

Abstract
Exposure to a stressor sensitizes behavioral and hormonal responses to future stressors. Stress-associated release of noradrenaline enhances the capacity of central synapses to exhibit plasticity (metaplasticity). We report noradrenaline-dependent metaplasticity at GABA synapses in the paraventricular nucleus of the hypothalamus in rat and mouse that control the hypothalamic-pituitary-adrenal axis. Previously, work indicates these GABA synapses become excitatory following acute stress. The current study finds that in vivo stress exposure is also required for these synapses to undergo activity-dependent long-term potentiation (LTPGABA). The activation of β-adrenergic receptors (β-ARs) during stress functionally upregulates metabotropic (mGluR1) glutamate receptors allowing for mGluR1-dependent LTPGABA during afferent bursts. LTPGABA is expressed postsynaptically and manifests as the emergence of new functional synapses. Our findings provide the first demonstration that noradrenaline release during an in vivo challenge alters information storage capacity at GABA synapses. These changes may contribute to neuroendocrine sensitization to stress.
Any threat to survival triggers rapid defense mechanisms known as the stress response1,2. In vertebrates, the hallmark of this response is the activation of the hypothalamic-pituitary-adrenal (HPA) axis culminating in an increase in circulating glucocorticoids1,2. Although a number of brain areas perceive and process diverse modalities of stress (e.g. psychological, physical and homeostatic stress), the neuroendocrine response requires that all stress-related signals eventually converge onto parvocellular neuroendocrine cells (PNCs) in the paraventricular nucleus of the hypothalamus (PVN)2,3. The stereotyped recruitment of these ‘stress effector’ cells is vital for managing impending challenges, but considerable evidence indicates that flexibility in this system is also important for tuning neuroendocrine output appropriately based on previous stress experience1,2,4. This may be achieved, in part, through plasticity in higher brain regions1,4, but emerging evidence showing that synapses in the PVN also undergo adaptations following stress3,5–11, suggests a key role for this structure.
Glutamate synapses onto PNCs undergo stress-dependent priming that manifests as short-term potentiation following tetanization9. Acute restraint stress also causes a depolarizing shift in the GABAA receptor (GABAAR) reversal potential (EGABA) that is sufficient to convert GABA synapses to excitatory both during and immediately after stress12,13. Given that GABA is the dominant synaptic input to PNCs (> 50% of all synapses)10,11, stress-dependent changes in efficacy and plasticity of GABA synapses, together with an excitatory conversion, would powerfully enhance the excitatory inputs to the PVN and consequently sensitize the neuroendocrine output to subsequent stressors.
In addition to GABA and glutamate, PNCs also receive noradrenaline inputs from A1/A2 cell groups in the caudal medulla14,15. noradrenaline rapidly excites PNCs and contributes to the activation of the system at stress onset15,16. In other systems, noradrenaline acting via β-adrenergic receptors (β-ARs) links behaviors/experience to subsequent synaptic plasticity1,17,18. We hypothesized that noradrenaline ensures specific information is extracted by PNCs during stress. Here we report that noradrenaline, through recruitment of β-ARs during stress, primes PNCs to be more sensitive to heterosynaptic glutamate signaling. This creates conditions that are permissive for the induction of activity-dependent potentiation at GABA synapses in the PVN.
Results
A single stress experience unmasks LTPGABA
Exposure to a single stressor alters neuroendocrine responses to future challenges4. This suggests the stress axis is capable of learning and provides an experimental model to study how stress affects subsequent information processing and storage. We asked whether a single stress alters the ability of GABA synapses onto PNCs to undergo activity-dependent plasticity. Using whole-cell voltage-clamp recordings we examined evoked inhibitory postsynaptic currents (eIPSCs). Synaptic efficacy was assessed in response to high-frequency stimulation (HFS, 100 Hz, 1 s repeated 4 times with 20 s interval, Vm = −80 mV) delivered to slices obtained either from rats subjected to 30 min immobilization stress (IMO) immediately prior to slice preparation, or from age-matched, naïve rats. In naïve rats, this protocol had no lasting effect on eIPSC amplitude (98 ± 9% of baseline, p = 0.4; Fig. 1a,c). By contrast, we noted a robust potentiation of eIPSC amplitude in slices from stressed rats (140 ± 6% of baseline, p = 0.0001; Fig. 1b,d). This potentiation persisted for the duration of the stable recording (> 25 min after HFS). We also tested different durations of HFS on synaptic strength (Fig. 1e). In slices from naive rats, shortening (0.5 s) or lengthening (4 s) the induction protocol had no effect on eIPSC amplitude (0.5 s, 110 ± 6.5% of baseline), p = 0.1; 4 s, 112 ± 9.8% of baseline, p = 0.2). Following stress, the potentiation in response to 0.5-s HFS was not statistically significant (139% ± 20% of baseline, p = 0.05) but the, 4-s protocol elicited LTPGABA (155.4 ± 19% of baseline,, p = 0.03). A cumulative probability distribution of normalized post-HFS (0.5–4 s) eIPSC shows that > 20% potentiation was observed in 28% of cells in naïve rats, and in 74% of cells following stress (Fig. 1g). In the majority of PNCs, a single stress exposure unmasks the ability of GABA synapses to potentiate in response to bursts of afferent activity.
Figure 1. Acute stress unmasks activity-dependent LTPGABA.

(a, b) Top, schematics for experiments, Bottom, raw (light) and averaged (solid) traces of eIPSCs recorded from PNCs before (black) and after (blue/red) HFS in slices from (a) naïve and (b) IMO stressed rats. Scale bars represent 50 pA and 20 ms. Amplitude values were assessed at the time points indicated by shadowed bands below. (c, d) Summary time-course of eIPSC amplitude recorded in slices from naïve (blue, (n = 8 (7 rats)) and IMO stressed (red, (n = 14 (14 rats)) rats. Arrows indicate time of HFS. Data are mean ± s.e.m. (e) Summary of the changes of eIPSC amplitude, in slices from naïve and IMO stressed rats following different durations of HFS (0.5, 1, 4 s; n=7, 10, 6 (5 rats each)). (f) Summary of eIPSC amplitude change in slices from IMO stressed rats following lower frequency stimulation (10 Hz for 10 s, n=5 (5 rats)), and in slices from rats exposed to predator odor (n=5 (5 rats) or forced swim (n=7 (5 rats) following HFS. (g) Cumulative probability distribution of normalized eIPSC amplitude from naïve (blue) and IMO-stressed (red) rats following HFS shown in (e). Bin size 10 %.
LTPGABA was also observed when lower frequency protocols (10 Hz, 10 s) that better recapitulate physiological synaptic inputs were used following stress (132 ± 10%, p = 0.03, Fig. 1f). The unmasking of LTPGABA is not limited to IMO stress, as predator odor stress (30-min exposure to a fox feces compound) also led to HFS-induced LTPGABA (133± 9%, p = 0.02). Forced swim stress (20-min inescapable swim), however, did not induce statistically significant LTPGABA (121 ± 12%, p = 0.08, Fig. 1f). These results argue that the priming of GABA synapses is not limited to a specific induction protocol (100 Hz) or a specific stressor (IMO), but also it may not be generalizable to all types of stressors. Below we use IMO stress and 100-Hz, 1-s protocol to study the mechanisms underlying LTPGABA.
