

Abstract
Wip1 (protein phosphatase Mg2+/Mn2+-dependent 1D, Ppm1d) is a nuclear serine/threonine protein phosphatase that is induced by p53 following the activation of DNA damage response (DDR) signaling. Ppm1d−/− mouse embryonic fibroblasts (MEFs) exhibit premature senescence under conventional culture conditions; however, little is known regarding the role of Wip1 in regulating cellular senescence. In this study, we found that even at a representative physiological concentration of 3% O2, Ppm1d−/− MEFs underwent premature cellular senescence that depended on the functional activation of p53. Interestingly, Ppm1d−/− MEFs showed increased H2AX phosphorylation levels without increased levels of reactive oxygen species (ROS) or DNA base damage compared with wild-type (Wt) MEFs, suggesting a decreased threshold for DDR activation or sustained DDR activation during recovery. Notably, the increased H2AX phosphorylation levels observed in Ppm1d−/− MEFs were primarily associated with S-phase cells and predominantly dependent on the activation of ATM. Moreover, these same phenotypes were observed when Wt and Ppm1d−/− MEFs were either transiently or chronically exposed to low levels of agents that induce replication-mediated double-stranded breaks. These findings suggest that Wip1 prevents the induction of cellular senescence at physiological oxygen levels by attenuating DDR signaling in response to endogenous double-stranded breaks that form during DNA replication.
Keywords: Wip1, p53, cellular senescence, DNA damage response, ATM, camptothecin
Introduction
In adult organisms, the capacity for tissue self-renewal is balanced against suppression of unregulated cellular proliferation in order to maximize fitness and reproductive success. In proliferating tissues, the progress of cells through the cell cycle is subject to an interrelated series of highly conserved checkpoint processes that guard against proliferation of cells with damaged genetic material. The checkpoint processes reduce the incidence of cancers, but do so at the cost of reduced cell numbers, such as through apoptosis, and reduced proliferation through temporary or permanent growth arrest. Cellular DNA is continuously exposed to environmental and endogenous mutagenic insults. In humans and in mice, DNA damage response (DDR) signaling is initiated by sensor kinases of the phosphatidylinositol 3-kinase-related kinase (PIKK) family, including ataxia telangiectasia-mutated (ATM), ataxia telangiectasia and Rad3-related (ATR), and DNA-dependent protein kinase (DNA-PK).1,2 These kinases phosphorylate multiple downstream target proteins and lead to checkpoint responses through the activation of p53.2 An important function of DDR signaling is to delay cell cycle progression until DNA damage is repaired. Upon successful repair of the damage, activated DDR signaling processes must be decommissioned to allow the resumption of normal cell cycle progression. Severe DNA damage that cannot be repaired successfully causes sustained activation of DDR signaling, leading to either apoptosis or the senescent state of permanent growth arrest and, consequently, a reduction in proliferative potential.
Wild-type p53-induced phosphatase 1 (Wip1) is a nuclear serine/threonine protein phosphatase encoded by the protein phosphatase Mg2+/Mn2+-dependent 1D (Ppm1d) gene and was originally identified as a gene induced in a p53-dependent manner following exposure to a genotoxic stress.3 Upon induction, Wip1 directly binds to and dephosphorylates several key protein targets associated with ATM-initiated DDR signaling, including ATM itself (Ser1981), p53 (Ser15), and the phosphorylated form of the histone 2A variant H2AX (γH2AX) (Ser139).4-8 Phosphorylation of H2AX occurs early in the cellular response to DNA damage and is important for amplification of DDR signaling through recruitment of DNA repair factors.9 Thus, Wip1 promotes proliferation by dampening DDR signaling and facilitating the return to homeostasis once the damage is repaired.
Consistent with its role in attenuating DDR signaling, Wip1 has been shown to cooperate with oncogenes in promoting tumorigenesis in several mouse models of cancer and, moreover, has been shown to be amplified or overexpressed in several types of human primary cancers.10-14 Previous studies demonstrated that the DDR signaling plays a crucial role as an anti-cancer barrier.15,16 Indeed, the activated DDR signaling observed in pre-neoplastic lesions of several human cancers is often lost in late-stage tumor tissues, indicating that the disabling of DDR signaling is a crucial prerequisite for cancer progression.15,16 In Wip1-overexpressing tumors, the resulting downregulation of DDR signaling and functional inactivation of several tumor suppressors accelerates tumorigenesis and suggests a role for Wip1 in supporting proliferation. Interestingly, while Wip1 knockout (Ppm1d−/−) mice are resistant to tumorigenesis,17,18 they exhibit senescent phenotypes such as reduced longevity, reduced fertility, and a progeroid appearance.18,19 Mouse embryonic fibroblasts (MEFs) derived from Ppm1d−/− mice exhibit markedly reduced proliferation rates in culture and rapidly develop the flattened and enlarged morphology characteristic of senescent cells.19
Cellular senescence is a metabolically active state of irreversible growth arrest that can develop in normal cells in response to many types of stress.20 Whereas replicative senescence is often used to describe the cellular response to critical shortening of telomeres, premature senescence is usually applied to the phenotypically identical state induced by stresses including expression of activated oncogenes, chronic oxidative stress, or unrepaired DNA damage.21-23 In human cells, the tumor suppressors p53 and pRb have been shown to play critical roles in the induction of cellular senescence.21 Premature senescence in MEFs relies predominantly on the p19Arf and p53 pathways.21-23 Indeed, the functional activation of p53 and increased p21 protein levels in Ppm1d−/− MEFs have been reported previously.19 However, little is known about how the loss of Wip1 induces premature cellular senescence in Ppm1d−/− MEFs. Interestingly, murine cells are more sensitive to oxidative stress than human cells,24 and the cultivation of mouse cells in atmospheric levels of oxygen leads to oxidative stress-induced activation of DDR signaling.25 Therefore, to investigate the role of Wip1 in forestalling the onset of premature senescence, we performed a detailed analysis of Ppm1d-deficient (Ppm1d−/−) MEFs cultured in a representative physiological oxygen level. We found, interestingly, that even when cells are cultured in 3% oxygen, Wip1 is necessary to prevent S phase-specific activation of ATM and DDR signaling that consequentially activates p53 and accelerates the onset of cellular senescence.
