

Abstract
Although a prominent role for the brain in glucose homeostasis was proposed by scientists in the nineteenth century, research throughout most of the twentieth century focused on evidence that the function of pancreatic islets is both necessary and sufficient to explain glucose homeostasis, and that diabetes results from defects of insulin secretion, action or both. However, insulin-independent mechanisms, referred to as ‘glucose effectiveness’, account for roughly 50% of overall glucose disposal, and reduced glucose effectiveness also contributes importantly to diabetes pathogenesis. Although mechanisms underlying glucose effectiveness are poorly understood, growing evidence suggests that the brain can dynamically regulate this process in ways that improve or even normalize glycaemia in rodent models of diabetes. Here we present evidence of a brain-centred glucoregulatory system (BCGS) that can lower blood glucose levels via both insulin-dependent and -independent mechanisms, and propose a model in which complex and highly coordinated interactions between the BCGS and pancreatic islets promote normal glucose homeostasis. Because activation of either regulatory system can compensate for failure of the other, defects in both may be required for diabetes to develop. Consequently, therapies that target the BCGS in addition to conventional approaches based on enhancing insulin effects may have the potential to induce diabetes remission, whereas targeting just one typically does not.
The escalating epidemic of obesity, metabolic syndrome and type 2 diabetes (T2D) represents one of the most pressing and costly biomedical challenges confronting modern society1,2. However, much about the pathogenesis of these disorders remains unknown. In this article, we review recent evidence for a BCGS that works in tandem with pancreatic islets to regulate blood glucose levels. Glucose lowering induced by BCGS activation can involve a variety of mechanisms, some of which depend on insulin whereas others are altogether independent of islet hormones. Although islet- and brain-centred systems are distinct entities, evidence suggests that they work cooperatively to maintain stable blood glucose levels across a range of homeostatic challenges. Moreover, each system seems to have the potential to compensate, at least partially, for the failure of the other. Consequently, defects in both systems may be required for diabetes to develop and/or progress. This redundancy of islet-and brain-centred glucoregulatory systems presumably ensures tight regulation of circulating glucose, the body's principal metabolic currency.
Historical perspective
On the basis of his observation in 1854 that diabetes could be induced in rabbits by puncturing the floor of the fourth-cerebral ventricle (‘piqûre diabetique’)3, the renowned physiologist Claude Bernard proposed a role for the brain in both glucose homeostasis and diabetes pathogenesis. This notion remained popular until the discovery of insulin in 1921, and the subsequent identification of liver, muscle and adipose tissue as principal targets of the powerful effects of insulin on glucose metabolism. Combined with evidence linking diabetes pathogenesis to defective insulin secretion and action4, the pancreatic islet quickly came to overshadow the brain as the focal point for understanding this disease (Box 1).
Box 1. Traditional glucose homeostasis model.
Box 1 Figure. The traditional, islet-centred model of normal and abnormal glucose homeostasis.
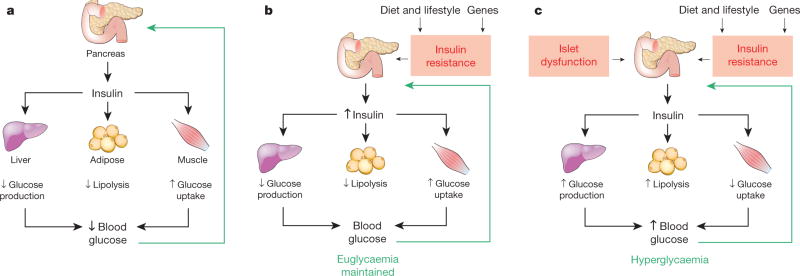
a, Under normal conditions, the islet-centred model proposes that glucose homeostasis is controlled primarily by the effect of rising blood glucose levels to stimulate insulin secretion. Insulin then acts on peripheral tissues such as the liver to suppress hepatic glucose production (HGP), and adipose tissue and muscle to stimulate glucose uptake. Not shown is the effect of the islet hormone glucagon, secretion of which is inhibited by rising glucose levels, and which acts to stimulate HGP. Thus, glucose has opposing actions on the secretion of insulin and glucagon, hormones that in turn have opposing effects on HGP. When blood glucose levels increase (for example, during a meal), therefore, the islet response effectively returns it to baseline. b, When individuals with normal islet function become insulin-resistant (for example, in association with dietary and/or genetic factors that cause obesity), the islet-centred model proposes that glucose homeostasis is preserved by the capacity of the islet to increase insulin secretion in a compensatory manner. c, If islet dysfunction precludes the increase of insulin secretion needed to overcome insulin resistance, glucose intolerance results. As islet dysfunction progresses, increased HGP and reduced tissue glucose uptake eventually cause overt hyperglycaemia and diabetes.
Current diabetes treatment options reflect this islet-centred view, consisting principally of recombinant human insulin preparations, insulin secretagogues (some of which also inhibit glucagon secretion), and drugs that increase insulin sensitivity. These drugs enjoy wide use and are effective in controlling hyperglycaemia, the hallmark of T2D, but they address the consequences of diabetes more than the underlying causes, and thus control rather than cure the disease.
Although insulin-independent mechanisms contribute nearly as much to glucose disposal as insulin does, little is known about how this type of glucose lowering works or what its therapeutic potential might be. Recent work indicates that BCGS activation can markedly improve glucose homeostasis in rodent models of diabetes via largely insulin-independent mechanisms5, and the possibility has been raised that a similar mechanism contributes to diabetes remission6,7 induced by bariatric surgical procedures such as Roux-en-Y gastric bypass8–11. Reconsideration of how glucose homeostasis is achieved by the body and the respective roles played by islet and brain in this process therefore seems justified.