β-AR signaling during stress unmasks LTPGABA
In other brain regions, noradrenaline, through activation of β-AR, modulates activity-dependent synaptic plasticity at glutamate synapses19–21. Since initiation of a neuroendocrine stress response requires local release of noradrenaline15,16 we asked whether β-AR activation during stress was necessary for unmasking subsequent LTPGABA. To test this, we injected rats with the β-AR antagonist propranolol (PRO, 10 mg kg−1, intraperitoneal) 30 min before IMO and repeated experiments as above. Propranolol pre-treatment prevented the stress-dependent unmasking of LTPGABA (93 ± 9% of baseline, p = 0.4; Fig. 2a), demonstrating that β-AR activation during IMO stress is necessary in order to enable GABA synapses to express subsequent activity-dependent LTP. We next asked if prior history of β-AR activation, specifically in the PVN in the absence of stress, was sufficient to mimic the effects of IMO. We treated naïve hypothalamic slices with β-AR agonist isoproterenol (ISO, 1 μM) for 10 min. First, we confirmed that ISO application alone caused no lasting change in eIPSCs (103 ± 4% of baseline after 20 min washout, p = 0.5). We did note a transient potentiation peaking approximately 15 min after application (118 ± 7% of baseline, n = 5 (4 rats), p = 0.06). In subsequent experiments ISO was washed out > 30 min between slice incubation and recording. Following ISO treatment, HFS induced robust LTPGABA (149 ± 15% of baseline, p = 0.01; Fig. 2a), suggesting that transient pharmacological activation of β-AB is sufficient to unmask subsequent LTPGABA.
Figure 2. Stress-induced β-AR signaling unmasks activity-dependent LTPGABA.

Left, schematics for the experiments. Middle, averaged traces before (grey) and after (colored) HFS. Right, summary time-course of normalized eIPSCs. (a) Recordings in slices from IMO rats treated with β-AR antagonist PRO (purple, n = 8 (6 rats)) and in slices from naïve rats treated ex vivo with β-AR agonist ISO (orange, n = 8 (6 rats)) (b) Recordings in slices from naïve TH-Cre/ChR2-eYFP mice without treatment (gray, n = 8 (7 mice)), ex vivo flushed with blue light in the absence (blue, n = 9 (8 mice)) and presence (orange, n = 6 (4 mice)) of PRO. Scale bars represent 50 pA and 20 ms. Data are mean ± s.e.m.
The primary noradrenaline projection to the PVN originates in the caudal medulla noradrenergic populations (A1/A2)14–16. To examine the contribution of noradrenergic inputs to GABA synapse priming, we used an optogenetic approach in which we stereotaxically injected Cre-recombinase-dependent adeno-associated viral vector (AAV) carrying channelrhodopsin-2 (ChR2) and eYFP or -mCherry into the caudal medulla of mice that expressed Cre under the control of tyrosine hydroxylase (TH) promoter (TH-Cre mice)22. Patch clamp recording from acute brain stem slices confirmed functional ChR2 expression in the caudal medulla neurons. eYFP-positive cells fired action potentials in response to 5 ms pulse of blue light at 5–50 Hz with 100% fidelity at 20 Hz and below (Supplementary Fig. 1). In the PVN, we observed efficient ChR2-eYFP expression in fibers (Supplementary Fig. 1). We stimulated ChR2-expressing noradrenergic fibers ex vivo in PVN slices prepared from naïve mice and asked whether the release of endogenous noradrenaline was sufficient to unmask subsequent LTPGABA. We delivered 5 ms blue light pulses at 10 Hz for 20 min (to mimic stress), and then rested the slices for > 30 min before recording. Following light stimulation, HFS induced robust LTPGABA (160 ± 18% of baseline, p = 0.004; Fig. 2c). HFS failed to induce LTPGABA in ChR2-expressing slices that did not receive light stimulation (107 ± 6% of baseline, p = 0.5; Fig. 2c). Incubation of the slice with PRO (50 μM) 10 min before and during light stimulation attenuated the priming (118 ± 5% of baseline, p = 0.03 vs no PRO; Fig. 2b), indicating the necessity of β-AR activation. These results collectively support the hypothesis that local release of noradrenaline in the PVN primes GABA synapses for subsequent activity-dependent plasticity. In addition to noradrenaline, other neuromodulators are also released in the PVN during the onset of stress. Specifically, CRH release during IMO stress primes glutamate synapses onto PNCs to express short-term potentiation (STP)9. Systemic administration of CRH receptor type 1 antagonist CP-154,526 (30 mg kg−1, intraperitoneal) 30 min before IMO, (a treatment that abolished IMO-dependent priming of glutamate STP9), failed to prevent HFS-induced LTPGABA (130 ± 9 % of baseline, n = 5 (3 rats), p = 0.03), indicating that CRHR1 activation is not required for LTPGABA. These results identify noradrenaline, through actions at β-ARs, as a stress-associative metaplastic signal23 that is necessary and sufficient for the subsequent expression of activity-dependent long-term plasticity at GABA synapses.
β-AR-induced priming requires PKA activation
β-ARs activation induces metaplasticity at glutamate synapses through downstream activation of protein kinase A (PKA)19–21, and PKA has been implicated in GABA synapse plasticity24. We examined the role of PKA in the priming of LTPGABA by first incubating slices from naïve rats with ISO in the presence of selective PKA inhibitor KT5720 (1 μM, applied 10 min before and during ISO). We observed no ISO-dependent priming of LTPGABA (102 ± 12% of baseline, p = 0.4; Fig. 3). Conversely, we found that direct activation of PKA with 8-bromo-cAMP (200 μM for 10 min, and washout > 30 min) was sufficient to unmask HFS-induced LTPGABA in slices from naïve rats (145 ± 14% of baseline, p = 0.02; Fig. 3). These data indicate that PKA activation is the key intracellular signaling mechanism downstream of β-AR activation.
Figure 3. β-AR-induced priming requires PKA activation.
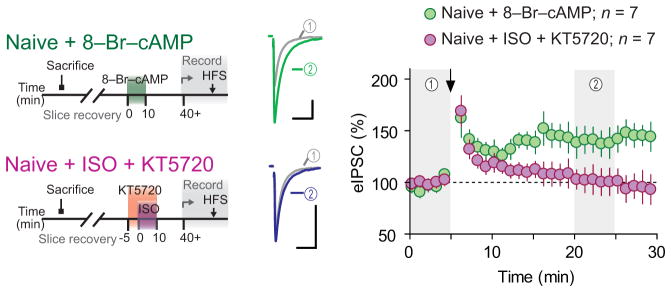
Recordings in slices from naïve rats treated ex vivo with PKA activator 8-Br-cAMP (green, n = 7 (4 rats)) and with ISO in the presence of PKA inhibitor KT5720 (purple, n = 7 (3 rats)). Scale bars represent 50 pA and 20 ms. Data are mean ± s.e.m.
Unmasking of LTPGABA requires stress- and β-AR-induced priming of mGluR1
We next investigated the mechanism by which IMO-induced β-AR signaling unmasks LTPGABA. Our data suggest that β-AR → PKA activation acts as a priming signal rather than an actual LTPGABA trigger as pharmacological activation of β-AR alone was insufficient to LTPGABA. Furthermore, LTPGABA induction required only a brief β-AR activation (by stress, pharmacological or optogenetic manipulation) prior to but not concomitant with HFS. In other brain areas, activity-dependent LTPGABA requires heterosynaptic signaling through group 1 metabotropic glutamate receptors (mGluR1 and 5)24,25, which are also expressed by PNCs26. Moreover, β-AR signaling and downstream PKA prevent agonist-induced internalization/desensitization of mGluR127, and PKA activation facilitates mGluR1-dependent LTPGABA24. Thus we hypothesized that mGluR1 signaling is required for LTPGABA, and that IMO-induced β-AR signaling enhances group 1 mGluRs responsiveness to glutamate released during the HFS protocol. To test this idea, we first asked whether activation of group 1 mGluRs is required for HFS-induced LTPGABA in PNCs by delivering HFS in the presence of subtype non-selective group 1/2 mGluR antagonist (S)-α-methyl-4-carboxyphenylglycine (MCPG, 200 μM) in slices from IMO stressed rats. MCPG abolished LTPGABA (92 ± 10% of baseline, p = 0.4; Fig. 4a,b). Two structurally unrelated mGluR1 subtype-specific antagonists 7-(hydroxyimino)-cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt, 100 μM) and JNJ16259685 (750 nM) also blocked (CPCCOEt, 93 ± 13% of baseline, p = 0.6; JNJ16259685, 105 ± 11, p = 0.6, Fig. 4b) while mGluR5 specific antagonist 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP, 10 μM) failed to inhibit LTPGABA (166 ± 19% of baseline, p = 0.03; Fig. 4b), indicating that heterosynaptic activation, specifically of mGluR1, is necessary for LTPGABA. The GABAAR antagonist bicuculline (100 μM; focally applied near the postsynaptic cell) prior to and during HFS failed to block LTPGABA (132 ± 11%, p = 0.04), suggesting that GABAARs do not participate in the induction of LTPGABA.
