

Abstract
Glycosylated β-endorphin analogues of various amphipathicity were studied in vitro and in vivo in mice. Opioid binding affinities of the O-linked glycopeptides (mono- or disaccharides) and unglycosylated peptide controls were measured in human receptors expressed in CHO cells. All were pan-agonists, binding to μ-, δ-, or κ-opioid receptors in the low nanomolar range (2.2–35 nM Ki’s). The glycoside moiety was required for intravenous (i.v.) but not for intracerebroventricular (i.c.v.) activity. Circular dichroism and NMR indicated the degree of helicity in H2O, aqueous trifluoroethanol, or micelles. Glycosylation was essential for activity after i.v. administration. It was possible to manipulate the degree of helicity by the alteration of only two amino acid residues in the helical address region of the β-endorphin analogues without destroying μ-, δ-, or κ-agonism, but the antinociceptive activity after i.v. administration could not be directly correlated to the degree of helicity in micelles.
Introduction
It is estimated that there are 1.5 billion people worldwide suffering at any given time from some type of central nervous system (CNS) disorder. Novel CNS drugs have the potential to further improve quality of life and to reduce the disease burden for these serious diseases and disorders.1 Since the discovery of the two endogenous pentapeptides, Met-enkephalin and Leu-enkephalin, in 1975, more than 250 endogenous neuropeptides have been identified, and there is now a broad vista for the application of these peptides in pharmacology, especially for the opioids that are so widespread throughout the CNS.2 Now in the fourth decade of research in this area, many potent and selective peptide agonists have been developed for the three cloned opioid receptors, but some crucial drawbacks still persist that dampen enthusiasm for the use of these compounds as peptide-based drugs, primarily instability in vivo and poor blood–brain barrier (BBB) penetration.
Numerous methods have been devised and successfully applied to overcome metabolic instability and high clearance of peptides.3 The principal problem remaining is poor penetration of the BBB.4 The BBB a component of the neurovascular unit, a structure consisting of endothelial cells of brain and spinal cord capillaries, astrocytes, basement membrane, pericytes, and neurons in physical proximity to the endothelium that varies in composition and function from site to site within the brain.5 It is known that the BBB has anatomic and neuroprotective functions because of the presence of oxidative enzymes and peptidases such as aminopeptidase, arylamidase, and enkephalinase.6 Thus, opioid peptides are generally degraded before they penetrate the CNS. The ability of drugs to diffuse passively across the BBB has been predicted by molecular size, charge, hydrogen bonding, and lipid solubility, but it is not clear how applicable Lipinski’s rules are to peptides.3,7,8 Several modifications have been studied in an effort to overcome the BBB penetration problem, including lipidization,9 structural modification to enhance stability,10 glycosylation,11 nutrient transporters,12 prodrugs,13 vector-based Trojan horses,14 cationization,15 and conjugation to or encapsulation by polymers.16 Glycosylation has been shown to improve antinociceptive potency and bioavailability of glycopeptides via higher metabolic stability,17 reduced clearance,18 and improved BBB transport.19 Some BBB penetration studies with glycopeptide agonists related to enkephalins have shown up to a 3-fold increase in the rate of brain delivery of these analogues compared with the unglycosylated parent peptides.20 Recent studies with glycopeptides in micelles indicate that amphipathicity of the glycopeptides is an important factor in BBB penetration.21 It has been suggested that glycosylation can alter tissue distribution patterns of glycopeptide drugs22 and affect interactions with receptors.21a,23
Endogenous Neuropeptide Conformations
The endogenous opioid β-endorphin has the Met-enkephalin peptide sequence YGGFM∼ at the N-terminus and consists of 31 residues. It binds preferentially to μ and δ receptors over κ-opioid receptors.24,25 Kaiser and co-workers synthesized several β-endorphins with the Met-enkephalin sequence, a hydrophilic linking segment, S-γ-amino-γ-hydroxymethylbutyric acid (HOMe-GABA), replacing residues 6–12, and an amphiphilic helical segment between the helix breaker residues Pro(13) and Gly(30). The circular dichroism (CD) spectra of all mimics, with minimal homology to the β-endorphin sequence, showed minima at 210 and 222 nm, indicative of α-helical structure.26 Compared to β-endorphin, the peptide mimics were 2 to 3 times more potent in μ- and κ-opioid receptor binding assays, about equipotent in the δ-receptor binding assay, and possessed strong resistance toward proteolytic enzymes.27 These findings strongly suggested that the amphipathic α-helical structure in the C-terminal region of β-endorphin plays a key role in receptor binding and opioid activity as well as resistance to proteolysis of mimic analogues. Kyle and co-workers designed and synthesized several conformationally constrained nociceptin (NC/ORL-1) analogues,28 where they exploited the α-helical-promoting residues α-aminoisobutyric acid (Aib) and N-methyl alanine (MeAla) as replacement(s) for Ala7, Ala11, or Ala15 in the native NC C-terminal sequence. The importance of α-helical address segments of peptides has also been demonstrated in the secretin family of peptide receptors, including CRF, secretin, and VIP. The interaction of PACAP1–38 with a phospholipid membrane has been shown to be involved in binding and receptor specificity, increasing peptide stability, and amplifying bioactivity in vivo.29
Glycopeptide Design Principles
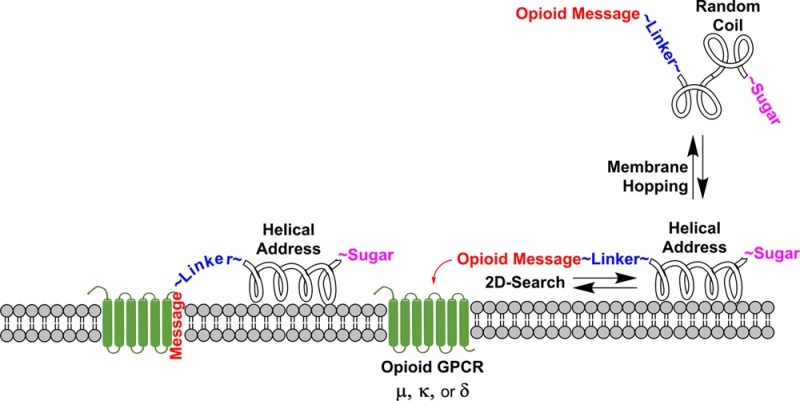
On the basis of the evidence that glycosylation decreases lipophilicity and on the hypothesis that amphipathic properties of the helix could help guide a membrane-associated peptide to its specific receptor, three generations of glycosylated β-endorphin analogues have now been synthesized for study:30 the first bearing longer helices21a too stable to engage in biousian behavior,30 the second being this study,21b and the third bearing more flexible linkages between the opioid message and the helix address, which will be published shortly. It is of interest that some of the longer endorphin glycopeptides analogues penetrated the BBB at higher rates than the shorter enkephalin glycopeptide analogues.21b The influence of the amphipathic helix in tandem with glycosylation on drug delivery is therefore of great interest and was further examined using a series of β-endorphin glycopeptide analogues of varying helicity and bearing different sugar moieties. To understand the conformation and dynamics of membrane-bound peptides or integral proteins, several model systems have been developed to mimic features of the membrane. It is generally accepted that the detergent sodium dodecyl sulfate (SDS) mimics the membrane-like environment and therefore its use has been reported in the literature extensively to study peptide–membrane interactions.21b,31 Schwyzer’s membrane compartment concept23 suggests that amphipathic helices32 will promote binding with the receptors via a 2D search of the membrane rather than a 3D search of the aqueous compartment (cf. reduction of dimensionality; Figure 1).33
Figure 1.