Results
Suppression of cellular senescence by Wip1 under atmospheric and physiological oxygen levels
Under conventional culture conditions (20% O2), Ppm1d−/− MEFs initially proliferated similarly to wild-type (Wt) MEFs, but showed markedly reduced proliferation after only a few days in culture (Fig. 1A), in agreement with previous observations.19 The general phenomenon of decreasing proliferation rates exhibited by Wt MEFs after extended culture conditions under atmospheric oxygen levels is considered to result from the high sensitivity of murine cells to oxidative damage in DNA.24 Cells in an organism are adapted to specific oxygen concentrations ranging from less than 1% to over 10%, dependent on the vascularization of the tissue.24,26 When cultured under a representative physiological condition of 3% oxygen, Wt MEFs proliferated steadily, but Ppm1d−/− MEFs still exhibited declining rates of growth. The reduced proliferation of Ppm1d−/− MEFs did not result from increased apoptosis, as staining with annexin-V and propidium iodide (PI) revealed no significant differences in the percentage of apoptotic cells between Wt and Ppm1d−/− MEFs cultured under either condition (Fig. S1). Cell cycle distributions were analyzed by bromodeoxyuridine (BrdU) incorporation combined with DNA-content analysis. Under both 20% and 3% oxygen levels, Ppm1d−/− MEFs showed reduced percentages of BrdU-positive cells as compared with the corresponding Wt MEFs (Fig. 1B). Interestingly, the percentages of cells in S phase were similar in Wt MEFs cultured in 20% O2 and in Ppm1d−/− MEFs cultured in 3% O2, consistent with the observed growth trajectories (Fig. 1A). For cells cultured in 20% O2, the percentage of G2/M phase cells was increased in Ppm1d−/− compared with Wt MEFs (Fig. 1B), in agreement with previous results.19 When cultured in 3% O2, Ppm1d−/− MEFs exhibited increased numbers of G0/G1 phase cells as compared with Wt MEFs (Fig. 1B), suggesting a block in entering S phase. In addition, under both culture conditions, Ppm1d−/− MEFs exhibited increased numbers of enlarged and flattened cells (Fig. 1C).20 The prevalence of senescence-associated β-galactosidase (SA-β-Gal)-positive cells was assessed quantitatively by flow cytometry.27,28 For both culture conditions, the proportion of SA-β-Gal-positive cells differed significantly between Wt and Ppm1d−/− MEFs. For cells grown in 20% O2, 32% ± 3% of Wt MEFs and 45% ± 5% of Ppm1d−/− MEFs were SA-β-Gal-positive (P < 0.05), whereas for cells grown in 3% O2, 19% ± 2% of Wt MEFs and 27% ± 4% of Ppm1d−/− MEFs were SA-β-Gal-positive (P < 0.05) (Fig. 1D). Thus, deletion of Ppm1d accelerated the onset of premature senescence, both when cultured in 20% O2, as reported previously,19 and when cultured under a more physiological oxygen level.

Figure 1. Premature cellular senescence associated with Ppm1d deficiency was observed in both 3% and 20% O2 conditions. (A) Growth curves of 3 Wt (solid line) and 3 Ppm1d−/− MEFs (dashed line) cultures in 3% (black) and 20% (gray) O2 were determined. Each culture was passed at 5 × 105 cells per 75 cm2 flask every 3 d. Cell numbers were determined, and cumulative population doubling levels were calculated at each passage. The averages of 3 independent cultures with SD are shown. (B) Cell cycle distributions of Wt and Ppm1d−/− MEFs at passage 2 as measured by BrdU incorporation and DNA content (PI) flow cytometry analysis. The upper gate shows the cells incorporating BrdU (S-phase), the lower-left gate shows G0/G1 cells, and the lower right indicates G2/M cells. The percentages of cells in each gate are indicated in the upper right of each diagram. The data are representative of 2 independent experiments. (C) Cell morphology at passage 3. Photographs are at the same magnification (× 100). (D) Flow cytometric detection of SA-β-Gal activity in Wt and Ppm1d−/− MEFs at passage 3. The upper right quadrant gate identifies cells with bright fluorescence for C12FDG, indicating SA-β-Gal-positive cells. The percentage of SA-β-Gal-positive cells is shown in the upper right of each diagram. The left diagram (Wt MEFs, passage 0) was used as a negative control. The data are representative of 3 independent experiments.
The involvement of p53 in premature cellular senescence elicited by Ppm1d deficiency
Premature senescence of MEFs relies principally on the p19Arf and p53 pathways.21-23 To investigate the involvement of these signaling pathways, we examined protein and phospho-protein levels by immunoblotting protein extracts from Wt and Ppm1d−/− MEFs cultured in 3% and 20% O2. The results showed no significant differences in total p53 protein levels in Ppm1d−/− compared with Wt MEFs in either 3% or 20% O2 conditions (Fig. 2A), consistent with an earlier report for MEFs grown in 20% oxygen.19 Although in some cells Wip1 has been shown to increase the stability and activity of MDM2, resulting in reduced p53 stability,29 this effect was not observed in our system. Levels of serine 15 phosphorylation of p53 (pSer15 p53), which serves as a marker of activated p53,30-32 were increased in Ppm1d−/− MEFs in both 3% and 20% O2 conditions (Fig. 2A). Furthermore, the levels of p21, a transcriptional target of p53 implicated in cellular senescence,33,34 were also higher in Ppm1d−/− MEFs as compared with Wt MEFs under both conditions (Fig. 2A). Moreover, the fact that p21 protein expression was not observed in Trp53−/− MEFs (Fig. 2A) indicates that the observed increased levels of p21 were dependent on p53. Thus, p53 protein in Ppm1d−/− MEFs exhibits increased functional activity compared with that of Wt MEFs in both 3% and 20% O2 conditions.

Figure 2. Premature cellular senescence of Ppm1d−/− MEFs is dependent on p53 activation. (A) Immunoblot analysis of Wip1, p53, p-p53 (pSer15), p21, p38 MAPK, p-p38 MAPK (pThr180/pTyr182), p16, p19, pRb, PCNA, and β-actin in Wt and Ppm1d−/− MEFs at passage 3. Trp53−/−, 5 Gy IR exposed Wt, and 2.5 mM H2O2 treated Wt MEFs were used as controls. PCNA and β-actin were used as proliferation and loading controls, respectively. (B) Ppm1d+/+Trp53+/+, Ppm1d−/−Trp53+/+, Ppm1d+/+Trp53−/− and Ppm1d−/−Trp53−/− MEFs were generated from the pups derived from matings of Ppm1d+/−Trp53+/− mice. Cultures of each strain were passed at 5 × 105 cells per 75 cm2 flask every 3 d in 20% O2 conditions. Cell numbers were determined and cumulative population doubling levels were calculated at each passage. The averages of 3 independent cultures with SD are shown. (C) Flow cytometric detection of SA-β-Gal activity in Ppm1d+/+Trp53+/+, Ppm1d−/−Trp53+/+, Ppm1d+/+Trp53−/−, and Ppm1d−/−Trp53−/− MEFs at passage 3 cultured in 20% oxygen. Experiments were done by the same method as shown in Figure 1D. The data are representative of 3 independent experiments.