Figure 4. LTPGABA requires priming of mGluR1 by β-AR activation.
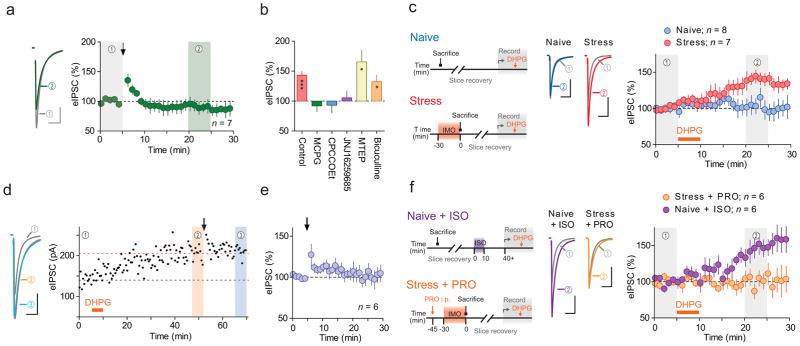
(a) Left, averaged traces before (grey) and after (green) HFS recorded in slices from IMO stressed rats, with bath application of MCPG (mGluR1/5 antagonist). Right, summary time-course. Arrow indicates HFS. (b) Summary of HFS-induced eIPSC amplitude changes in control (without antagonist, adapted from experiment shown in Fig. 1d) adapted from experiment shown in Fig. 1d, and with MCPG, n = 6 (4 rats), CPCCOEt (mGluR1 antagonist, n = 7 (5 rats)), JNJ16259685 (mGluR1 antagonist, n = 7 (5 rats)), MTEP (mGluR5 antagonist, n = 5 (4 rats)) and bicuculline (GABAAR antagonist, n = 5 (3rats)). * p < 0.03, *** p = 0.0001. (c) Left, schematics for the experiments. Middle, averaged traces of eIPSC before (grey) and after DHPG (mGluR1/5 agonist, blue/red) recorded in slices from naïve (blue, n = 8 (5 rats)) and IMO (red, n = 7 (5 rats)) rats. Right, summary time-course. (d) Left, averaged traces of eIPSC from time periods 1–3 indicated in the right graph. Right, time course of eIPSC amplitude recorded in a slice from IMO stressed rat. DHPG (orange bar) application was followed by HFS (arrow). Broken lines indicate the average amplitude before DHPG (black) and HFS (orange). (e) Summary of the effects of HFS on eIPSC amplitude after DHPG-induced potentiation, n = 6 (5 rats). (f) Left, schematics for the experiments. Middle, averaged traces of eIPSC before (grey) and after DHPG (orange/purple) recorded in slices from IMO rats injected with PRO (orange, n = 6 (6 rats)) and in slices from naïve rats treated ex vivo with ISO (purple, n = 9 (7 rats)). Right, summary time-course. Arrow and orange horizontal bars represent the time of HFS and DHPG application, respectively. Data are mean ± s.e.m. Scale bars represent 50 pA and 20 ms.
If IMO/β-AR enhancement of mGluR1 signaling is necessary for LTPGABA induction following stress, then pharmacological activation of mGluR1, specifically following stress, should be sufficient to induce LTPGABA independent of HFS. Bath application of mGluR1/5 agonist (S)-3,5-dihydroxyphenylglycine (DHPG, 5 μM, 5 min) to slices from stressed rats elicited long-lasting potentiation of eIPSCs in slices from stressed (141 ± 8% of baseline, p = 0.003; Fig. 4e), but not naïve rats (94 ± 11% of baseline, p = 0.6; Fig. 4e). Next, we asked whether DHPG-mediated LTPGABA and HFS-induced LTPGABA share a common underlying mechanism. We pretreated slices obtained from stressed animals with DHPG and washed out the drug > 30 min to allow LTPGABA to develop. Subsequent delivery of HFS failed to induce LTPGABA (102 ± 9% of post-DHPG level, p = 0.8; Fig. 4d,e). Finally, we examined whether β-AR signaling causes the functional upregulation of mGluR1 by IMO. Blockade of β-AR with PRO during IMO stress challenge (same treatment as above) abolished DHPG-induced LTPGABA (98 ± 9% of baseline, p = 0.9; Fig. 4f). Conversely, ex vivo stimulation of β-AR with ISO in PVN slices from naïve rats unmasked DHPG-induced LTPGABA (137 ± 11% of baseline, p = 0.02; Fig. 4f). Importantly, β-AR activation needs to occur prior to mGluR1 activation as reversing the temporal sequence of ISO → DHPG treatment (e.g. DHPG → ISO) did not induce LTPGABA (108 ± 7%, n = 7 (4 rats), p = 0.3, Supplementary Fig. 2). These data support our hypothesis that β-AR activation functionally upregulates mGluR1 to enable GABA synapses to express activity-dependent LTP.
Number of functional GABA synapses increases during LTPGABA
We next examined the mechanisms underlying the expression of LTPGABA. To this end, we analyzed the paired-pulse ratio (PPR) and coefficient of variation (CV) of the eIPSCs shown in Fig. 1c-d (see Methods for details of analysis). Alterations of PPR indicate changes to probability of neurotransmitter release from the presynaptic terminals (pr), while alterations to 1/CV2 suggest changes to either pr, the number of functional synapses (N), or both28. Baseline PPR or 1/CV2 prior to HFS were similar between naïve and IMO rats (PPR: 0.8 ± 0.04 and 0.7 ± 0.03 for naïve and IMO, p = 0.2; 1/CV2: 22 ± 6 and 34 ± 13, p = 0.5), suggesting that stress alone did not affect these synaptic properties. We found no change in PPR during LTPGABA following stress (97 ± 3% of baseline, p = 0.4; Fig. 5a), suggesting that LTPGABA occurs without pr increase. By contrast, we observed an increase in 1/CV2 (234 ± 28% of baseline, p = 0.0004; Fig. 5a), suggesting an increase in N during HFS-induced LTPGABA. In naïve rats, HFS failed to alter PPR (104 ± 7% of baseline, p = 0.6) or 1/CV2 (122 ± 29% of baseline, p = 0.5). To more closely examine the relationship between PPR, CV and LTPGABA, we plotted the changes in PPR or 1/CV2 against eIPSC amplitude changes for individual cells. This revealed a linear, positive correlation between 1/CV2 and eIPSC changes (r = 0.7, p = 0.0009; Fig. 5b), but not between eIPSC and PPR (r = 0.4, p = 0.06; Fig. 5c). These results indicate that LTPGABA is primarily attributable to an increase in N.
Figure 5. LTPGABA is accompanied by changes relevant to an increase of synapse number.