Brain control of glucose homeostasis
A large literature documents glucoregulatory effects of pharmacological or genetic interventions targeting neurons in any of several areas of the hypothalamus (arcuate, ventromedial and paraventricular hypothalamic nuclei) and brainstem. Although uncertainty surrounds both the molecular identity and the role in glucose homeostasis played by many of these neuronal groups12–14, injection of insulin or glucose into discrete hypothalamic areas can lower blood glucose levels and increase liver insulin sensitivity15,16, and similar effects are achieved by restoring functional leptin receptors to specific hypothalamic nuclei of animals that otherwise lack them17,18. Conversely, deletion of receptors for either insulin or leptin (or their downstream signalling intermediates) from defined hypothalamic neurons causes glucose intolerance and systemic insulin resistance, indicating a physiological role for these neurons in the control of glucose metabolism19,20. These and many other observations highlight how the brain can influence glucose homeostasis in response to afferent input from peripheral signals, but they have yet to establish the extent to which such responses participate in the physiological control of circulating glucose levels.
Indirect control of hepatic glucose production
Although there is little doubt that insulin regulates hepatic glucose production (HGP) through a direct action on hepatocytes, insulin has also been proposed to regulate HGP via an indirect mechanism involving insulin action at a remote site21. As aberrant control of HGP is fundamental to diabetic hyperglycaemia21,22, its regulation has important clinical implications. The direct action of insulin on hepatocytes and other cell types involves its binding to insulin receptors and activation of signal transduction cascades that regulate a wide range of cellular processes. Of particular relevance to glycaemic control is the canonical insulin receptor substrate– phosphatidylinositol-3-OH kinase (IRS–PI(3)K) pathway (Fig. 1), which mediates insulin inhibition of both glycogenolysis and gluconeogenesis, the two primary determinants of HGP21.
Figure 1. Insulin signal transduction.

In hepatocytes, insulin regulates HGP by activating the IRS–PI(3)K pathway, which inhibits the transcription factor FOXO1. FOXO1 activation increases gluconeogenesis, leading to increased hepatic glucose production (HGP). PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-triphosphate.
In the fasted state, when the intestine is not absorbing nutrients and insulin levels are low, the liver is the primary source of circulating glucose and the rate of HGP is high. After a meal, nutrient-induced insulin secretion and subsequent activation of hepatic IRS–PI(3)K signalling inhibits HGP. The cellular basis for this effect involves PI(3)K-mediated activation of Akt, a serine-threonine kinase that, among other actions, inhibits the transcription factor FOXO1. FOXO1 stimulates gluconeogenesis in hepatocytes, and its inhibition is mandatory for insulin suppression of HGP21. Insulin activation of the canonical IRS–PI(3)K pathway in hepatocytes is implicated in the control of HGP by insulin under physiological conditions, and previous studies23 offer clear evidence in support of this hypothesis.
The concept that HGP can also be controlled by insulin action at a remote site was first proposed more than 15 years ago24 and received compelling support in a recent study25 of ‘TLKO’ mice with hepatocytes unresponsive to insulin owing to liver-specific deletion of key signal transduction molecules (the two Akt isoforms as well as FOXO1). In these animals, insulin cannot directly regulate HGP via the Akt–FOXO1 pathway. However, rather than exhibiting the expected loss of regulation, both HGP and systemic glucose homeostasis are controlled normally in these mice, even in response to exogenous insulin.
These data are among several observations21 that point to the existence of an indirect pathway through which insulin and nutrients can regulate HGP even when hepatocytes themselves are insensitive to direct insulin action. An intriguing question is what mechanism mediates the indirect control of HGP by insulin. Although other explanations are possible26,27, the BCGS is both activated by insulin and capable of regulating HGP in humans28 as well as rodent models19,20.
Comparison of the phenotypes of the previously reported TLKO mice25 with liver-specific insulin receptor knockout (LIRKO) mice, in which the liver is also unable to respond to insulin29, is informative. Unlike TLKO mice that have generally normal glycaemic regulation, LIRKO mice are severely glucose intolerant and insulin resistant. This is because LIRKO mice have unrestrained and unregulated HGP because FOXO1 is constitutively active in the absence of insulin-stimulated Akt activation. In LIRKO mice, therefore, increased HGP results from excessive FOXO1 activity, removal of which enables normal control of HGP via the indirect pathway. From this conclusion we infer that although inhibition of HGP by the indirect insulin pathway involves a FOXO1-independent mechanism, it can be blocked by excessive FOXO1 signalling.
Insulin–independent glucose disposal by the BCGS
Although a large literature has established the brain's capacity to affect glucose homeostasis, it has become clear only recently that this can involve mechanisms that are independent of insulin. Studies in which leptin was infused directly into brain ventricles—at doses too low to have any effect outside the brain—of rats and mice with insulin-deficient diabetes clearly demonstrate the ability of central leptin action to normalize markedly increased blood glucose levels30,31 despite persistent, severe insulin deficiency. This surprising outcome is incompatible with a strictly islet-centric model of glucose homeostasis. Similar findings have been reported in other rodent models of insulin-deficient diabetes using systemic (rather than central) administration of leptin at supraphysiological doses32,33.