Reduction of dimensionality. The calculated hydrophilic (red) and hydrophobic (blue) Connolly surfaces are illustrated for glycopeptide L1 in side view (a) and down the axis of the helical address region (b). The 9th and 12th residues were replaced by α-aminoisobutyric acid (Aib), alanine, or glycine to adjust the helicity by increments. (c) The sugar can play an important role in drug transport by pulling the glycopeptide away from membranes into the aqueous milieu to enable membrane hopping.34,35 The amphipathic helix promotes 2D searching of the membrane in order to facilitate receptor binding.64,65
According to detailed analysis of globular proteins physical-chemical and structural properties, Segrest and co-workers grouped amphipathic helices into different classes (A, H, L, G, K, C, and M)34 according to the geometric distribution of lipophilic and hydrophilic residues. Class H, polypeptide hormones, typically contain two distinct functional domains, a short N-terminal domain including a specific message segment that binds to the transmembrane portion of the receptor and a much longer amphipathic helix, or address segment, located at the C-terminal portion of the peptide. The helical domain may provide enhanced receptor targeting in a relatively nonspecific manner by increasing affinity for the hydrophobic membrane that contains the receptor. It has been proposed that peptide–lipid interaction leading to cell penetration plays a major role in their activity by one of two general mechanisms: (1) transmembrane pore formation via a barrel-stave mechanism and (2) membrane destruction/solubilization via a carpet mechanism.35 It is proposed that exploitation of the first step of these mechanisms by class H glycopeptides will allow for reversible adsorption to biological membranes without membrane disruption. This putative glycopeptide–lipid interaction allows peptides with amphipathic helix conformation to float in the cell membrane, exposing the hydrophobic side to the hydrophobic membrane and the hydrophilic side to the aqueous exterior of the cell. Furthermore, it is proposed that this transient interaction with biological membranes is essential for crossing cellular barriers (not membranes), such as the endothelial layer of cells that compose an important part of the BBB.
The peptides and glycopeptides examined in the current studies were designed in accordance with Kaiser’s classic studies of β-endorphin24−27 combined with a simple Edmundson wheel approach to introduce amphipathicity per Segrest’s insights.36 Molecular mechanics calculations confirmed the potential for helical amphipathic structures for the glycopeptides. In this study, all of the glycopeptides shared the same message segment, YtGFL∼, used in previously published studies.21 The helix-breaking residue Pro6 was used to link the N-terminal message domain and C-terminal helix. The three series (Table 1) bore either an unglycosylated l-serine residue (U series), an l-serine monosaccharide bearing a β-O-glucose (G series), or an l-serine disaccharide bearing a β-O-lactose (L series). Each series of seven ligands differed in the address domain sequence residues at position 9 or 12, where Aib, Ala, and Gly were introduced into the C-terminal address to obtain different helix propensities.21b,36 All of the helical conformations were presumably stabilized by a salt bridge between Glu10 and Lys14 (i → i + 4).37
Table 1. Peptide/Glycopeptide Sequencesa.
|
S° = S OH |
S° = S* β-glucose |
S° = S** β-lactose |
helix determinant | message∼Pro6∼helix-amide |
|---|---|---|---|---|
| U1 | G1 | L1 | ∼B∼B∼ | YtGFL-P6-NLB9EKB12LKS°L-NH2 |
| U2 | G2 | L2 | ∼A∼B∼ | YtGFL-P6-NLA9EKB12LKS°L-NH2 |
| U3 | G3 | L3 | ∼B∼A∼ | YtGFL-P6-NLB9EKA12LKS°L-NH2 |
| U4 | G4 | L4 | ∼A∼A∼ | YtGFL-P6-NLA9EKA12LKS°L-NH2 |
| U5 | G5 | L5 | ∼A∼G∼ | YtGFL-P6-NLA9EKG12LKS°L-NH2 |
| U6 | G6 | L6 | ∼G∼A∼ | YtGFL-P6-NLG9EKA12LKS°L-NH2 |
| U7 | G7 | L7 | ∼G∼G∼ | YtGFL-P6-NLG9EKG12LKS°L-NH2 |
S = l-serine, S* = β-O-glucosyl-l-serine, S** = β-O-lactosyl-l-serine, and B = α-aminoisobutyric acid (Aib).
Experimental Details
Materials
The Fmoc-protected amino acids and the Rink amide MBHA resin (4-(2′,4′-dimethoxyphenyl-fmoc-aminomethyl)-phenoxy-acetamido MBHA, grain size: 100–200 mesh, substitution 0.83 mmol/g) were obtained from Chem-Impex International. Sodium dodecyl-d25 used in NMR experiments was purchased from CDN Isotopes Inc., Canada. All other reagents and solvents were purchased from Aldrich Co. and used without further purification.
Peptide Synthesis and Purification
The Fmoc-protected serine glycosides were prepared using published procedures.38−41 The glycopeptides were synthesized manually using established solid-phase Fmoc-chemistry methodology with Rink amide MBHA resin (substitution: 0.83 meq/g, 1% DVB).42,43 The side-chain-protected amino acids used in the synthesis were Fmoc-Lys(Boc)-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-d-Thr(But)-OH, and Fmoc-Tyr(But)-OH. Coupling of all the FMOC-protected amino acids was performed in a sealed tube heated by an Emerson 900 W microwave oven at power level 1 for 10 consecutive minutes. Coupling was performed (2.0 equiv Fmoc-AA compared to resin) using 1-hydroxybenzotriazole (HOBt, 2.0 equiv) and N,N′-diisopropylcarbodiimide (DIC, 2.0 equiv) in a 1:1 mixture of dimethylformamide (DMF) and N-methylpyrrolidone (NMP) (Scheme 1). Coupling was monitored using Kaiser’s ninhydrin test. The Fmoc groups were removed using a mixture of 3% piperidine and 2% diaza-1,3-bicyclo[5.4.0]-undecane (DBU) in DMF for 10 min with argon bubbling as agitation. The final Fmoc deprotection as well as the acetyl protecting groups of sugar moiety were removed by 80% hydrazine hydrate (H2NNH2·H2O) in CH3OH with argon agitation 3× for 2 h. The glycopeptides were cleaved from the resin with a F3CCOOH/Et3SiH/H2O/PhOCH3/CH2Cl2 (8:0.5:0.5:0.05:1) cocktail, which simultaneously removed the side chain protecting groups. The crude glycopeptides were precipitated in cold Et2O, redissolved in a minimal amount of distilled H2O, and then lyophilized. The crude glycopeptides were purified by RP-HPLC on a preparative C-18 Phenomenex (250 × 21.9 mm) column using CH3CN–H2O gradient system containing 0.1% CF3COOH. Homogeneity of the pure glycopeptides (≥95%) was confirmed by analytical RP-HPLC and high-resolution mass spectrometry.
Scheme 1. Glycopeptide Synthesis.

Fmoc construction of the glycopeptides using DIC/HOBt coupling with microwave heating was applied to MBHA-functionalized Rink polystyrene resin. Fmoc deprotection was accomplished with 3% piperidine/2% DBU in DMF. Treatment with H2NNH2·H2O was required to remove the acetates from the glycoside moiety prior to cleavage from the Rink resin using TFA/PhOCH3/Et3Si/H2O in CH2Cl2 to provide the C-terminal amides.