(a) Summary time-course of normalized PPR (green) and 1/CV2 (orange) of eIPSCs recorded in slices from IMO stressed rats. Arrow indicates the time of HFS. (b, c) Plots of normalized post-HFS 1/CV2 (b) and PPR (c) against amplitude, recorded in slices from naïve (blue, n=16) and IMO stressed (red, n=21) rats. Smaller symbols represent individual data and larger ones mean ± s.e.m. Lines represent least square linear best fit. (d–f) Sample traces (d) and cumulative probability plots of sIPSC frequency (e) and amplitude (f) before (grey) and after (red) HFS recorded in a slice from IMO rats. Scale bars represent 20 pA and 500 ms. (g) Summary time-course of normalized sIPSC frequency (purple) and amplitude (blue) recorded in IMO slices. (h, i) Plots of normalized post-HFS sIPSC frequency (h) and sIPSC amplitude (i) against eIPSC amplitude recorded in slices from naïve (blue) and IMO stressed (red) rats.
If recruitment of new synapses underlies LTPGABA, such changes may be reflected as an increase in the frequency of spontaneous IPSCs (sIPSCs). Consistent with this, we found an increase in sIPSC frequency during HFS-induced LTPGABA following stress (158 ± 14% of baseline, p = 0.002; Fig. 5d–g). Mean sIPSC amplitude did not change (110 ± 7% of baseline, p = 0.07; Fig. 5d–g), although this does not exclude the possibility that HFS expands the size of pre-existing receptor clusters only in a subset of synapses contributing to the eIPSC, as sIPSC amplitude represents the average of entire population of GABAergic inputs; consequently, subpopulation-specific changes to synapses could be underestimated. The rise in sIPSC frequency can be primarily attributed to N but not pr increase, as linear regression analysis found that sIPSC frequency change strongly correlated with the change in eIPSC 1/CV2 (r = 0.7, p = 0.0007; Supplementary Fig. 2) but not PPR (r = 0.3, p = 0.3). Consistent with our observations for eIPSCs, HFS failed to affect either the frequency or amplitude of sIPSCs in the naïve rats (frequency, 87 ± 7% of baseline, p = 0.07; amplitude, 100 ± 4 %; p = 0.9), suggesting that HFS-induced sIPSC frequency facilitation is stress-dependent. Linear regression analysis revealed that the increase in sIPSC frequency positively correlated with the increase in amplitude of eIPSC (r = 0.6, p = 0.01; Fig. 5h), while sIPSC amplitude did not (r = 0.4, p = 0.2; Fig. 5i). This indicates that, similar to the conclusion derived from 1/CV2 and PPR analysis, LTPGABA can be primarily explained by an increase in N.
LTPGABA requires postsynaptic mechanisms
We next examined biochemical mechanisms responsible for LTPGABA. In the hippocampus and cerebellum, glutamatergic signaling that leads to heterosynaptic LTPGABA requires elevations in postsynaptic Ca2+ concentration, activation of Ca2+/calmodulin-dependent kinase II (CaMKII) and surface trafficking of GABAARs29–31. In support of a requirement for postsynaptic Ca2+ signaling during LTPGABA in our experiments, postsynaptic loading with Ca2+ chelator 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 10 mM) completely blocked HFS-induced LTPGABA (95 ± 7% of baseline, p = 0.4; Fig. 6a). Elevated intracellular Ca2+ triggers calmodulin binding to CaMKII, autophosphorylation of a specific threonine residue (Thr286) and generation of autonomous (Ca2+/calmodulin-independent) CaMKII activity. Phosphorylation of Thr286 is required for GABAAR surface trafficking in hippocampal neurons32 and is enhanced in parvocellular subdivision of the PVN following stress33. Inclusion of the specific CaMKII inhibitor autocamtide-2-related inhibitory peptide (AIP, 1 μM) in the patch pipette abolished HFS-induced LTPGABA (97 ± 5% of baseline, p = 0.7; Fig. 6b). Next we evaluated the involvement of SNARE-dependent exocytosis in the postsynaptic cell, a process required for the membrane GABAAR insertion. Inclusion of botulinum toxin C (BoNT/C, 5 μg/ml) in the patch pipette, which blocks SNARE-dependent exocytosis, prevented LTPGABA (116 ± 8% of baseline, p = 0.2; Fig. 6c). Compared to BAPTA and AIP, however, BoNT/C was less effective in preventing the early phase of LTP (~ 10 min after HFS), suggesting that there are additional mechanisms contributing to the onset of potentiation independent of BoNT/C-sensitive exocytosis. In addition to blocking eIPSC potentiation, the infusion of BAPTA, AIP, or BoNT/C into the postsynaptic cells completely blocked the increase in sIPSC frequency (BAPTA, 98 ± 12% of baseline, p = 0.9; AIP, 102 ± 19%, p = 0.8; BoNT/C, 71 ± 9%, p = 0.02; Fig. 6d), suggesting that the increase in N requires similar postsynaptic mechanisms. Interestingly, BoNT/C, but not BAPTA or AIP, decreased sIPSC frequency to below pre-HFS levels, without altering the amplitude (102 ± 6% of baseline, p = 0.8; Fig. 6e), implying that the blockade of exocytosis unmasked an opposing elimination of functional synapses presumably downstream of Ca2+ and CaMKII. BAPTA and AIP did not change sIPSC amplitude (BAPTA, 96 ± 6% of baseline, p = 0.6; AIP, 107 ± 5% of baseline, p = 0.3; Fig. 6e).
Figure 6. LTPGABA requires postsynaptic mechanisms.

(a–c) Left, average sample traces of eIPSC before (grey) and after (colored) HFS recorded in slices from IMO stressed rats where BAPTA (green, n = 9 (7 rats)), AIP (purple, n = 6 (3 rats)), or BoNT/C (blue, n = 6 (6 rats)) was included in the patch pipette. Without inhibitor group (pink, adopted from experiment shown in Fig. 1d) is shown for comparison. Right, summary time-course of normalized eIPSC amplitude. Arrows indicate the time of HFS. Scale bars represent 50 pA and 20 ms. (d–e) Summary of the effects of HFS on sIPSC frequency (d) and amplitude (e) where BAPTA, AIP or BoNT/C were included in the pitch pipette. Data represent mean ± s.e.m. * p = 0.02, *** p = 0.002.
Increase in functional synapses requires postsynaptic mechanisms
Our data strongly implicate both increase in N and postsynaptic vesicle exocytosis in the potentiation of eIPSCs (i.e. LTPGABA). To directly examine the alterations in the quantal events composing eIPSCs and their causality with postsynaptic exocytosis, we examined the frequency and amplitude of Sr2+-induced asynchronous eIPSCs before and after LTPGABA and their sensitivity to postsynaptic infusion of BoNT/C. Substitution of Ca2+ by equimolar amount of Sr2+ in aCSF alters fast-onset, synchronous synaptic responses into slow-onset asynchronous quantal events. This approach has been used to determine whether synaptic plasticity involves an increase in the number (frequency) and/or size (amplitude) of the quantal events34,35. We first validated that Sr2+ substitution indeed produced asynchronous synaptic events identical to miniature events in GABA synapses onto PNCs (Supplementary Fig. 4). To compare the changes in asynchronous events associated with LTPGABA, we next replaced Ca2+-ACSF with Sr2+-ACSF before and after the induction of LTPGABA in slices from IMO stressed rats. Figure 6a–c shows the expression of LTPGABA similar to that observed in Fig. 1d and asynchronous synaptic events before and after LTPGABA induction. In line with our previous results, we found significant increase in the frequency of asynchronous eIPSCs (152 ± 18% of baseline, p = 0.03, Fig. 7b) with no change in amplitude (106 ± 10%, p = 0.6, Fig. 7c). We next examined the role of exocytosis by repeating the same experiment but with BoNT/C included in the patch pipette. As observed in the previous experiment (Fig. 6c), BoNT/C prevented LTPGABA (Fig. 7d). In line with the blockade of LTPGABA, BoNT/C effectively blocked increase of asynchronous IPSCs frequency (131 ± 17%, p = 0.1, Fig. 7e). The amplitude of asynchronous IPSC was not affected (112 ± 8%, p = 0.2, Fig. 7f). These results provide further support for our hypothesis that N increase contributes to LTPGABA through BoNT/C-sensitive postsynaptic mechanisms.