Normal glucose tolerance, the ability to clear glucose from the bloodstream after a systemic glucose load, is believed to involve wide-ranging and highly coordinated effects of insulin across many different tissues. Thus, it seems surprising that in addition to normalizing fasting plasma glucose levels, intracerebroventricular (ICV) leptin infusion also restores glucose tolerance to nearly normal levels in rats with uncontrolled insulin-deficient diabetes30. Leptin action in the brain can therefore orchestrate complex and interconnected processes across several tissues to lower blood glucose despite the absence of insulin signalling. Although mechanisms mediating this effect are still under investigation, normalization of HGP, along with increased glucose uptake in tissues such as skeletal muscle, heart and brown adipose tissue, have a role30.
If activation of the BCGS by exogenous leptin is sufficient to correct diabetes without the need for insulin, why does severe insulin deficiency cause uncontrolled diabetes if the BCGS is left undamaged? The answer may lie in the extensive overlap between peripheral and central glucoregulatory systems. Insulin is required for the proper functioning of many cells and organ systems, including adipose tissue, and states of severe insulin deficiency undermine the ability of adipocytes both to store calories as fat and to secrete leptin. Consequently, severe insulin deficiency begets severe leptin deficiency34, depriving the BCGS of two key inputs and thereby undermining its function. Importantly, this defect is reversible by activating leptin receptors exclusively in the brain, as evidenced by the ability of central leptin infusion to restore normal glucose homeostasis to animals with uncontrolled, insulin-deficient diabetes30. Of course, uncontrolled diabetes can also be reversed by systemic insulin treatment, but this normalizes plasma levels of leptin as well as insulin34. Restoring normal leptin levels is important, because in the absence of a leptin signal (for example, in lipodystrophy or other leptin-deficient conditions), control of hyperglycaemia is much more difficult than in other forms of diabetes35.
Although physiological leptin replacement blocks or attenuates many neuroendocrine responses induced by insulin-deficient diabetes, it does not normalize hyperglycaemia36. This finding suggests that in the absence of insulin, supraphysiological activation of the BCGS is necessary to restore euglycaemia, and delivery of leptin to the brain in supraphysiological amounts achieves this effect. Thus, just as compensation for leptin deficiency requires insulin concentrations well above the normal physiological range (for example, in lipodystrophy or ob/ob mice, in which diabetes develops despite profound hyperinsulinaemia), high leptin levels are required to compensate for severe insulin deficiency. Stated differently, although islet- and brain-centred control systems are each able to compensate for the failure of each other, the activity of either system must be amplified for full compensation to occur. Unfortunately, islet failure does not trigger compensatory BCGS activation, but rather has the opposite effect, leading to a vicious cycle that ends in hyperglycaemia.
The use of the term ‘insulin independent’ to refer to actions mediated by the BCGS that become dysfunctional in the face of islet failure is a potential source of confusion. This is because if we accept that normal operation of the BCGS (including production of leptin by adipocytes) depends on insulin, one can argue that the entirety of glucose homeostasis is ‘insulin dependent’, even those effects mediated by the BCGS that do not involve a direct effect of insulin to stimulate glucose uptake. To avoid this confusion, we use ‘insulin independent’ hereafter to refer to effects on tissue glucose metabolism that do not involve direct, insulin-mediated signal transduction.
Recent evidence indicates that hormones other than leptin can also act in the brain to promote insulin-independent glucose lowering. Like insulin, the gastrointestinal hormone FGF19 (or its rodent homologue, FGF15) is secreted in response to meals, and, when given at pharmacological doses, exerts potent anti-diabetic effects37. Glucose-lowering by FGF19 involves actions in liver and adipose tissue, but the brain is also implicated, as ICV administration of FGF19 improves glucose tolerance in obese rats38. To investigate the mechanism underlying centrally mediated glucose lowering by FGF19, a study was recently performed in genetically obese, leptin-deficient ob/ob mice5. Within 2 h of a single ICV injection of FGF19 (at a dose causing no glucose lowering when given peripherally), ob/ob mice displayed markedly improved glucose tolerance, despite no change in insulin secretion or sensitivity5. Instead, the glucose-lowering effect of ICV FGF19 resulted from a selective, threefold increase in the insulin-independent component of glucose disposal. In response to diverse hormonal stimuli, therefore, the brain has the inherent capacity to remedy diabetic hyperglycaemia and glucose intolerance via potent, insulin-independent mechanisms5, as well as through enhanced insulin sensitivity12–20.
Glucose effectiveness
The term glucose effectiveness (GE) refers to the effect of an increased concentration of glucose to promote its own disposal, independent of insulin action39. Insulin-independent glucose disposal also occurs at basal glucose levels, but our understanding of the underlying mechanisms is insufficiently advanced to know whether the same or distinct processes contribute when plasma glucose levels are high versus in the basal state. In accord with convention, therefore, we use the term ‘insulin-independent glucose disposal’ to refer to the overall process, including those that operate at basal glucose levels, and reserve the use of ‘GE’ to refer to insulin-independent glucose disposal when blood glucose levels are increased.
A key point is that insulin-independent glucose disposal makes a large contribution to overall glucose homeostasis, roughly comparable to that of insulin39. Combined with the fact that reduced GE is both a major contributor to obesity-associated glucose intolerance39,40 and a strong risk factor for the future development of T2D (ref. 40), it is surprising how little is known about it. Unlike the dynamic and physiologically important regulation that characterizes insulin secretion and action, insulin-independent glucose disposal has traditionally been viewed as the fixed and unregulated process through which insulin-independent tissues obtain glucose to meet their needs39,41. The mechanism typically invoked to explain insulin-independent glucose disposal involves the passive effect of an increased glucose level to drive its movement down a concentration gradient and into cells (termed glucose mass action), but it is now clear that other mechanisms also exist—mechanisms that are subject to rapid regulation and can profoundly affect glucose homeostasis.