Receptor Binding Studies
To determine the affinity and selectivity of the peptides for the μ-, δ-, and κ-opioid receptors, Chinese hamster ovary (CHO) cells that stably expressed one type of human opioid receptor were used as previously described.44 Cell membranes were incubated at 25 °C with the radiolabeled ligands in a final volume of 1 mL of 50 mM Tris-HCl, pH 7.5. Incubation times of 60 min were used for the μ-selective peptide [3H]DAMGO and the κ-selective ligand [3H]U69,593, and a 3 h incubation was used with the δ-selective antagonist [3H]naltrindole. The final concentrations of [3H]DAMGO, [3H]naltrindole, and [3H]U69,593 were 0.25, 0.2, and 1 nM, respectively. Nonspecific binding was measured by inclusion of 10 μM naloxone for the μ- and κ-opioid receptors and 100 μM naloxone for the δ-opioid receptors. The binding was terminated by filtering the samples through Schleicher & Scheull no. 32 glass-fiber filters using a Brandel 48-well cell harvester. The filters were washed 3× with 3 mL of cold 50 mM Tris-HCl, pH7.5, and were counted in 2 mL of ScintiSafe 30% scintillation fluid (Fisher Scientific, Fair Lawn, NJ). For [3H]U69,593 binding, the filters were soaked in 0.1% polyethylenimine for at least 30 min before use. Each experiment was performed in triplicate and included 12 different concentrations of the competing compound. Each experiment was repeated three times. IC50 values were calculated by least-squares fit to a logarithm-probit analysis. Ki values of unlabeled compounds were calculated from the equation Ki = (IC50)/1 + S, where S = (concentration of radioligand)(Kd of radioligand).45
Circular Dichroism
All circular dichroism (CD) spectra were obtained on OLIS DSM-20 automatic recording spectrophotometer equipped with temperature controller. The glycopeptide stock solutions were prepared by weighing the lyophilized powder using a Cahn/Ventron Instruments model 21 automatic analytical electrobalance. The samples were prepared by diluting the stock solution to 30 μM. All CD spectra were the average of three scans recorded with baseline correction between 190 and 250 nm using an integration time of three seconds and a scan step of 0.5 nm in a cell with a path length of 0.1 cm at 20 °C. All spectra were smoothed by KaleidaGraph software (Synergy Software, USA). The molar ellipticities were calculated using the equation [θ] = [θ]obs(MRW)/10lC, where [θ]obs is the observed ellipticity in millidegrees, MRW is the mean residue weight, l is the cell path length in centimeters, and C is the glycopeptide concentration in milligrams per milliliter. The percent α-helicity was determined using the equation % helix = [θ]n → p*/–40 000(1 – 2.5/n)100, where n represents the number of amide bonds (including the C-terminal amide) in the glycopeptides and [θ]n → p* is the molar ellipticity of the n → p* transition band at 222 nm.46
NMR Spectroscopy
All NMR spectra were obtained from a Bruker DRX600 600 MHz spectrometer. The concentration of glycopeptide samples for the NMR experiments varied from 2.5 to 3 mM. The micelle samples were prepared by dissolving the peptide and 50 equiv of perdeuterated SDS in 0.5 mL of phosphate buffer (10 mM)/D2O (9:1 ratio by volume). The acidity of the each sample was adjusted to pH 5.5 using NaOH as necessary. Internal standard 3-(trimethylsilyl)-d4-propionic acid (TSP) was added as a reference peak, δ = 0. Rotating-frame Overhauser enhancement (ROESY),47 nuclear Overhauser enhancement (NOESY), and total correlation spectra (TOCSY)48 were acquired using standard pulse sequences and processed using XWINNMR (Bruker Inc.) and FELIX2000 (Accelrys Inc., San Diego, CA). Mixing times were 100 ms for TOCSY spectra and 300 ms for ROESY and NOESY spectra. All NMR experiments were 750 increments in t1, 24/32/32 scans each, and 1.5 s relaxation delay. The WATERGATE pulse sequence was employed to suppress the H2O/HOD signal.49
Conformational Analysis
Molecular distance constraints for the structure calculation were obtained from integral volumes of the ROESY or NOESY peaks with using software FELIX2000, and the NOE integral volumes were classified into strong, medium, and weak with 1.0, 2.5, and 3.5 Å as upper-bound distance. Molecular dynamics simulation was performed with the MOE software (Molecular Operating Environment, Chemical Computing Group, Canada) using a standard protocol available within the system.50 Distance constraints are placed between protons identified through NMR-determined NOE-corresponding upper-boundary distances of 3 (strong), 4 (medium), and 5 Å (weak). A 25 kcal/mol energy penalty was used for the constraints. The structure was minimized initially using steepest descent followed by the conjugate gradient algorithm.
Antinociceptive Potency and Efficacy Studies
Adult male CD-1 mice (25–35 g) were obtained from Charles River Laboratories and housed in groups of 4 to 5 animals/cage. Animals were kept on a 12 h light–dark cycle (lights on 0700 h) with food and water available ad libitum until the time of formal testing/drug administration. They were maintained under standard housing conditions (temperature 22 ± 2 °C and relative humidity between 55 and 60%). All experimental procedures were approved by the University of New England Institutional Animal Care and Use Committee (IACUC) and were conducted in compliance with the NIH Guide for the Care and Use of Laboratory Animals. The warm-water tail-flick assay was used to assess potency and efficacy of the test compounds. The assay used is a modified version51 of the classic tail-flick test developed by D’Amour and Smith.52 Mice were lightly but firmly grasped by the nape of the neck with the evaluators thumb and fingers, and the distal half of the tail was then dipped into a bath of circulating water thermostatically controlled at 55 °C (Neslab circulator). Latency to respond to the heat stimulus with a vigorous flexion of the tail was measured to the nearest 0.1 s. A baseline determination was made followed by testing at various times after drug injection (10, 20, 30, 45, 60, 90, and 120 min). A 10 s cutoff was used to prevent tissue damage to the tail. Antinociception was calculated by the following formula: % antinociception = [(test latency – baseline latency)/(10 – baseline latency)]100. For graphing purposes, mean and SEM values were calculated in Excel for each treatment group and time point. The A50 values and 95% confidence intervals (CI) were calculated using linear regression software from the dose–response curves (FlashCalc software; Dr. Michael Ossipov, University of Arizona, Tucson, AZ). Because latencies in this test are affected by tail skin temperature,53 careful attention was paid to ensure that the ambient room temperature was maintained at 22 to 23 °C. All drugs were dissolved in distilled H2O for intracerebroventricular (i.c.v.) injections and in physiological saline (0.9% NaCl) for systemic injections. The i.c.v. injections were performed as previously described.54 Briefly, mice were lightly anesthetized with ether, and a 5 mm incision was made along the midline of the scalp. An injection was made using a 25 μL Hamilton syringe at a point 2 mm caudal and 2 mm lateral from bregma. All systemic injections were given in a volume based on the weight of the animal (0.1 mL/10 g bodyweight). For intravenous (i.v.) injections, mice were restrained in a Plexiglas holder, and the distal portion of the tail was dipped into 40 °C warm water for approximately 10 s to dilate the tail vein. The injection was made into the tail vein using a 30 gauge needle and a 1 mL syringe. Ten mice were used for each dose, i.c.v. and i.v., in order to construct dose–response curves.
Results
Conformational Analysis by Circular Dichroism
Because circular dichroism (CD) spectra reflect the peptide ensemble average of the alignment of the dipoles of the helix backbone, this simple but powerful technique can be used to obtain the secondary structures in both peptides and proteins quantitatively.55 The CD spectra provide overall conformation but do not yield residue-specific information.56 The helicity of the peptide sequences were measured in the three different solvent systems: H2O buffer, H2O/CF3CH2OH, and H2O/SDS micelles. See the Supporting Information for details of the CD data collection and interpretation.
