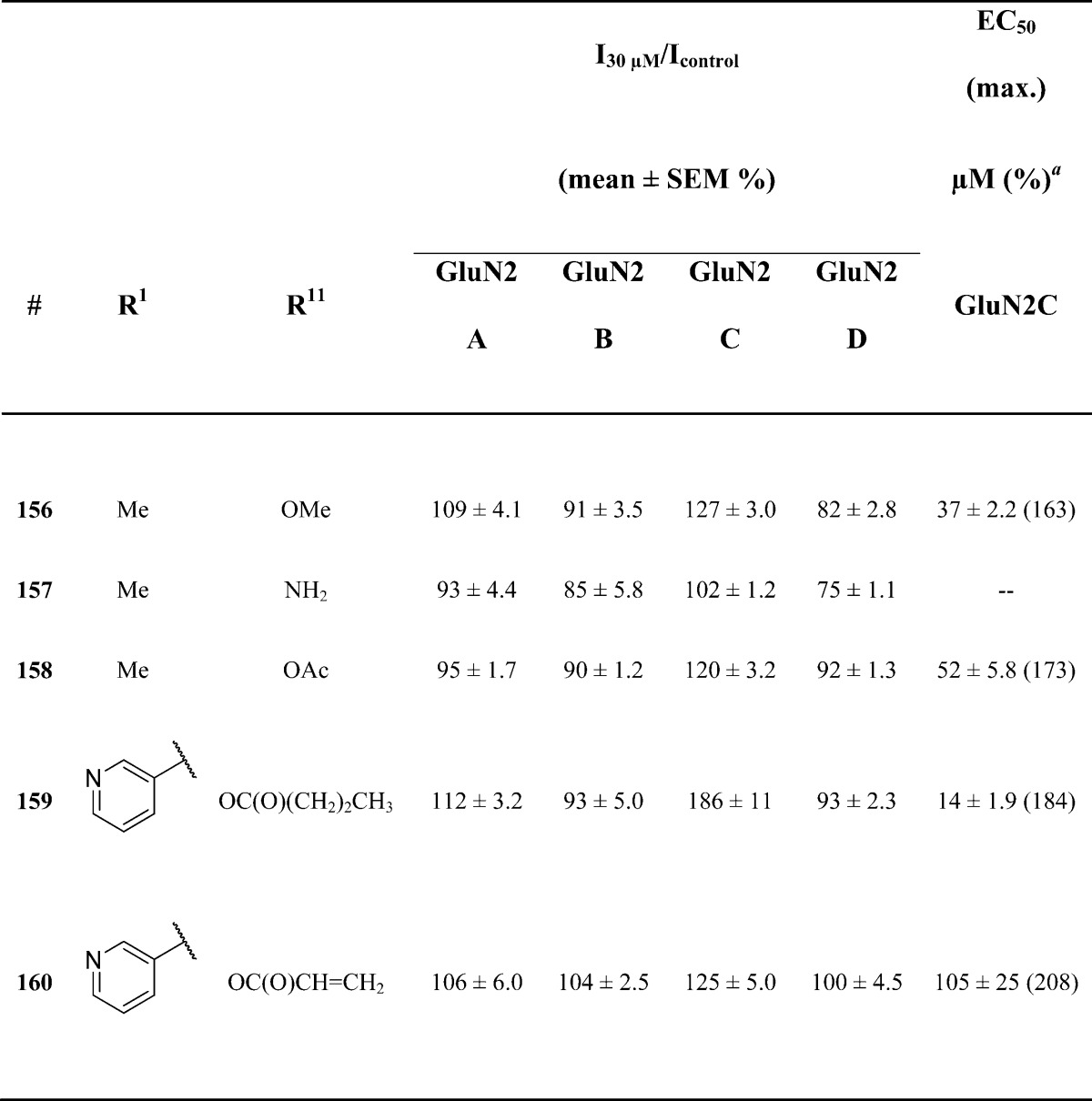
Fitted EC50 values are shown for GluN1/GluN2C to two significant digits when potentiation at 30 μM of the test compound exceeded 120% of control; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) response. Data for active compounds at GluN1/GluN2C are from between 5 and 9 oocytes from 2–3 frogs for each compound. The Hill slope varied between 1.2 and 1.8 and was fixed to be 1.5 for less potent analogues (157, 159). When no effect was found (n = 3–9 oocytes), the lack of effect was confirmed by testing at 100 μM (data not shown, n ≥ 4 oocytes for all compounds).
Effect of Absolute Configuration on Potency at GluN2C-Containing Receptors
To enable evaluation of potential stereoselectivity for pyrrolidinone analogues at GluN1/GluN2C, we separated the enantiomers of 106 using a semipreparatory OD-RH chiral HPLC column (see Chemistry Experimentals). Each enantiomer was subjected to two-electrode voltage clamp analysis in Xenopus laevis oocytes. The results, illustrated in Figure 3, indicate that only one enantiomer (106a) is active and may account for the activity of 106. Compound 106a potentiated GluN2C response by 259 ± 7.8% with an estimated EC50 value of 18 ± 0.6 μM (n = 6). In contrast, no activity was observed for the other enantiomer (106b) (n = 6). The active analogue demonstrated weak inhibition at GluN2D-containing receptors and had no effect at GluN2A- or GluN2B-containing receptors. Compound 106b was inactive at all other subunits. These data suggest that the activity of pyrrolidinone analogues may rely on a single enantiomer and that the binding pocket can distinguish between the enantiomers.
Figure 3.

Composite concentration–effect curves for 106 enantiomers. Concentration–effect curves for the enantiomers of 106 demonstrate that only one enantiomer, 106a, is active, potentiating the GluN1/GluN2C receptor to a fitted maximum of 259 ± 8% of control with an EC50 of 18 ± 0.6 μM (n = 6).
Conclusion
The incorporation of a methyl group at the C-7 position of the indole of initial screening hit 1 afforded 111, which selectively potentiates GluN2C-containing NMDA receptors with a potency of 4.3 ± 0.3 μM. In addition, the activity of this series appears to originate from one enantiomer. These compounds represent the first class of allosteric potentiators selective for diheteromeric GluN1/GluN2C receptors over receptors containing GluN2A-, GluN2B-, and GluN2D subunits. Future studies will address the activity of this series of modulators on triheteromeric GluN2C-containing NMDA receptors containing two different GluN2 subunits (e.g., GluN1/GluN2A/GluN2C). This series of molecules may serve as a pharmacological tool to evaluate the role of the GluN2C subunit in normal and neuropathological function.
Experimental Methods
Biology Experimentals
All protocols involving Xenopus laevis were approved by the Emory University Institutional Animal Care and Use Committee. Two-electrode voltage-clamp recordings were made from Xenopus laevis oocytes expressing recombinant GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, GluN1/GluN2D, GluA1, or GluK2 receptors following injection of cRNA. cDNAs for rat GluN1–1a (GenBank accession numbers U11418 and U08261; hereafter GluN1), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), GluN2D (L31611), GluA1 (X17184), and GluK2 (Z11548) were provided by Drs. S. Heinemann (Salk Institute), S. Nakanishi (Kyoto University), and P. Seeburg (University of Heidelberg). Oocyte isolation, cRNA synthesis, and cRNA injection have been previously described;15 some experiments were performed with oocytes obtained from Ecocyte (Austin, TX). Voltage-clamp recordings from oocytes were made during perfusion with recording solution containing 90 mM NaCl, 1.0 mM KCl, 0.5 mM BaCl2, 0.005 mM EDTA, and 10 mM HEPES at pH 7.4 (23 °C). Glass microelectrodes had resistances of 0.3–1.0 MΩ and were filled with 0.3–3.0 M KCl; the membrane potential was held at −40 mV for all recordings. Compounds were made as 20 mM stock solutions in DMSO and diluted to the final concentration in recording solution; final DMSO content was 0.05–0.5% (v/v). Oocytes expressing GluK2 receptors were pretreated with 10 μM concanavalin A for 10 min. NMDA receptors were activated by 100 μM glutamate plus 30 μM glycine; GluA1 and GluK2 receptors were activated by 100 μM glutamate. To prevent a gradual increase in current response over the course of the experiment of GluN1/GluN2A receptor responses in oocytes, some oocytes expressing GluN1/GluN2A were injected with 20–50 nL of 2 mM K-BAPTA (potassium 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid). When the response to agonist in the presence of 30 μM of a test compound exceeded 120% of control, the response to glutamate and glycine in the absence and presence of 5–7 concentrations of active analogues were recorded in multiple oocytes obtained from two or more different frogs for all experiments. The EC50 (half-maximally effective concentration of potentiator) was determined by fitting the equation
| 1 |
to the concentration–response data normalized to the current in the absence of potentiator (100%) for each oocyte, and the mean (±SEM) presented. N is the Hill slope, which ranged between 1 and 2 and is not reported; maximum is the fitted maximal response expressed as a percent of control to a saturating concentration of potentiator. When responses were inhibited by test compound at 30 μM to less than 60% of control, the IC50 value was determined by fitting the equation
| 2 |
to the concentration–response data normalized to the current. For some compounds, visual detection of precipitation led to inclusion of 1–10 mM 2-hydroxypropyl-β-cyclodextrin in the recording solution to enhance solubility and enable generation of the full concentration–response data.
To generate a cell line with inducible NMDA receptor expression, we used a previously described Tet-On (tetracycline-inducible promoter; Clontech, Mountain View, CA) baby hamster kidney (BHK-21, ATCC CCL-10) cell line.16 The BHK-21 Tet-On cell line was maintained at 37 °C, 5% CO2, and 95% relative humidity in culture medium composed of Dulbecco’s Modified Eagle Medium (DMEM) containing GlutaMAX-I, 4500 mg/L glucose, and 110 mg/L sodium pyruvate (Invitrogen, Carlsbad, CA) supplemented with penicillin (100 units/mL), streptomycin (100 μg/mL), (Invitrogen, Carlsbad, CA), 10% dialyzed fetal bovine serum (Invitrogen, Carlsbad, CA), and 1 mg/mL G418 (Invitrogen, Carlsbad, CA). The selection marker G418 was always included to provide continuous selection of Tet-On-compatible BHK-21 cells. The cells were cotransfected with rat GluN1-1a (GenBank accession no. U11418) in the inducible pTRE2 vector and rat GluN2C (GenBank accession no. D13212) in the pCI-IRES-bla vector (see ref (16) for details on this vector) using Fugene 6 transfection reagent (Promega, Madison, WI). The ratio of GluN1 and GluN2C DNA used for transfection was 10:1. The NMDA receptor antagonists dl-2-amino-5-phosphonopentanoate (AP5) (200 μM; Abcam, Cambridge, MA) and 7-chloro-kynurenate (7-CKA) (200 μM; Abcam, Cambridge, MA) were added to the culture medium to prevent NMDA receptor-mediated cell death. The following day, the cells were diluted 1:1000 and 1:10,000 and seeded in 144 mm dishes. The next day (e.g., two days after transfection), 10 μg/mL blasticidin S (Invivogen, San Diego, CA) was added to the culture medium to select for transfected cells. Unless otherwise stated, the culture medium for the cell lines always contained 1 mg/mL G418 and 10 μg/mL blasticidin S for selection as well as 200 μM AP5 and 200 μM DCKA to prevent NMDA receptor-mediated cell death. The media was changed every 2–3 days, and blasticidin S-resistant clones were isolated 10–20 days after transfection and evaluated for their response properties. Fluorescence-based assays were conducted as previously described,17 and test compounds were screened at 10 μM.
Chemistry Experimentals
Compounds for which synthesis is not described were purchased from commercial vendors. Purity of purchased compounds was greater than 90%, as determined by the suppliers, via HPLC or NMR.
All dry solvents were obtained from a Glass Contour System. Reagents used were acquired from commercial suppliers and utilized without additional purification. Precoated glass plates (silica gel 60 F254, 0.25 mm) were used to monitor the progress of reactions by thin layer chromatography (TLC). Purification by flash column chromatography was performed on a Teledyne ISCO Combiflash Companion using prepackaged Teledyne RediSep disposable normal phase silica columns. Melting temperatures were determined on a Mel-Temp apparatus and are uncorrected. 1H and 13C NMR experiments were each carried out on an INOVA-400 (400 MHz), VNMR 400 (400 MHz), INOVA-600 (600 MHz), Unity-600 (600 MHz), or Mercury 300 Vx (300 MHz). All chemical shifts are reported in parts per million and referenced to the residual solvent peak. All coupling constants are reported in hertz (Hz). The IR spectra were acquired with a Nicolet Avatar 370 DTGS. Mass spectra were performed by the Emory University Mass Spectrometry Center on a VG 70-S Nier Johnson or JEOL instrument. Purity of all final compounds was found to be ≥95% by LC/MS analysis unless otherwise noted.
Separation of Enantiomers
The separation of the enantiomers of 106 was obtained using a ChiralPak OD-RH 30 mm × 250 mm, 5 μm column with the following conditions: flow rate 10 mL/min, injection volume 1–2 mL (5 mg/mL), 44% ACN/66% water with 0.1% formic acid; 106atR = 121.3 min; 106btR = 129.3 min. Enantiomeric excess (ee) of both enantiomers 106a and 106b was determined using a ChiralPak OD-RH 4.6 mm × 150 mm, 5 μm column with the following conditions: flow rate 0.5 mL/min, injection volume 10 μL, 44% ACN/66% water with 0.1% formic acid; 106a [α]D20 −18 (c = 0.10, methanol), tR = 26.1 min, 98% ee; 106b [α]D + 9 (c = 0.10, methanol), tR = 29.1 min, 96% ee. A Perkin-Elmer 314 instrument was used to obtain optical rotation data.
General Procedure for Synthesis of Pyrrolidinone Compounds (Procedure I: 1, 62–82, 84–122, 161–180)
To a stirred solution of aldehyde (1.0 mmol) in dioxane (1.0 M) was added tryptamine (1.0 equiv) and 10 mol % pyridinium 4-methylbenzenesulfonate. Upon the formation of a slurry, methyl acetopyruvate (1.0 equiv) was added. The resulting mixture was allowed to stir at rt for up to 12 h. In most instances, a precipitate was visible, which was collected via filtration and washed with Et2O. The solid was dissolved in an appropriate solvent and washed with saturated ammonium chloride and brine before being dried over MgSO4, filtered, and concentrated in vacuo. If a precipitate did not form, the mixture was concentrated in vacuo before being subjected to the workup as described above. Purification was achieved via flash column chromatography on SiO2 (MeOH/DCM) to afford the desired pyrrolidinone. Additional purification was obtained by HPLC (85% ACN/15% water with 0.1% formic acid) as needed.
General Preparation of Pyruvate Compounds (Procedure II: 3–20, 144)
To a solution of sodium ethanolate (1.0 equiv) in EtOH (0.72 M) at 0 °C was added a mixture of diethyl oxalate (1.0 equiv) and ethanone (1.0 mmol) over 20 min. The mixture was allowed to stir at rt for 4 h. In most instances, a precipitate had formed which was collected via filtration and washed with absolute EtOH. If no precipitate was evident, a minimal amount of water was added and the mixture was concentrated in vacuo. The residue was dissolved in water, neutralized with acetic acid, and extracted with Et2O (3×). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification was achieved as needed via flash column chromatography on SiO2 (hexanes/EtOAc: 4/1) to obtain the product.
General Preparation of Methyl Benzoate Compounds (Procedure III: 184–186)
To a solution of 4-bromobenzoic acid (1.0 mmol) in THF:MeOH (4:1, 0.3 M) at 0 °C was added (diazomethyl)trimethylsilane (2.4 equiv). The reaction was allowed to warm to rt over the period of 1 h. At this time, the mixture was concentrated in vacuo and 1.0 M HCl was added. The mixture was extracted with EtOAc (2×), dried over MgSO4, filtered, and concentrated in vacuo to afford the product.