In the presence of activated oncogenes, deficiency of Ppm1d resulted in the rapid establishment of p53-dependent premature senescence through increased activation of p38 MAPK, as indicated by increased levels of phosphorylated kinase (pp38).17 In contrast, we observed that in early passage, nontransformed MEFs, the levels of p38 MAPK and pp38 did not differ between Wt and Ppm1d−/− MEFs cultured under either 3% or 20% O2 (Fig. 2A). Interestingly, the levels of p16Ink4a and p19Arf were increased in Ppm1d−/− MEFs compared with Wt MEFs for both culture conditions.
To further investigate the involvement of p53 in the onset of cellular senescence, we generated MEFs with genetic alterations in both Ppm1d and Trp53 genes by crossing Ppm1d+/−Trp53+/− mice. We analyzed cell proliferation and SA-β-Gal activity in cultures of Ppm1d+/+Trp53+/+, Ppm1d−/−Trp53+/+, Ppm1d+/+Trp53−/−, and Ppm1d−/−Trp53−/− MEFs. Similar to previous findings,35 Ppm1d+/+Trp53−/− MEFs proliferated vigorously (Fig. 2B) and showed fewer SA-β-Gal-positive cells (Fig. 2C) than Ppm1d+/+Trp53+/+ MEFs. Specifically, 38% ± 1% of Ppm1d+/+Trp53+/+ MEFs and 7.3% ± 1.1% of Ppm1d+/+Trp53−/− MEFs were SA-β-Gal-positive (P < 0.01). Notably, Ppm1d−/−Trp53−/− MEFs grew similarly to Ppm1d+/+Trp53−/− MEFs (Fig. 2B), in agreement with previous results.17 The proportion of SA-β-Gal-positive cells in Ppm1d−/−Trp53−/− MEFs, 6.9% ± 0.3%, (Fig. 2C) did not differ significantly from that in Ppm1d+/+Trp53−/− MEFs. As expected, Ppm1d−/−Trp53+/+ MEFs showed a profoundly reduced proliferation rate (Fig. 2B) and significantly increased numbers of SA-β-Gal-positive cells (49% ± 4%) compared with Ppm1d+/+Trp53+/+ MEFs (P < 0.01) (Fig. 2C). Thus, the quantitative differences in the prevalence of senescent phenotypes observed between Ppm1d+/+Trp53+/+ and Ppm1d−/−Trp53+/+ MEFs were eliminated by abrogation of the Trp53 gene. These results therefore suggest that functional activation of p53 is required for the onset of premature cellular senescence elicited by Ppm1d deficiency.
Early passage Ppm1d−/− MEFs cultured in 3% O2 do not show increased accumulation of ROS and DNA damage
Persistence of DNA damage strongly induces cellular senescence.36 More particularly in murine than in human cells, the accumulation of oxidative DNA damage is a major cause of premature senescence under standard culture conditions.24 Wt MEFs cultured in the more physiological condition of 3% O2 avoid activation of DNA damage signaling and the development of premature senescence.24 However, even under the condition of 3% O2, defects in DNA repair genes or other genetic imbalances may lead to the accumulation of DNA damage, activation of DDR signaling, and the onset of premature senescence, as was observed in Ku80−/− MEFs.24 The finding that Ppm1d−/− MEFs exhibited features of premature cellular senescence even under the condition of 3% O2 (Fig. 1A–D) suggested that Ppm1d−/− MEFs may accumulate more DNA damage compared with Wt MEFs. Furthermore, the induction of senescence by oxygen-induced DNA damage in MEFs has been shown to be mediated through the p53/p21 pathway,21,22 and we observed increased activation of p53 and increased p53-dependent p21 protein expression in Ppm1d−/− MEFs from both 3% and 20% O2 culture conditions (Fig. 2A). Therefore, we hypothesized that Ppm1d−/− MEFs may accumulate increased levels of oxidative DNA damage compared with Wt MEFs even when cultured in 3% O2. Since a major source of DNA oxidation is through reaction with ROS,37 we first analyzed the levels of intracellular ROS in Wt and Ppm1d−/− MEFs cultured in either 3% or 20% O2 using the reporter 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCFDA). Early passage Ppm1d−/− MEFs cultured in atmospheric levels of oxygen showed higher levels of intracellular ROS compared with Wt MEFs, but, interestingly, Wt and Ppm1d−/− MEFs cultured in 3% O2 condition exhibited indistinguishable levels of intracellular ROS (Fig. 3A).
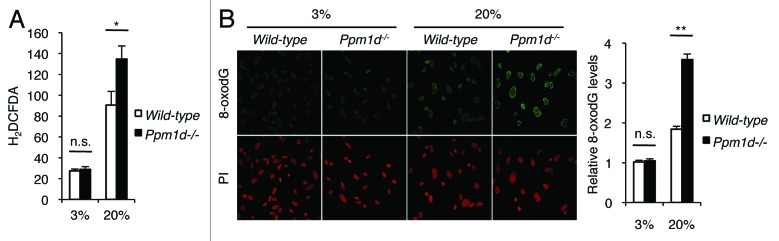
Figure 3.Ppm1d−/− MEFs did not show an increased accumulation of ROS and oxidative DNA damage when cultured in 3% O2. (A) Intracellular ROS levels in Wt and Ppm1d−/− MEFs at passage 2. Cumulative data from 3 independent experiments are presented as mean H2DCFDA fluorescence ± SD (*, P < 0.05, Student t test; n.s.,non significant) (B) Immunofluorescent staining for 8-oxodG in Wt and Ppm1d−/− MEFs at passage 2. The ratio of green (8-oxodG) to red (PI) staining was quantified for at least 100 cells in each group. The ratios shown have been normalized to the ratio determined for Wt MEFs cultured in 3% O2. Cumulative data are shown as the averages of the relative 8-oxodG levels ± standard error of the mean (SEM). (**P < 0.01, Student t test; n.s., non significant)
Next, we compared the levels of 8-hydroxy-2′-deoxyguanosine (8-oxodG), a stable and sensitive marker of oxidative DNA damage,38,39 between Wt and Ppm1d−/− MEFs cultured in either 3% or 20% O2. Staining for 8-oxodG revealed that for cells cultured in 20% O2, Ppm1d−/− MEFs showed approximately 2-fold higher levels of 8-oxodG as compared with Wt MEFs; however, for early passage cells cultured in 3% O2, the levels of DNA oxidation were indistinguishable between Wt and Ppm1d−/− MEFs (Fig. 3B). Thus, in 3% O2 conditions, Ppm1d−/− MEFs did not show an increased accumulation of ROS or DNA damage compared with Wt MEFs.