Perhaps the best-documented and most obvious example of rapid regulation of insulin-independent glucose disposal is in response to physical exercise, with the heightened metabolic demands of exercising muscle stimulating glucose uptake in the presence of stable ambient insulin and glucose levels. In addition to exercise, rapid regulation of GE has been reported in response to hormonal stimulation, for example, during intravenous infusion of glucagon-like-peptide-1 (GLP-1). Although GLP-1 improves glucose tolerance by enhancing insulin secretion, it also increases GE via mechanisms that have yet to be studied42. Interestingly, GLP-1 action in the hypothalamic arcuate nucleus also improves glucose tolerance43, raising the untested possibility that its effects on GE (like those of leptin and FGF19) are centrally mediated.
Extending this reasoning, it is noteworthy that, by definition, GE increases in response to rising blood glucose levels, and that glucose action on arcuate nucleus neurons has a rapid glucose-lowering effect16. Collectively, these observations support a model in which, by increasing plasma concentrations of insulin, GLP-1, FGF19 and glucose, consuming a meal generates diverse signals that activate the BCGS. This BCGS activation then contributes to glucose disposal via stimulation of both insulin-dependent and -independent mechanisms that, together with islet responses, are essential for proper glucose handling by the body (Fig. 2).
Figure 2. Schematic illustrations of brain- and islet-centred glucoregulatory systems.

The BCGS is proposed to regulate tissue glucose metabolism and plasma glucose levels via mechanisms that are both insulin dependent (for example, by regulating tissue insulin sensitivity) and insulin independent. Because of extensive redundancy between islet- and brain-centred pathways, dysfunction of both may be required for T2D to develop, and diabetes remission may be possible with therapies that target both pathways.
If insulin-independent glucose disposal is subject to rapid and potent regulation by the brain, it is not clear why neural control of GE has not been detected previously. One explanation may be that previous studies have relied on methods that are not optimized to detect GE. Chief among these is the euglyaemic–hyperinsulinaemic clamp method, considered by many to be the gold standard for quantitative, in vivo assessment of glucose metabolism. With this method, insulin sensitivity is measured as the amount of exogenous glucose that must be infused to maintain stable (or ‘clamped’) blood glucose concentrations when insulin levels are raised. Consequently, experimental interventions that change the amount of glucose required during the clamp are interpreted as having changed insulin sensitivity, despite the fact that some of the infused glucose could have been disposed of by insulin-independent mechanisms. Thus, one cannot know with certainty the extent to which observations based on the clamp method are due to changes in insulin-independent glucose disposal instead of, or in addition to, changes of insulin sensitivity. This limitation can be addressed using a complementary approach based on minimal model analysis of glucose and insulin kinetics during an intravenous glucose tolerance test. This method has seen broad use in clinical research39,42,44 and was recently used to reveal the potent stimulatory effect of centrally infused FGF19 on GE in ob/ob mice5.
A physiological role for the BCGS
Although there is little question that the brain participates in the glucoregulatory response to emergent or stressful conditions (for example, hypoglycaemia), the notion that the BCGS acts together with the islet to control glucose homeostasis under physiological conditions has yet to gain broad acceptance. A common and appropriate criticism is that although brain-directed interventions can affect glucose homeostasis, this should not be taken as evidence that the brain has a physiological role. Although the question of whether the BCGS is vital for normal, day-to-day control of blood glucose levels remains unanswered, several recent observations—that an indirect pathway controlling HGP exists and that this pathway can support normal glucose homeostasis even when the liver cannot respond to insulin directly25, that BCGS activation can be rapidly and potently engaged to increase insulin-independent glucose disposal, and that doing so has the potential to treat diabetic hyperglycaemia—justify reconsideration of this hypothesis and call for studies that offer a definitive test.
Among factors that engender scepticism of the part played by the BCGS is the notion that an islet-centred model (Box 1) is sufficient to explain the physiological control of glucose homeostasis under usual circumstances, because the capacity of the islet to secrete insulin in response to rising glucose levels often compensates for centrally mediated effects. For example, although even subtle disruption of the BCGS (for example, deletion of insulin receptors from a distinct subset of hypothalamic neurons in mice19,20) can reduce liver insulin sensitivity, the effect on glucose homeostasis is minimal because the tendency for blood glucose levels to rise is offset by a compensatory increase of insulin secretion. However, the logic of this argument weakens if BCGS dysfunction is more advanced. As an example, leptin-deficient states such as lipodystrophy increase both HGP and blood glucose levels despite marked hyperinsulinaemia45. Thus, impairments of both BCGS and islet function exist along a spectrum that ranges from mild to severe and, although the capacity of the islet to compensate for BCGS impairment is substantial, it has its limits. Normal BCGS function can therefore be seen as being permissive for normal glucose homeostasis, with islet compensation limiting the effect of BCGS dysfunction when it is mild, but not when it is more advanced.
A paucity of mechanistic information is another factor that has limited acceptance of a physiological role for the BCGS in glucose homeostasis. Whereas a great deal is known about how cellular insulin action affects glucose metabolism in hepatocytes (Fig. 1), for example, much less is known about how the brain controls HGP. Although a role for hepatic vagal innervation has been suggested12–14, it is premature to invoke the vagus nerve as the predominant mediator of this effect41. Another mechanism that may be relevant involves the islet hormone glucagon, which stimulates hepatic gluconeogenesis and gycogenolysis and hence raises HGP. Increased glucagon levels are implicated in the hyperglycaemia of uncontrolled, insulin-deficient diabetes, because both HGP and plasma glucagon levels are raised in this setting.