In distilled H2O buffered to pH 5.5, the peptides and glycopeptides were largely unstructured. In the presence of SDS, the degree of helicity was as high as 57% for the glucosides (G1–G7) and mainly depended on the amino acids present in the address segment (Figure 2). Even higher helicities were observed for the unglycosylated peptides (U1–U7), the most helical of which (U1, 70%) was not soluble in water in the absence of SDS. The lactosides (L1–L7) were less helical than the glucosides (G1–G7). The highest helicities were observed with sequences bearing two helicogenic α-aminoisobutyric acid (Aib) residues (U1, G1, and L1) and the lowest, sequences bearing two glycine residues in the address segment (U7, G7 and L7), with the alanine-bearing sequences showing intermediate helicities. In each case, increasing the degree of glycosylation (Ser[OH] → Ser[β-Glc] → Ser[β-Lact]) reduced the degree of helicity. However, the identity of the sugar moieties (glucoside vs lactoside) did not significantly affect the conformation of the micelle-bound glycopeptides, causing only slight changes in the observed NOE’s (see NMR results later). The CD spectra in the 30% TFE/H2O solvent mixture showed very similar trend as the micelle-bound compounds but with somewhat reduced helicities.
Figure 2.
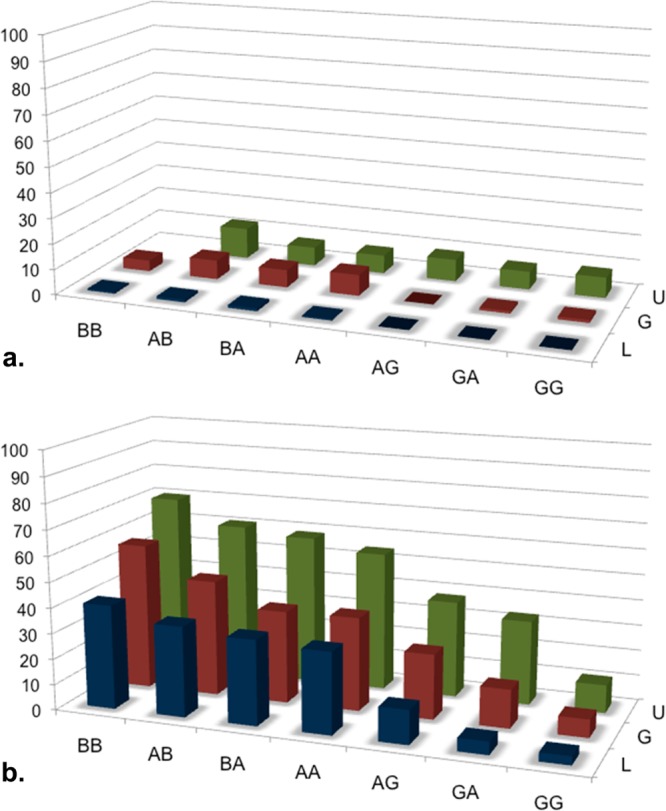
Helicity in the presence of H2O and SDS micelles. (a) Only small degrees of helicity (12% max) were observed in H2O buffer (pH 5.5) and only with the unglycosylated peptides, U. (b) In the presence of micelles, increased methylation at positions 9 and 12 in the address sequence (Gly → Ala → Aib) led to increased helicity (70% max). Increased glycosylation (-OH → glucose → lactose) led to reduced helicity.
Conformational Analysis by NMR
Circular dichroism reflects general information on the overall molecular structure of glycopeptides in different solvents. In contrast, NMR spectroscopy is well-suited to study the local structure at a residue-specific level. All of the peptides and glycopeptides were characterized for their conformation in aqueous buffer and in deuterated SDS micelles (peptide/SDS ≈ 1:100) using 2D 1H NMR (600 MHz). The spin systems were identified with TOCSY, and sequential assignments were made by the combined use of TOCSY and NOESY for experiments done in d25SDS/D2O/H2O and ROESY for experiments done in D2O/H2O. Although a few overlapping peaks were observed, unambiguous 1H chemical shift assignments of all glycopeptides were completed on the basis of the sequential NOE measurements that were made, for example, dNN (i, i + 1), dαN (i, i + 1), and dβN (i, i + 1).57 The complete chemical shift values of the amino acid residues and coupling constants for all glycopeptides are provided in the Supporting Information along with a complete description of the NMR experiments.
Proton chemical shift indices (CSI) for the α positions were consistent with the helix assignments made by NOE data.58 The observed chemical shift differences between the ideal helix and random coil CSI values expected and the observed CSI values were consistent with the CD data for each molecule. Almost all of the αCH resonances showed negative deviations except for residues Leu5 and Ser.15 This trend was observed for all the glycopeptides; however, it should be noted that there is no random-coil reference standard available for glycosylated serine. However, it seems reasonable to make a qualitative comparison of helix content between closely related peptides.
Methods described by Gierasch and co-workers59 were applied. First, the average conformational shift was calculated for each peptide by adding all upfield shifts in the helical regions and dividing by the total number of peptide bonds. Then, to obtain the overall helical contents for each peptide, the average conformational shift was divided by 0.35 ppm, which was assigned for 100% helicity. Because there are no random-coil values available for the serine glycosides60 and α-amino isobutyric acid has no α-protons, these residues were not included in the calculation.
All of the peptide backbones exhibited strong consecutive dαN (i, i + 1) NOEs in H2O/D2O and in the presence of SDS micelles. Unlike what was observed in H2O/D2O, a continuous stretch of sequential, strong dNN (i, i + 1) NOEs were observed throughout all sequences in the presence of SDS micelles. In H2O/D2O, the dNN (i, i + 1) NOEs were too weak to be observed, and no other long-range NOEs were observable, suggesting that random-coil conformational ensembles exist in this solvent, as suggested by the CD data, or only nascent helix formation at best. CD reflects an instantaneous snapshot of the entire ensemble, whereas the nuclear Overhauser effects (NOEs) take 50–100 μs to build, depending on the peptide backbone to hold a particular conformation for a relatively long period of time. Thus, the helical content obtained by NMR for a typical case, glycopeptide G1 (β-glucoside), was less than 20% of CD results. Because the NOEs were obtained by true NOESY experiments (not ROESY), the NMR structures are biased toward the more static, micelle-bound structures at the expense of the more dynamic, random-coil structures found in solution. Thus, the NMR data is useful insofar as it confirms the existence of helices, but it is not useful for quantifying the degree of helicity, which is more reliably predicted by the CD experiments.
Receptor Binding Studies
The peptides (U1–U7) and glycopeptides (L1–L7 and G1–G7) all showed low nanomolar affinities (Figure 3 and Table 2) for the three classical opioid receptor subtypes, μ (hMOR), δ (hDOR), and κ (hKOR), using published methods.61
Figure 3.

Opioid binding.46,47 Binding was determined in membranes from Chinese hamster ovary (CHO) cells that stably expressed either the human μ-, κ-, or δ-opioid receptors. Each membrane preparation was incubated with 12 different concentrations of each peptide/glycopeptide. Each measurement was performed in triplicate, and each experiment was replicated three times.