The finding that for cells cultured in 20% O2, Ppm1d−/− MEFs exhibited increased levels of intracellular ROS and DNA damage compared with Wt MEFs may be explained by the increased numbers of senescent cells present in the Ppm1d−/− MEF population. When we analyzed Wt and Ppm1d−/− MEFs cultured in 20% O2 by flow cytometry, Ppm1d−/− MEFs exhibited a broader range of light-scattering characteristics compared with Wt MEFs (Fig. S2A). In particular, the number of cells exhibiting high levels of both forward scatter (FSC) and side scatter (SSC) (Gate R2) was increased in Ppm1d−/− MEFs (Fig. S2A). Furthermore, the number of Gate R2 cells increased in bleomycin-treated Wt MEFs compared with early passage (P0) untreated Wt MEFs (Fig. S2B), suggesting that the highly scattering cells in Gate R2 are mostly large, senescent cells. Therefore, the scattering characteristics of Ppm1d−/− MEFs cultured in 20% O2 indicate an increased proportion of senescent cells. Moreover, while the distributions of intracellular ROS levels in R1-gated Wt and R1-gated Ppm1d−/− MEFs were similar, R2-gated Ppm1d−/− MEFs have a higher mean intracellular ROS level compared with R2-gated Wt MEFs (Fig. S2C). Given that senescent cells have been reported to have higher levels of ROS than younger cells,40 the increased ROS levels observed in R2-gated Ppm1d−/− MEFs may be attributed to an increased prevalence of senescent cells. Increased ROS levels in R2-gated cells will contribute to the mean fluorescent intensity when the entire population is analyzed, thus explaining the observed increase in mean H2DCFDA fluorescent levels in Ppm1d−/− MEFs compared with Wt MEFs cultured in 20% O2. In addition, since DNA oxidation is primarily caused by intracellular ROS,37 the increased 8-oxodG levels observed in Ppm1d−/− MEFs cultured in 20% O2 is consistent with increased numbers of senescent cells.
Ppm1d deficiency results in activation of DDR signaling during S phase
In cells cultured in 3% O2, Ppm1d−/− MEFs did not show increased levels of intracellular ROS or DNA damage compared with Wt MEFs (Fig. 3A and B). They did, however, show increased activation of p53, expression of p21 protein (Fig. 2A), and prevalence of premature senescence markers (Fig. 1A–D). Previous work has demonstrated that the presence of activated DNA damage signaling, either independent of or resulting from DNA damage, is sufficient for the initiation of cellular senescence and accelerated organismal aging.31,35,41-43 Therefore, we hypothesized that DDR signaling may be activated in Ppm1d−/− MEFs compared with Wt MEFs. To address this hypothesis, we determined the number of foci containing γH2AX, an early marker of the cellular response to DNA damage,9 in Wt and Ppm1d−/− MEFs cultured in either 3% or 20% O2. For cells cultured in 20% O2, Ppm1d−/− MEFs exhibited significantly higher (P < 0.01) numbers of γH2AX foci per cell (5.9 ± 0.7) compared with Wt MEFs (2.7 ± 0.4) (Fig. 4A). Notably, even for cells cultured in 3% O2, significantly increased (P < 0.01) numbers of γH2AX foci were observed in Ppm1d−/− MEFs (3.1 ± 0.4) compared with Wt MEFs (1.4 ± 0.2) (Fig. 4A). Thus, even in 3% O2, Ppm1d−/− MEFs exhibited increased activation of DDR signaling compared with Wt MEFs.
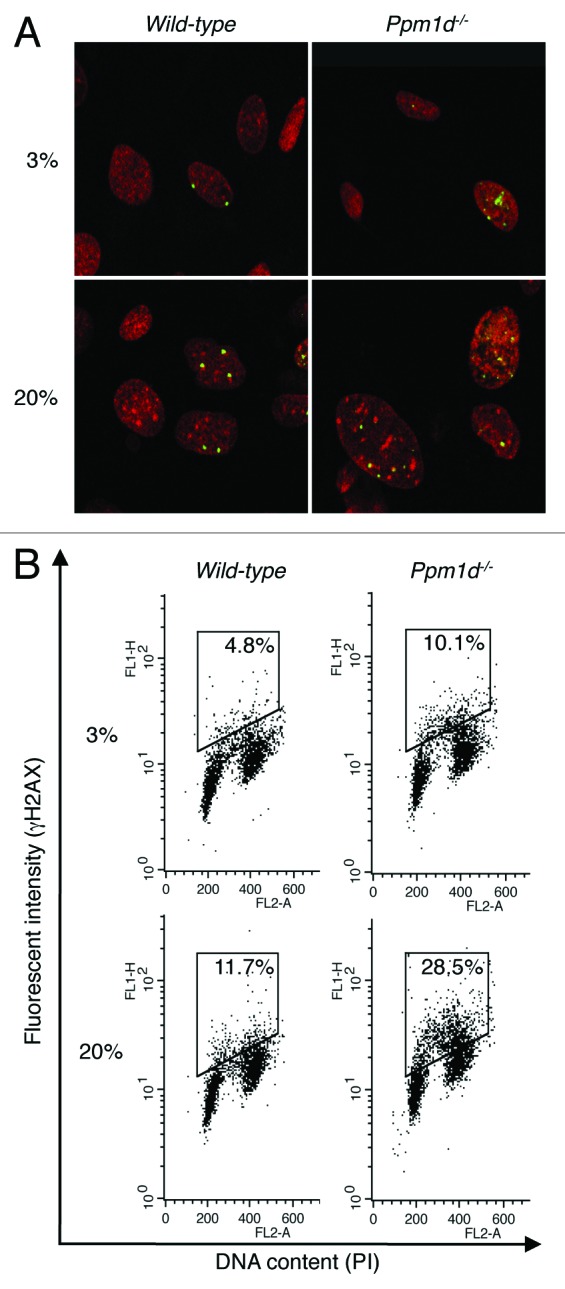
Figure 4. Increased H2AX phosphorylation associated with Ppm1d deficiency occurred predominantly in S phase. (A) Wt and Ppm1d−/− MEFs at passage 3 cultured in 3% or 20% O2 conditions were stained with anti-γH2AX antibody (Green) and PI (red). The number of γH2AX foci per cell was visually counted in at least 200 cells per group. Representative image from each sample is shown. Photographs are at the same magnification (× 400). (B) Cell cycle flow cytometric detection of γH2AX. Wt and Ppm1d−/− MEFs at passage 2 cultured in 3% or 20% O2 were stained with anti-γH2AX antibody and PI and then analyzed by dual parameter flow cytometry. The percentage of γH2AX-positive cells is indicated in the upper right of each diagram. The gate was set up to exclude background fluorescence levels by using H2AX−/− MEFs treated with the same primary antibody. The data are representative of 3 independent experiments.