Figure 7. Increase of active synapse number during LTPGABA requires postsynaptic mechanism.

(a, d) Top, sample traces of asynchronous eIPSC in Sr2+ aCSF before (black) and after (red/blue) LTPGABA from the same neuron recorded in a slice from IMO-stressed rat. Scale bars represent 50 pA and 20 ms. Arrowheads indicate the time of afferent stimulation. Bottom, summary time course of eIPSC, n = 6 (4 rats). Ca2+ was replaced with equimolar Sr2+ during the time window indicated by shadows. Cumulative histograms of interevent interval (b, e) and amplitude (c, f) of the Sr2+-induced asynchronous eIPSCs before (grey) and after LTPGABA (red/blue). Experiments were conducted in the absence (a–c) and presence (d–f) of BoNT/C in the patch pipette, n = 6 (4 rats). (g) Left, raw (thin) and averaged (thick) traces of whole-cell current observed in response to focal pressure application of GABAAR agonist muscimol in slices from IMO-stressed rat. Traces are from the time windows indicated by shadows before (black) and after (red/blue) DHPG treatment. Right, summary time course of normalized muscimol-induced whole-cell current in the presence (blue, n = 6 (4 rats)), or absence n = 7 (5 rats), of BoNT/C in the patch pipette or not (red). Scale bars represent 100 pA and 1 s. Data are mean ± s.e.m.
Although BoNT/C is commonly used to block insertion of GABAARs in postsynaptic membrane30, it can also block the release of vesicularly packaged retrograde signaling molecule(s) which in turn acts on the presynaptic terminals9. To determine specific contribution of postsynaptic GABAAR insertion to LTPGABA, we directly activated postsynaptic GABAARs in slices from stressed rats with GABAAR agonist muscimol and assessed the effects of DHPG (5 μM) application on the whole-cell current. If mGluR1 activation following stress elicits the insertion of GABAARs, we predict that more receptors would be available for activation after exposure to DHPG and the whole-cell current induced by direct GABAAR activation would become larger. As predicted, following DHPG treatment, we observed an increase in the amplitude of whole-cell current in response to muscimol (145 ± 18 %, p = 0.008, Fig. 7g). Inclusion of BoNT/C in the patch pipette abolished this potentiation (99 ± 8 %, p = 0.5). These results argue that SNARE-dependent postsynaptic GABAARs insertion contributes to LTPGABA and that the same mechanism is required for the increase of quantal number during LTPGABA.
Discussion
Our data demonstrate that, immediately after stress, GABA synapses can undergo activity-dependent LTP. This requires (1) noradrenaline as an obligatory instructive signal during stress (2) activation of mGluR1 by heterosynaptically released glutamate (3) postsynaptic insertion of GABAARs. These results further support emerging evidence that the hypothalamic PVN is a critical site for stress memory formation3,5–11 and identify noradrenaline as a key associative signal that allows this stress effector circuit to undergo experience-dependent plasticity.
In addition to directly altering synaptic efficacy, neuromodulators including neurotransmitters19–21, neuropeptides9, steroids8,36 and lipid mediators37 are important for more flexible tuning of neural circuits. Specifically, our data provide evidence for stress-induced noradrenaline regulating metaplasticity of GABA synapses. That is, a history of β-AR activation in PNCs during stress provides a higher-order (meta) control over the ability of the synapses to express plasticity23. Our findings provide, to the best of our knowledge, the first demonstration of β-AR-dependent metaplasticity at GABA synapses, and add to the previous description made at glutamate synapses in the hippocampus and amygdala where β-AR signaling prior to or concomitant with afferent stimulation facilitates the induction of LTP19–21. Mechanistically, we demonstrate that two types of mGluR1 antagonists completely blocked LTPGABA, and that priming by β-AR was necessary and sufficient for both mGluR1 agonist-induced and afferent stimulation (which triggers endogenous glutamate release)-induced LTPGABA. Moreover, β-AR activation needs to precede mGluR1 stimulation. In effect, we propose that β-AR priming involves functional upregulation of mGluR1, whose activation in turn induces LTPGABA. Although the exact mechanisms mediating mGluR1 functional upregulation in PNCs remain unclear, several lines of evidence suggest a key role for PKA. We provide evidence that PKA activation is necessary and sufficient for the priming of LTPGABA. PKA activation in turn facilitates LTPGABA via multiple mechanisms including (1) inhibition of agonist-induced mGluR1 internalization/desensitization27 (2) facilitation of CaMKII activation by inhibiting protein phosphatases that negatively regulate CaMKII24 (3) upregulation of translational process20. Interestingly, glucocorticoids, which function as stress feedback signals to the brain, also unmask mGluR1-36 and mGluR5-dependent (our coordinated submission by Wamsteeker et al.) plasticity following in vivo stress. In these cases, however, the priming of mGluRs was evident 90 min (0 min for the current study) after the termination of stress, suggesting that multiple mechanisms exist for mGluR1/5 modulation by stress that vary in time and context.
Collectively, our data suggest that, following acute stress, mGluR1 activation and subsequent SNARE-dependent exocytosis in the postsynaptic cells lead to LTPGABA that results from an increase in the number of functional GABA synapses. SNARE-dependent exocytosis serves two mutually nonexclusive mechanisms (1) receptor insertion (2) vesicular release of retrograde messengers that in turn may facilitate presynaptic terminals. Our results argue for the substantial contribution of the former mechanism, although do not exclude the recruitment of the former, as mGluR1-induced exocytosis leads to persistent augmentation of the whole-cell postsynaptic currents in response to focal application of muscimol that reflects the amount of total (synaptic and extrasynaptic) surface GABAARs. Interestingly, although postsynaptic infusion of BoNT/C completely abolished the augmentation of muscimol-induced total GABAAR current, it did not effectively block the early phase of LTPGABA. Given that GABAARs in postsynaptic membranes are dynamic, with continuous turnover between synaptic and extrasynaptic loci38,39, our results point to an intriguing possibility that LTPGABA may initially rely on recruitment and clustering of GABAARs from the extrasynaptic pool and that exocytotic GABAAR insertion may be necessary for its stable expression. Although the exact mechanisms await further characterization, our observations collectively suggest that activity-dependent remodeling of GABAARs in postsynaptic membranes plays a deterministic role in controlling the number of functional synapses, and thereby the strength of synaptic inhibition on PNCs. This new form of GABA synapse plasticity is reminiscent of ‘silent synapses’ described at glutamate synapses where NMDAR only (silent) synapses before LTP acquire functional AMPARs following afferent stimulation40. Indeed, recent imaging studies showing that nascent GABAergic synapses are formed with presynaptic changes preceding postsynaptic appearance of GABAAR clusters in hippocampal cultures41,42, support the idea that GABA presynaptic terminals lacking postsynaptic receptor clusters do exist. Intriguingly, a recent study demonstrating that chronic stress causes an anatomical reorganization and increase of GABA synapse contacts onto PNCs11, may hint at the functional end point for the plasticity we described here.