Table 2. Opioid Receptor Binding Determined by Radioligand Displacement in Membrane Preparations with hMOR, hDOR, and hKOR Receptors Expressed in CHO Cells45,46.
| helix series |
1 ∼B∼B∼ |
2 ∼A∼B∼ |
3 ∼B∼A∼ |
4 ∼A∼A∼ |
5 ∼A∼G∼ |
6 ∼G∼A∼ |
7 ∼G∼G∼ |
S° |
|---|---|---|---|---|---|---|---|---|
| μ-Binding (Ki’s in nM) vs [3H]DAMGO | ||||||||
| L | 21 ± 0.90 | 8.2 ± 0.60 | 13 ± 0.54 | 11 ± 1.4 | 9.6 ± 0.18 | 9.8 ± 0.97 | 5.0 ± 0.37 | S** |
| G | 9.1 ± 0.39 | 2.6 ± 0.29 | 6.1 ± 0.31 | 6.2 ± 0.47 | 2.2 ± 0.30 | 2.9 ± 0.32 | 3.5 ± 0.55 | S* |
| U | 29 ± 3.1 | 29 ± 0.67 | 21 ± 2.6 | 14 ± 2.3 | 13 ± 0.97 | 13 ± 1.2 | 12 ± 1.8 | S |
| δ-Binding (Ki’s in nM) vs [3H]naltrindole | ||||||||
| L | 35 ± 3.0 | 12 ± 0.94 | 19 ± 0.67 | 12 ± 0.99 | 12 ± 1.0 | 8.2 ± 0.29 | 7.0 ± 0.57 | S** |
| G | 11 ± 1.2 | 3.6 ± 0.23 | 14 ± 2.1 | 7.9 ± 1.0 | 6.3 ± 0.094 | 5.7 ± 0.35 | 7.1 ± 1.0 | S* |
| U | 23 ± 2.8 | 15 ± 0.79 | 25 ± 1.6 | 16 ± 1.9 | 15 ± 0.98 | 11 ± 0.91 | 13 ± 2.2 | S |
| κ-Binding (Ki’s in nM) vs [3H]U69,593 | ||||||||
| L | 17 ± 1.9 | 5.7 ± 0.13 | 17 ± 1.6 | 15 ± 0.39 | 13 ± 0.76 | 23 ± 2.3 | 8.4 ± 1.2 | S** |
| G | 6.9 ± 0.58 | 2.3 ± 0.31 | 12 ± 0.94 | 8.9 ± 1.0 | 6.3 ± 0.41 | 11 ± 0.26 | 4.4 ± 0.47 | S* |
| U | 14 ± 1.4 | 16 ± 0.16 | 26 ± 1.6 | 14 ± 2.1 | 15 ± 1.3 | 20 ± 1.4 | 15 ± 1.9 | S |
In Vivo Antinociception
Figures 4 and 5 include the A50 values of each peptide in the 55 °C tail-flick test following i.c.v. or i.v. administration. All compounds were full and potent agonists following i.c.v. administration, with A50 values falling in the 0.1–2.5 nmol/mouse range. The lactosylated peptides tended to be more potent than both the glucosylated and unglycosylated compounds, with A50 scores ranging between 0.1 and 0.9 nmol/mouse (Figure 4). Peptide A50 values following i.v. administration displayed more variability than those obtained from i.c.v. administration. The unglycosylated peptides failed to produce >20% antinociception following i.v. administration in the 55 °C tail-flick assay at the doses tested (10 or 32 mg/kg, Figure 5). In contrast, most of the glycosylated analogues produced potent full-agonist effects, with A50 values for the disaccharides ranging from <1 to 5.3 μmol/kg. The one exception was L7 (lactosylated GG), which produced <40% antinociception at 32 mg/kg. Higher doses of L7 were not tested because there were insufficient amounts of compound available. The role of multiple receptor subtype activation62 is not considered here.
Figure 4.
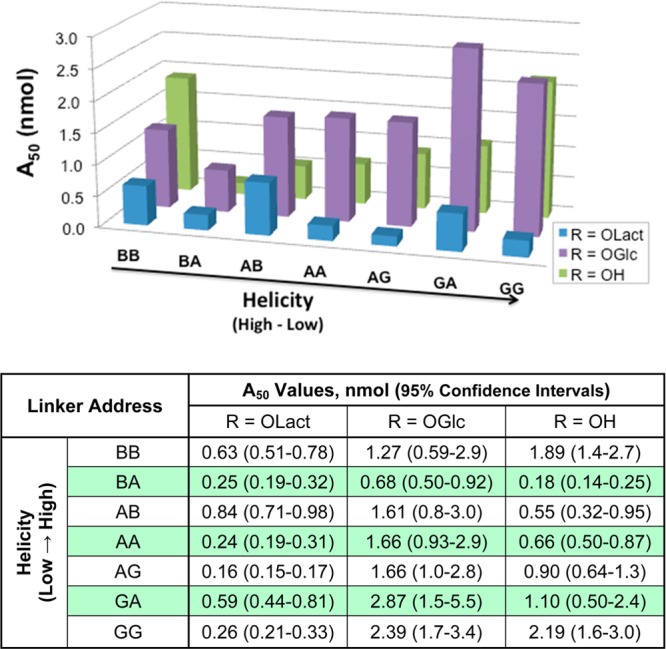
Potency estimates after i.c.v. administration. Mouse tail-flick studies were performed at 55 °C. The vertical axis (A50 values) is marked in nanomoles per mouse. The A50 values were calculated using linear regression software (FlashCalc), and 95% confidence intervals are included in the table.
Figure 5.
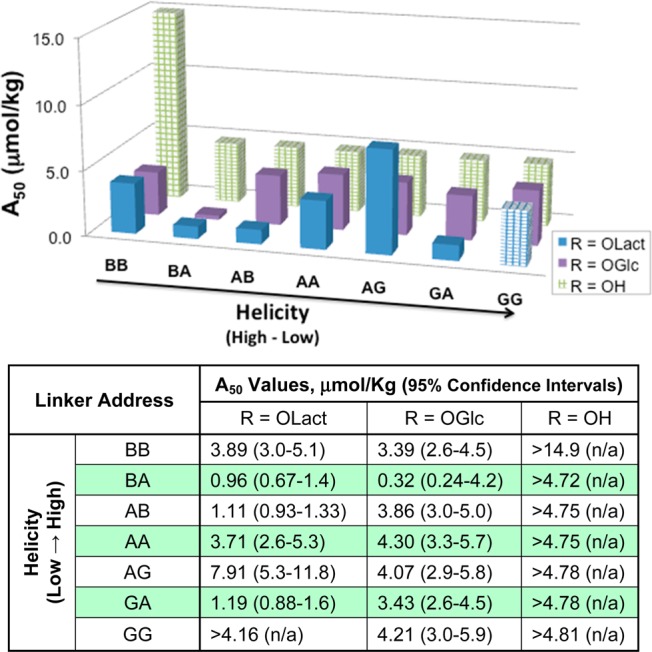
Potency estimates after i.v. administration. Mouse tail-flick studies were performed at 55 °C. The vertical axis is marked in μmol/kg. The A50 values were calculated using linear regression software (FlashCalc), and 95% confidence intervals are included in the table. Note that the checked bars indicate maximum doses tested, not A50 values. No significant antinociception was observed for the unglycosylated peptides nor for the least helical lactoside (GG).
Discussion
It was hypothesized that modulation of membrane affinity (via alterations in the degree of amphipathicity) is important for BBB penetration rates,30 for drug distribution properties,21 and for receptor affinity.63 The amphipathic character of the helix can facilitate a drug or hormone to bind its specific receptor by narrowing the receptor search from an inefficient 3D search of the extracellular milieu to a much more rapidly converging 2D search along the membrane surface. Second, membrane insertion of the helical address might allow the pharmacophore or message to be fixed in a specific geometry relative to the membrane.64 It is known that simply producing highly amphipathic sequences is insufficient to facilitate systemic delivery and penetration of the BBB.21b
The α-methylation of amino acids is well-known to stabilize helix formation8,65 and was used to produce a series of helical address regions with increasing intrinsic stability by increasing methylation (glycine → alanine → α-aminoisobutyric acid). In all three series of opioid agonists, U1-7, G1-7, and L1-7 (unglycosylated, glucosylated, and lactosylated, respectively), the CD and NMR data showed increasing helicity in the presence of TFE or SDS. Increasing glycosylation (no sugar → glycoside → lactoside) decreased the observed helicity in every case. We hypothesize that this occurs not because the helix is destabilized but by further stabilizing the random-coil structures by increasing their water solubility. Although we can gauge the overall ratio of bound versus unbound helices, we do not know the on and off rates for the helix–micelle binding event (a surrogate for helix–membrane binding). It would be surprising if these rates were not affected by the glycosylation state. We hypothesize that the glycopeptides gain entry to the CNS by transcytosis at the BBB and that a minimal level of membrane interaction (residence time?) is required for efficient BBB penetration. It is tempting to speculate that increased rates of membrane adsorption–desorption might lead to enhanced BBB penetration rates, either by affecting the biophysics of the initial endocytotic event or by promoting subsequent endosomal escape of the glycopeptides.