Cells in S and M phases are more sensitive to DNA damage agents.44 To investigate the relationship between cell cycle phase and the levels of H2AX phosphorylation, we performed flow cytometry of fixed cells stained with anti-γH2AX antibody and propidium iodide (PI). In agreement with the above results, for cells cultured in 20% O2, a significantly larger (P < 0.05) number of Ppm1d−/− MEFs exhibited high anti-γH2AX antibody fluorescence (28% ± 5%) compared with Wt MEFs (12% ± 2%), and for cells cultured in 3% O2, a significantly larger (P < 0.01) proportion of Ppm1d−/− MEFs exhibited high anti-γH2AX antibody fluorescence (10% ± 2%) compared with Wt MEFs (5% ± 1%) (Fig. 4B). It is especially interesting that for both culture conditions, a large proportion of the Ppm1d−/− MEFs with high levels of H2AX phosphorylation have an intermediate DNA content between 2N and 4N, suggesting that Wip1 specifically reduces H2AX phosphorylation during S phase.
Increased H2AX phosphorylation levels observed in Ppm1d−/− MEFs are primarily dependent on the activation of ATM
ATR/checkpoint kinase 1 (Chk1) signaling plays an important role in maintaining genomic integrity during S phase.45-50 Furthermore, H2AX can be directly phosphorylated by ATR in response to replicative stress.51 Therefore, to test whether ATR and Chk1 signaling was differentially activated by the loss of Wip1, we determined the levels of ATR phosphorylated on Ser428 and Chk1 phosphorylated on Ser345 by immunoblotting.52,53 As shown in Figure 5A, the phosphorylation levels of ATR and Chk1 did not differ between Wt and Ppm1d−/− MEFs cultured in 3% O2. These results suggest that ATR/Chk1 signaling is not responsible for the increased levels of H2AX phosphorylation detected in Ppm1d−/− MEFs cultured in 3% O2.

Figure 5. Increased H2AX phosphorylation levels observed in Ppm1d−/− MEFs are primarily due to the activation of ATM. (A) Immunoblot analysis of ATR, p-ATR (pSer428), Chk1, p-Chk1 (pSer345), and β-actin in Wt and Ppm1d−/− MEFs at passage 2 cultured in 3% O2. Immunoprecipitation was performed with an antibody specific for Chk1, followed by immunoblotting with anti-p-Chk1 (pSer345) and anti-Chk1 antibodies. The band intensity of p-Chk1 (pSer345) was quantified using NIH ImageJ software (National Institutes of Health, USA) and normalized to the total Chk1 band. The data are representative of 3 independent experiments. (B) Wt and Ppm1d−/− MEFs at passage 3 cultured in 3% O2 were stained with anti-p-ATM (pSer1981) antibody (Green) and PI (red). The number of p-ATM foci per cell was visually counted in at least 200 cells per group. A representative image from each sample is shown. Photographs are at the same magnification (× 400). (C) Immunoblot analysis of ATM, p-ATM (pSer1981), p53, p-p53 (pSer15), p21, γH2AX and β-actin in Wt and Ppm1d−/− MEFs at passage 2 cultured in 3% O2 condition. Cells were treated with 10 µM ATM kinase inhibitor KU55933 (Tocris Bioscience) (ATMi) or 0.1% DMSO in complete medium for 3 h. (D) Cell cycle flow cytometric detection of γH2AX. Wt and Ppm1d−/− MEFs at passage 2 cultured in 3% O2 condition were treated with 10 µM ATMi or 0.1% DMSO for 3 h. Fixed cells were stained with anti-γH2AX antibody and PI and then analyzed by dual parameter flow cytometry. γH2AX intensity is represented on the y-axis, while PI staining (DNA content) is plotted along the x-axis. The percentage of γH2AX-positive cells is indicated in the upper right of each gate. The data are representative of 3 independent experiments.
For MEFs propagated under conventional culture conditions, the induction of premature senescence has been characterized as a DDR-dependent process mediated by ATM kinase.25 H2AX also can be phosphorylated by ATM,2 and ATM, itself, has been shown to be a target of Wip1 through dephosphorylation of Ser1981, an important autophosphorylation site.8,54 Phosphorylation of ATM Ser1981 occurs rapidly in response to the formation of double-stranded breaks (DSBs) in DNA and is generally used as an indicator of activated ATM.54 Although Ser1981 may not be required for the activation of ATM under all circumstances, its phosphorylation has been shown to be important for the stabilization of ATM near DSBs and, moreover, dephosphorylation of ATM p-Ser1981 by Wip1 results in reduced ATM activity.8,55,56 Interestingly, accumulation of DNA damage and activation of DDR signaling, including formation of phosphorylated Ser1981 foci, were not observed in Wt MEFs cultured in 3% O2.24,25 Thus, the increased H2AX phosphorylation levels observed in Ppm1d−/− MEFs, even when cultured in 3% O2, (Fig. 4A and B), suggested that the level of activation of ATM may be higher in Ppm1d−/− MEFs compared with Wt MEFs. To address this hypothesis, we determined the number of ATM p-Ser1981 foci in Wt and Ppm1d−/− passage 3 MEFs cultured in 3% O2. Notably, significantly increased numbers (P < 0.01) of p-ATM foci were observed in Ppm1d−/− MEFs (0.46 ± 0.08) compared with Wt MEFs (0.18 ± 0.04) (Fig. 5B). Thus, even in the condition of 3% O2, Ppm1d−/− MEFs exhibited increased activation of ATM compared with Wt MEFs.