Various types of stress, including IMO, trigger noradrenaline release which in turn increases the firing of PNCs and drives release of CRH15,16. Although previous studies identified the expression and activities of both α1-, α2- and β-ARs in PNCs16,43–45, extensive evidence shows that the noradrenaline stimulation of neuroendocrine response is primarily mediated by α1-ARs15,16,44,46. In line with this, we previously reported that α1-ARs activation causes, through down-regulation of K+ Cl− co-transporter KCC2, a depolarizing shift in EGABA and conditional GABA-mediated excitation of PNCs13. These findings clarify a synaptic mechanism by which α1-ARs activation initiates and sustain neuroendocrine response. The present study, on the other hand, reveals a previously unknown consequence of noradrenaline action in the PVN where a history of β-AR activation primes GABA synapses for subsequent LTP. Because GABA becomes excitatory immediately after stress onset, we speculate that this priming mechanism of LTPGABA may contribute to stress-induced sensitization of the HPA axis. This priming for GABA potentiation, however, is likely to have a “refractory period” when an increased levels of glucocorticoids following stress signal back to the PVN. Our accompanying paper demonstrates that in response to glucocorticoid feedback signaling, the same GABA synapses can be depressed through retrograde opioid signals that decrease the presynaptic release of the transmitter following pro-longed afferent activation. These two opposing mechanisms likely work in a coordinated manner, during temporally distinct windows, to prevent excessive activation of stress axis.
More generally our data are consistent with extensive behavioral evidence that noradrenergic activities, in particular through β-ARs, have been implicated in learning and memory formation associated with emotional arousal and stress challenges18. Importantly, noradrenaline release in the PVN is not universal but varies as a function of stressor modality15,47. Consistent with this idea, we observed stress-specificity with respect to priming of LTPGABA. Thus, this type of plasticity would be particularly useful for sculpting neuroendocrine output based on the modality/intensity of prior stress experience. It is intriguing that β-AR has been proposed as a potential target for stress-related disorders such as posttraumatic stress disorders48. Understanding the neurobiological mechanisms of long-term changes in stress-relevant neurocircuitry will provide potential treatment targets for mental health disorders.
Methods
Animal Handling and Stress Procedure
All experiments were approved by the University of Calgary Animal Care and Use Committee in accordance with Canadian Council on Animal Care guidelines. Male Sprague-Dawley rats (postnatal day 22–35, Charles Rivers) were group-housed on a 12 h:12 h light:dark cycle (lights on 06:00) and had free access to food and water. Animals were randomly assigned to naïve or stress groups. For IMO stress procedure, rats were confined and cervically immobilized in a plexiglass restrainer for 30 min. In some experiments, rats were injected with (±)-propranolol (PRO; 10 mg kg−1, intraperitoneal, Sigma-Aldrich) or CP-154,526 (30 mg kg−1, intraperitoneal, Tocris Bioscience) dissolved in saline 30 min before IMO. For predator odor stress, rats were exposed to 2,5-diydro-2,4,5-trimethyulthiazoline (TMT, Contech), a compound isolated from fox feces. A single rat was placed in a testing cage and exposed to TMT (150 μl, absorbed in Kimwipe in a scintillation vial) placed in one corner of the cage for 30 min. For forced swim stress, a single rat was placed in a plastic cylinder (40 cm diameter) filled with water (> 30 cm depth, 30°C) for 20 min. All stress procedures were performed between 10:00 and 12:00. Immediately after stress, rats were anesthetized and brain slices were prepared as described below.
Slice Preparation and Electrophysiology
Animals were anesthetised with isoflurane and decapitated. The brain was quickly removed; it was submerged and coronally sectioned on a vibratome (Leica, Nussloch, Germany) to 300 μm in slicing solution (0°C, 95% O2/5% CO2 saturated) containing (in mM): 87 NaCl, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 25 NaHCO3, 25 D-glucose, 1.25 NaH2PO4, 75 sucrose. After placement into aCSF (30°C, 95% O2/5% CO2 saturated) containing (in mM):126 NaCl, 2.5 KCl, 26 NaHCO3, 2.5 CaCl2, 1.5 MgCl2, 1.25 NaH2PO4, 10 glucose, hypothalamic slices recovered for at least 1 h. Once transferred to a recording chamber superfused with aCSF (1 ml/min; 30–32°C; 95% O2/5% CO2), slices were visualized using an upright microscope (BX51WI, Olympus) fitted with infrared differential interference contrast optics. Pulled borosilicate glass pipettes (3–6 M3) were filled with a solution containing (in mM) 108 K-gluconate, 2 MgCl2, 8 Na-gluconate, 8 KCl, 1 K2-EGTA, 4 K2-ATP, 0.3 Na3-GTP, and 10 HEPES. In some experiments, BAPTA (10 mM, Sigma-Aldrich), AIP (1 μM, Tocris Bioscience) or the light chain BoNT/C (the proteinase subunit which lacks membrane permeability, 5 μg/ml, List Biological Laboratories) were included in the internal solution. For Sr2+ experiment, recording chamber was superfused with regular aCSF or Sr2+-ACSF (in which Ca2+ was replaced with equimolar concentration of Sr2+) at a flow rate of 2 ml/min to achieve fast buffer exchange.
Whole-cell patch-clamp recordings were performed from PNCs identified by location, morphology, and current clamp fingerprint49. PNCs were voltage-clamped at −80 mV with constant perfusion of 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μM, Tocris Bioscience); other drugs were bath applied by perfusion pump. Pairs of GABAAR-mediated IPSCs were evoked 50 ms apart at 0.2 Hz intervals using a monopolar aCSF-filled glass electrode placed in surrounding neuropil. For HFS, afferents were stimulated at 100 Hz for 1 s (or for 0.5 or 4 s where indicated), repeated four times with an interval of 20 s while postsynaptic cells were held at −80 mV in voltage clamp configuration. In some experiment, lower frequency stimulation (10 Hz for 10 s) was also used. In the experiments where HFS was delivered after DHPG application, half of the recordings (three out of six cells) were obtained in slices pre-treated with DHPG (followed by > 30 min wash) without recording to confirm that the lack of HFS-induced LTPGABA is not due to intracellular dialysis by long (> 45 min) whole cell recording prior to HFS. Another three recordings were obtained from single cells treated with DHPG and then HFS as shown in (Fig. 2c). To measure the changes in postsynaptic GABAAR surface expression, we monitored inward current induced by focal application of GABAAR agonist. Muscimol (100 μM dissolved in aCSF) was pressure applied from a glass pipette whose tip was placed 20–30 μm away from the soma of the postsynaptic cell. Three applications (10 ms, 4 s apart) was delivered every 1 min by using a Picospritzer III (Science Products). The responses to the three successive puffs were averaged for each epoch. Access resistance was continuously monitored; recordings in which values exceeded 20 M3 or 15% change were excluded from analysis. DL-isoproterenol was obtained from Sigma-Aldrich. 8-Br-cAMP, KT5720, (s)-MCPG, CPCCOEt, JNJ16259685, MTEP, bicuculline, (s)-DHPG and muscimol were purchased from Tocris Bioscience.
Optogenetics
Tyrosine hydroxylase (TH)-Cre transgenic mice22 were obtained from the Jackson Laboratory (B6.Cg-Tg(Th-cre)1Tmd/J, Stock Number:008601), and were maintained on a C57BL/6J background. Cre transgenes were genotyped with standard PCR protocols using primers 5′-GCG GTC TGG CAG TAA AAA CTA TC-3′ and 5′-GTG AAA CAG CAT TGC TGT CAC TT-3′ to produce a 100 bp transgenic fragment. Mice were single housed in a cage on a 12 h:12 h light:dark cycle (lights on 06:00) and had free access to food and water. 7 week old mice were anaesthetized using isofluorane (5% at induction, maintained with 1–2% during surgery) with a nose cone, and were given buprenorphine (0.1 mg kg−1) for post-surgery analgesia. The heads of the mice were positioned in a stereotaxic apparatus with bregma and lambda in the horizontal plane. Through a burr hole in the skull, glass capillaries were lowered into the brain at stereotaxic coordinates corresponding to the nucleus of the solitary tract (NTS). Recombinant AAV carrying ChR2-eYFP (Addgene plasmid 20298, pAAV-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-HGHpA)50 and ChR2-mCherry (Addgene plasmid 20297, pAAV-EF1a-double floxed-hChR2(H134R)-mCherry-WPRE-HGHpA)50 was pressure injected with Nanoject II apparatus (Drummond Scientific Company) in a total volume of 350 nl (3.4 × 1013 GC ml−1). Mice were then allowed to recover > 14 days before ex vivo optogenetic/electrophysiological experiments. The age of mice at the time of electrophysiological experiments was 9–11 weeks old. Both male and females were used. Coronal brainstem and PVN slices (250 μm) were prepared using a vibratome as described above, and transduced neurons (in the caudal medulla) and fibres (in the PVN) were identified by the expression of fluorescent proteins. Photostimulation experiments were conducted in PVN slices from both male and female mice. In a preliminary experiment we did not observe any gender specificity in the expression of LTPGABA. The fiber optic cable (105 μm core diameter) was placed 1–2 mm above the slice using a manipulator, and a blue-light laser (473 nm, OptoGeni 473, IkeCool corporation) was controlled via Digidata 1440A (Molecular Devices). Light intensity on tissue was 2.5 mW as measured by Photodiode Power Sensor (Thorlabs).