On the basis of the receptor binding results and intracerebroventricular (i.c.v.) (Figure 4) and intravenous (i.v.) tail-flick studies (Figure 5), only the glucoside G3 (BA) displayed somewhat greater receptor binding affinity and in vivo potency compared to the compounds that had similar bioactivities. On the basis of the i.v. A50 value calculations, the compound presumably exhibited efficient penetration across the BBB in mice. All lactosides (L series) had higher binding affinities with μ-, δ-, and κ-receptors than the corresponding glucosides (G series) after i.c.v injection. Nevertheless, the more flexible lactoside L7 (GG) could not, apparently, cross the BBB at all after i.v. injection. (Figure 5). On the basis of the i.c.v. results, the lactosides were 2–10 times more potent than the glucosides, presumably because of increased water solubility within the CNS. Compared to tail flick results after i.v. injection, the more hydrophilic peptide L2 was ∼3 times more potent than peptide G2, but L3 showed similar potency to G3 after peripheral administration. Lactoside L7 with random-coil conformations in TFE and SDS micelles likely does not penetrate the BBB after i.v. injection, even though the compound has a greater antinociceptive potency than G7 after i.c.v. injection. Presumably, it is too water-soluble and does not bind to membranes strongly enough to undergo endocytosis to penetrate the BBB. None of the unglycosylated peptides showed antinociceptive properties when administered peripherally (Figure 5).
Conclusions
All of the glycopeptides and peptides had relatively high affinity for the three cloned opioid receptors, (Table 2) and displayed good in vivo antinociception following i.c.v. administration (Figure 4). Not all of the glycopeptides showed good activity after peripheral administration by i.v. injection (Figure 5). This supports earlier conclusions that glycosylation of smaller enkephalin-based peptides21a,44,45 increases bioactivities and penetration of the BBB.19,20 Both CD and NMR studies confirmed that all the glycopeptides displayed random-coil conformational ensembles in aqueous solution and increasing degrees of helicity in TFE and SDS solution predicted by increasing substitution of residues 9 and 12, Gly → Ala → Aib.8,21,30,34,67 If there were no helix-destabilizing glycine residues on the address segment, then glycopeptides showed clear amphipathic α-helical structures by CD and by NMR. None of the unglycosylated peptides showed antinociceptive properties when administered peripherally (Figure 5). The helix of peptide U1 was so stable that it was not even soluble in aqueous media in the absence of SDS.
The results suggest that simply introducing highly helical sequences on the address segment is not by itself sufficient to promote stability and penetration of the BBB. It is hypothesized that the helix must also be capable of assuming a water-soluble random-coil conformation and that the energy barrier between random coil and helical states must be low enough to permit rapid interconversion between the two states; this characteristic was termed biousian, denoting two (bi) essences (ousia), a water-soluble conformational ensemble and a membrane-bound conformation.30 The biousian nature of a glycopeptide permits high-affinity receptor binding, allows membrane hopping to impart drug-like characteristics, and promotes penetration of the BBB. This is confirmed by comparing glucoside G2 and lactoside L2 (Table 2). The CD spectra of G2 indicates a strong helix but a relatively low affinity for opioid receptors and a weak penetration of the BBB. The increased hydrophilicity of the lactose-bearing L2 (decreased energy barrier between random coil and helical states) allows L2 to bind more strongly than G2 and to penetrate the BBB after i.v. injection. Unglycosylated peptide U1 showed excellent opioid binding, yet it was less antinociceptive than U2 or U3 after i.c.v. administration (Figure 4) and was not water-soluble in the absence of SDS. Further studies with other opioid messages (e.g., μ- and δ-selective agonists) and replacement of the linkage element l-proline with more flexible linkers will be discussed elsewhere.
Acknowledgments
We thank the Office of Naval Research (N00014-05-1-0807 and N00014-02-1-0471), the National Science Foundation (CHE-9526909), the National Institute of Neurological Disorders and Stroke (R01NS52727), and the National Institute of General Medical Sciences (P20 GM103643-01A1) for financial support. In addition, the expert technical assistance of Drs. Lajos Szabò (HPLC analyses) and Chad Park (CD measurements) is gratefully acknowledged.
Glossary
Abbreviations Used
- TFE
trifluoroethanol
- A50
analgesic potency (50% activity in the 55 °C mouse tail-flick assay)
Supporting Information Available
Experimental details for the synthesis, HPLC, MS, NMR, and CD of the peptides and glycopeptides. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brasnjevic I.; Steinbusch H. W. M.; Schmitz C.; Martinez-Martinez P. Delivery of peptide and protein drugs over the blood-brain barrier. Prog. Neurobiol. 2009, 87, 212–251. [DOI] [PubMed] [Google Scholar]
- Hughes J.; Smith T. W.; Kosterlitz H. W.; Fothergill L. A.; Morgan B. A.; Morris H. R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975, 258, 577–580. [DOI] [PubMed] [Google Scholar]
- a Adessi C.; Soto C. Converting a peptide into a drug: Strategies to improve stability and bioavailability. Curr. Med. Chem. 2002, 9, 963–978. [DOI] [PubMed] [Google Scholar]; b Peptide and Protein Delivery; Van der Walle C., Ed.; Academic Press: London, 2011. [Google Scholar]
- Witt K.; Davis T. CNS drug delivery: Opioid peptides and the blood-brain barrier. AAPS J. 2006, 8, E76–E88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo G. J. Stroke and neurovascular protection. New Engl. J. Med. 2006, 354, 553–555. [DOI] [PubMed] [Google Scholar]
- Mercadante S.; Arcuri E. Delivery of opioid analgesics to the brain: The role of blood-brain barrier. Gene Ther. Mol. Biol. 2009, 13A, 82–90. [Google Scholar]
- Habgood M. D.; Begley D. J.; Abbott N. J. Determinants of passive drug entry into the central nervous system. Cell. Mol. Neurobiol. 2000, 20, 231–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polt R.; Dhanasekaran M.; Keyari C. Glycosylated neuropeptides: A new vista for neuropsychopharmacology?. Med. Res. Rev. 2005, 25, 557–585. [DOI] [PubMed] [Google Scholar]
- Hansen D. W.; Stapelfeld A.; Savage M. A.; Reichman M.; Hammond D. L.; Haaseth R. C.; Mosberg H. I. Systemic analgesic activity and delta-opioid selectivity in [2,6-dimethyl-Tyr1,D-Pen2,D-Pen5]enkephalin. J. Med. Chem. 1992, 35, 684–687. [DOI] [PubMed] [Google Scholar]