To investigate further the relationship between activation of ATM and the levels of DDR proteins in 3% O2, we performed immunoblotting of protein extracts from passage 2 Wt and Ppm1d−/− MEFs cultured in 3% O2 and treated with or without KU55933, a selective ATM inhibitor (ATMi).57 The results showed that the increased expression of ATM pSer1981, p53 pSer15, p21, and γH2AX proteins observed in Ppm1d−/− compared with Wt MEFs in the absence of ATMi was reduced in the presence of ATMi (Fig. 5C). Moreover, we performed flow cytometry for γH2AX and DNA content under these same conditions. Co-staining for γH2AX and PI revealed that the percentages of γH2AX-positive cells detected for each genotype in the absence of ATMi were significantly reduced (P < 0.01 for each genotype) in the presence of the ATMi (Fig. 5D). Note that ATMi-treated Ppm1d−/− MEFs still showed increased numbers of γH2AX-positive cells compared with ATMi-treated Wt MEFs. Both ATM and DNA-PK have been reported to contribute to the phosphorylation of H2AX when the abundance of Wip1 is reduced or the phosphatase activity of the Wip1 protein is impaired,6 thus suggesting that DNA-PK also may have contributed to the observed increase in γH2AX levels in Ppm1d−/− MEFs. However, the marked reduction in H2AX phosphorylation levels in both genotypes resulting from addition of KU55933 (Fig. 5C and D) suggests that the increased H2AX phosphorylation levels observed in Ppm1d−/− MEFs cultured in 3% O2 are primarily dependent on the activation of ATM.
To test whether inhibition of ATM affected the onset of cellular senescence in Ppm1d−/− MEFs cultured in 3% O2 conditions, we treated both genotypes of MEFs without or with 10 µM ATMi and analyzed cell proliferation and SA-β-Gal activity. Notably, Wt and Ppm1d−/− MEFs treated with ATMi showed lower proliferation rates compared with the respective control-treated (0.1% DMSO) cells (Fig. S3A). Moreover, ATMi-treated Ppm1d−/− MEFs showed a reduced proliferation rate compared with ATMi-treated Wt cells (Fig. S3A). In addition, quantitative flow cytometry revealed that ATMi-treated Wt and Ppm1d−/− MEFs showed increased numbers of SA-β-Gal-positive cells compared with the respective control-treated cells, both for Wt (P < 0.01) and Ppm1d−/− (P < 0.01) MEFs (Fig. S3B). In addition, ATMi-treated Ppm1d−/− MEFs showed more SA-β-Gal-positive cells compared with ATMi-treated Wt cells (P < 0.05) (Fig. S3B). Notably, prolonged treatment with ATMi accelerated premature cellular senescence in Wt and Ppm1d−/− MEFs cultured in 3% O2. Interestingly, fibroblasts derived from ataxia telangiectasia patients displayed an apparent premature senescence.58 The observation that ATM−/− MEFs and KU55933-treated Wt MEFs exhibited similar defects in DSBs repair59 suggests that cells treated with ATMi may accumulate DSBs, thus promoting cellular senescence. Thus, the increased levels of premature senescence observed in ATMi-treated Ppm1d−/− MEFs may result from the activation of DDR signaling independent of ATM/p53 pathways such as increased activation of DNA-PK.
Ppm1d−/− MEFs show prolonged H2AX phosphorylation levels during recovery from an acute exposure to oxidative stress
Cells explanted from an organism for culturing in vitro experience various stresses from the culture environment.20 During conventional culture, oxidative stress to the cells is a major environmental insult that results from the differences between atmospheric and physiological oxygen concentrations.60 In addition, DNA in S phase is the most susceptible to oxidative stress.44 Indeed, Wt MEFs cultured in 3% O2 but transiently exposed to 100 µM H2O2 exhibited a temporary growth arrest state30 and increased H2AX phosphorylation during S phase (Fig. S4). The resemblance of the cell cycle-dependent patterns in H2AX phosphorylation between H2O2-treated Wt MEFs and untreated Ppm1d−/− MEFs (Fig. S4; Fig. 4B) suggested that Wip1 may regulate the response to oxidative stress even in 3% O2. To further investigate this idea, we used immunoblotting to detect the generation and removal of induced H2AX phosphorylation in cells cultured at 3% O2 conditions following a brief exposure to 100 µM H2O2. Thirty minutes after a brief exposure to H2O2, similarly increased levels of γH2AX were observed in both genotypes (Fig. 6). However, 8 h after treatment, the γH2AX level in Wt MEFs had returned nearly to the level in untreated cells, but the γH2AX level was still markedly elevated in Ppm1d−/− MEFs (Fig. 6). Furthermore, 24 h after treatment, γH2AX levels had returned to the background level in Wt MEFs, but remained high in Ppm1d−/− MEFs (Fig. 6). Thus, Ppm1d−/− MEFs were exposed to the same amount of oxidative stress as Wt MEFs, but exhibited prolonged elevation of γH2AX levels during recovery. These results suggest that increased phosphorylation levels of H2AX during S phase observed in Ppm1d−/− MEFs may be due to sustained activation of DDR signaling.
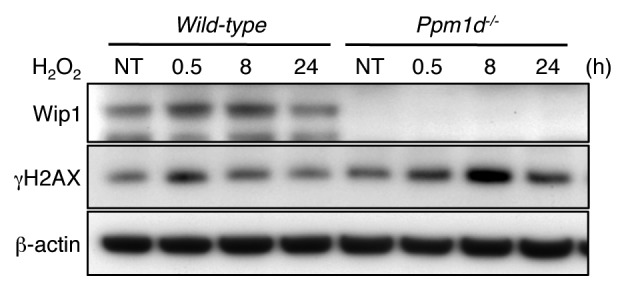
Figure 6. Prolonged H2AX phosphorylation induced by brief H2O2 exposure in Ppm1d−/− MEFs. Wt and Ppm1d−/− MEFs at passage 1 cultured in 3% O2 were treated with 100 µM H2O2 for 15 min. Cells were then washed with PBS and incubated in fresh complete medium. Cells were collected at indicated times (0.5, 8, and 24 h) after H2O2 treatment, and blotted proteins were probed with Wip1, γH2AX and β-actin antibodies. β-actin was used as loading control. NT, no treatment. The data are representative of 3 independent experiments.