Analysis and Statistics
Signals were amplified (Multiclamp 700B, Molecular Devices), low pass filtered at 1 kHz, digitized at 10 kHz (Digidata 1440, Molecular Devices), and recorded (pClamp 10.1, Molecular Devices) for offline analysis. Evoked IPSCs (eIPSC) were calculated by subtraction of peak synaptic current from pre-stimulation baseline current. Spontaneous IPSC (sIPSC) events, with eIPSCs and stimulus artifacts removed, were detected using variable thresholds and confirmed by eye (MiniAnalysis, Synaptosoft). Data were normalized and expressed as percentage of the baseline values (0–5 min before treatment) within each cell. Post-treatment values were evaluated 15–20 min after treatment unless otherwise specified. Paired-pulse ratio (2nd evoke/1st evoke), sIPSC event frequency/amplitude, and coefficient of variation (CV, standard deviation/mean) were analyzed with 5 minute intervals. Asynchronous IPSCs in Sr2+-aCSF were evoked by a single pulse delivered at 0.2 Hz intervals. Amplitudes and frequencies of the asynchronous eIPSCs were measured during a 150 ms period, starting 5 ms after stimulus to exclude the initial synchronous synaptic events. Results were presented as mean ± s.e.m. To ensure physiological significance, recordings for each group were obtained from minimum of three animals. 4 slices were prepared from one animal and the slices were randomly assigned to drug/no-drug treatments. Experimenters were not blinded to the treatment. Sample sizes were determined post-hoc based on those used in previous studies8,9,13. Because the effects of HFS or drugs were compared to the baseline within cells, one sample parametric t-test (two tailed) was used for all experiment, except for one experiment (TH-cre light stimulation, Fig. 2b) when sample distribution was not normal (used Wilcoxon ranked test). Gaussian distribution of the data was examined by a D’Agostino & Pearson omnibus normality test (GraphPad Prism 4). In one experiment (Fig. 2b, TH-cre light vs. TH-cre light + PRO), the values between groups were compared using non-parametric Mann Whitney test. Correlation between two variables was analyzed using linear regression analysis. p < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank members of Bains laboratory for comments and discussion regarding the manuscript and Cheryl Sank for technical assistance. W.I. and T.F are supported by a postdoctoral fellowship, and J.I.W and K.K by a PhD scholarship from the Alberta Innovates for Health Solution (AI-HS). W.I., K.K. and J.I.W. also received fellowship/scholarship support from the Hotchkiss Brain Institute. J.S.B and P.J.W are AI-HS Senior Scholars. Q.J.P. is an AI-HS Scientist. This work was funded by operating grants from Canadian Institute for Health Research (J.S.B., P.J.W. and Q.J.P). We are grateful to the Hotchkiss Brain Institute for providing optogenetics core resources. AAV vectors carrying the ChR2 gene were developed by Dr. Karl Deisseroth and distributed by Addgene.
Footnotes
Author Contributions
W.I. designed and conducted the experiments, analyzed the data, prepared and wrote the manuscript. D.V.B., T.F. and J.I.W. designed and conducted the experiments and analyzed the data. T.F., K.K. and P.J.W. developed AAV-ChR2/TH-Cre mouse tools. Q.J.P. supervised the project and edited the manuscript. J.S.B. designed the experiments, prepared the manuscript and supervised the project.
References
- 1.Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cullinan WE, Ziegler DR, Herman JP. Functional role of local GABAergic influences on the HPA axis. Brain Struct Funct. 2008;213:63–72. doi: 10.1007/s00429-008-0192-2. [DOI] [PubMed] [Google Scholar]
- 4.Armario A, Vallès A, Dal-Zotto S, Márquez C, Belda X. A single exposure to severe stressors causes long-term desensitisation of the physiological response to the homotypic stressor. Stress. 2004;7:157–172. doi: 10.1080/10253890400010721. [DOI] [PubMed] [Google Scholar]
- 5.Flak JN, Ostrander MM, Tasker JG, Herman JP. Chronic stress-induced neurotransmitter plasticity in the PVN. J Comp Neurol. 2009;517:156–165. doi: 10.1002/cne.22142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verkuyl JM, Hemby SE, Joëls M. Chronic stress attenuates GABAergic inhibition and alters gene expression of parvocellular neurons in rat hypothalamus. Eur J Neurosci. 2004;20:1665–1673. doi: 10.1111/j.1460-9568.2004.03568.x. [DOI] [PubMed] [Google Scholar]
- 7.Verkuyl JM, Karst H, Joëls M. GABAergic transmission in the rat paraventricular nucleus of the hypothalamus is suppressed by corticosterone and stress. Eur J Neurosci. 2005;21:113–121. doi: 10.1111/j.1460-9568.2004.03846.x. [DOI] [PubMed] [Google Scholar]
- 8.Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30:11188–11196. doi: 10.1523/JNEUROSCI.1046-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuzmiski JB, Marty V, Baimoukhametova DV, Bains JS. Stress-induced priming of glutamate synapses unmasks associative short-term plasticity. Nat Neurosci. 2010;13:1257–1264. doi: 10.1038/nn.2629. [DOI] [PubMed] [Google Scholar]
- 10.Miklós IH, Kovács KJ. GABAergic innervation of corticotropin-releasing hormone (CRH)-secreting parvocellular neurons and its plasticity as demonstrated by quantitative immunoelectron microscopy. Neuroscience. 2002;113:581–592. doi: 10.1016/s0306-4522(02)00147-1. [DOI] [PubMed] [Google Scholar]
- 11.Miklós IH, Kovács KJ. Reorganization of synaptic inputs to the hypothalamic paraventricular nucleus during chronic psychogenic stress in rats. Biol Psychiatry. 2012;71:301–308. doi: 10.1016/j.biopsych.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar J, Wakefield S, MacKenzie G, Moss SJ, Maguire J. Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J Neurosci. 2011;31:18198–18210. doi: 10.1523/JNEUROSCI.2560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- 14.Sawchenko PE, Swanson LW. The organization of noradrenergic pathways from the brainstem to the paraventricular and supraoptic nuclei in the rat. Brain Res. 1982;257:275–325. doi: 10.1016/0165-0173(82)90010-8. [DOI] [PubMed] [Google Scholar]
- 15.Pacák K, Palkovits M. Stressor Specificity of Central Neuroendocrine Responses: Implications for Stress-Related Disorders. Endocr Rev. 2001;22:502–548. doi: 10.1210/edrv.22.4.0436. [DOI] [PubMed] [Google Scholar]
- 16.Saphier D, Feldman S. Catecholaminergic projections to tuberoinfundibular neurones of the paraventricular nucleus: III. Effects of adrenoceptor agonists and antagonists. Brain Res Bull. 1991;26:863–870. doi: 10.1016/0361-9230(91)90250-n. [DOI] [PubMed] [Google Scholar]