General Preparation of Methyl 4-Formylbenzoate Compounds (Procedure IV: 37–40)
To a solution of methyl 4-bromobenzoate (1.0 mmol) in DMF (0.6 M) was added 17 mol % bis(triphenylphosphine)palladium(II) dichloride and sodium formate (1.5 equiv). The reaction mixture was stirred at 110 °C under a steady stream of CO(g) for 2 h. At this time, the mixture was cooled to rt, diluted with saturated sodium carbonate, and extracted with EtOAc (2×). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification was achieved via flash column chromatography on SiO2 (hexanes/EtOAc: 3/1) to yield the desired product, which was taken on without further purification.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (1)
Compound 1 was prepared via procedure I from methyl 4-formylbenzoate (3.0 g, 18 mmol), tryptamine (2.9 g, 18 mmol), and methyl acetopyruvate (2.6 g, 18 mmol) to yield a cream-colored solid (5.5 g, 72%). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 7.88 (d, J = 8.0 Hz, 2H), 7.33–7.24 (m, 4H), 7.12–7.03 (m, 2H), 6.91 (t, J = 7.6 Hz, 1H), 5.17 (s, 1H), 3.83 (s, 3H), 3.83–3.77 (m, 1H), 3.00–2.90 (m, 1H), 2.87–2.80 (m, 1H), 2.74–2.67 (m, 1H), 2.27 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 165.6, 165.1, 142.5, 136.3, 129.3, 128.1, 126.9, 125.5, 122.9, 121.2, 121.1, 118.4, 118.3, 118.1, 111.6, 111.5, 110.8, 66.4, 59.8, 52.2, 40.8, 23.6; mp 99–105 °C. HMS (APCI) calcd for C24H22N2O5 419.1607; found 419.1606 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-5-oxo-3-propionyl-2,5-dihydro-1H-pyrrol-2-yl)benzoate (62)
Compound 62 was prepared via procedure I from methyl 4-formylbenzoate (0.095 g, 0.58 mmol), tryptamine (0.93 g, 0.58 mmol), and 3 (0.10 g, 0.58 mmol) to yield a cream-colored solid (0.16 g, 64%). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 7.87 (d, J = 8.4 Hz, 2H), 7.33–7.23 (m, 4H), 7.09 (d, J = 2.4 Hz, 1H), 7.05 (t, J = 8.0 Hz, 1H), 6.91 (t, J = 6.8 Hz, 1H), 5.17 (s, 1H), 3.83–3.76 (m, 4H), 2.96–2.89 (m, 1H), 2.86–2.79 (m, 1H), 2.75–2.57 (m, 3H), 0.85 (t, J = 7.6 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 165.9, 165.1, 142.5, 136.2, 129.7, 129.3, 128.1, 126.9, 125.5, 122.9, 121.0, 118.2, 118.1, 111.5, 110.7, 59.8, 54.9, 52.2, 40.8, 40.0, 23.6 (note: carbons 1 and 2 are absent); mp 175–180 °C. HMS (APCI) calcd C25H24N2O5 433.1758; found 433.1756 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-isobutyryl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (63)
Compound 63 was prepared via procedure I from methyl 4-formylbenzoate (0.088 g, 0.54 mmol), tryptamine (0.086 g, 0.54 mmol), and 4 (0.10 g, 0.54 mmol) to yield a light-brown, amorphous solid (0.067 g, 28%). 1H NMR (600 MHz, DMSO-d6, 70 °C) δ 10.65 (s, 1H), 7.84 (d, J = 7.8 Hz, 2H), 7.32 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 7.8 Hz, 2H), 7.06–7.03 (m, 2H), 6.92 (t, J = 8.4 Hz, 1H), 5.18 (s, 1H), 3.83 (s, 3H), 3.79–3.75 (m, 1H), 3.40–3.20 (m, 2H), 2.98–2.93 (m, 1H), 2.91–2.86 (m, 1H), 2.72–2.67 (m, 1H), 0.86 (d, J = 4.8 Hz, 6H). 13C NMR (150 MHz, DMSO-d6) δ 182.1, 166.0, 136.2, 129.2, 128.1, 128.0, 126.9, 125.5, 123.4, 122.8, 121.1, 121.0, 118.5, 118.2, 118.1, 111.6, 111.5, 110.9, 109.5, 60.0, 52.1, 40.9, 23.7, 23.2, 18.5, 17.7. HMS (APCI) calcd for C26H26N2O5 447.1915; found 447.1916 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-5-oxo-3-pivaloyl-2,5-dihydro-1H-pyrrol-2-yl)benzoate (64)
Compound 64 was prepared via procedure I from methyl 4-formylbenzoate (0.082 g, 0.50 mmol), tryptamine (0.080 g, 0.50 mmol), and 5 (0.10 g, 0.50 mmol) to yield an orange oil (0.092 g, 40%). 1H NMR (600 MHz, CDCl3) δ 8.17 (br s, 1H), 7.93 (d, J = 7.8 Hz, 2H), 7.40–7.35 (m, 2H), 7.20 (t, J = 7.2 Hz, 1H), 7.09 (t, J = 7.2 Hz, 1H), 7.04 (dd, J = 1.2 Hz, J = 7.8 Hz, 2H), 6.96 (s, 1H), 5.04 (s, 1H), 4.02–3.99 (m, 1H), 3.91 (s, 3H), 3.09–3.00 (m, 2H), 2.95–2.91 (m, 1H), 1.06 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 202.2, 181.2, 166.7, 141.2, 136.5, 130.6, 130.2, 128.0, 127.2, 122.5, 122.2, 119.8, 118.8, 118.7, 111.6, 111.4, 63.0, 52.4, 41.7, 27.7, 25.3, 24.5 (note: either carbon 1 or 2 is absent). HMS (APCI) calcd for C27H28N2O5 461.2071; found 461.2077 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-benzoyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (65)
Compound 65 was prepared via procedure I from 6 (0.15 g, 0.68 mmol), tryptamine (0.11 g, 0.68 mmol), and methyl 4-formylbenzoate (0.11 g, 0.68 mmol) to yield a cream-colored solid (0.031 g, 9%). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 7.79 (d, J = 7.2 Hz, 2H), 7.59–7.48 (m, 2H), 7.40–7.28 (m, 3H), 7.24–7.21 (m, 3H), 7.11–7.04 (m, 2H), 6.92 (t, J = 7.6 Hz, 1H), 6.74 (s, 1H), 5.25 (s, 1H), 3.80–3.74 (m, 4H), 3.00–2.90 (m, 1H), 2.82–2.67 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 190.1, 188.9, 165.9, 165.4, 142.1, 138.2, 136.4, 132.4, 129.6, 129.5, 128.7, 128.3, 128.1, 127.0, 123.0, 121.1, 118.9, 118.4, 118.2, 111.6, 110.9, 60.8, 52.1, 41.2, 23.8; mp 200–205 °C. HMS (APCI) calcd for C29H24N2O5 481.1771; found 481.1765 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-(3-hydroxybenzoyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (66)
Compound 66 was prepared via procedure I from methyl 4-formylbenzoate (0.35 g, 2.1 mmol), tryptamine (0.34 g, 2.1 mmol), and 21 (0.50 g, 2.1 mmol) to yield a cream-colored solid (1.0 g, 96%). 1H NMR (600 MHz, DMSO-d6) δ 10.77 (s, 1H), 7.83 (d, J = 7.8 Hz, 2H), 7.37–7.29 (m, 6H), 7.13–7.12 (m, 1H), 7.07–7.03 (m, 3H), 6.91 (t, J = 7.8 Hz, 1H), 5.36 (s, 1H), 3.87–3.81 (m, 4H), 3.00–2.93 (m, 2H), 2.78–2.73 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 198.0, 166.0, 157.1, 136.40, 136.3, 129.4, 128.4, 128.2, 125.6, 123.4, 122.9, 121.2, 121.1, 119.5, 118.6, 118.4, 118.25, 118.19, 115.2, 111.7, 111.6, 110.9, 109.7, 66.5, 52.2, 41.2, 23.8; mp 78–80 °C. HMS (APCI) calcd for C29H24N2O6 497.1707; found 497.1707 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-(3-methoxybenzoyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (67)
Compound 67 was prepared via procedure I from methyl 4-formylbenzoate (0.066 g, 0.40 mmol), tryptamine (0.064 g, 0.40 mmol), and 7 (0.10 g, 0.40 mmol) to yield a pale-yellow, amorphous solid (0.046 g, 23%). 1H NMR (600 MHz, DMSO-d6, 80 °C) δ 10.64 (s, 1H), 7.89–7.82 (m, 3H), 7.35–7.22 (m, 6H), 7.07–7.05 (m, 2H), 6.98–6.91 (m, 2H), 5.34 (s, 1H), 4.26 (m, 1H), 3.82 (s, 3H), 3.74 (s, 3H), 3.01–2.90 (m, 2H), 2.78–2.75 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 188.6, 165.9, 165.1, 158.9, 141.9, 139.3, 136.3, 129.6, 129.4, 129.3, 128.3, 128.1, 126.9, 125.5, 122.9, 121.2, 121.1, 119.0, 118.3, 118.1, 113.5, 111.5, 110.8, 60.7, 55.3, 52.2, 41.1, 23.8. HMS (APCI) calcd for C30H26N2O6 511.1877; found 511.1871 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-(3-methylbenzoyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (68)
Compound 68 was prepared via procedure I from methyl 4-formylbenzoate (0.17 g, 1.0 mmol), tryptamine (0.17 g, 1.0 mmol), and 8 (0.24 g, 1.0 mmol) to yield a pale-yellow, amorphous solid (0.029 g, 6%). 1H NMR (600 MHz, DMSO-d6, 80 °C) δ 10.67 (s, 1H), 7.72 (m, 2H), 7.36–7.33 (m, 2H), 7.28–7.21 (m, 2H), 7.11–7.04 (m, 6H), 6.93 (t, J = 7.2 Hz, 1H), 5.29 (s, 1H), 3.86–3.78 (m, 4H), 3.01–2.96 (m, 1H), 2.91–2.86 (m, 1H), 2.76–2.72 (m, 1H), 2.23 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 190.2, 190.0, 182.1, 182.0, 166.0, 145.4, 144.7, 136.2, 128.8, 127.9, 126.9, 122.9, 122.8, 121.0, 118.3, 118.1, 111.5, 111.1, 109.2, 52.0, 48.6, 41.1, 23.6, 20.9 (note: carbons 1, 2, 3, and 4 are absent). HMS (APCI) calcd for C30H26N2O5 493.1769; found 493.1768 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-(3-chlorobenzoyl)-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (69)
Compound 69 was prepared via procedure I from methyl 4-formylbenzoate (0.064 g, 0.39 mmol), tryptamine (0.063 g, 0.39 mmol), and 9 (0.10 g, 0.39 mmol) to yield a pale-yellow, amorphous solid (0.045 g, 22%). 1H NMR (600 MHz, DMSO-d6, 80 °C) δ 10.69 (s, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.69 (s, 1H), 7.58 (d, J = 6.0 Hz, 1H), 7.35–7.32 (m, 3H), 7.29–7.26 (m, 3H), 7.07–7.05 (m, 2H), 6.93 (t, J = 7.2 Hz, 1H), 5.28 (s, 1H), 3.82–3.79 (m, 4H), 2.98–2.94 (m, 1H), 2.88–2.84 (m, 1H), 2.74–2.68 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 182.1, 166.1, 145.4, 137.9, 136.4, 136.3, 129.2, 128.2, 127.0, 126.8, 125.5, 123.44, 123.43, 122.8, 121.2, 121.0, 118.5, 118.3, 118.1, 111.6, 111.5, 111.0, 109.5, 60.5, 52.1, 23.2, 20.8. HMS (APCI) calcd for C29H23ClN2O5 513.1223; found 513.1219 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-(3-fluorobenzoyl)-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (70)
Compound 70 was prepared via procedure I from methyl 4-formylbenzoate (0.069 g, 0.42 mmol), tryptamine (0.067 g, 0.42 mmol), and 10 (0.10 g, 0.42 mmol) to yield a yellow, amorphous solid (0.028 g, 14%). 1H NMR (600 MHz, DMSO-d6, 80 °C) δ 10.66 (s, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.53–7.45 (m, 2H), 7.34 (dd, J = 3.0 Hz, J = 8.4 Hz, 2H), 7.29–7.26 (m, 1H), 7.22 (d, J = 7.8 Hz, 2H), 7.13–7.10 (m, 1H), 7.07–7.04 (m, 2H), 6.93 (t, J = 7.8 Hz, 1H), 5.32 (s, 1H), 3.84–3.79 (m, 4H), 3.01–2.96 (m, 1H), 2.92–2.88 (m, 1H), 2.76–2.71 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 191.7, 167.0, 161.5 (d, J = 241.7 Hz), 143.0, 137.6, 136.2, 129.3, 128.9, 128.6, 128.1, 127.9, 126.9, 125.5, 124.1, 122.8, 121.0, 118.3, 118.1, 116.6, 116.5, 111.5, 110.9, 61.0, 52.0, 41.2, 23.5 (note: carbon 3 is absent). HMS (APCI) calcd for C29H23FN2O5 497.1510; found 497.1513 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-5-oxo-3-picolinoyl-2,5-dihydro-1H-pyrrol-2-yl)benzoate (71)
Compound 71 was prepared via procedure I from methyl 4-formylbenzoate (0.074 g, 0.45 mmol), tryptamine (0.072 g, 0.45 mmol), and 18 (0.10 g, 0.45 mmol) to yield a yellow, amorphous solid (0.037 g, 17%). 1H NMR (600 MHz, CDCl3) δ 8.66 (d, J = 4.2 Hz, 1H), 8.21 (s, 1H), 8.14 (d, J = 7.8 Hz, 1H), 8.07 (dt, J = 1.2 Hz, J = 7.8 Hz, 1H), 7.94 (d, J = 8.4 Hz, 2H), 7.71 (dt, J = 0.6 Hz, J = 6.0 Hz, 1H), 7.39 (d, J = 7.2 Hz, 1H), 7.36 (d, J = 7.8 Hz, 1H), 7.21–7.17 (m, 3H), 7.07 (t, J = 8.4 Hz, 1H), 7.01 (d, J = 1.8 Hz, 1H), 5.14 (s, 1H), 4.15–4.10 (m, 1H), 3.89 (s, 3H), 3.11–2.95 (m, 3H). 13C NMR (150 MHz, CDCl3) δ 181.8, 173.2, 166.8, 165.7, 151.7, 145.3, 142.5, 140.6, 136.5, 130.3, 130.0, 128.3, 128.1, 127.3, 125.2, 122.3, 119.6, 118.7, 112.5, 111.5, 109.6, 61.6, 41.4, 29.9, 24.3 (note: carbon 3 is absent). HMS (APCI) calcd for C28H23N3O5 482.1710; found 482.1708 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (72)
Compound 72 was prepared via procedure I from methyl 4-formylbenzoate (0.079 g, 0.48 mmol), tryptamine (0.077 g, 0.45 mmol), and 19 (0.10 g, 0.48 mmol) to yield a yellow solid (0.11 g, 49%). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 8.80 (s, 1H), 8.69 (d, J = 4.4 Hz, 1H), 8.00 (d, J = 7.6 Hz, 1H), 7.88 (d, J = 7.2 Hz, 2H), 7.43–7.42 (m, 2H), 7.34–7.29 (m, 2H), 7.14 (d, J = 8.0 Hz, 2H), 7.06 (t, J = 6.8 Hz, 1H), 6.92 (t, J = 7.2 Hz, 1H), 5.42 (s, 1H), 3.87–3.82 (m, 4H), 3.02–2.89 (m, 2H), 2.79–2.74 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 186.8, 165.9, 165.0, 152.1, 149.1, 142.1, 136.3, 133.9, 129.5, 129.4, 128.3, 128.1, 126.9, 125.5, 123.5, 122.9, 121.0, 118.3, 118.1, 111.5, 110.7, 109.5, 60.4, 52.2, 41.1, 23.7; mp 199–205 °C. HMS (APCI) calcd for C28H23N3O5 482.1711; found 482.1707 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-isonicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (73)
Compound 73 was prepared via procedure I from methyl 4-formylbenzoate (0.15 g, 0.9 mmol), tryptamine (0.15 g, 0.9 mmol), and 20 (0.20 g, 0.9 mmol) to yield a yellow solid (0.13 g, 30%). 1H NMR (600 MHz, DMSO-d6, 80 °C) δ 10.63 (s, 1H), 8.47 (d, J = 2.8 Hz, 1H), 7.76–6.92 (m, 12H), 5.27 (s, 1H), 3.82–3.78 (m, 4H), 3.00–2.95 (m, 1H), 2.91–2.86 (m, 1H), 2.74–2.72 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 191.3, 166.0, 149.5, 149.0, 146.6, 136.3, 128.9, 128.5, 128.0, 127.0, 122.8, 122.3, 121.1, 120.7, 118.3, 118.1, 111.5, 111.0, 52.0, 48.6, 41.1, 23.5 (note: carbons 1 and 2 are absent); mp >250 °C. HMS (APCI) calcd for C28H23N3O5 480.1570; found 480.1568 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-(2-methoxybenzoyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (74)
Compound 74 was prepared via procedure I from methyl 4-formylbenzoate (0.13 g, 0.80 mmol), tryptamine (0.13 g, 0.80 mmol), and 11 (0.20 g, 0.80 mmol) to yield a pink, amorphous solid (0.028 g, 7%). 1H NMR (600 MHz, DMSO-d6) δ 10.83 (s, 1H), 7.89 (s, 1H), 7.50 (d, J = 6.0 Hz, 1H), 7.34–7.18 (m, 5H), 7.09–7.04 (m, 3H), 6.96–6.88 (m, 3H), 5.27 (s, 1H), 3.83–3.74 (m, 6H), 2.99–2.87 (m, 2H), 2.73–2.65 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 190.1, 182.1, 166.2, 156.0, 146.1, 137.3, 132.7, 129.1, 128.8, 128.4, 128.1, 128.0, 127.7, 127.0, 125.6, 122.8, 121.0, 119.7, 118.2, 118.1, 111.5, 111.1, 111.0, 60.3, 56.1, 55.2, 52.1, 40.9, 23.6, 18.6. HMS (APCI) calcd for C30H26N2O6 509.1710; found 509.1711 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-(2-methylbenzoyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (75)
Compound 75 was prepared via procedure I from methyl 4-formylbenzoate (0.14 g, 0.85 mmol), tryptamine (0.14 g, 0.85 mmol), and 12 (0.20 g, 0.85 mmol) to yield a pale-yellow, amorphous solid (0.040 g, 9%). 1H NMR (600 MHz, DMSO-d6, 70 °C) δ 10.71 (br s, 1H), 7.67 (d, J = 7.2 Hz, 2H), 7.37 (d, J = 9.0 Hz, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.13–6.99 (m, 4H), 6.94–6.91 (m, 2H), 6.84–6.74 (m, 3H), 4.96 (s, 1H), 3.82–3.76 (m, 4H), 2.97–2.92 (m, 1H), 2.82–2.71 (m, 2H), 1.78 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 190.1, 182.1, 182.03, 182.01, 165.9, 144.4, 139.4, 136.3, 134.2, 129.3, 128.8, 128.3, 127.9, 127.0, 124.7, 123.0, 122.7, 121.1, 118.3, 118.1, 111.6, 111.2, 108.9, 60.3, 52.0, 48.6, 41.1, 23.6. HMS (APCI) calcd for C30H26N2O5 493.1761; found 493.1763 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-(2-chlorobenzoyl)-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (76)