Ppm1d−/− MEFs exhibit accelerated premature senescence in response to chronic induction of replication-mediated DSBs
ATM has been characterized as the apical protein kinase in the complex network of signaling interactions that characterize the cellular response to DSBs.2 Here, we found that early passage Ppm1d−/− MEFs cultured in physiological levels of O2 exhibited increased H2AX phosphorylation levels during S phase that were primarily dependent on the activation of ATM, compared with similarly cultured Wt MEFs (Figs. 4B and 5C and D). These results suggested that Wip1 attenuates DDR signaling activated in response to endogenous DSBs generated during S phase. Although the Ppm1d−/− and Wt MEFs exhibited indistinguishable levels of intracellular ROS and oxidized DNA base damage (Fig. 3A and B), there remains the possibility that some aspect of metabolism differs between the 2 genotypes, resulting in increased numbers of endogenous, S phase-specific DSBs. To further investigate the role of Wip1 during S phase, we used a low concentration of camptothecin (CPT), a topoisomerase 1 inhibitor known to induce replication-mediated DSBs,61 to elicit DSBs during S phase in MEFs of both genotypes cultured in 3% O2. First, we performed flow cytometry for γH2AX and PI staining to detect the generation of induced H2AX phosphorylation following exposure to 20 nM CPT. In CPT-treated MEFs, the proportion of cells with high H2AX phosphorylation levels was significantly higher (P < 0.01) for Ppm1d−/− MEFs (26% ± 5%) than for Wt MEFs (15% ± 4%) (Fig. 7A). In agreement with previous results, control-treated (0.1% DMSO) Ppm1d−/− MEFs also exhibited a significantly increased (P < 0.01) proportion of cells with high H2AX phosphorylation levels (7.2% ± 0.1%) compared with Wt MEFs (2.0% ± 0.4%). Interestingly, most of the Ppm1d−/− cells exhibiting high levels of H2AX phosphorylation had an intermediate DNA content, as indicated by PI staining, suggesting S phase-specific induction of γH2AX. Next, immunoblotting revealed that CPT treatment induced ATM phosphorylation in both Wt and Ppm1d−/− MEFs compared with the respective control-treated MEFs (Fig. 7B). More particularly, Ppm1d−/− MEFs showed increased ATM phosphorylation compared with Wt MEFs following CPT addition (Fig. 7B). In addition, p53 pSer,15 p53, and p21 protein levels were also increased in Ppm1d−/− MEFs in the presence of CPT (Fig. 7B). Pre-treatment of both genotypes with the ATM inhibitor KU55933 suppressed the CPT-induced increases in p53 pSer15, p53, p21 and γH2AX protein levels under conditions in which ATM phosphorylation was suppressed (Fig. 7B). Thus, although addition of a low concentration of CPT to Wt or Ppm1d−/− MEFs, cultured in 3% O2, would be expected to generate the same numbers of replication-mediated DSBs, Ppm1d−/− MEFs exhibited increased ATM-dependent DDR signaling.

Figure 7.Ppm1d−/− MEFs showed premature senescence in the response to camptothecin-induced DNA damage. (A) Dual parameter flow cytometric detection of γH2AX and DNA content. Wt and Ppm1d−/− MEFs at passage 1 cultured in 3% O2 condition were treated with or without 20 nM camptothecin (CPT) for 2 h. As a control for CPT treatment, 0.1% DMSO was added. γH2AX intensity is represented on the y-axis, while PI staining (DNA content) is plotted along the x-axis. The percentage of γH2AX-positive cells is indicated in the upper right of each gate. The data are representative of 3 independent experiments. (B) Immunoblot analysis of ATM, p-ATM (pSer1981), p53, p-p53 (pSer15), p21, γH2AX, and β-actin in cells treated with or without 10 µM ATMi for 1 h before the addition of 20 nM CPT for 2 h. (C) Cells were cultured in 3% O2 in medium with or without 20 nM CPT and passaged at 5 × 105 cells per 75 cm2 flask every 2 d. As a control for CPT treatment, medium containing 0.1% DMSO was used. Cell numbers were determined, and cumulative population doubling levels were calculated at each passage. The averages of 3 independent cultures with SD are shown. (D) Flow cytometric detection of SA-β-Gal activity in cells at passage 3, cultured as described in (C). The percentage of SA-β-Gal-positive cells is shown in the upper right of each diagram. The data are representative of 3 independent experiments.
To determine whether increased ATM-dependent DDR signaling observed in CPT-treated Ppm1d−/− MEFs accelerated the induction of cellular senescence, we analyzed cell proliferation, cell morphology, and SA-β-Gal activity of both genotypes of MEFs in the presence or absence of CPT. Wt and Ppm1d−/− MEFs treated with CPT each showed reduced proliferation rates compared with the respective DMSO-treated MEFs (Fig. 7C). Furthermore, in the presence of CPT, Ppm1d−/− MEFs showed a markedly reduced proliferation rate compared with Wt MEFs (Fig. 7C). The reduced proliferation rate observed in CPT-treated Ppm1d−/− MEFs resulted from a lower replicative capacity of these cells (Fig. S5A), not an increase in apoptotic cells (Fig. S5B), compared with CPT-treated Wt MEFs. Moreover, CPT-treated Ppm1d−/− MEFs showed increased numbers of cells with enlarged and flattened morphologies (data not shown). In addition, quantitative flow cytometry analysis revealed that the prevalence of SA-β-Gal-positive cells was significantly higher in Ppm1d−/− MEFs compared with Wt MEFs, both for CPT-treated cells (P < 0.05) and control-treated (P < 0.01) cells (Fig. 7D). Specifically, the proportions of SA-β-Gal-positive cells detected in CPT-treated Wt MEFs and Ppm1d−/− MEFs were 32% ± 2% and 43% ± 4%, respectively, and in control-treated Wt MEFs and Ppm1d−/− MEFs, the respective proportions were 6% ± 2% and 14% ± 1%. Notably, these senescent phenotypes correlated with the extent of DDR signaling indicated by increased levels of the phosphorylated forms of ATM and p53. Thus, these results indicate that Ppm1d−/− MEFs develop premature senescence in response to induced replication-mediated DSBs, similar to the observations for endogenous, replication-mediated DSBs.
Discussion
In the present study, we performed a detailed analysis of Ppm1d−/− MEFs cultured in 3% O2 to investigate the role of Wip1 in the regulation of premature senescence under conditions of reduced oxidative stress. We found that Wip1 forestalls premature senescence at physiological oxygen levels by attenuating DDR signaling from endogenous DSBs that form during DNA replication.