- Bewley T. A.; Li C. H. Evidence for tertiary structure in aqueous-solutions of human beta-endorphin as shown by difference absorption-spectroscopy. Biochemistry 1983, 22, 2671–2675. [DOI] [PubMed] [Google Scholar]
- a Jakas A.; Horvat S. The effect of glycation on the chemical and enzymatic stability of the endogenous opioid peptide, leucine-enkephalin, and related fragments. Bioorg. Chem. 2004, 32, 516–526. [DOI] [PubMed] [Google Scholar]; b Poduslo J. F.; Curran G. L. Glycation increases the permeability of proteins across the blood nerve and blood-brain barriers. Mol. Brain Res. 1994, 23, 157–162. [DOI] [PubMed] [Google Scholar]
- a Tsuji A.; Tamai I. Carrier-mediated or specialized transport of drugs across the blood-brain barrier. Adv. Drug Delivery Rev. 1999, 36, 277–290. [DOI] [PubMed] [Google Scholar]; b Begley D. J. The blood-brain barrier: Principles for targeting peptides and drugs to the central nervous system. J. Pharm. Pharmacol. 1996, 48, 136–146. [DOI] [PubMed] [Google Scholar]
- Ghosh M. K.; Mitra A. K. Brain parenchymal metabolism of 5-iodo-2′-deoxyuridine and 5′-ester prodrugs. Pharm. Res. 1992, 9, 1048–1052. [DOI] [PubMed] [Google Scholar]
- Bickel U.; Kang Y. S.; Pardridge W. M. In-vivo cleavability of a disulfide-based chimeric opioid peptide in rat-brain. Bioconj. Chem. 1995, 6, 211–218. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S.; Sima A. A. F. The presence of anionic sites in basement-membranes of cerebral capillaries. Microvasc. Res. 1990, 39, 123–127. [DOI] [PubMed] [Google Scholar]
- Tosi G.; Costantino L.; Ruozi B.; Forni F.; Vandelli M. A. Polymeric nanoparticles for the drug delivery to the central nervous system. Expert Opin. Drug Delivery 2008, 5, 155–174. [DOI] [PubMed] [Google Scholar]
- Powell M. F.; Stewart T.; Otvos L.; Urge L.; Gaeta F. C. A.; Sette A.; Arrhenius T.; Thomson D.; Soda K.; Colon S. M. Peptide stability in drug development 0.2. Effect of single amino-acid substitution and glycosylation on peptide reactivity in human serum. Pharm. Res. 1993, 10, 1268–1273. [DOI] [PubMed] [Google Scholar]
- Fisher J. F.; Harrison A. W.; Bundy G. L.; Wilkinson K. F.; Rush B. D.; Ruwart M. J. Peptide to glycopeptide - glycosylated oligopeptide renin inhibitors with attenuated invivo clearance properties. J. Med. Chem. 1991, 34, 3140–3143. [DOI] [PubMed] [Google Scholar]
- Egleton R. D.; Mitchell S. A.; Huber J. D.; Janders J.; Stropova D.; Polt R.; Yamamura H. I.; Hruby V. J.; Davis T. P. Improved bioavailability to the brain of glycosylated Met-enkephalin analogs. Brain Res. 2000, 881, 37–46. [DOI] [PubMed] [Google Scholar]
- Egleton R. D.; Mitchell S. A.; Huber J. D.; Palian M. M.; Polt R.; Davis T. P. Improved blood-brain barrier penetration and enhanced analgesia of an opioid peptide by glycosylation. J. Pharm. Exp. Ther. 2001, 299, 967–972. [PubMed] [Google Scholar]
- a Palian M. M.; Boguslavsky V. I.; O’Brien D. F.; Polt R. Glycopeptide-membrane interactions: Glycosyl enkephalin analogues adopt turn conformations by NMR and CD in amphipathic media. J. Am. Chem. Soc. 2003, 125, 5823–5831. [DOI] [PubMed] [Google Scholar]; b Dhanasekaran M.; Palian M.; Alves I.; Yeomans L.; Keyari C.; Davis P.; Bilsky E.; Egleton R.; Yamamura H.; Jacobsen N.; Tollin G.; Hruby V.; Porreca F.; Polt R. Glycopeptides related to beta-endorphin adopt helical amphipathic conformations in the presence of lipid bilayers. J. Am. Chem. Soc. 2005, 127, 5435–5448. [DOI] [PubMed] [Google Scholar]
- a Suzuki K.; Susaki H.; Okuno S.; Yamada H.; Watanabe H. K.; Sugiyama Y. Specific renal delivery of sugar-modified low-molecular-weight peptides. J. Pharm. Exp. Therap. 1999, 288, 888–897. [PubMed] [Google Scholar]; b Suzuki K.; Susaki H.; Okuno S.; Sugiyama Y. Renal drug targeting using a vector “alkylglycoside”. J. Pharm. Exp. Ther. 1999, 288, 57–64. [PubMed] [Google Scholar]
- Gysin B.; Schwyzer R. Head group and structure specific interactions of enkephalins and dynorphin with liposomes – investigation by hydrophobic photolabeling. Arch. Biochem. Biophys. 1983, 225, 467–474. [DOI] [PubMed] [Google Scholar]
- Taylor J. W.; Kaiser E. T. Opioid receptor selectivity of peptide models of beta-endorphin. Int. J. Pept. Protein Res. 1989, 34, 75–80. [PubMed] [Google Scholar]
- Taylor J. W.; Miller R. J.; Kaiser E. T. Characterization of an amphiphilic helical structure in beta-endorphin through the design, synthesis, and study of model peptides. J. Biol. Chem. 1983, 258, 4464–4471. [PubMed] [Google Scholar]
- Taylor J. W.; Miller R. J.; Kaiser E. T. Structural characterization of beta-endorphin through the design, synthesis, and study of model peptides. Mol. Pharmacol. 1982, 22, 657–666. [PubMed] [Google Scholar]
- Taylor J. W.; Osterman D. G.; Miller R. J.; Kaiser E. T. Design and synthesis of a model peptide with beta-endorphin-like properties. J. Am. Chem. Soc. 1981, 103, 6965–6966. [Google Scholar]
- Zhang C. W.; Miller W.; Valenzano K. J.; Kyle D. J. Novel, potent ORL-1 receptor agonist peptides containing alpha-helix-promoting conformational constraints. J. Med. Chem. 2002, 45, 5280–5286. [DOI] [PubMed] [Google Scholar]
- Krishnadas A.; Onyuksel H.; Rubinstein I. Interactions of VIP, secretin and PACAP(1–38) with phospholipids: A biological paradox revisited. Curr. Pharm. Des. 2003, 9, 1005–1012. [DOI] [PubMed] [Google Scholar]
- Egleton R. D.; Bilsky E. J.; Tollin G.; Dhanasekaran M.; Lowery J.; Alves I.; Davis P.; Porreca F.; Yamamura H. I.; Yeomans L.; Keyari C. M.; Polt R. Biousian glycopeptides penetrate the blood-brain barrier. Tetrahedron: Asymmetry 2005, 16, 65–75. [Google Scholar]
- Buck M. Trifluoroethanol and colleagues: Cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 1998, 31, 295–355. [DOI] [PubMed] [Google Scholar]
- Cornette J. L.; Cease K. B.; Margalit H.; Spouge J. L.; Berzofsky J. A.; Delisi C. Hydrophobicity scales and computational techniques for detecting amphipathic structures in proteins. J. Mol. Biol. 1987, 195, 659–685. [DOI] [PubMed] [Google Scholar]
- a Adam G.; Delbruck M.. Reduction of dimensionality in biological diffusion processes. In Structural Chemistry and Molecular Biology; Rich A., Davidson N., Eds.; W. H. Freeman: San Francisco, CA, 1968; pp 198–215. [Google Scholar]; b Taylor J. W.; Kaiser E. T. The structural characterization of β-endorphin and related peptide hormones and neurotransmitters. Pharm. Rev. 1986, 38, 291–319. [PubMed] [Google Scholar]
- Lowery J. J.; Yeomans L.; Keyari C. M.; Davis P.; Porreca F.; Knapp B. I.; Bidlack J. M.; Bilsky E. J.; Polt R. Glycosylation improves the central effects of DAMGO. Chem. Biol. Drug Des. 2007, 69, 41–47. [DOI] [PubMed] [Google Scholar]