- 17.Seidenbecher T, Reymann KG, Balschun D. A post-tetanic time window for the reinforcement of long-term potentiation by appetitive and aversive stimuli. Proc Natl Acad Sci USA. 1997;94:1494–1499. doi: 10.1073/pnas.94.4.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berridge CW, Waterhouse BD. The locus coeruleus–noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- 19.Huang YY, Martin KC, Kandel ER. Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci. 2000;20:6317–6325. doi: 10.1523/JNEUROSCI.20-17-06317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tenorio G, et al. ‘Silent’ priming of translation-dependent LTP by β-adrenergic receptors involves phosphorylation and recruitment of AMPA receptors. Learn Mem. 2010;17:627–638. doi: 10.1101/lm.1974510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas MJ, Moody TD, Makhinson M, O’Dell TJ. Activity-Dependent β-Adrenergic Modulation of Low Frequency Stimulation Induced LTP in the Hippocampal CA1 Region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- 22.Savitt JM, Jang SS, Mu W, Dawson VL, Dawson TM. Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci. 2005;25:6721–6728. doi: 10.1523/JNEUROSCI.0760-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- 24.Sugiyama Y, Kawaguchi S, Hirano T. mGluR1-mediated facilitation of long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. Eur J Neurosci. 2008;27:884–896. doi: 10.1111/j.1460-9568.2008.06063.x. [DOI] [PubMed] [Google Scholar]
- 25.Patenaude C, Chapman CA, Bertrand S, Congar P, Lacaille JC. GABAB receptor- and metabotropic glutamate receptor-dependent cooperative long-term potentiation of rat hippocampal GABAA synaptic transmission. J Physiol (Lond) 2003;553:155–167. doi: 10.1113/jphysiol.2003.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kocsis K, Kiss J, Görcs T, Halász B. Metabotropic glutamate receptor in vasopressin, CRF and VIP hypothalamic neurones. Neuroreport. 1998;9:4029–4033. doi: 10.1097/00001756-199812210-00008. [DOI] [PubMed] [Google Scholar]
- 27.Mundell SJ, et al. Activation of cyclic AMP-dependent protein kinase inhibits the desensitization and internalization of metabotropic glutamate receptors 1a and 1b. Mol Pharmacol. 2004;65:1507–1516. doi: 10.1124/mol.65.6.1507. [DOI] [PubMed] [Google Scholar]
- 28.Korn H, Faber DS. Quantal analysis and synaptic efficacy in the CNS. Trends Neurosci. 1991;14:439–445. doi: 10.1016/0166-2236(91)90042-s. [DOI] [PubMed] [Google Scholar]
- 29.Kano M, Kano M, Fukunaga K, Konnerth A. Ca(2+)-induced rebound potentiation of gamma-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proc Natl Acad Sci USA. 1996;93:13351–13356. doi: 10.1073/pnas.93.23.13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsden KC, Beattie JB, Friedenthal J, Carroll RC. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABA(A) receptors. J Neurosci. 2007;27:14326–14337. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ouardouz M, Sastry BR. Mechanisms underlying LTP of inhibitory synaptic transmission in the deep cerebellar nuclei. J Neurophysiol. 2000;84:1414–1421. doi: 10.1152/jn.2000.84.3.1414. [DOI] [PubMed] [Google Scholar]
- 32.Marsden KC, Shemesh A, Bayer KU, Carroll RC. Selective translocation of Ca2+/calmodulin protein kinase IIalpha (CaMKIIalpha) to inhibitory synapses. Proc Natl Acad Sci USA. 2010;107:20559–20564. doi: 10.1073/pnas.1010346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon MS, et al. The effect of single or repeated restraint stress on several signal molecules in paraventricular nucleus, arcuate nucleus and locus coeruleus. Neuroscience. 2006;142:1281–1292. doi: 10.1016/j.neuroscience.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 34.Oliet SH, Malenka RC, Nicoll RA. Bidirectional control of quantal size by synaptic activity in the hippocampus. Science. 1996;271:1294–1297. doi: 10.1126/science.271.5253.1294. [DOI] [PubMed] [Google Scholar]
- 35.Zhao JP, Phillips MA, Constantine-Paton M. Long-term potentiation in the juvenile superior colliculus requires simultaneous activation of NMDA receptors and L-type Ca2+ channels and reflects addition of newly functional synapses. J Neurosci. 2006;26:12647–12655. doi: 10.1523/JNEUROSCI.3678-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaouloff F, Hémar A, Manzoni O. Acute stress facilitates hippocampal CA1 metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2007;27:7130–7135. doi: 10.1523/JNEUROSCI.1150-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crosby KM, Inoue W, Pittman QJ, Bains JS. Endocannabinoids gate state-dependent plasticity of synaptic inhibition in feeding circuits. Neuron. 2011;71:529–541. doi: 10.1016/j.neuron.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vithlani M, Terunuma M, Moss SJ. The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol Rev. 2011;91:1009–1022. doi: 10.1152/physrev.00015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tyagarajan SK, et al. Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc Natl Acad Sci USA. 2011;108:379–384. doi: 10.1073/pnas.1011824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- 41.Dobie FA, Craig AM. Inhibitory synapse dynamics: coordinated presynaptic and postsynaptic mobility and the major contribution of recycled vesicles to new synapse formation. J Neurosci. 2011;31:10481–10493. doi: 10.1523/JNEUROSCI.6023-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wierenga CJ, Becker N, Bonhoeffer T. GABAergic synapses are formed without the involvement of dendritic protrusions. Nat Neurosci. 2008;11:1044–1052. doi: 10.1038/nn.2180. [DOI] [PubMed] [Google Scholar]
- 43.Day HE, Campeau S, Watson SJ, Jr, Akil H. Expression of alpha(1b) adrenoceptor mRNA in corticotropin-releasing hormone-containing cells of the rat hypothalamus and its regulation by corticosterone. J Neurosci. 1999;19:10098–10106. doi: 10.1523/JNEUROSCI.19-22-10098.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daftary SS, Boudaba C, Tasker JG. Noradrenergic regulation of parvocellular neurons in the rat hypothalamic paraventricular nucleus. Neuroscience. 2000;96:743–751. doi: 10.1016/s0306-4522(00)00003-8. [DOI] [PubMed] [Google Scholar]
- 45.Takano T, et al. Beta-adrenergic receptors in the vasopressin-containing neurons in the paraventricular and supraoptic nucleus of the rat. Brain Res. 1989;499:174–178. doi: 10.1016/0006-8993(89)91149-9. [DOI] [PubMed] [Google Scholar]
- 46.Itoi K, et al. Microinjection of norepinephrine into the paraventricular nucleus of the hypothalamus stimulates corticotropin-releasing factor gene expression in conscious rats. Endocrinology. 1994;135:2177–2182. doi: 10.1210/endo.135.5.7956940. [DOI] [PubMed] [Google Scholar]
- 47.Pacák K, et al. Effects of various stressors on in vivo norepinephrine release in the hypothalamic paraventricular nucleus and on the pituitary-adrenocortical axis. Ann N Y Acad Sci. 1995;771:115–130. doi: 10.1111/j.1749-6632.1995.tb44675.x. [DOI] [PubMed] [Google Scholar]
- 48.Strawn JR, Geracioti TD., Jr Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depress Anxiety. 2008;25:260–271. doi: 10.1002/da.20292. [DOI] [PubMed] [Google Scholar]
- 49.Luther JA, et al. Neurosecretory and non-neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J Neuroendocrinol. 2002;14:929–932. doi: 10.1046/j.1365-2826.2002.00867.x. [DOI] [PubMed] [Google Scholar]
- 50.Zhang F, et al. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc. 2010;5:439–456. doi: 10.1038/nprot.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.