Compound 76 was prepared via procedure I from methyl 4-formylbenzoate (0.13 g, 0.79 mmol), tryptamine (0.13 g, 0.79 mmol), and 13 (0.20 g, 0.79 mmol) to yield a cream-colored solid (0.27 g, 66%). 1H NMR (600 MHz, DMSO-d6, 80 °C) δ 10.64 (s,1H), 7.67 (m, 2H), 7.35 (d, J = 8.4 Hz, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.24–7.18 (m, 2H), 7.10–7.03 (m, 4H), 6.92 (t, J = 7.2 Hz, 2H), 6.78 (m, 1H), 5.04 (s, 1H), 3.86–3.75 (m, 4H), 2.97–2.92 (m, 1H), 2.85–2.83 (m, 1H), 2.73–2.68 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 185.2, 165.9, 144.1, 136.3, 130.0, 129.5, 129.1, 128.8, 128.7, 128.6, 128.1, 128.0, 126.9, 126.5, 123.0, 121.0, 118.3, 118.1, 111.5, 111.0, 67.1, 52.1, 40.0, 25.2 (note: carbons 1, 2, and 3 are absent); mp 248–253 °C. HMS (APCI) calcd for C29H23ClN2O5 513.1214; found 513.1215 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-(2-fluorobenzoyl)-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (77)
Compound 77 was prepared via procedure I from methyl 4-formylbenzoate (0.19 g, 0.84 mmol), tryptamine (0.14 g, 0.84 mmol), and 14 (0.20 g, 0.84 mmol) to yield a brown, amorphous solid (0.070 g, 17%). 1H NMR (600 MHz, DMSO-d6, 70 °C) δ 10.68 (s, 1H), 7.80–7.54 (m, 2H), 7.35–7.30 (m, 4H), 7.11–6.78 (m, 7H), 5.07 (s, 1H), 3.81–3.74 (m, 4H), 2.97–2.92 (m, 1H), 2.87–2.82 (m, 1H), 2.72–2.67 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 172.6, 165.9, 161.2, 156.0, 155.1, 139.5, 136.3, 135.0, 130.0, 129.6, 128.6, 126.8, 126.7, 126.3, 125.3, 124.8, 123.1, 121.1, 119.3, 118.3, 118.0, 111.5, 110.6, 59.1, 52.3, 41.2, 23.8. HMS (APCI) calcd for C29H23FN2O5 497.1510; found 497.1513 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-(furan-2-carbonyl)-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (78)
Compound 78 was prepared via procedure I from methyl 4-formylbenzoate (0.42 g, 2.6 mmol), tryptamine (0.41 g, 2.6 mmol), and 15 (0.50 g, 2.6 mmol) to yield an orange solid (0.079 g, 7%). 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 8.05 (s, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.43–7.41 (m, 2H), 7.32 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 8.4 Hz, 2H), 7.16 (t, J = 7.2 Hz, 1H), 7.06 (t, J = 7.2 Hz, 1H), 6.94 (s, 1H), 6.39 (dd, J = 1.8 Hz, J = 3.6 Hz, 1H), 5.39 (s, 1H), 4.04–3.99 (m, 1H), 3.88 (s, 3H), 3.07–2.96 (m, 2H), 2.93–2.89 (m, 1H). 13C NMR (150 MHz, CDCl3) δ 173.9, 167.1, 153.1, 145.1, 136.5, 129.8, 129.7, 129.2, 128.4, 128.2, 127.4, 122.3, 122.2, 119.5, 118.9, 117.1, 114.2, 112.7, 111.7, 111.4, 111.3, 61.3, 52.2, 41.3, 24.3; mp 60–65 °C. HMS (APCI) calcd for C27H22N2O6 471.1551; found 471.1547 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-5-oxo-3-(thiophene-2-carbonyl)-2,5-dihydro-1H-pyrrol-2-yl)benzoate (79)
Compound 79 was prepared via procedure I from methyl 4-formylbenzoate (0.073 g, 0.44 mmol), tryptamine (0.071 g, 0.44 mmol), and 16 (0.10 g, 0.44 mmol) to yield a yellow, amorphous solid (0.043 g, 20%). 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 4.0 Hz, 1H), 7.34–7.29 (m, 4H), 7.06–7.02 (m, 3H), 6.92 (t, J = 7.2 Hz, 1H), 5.80–5.70 (m, 1H), 5.28 (s, 1H), 3.86–3.74 (m, 4H), 3.00–2.89 (m, 1H), 2.85–2.78 (m, 1H), 2.73–2.67 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 165.8, 144.7, 136.2, 129.0, 128.2, 126.9, 126.8, 123.4, 122.9, 121.2, 121.0, 118.5, 118.3, 118.1, 111.6, 111.5, 111.1, 109.5, 52.0, 48.6, 40.0, 23.5 (note: carbons 1, 2, and 4 are absent); mp 190–195 °C. HMS (APCI) calcd for C27H22N2O5S 485.1177; found 485.1173 [M – H]−.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-5-oxo-3-(thiophene-3-carbonyl)-2,5-dihydro-1H-pyrrol-2-yl)benzoate (80)
Compound 80 was prepared via procedure I from methyl 4-formylbenzoate (0.36 g, 2.2 mmol), tryptamine (0.35 g, 2.2 mmol), and 17 (0.50 g, 2.2 mmol) to yield a cream-colored solid (0.70 g, 65%). 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 8.38 (d, J = 2.8 Hz, 1H), 7.85 (d, J = 7.6 Hz, 2H), 7.49 (t, J = 4.0 Hz, 1H), 7.37–7.31 (m, 4H), 7.13 (s, 1H), 7.07 (t, J = 7.6 Hz, 1H), 6.93 (t, J = 6.8 Hz, 1H), 5.76 (d, J = 1.2 Hz, 1H), 5.44 (s, 1H), 3.89–3.81 (m, 4H), 3.02–2.90 (m, 2H), 2.80–2.74 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 181.8, 165.9, 142.1, 141.6, 136.3, 134.2, 129.5, 129.4, 128.3, 128.1, 127.3, 126.9, 126.3, 125.5, 122.9, 121.1, 118.3, 118.1, 111.5, 110.8, 60.7, 52.1, 41.1, 23.8; mp 189–192 °C. HMS (APCI) calcd for C27H22N2O5S 487.1336; found 487.1335 [M + H]+.
Methyl 3-(1–2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (81)
Compound 81 was prepared via procedure I from methyl 3-formylbenzoate (0.50 g, 3.1 mmol), tryptamine (0.49 g, 3.1 mmol), and methyl acetopyruvate (0.44 g, 3.1 mmol) to yield a pale-pink solid (0.77 g, 60%). 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.73 (s, 1H), 7.45 (t, J = 7.2 Hz, 1H), 7.39–7.26 (m, 3H), 7.10 (d, J = 2.0 Hz, 1H), 7.05 (t, J = 7.2 Hz, 1H), 6.89 (t, J = 8.0 Hz, 1H), 5.24 (s, 1H), 3.85–3.76 (m, 4H), 2.98–2.91 (m, 1H), 2.87–2.80 (m, 1H), 2.72–2.65 (m, 1H), 2.72 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 191.5, 166.0, 165.1, 154.8, 137.8, 136.3, 132.5, 129.9, 129.1, 129.0, 128.4, 126.9, 122.9, 121.1, 119.8, 118.3, 118.1, 111.5, 110.8, 59.8, 52.3, 40.8, 29.8, 23.7; mp 218–220 °C. HMS (APCI) calcd for C24H22N2O5 419.1593; found 419.1596 [M + H]+.
Methyl 2-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (82)
Compound 82 was prepared via procedure I from methyl 2-formylbenzoate (0.10 g, 0.61 mmol), tryptamine (0.098 g, 0.61 mmol), and methyl acetopyruvate (0.088 g, 0.61 mmol) to yield a white solid (0.18 g, 72%). 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.72 (s, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.32–7.25 (m, 2H), 7.10 (d, J = 1.6 Hz, 1H), 7.02 (t, J = 7.6 Hz, 1H), 6.89 (t, J = 7.6 Hz, 1H), 5.24 (s, 1H), 3.86 (s, 3H), 3.82–3.75 (m, 1H), 2.97–2.90 (m, 1H), 2.86–2.79 (m, 1H), 2.71–2.66 (m, 1H), 2.27 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.6, 166.0, 165.0, 154.5, 137.8, 136.2, 132.5, 129.8, 129.0, 128.9, 128.3, 126.8, 122.9, 121.0, 119.9, 118.2, 118.0, 111.4, 110.7, 59.7, 52.2, 40.8, 29.8, 23.6; mp 210–218 °C. HMS (APCI) calcd for C24H22N2O5 419.1602; found 419.1599 [M + H]+.
4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoic Acid (83)
To a suspension of 1 (0.10 g, 0.24 mmol) in EtOH (3.5 mL) was added a 2N aq sodium hydroxide solution (0.8 mL). The reaction was heated at reflux for 2 h. The solution was then cooled to 0 °C, and concentrated HCl was added carefully. The precipitate was filtered off, rinsed with cold water, and dried under vacuum for 12 h to afford a white powder (0.090 g, 93%). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (br s, 1H), 10.79 (br s, 1H), 8.09 (d, J = 8.0 Hz, 2H), 7.30–7.28 (m, 2H), 7.23 (d, J = 8.0 Hz, 2H), 7.03–7.01 (m, 2H), 6.90 (app t, J = 8.0 Hz, 1H), 5.56 (s, 1H), 3.77–3.74 (m, 1H), 2.89–2.71 (m, 1H), 2.71–2.61 (m, 2H), 2.10 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 196.5, 179.9, 169.3, 164.3, 146.9, 136.5, 130.1 (×2), 128.5, 127.8 (×2), 127.4, 123.0, 119.1, 121.7, 119.8, 118.8, 113.0, 111.1, 54.7, 49.1, 26.6, 24.3; mp >250 °C. HMS (ESI+) calcd For C23H20N2NaO5 427.1270; found 427.1263 [M + Na]+.
Ethyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (84)
Compound 84 was prepared via procedure I from 45 (0.20 g, 1.1 mmol), tryptamine (0.18 g, 1.1 mmol), and methyl acetopyruvate (0.16 g, 1.1 mmol) to yield a pale-pink solid (0.20 g, 42%). 1H NMR (400 MHz, DMSO-d6) δ 12.48 (br s, 1H), 10.82 (s, 1H), 7.87 (d, J = 8.4 Hz, 2H), 7.33–7.24 (m, 4H), 7.09 (d, J = 2.4 Hz, 1H), 7.05 (t, J = 7.2 Hz, 1H), 6.91 (t, J = 7.6 Hz, 1H), 5.18 (s, 1H), 4.30 (q, J = 7.2 Hz, 2H), 3.83–3.76 (m, 1H), 2.97–2.89 (m, 1H), 2.87–2.80 (m, 1H), 2.74–2.67 (m, 1H), 2.26 (s, 3H), 1.30 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.6, 165.4, 165.1, 142.4, 136.2, 129.6, 129.3, 128.1, 126.9, 122.9, 121.0, 118.3, 118.1, 111.5, 110.7, 60.7, 59.8, 40.8, 39.9, 23.6, 14.2 (note: carbon 3 and either carbon 1 or 2 are absent); mp 180–183 °C. HMS (APCI) calcd for C25H24N2O5 433.1771; found 433.1765 [M + H]+.
Isopropyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (85)
Compound 85 was prepared via procedure I from 46 (0.20 g, 1.0 mmol), tryptamine (0.17 g, 1.0 mmol), and methyl acetopyruvate (0.15 g, 1.0 mmol) to yield an orange, amorphous solid (0.028 g, 6%). 1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.33–7.23 (m, 3H), 7.11–7.00 (m, 3H), 6.91 (t, J = 7.6 Hz, 1H), 5.16–5.11 (m, 2H), 3.82–3.77 (m, 1H), 3.02–2.85 (m, 2H), 2.76–2.71 (m, 1H), 2.26 (s, 3H), 1.32 (s, 3H), 1.31 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 190.0, 164.9, 136.2, 129.2, 128.1, 126.8, 125.5, 123.5, 122.8, 121.2, 121.0, 118.5, 118.3, 118.1, 111.5, 110.8, 68.1, 59.8, 40.9, 39.9, 23.6, 21.6 (note: carbons 1 and 2 are absent). HMS (APCI) calcd for C26H26N2O5 447.1928; found 447.1922 [M + H]+.
tert-Butyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2, 5-dihydro-1H-pyrrol-2-yl) benzoate (86)
Compound 86 was prepared via procedure I from 47 (0.50 g, 2.4 mmol), tryptamine (0.39 g, 2.4 mmol), and methyl acetopyruvate (0.35 g, 2.4 mmol) to yield a pale-yellow solid (0.92 g, 83%). 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 7.81 (d, J = 8.4 Hz, 2H), 7.33–7.22 (m, 4H), 7.10 (d, J = 2.0 Hz, 1H), 7.05 (t, J = 7.2 Hz, 1H), 6.91 (t, J = 7.6 Hz, 1H), 5.17 (s, 1H), 3.83–3.76 (m, 1H), 2.96–2.89 (m, 1H), 2.86–2.81 (m, 1H), 2.79–2.67 (m, 1H), 2.26 (s, 3H), 1.53 (s, 9H). 13C NMR (150 MHz, DMSO-d6) δ 191.5, 165.1, 164.6, 154.4, 142.0, 131.1, 129.1, 127.9, 126.9, 126.2, 122.8, 121.0, 119.8, 118.2, 118.1, 111.4, 110.7, 80.7, 59.8, 40.8, 29.7, 27.8, 23.6; mp 145–150 °C. HMS (APCI) calcd for C27H28N2O5 461.2063; found 461.2065 [M + H]+.
4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzonitrile (87)
Compound 87 was prepared via procedure I from 4-formylbenzonitrile (0.15 g, 1.1 mmol), tryptamine (0.18 g, 1.1 mmol), and 19 (0.25 g, 1.1 mmol) to yield a yellow solid (0.084 g, 17%). 1H NMR (600 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.81 (s, 1H), 8.69 (d, J = 3.6 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 2H), 7.50–7.47 (m, 3H), 7.33 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.13 (s, 1H), 7.07 (t, J = 7.8 Hz, 1H), 6.94 (t, J = 7.2 Hz, 1H), 5.44 (s, 1H), 3.88–3.83 (m, 1H), 3.01–2.90 (m, 2H), 2.79–2.75 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 189.3, 165.1, 152.1, 150.2, 149.2, 142.5, 136.3, 133.9, 132.4, 129.0, 126.9, 123.9, 123.4, 122.9, 121.1, 118.6, 118.3, 118.1, 117.7, 111.5, 111.0, 110.7, 60.3, 41.2, 23.7; mp >250 °C. HMS (APCI) calcd for C27H20N4O3 449.1608; found 449.1607 [M + H]+.
1-(2-(1H-Indol-3-yl)ethyl)-4-acetyl-3-hydroxy-5-(4-nitrophenyl)-1H-pyrrol-2(5H)-one (88)
Compound 88 was prepared via procedure I from 4-nitrobenzaldehyde (0.50 g, 3.3 mmol), tryptamine (0.53 g, 3.3 mmol), and methyl acetopyruvate (0.48 g, 3.3 mmol) to yield a pale-yellow solid (1.0 g, 75%). 1H NMR (400 MHz, DMSO-d6) δ 12.62 (br s, 1H), 10.82 (s, 1H), 8.10 (d, J = 8.8 Hz, 2H), 7.37 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 1.6 Hz, 1H), 7.04 (t, J = 8.0 Hz, 1H), 6.91 (t, J = 7.6 Hz, 1H), 5.26 (s, 1H), 3.85–3.78 (m, 1H), 2.97–2.84 (m, 2H), 2.80–2.77 (m, 1H), 2.27 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.6, 165.2, 154.7, 147.1, 144.9, 136.2, 129.0, 126.8, 123.5, 122.9, 121.0, 118.2, 118.0, 111.4, 110.7, 59.4, 41.0, 29.8, 23.5 (note: carbon 3 is absent); mp 142–150 °C. HMS (APCI) calcd for C22H19N3O5 406.1398; found 406.1395 [M + H]+.
4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzamide (89)
Compound 89 was prepared via procedure I from 42 (0.29 g, 2.0 mmol), tryptamine (0.32 g, 2.0 mmol), and 19 (0.44 g, 2.0 mmol) to yield a yellow solid (0.70 g, 76%). 1H NMR (600 MHz, DMSO-d6) δ 10.65 (s, 1H), 8.81 (s, 1H), 8.66 (s, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 8.4 Hz, 2H), 7.45–7.32 (m, 6H), 7.12–7.06 (m, 3H), 6.95 (t, J = 7.2 Hz, 1H), 5.39 (s, 1H), 3.91–3.86 (m, 1H), 3.04–2.98 (m, 2H), 2.83–2.79 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 186.1, 175.4, 167.3, 165.0, 148.7, 139.8, 136.1, 135.7, 135.3, 134.0, 133.9, 127.4, 127.2, 126.7, 122.8, 122.7, 122.4, 120.6, 117.7, 111.1, 110.7, 108.5, 60.2, 39.9, 13.7; mp 175–180 °C. HMS (APCI) calcd for C27H22N4O4 467.17138; found 467.17160 [M + H]+.
4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-N-methylbenzamide (90)
Compound 90 was prepared via procedure I from 43 (0.15 g, 0.93 mmol), tryptamine (0.15 g, 0.93 mmol), and 19 (0.21 g, 0.93 mmol) to yield a yellow solid (0.33 g, 73%). 1H NMR (600 MHz, DMSO-d6) δ 10.72 (s, 1H), 8.79 (s, 1H), 8.67 (d, J = 4.8 Hz, 1H), 8.24 (d, J = 3.6 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 7.8 Hz, 2H), 7.45 (t, J = 5.4 Hz, 1H), 7.39–7.34 (m, 4H), 7.11–7.05 (m, 2H), 6.94 (t, J = 7.2 Hz, 1H), 5.38 (s, 1H), 3.88–3.84 (m, 1H), 3.11–3.09 (m, 1H), 3.02–2.94 (m, 2H), 2.76 (d, J = 4.8 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 166.3, 165.0, 152.3, 149.3, 136.3, 136.1, 134.5, 128.1, 127.9, 127.3, 125.5, 123.4, 122.4, 122.9, 121.0, 118.5, 118.3, 118.14, 118.1, 111.6, 110.8, 60.4, 41.0, 26.2, 23.7 (note: carbon 4 is absent); mp 240–245 °C. HMS (APCI) calcd for C28H24N4O4 481.1876; found 481.1879 [M + H]+.