- Varamini P.; Mansfeld F. M.; Blanchfield J. T.; Wyse B. D.; Smith M. T.; Toth I. Synthesis and biological evaluation of an orally active glycosylated endomorphin-1. J. Med. Chem. 2012, 55, 5859–5867. [DOI] [PubMed] [Google Scholar]
- Segrest J. P.; Deloof H.; Dohlman J. G.; Brouillette C. G.; Anantharamaiah G. M. Amphipathic helix motif – classes and properties. Proteins: Struct., Funct., Genet. 1990, 8, 103–117. [DOI] [PubMed] [Google Scholar]
- Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [DOI] [PubMed] [Google Scholar]
- a Costantini S.; Colonna G.; Facchiano A. M. Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem. Biophys. Res. Commun. 2006, 342, 441–451. [DOI] [PubMed] [Google Scholar]; b Wako H.; Blundell T. L. Use of amino-acid environment-dependent substitution tables and conformational propensities in structure prediction from aligned sequences of homologous proteins 0.2. Secondary structures. J. Mol. Biol. 1994, 238, 693–708. [DOI] [PubMed] [Google Scholar]
- a Su J. Y.; Hodges R. S.; Kay C. M. Effect of chain-length on the formation and stability of synthetic alpha-helical coiled coils. Biochemistry 1994, 33, 15501–15510. [DOI] [PubMed] [Google Scholar]; b Kinnear B. S.; Hartings M. R.; Jarrold M. F. Helix unfolding in unsolvated peptides. J. Am. Chem. Soc. 2001, 123, 5660–5667. [DOI] [PubMed] [Google Scholar]
- Seibel J.; Hillringhaus L.; Moraru R. Microwave-assisted glycosylation for the synthesis of glycopeptides. Carbohydr. Res. 2005, 340, 507–511. [DOI] [PubMed] [Google Scholar]
- Keyari C. M.; Polt R. Serine and threonine schiff base esters react with beta-anomeric peracetates in the presence of BF3·Et2O to produce β-glycosides. J. Carbohydr. Chem. 2010, 29, 181–206. [Google Scholar]
- Lefever M. R.; Szabò L. Z.; Anglin B.; Ferracane M.; Hogan J.; Cooney L.; Polt R. Glycosylation of α-amino acids by sugar acetate donors with InBr3. Minimally competent Lewis acids. Carbohydr. Res. 2012, 351, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coss C.; Carrocci T.; Maier R. M.; Pemberton J. E.; Polt R. Minimally competent lewis acid catalysts: Indium(III) and bismuth(III) salts produce rhamnosides (=6-deoxymannosides) in high yield and purity. Helv. Chim. Acta 2012, 95, 2652–2659. [Google Scholar]
- Polt R.; Szabò L. Z.; Treiberg J.; Li Y.; Hruby V. J. General methods for α- or β-O-Ser/Thr glycosides and glycopeptides. Solid-phase synthesis of O-glycosyl cyclic enkephalin analogues. J. Am. Chem. Soc. 1992, 114, 10249–10258. [Google Scholar]
- Mitchell S. A.; Pratt M. R.; Hruby V. J.; Polt R. Solid-phase synthesis of O-linked glycopeptide analogues of enkephalin. J. Org. Chem. 2001, 66, 2327–2342. [DOI] [PubMed] [Google Scholar]
- Parkhill A. L.; Bidlack J. M. Several δ-opioid receptor ligands display no subtype selectivity to the human δ-opioid receptor. Eur. J. Pharmacol. 2002, 451, 257–264. [DOI] [PubMed] [Google Scholar]
- Cheng Y. C.; Prusoff W. H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which casues 50% inhibition (150) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Scholtz J. M.; Marqusee S.; Baldwin R. L.; York E. J.; Stewart J. M.; Santoro M.; Bolen D. W. Calorimetric determination of the enthalpy change for the alpha-helix to coil transition of an alanine peptide in water. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 2854–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rance M. Improved techniques for homonuclear rotating-frame and isotropic mixing experiments. J. Magn. Reson. 1987, 74, 557–564. [Google Scholar]
- Davis D. G.; Bax A. Assignment of complex 1H NMR spectra via two-dimensional homonuclear Hartmann-Hahn spectroscopy. J. Am. Chem. Soc. 1985, 107, 2820–2821. [Google Scholar]
- Piotto M.; Saudek V.; Sklenar V. Gradient-tailored excitation for single-quantum nmr-spectroscopy of aqueous-solutions. J. Biomol. NMR 1992, 2, 661–665. [DOI] [PubMed] [Google Scholar]
- Ogawa H.; Nakano M.; Watanabe H.; Starikov E. B.; Rothstein S. M.; Tanaka S. Molecular dynamics simulation study on the structural stabilities of polyglutamine peptides. Comput. Biol. Chem. 2008, 32, 102–110. [DOI] [PubMed] [Google Scholar]
- Janssen P. A. J.; Niemegeers C. J. C.; Dony J. G. H. Inhibitory effect of fentanyl and other morphine-like analgesics on warm water induced tail withdrawal reflex in rats. Arzneim. Forsch. 1963, 13, 502–ff. [PubMed] [Google Scholar]
- D’Amour F. E.; Smith D. L. A method for determining loss of pain sensation. J. Pharm. Exp. Ther. 1941, 72, 74–79. [Google Scholar]
- Hole K.; Tjolsen A. The tail-flick and formalin tests in rodents – changes in skin temperature as a confounding factor. Pain 1993, 53, 247–254. [DOI] [PubMed] [Google Scholar]
- Porreca F.; Mosberg H. I.; Hurst R.; Hruby V. J.; Burks T. F. Roles of mu-receptors, delta-receptors and kappa-opioid receptors in spinal and supraspinal mediation of gastrointestinal transit effects and hot-plate analgesia in the mouse. J. Pharm. Exp. Ther 1984, 230, 341–348. [PubMed] [Google Scholar]
- Woody R. W. Circular dichroism. Methods Enzymol. 1995, 246, 34–71. [DOI] [PubMed] [Google Scholar]
- Merutka G.; Morikis D.; Bruschweiler R.; Wright P. E. NMR evidence for multiple conformations in a highly helical model peptide. Biochemistry 1993, 32, 13089–13097. [DOI] [PubMed] [Google Scholar]
- Wuthrich K.NMR of Proteins and Nucleic Acids; Wiley Press: New York, 1986. [Google Scholar]
- Merutka G.; Dyson H. J.; Wright P. E. Random coil 1H chemical-shifts obtained as a function of temperature and trifluoroethanol concentration for the peptide series GGXGG. J. Biomol. NMR 1995, 5, 14–24. [DOI] [PubMed] [Google Scholar]
- Rizo J.; Blanco F. J.; Kobe B.; Bruch M. D.; Gierasch L. M. Conformational behavior of Escherichia coli OmpA signal peptides in membrane mimetic environments. Biochemistry 1993, 32, 4881–4894. [DOI] [PubMed] [Google Scholar]
- Palian M. M.; Jacobsen N. E.; Polt R. O-linked glycopeptides retain helicity in water. J. Peptide Res. 2001, 58, 180–189. [DOI] [PubMed] [Google Scholar]
- Lowery J. J.; Raymond T. J.; Giuvelis D.; Bidlack J. M.; Polt R.; Bilsky E. J. In vivo characterization of MMP-2200, a mixed δ/μ opioid agonist, in mice. J. Pharm. Exp. Ther. 2011, 336, 767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller P. W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Lefever M. R.; Muthu D.; Bidlack J. M.; Bilsky E. J.; Polt R. Opioid glycopeptide analgesics derived from endogenous enkephalins and endorphins. Future Med. Chem. 2012, 4, 205–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu P. C.; Sherman J. C.; Chen A.; Kallenbach N. R. Alpha-helix stabilization by natural and unnatural amino acids with alkyl side chains. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 5317–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.