In this context, the interaction between leptin and glucagon is of interest. First, because leptin normalizes both HGP and increased glucagon levels in rodents with uncontrolled diabetes30, leptin-mediated inhibition of HGP may involve normalization of increased glucagon levels. Interestingly, leptin-mediated inhibition of glucagon secretion seems to be centrally mediated, because the effect is observed regardless of whether leptin is given systemically (at a high dose) or by ICV injection (at a low dose)30,33. Furthermore, the effect of uncontrolled diabetes to increase both glucagon secretion and HGP seems to be triggered, at least in part, by leptin deficiency, because both are reversed by leptin treatment30,32,33. However, increased plasma glucagon levels were normalized by systemic administration of a physiological dose of leptin to rats with streptozotocin-induced diabetes mellitus, and yet hyperglycaemia did not substantially improve36. Thus, the extent to which leptin-mediated normalization of circulating glucagon levels mediates its glucose-lowering effects in this setting awaits further study.
Further insight into the physiological role the BCGS has in glucose homeostasis can be gleaned from the hepatic response to a nutrient challenge. After a meal (or in response to a glucose load), the liver switches from being a net producer to a net consumer of glucose, and a surprisingly large fraction of the glucose absorbed during a meal is taken up into the liver41. This response is triggered by rising glucose concentrations in the hepatic portal vein (the vessel into which ingested nutrients enter before gaining access to the systemic circulation), which seems to be sensed by the BCGS41. Activation of the BCGS in turn strongly enhances liver glucose uptake via a mechanism that is augmented by insulin action in the brain27.
Several key questions remain to be addressed. One is whether the regulated component of insulin-independent glucose disposal is required for normal glucose homeostasis (a possibility that seems likely, given its considerable involvement), and if so, another is whether intact BCGS function is required for normal GE. Affirmative answers to both questions would constitute indisputable evidence that the brain has a physiological role in glucose homeostasis—perhaps comparable to that played by the islet, which itself is subject to regulation by the brain12–20,46.
Two–system control of glucose homeostasis
On the basis of the above reasoning, we propose that in response to a meal, both islet- and brain-centred systems are engaged and have important roles to restore homeostasis (Box 2). As ingested nutrients are absorbed into the circulation, increased insulin secretion and its canonical action on muscle, fat and liver both promote glucose disposal and inhibit its endogenous production. At the same time, the recruitment of insulin-independent mechanisms, in part through BCGS activation, makes a contribution to the overall process comparable to that of insulin. Like the action of insulin, these insulin-independent effects serve to both enhance glucose disposal (for example, through increased liver glucose uptake) and inhibit glucose production.
Box 2. BCGS and islet-centred glucose homeostasis model.
Box 2 Figure. Model integrating the BCGS and islet-centred system in normal and abnormal glucose homeostasis.

a, Under normal conditions, glucose homeostasis is controlled by complex and highly coordinated interactions between brain- and islet-centred systems. Like islets, the BCGS senses a variety of humoral signals, and in response to these inputs, BCGS activation increases glucose disposal by both insulin-dependent (for example, by increasing tissue insulin sensitivity) and insulin-independent (by increasing GE, which accounts for ∼50% of overall glucose disposal39) mechanisms. b, Although insulin normally inhibits HGP through its direct action on the liver, an indirect pathway also exists through which insulin can preserve normal HGP and blood glucose levels even when hepatocytes cannot respond to insulin directly25. We propose that this is among the effects mediated by the BCGS. c, Obesity is associated with reduced GE39 and with insulin resistance, and BCGS dysfunction contributes to both. When BCGS dysfunction is mild, the resulting tendency for blood glucose levels to increase stimulates insulin secretion, such that glucose homeostasis is preserved (at the expense of higher insulin levels). When BCGS dysfunction is more severe, however, even marked hyperinsulinaemia cannot preserve normal glucose homeostasis35, owing in part to the inability of reduced GE to be compensated by increased insulin secretion. Thus, intact BCGS function is required for normal glucose homeostasis. d, Islet dysfunction is not compensated by BCGS activation; to the contrary, impaired islet function can itself impair BCGS function (by reducing secretion of leptin as well insulin, when islet damage is severe) creating a vicious cycle that results initially in glucose intolerance. As both BCGS and islet dysfunction progress, overt hyperglycaemia and T2D result. e, Islet dysfunction can be compensated for by supraphysiological BCGS activation, which can achieve near-normal glucose homeostasis in rodent models of diabetes via insulin-independent mechanisms. Thus, therapeutic interventions targeting the BCGS as well as the traditional islet-based system may achieve diabetes remission, whereas targeting just one system typically does not.
After a meal, the contributions made by insulin-dependent and -independent mechanisms to the overall process are roughly equal, reflecting a partnership between direct, peripheral tissue effects of insulin and BCGS activation that ensures the efficient return of increased plasma glucose levels to basal values (Fig. 2). This two-system model incorporates interactions between the BCGS and islet-based systems into physiological glucose homeostasis via coordinate regulation of insulin-dependent and -independent mechanisms.
Is diabetes a failure of two systems?