4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-N,N-dimethylbenzamide (91)
Compound 91 was prepared via procedure I from 19 (0.38 g, 1.7 mmol), tryptamine (0.27 g, 1.7 mmol), and 44 (0.30 g, 1.7 mmol) to yield a yellow solid (0.33 g, 39%). 1H NMR (600 MHz, DMSO-d6) δ 10.85 (s, 1H), 8.81 (s, 1H), 8.69 (d, J = 4.8 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.49–7.47 (m, 2H), 7.36–7.29 (m, 4H), 7.14–7.10 (m, 2H), 7.06 (t, J = 7.8 Hz, 1H), 6.92 (t, J = 7.2 Hz, 1H), 5.42 (s, 1H), 3.88–3.82 (m, 1H), 3.02–2.95 (m, 5H), 2.84 (s, 3H), 2.77–2.71 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.71, 164.9, 152.3, 149.3, 136.4, 136.3, 136.2, 128.1, 127.8, 127.2, 125.5, 123.4, 122.9, 121.2, 121.0, 118.5, 118.3, 118.1, 111.6, 111.5, 110.8, 60.5, 41.0, 40.0, 23.8, 23.2 (note: carbon 4 is absent); mp 210–212 °C. HMS (APCI) calcd for C29H26N4O4 495.2040; found 495.2036 [M + H]+.
4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5,-dihydro-1H-pyrrol-2-yl)benzenesulfonamide (92)
Compound 92 was prepared via procedure I from 4-formylbenzenesulfonamide (0.19 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield a pale-orange solid (0.024 g, 5%). 1H NMR (600 MHz, DMSO-d6) δ 10.86 (s, 1H), 8.81 (s, 1H), 8.69 (d, J = 4.2 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.76 (d, J = 9.0 Hz, 2H), 7.50–7.47 (m, 3H), 7.34 (d, J = 7.8 Hz, 2H), 7.32 (s, 2H), 7.14 (d, J = 1.8 Hz, 1H), 7.07 (t, J = 8.4 Hz, 1H), 6.96 (t, J = 7.2 Hz, 1H), 5.41 (s, 1H), 3.91–3.86 (m, 1H), 3.03–2.98 (m, 1H), 2.92–2.88 (m, 1H), 2.81–2.76 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 181.3, 165.0, 149.3, 149.1, 143.9, 140.6, 136.3, 133.9, 131.8, 128.6, 126.9, 125.9, 123.4, 122.9, 121.1, 118.4, 118.2, 111.5, 110.8, 60.1, 41.0, 23.7 (note: two of either carbons 1, 2, or 3 are absent); mp >250 °C. HMS (APCI) calcd for C26H22N4O5S 503.1384; found 503.1381 [M + H]+.
4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-N-methylbenzenesulfonamide (93)
Compound 93 was prepared via procedure I from 4-formyl-N-methylbenzenesulfonamide (0.20 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield an orange-yellow solid (0.11 g, 21%). 1H NMR (600 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.80 (s, 1H), 8.50 (d, J = 4.8 Hz, 1H), 8.02–8.00 (m, 2H), 7.71 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.40 (d, J = 4.8 Hz, 1H), 7.34–7.29 (m, 3H), 7.09–7.05 (m, 2H), 6.94 (t, J = 7.8 Hz, 1H), 3.84–3.79 (m, 1H), 3.01–2.95 (m, 1H), 2.89–2.85 (m, 1H), 2.70–2.65 (m, 1H), 2.39 (d, J = 4.8 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 169.7, 149.9, 149.6, 149.5, 146.1, 138.0, 136.4, 136.3, 135.9, 128.5, 128.1, 127.0, 126.5, 125.6, 123.4, 122.7, 122.5, 121.0, 118.5, 118.3, 118.1, 111.5, 111.1, 60.3, 41.1, 28.7, 23.6; mp >220 °C. HMS (APCI) calcd for C27H24N4O5S 517.1540; found 517.1545 [M + H]+.
1-(2-(1H-Indol-3-yl)ethyl)-3-hydroxy-4-nicotinoyl-5-(4-(trifluoromethyl)phenyl)-1H-pyrrol-2(5H)-one (94)
Compound 94 was prepared via procedure I from 4-(trifluoromethyl)benzaldehyde (0.17 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield a yellow solid (0.24 g, 49%). 1H NMR (600 MHz, DMSO-d6) δ 10.94 (s, 1H), 8.83 (d, J = 1.2 Hz, 1H), 8.70 (dd, J = 1.8 Hz, J = 4.8 Hz, 1H), 8.20 (br s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.66 (d, J = 7.8 Hz, 2H), 7.54–7.48 (m, 3H), 7.34 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 7.13 (s, 1H), 7.05 (t, J = 7.8 Hz, 1H), 6.91 (t, J = 7.2 Hz, 1H), 5.46 (s, 1H), 3.88–3.83 (m, 1H), 3.02–2.92 (m, 2H), 2.78–2.73 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 181.2, 165.3, 151.8, 149.0, 141.9, 136.6, 136.3, 134.2, 128.9, 128.2, 126.9, 125.5, 125.4, 123.5, 122.9, 121.2, 121.1, 118.2, 118.0, 111.5, 110.7, 60.3, 41.2, 23.7 (note: either carbon 1 or 2 is absent); mp 235–240 °C. HMS (APCI) calcd for C27H20F3N3O3 492.1535; found 492.1532 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-2-hydroxybenzoate (95)
Compound 95 was prepared via procedure I from 35 (0.15 g, 0.83 mmol), tryptamine (0.13 g, 0.83 mmol), and methyl acetopyruvate (0.12 g, 0.83 mmol) to yield a brown solid (0.10 g, 27%). 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 10.49 (s, 1H), 7.68 (dd, J = 8.0 Hz, J = 2.4 Hz, 1H), 7.34–7.30 (m, 2H), 7.10 (s, 1H), 7.05 (t, J = 6.8 Hz, 1H), 6.92 (t, J = 6.8 Hz, 1H), 6.84 (s, 1H), 6.64 (d, J = 8.0 Hz, 1H), 5.12 (s, 1H), 3.86 (s, 3H), 3.83–3.76 (m, 1H), 2.98–2.83 (m, 2H), 2.75–2.68 (m, 1H), 2.27 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.7, 168.8, 165.0, 159.8, 154.4, 154.1, 136.0, 130.3, 126.9, 122.8, 121.0, 119.6, 118.3, 118.2, 118.1, 117.0, 112.8, 111.4, 110.7, 59.6, 52.4, 40.9, 29.8, 23.5; mp 188–190 °C. HMS (APCI) calcd for C24H22N2O6 435.1551; found 435.1549 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-2-methoxybenzoate (96)
Compound 96 was prepared via procedure I from 33 (0.1 g, 0.52 mmol), tryptamine (0.083 g, 0.52 mmol), and methyl acetopyruvate (0.074 g, 0.52 mmol) to yield a cream-colored solid (0.17 g, 74%). 1H NMR (600 MHz, DMSO-d6) δ 10.83 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.34–7.31 (m, 2H), 7.11 (s, 1H), 7.06 (t, J = 6.8 Hz, 1H), 6.94–6.91 (m, 2H), 6.70 (d, J = 8.0 Hz, 1H), 5.14 (s, 1H), 3.83–3.76 (m, 7H), 2.99–2.84 (m, 2H), 2.75–2.69 (m, 1H), 2.28 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 165.9, 165.0, 158.1, 136.2, 130.9, 128.1, 126.9, 125.5, 125.0, 122.9, 121.0, 119.6, 118.6, 118.3, 118.1, 111.5, 110.8, 60.0, 55.9, 51.9, 40.9, 40.0, 23.6 (note: carbons 3 and 4 are absent); mp 130–135 °C. HMS (APCI) calcd for C25H24N2O6 449.1707; found 449.1709 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-2-methylbenzoate (97)
Compound 97 was prepared via procedure I from 40 (0.10 g, 0.56 mmol), tryptamine (0.090 g, 0.56 mmol), and methyl acetopyruvate (0.081 g, 0.56 mmol) to yield a pale-orange solid (0.13 g, 53%). 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.40–7.36 (m, 2H), 7.23 (t, J = 7.6 Hz, 1H), 7.11 (t, J = 7.6 Hz, 1H), 7.00 (s, 1H), 6.83 (d, J = 7.6 Hz, 1H), 6.74 (s, 1H), 4.76 (s, 1H), 4.07–4.02 (m, 1H), 3.90 (s, 3H), 3.11–2.93 (m, 3H), 2.51 (s, 3H), 1.98 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 194.6, 167.6, 164.6, 159.8, 141.4, 139.0, 136.5, 131.5, 131.3, 130.5, 127.2, 125.2, 122.6, 122.3, 119.8, 119.5, 118.7, 112.4, 111.6, 61.4, 52.2, 41.3, 28.2, 24.4, 21.9; mp 40–43 °C. HMS (APCI) calcd for C25H24N2O5 433.1745; found 433.1745 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-2-chlorobenzoate (98)
Compound 98 was prepared via procedure I from 39 (0.09 g, 0.45 mmol), tryptamine (0.073 g, 0.45 mmol), and methyl acetopyruvate (0.065 g, 0.45 mmol) to yield a pale-orange, amorphous solid (0.14 g, 68%). 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.40–7.23 (m, 4H), 7.14–7.03 (m, 2H), 6.93 (t, J = 7.6 Hz, 1H), 5.13 (s, 1H), 3.83–3.78 (m, 4H), 2.98–2.82 (m, 2H), 2.75–2.68 (m, 1H), 2.27 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 190.8, 165.2, 142.8, 136.2, 131.8, 131.2, 130.5, 129.4, 128.0, 126.8, 126.0, 122.9, 121.0, 118.3, 118.0, 111.4, 110.7, 59.1, 52.5, 48.6, 40.8, 23.6 (note: carbons 1 and 2 are absent). HMS (APCI) calcd for 453.1225; found 453.1219 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-2-fluorobenzoate (99)
Compound 99 was prepared via procedure I from 38 (0.08 g, 0.44 mmol), tryptamine (0.070 g, 0.44 mmol), and methyl acetopyruvate (0.063 g, 0.44 mmol) to yield a pale-orange solid (0.096 g, 50%). 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 7.81 (t, J = 7.6 Hz, 1H), 7.40–7.27 (m, 2H), 7.20 (t, J = 7.2 Hz, 1H), 7.14–7.06 (m, 1H), 6.95 (d, J = 1.2 Hz, 1H), 6.78 (dd, J = 1.2 Hz, J = 7.6 Hz, 1H), 6.70 (dd, J = 1.2 Hz, J = 10.8 Hz, 1H), 4.80 (s, 1H), 4.09–4.03 (m, 1H), 3.92 (s, 3H), 3.10–2.96 (m, 3H), 2.19 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 198.1, 193.6, 165.5, 162.0 (d, J = 260.4 Hz), 142.8 (d, J = 7.4 Hz), 136.5, 132.8, 127.0, 123.5, 123.4 (d, J = 26.0 Hz), 122.5, 122.3, 122.1, 120.1, 119.7, 118.7, 118.5, 111.9, 111.7, 61.2, 52.6, 41.6, 29.2, 24.4; mp 40–45 °C. HMS (APCI) calcd for C24H21FN2O5 437.1512; found 437.1512 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl) ethyl)-3-acetyl-4-hydroxy-5-oxo-2, 5-dihydro-1H-pyrrol-2-yl)-3-hydroxybenzoate (100)
Compound 100 was prepared via procedure I from 34 (0.1 g, 0.56 mmol), tryptamine (0.089 g, 0.52 mmol), and methyl acetopyruvate (0.080 g, 0.56 mmol) to yield an off-white solid (0.020 g, 8%). 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 10.07 (br s, 1H), 7.47 (s, 1H), 7.35–7.29 (m, 3H), 7.05–7.02 (m, 3H), 6.91 (t, J = 10.8 Hz, 1H), 5.58 (s, 1H), 3.83 (s, 3H), 3.80–3.74 (m, 1H), 3.02–2.88 (m, 2H), 2.76–2.68 (m, 1H), 2.27 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 196.6, 190.2, 163.6, 151.5, 136.4, 136.2, 127.0, 126.9, 123.5, 123.0, 122.5, 121.0, 118.32, 118.25, 118.0, 111.4, 111.2, 110.4, 94.5, 52.7, 52.2, 44.8, 29.3, 26.7; mp 194–197 °C. HMS (APCI) calcd for C24H22N2O6 435.1551; found 435.1552 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-3-methoxybenzoate (101)
Compound 101 was prepared via procedure I from 41 (0.20 g, 1.0 mmol), tryptamine (0.17 g, 1.0 mmol), and methyl acetopyruvate (0.15 g, 1.0 mmol) to yield an off-white solid (0.16 g, 36%). 1H NMR (600 MHz, DMSO-d6, 56 °C) δ 10.68 (s, 1H), 7.53 (s, 1H), 7.48 (dd, J = 1.6 Hz, J = 8.0 Hz, 1H), 7.32–7.28 (m, 2H), 7.06–7.02 (m, 3H), 6.92 (td, J = 7.2 Hz, J = 0.8 Hz, 1H), 5.59 (s, 1H), 3.86 (s, 6H), 3.81–3.72 (m, 1H), 2.97–2.85 (m, 2H), 2.84–2.69 (m, 1H), 2.27 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.5, 165.9, 165.2, 157.9, 154.8, 136.2, 130.6, 126.9, 126.6, 122.7, 121.9, 121.0, 118.3, 117.8, 111.7, 111.5, 110.7, 56.0, 52.3, 41.0, 40.1, 29.7, 23.4 (note: carbon 3 and either carbon 1 or 2 are absent); mp 103–107 °C. HMS (APCI) calcd for C25H24N2O6 449.1707; found 449.1704 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-3-methylbenzoate (102)
Compound 102 was prepared via procedure I from 36 (0.10 g, 0.56 mmol), tryptamine (0.090 g, 0.56 mmol), and methyl acetopyruvate (0.081 g, 0.56 mmol) to yield an orange solid (0.12 g, 50%). 1H NMR (600 MHz, DMSO-d6) δ 10.83 (s, 1H), 7.75 (s, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.09 (d, J = 1.8 Hz, 1H), 7.04 (t, J = 7.2 Hz, 1H), 6.90 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 5.27 (s, 1H), 3.81 (s, 3H), 3.75 (dt, J = 8.4 Hz, J = 13.8 Hz, 1H), 2.94–2.89 (m, 1H), 2.76–2.69 (m, 2H), 2.31 (s, 3H), 2.25 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 166.0, 165.5, 140.6, 138.2, 136.3, 131.0, 128.8, 127.0, 126.8, 125.4, 122.9, 121.1, 118.3, 117.7, 111.5, 110.7, 55.6, 41.5, 40.1, 23.6, 18.5, 14.1 (note: carbons 3 and 4, and either carbons 1 or 2 are absent); mp 60–70 °C. HMS (APCI) calcd for C25H24N2O5 433.1763; found 433.1764 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-3-chlorobenzoate (103)
Compound 103 was prepared via procedure I from 32 (0.10 g, 0.50 mmol), tryptamine (0.081 g, 0.50 mmol), and methyl acetopyruvate (0.073 g, 0.50 mmol) to yield a pale-yellow solid (0.080 g, 35%). 1H NMR (400 MHz, DMSO-d6) δ 12.50 (br s, 1H), 10.81 (s, 1H), 7.95 (d, J = 1.6 Hz, 1H), 7.76 (dd, J = 1.2 Hz, J = 8.0 Hz, 1H), 7.29 (d, J = 8.4 Hz, 2H), 7.09–7.01 (m, 3H), 6.91 (t, J = 7.6 Hz, 1H), 5.68 (s, 1H), 3.85 (s, 3H), 3.80–3.73 (m, 1H), 2.97–2.84 (m, 2H), 2.77–2.72 (m, 1H), 2.28 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 192.5, 170.4, 165.5, 164.8, 139.8, 136.2, 134.7, 130.8, 129.8, 128.2, 127.8, 126.9, 122.8, 121.0, 118.3, 117.7, 111.5, 110.4, 59.8, 55.6, 52.6, 41.5, 23.5 (note: carbon 3 is absent); mp 55–60 °C. HMS (APCI) calcd for C24H21ClN2O5 453.1225; found 453.1222 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-3-fluorobenzoate (104)
Compound 104 was prepared via procedure I from 37 (0.12 g, 0.67 mmol), tryptamine (0.11 g, 0.67 mmol), and methyl acetopyruvate (0.097 g, 0.67 mmol) to yield an orange, amorphous solid (0.033 g, 11%). 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 7.65 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 7.6 Hz, 1H), 7.31–7.27 (m, 2H), 7.11 (t, J = 7.2 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 6.93 (s, 1H), 5.52 (s, 1H), 4.02–3.97 (m, 1H), 3.88 (s, 3H), 3.02–2.87 (m, 3H), 2.35 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 192.9, 191.4, 171.1, 167.1 (d, J = 186.7 Hz), 162.7, 136.4, 132.6, 130.9, 127.4, 125.4, 122.3, 121.9, 119.2, 118.7, 116.9 (d, J = 23.8), 114.0, 112.5, 111.3, 102.3, 52.4, 45.9, 41.3, 28.4, 24.1. HMS (APCI) calcd for C24H21FN2O5 437.1507; found 437.1509 [M + H]+.
Methyl 4-(4-Hydroxy-1-(2-(1-methyl-1H-Indol-3-yl)ethyl)-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (105)
Compound 105 was prepared via procedure I from methyl 4-formylbenzoate (0.16 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and 2-(1-methyl-1H-indol-3-yl)ethanamine (0.17 g, 1.0 mmol) to yield cream-colored solid (0.21 g, 43%). 1H NMR (600 MHz, DMSO-d6) δ 8.81 (d, J = 1.8 Hz, 1H), 8.70 (dd, J = 1.8 Hz, J = 4.8, 1H), 8.01 (dt, J = 1.8 Hz, 8.4 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.50–7.46 (m, 3H), 7.36 (d, J = 9.0 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.14–7.10 (m, 2H), 6.96 (t, J = 7.8 Hz, 1H), 5.50 (s, 1H), 3.85–3.80 (m, 4H), 3.70 (s, 3H), 3.00–2.92 (m, 2H), 2.76–2.72 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 181.2, 165.9, 165.0, 153.9, 152.2, 149.1, 142.0, 136.6, 136.3, 133.9, 129.5, 129.4, 128.4, 127.3, 127.2, 123.5, 121.2, 118.4, 118.3, 110.1, 109.7, 60.4, 52.2, 41.3, 32.2, 23.5 (note: either carbon 1 or 2 is absent); mp 215–218 °C. HMS (APCI) calcd for C29H25N3O5 496.1867; found 496.1872 [M + H]+.
Methyl 4-(3-Acetyl-4-hydroxy-1-(2-(2-methyl-1H-indol-3-yl)ethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (106)
Compound 106 was prepared via procedure I from methyl 4-formylbenzoate (0.094 g, 0.57 mmol), 2-(2-methyl-1H-indol-3-yl)ethanamine (0.10 g, 0.57 mmol), and methyl acetopyruvate (0.083 g, 0.57 mmol) to yield a cream-colored solid (0.18 g, 73%). 1H NMR (600 MHz, DMSO-d6) δ 10.73 (s, 1H), 7.85 (d, J = 9.0 Hz, 2H), 7.21 (d, J = 7.8 Hz, 1H), 7.18–7.15 (m, 3H), 6.97 (t, J = 7.8 Hz, 1H), 6.86 (t, J = 7.8 Hz, 1H), 5.03 (s, 1H), 3.83 (s, 3H), 3.64–3.59 (m, 1H), 2.92–2.87 (m, 1H), 2.77–2.72 (m, 1H), 2.60–2.56 (m, 1H), 2.26 (s, 3H), 2.17 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.1, 165.9, 165.0, 142.4, 135.1, 132.2, 129.8, 129.7, 129.3, 128.1, 127.9, 125.6, 120.1, 118.2, 117.0, 110.5, 106.5, 60.1, 52.1, 40.9, 39.9, 24.5, 10.9; mp 182–187 °C. HMS (APCI) calcd for C25H24N2O5 433.1758; found 433.1759 [M + H]+.
Methyl 4-(4-Hydroxy-1-(2-(4-methyl-1H-indol-3-yl)ethyl)-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (107)
Compound 107 was prepared via procedure I from methyl 4-formylbenzoate (0.16 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and 2-(4-methyl-1H-indol-3-yl)ethanamine (0.17 g, 1.0 mmol) to yield a pale-yellow solid (0.05 g, 11%). 1H NMR (600 MHz, DMSO-d6) δ 10.84 (d, J = 1.8 Hz, 1H), 8.82 (d, J = 1.8 Hz, 1H), 8.70 (dd, J = 1.8 Hz, J = 4.8 Hz, 1H), 8.01 (dt, J = 1.8 Hz, J = 8.4 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.49–7.44 (m, 3H), 7.15 (d, J = 8.4 Hz, 1H), 7.04 (d, J = 2.4 Hz, 1H), 6.92 (t, J = 6.6 Hz, 1H), 6.67 (d, J = 7.2 Hz, 1H), 5.48 (s, 1H), 3.89–3.81 (m, 4H), 3.14–3.09 (m, 1H), 2.98–2.87 (m, 2H), 2.44 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 186.9, 165.9, 165.1, 152.3, 149.3, 142.2, 136.7, 136.2, 133.9, 129.5, 129.4, 128.3, 125.4, 123.4, 123.1, 122.9, 121.1, 120.1, 119.9, 118.2, 111.5, 109.6, 66.4, 60.4, 52.2, 25.5, 19.7; mp 170–175 °C. HMS (APCI) calcd for C29H25N3O5 496.1845; found 496.1850 [M + H]+.
Methyl 4-(4-Hydroxy-1-(2-(5-methoxy-1H-indol-3-yl)ethyl)-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (108)
Compound 108 was prepared via procedure I from methyl 4-formylbenzoate (0.16 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and 2-(5-methoxy-1H-indol-3-yl)ethanamine (0.19 g, 1.0 mmol) to yield a yellow solid (0.35 g, 68%). 1H NMR (600 MHz, DMSO-d6) δ 10.72 (s, 1H), 8.79 (d, J = 1.8 Hz, 1H), 8.68 (dd, J = 1.8 Hz, J = 4.8 Hz, 1H), 7.99 (dt, J = 1.8 Hz, J = 7.8 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.48–7.46 (m, 1H), 7.44 (d, J = 7.8 Hz, 2H), 7.22–7.21 (m, 2H), 7.09 (d, J = 2.4 Hz, 1H), 6.75 (d, J = 2.4 Hz, 1H), 6.70 (dd, J = 2.4 Hz, J = 9.0 Hz, 1H), 5.44 (s, H), 3.85–3.80 (m, 4H), 2.68 (s, 3H), 2.98–2.89 (m, 2H), 2.73–2.70 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 186.7, 181.1, 165.9, 165.2, 153.0, 152.0, 149.1, 142.4, 136.3, 134.1, 131.4, 129.5, 129.4, 128.3, 127.2, 123.6, 123.5, 117.9, 112.2, 111.3, 110.5, 99.7, 60.4, 55.2, 52.2, 41.1, 23.8; mp 222–225 °C. HMS (APCI) calcd for C29H25N3O6 512.1816; found 512.1822 [M + H]+.
Methyl 4-(1-(2-(6-Fluoro-1H-indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (109)
Compound 109 was prepared via procedure I from methyl 4-formylbenzoate (0.16 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and 3-(6-fluoro-1H-indol-3-yl)ethanamine (0.18 g, 1.0 mmol) to yield a pale-orange solid (0.44 g, 87%). 1H NMR (600 MHz, DMSO-d6) δ 10.94 (s, 1H), 8.82 (d, J = 0.6 Hz, 1H), 8.69 (d, J = 4.8 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.80 (br s, 1H), 7.49–7.47 (m, 1H), 7.43 (d, J = 7.8 Hz, 2H), 7.33–7.30 (m, 1H), 7.31–7.10 (m, 2H), 6.79 (td, J = 9.6 Hz, J = 1.8 Hz, 1H), 5.44 (s, 1H), 3.89–3.82 (m, 4H), 2.98–2.91 (m, 2H), 2.80–2.76 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 186.9, 181.1, 165.9, 165.1, 159.7, 158.1, 152.3, 142.2, 136.3, 136.1, 133.9, 129.5, 129.3, 128.3, 125.5, 123.8, 123.4, 119.1, 118.1, 111.1, 106.7, 106.6, 60.4 (d, J = 14.4 Hz), 52.1, 41.1, 23.1; mp 162–167 °C. HMS (APCI) calcd for C28H22FN3O5 500.1595; found 500.1600 [M + H]+.
Methyl 4-(1-(2-(6-Chloro-1H-indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (110)
Compound 110 was prepared via procedure I from methyl 4-formylbenzoate (0.16 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and 2-(6-chloro-1H-indol-3-yl)ethanamine (0.20 g, 1.0 mmol) to yield a yellow solid (0.44 g, 85%). 1H NMR (600 MHz, DMSO-d6) δ 11.00 (s, 1H), 8.80 (s, 1H), 8.68 (d, J = 3.6 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 7.8 Hz, 2H), 7.77 (br s, 1H), 7.48–7.41 (m, 3H), 7.37 (d, J = 1.2 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.17 (s, 1H), 6.94 (dd, J = 1.2 Hz, J = 8.4 Hz, 1H), 3.87–3.82 (m, 4H), 2.97–2.89 (m, 2H), 2.79–2.76 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 181.2, 165.9, 165.3, 149.2, 136.6, 136.2, 129.44, 129.36, 128.3, 128.1, 125.8, 125.7, 125.5, 124.7, 124.1, 123.3, 119.5, 118.8, 118.6, 111.2, 111.1, 109.8, 60.3, 52.1, 41.0, 23.4; mp 184–189 °C. HMS (APCI) calcd for C28H22ClN3O5 516.1321; found 516.1324 [M + H]+.
Methyl 4-(4-Hydroxy-1-(2-(6-methyl-1H-indol-3-yl)ethyl)-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (111)
Compound 111 was prepared via procedure I from methyl 4-formylbenzoate (0.16 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and 2-(6-methyl-1H-indol-3-yl)ethanamine (0.17 g, 1.0 mmol) to yield a cream-colored solid (0.10 g, 20%). 1H NMR (600 MHz, DMSO-d6) δ 10.71 (s, 1H), 8.79 (s, 1H), 8.68 (d, J = 3.6 Hz, 1H), 7.99 (d, J = 6.6 Hz, 1H), 7.87 (d, J = 7.8 Hz, 2H), 7.47 (t, J = 6.0 Hz, 1H), 7.42 (d, J = 7.8 Hz, 2H), 7.17 (d, J = 7.8 Hz, 1H), 7.11 (s, 1H), 7.02 (s, 1H), 6.75 (d, J = 7.8 Hz, 1H), 5.40 (s, 1H), 3.90–3.82 (m, 4H), 2.99–2.89 (m, 2H), 2.74–2.71 (m, 1H), 2.37 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 186.7, 165.9, 165.1, 152.1, 149.1, 142.3, 136.7, 136.2, 134.0, 130.0, 129.5, 129.4, 128.4, 128.1, 125.5, 124.9, 123.4, 122.2, 120.0, 117.8, 111.3, 110.6, 60.4, 52.2, 41.1, 23.8, 21.4; mp 202–207 °C. HMS (APCI) calcd for C29H25N3O5 496.1867; found 496.1868 [M + H]+.
Methyl 4-(4-Hydroxyl-1-(2-(6-methyoxy-1H-indol-3-yl)ethyl)-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (112)
Compound 112 was prepared via procedure I from methyl 4-formylbenzoate (0.16 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and 2-(6-methoxy-1H-indol-3-yl)ethanamine (0.19 g, 1.0 mmol) to yield a pale-orange solid (0.39 g, 76%). 1H NMR (600 MHz, DMSO-d6) δ 10.64 (s, 1H), 8.78 (s, 1H), 8.67 (s, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.87 (d, J = 7.8 Hz, 2H), 7.55 (d, J = 7.2 Hz, 1H), 7.41 (d, J = 7.8 Hz, 2H), 7.11 (d, J = 7.8 Hz, 1H), 6.96 (s, 1H), 6.83 (s, 1H), 6.58 (d, J = 7.8 Hz, 1H), 5.39 (s, 1H), 3.82 (s, 3H), 3.75–3.70 (m, 4H), 2.94–2.90 (m, 2H), 2.71–2.69 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 165.8, 155.6, 145.7, 137.6, 137.4, 137.0, 130.8, 130.4, 129.5, 129.4, 128.3, 128.11, 128.07, 125.5, 121.3, 118.7, 110.8, 108.5, 107.5, 94.5, 60.4, 55.2, 40.9, 23.8, 20.8 (note: two of either carbon 1, 2, or 3 are absent); mp 195–200 °C. HMS (APCI) calcd for C29H25N3O6 512.1816; found 512.1821 [M + H]+.
Methyl 4-(1-(2-(7-Fluoro-1H-indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (113)
To a slurry of 2-(7-fluoro-1H-indol-3-yl)ethanamine hydrochloride (0.05 g, 0.23 mmol) in MeOH (0.026 mL) was added triethylamine (0.039 mL, 0.28 mmol). Ether (1.3 mL) was added, and the mixture was stirred at −10 °C for 1 h. The resulting triethylamine hydrochloride salt was filtered off, and the filtrate was concentrated in vacuo to afford a white solid (0.040 g, >99%) which was carried on immediately. Compound 113 was prepared via procedure I from methyl 4-formylbenzoate (0.039 g, 0.24 mmol), 19 (0.052 g, 0.24 mmol), and 2-(7-fluoro-1H-indol-3-yl)ethanamine hydrochloride (0.040 g, 0.24 mmol) to yield a pale-yellow solid (0.11 g, 97%). 1H NMR (600 MHz, DMSO-d6) δ 11.30 (s, 1H), 10.11 (s, 1H), 8.74 (m, 1H), 8.46 (m, 1H), 7.98–7.97 (m, 1H), 7.87–7.86 (m, 1H), 7.37–7.33 (m, 2H), 7.28 (m, 1H), 7.16–7.14 (m, 2H), 6.89–6.87 (m, 2H), 5.23 (s, 1H), 3.84–3.76 (m, 4H), 2.92 (m, 1H), 2.79 (m, 1H), 2.73–2.68 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 193.0 (d, J = 13.4 Hz), 169.4, 166.2, 165.5, 150.0, 149.5, 148.4, 147.7, 139.1, 136.6, 134.3, 131.1, 129.7, 129.0, 128.7, 128.3, 127.7, 124.0, 118.6, 114.7, 112.4, 105.8, 60.0 (d, J = 45.5 Hz), 52.0 (d, J = 41.2 Hz), 40.1 (d, J = 21.8 Hz), 23.3; mp 65–69 °C. HMS (APCI) calcd for C28H22FN3O5 500.1616; found 500.1616 [M + H]+.
Methyl 4-(1-(2-(7-Chloro-1H-indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (114)
To a slurry of 2-(7-chloro-1H-indol-3-yl)ethanamine hydrochloride (0.15 g, 0.65 mmol) in MeOH (0.072 mL) was added triethylamine (0.11 mL, 0.78 mmol). Ether (3.62 mL) was added, and the mixture was stirred at −10 °C for 1 h. The resulting triethylamine hydrochloride salt was filtered off, and the filtrate was concentrated in vacuo to afford a white solid (0.074 g, 58%) which was carried on immediately. Compound 114 was prepared via procedure I from methyl 4-formylbenzoate (0.064 g, 0.39 mmol), 19 (0.086 g, 0.39 mmol), and 2-(7-chloro-1H-indol-3-yl)ethanamine hydrochloride (0.074 g, 0.39 mmol) to yield a yellow solid (0.009 g, 5%). 1H NMR (600 MHz, DMSO-d6) δ 11.22 (s, 1H), 8.80 (s, 1H), 8.69 (d, J = 4.2 Hz, 1H), 8.00 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.49–7.44 (m, 3H), 7.31 (d, J = 7.8 Hz, 1H), 7.22 (d, J = 1.8 Hz, 1H), 7.14 (d, J = 7.2 Hz, 1H), 6.94 (t, J = 7.2 Hz, 1H), 5.49 (s, 1H), 3.89–3.82 (m, 4H), 3.00–2.91 (m, 2H), 2.83–2.78 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 181.3, 165.9, 165.0, 152.4, 149.3, 142.0, 136.2, 133.9, 133.0, 129.5, 129.4, 129.0, 128.3, 124.4, 123.5, 120.6, 119.4, 118.2, 117.3, 115.9, 112.3, 60.2, 52.2, 41.0, 23.6 (note: one of either carbon 1, 2, or 3 are absent); mp 246–249 °C. HMS (APCI) calcd for C28H22ClN3O5 516.1321; found 516.1325 [M + H]+.
Methyl 4-(3-Acetyl-4-hydroxy-1-(2-(7-methyl-1H-indol-3-yl)ethyl)-5-oxo-2, 5-dihydro-1H-pyrrol-2-yl) benzoate (115)
Compound 115 was prepared via procedure I from methyl 4-formylbenzoate (0.094 g, 0.57 mmol), 2-(7-methyl-1H-indol-3-yl) ethanamine (0.10 g, 0.57 mmol), and methyl acetopyruvate (0.083 g, 0.57 mmol) to yield a white solid (0.16 g, 66%). 1H NMR (400 MHz, DMSO-d6) δ 12.50 (br s, 1H), 10.78 (s, 1H), 7.88 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.12–7.07 (m, 2H), 6.83–6.82 (m, 2H), 5.20 (s, 1H), 3.83–3.76 (m, 4H), 2.98–2.80 (m, 2H), 2.80–2.66 (m, 1H), 2.41 (s, 3H), 2.26 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 165.9, 165.0, 142.4, 135.7, 129.3, 128.1, 126.5, 125.5, 122.5, 121.5, 120.5, 118.5, 115.7, 111.1, 109.7, 59.7, 52.1, 40.8, 40.0, 23.7, 16.7 (note: carbons 3 and 4 are absent); mp 220–227 °C. HMS (APCI) calcd from C25H24N2O5 433.1758; found 433.1758 [M + H]+.
Ethyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (116)
Compound 116 was prepared via procedure I from 19 (0.15 g, 0.68 mmol), tryptamine (0.11 g, 0.68 mmol), and 35 (0.12 g, 0.68 mmol) to yield a yellow solid (0.027 g, 8%). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.75 (s, 1H), 8.48 (d, J = 3.2 Hz, 1H), 7.94 (d, J = 7.2 Hz, 1H), 7.83 (d, J = 8.0 Hz, 2H), 7.34–7.27 (m, 5H), 7.08–7.04 (m, 2H), 6.92 (t, J = 7.2 Hz, 1H), 5.33 (s, 1H), 4.28 (q, J = 7.6 Hz, 2H), 3.84–3.77 (m, 1H), 3.00–2.93 (m, 1H), 2.87–2.80 (m, 1H), 2.73–2.66 (m, 1H), 1.28 (t, J = 6.8 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 186.8, 182.1, 162.9, 155.0, 148.3, 148.1, 136.4, 136.3, 127.0, 123.8, 123.4, 123.2, 123.1, 121.5, 121.3, 120.8, 118.5, 118.2, 117.8, 111.8, 111.2, 109.5, 60.6, 45.3, 23.6, 23.3, 14.0; mp >250 °C. HMS (APCI) calcd for C29H25N3O5 496.1867; found 496.1872 [M + H]+.
Ethyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-hydroxy-3-isonicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (117)
Compound 117 was prepared via procedure I from 45 (0.40 g, 2.3 mmol), tryptamine (0.36 g, 2.3 mmol), and 20 (0.50 g, 2.3 mmol) to yield a yellow solid (0.019 g, 2%). 1H NMR (600 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.68 (d, J = 5.4 Hz, 2H), 7.88 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 5.4 Hz, 2H), 7.40 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.12 (d, J = 1.8 Hz, 1H), 7.06 (t, J = 7.8 Hz, 1H), 6.92 (t, J = 7.2 Hz, 1H), 5.40 (s, 1H), 4.29 (q, J = 7.2 Hz, 2H), 3.88–3.82 (m, 1H), 3.00–2.90 (m, 2H), 2.78–2.73 (m, 1H), 1.29 (7.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 189.0, 165.4, 165.0, 149.6, 145.8, 142.2, 136.2, 129.8, 129.7, 129.3, 128.3, 126.9, 122.9, 121.8, 121.1, 118.3, 118.1, 117.35, 111.5, 110.7, 60.7, 60.3, 41.1, 23.6, 14.2; mp 169–172 °C. HMS (APCI) calcd for C29H25N3O5 496.1880; found 496.1877 [M + H]+.
Methyl 4-(4-Hydroxy-1-(2-(2-methyl-1H-indol-3-yl)ethyl)-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (118)
Compound 118 was prepared via procedure I from 19 (0.15 g, 0.68 mmol), 2-(2-methyl-1H-indol-3-yl)ethanamine (0.12 g, 0.68 mmol), and methyl 4-formylbenzoate (0.11 g, 0.68 mmol) to yield an orange solid (0.046 g, 14%). 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.73 (s, 1H), 8.48 (d, J = 4.4 Hz, 1H), 7.93 (s, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.30–7.17 (m, 5H), 6.97 (t, J = 6.4 Hz, 1H), 6.86 (t, J = 7.2 Hz, 1H), 5.23 (s, 1H), 3.81 (s, 3H), 3.63–3.56 (m, 1H), 2.96–2.88 (m, 1H), 2.76–2.69 (m, 1H), 2.57–2.50 (m, 1H), 2.18 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 182.6, 166.0, 150.4, 150.1, 149.2, 136.8, 136.1, 135.5, 135.2, 132.2, 129.0, 128.6, 128.0, 123.6, 122.6, 120.1, 118.2, 117.0, 110.6, 106.7, 61.2, 52.0, 41.3, 22.6, 11.0 (note: two of carbons 1, 2, 3, or 4 are absent); mp >250 °C. HMS (APCI) calcd for C29H25N3O5 496.1867; found 496.1872 [M + H]+.
Ethyl 4-(3-Acetyl-4-hydroxy-1-(2-(2-methyl-1H-indol-3-yl)ethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (119)
Compound 119 was prepared via procedure I from 45 (0.15 g, 0.84 mmol), 2-(2-methyl-1H-indol-3-yl)ethanamine (0.15 g, 0.84 mmol), and methyl acetopyruvate (0.12 g, 0.84 mmol) to yield a cream-colored solid (0.078 g, 21%). 1H NMR (600 MHz, DMSO-d6, 80 °C) δ 10.5 (br s, 1H), 7.83 (d, J = 7.2 Hz, 2H), 7.23–7.18 (m, 4H), 6.95 (t, J = 7.2 Hz, 1H), 6.86 (t, J = 7.2 Hz, 1H), 5.09 (s, 1H), 4.30 (q, J = 7.2 Hz, 2H), 3.61–3.56 (m, 1H), 2.92–2.87 (m, 1H), 2.80–2.75 (m, 1H), 2.57–2.53 (m, 1H), 2.19 (s, 3H), 2.05 (s, 3H), 1.30 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.3, 189.3, 165.5, 135.1, 132.1, 129.1, 127.9, 120.1, 118.1, 117.0, 112.1, 110.5, 108.9, 106.6, 98.5, 90.2, 54.9, 48.7, 48.6, 40.0, 29.0, 14.2, 11.0 (note: one of either carbon 1, 2, or 4 is absent); mp 190–195 °C. HMS (APCI) calcd for C26H26N2O5 447.1928; found 447.30 [M + H]+.
Ethyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)-3-hydroxybenzoate (120)
Compound 120 was prepared via procedure I from 189 (0.15 g, 0.77 mmol), tryptamine (0.12 g, 0.77 mmol), and methyl acetopyruvate (0.11 g, 0.77 mmol) to yield a cream-colored solid (0.20 g, 57%). 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 10.42 (s, 1H), 7.59–7.46 (m, 1H), 7.34–7.23 (m, 3H), 7.12–7.02 (m, 2H), 6.97–6.89 (m, 2H), 5.76 (s, 1H), 4.28 (q, J = 7.2 Hz, 2H), 3.78–3.74 (m, 1H), 2.98–2.85 (m, 2H), 2.73–2.68 (m, 1H), 2.28 (s, 3H), 1.29 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.7, 165.5, 165.1, 156.2, 154.8, 136.2, 130.5, 128.1, 127.0, 126.7, 125.5, 122.7, 121.0, 120.2, 118.3, 118.0, 116.1, 111.4, 110.8, 60.7, 41.0, 40.0, 29.7, 23.4, 14.2; mp 200–205 °C. HMS (APCI) calcd for C25H24N2O6 449.1721; found 449.1723 [M + H]+.
Methyl 4-(1-((1H-Indol-3-yl)methyl)-4-hydroxy-3-isonicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (121)
Compound 121 was prepared via procedure I from methyl 4-formylbenzoate (0.28 g, 1.7 mmol), (1H-indol-3-yl)methanamine (0.25 g, 1.7 mmol), and 20 (0.38 g, 1.7 mmol) to yield an orange solid (0.78 g, 98%). 1H NMR (400 MHz, DMSO-d6) δ 11.08 (s, 1H), 8.66 (d, J = 5.6 Hz, 2H), 7.92 (d, J = 8.0 Hz, 2H), 7.51–7.37 (m, 6H), 7.13–7.08 (m, 2H), 6.97 (t, J = 7.2 Hz, 1H), 5.11–5.07 (m, 2H), 3.84 (s, 3H), 3.80 (d, J = 14.8 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 166.0, 165.1, 149.2, 129.45, 129.36, 128.2, 128.1, 126.2, 125.5, 125.1, 122.0, 121.7, 121.5, 119.0, 118.9, 118.6, 118.2, 111.7, 109.3, 59.2, 52.2, 35.5 (note: carbon 4 is absent); mp 240–243 °C. HMS (APCI) calcd for C27H21N3O5 468.1567; found 468.1566 [M + H]+.
Methyl 4-(1-(3-(1H-Indol-3-yl)propyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (122)
Compound 122 was prepared via procedure I from methyl 4-formylbenzoate (0.16 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and 3-(1H-indol-3-yl)propan-1-amine (0.17 g, 1.0 mmol) to yield a yellow solid (0.35 g, 71%). 1H NMR (600 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.82 (s, 1H), 8.69 (d, J = 4.8 Hz, 1H), 8.02 (d, J = 7.8 Hz, 1H), 7.87 (d, J = 8.4 Hz, 2H), 7.49–7.47 (m, 3H), 7.37 (d, J = 7.8 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 7.06–7.03 (m, 2H), 6.92 (t, J = 7.2 Hz, 1H), 5.54 (s, 1H), 3.62–3.55 (m, 4H), 2.83–2.79 (m, 1H), 2.62–2.55 (m, 2H), 1.84–1.80 (m, 1H), 1.73–1.69 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 186.9, 165.9, 165.2, 154.4, 152.1, 149.2, 142.4, 136.3, 134.0, 129.5, 128.3, 127.0, 123.5, 122.4, 122.3, 120.8, 118.3, 118.0, 113.4, 111.4, 60.4, 52.3, 40.7, 28.0, 22.1 (note: either carbon 1 or 2 and carbon 3 are absent); mp 221–226 °C. HMS (APCI) calcd for C29H25N3O5 496.1845; found 496.1852 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (156)
To a solution of 1 (0.50 g, 1.2 mmol) in DCM:MeOH (1:1, 0.13M) was added (diazomethyl)trimethylsilane (0.72 mL, 1.4 mmol). The reaction mixture continued to stir at rt for 5 h before being concentrated in vacuo. The crude residue was then purified using flash column chromatography on SiO2 (3% MeOH/DCM) to yield a pale-yellow solid (0.24 g, 46%). 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 7.88 (dd, J = 1.6 Hz, J = 8.0 Hz, 2H), 7.33–7.24 (m, 4H), 7.09–7.03 (m, 2H), 6.92 (t, J = 7.6 Hz, 1H), 5.20 (s, 1H), 4.36 (s, 3H), 3.84 (s, 3H), 3.80–3.71 (m, 1H), 2.96–2.89 (m, 1H), 2.84–2.77 (m, 1H), 2.72–2.63 (m, 1H), 2.25 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 191.9, 165.9, 164.2, 154.0, 141.7, 136.2, 129.5, 129.3, 128.2, 126.8, 126.2, 122.9, 121.0, 118.3, 118.0, 111.5, 110.7, 59.5, 59.1, 52.2, 40.9, 30.3, 23.6; mp 40–45 °C. HMS (APCI) calcd for C25H24N2O5 433.1763; found 433.1761 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-3-acetyl-4-amino-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (157)
To a solution of 1 (0.50 g, 1.2 mmol) in 2-methoxyethanol (8.36 mL, 0.14 M) was added ammonium formate (0.11 mL, 2.2 mmol, 1.8 equiv). The reaction mixture was refluxed for 3 h before being concentrated in vacuo, ground with a mortar and pestle, and triturated with Et2O. Further purification was achieved via flash column chromatography on SiO2 (10% MeOH/DCM) to yield a pale-yellow solid (0.070 g, 14%). 1H NMR (400 MHz, CDCl3, 56 °C) δ 10.02 (br s, 1H), 8.36 (br s, 1H), 7.92 (d, J = 8.0 Hz, 2H), 7.39–7.34 (m, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.08–7.02 (m, 3H), 6.97 (s, 1H), 6.44 (br s, 1H), 4.77 (s, 1H), 4.04–3.97 (m, 1H), 3.91 (s, 3H), 3.09–2.99 (m, 2H), 2.92–2.85 (m, 1H), 1.56 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 177.8, 165.9, 164.4, 163.2, 144.7, 136.2, 129.7, 129.5, 128.3, 126.9, 122.7, 121.0, 118.2, 118.0, 111.4, 110.8, 105.6, 58.0, 52.1, 48.6, 41.0, 23.2; mp 50–54 °C. HMS (APCI) calcd for C24H23N3O4 418.1766; found 418.1766 [M + H]+.
Methyl 4-(1-(2-(1H-Indol-3-yl)ethyl)-4-acetoxy-3-acetyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (158)
To a solution of 1 (0.50 g, 1.2 mmol) in DCM (11 mL, 0.11 M) was added acetic anhydride (0.14 mL, 1.4 mmol, 1.2 equiv) and pyridine (0.14 mL, 1.8 mmol, 1.5 equiv). The reaction mixture was stirred at rt for 61/2 h before being concentrated in vacuo. The crude material was then purified by flash column chromatography on SiO2 (3% MeOH/DCM). Additional purification was achieved using HPLC (75% ACN with 0.1% formic acid) to give a yellow oil (0.038 g, 7%). 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.94–7.92 (m, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.21 (t, J = 8.0 Hz, 1H), 7.10–7.05 (m, 3H), 6.98 (d, J = 2.0 Hz, 1H), 4.93 (s, 1H), 4.07–4.00 (m, 1H), 3.91 (s, 3H), 3.08–2.89 (m, 3H), 2.47 (s, 3H), 2.26 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 191.5, 167.0, 166.6, 163.8, 147.7, 139.2, 137.0, 136.6, 131.0, 130.4, 128.1, 127.4, 122.6, 122.2, 119.9, 118.7, 112.6, 111.5, 62.3, 62.4, 41.5, 30.1, 24.4, 20.8. HMS (APCI) calcd for C26H24N2O6 461.1721; found 461.1717 [M + H]+.
Methyl 4-(3-Acetyl-4-(butyryloxy)-1-(2-(2-methyl-1H-indol-3-yl)ethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (159)
To a solution of 106 (0.30 g, 0.69 mmol) and triethylamine (0.19 mL, 1.4 mmol, 2.0 equiv) in THF (0.69 mL, 1.0 M) at −30 °C was added butyryl chloride (0.072 mL, 0.69 mmol, 1.0 equiv) dropwise over 20 min. The mixture was allowed to stir at −30 °C for 2 h before being concentrated in vacuo. The crude material was dissolved in EtOAc, washed with water and brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification was achieved via flash column chromatography on SiO2 (10% EtOAc:DCM) to afford a yellow, amorphous solid (0.12 g, 35%). 1H NMR (600 MHz, CDCl3) δ 7.96 (s, 1H), 7.88 (d, J = 7.8 Hz, 2H), 7.25 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 7.2 Hz, 1H), 7.11 (t, J = 7.2 Hz, 1H), 7.01 (t, J = 7.2 Hz, 1H), 6.94 (d, J = 7.8 Hz, 2H), 4.78 (s, 1H), 3.89 (s, 3H), 3.87–3.82 (m, 1H), 3.02 (dt, J = 13.8 Hz, J = 8.4 Hz, 1H), 2.91 (dt, J = 13.8 Hz, J = 7.8 Hz, 1H), 2.82–2.78 (m, 1H), 2.70 (t, J = 7.8 Hz, 2H), 2.24 (s, 3H), 2.23 (s, 3H), 1.85 (sextet, 7.8 Hz, 2H), 1.09 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 191.6, 170.0, 166.6, 163.7, 147.9, 139.1, 136.9, 135.4, 132.1, 130.8, 130.2, 128.3, 128.0, 121.5, 119.7, 117.8, 110.6, 108.0, 62.5, 52.4, 41.4, 35.9, 30.2, 23.3, 18.4, 13.7, 11.5. HMS (APCI) calcd for C29H30N2O6 503.2168; found 503.2172 [M + H]+.
Methyl 4-(3-Acetyl-4-(acryloyloxy)-1-(2-(2-methyl-1H-indol-3-yl)ethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (160)
To a solution of 106 (3.0 g, 6.9 mmol) and triethylamine (1.9 mL, 14 mmol, 2.0 equiv) in THF (6.9 mL, 1.0 M) at −30 °C was added acryloyl chloride (0.72 mL, 6.9 mmol, 1.0 equiv) dropwise over 20 min. The mixture was allowed to stir at −30 °C for 2 h before being concentrated in vacuo. The crude material was dissolved in EtOAc, washed with water and brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification was achieved via flash column chromatography on SiO2 (10% EtOAc:DCM) to afford a yellow, amorphous solid (0.68 g, 20%). 1H NMR (300 MHz, CDCl3) δ 8.00 (s, 1H), 7.89 (d, J = 8.7 Hz, 2H), 7.27–7.20 (m, 2H), 7.11 (t, J = 7.2 Hz, 1H), 7.03–6.94 (m, 3H), 6.71 (d, J = 0.9 Hz, 1H), 6.43 (d, J = 10.2 Hz, 1H), 6.20 (d, J = 0.6 Hz, 1H), 4.81 (s, 1H), 3.89–3.81 (m, 4H), 3.08–2.76 (m, 3H), 2.24 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 196.1, 191.6, 186.7, 166.6, 163.6, 162.2, 147.6, 139.0, 137.3, 135.7, 135.4, 132.1, 130.8, 130.2, 128.3, 128.0, 126.0, 121.5, 119.6, 117.7, 110.7, 107.9, 62.6, 52.4, 41.5, 30.2, 23.3, 11.5. HMS (APCI) calcd for C28H26N2O6 487.1837; found 487.1860 [M + H]+.
Methyl 4-(3-Acetyl-4-hydroxy-5-oxo-1-phenethyl-2,5-dihydro-1H-pyrrol-2-yl)benzoate (161)
Compound 161 was prepared via procedure I from methyl 4-formylbenzoate (0.34 g, 2.1 mmol), ethyl acetopyruvate (0.33 g, 2.1 mmol), and 2-phenylethanamine (0.25 g, 2.1 mmol) to yield a white solid (0.73 g, 93%). 1H NMR (400 MHz, DMSO-d6) δ 7.92 (d, J = 8.4 Hz, 2H), 7.31–7.24 (m, 4H), 7.19 (d, J = 7.2 Hz, 1H), 7.10 (d, J = 6.8 Hz, 2H), 5.15 (s, 1H), 3.83–3.76 (m, 4H), 2.82–2.60 (m, 3H), 2.26 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 190.9, 165.9, 165.0, 142.4, 138.5, 129.4, 129.4, 128.6, 128.5, 128.1, 126.4, 125.6, 119.7, 66.4, 59.7, 52.2, 41.5, 33.5; mp 123–128 °C. HMS (APCI) calcd for C22H21NO5 380.1493; found 380.1494 [M + H]+.