In addition to establishing that the brain can potently increase GE, the observation that ICV leptin administration normalizes hyperglycaemia in rodents with uncontrolled diabetes indicates that BCGS activation can compensate effectively for severe insulin deficiency. This conclusion in turn suggests that disorders of both islet- and brain-centred systems may be necessary for T2D to occur (Fig. 3). This hypothesis is compatible with the observation that loss of canonical insulin action in specific tissues (for example, liver) has little effect on glucose homeostasis24, and that reduced GE contributes importantly to hyperglycaemia in T2D (ref. 39). But what is the evidence that BCGS function is impaired in individuals with diabetes? To our knowledge, there are no established examples in which diabetes occurs in the absence of BCGS dysfunction. Diabetes and BCGS dysfunction are tightly coupled to one another because (1) proper BCGS function depends on normal islet function, relying on inputs from insulin as well as other hormones whose secretion is either dependent on islet function (for example, leptin) or defective in diabetes (for example, GLP-1), and (2) rodent models of obesity and T2D are associated with hypothalamic injury and gliosis, a potentially important cause of BCGS dysfunction47–51. These hypothalamic alterations are proposed to reduce the ability of the BCGS to respond to relevant humoral signals (including insulin as well as leptin), and hence contribute to the associated fall of GE and onset of systemic insulin resistance that places an increased demand on islets in the lead up to T2D (Fig. 3). Whether this form of hypothalamic injury also occurs in human hypothalamus is under investigation, and early data support this possibility47,52. Thus, hypothalamic injury or inflammation offers a plausible mechanism linking impairment of the BCGS to T2D pathogenesis, and studies to test this hypothesis critically are warranted.
Figure 3. Proposed contributions of defective brain- and islet-centred glucoregulatory systems to T2D pathogenesis.

The traditional view holds that diabetes arises as a consequence of damage to, and ultimately failure of, beta-cell function. We propose a two-component model in which failure of glucose homeostasis can begin after initial impairment of either pancreatic islets or the BCGS. Malfunction of either of the two systems can initiate a cascade that drives the remaining glucoregulatory system into failure over time. Only when both systems are compromised does diabetes develop. Consequently, interventions that target both systems have greater therapeutic potential than those that target just one system.
Prospects for diabetes remission
Beyond causing weight loss, bariatric surgery induces diabetes remission in a far higher percentage of cases than can be achieved with conventional medical therapy6,7,10. The mechanism underlying metabolic benefit conferred by bariatric procedures is incompletely understood but may involve improvements of both islet- and brain-centred glucoregulatory systems. A previous study in a model of bariatric surgery (‘duodenal exclusion’) showed that blood glucose levels could be normalized in diabetic rats via insulin-independent activation of a neural circuit that inhibits HGP8. Using a similar surgical model, another study53 demonstrated that regulation of HGP after this procedure requires neuronal glucose sensing in the hepatic portal bed, and recent work indicates that despite having no effect on weight loss, body composition, food intake or energy expenditure54, sub-diaphragmatic vagotomy blocked the effect of bariatric surgery to reduce HGP in a rat model of obesity9. Furthermore, recent work suggests that insulin signalling in the ventromedial hypothalamus is required for the effect of bariatric surgery to inhibit HGP in an obese rat model55. Although mechanisms underlying BCGS activation by bariatric surgery await further study, recent evidence offers a link between enhanced secretion of FGF19, the nervous system and the gastrointestinal tract56. The larger point is that, should metabolic benefit arising from bariatric procedures be shown to involve BCGS activation, this would in turn suggest that diabetes remission may be achievable through interventions that activate both islet- and brain-centred glucoregulatory systems, whereas targeting just one does not. In principle, achieving this goal should not require surgical manipulation of the gastrointestinal tract.
Conclusion
When Claude Bernard proposed a dominant role for the brain in glucose homeostasis and diabetes pathogenesis, it was not the radical notion that it seems to be today. After all, the brain is implicated in the homeostatic control of most physiological processes that are essential for survival, ranging from body fuel stores (for example, fat mass) and body temperature to blood pressure and many endocrine systems. From this perspective, it seems surprising that control of glucose homeostasis should be governed entirely by peripheral mechanisms, despite some 90 years of research that has focused more or less exclusively on this view. One wonders how this area of science would have developed if leptin and its ability to normalize glucose levels in uncontrolled diabetes had been discovered in 1921, rather than insulin.
The surprise with which recent demonstrations of brain-mediated, insulin-independent correction of diabetes have been greeted brings into bold relief how, in the years since the discovery of insulin, metabolic research has been focused on only one part of the system governing glucose homeostasis —the part involving pancreatic islets. Despite having a role that may be comparable to that of insulin, insulin-independent glucose disposal has until now been seen as phenomenological and less worthy of study, and the notion that it might be regulated by the brain has not been previously considered.
Looking to the future, there are several important fundamental questions to address. Before the broader scientific community can (or should) be expected to embrace a role for the brain comparable to that of the islet in the day-to-day control of blood glucose levels, studies are needed to determine whether the maintenance of normal GE, which is known to be required for normal glucose tolerance, is dependent on a properly functioning BCGS. A related and equally important question is whether the link between reduced GE and the development of T2D (ref. 40) is explained by BCGS dysfunction. Such a finding would offer direct evidence that failure of both the BCGS and the islet is integral to diabetes pathogenesis.
Lastly, the observation that hormones such as FGF19 can act in the brain to improve glucose homeostasis in animal models of diabetes identifies new avenues for diabetes drug development. To expand on this specific example, the mechanism underlying the central action of FGF19 is proposed to involve a specific FGF receptor subtype, FGFR1c, that is widely expressed in the brain. In principle, there is no reason why synthetic agonists of this receptor should not prove effective for glucose lowering in patients with diabetes. Indeed, the efficacy of such drugs may not rely entirely on their central action, because activating this receptor in peripheral tissues seems to also be beneficial57. The larger point is that drugs that target the BCGS have important potential to synergize with current, islet-based approaches in ways that may fundamentally improve the management of what is among the most common, costly and debilitating diseases afflicting Western society.
Acknowledgments
The authors would like to thank A. G. Bell for inspiration and D. Porte Jr for comments. This work was partly funded by National Institutes of Health (NIH) grants DK083042 (M.W.S.), DK093848 (R.J.S.) and DK089053 (G.J.M.), and the Nutrition Obesity Research Centre and Diabetes Research Centre at the University of Washington, and the Helmholtz Alliance ICEMED (Imaging and Curing Environmental Metabolic Diseases), through the Initiative and Networking Fund of the Helmholtz Association.
Footnotes
Author Contributions All of the authors contributed to the ideas presented in and the writing of this manuscript.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Ogden CL, et al. Prevalence of overweight and obesity in the United States, 1999– 2004. J Am Med Assoc. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 2.Cowie CC, et al. Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National Health And Nutrition Examination Survey 1999– 2002. Diabetes Care. 2006;29:1263–1268. doi: 10.2337/dc06-0062. [DOI] [PubMed] [Google Scholar]
- 3.Bernard C. Leçons de Ohysiologie Experimentale Appliqués á lá Medecine. Paris: J.-B. Baillière; 1854. [Google Scholar]
- 4.Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- 5.Morton GJ, et al. FGF19 action in the brain induces insulin-independent glucose lowering. J Clin Invest. doi: 10.1172/JCI70710. http://dx.doi.org/10.1172/JCI70710 (1 October 2013) Administration of a low dose of the hormone FGF19 directly into the brain of leptin-deficient ob/ob mice ameliorated glucose intolerance by rapidly, potently and selectively increasing glucose effectiveness. [DOI] [PMC free article] [PubMed]
- 6.Mingrone G, et al. Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med. 2012;366:1577–1585. doi: 10.1056/NEJMoa1200111. [DOI] [PubMed] [Google Scholar]
- 7.Schauer PR, et al. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med. 2012;366:1567–1576. doi: 10.1056/NEJMoa1200225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breen DM, et al. Jejunal nutrient sensing is required for duodenal–jejunal bypass surgery to rapidly lower glucose concentrations in uncontrolled diabetes. Nature Med. 2012;18:950–955. doi: 10.1038/nm.2745. [DOI] [PubMed] [Google Scholar]
- 9.Jiao J, et al. Restoration of euglycemia after duodenal bypass surgery is reliant on central and peripheral inputs in zucker fa/fa rats. Diabetes. 2013;62:1074–1083. doi: 10.2337/db12-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cummings DE, Flum DR. Gastrointestinal surgery as a treatment for diabetes. J Am Med Assoc. 2008;299:341–343. doi: 10.1001/jama.299.3.341. [DOI] [PubMed] [Google Scholar]
- 11.Kashyap SR, et al. Metabolic effects of bariatric surgery in patients with moderate obesity and type 2 diabetes: analysis of a randomized control trial comparing surgery with intensive medical treatment. Diabetes Care. 2013;36:2175–2182. doi: 10.2337/dc12-1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz MW, Porte D., Jr Diabetes, obesity, and the brain. Science. 2005;307:375–379. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- 13.Sandoval D, Cota D, Seeley RJ. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annu Rev Physiol. 2008;70:513–535. doi: 10.1146/annurev.physiol.70.120806.095256. [DOI] [PubMed] [Google Scholar]
- 14.Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol. 2005;493:63–71. doi: 10.1002/cne.20786. [DOI] [PubMed] [Google Scholar]
- 15.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nature Med. 2002;8:1376–1382. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 16.Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science. 2005;309:943–947. doi: 10.1126/science.1112085. [DOI] [PubMed] [Google Scholar]
- 17.Coppari R, et al. The hypothalamic arcuate nucleus: a key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Morton GJ, et al. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–420. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Jordan SD, Konner AC, Bruning JC. Sensing the fuels: glucose and lipid signaling in the CNS controlling energy homeostasis. Cell Mol Life Sci. 2010;67:3255–3273. doi: 10.1007/s00018-010-0414-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill JW, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14:9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeFronzo RA. Banting Lecture. From the triumvirate to the ominous octet:a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgerton DS, et al. Effects of insulin on the metabolic control of hepatic gluconeogenesis in vivo. Diabetes. 2009;58:2766–2775. doi: 10.2337/db09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mittelman SD, Fu YY, Rebrin K, Steil G, Bergman RN. Indirect effect of insulin to suppress endogenous glucose production is dominant, even with hyperglucagonemia. J Clin Invest. 1997;100:3121–3130. doi: 10.1172/JCI119867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu M, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nature Med. 2012;18:388–395. doi: 10.1038/nm.2686. Systemic insulin administration was shown to suppress HGP effectively in mice with livers that were genetically modified to be unable to respond to the direct effect of insulin, thus establishing the existence of an indirect mechanism for control of HGP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Z, White MF. The AKTion in non-canonical insulin signaling. Nature Med. 2012;18:351–353. doi: 10.1038/nm.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramnanan CJ, et al. Brain insulin action augments hepatic glycogen synthesis without suppressing glucose production or gluconeogenesis in dogs. J Clin Invest. 2011;121:3713–3723. doi: 10.1172/JCI45472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kishore P, et al. Activation of KATP channels suppresses glucose production in humans. J Clin Invest. 