Methyl 4-(4-Hydroxy-3-isonicotinoyl-1-(2-(naphthalen-1-yl)ethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (162)
Compound 162 was prepared via procedure I from methyl 4-formylbenzoate (0.40 g, 2.4 mmol), 188 (0.41 g, 2.4 mmol), and 20 (0.53 g, 2.4 mmol) to yield a yellow solid (0.67 g, 56%). 1H NMR (600 MHz, DMSO-d6) δ 8.70 (d, J = 3.6 Hz, 2H), 7.93–7.84 (m, 4H), 7.81 (d, J = 7.8 Hz, 1H), 7.57–7.40 (m, 7H), 7.30 (d, J = 6.6 Hz, 1H), 5.40 (s, 1H), 3.84–3.80 (m, 4H), 3.37–3.34 (m, 1H), 3.13–3.08 (m, 1H), 3.03–3.00 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 196.1, 165.9, 162.1, 160.1, 136.4, 136.3, 130.7, 129.5, 128.2, 128.0, 125.5, 123.4, 122.8, 121.2, 120.8, 118.48, 118.45, 118.2, 118.1, 115.2, 111.6, 111.3, 61.5, 52.2, 48.6, 26.2; mp 221–224 °C. HMS (APCI) calcd for C30H24N2O5 493.1758; found 493.1756 [M + H]+.
Methyl 4-(1-(2-(Benzofuran-3-yl)ethyl)-4-hydroxy-3-nicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (163)
Compound 163 was prepared via procedure I from methyl 4-formylbenzoate (0.20 g, 1.2 mmol), 19 (0.27 g, 1.2 mmol), and 2-(benzofuran-3-yl)ethanamine (0.20 g, 1.2 mmol) to yield a pale-yellow solid (0.028 g, 4.7%). 1H NMR (600 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.57 (s, 1H), 7.99 (d, J = 7.2 Hz, 1H), 7.86 (d, J = 7.8 Hz, 2H), 7.77 (s, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.47–7.42 (m, 3H), 7.37 (t, J = 5.4 Hz, 1H), 7.29 (t, J = 7.2 Hz, 1H), 7.20 (t, J = 7.2 Hz, 1H), 5.44 (s, 1H), 3.87–3.82 (m, 4H), 2.97–2.92 (m, 2H), 2.76–2.75 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 189.3, 181.3, 166.0, 154.6, 150.6, 149.5, 142.4, 135.9, 129.1, 128.8, 128.1, 127.5, 124.4, 122.7, 122.4, 119.6, 117.0, 115.6, 111.3, 60.2, 52.1, 40.0, 21.7 (note: carbons 1, 2, and 3 are absent); mp 225–230 °C. HMS (APCI) calcd for C28H22N2O6 481.1397; found 481.1396 [M – H]−.
Methyl 4-(1-(2-(1H-Benzo[d]imidazol-1-yl)ethyl)-4-hydroxy-3-isonicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (164)
Compound 164 was prepared via procedure I from methyl 4-formylbenzoate (0.19 g, 1.1 mmol), 2-(1H-benzo[d]imidazol-1-yl)ethanamine (0.18 g, 1.1 mmol), and 20 (0.25 g, 1.1 mmol) to yield a yellow solid (0.40 g, 73%). 1H NMR (400 MHz, DMSO-d6) δ 8.66 (d, J = 1.3 Hz, 2H), 8.59 (s, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.67 (t, J = 5.2 Hz, 1H), 7.61 (t, J = 5.2 Hz, 1H), 7.52 (d, J = 5.6 Hz, 2H), 7.33–7.29 (m, 4H), 5.37 (s, 1H), 4.61–4.54 (m, 1H), 4.48–4.44 (m, 1H), 4.03–3.98 (m, 1H), 3.82 (s, 3H), 3.08–3.04 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 186.2, 166.4, 165.9, 149.2, 146.4, 143.4, 142.6, 139.6, 132.9, 129.4, 128.4, 128.14, 128.06, 125.5, 123.5, 123.1, 122.1, 118.2, 111.0, 60.08, 52.2, 43.1 (note: one sp3 carbon is under the DMSO peak); mp 135–140 °C. HMS (APCI) calcd for C27H22N4O5 483.1676; found 483.1674 [M + H]+.
Methyl 4-(1-(2-(1H-Imidazol-4-yl)ethyl)-4-hydroxy-3-isonicotinoyl-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (165)
Compound 165 was prepared via procedure I from methyl 4-formylbenzoate (0.37 g, 2.3 mmol), 2-(1H-imidazol-4-yl)ethanamine (0.25 g, 2.3 mmol), and 20 (0.50 g, 2.3 mmol) to yield an orange solid (0.031 g, 3%). 1H NMR (400 MHz, DMSO-d6) δ 8.61 (s, 1H), 8.50 (d, J = 5.2 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 5.2 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 7.26 (s, 1H), 7.11 (s, 1H), 5.27 (s, 1H), 3.90–3.82 (m, 4H), 2.83–2.76 (m, 3H). 13C NMR (150 MHz, DMSO-d6) δ 182.5, 169.1, 166.1, 148.8, 148.0, 134.0, 131.2, 129.0, 128.5, 128.1, 127.8, 127.6, 122.4, 116.4, 111.6, 66.3, 52.0, 40.0, 22.3; mp 64–70 °C. HMS (APCI) calcd for C23H20N4O5 433.1512; found 433.1513 [M + H]+.
Methyl 4-(3-Acetyl-4-hydroxy-1-(3-methylphenethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (166)
Compound 166 was prepared via procedure I from methyl 4-formylbenzoate (0.30 g, 1.8 mmol), ethyl acetopyruvate (0.29 g, 1.8 mmol), and 2-(m-tolyl)ethanamine (0.25 g, 1.8 mmol) to yield a white solid (0.56 g, 76%). 1H NMR (400 MHz, DMSO-d6) δ 7.90 (d, J = 7.6 Hz, 2H), 7.28 (d, J = 7.6 Hz, 2H), 7.12 (t, J = 8.0 Hz, 1H), 6.99 (d, J = 7.6 Hz, 1H), 6.88–6.86 (m, 2H), 5.13 (s, 1H), 3.83 (s, 3H), 3.79–3.73 (m, 1H), 2.77–2.70 (m, 2H), 2.61–2.57 (m, 1H), 2.26 (s, 3H), 2.22 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 191.5, 166.0, 165.1, 142.4, 138.4, 137.5, 129.4, 129.2, 128.4, 128.1, 127.1, 126.9, 125.62, 125.56, 119.7, 66.4, 59.7, 52.2, 41.6, 33.4, 21.0; mp 118–123 °C. HMS (APCI) calcd for C23H23NO5 394.1649; found 394.1651 [M + H]+.
Methyl 4-(3-Acetyl-1-(3-chlorophenethyl)-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (167)
Compound 167 was prepared via procedure I from methyl 4-formylbenzoate (0.26 g, 1.6 mmol), ethyl acetopyruvate (0.25 g, 1.6 mmol), and 2-(3-chlorophenyl)ethanamine (0.25 g, 1.6 mmol) to yield a pale-yellow solid (0.50 g, 76%). 1H NMR (400 MHz, DMSO-d6) δ 7.90 (d, J = 8.4 Hz, 2H), 7.32–7.20 (m, 5H), 7.08 (d, J = 7.2 Hz, 1H), 5.22 (s, 1H), 3.83–3.79 (m, 4H), 2.79–2.69 (m, 3H), 2.26 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 165.1, 142.3, 141.2, 133.0, 130.5, 130.2, 129.4, 128.5, 128.1, 127.6, 127.4, 126.4, 125.5, 59.5, 52.2, 41.1, 32.9, 29.7 (note: carbon 4 is absent); mp 118–122 °C. HMS (APCI) calcd for C22H20ClNO5 414.1108; found 414.1109 [M + H]+.
Methyl 4-(3-Acetyl-1-(3-fluorophenethyl)-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (168)
Compound 168 was prepared via procedure I from methyl 4-formylbenzoate (0.30 g, 1.8 mmol), ethyl acetopyruvate (0.28 g, 1.8 mmol), and 2-(3-fluorophenyl)ethanamine (0.25 g, 1.8 mmol) to yield an off-white solid (0.57 g, 79%). 1H NMR (400 MHz, DMSO-d6) δ 7.90 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.0 Hz, 2H), 7.29–7.25 (m, 1H), 7.00–6.94 (m, 3H), 5.22 (s, 1H), 3.83–3.79 (m, 4H), 2.81–2.69 (m, 3H), 2.26 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 165.1, 162.2 (d, J = 241.8 Hz), 142.3, 141.6, 141.5, 130.3 (d, J = 8.1 Hz), 129.44, 129.40, 128.1, 125.6, 124.8, 115.3 (d, J = 20.9 Hz), 113.2 (d, J = 20.9 Hz), 59.5, 52.2, 41.1, 33.0, 15.2 (note: carbon 4 is absent); mp 114–119 °C. HMS (APCI) calcd for C22H20FNO5 398.1403; found 398.1404 [M + H]+.
Methyl 4-(3-Acetyl-4-hydroxy-1-(3-methoxyphenethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (169)
Compound 169 was prepared via procedure I from methyl 4-formylbenzoate (0.27 g, 1.7 mmol), ethyl acetopyruvate (0.26 g, 1.7 mmol), and 2-(3-methoxyphenyl)ethanamine (0.25 g, 1.7 mmol) to yield an off-white solid (0.52 g, 76%). 1H NMR (400 MHz, DMSO-d6) δ 7.90 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 7.16 (t, J = 8.0 Hz, 1H), 6.75 (d, J = 7.6 Hz, 1H), 6.68–6.66 (m, 2H), 5.15 (s, 1H), 3.83–3.76 (m, 4H), 3.69 (s, 3H), 2.77–2.70 (m, 2H), 2.65–2.60 (m, 1H), 2.26 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.0, 165.0, 159.3, 145.8, 142.4, 140.1, 129.5, 129.42, 129.38, 128.1, 125.5, 120.8, 114.1, 111.9, 59.6, 54.9, 52.2, 41.4, 33.4, 29.7 (note: carbon 4 is absent); mp 150–153 °C. HMS (APCI) calcd for C23H23NO6 410.1603; found 410.1604 [M + H]+.
Methyl 4-(3-Acetyl-4-hydroxy-1-(3-hydroxyphenethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate (170)
Compound 170 was prepared via procedure I from methyl 4-formylbenzoate (0.55 g, 3.4 mmol), ethyl acetopyruvate (0.53 g, 3.4 mmol), and 190 (0.46 g, 3.4 mmol) to yield a cream-colored solid (1.0 g, 78%). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br s, 1H), 7.93 (d, J = 8.8 Hz, 2H), 7.33 (d, J = 8.8 Hz, 2H), 7.08 (t, J = 7.6 Hz, 1H), 6.61 (dt, J = 1.6 Hz, J = 6.8 Hz, 1H), 6.54–6.52 (m, 2H), 5.16 (s, 1H), 3.87–3.83 (m, 4H), 2.74–2.69 (m, 2H), 2.56–2.52 (m, 1H), 2.30 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 193.0, 166.0, 165.0, 157.5, 142.4, 139.9, 129.9, 129.7, 129.5, 129.2, 128.1, 127.0, 119.2, 115.4, 113.5, 59.7, 52.2, 48.7, 41.6, 33.6; mp 98–104 °C. HMS (APCI) calcd for C22H21NO6 396.1447; found 396.1446 [M + H]+.
Methyl 4-(4-Hydroxy-3-isonicotinoyl-5-oxo-1-(2-(pyridin-4-yl)ethyl)-2,5-dihydro-1H-pyrrol-2-yl)benzoate (171)
Compound 171 was prepared via procedure I from methyl 4-formylbenzoate (0.19 g, 1.1 mmol), 2-(pyridin-4-yl)ethanamine (0.14 g, 1.1 mmol), and 20 (0.25 g, 1.1 mmol) to yield a yellow solid (0.47 g, 95%). 1H NMR (400 MHz, DMSO-d6) δ 8.65 (d, J = 6.0 Hz, 2H), 8.49–8.48 (m, 2H), 7.92 (d, J = 8.0 Hz, 2H), 7.56–7.53 (m, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.29–7.26 (m, 2H), 5.47 (s, 1H), 3.91–3.83 (m, 4H), 2.88–2.78 (m, 3H). 13C NMR (150 MHz, DMSO-d6) δ 190.1, 185.8, 182.0, 166.3, 166.0, 149.5, 149.2, 148.3, 146.6, 143.4, 129.4, 128.2, 124.6, 122.2, 115.6, 59.9, 52.2, 40.6, 32.7; mp 126–129 °C. HMS (APCI) calcd for C25H21N3O5 444.1567; found 444.1566 [M + H]+.
Methyl 4-(4-Hydroxy-3-isonicotinoyl-5-oxo-1-(2-(pyridin-3-yl)ethyl)-2,5-dihydro-1H-pyrrol-2-yl)benzoate (172)
Compound 172 was prepared via procedure I from methyl 4-formylbenzoate (0.19 g, 1.1 mmol), 2-(pyridin-3-yl)ethanamine (0.14 g, 1.1 mmol), and 20 (0.25 g, 1.1 mmol) to yield a yellow solid (0.47 g, 93%). 1H NMR (400 MHz, DMSO-d6) δ 8.67 (d, J = 6.0 Hz, 2H), 8.44 (dd, J = 1.2 Hz, J = 4.8 Hz, 1H), 8.41 (d, J = 1.6 Hz, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 7.6 Hz, 1H), 7.55 (d, J = 6.0 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.35 (dd, J = 4.8 Hz, J = 7.6 Hz, 1H), 5.51 (s, 1H), 3.91–3.83 (m, 4H), 2.91–2.77 (m, 3H). 13C NMR (100 MHz, DMSO-d6) δ 206.6, 186.6, 165.9, 165.6, 149.3, 148.9, 146.9, 146.1, 142.7, 137.3, 134.7, 129.5, 128.2, 128.1, 123.8, 122.0, 116.6, 59.9, 52.2, 41.3, 30.5; mp 160–163 °C. HMS (APCI) calcd for C25H21N3O5 444.1567; found 444.1565 [M + H]+.
Methyl 4-(4-Hydroxy-3-isonicotinoyl-5-oxo-1-(2-(pyridin-2-yl)ethyl)-2,5-dihydro-1H-pyrrol-2-yl)benzoate (173)
Compound 173 was prepared via procedure I from methyl 4-formylbenzoate (0.19 g, 1.1 mmol), 2-(pyridin-2-yl)ethanamine (0.14 g, 1.1 mmol), and 20 (0.25 g, 1.1 mmol) to yield a yellow solid (0.50 g, >99%). 1H NMR (400 MHz, DMSO-d6) δ 8.67 (d, J = 6.0 Hz, 2H), 8.48 (dd, J = 1.6 Hz, J = 5.2 Hz, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.73 (td, J = 2.0 Hz, J = 7.6 Hz, 1H), 7.53 (dd, J = 1.2 Hz, J = 4.4 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.27 (s, 1H), 7.25 (t, J = 3.6 Hz, 1H), 5.44 (s, 1H), 4.00–3.93 (m, 1H), 3.82 (s, 3H), 3.07–2.97 (m, 2H), 2.92–2.84 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 206.6, 186.7, 165.9, 165.5, 158.1, 156.6, 149.4, 148.7, 146.1, 142.6, 137.3, 129.5, 128.2, 125.6, 123.6, 122.0, 116.9, 60.1, 52.2, 40.3, 35.4; mp 187–190 °C. HMS (APCI) calcd for C25H21N3O5 444.1567; found 444.1564 [M + H]+.
1-(2-(1H-Indol-3-yl(ethyl)-3-hydroxy-4-nicotinoyl-5-(pyridine-4-yl)-1H-pyrrol-2(5H)-one (173)
Compound 174 was prepared via procedure I from isonicotinaldehyde (0.11 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield a mustard-colored solid (0.32 g, 76%). 1H NMR (600 MHz, DMSO-d6) δ 10.93 (s, 1H), 8.84 (d, J = 1.8 Hz, 1H), 8.67 (dd, J = 1.8 Hz, J = 4.8 Hz, 1H), 8.53 (d, J = 5.4 Hz, 2H), 8.05 (dt, J = 2.4 Hz, J = 8.4 Hz, 1H), 7.49–7.47 (m, 2H), 7.42–7.34 (m, 3H), 7.30 (br s, 1H), 7.13 (d, J = 1.8 Hz, 1H), 7.06 (t, J = 7.8 Hz, 1H), 6.94 (t, J = 8.4 Hz, 1H), 5.34 (s, 1H), 3.92–3.87 (m, 1H), 3.02–2.91 (m, 2H), 2.82–2.77 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 185.5, 166.1, 151.3, 148.9, 148.4, 136.7, 136.3, 134.5, 126.9, 125.5, 123.6, 123.4, 123.0, 121.1, 118.3, 118.1, 116.1, 111.6, 110.8, 59.7, 41.2, 23.7 (note: either carbon 1 or 2 is absent); mp 225–230 °C. HMS (APCI) calcd for C25H20N4O3 425.1608; found 425.1610 [M + H]+.