2011;121:4916–4920. doi: 10.1172/JCI58035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michael MD, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 30.German JP, et al. Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology. 2011;152:394–404. doi: 10.1210/en.2010-0890. Hyperglycaemia was normalized by direct infusion of leptin into the brain of rats with uncontrolled, insulin-deficient diabetes, establishing the inherent capacity of the brain to maintain glucose homeostasis without the need for insulin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev. 2011;91:389–411. doi: 10.1152/physrev.00007.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu X, Park BH, Wang MY, Wang ZV, Unger RH. Making insulin-deficient type 1 diabetic rodents thrive without insulin. Proc Natl Acad Sci USA. 2008;105:14070–14075. doi: 10.1073/pnas.0806993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruger AJ, et al. Leptin treatment confers clinical benefit at multiple stages of virally induced type 1 diabetes in BB rats. Autoimmunity. 2011;44:137–148. doi: 10.3109/08916934.2010.482116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Havel PJ, et al. Marked and rapid decreases of circulating leptin in streptozotocin diabetic rats: reversal by insulin. Am J Physiol. 1998;274:R1482–R1491. doi: 10.1152/ajpregu.1998.274.5.R1482. [DOI] [PubMed] [Google Scholar]
- 35.Semple RK, Savage DB, Cochran EK, Gorden P, O'Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev. 2011;32:498–514. doi: 10.1210/er.2010-0020. [DOI] [PubMed] [Google Scholar]
- 36.German JP, et al. Leptin deficiency causes insulin resistance induced by uncontrolled diabetes. Diabetes. 2010;59:1626–1634. doi: 10.2337/db09-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaap FG. Role of fibroblast growth factor 19 in the control of glucose homeostasis. Curr Opin Clin Nutr Metab Care. 2012;15:386–391. doi: 10.1097/MCO.0b013e3283547171. [DOI] [PubMed] [Google Scholar]
- 38.Ryan KK, et al. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology. 2013;154:9–15. doi: 10.1210/en.2012-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Best JD, et al. Role of glucose effectiveness in the determination of glucose tolerance. Diabetes Care. 1996;19:1018–1030. doi: 10.2337/diacare.19.9.1018. A definitive review of glucose effectiveness and its role in both normal glucose homeostasis and diabetes. [DOI] [PubMed] [Google Scholar]
- 40.Martin BC, et al. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet. 1992;340:925–929. doi: 10.1016/0140-6736(92)92814-v. [DOI] [PubMed] [Google Scholar]
- 41.Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr. 2012;3:286–294. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D'Alessio DA, Kahn SE, Leusner CR, Ensinck JW. Glucagon-like peptide 1 enhances glucose tolerance both by stimulation of insulin release and by increasing insulin-independent glucose disposal. J Clin Invest. 1994;93:2263–2266. doi: 10.1172/JCI117225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandoval DA, Bagnol D, Woods SC, D'Alessio DA, Seeley RJ. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes. 2008;57:2046–2054. doi: 10.2337/db07-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kahn SE, et al. Treatment with a somatostatin analog decreases pancreatic B-cell and whole body sensitivity to glucose. J Clin Endocrinol Metab. 1990;71:994–1002. doi: 10.1210/jcem-71-4-994. [DOI] [PubMed] [Google Scholar]
- 45.Petersen KF, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osundiji MA, Evans ML. Brain control of insulin and glucagon secretion. Endocrinol Metab Clin North Am. 2013;42:1–14. doi: 10.1016/j.ecl.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 47.Thaler JP, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai D. One step from prediabetes to diabetes: hypothalamic inflammation? Endocrinology. 2012;153:1010–1013. doi: 10.1210/en.2011-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Posey KA, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296:E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milanski M, et al. Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes. 2012;61:1455–1462. doi: 10.2337/db11-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horvath TL, et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci USA. 2010;107:14875–14880. doi: 10.1073/pnas.1004282107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alkemade A, et al. AgRP and NPY expression in the human hypothalamic infundibular nucleus correlate with body mass index, whereas changes in aMSH are related to type 2 diabetes. J Clin Endocrinol Metab. 2012;97:E925–E933. doi: 10.1210/jc.2011-3259. [DOI] [PubMed] [Google Scholar]
- 53.Troy S, et al. Intestinal gluconeogenesis is a key factor for early metabolic changes after gastric bypass but not after gastric lap-band in mice. Cell Metab. 2008;8:201–211. doi: 10.1016/j.cmet.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 54.Shin AC, Zheng H, Berthoud HR. Vagal innervation of the hepatic portal vein and liver is not necessary for Roux-en-Y gastric bypass surgery-induced hypophagia, weight loss, and hypermetabolism. Ann Surg. 2012;255:294–301. doi: 10.1097/SLA.0b013e31823e71b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paranjape SA, et al. Improvement in hepatic insulin sensitivity after Roux-en-Y gastric bypass in a rat model of obesity is partially mediated by hypothalamic insulin action. Diabetologia. 2013;56:2055–2058. doi: 10.1007/s00125-013-2952-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gerhard GS, et al. A role for fibroblast growth factor 19 and bile acids in diabetes remission after Roux-en-Y gastric bypass. Diabetes Care. 2013;36:1859–1864. doi: 10.2337/dc12-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ai-Luen W, et al. Amelioration of type 2 diabetes by antibody-mediated activation of fibroblast growth factor receptor 1. Sci Transl Med. 2011;3:113ra26. doi: 10.1126/scitranslmed.3002669. [DOI] [PubMed] [Google Scholar]