1-(2-(1H-Indol-3-yl)ethyl)-3-hydroxy-4-nicotinoyl-5-(pyridine-3-yl)-1H-pyrrol-2(5H)-one (175)
Compound 175 was prepared via procedure I from nicotinaldehyde (0.11 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield an orange solid (0.29 g, 67%). 1H NMR (600 MHz, DMSO-d6) δ 10.90 (s, 1H), 8.83 (d, J = 2.4 Hz, 1H), 8.68 (dd, J = 1.8 Hz, J = 4.8 Hz, 1H), 8.57 (d, J = 1.8 Hz, 1H), 8.49 (dd, J = 2.4 Hz, J = 4.8 Hz, 1H), 8.04 (dt, J = 1.8 Hz, J = 7.8 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.49–7.47 (m, 1H), 7.36–7.32 (m, 3H), 7.24 (s, 1H), 7.13 (d, J = 1.8 Hz, 1H), 7.06 (t, J = 7.8 Hz, 1H), 6.94 (t, J = 8.4 Hz, 1H), 5.39 (s, 1H), 3.89–3.85 (m, 1H), 3.02–2.93 (m, 2H), 2.79–2.75 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 165.3, 151.9, 149.3, 149.1, 148.9, 136.4, 136.3, 135.7, 134.1, 128.1, 126.9, 125.5, 124.0, 123.4, 122.9, 121.1, 118.3, 118.1, 111.5, 110.7, 58.4, 41.0, 23.7 (note: carbons 1 and 2 are absent); mp 225–227 °C. HMS (APCI) calcd for C25H20N4O3 425.1608; found 425.1610 [M + H]+.
1-(2-(1H-Indol-3-yl)ethyl)-3-hydroxy-4-nicotinoyl-5-(pyridine-2-yl)-1H-pyrrol-2(5H)-one (176)
Compound 176 was prepared via procedure I from picolinaldehyde (0.11 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield a yellow solid (0.30 g, 71%). 1H NMR (600 MHz, DMSO-d6) δ 10.91 (s, 1H), 8.80 (d, J = 1.2 Hz, 1H), 8.69 (dd, J = 1.2 Hz, J = 4.8 Hz, 8.55 (d, J = 4.2 Hz, 1H), 8.00 (d, J = 7.2 Hz, 1H), 7.77 (td, J = 1.2 Hz, J = 7.2 Hz, 1H), 7.50–7.46 (m, 2H), 7.38–7.30 (m, 4H), 7.12 (d, J = 1.8 Hz, 1H), 7.05 (t, J = 7.8 Hz, 1H), 6.95 (t, J = 7.8 Hz, 1H), 5.54 (s, 1H), 3.87–3.82 (m, 1H), 3.01–2.91 (m, 2H), 2.72–2.67 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 189.2, 165.4, 156.1, 151.9, 149.4, 149.0, 137.2, 136.3, 134.2, 128.2, 126.9, 125.5, 123.8, 123.4, 122.8, 121.0, 118.3, 118.1, 111.5, 110.8, 62.1, 41.4, 23.6 (note: either carbon 1 or 2 and carbon 3 are absent); mp 218–223 °C. HMS (APCI) calcd for C25H20N4O3 425.1613; found 425.1613 [M + H]+.
1-(2-(1H-Indol-3-yl)ethyl)-5-(furan-3-yl)-3-hydroxy-4-nicotinoyl-1H-pyrrol-2(5H)-one (177)
Compound 177 was prepared via procedure I from furan-3-carbaldehyde (0.10 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield an orange solid (0.12 g, 28%). 1H NMR (600 MHz, DMSO-d6) δ 10.86 (s, 1H), 8.85 (d, J = 1.8 Hz, 1H), 8.71 (dd, J = 1.2 Hz, J = 4.8 Hz, 1H), 8.05 (dt, J = 1.8 Hz, J = 7.8 Hz, 1H), 7.74 (s, 1H), 7.50–7.48 (m, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 7.8 Hz, 1H), 7.18 (s, 1H), 7.15 (d, J = 1.8 Hz, 1H), 7.10 (s, 1H), 7.07 (t, J = 7.2 Hz, 1H), 6.98 (t, J = 7.8 Hz, 1H), 6.48 (d, J = 1.2 Hz, 1H), 5.40 (s, 1H), 3.89–3.84 (m, 1H), 3.12–3.00 (m, 2H), 2.84–2.80 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 164.4, 152.4, 149.5, 144.1, 142.7, 136.3, 133.8, 127.0, 125.5, 123.4, 122.9, 121.1, 120.8, 118.3, 118.2, 117.5, 111.5, 110.9, 108.6, 52.4, 40.7, 23.7 (note: carbon 1 and 2 are absent); mp 211–217 °C. HMS (APCI) calcd for C24H19N3O4 414.1448; found 414.1449 [M + H]+.
1-(2-(1H-Indol-3-yl)ethyl)-5-(furan-2-yl)-3-hydroxy-4-nicotinoyl-1H-pyrrol-2(5H)-one (178)
Compound 178 was prepared via procedure I from furfural (0.10 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield a mustard-colored solid (0.06 g, 14%). 1H NMR (600 MHz, DMSO-d6) δ 10.87 (s, 1H), 8.82 (d, J = 1.2 Hz, 1H), 8.72 (dd, J = 1.8 Hz, J = 4.8 Hz, 1H), 8.03 (dt, J = 1.8 Hz, J = 5.4 Hz, 1H), 7.62 (t, J = 0.6 Hz, 1H), 7.53–7.52 (m, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 7.8 Hz, 1H), 7.18 (s, 1H), 7.15 (d, J = 2.4 Hz, 1H), 7.10–7.06 (m, 2H), 6.99 (t, J = 6.6 Hz, 1H), 6.52 (d, J = 3.0 Hz, 1H), 6.433–6.425 (m, 1H), 3.82–3.77 (m, 1H), 3.19–3.14 (m, 1H), 3.00–2.95 (m, 1H), 2.66–2.61 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 186.9, 164.5, 152.3, 149.1, 148.9, 143.4, 143.2, 136.3, 134.0, 127.0, 123.5, 122.9, 122.8, 121.0, 118.4, 118.3, 118.1, 115.0, 111.5, 110.8, 110.2, 54.5, 41.4, 23.7; mp 211–216 °C. HMS (APCI) calcd for C24H19N3O4 414.1448; found 414.1452 [M + H]+.
1-(2-(1H-Indol-3-yl)ethyl)-3-hydroxy-4-nicotinoyl-5-(thiophen-2-yl)-1H-pyrrol-2(5H)-one (179)
Compound 179 was prepared via procedure I from thiophene-2-carbaldehyde (0.11 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield an orange solid (0.07 g, 16%). 1H NMR (600 MHz, DMSO-d6) δ 10.87 (s, 1H), 8.81 (s, 1H), 8.72 (d, J = 4.8 Hz, 1H), 8.02 (dd, J = 1.8 Hz, J = 6.0 Hz, 1H), 7.53-7.51 (m, 1H), 7.48–7.44 (m, 2H), 7.34 (d, J = 7.8, 1H), 7.20 (d, J = 3.0 Hz, 1H), 7.15 (d, J = 1.8 Hz, 1H), 7.07 (t, J = 7.8 Hz, 2H), 7.00–6.96 (m, 2H), 5.76 (s, 1H), 3.87–3.83 (m, 1H), 3.14–3.09 (m, 1H), 3.05–3.00 (m, 1H), 2.77–2.72 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 164.2, 152.4, 149.2, 140.2, 136.3, 133.8, 130.3, 128.7, 127.02, 126.96, 126.5, 123.6, 122.9, 121.1, 118.3, 118.2, 111.5, 110.8, 56.1, 40.9, 23.6 (note: carbons 1, 2, and 3 are absent); mp 150–155 °C. HMS (APCI) calcd for C24H19N3O3S 430.1226; found 430.1223 [M + H]+.
1-(2-(1H-Indol-3-yl)ethyl)-3-hydroxy-4-nicotinoyl-5-(thiophen-3-yl)-1H-pyrrol-2(5H)-one (180)
Compound 180 was prepared via procedure I from thiophene-3-carbaldehyde (0.11 g, 1.0 mmol), 19 (0.22 g, 1.0 mmol), and tryptamine (0.16 g, 1.0 mmol) to yield a yellow solid (0.11 g, 25%). 1H NMR (600 MHz, DMSO-d6) δ 10.87 (s, 1H), 8.86 (s, 1H), 8.71 (d, J = 4.8 Hz, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 1.8 Hz, 1H), 7.50–7.47 (m, 2H), 7.40 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 7.8 Hz, 1H), 7.25 (s, 1H), 7.14 (s, 1H), 7.08–7.06 (m, 2H), 6.96 (t, J = 7.8 Hz, 1H), 5.55 (s, 1H), 3.86–3.80 (m, 1H), 3.06–2.98 (m, 2H), 2.76–2.70 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 187.4, 181.2, 164.5, 152.5, 137.2, 136.3, 133.8, 127.05, 126.98, 126.2, 126.0, 125.4, 123.5, 122.9, 122.8, 121.1, 118.4, 118.2, 118.0, 111.5, 110.9, 55.0, 41.2, 23.9; mp 238–242 °C. HMS (APCI) calcd for C24H19N3O3S 430.1198; found 430.1201 [M + H]+.
Acknowledgments
We thank Kimberly Vellano, Phuong Le, and Jing Zhang for excellent technical assistance. This work was supported by the NIH (NS065371, S.F.T.; MH094525, S.F.T.; ES012870, NS078873, A.K.) and a research grant from Lundbeck (S.F.T.).
Glossary
Abbreviations Used
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- NMDA
N-methyl d-aspartate
- ATD
amino-terminal domain
- LBD
ligand-binding domain
- TMD
transmembrane domain
- CTD
carboxyl-terminal domain
- GABA
γ-aminobutyric acid
- DMF
dimethylformamide
- SEM
standard error of the mean
Supporting Information Available
Experimental information and the generic formula for experimental test compounds described in Tables 1–8. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written by all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Some authors are coinventors on Emory owned IP (S.S.Z., E.G.A., S.F.T., D.C.L.), have an equity position (S.F.T, D.C.L.), are Board members (D.C.L.), or paid consultants (S.F.T., K.B.H) for companies developing NMDA receptor modulators.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Furukawa H.; Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003, 22, 2873–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Furukawa H.; Singh S. K.; Mancusso R.; Gouaux E. Subunit arrangement and function in NMDA receptors. Nature 2005, 4387065185–192. [DOI] [PubMed] [Google Scholar]
- Traynelis S.; Wollmuth L.; McBain C.; Menniti F.; Vance K.; Ogden K.; Hansen K.; Yuan H.; Myers S.; Dingledine R. Glutamate Receptor Ion Channels: Structure, Regulation and Function. Pharmacol. Rev. 2010, 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Akazawa C.; Shigemoto R.; Bessho Y.; Nakanishi S.; Mizuno N. Differential expression of five N-methyl-d-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994, 347, 150–160. [DOI] [PubMed] [Google Scholar]; b Monyer H.; Burnashev N.; Laurie D. J.; Sakmann B.; Seeburg P. H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [DOI] [PubMed] [Google Scholar]
- a Vicini S.; Wang J. F.; Li J. H.; Zhu W. J.; Wang Y. H.; Luo J. H.; Wolfe B. B.; Grayson D. R. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998, 72, 555–566. [DOI] [PubMed] [Google Scholar]; b Yuan H.; Hansen K. B.; Vance K. M.; Ogden K. K.; Traynelis S. F. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 2009, 29, 12045–12058. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Paoletti P.; Neyton J. NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [DOI] [PubMed] [Google Scholar]
- a Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science 1992, 258, 597–603. [DOI] [PubMed] [Google Scholar]; b Nicoll R. A.; Malenka R. C. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann. N. Y. Acad. Sci. 1999, 868, 515–525. [DOI] [PubMed] [Google Scholar]
- Choi D. W.; Rothman S. M. The Role of Glutamate Neurotoxicity in Hypoxic-Ischemic Neuronal Death. Annu. Rev. Neurosci. 1990, 13, 171–182. [DOI] [PubMed] [Google Scholar]
- Hu N.-W.; Ondrejcak T.; Rowan M. J. Glutamate receptors in preclinical research on Alzheimer’s disease: update on recent advances. Pharmacol., Biochem. Behav. 2012, 100, 855–862. [DOI] [PubMed] [Google Scholar]
- Coyle J. T.; Basu A.; Benneyworth M.; Balu D.; Konopaske G. Glutamatergic synaptic disregulation in schizophrenia: therapeutic implications. Handb. Exp. Pharmacol. 2012, 213, 267–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew S. J.; Shah A.; Lapidus K.; Clark C.; Jarun N.; Ostermeyer B.; Murrough J. W. Ketamine for Treatment-Resistant Unipolar Depression. CNS Drugs 2012, 26, 189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hallett P. J.; Spoelgen R.; Hyman B. T.; Standaert D. G.; Dunah A. W. Dopamine D1 Activation Potentiates Striatal NMDA Receptors by Tyrosine Phosphorylation-Dependent Subunit Trafficking. J. Neurosci. 2006, 26, 4690–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hallett P. J.; Standaert D. G. Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacol. Ther. 2004, 102, 155–174. [DOI] [PubMed] [Google Scholar]; c Hallett P. J.; Dunah A. W.; Ravenscroft P.; Zhou S.; Bezard E.; Crossman A. R.; Brotchie J. M.; Standaert D. G. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology 2005, 48, 503–516. [DOI] [PubMed] [Google Scholar]; d Duty S. Targeting Glutamate Receptors to Tackle the Pathogenesis, Clinical Symptoms and Levodopa-Induced Dyskinesia Associated with Parkinson’s Disease. CNS Drugs 2012, 26, 1017–1032. [DOI] [PubMed] [Google Scholar]; e Nakamura T.; Lipton S. A. Excitatory amino acids, S-nitrosylation, and protein misfolding in neurodegenerative disease protection by memantine and nitromemantine at NMDA-gated channels. Oxid. Stress Dis. 2010, 26Micronutrients and Brain Health59–78. [Google Scholar]
- a Mullasseril P.; Hansen K.; Vance K.; Ogden K.; Yuan H.; Kurtkaya N.; Santangelo R.; Orr A.; Le P.; Vellano K.; Liotta D.; Traynelis S. A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nature Commun. 2010, 1, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mosley C. A.; Acker T. M.; Hansen K. B.; Mullasseril P.; Andersen K. T.; Le P.; Vellano K. M.; Bräuner-Osborne H.; Liotta D. C.; Traynelis S. F. Quinazolin-4-one Derivatives: A Novel Class of Noncompetitive NR2C/D Subunit-Selective N-Methyl-d-aspartate Receptor Antagonists. J. Med. Chem. 2010, 53, 5476–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bettini E.; Sava A.; Griffante C.; Carignani C.; Buson A.; Capelli A. M.; Negri M.; Andreeta F.; Senar-Sancho S. A.; Guiral L.; Cardullo F. Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J. Pharmacol. Exp. Ther. 2010, 335, 636–644. [DOI] [PubMed] [Google Scholar]; d Costa B. M.; Irvine M. W.; Fang G.; Eaves R. J.; Mayo-Martin M. B.; Skifter D. A.; Jane D. E.; Monaghan D. T. A Novel Family of Negative and Positive Allosteric Modulators of NMDA Receptors. J. Pharmacol. Exp. Ther. 2010, 335, 614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Acker T. M.; Yuan H.; Hansen K. B.; Vance K. M.; Ogden K. K.; Jensen H. S.; Burger P. B.; Mullasseril P.; Snyder J. P.; Liotta D. C.; Traynelis S. F. Mechanism for Noncompetitive Inhibition by Novel GluN2C/D N-Methyl-d-aspartate Receptor Subunit-Selective Modulators. Mol. Pharmacol. 2011, 80, 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Santangelo Freel R. M.; Ogden K. K.; Strong K. L.; Khatri A.; Chepiga K. M.; Jensen H. S.; Traynelis S. F.; Liotta D. C. Synthesis and Structure–Activity Relationship of Tetrahydroisoquinoline-Based Potentiators of GluN2C and GluN2D Containing N-Methyl-d-aspartate Receptors. J. Med. Chem. 2013, 56, 5351–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Acker T. M.; Khatri A.; Vance K. M.; Slabber C.; Bacsa J.; Snyder J. P.; Traynelis S. F.; Liotta D. C. Structure–Activity Relationships and Pharmacophore Model of a Noncompetitive Pyrazoline Containing Class of GluN2C/GluN2D Selective Antagonists. J. Med. Chem. 2013, 56, 6434–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Hansen B. K.; Ogden K. K.; Traynelis S. F. Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. J. Neurosci. 2012, 32, 6197–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauuan P. J. F.; Trova M. P.; Gregor-Boros L.; Bocckino S. B.; Crapo J. D.; Day B. J. Superoxide dismutase mimetics: synthesis and structure–activity relationship study of MnTBAP analogues. Bioorg. Med. Chem. 2002, 10, 3013–3021. [DOI] [PubMed] [Google Scholar]
- Kelly M. G.; Kincaid J.; Duncton M.; Sahasrabudhe K.; Janagani S.; Upasani R. B.; Wu G.; Fang Y.; Weig Z.-L.. Amide derivatives as ion-channel ligands and pharmaceutical compositions and methods of using same. US 2006/0194801, 2006.
- Widmer U. A convenient preparation of t-butyl esters. Synthesis 1983, 135–136. [Google Scholar]
- Hansen K. B.; Tajima N.; Risgaard R.; Perszyk R. E.; Jørgensen L.; Vance K. M.; Ogden K. K.; Clausen R. P.; Furukawa H.; Traynelis S. F. Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-d-aspartate receptors. Mol. Pharmacol. 2013, 84, 114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K. B.; Brauner-Osborne H.; Egebjerg J. Pharmacological characterization of ligands at recombinant NMDA receptor subtypes by electrophysiological recordings and intracellular calcium measurements. Comb. Chem. High Throughput Screening 2008, 11, 304–315. [DOI] [PubMed] [Google Scholar]
- Hansen K. B.; Mullasseril P.; Dawit S.; Kurtkaya N.; Yuan H.; Vance K.; Orr A.; Kvist T.; Ogden K.; Le P. Implementation of a fluorescence-based screening assay identifies histamine H3 receptor antagonists clobenpropit and iodophenpropit as subunit-selective N-methyl-d-aspartate receptor antagonists. J. Pharmacol. Exp. Ther. 2010, 333, 650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.