Ppm1d−/− MEFs have been previously reported to exhibit accelerated premature senescence under conventional culture conditions or in the presence of activated oncogenes.10,19 Mouse cells are more sensitive to oxidative stress than human cells.24 We found that even when cultured in 3% O2, Ppm1d−/− MEFs exhibited reduced rates of proliferation and increased numbers of γH2AX foci compared with Wt MEFs, despite having indistinguishable levels of intracellular ROS and oxidized DNA bases. The cells that exhibited high levels of H2AX phosphorylation were predominantly in S phase. DNA in S phase is the most susceptible to oxidative stress.44 In unstressed cells, the presence of γH2AX foci is presumed to mark DSBs that result from DNA replication-induced conversion of endogenous single-stranded breaks (SSBs) produced by metabolically generated ROS or other endogenous processes.62,63 It has been estimated that in cultured human fibroblasts, approximately 1% of endogenous SSBs are converted to DSBs, leading to the generation, on average, of 50 DSBs over the duration of S phase.64 Human fibroblasts cultured in 20% O2 and MEFs cultured in 3% O2 exhibit similar numbers of oxidized bases in DNA, and Wt MEFs cultured in 3% O2 proliferate steadily without overt activation of DDR signaling.24 In contrast, we observed that a fraction of Ppm1d−/− MEFs exhibited high levels of H2AX phosphorylation, and, moreover, that most of these cells were in S phase. Therefore, our results suggest that the absence of Wip1 leads to increased DDR signaling activity resulting from endogenous DSBs produced during S phase. Interestingly, a recent report demonstrated that in several human tumor cell lines the levels of Wip1 protein progressively increased from G1 through G2 phases before declining during mitosis.65
Wip1 facilitates the restoration of cellular homeostatsis following exposure to genotoxic stress through its dephosphorylation of several key DDR proteins.66,67 The marked reduction of H2AX phosphorylation levels observed in both genotypes in the presence of the ATM inhibitor KU55933 suggests that ATM plays an important role in the induction of H2AX phosphorylation observed in this study. Specifically, the increased H2AX phosphorylation levels observed during S phase in Ppm1d−/− MEFs were primarily dependent on the activation of ATM. The increased phosphorylation of ATM Ser1981 observed in Ppm1d−/− MEFs, even when cultured in 3% O2, supports this idea. Wip1 directly binds to ATM and dephosphorylates pSer1981 both in vivo and in vitro.8 Furthermore, Wip1 directly binds and dephosphorylates γH2AX at serine 139 and facilitates the restoration of the cell to a pre-stress state after repair of DSBs.4,6,7 Notably, the numbers of γH2AX foci initially formed following exposure to IR are not affected by the presence or absence of Wip1,4,7 whereas the rate of foci disappearance is increased in the presence of Wip1.6 Consistent with this, we observed sustained elevation in γH2AX levels during recovery of Ppm1d−/− MEFs following a brief exposure to H2O2.
The activation of ATM is regulated by phosphatases.68,69 In unstressed cells, ATM is found as an inactive dimer with low levels of phosphorylation of its autophosphorylation sites, including Ser1981.54 In an early step of ATM activation, phosphorylation of Ser1981, which is located in the kinase activation loop, is increased. Full activation of ATM, including dissociation of the dimer into active monomers, requires interaction with ROS or with a DSB-associated MRN complex.54,70 Interestingly, ATM is maintained in an inactive state by its association with the phosphatases PP2A and Wip1 in unstressed cells. Both phosphatases have been shown to be associated with ATM in unstressed cells, to dephosphorylate pSer1981, and to regulate ATM activity toward downstream targets.8,71,72 The existence of a threshold for ATM activation has also been proposed to explain the correlated reductions in cellular survival and γH2AX induction following exposure to IR delivered at a low dose rate compared with the same dose delivered at a high dose rate.73 Thus, the association of ATM with Wip1 during S phase may increase the threshold for its activation in response to endogenous DSBs.
Although ATM and DNA-PK have overlapping functions in the repair of most DSBs, ATM is uniquely required for the slow phase of DSB repair, for the suppression of radiosensitivity, and for the rapid amplification of DDR signaling.74,75 Following exposure to IR, H2AX becomes rapidly phosphorylated by ATM in megabase regions flanking DSBs.9 In the absence of ATM, γH2AX foci form more slowly after exposure to IR, but similar numbers of foci are formed either in the presence or absence of ATM by 30 min after irradiation.74,75 Phosphorylated H2AX forms a docking site for binding of MDC1 (mediator of DNA damage checkpoint 1) and induces sustained recruitment of other DNA repair factors that mediate the repair of DSBs.76 ATM is specifically required for the propagation of H2AX phosphorylations over large distances from the DSB site and for the generation of a high density of γH2AX that is associated with efficient recruitment of repair factors.77 The formation of large, persistent foci containing γH2AX, ATM pSer1981, and activated p53 was associated with the onset of cellular senescence in cultured human cells.78 Recently, reduced expression of Wip1 and consequential increase in p53 activity were shown to be necessary for chemotherapeutic drug-induced senescence in human cancer cells.79 Induction of p53 is pivotal for the establishment of senescence, usually resulting from its activation by persistent DDR signaling.25,80,81 Even in the absence of overt DNA damage, persistent activation of DDR signaling is sufficient to induce senescence.42,82 Moreover, the state of senescence itself reinforces activation of DDR signaling.83
The loss of Wip1 activity may result in increased cellular senescence and contribute to a decrease in organismal fitness. Notably, Ppm1d−/− mice showed atrophied splenic white pulp compared with Wt mice.19 Since atrophy is recognized as a phenotype of aging,84 the atrophied splenic structure observed in Ppm1d−/− mice may contribute to the observed premature aging phenotype. Interestingly, Ppm1d−/− mice showed increased γH2AX protein levels in the spleen compared with Wt mice, even without additional stress.7 γH2AX has been proposed as a potential biomarker for senescent cells, as its levels correlate with those of SA-β-Gal in mouse tissues.85 Moreover, the frequency of γH2AX-positive cells in the lymphocytes of splenic white pulp of C57BL/6 mice increased significantly with age.85 Considering that most splenic lymphocytes are present within the white pulp, the atrophied splenic structures observed in Ppm1d−/− mice may result from increased cellular senescence of splenic lymphocytes. In addition, both B- and T-cells from spleen of Ppm1d−/− mice showed reduced proliferative response against mitogenic stimulation,19 indicating reduced proliferative potential. Together, these observations suggest that increased cellular senescence induced by loss of Wip1 may accelerate the aging of organs through increased activation of the DDR.
Therefore, we suggest that the acceleration of premature senescence observed in Ppm1d−/− MEFs cultured in physiological O2 levels may result from the combined effects of a lower threshold for activation of ATM, increased ATM-dependent amplification of DDR signaling, and sustained activation of DDR signaling. In conclusion, our study found that Wip1 prevents cellular senescence at physiological oxygen levels by regulating DDR signaling activated during DNA replication.
Material and Methods
Mice and MEFs
Wip1 knockout (Ppm1d−/−) mice have been described previously.19 All mice were backcrossed with inbred C57BL/6NCr mice for at least 9 generations to yield Ppm1d−/− mice congenic in the C57BL/6NCr genetic background. Wt C57BL/6NCr control mice were originally purchased from Division of Cancer Treatment/National Cancer Institute (Frederick, MD)86 and were maintained in our animal facility. All mice were bred and kept under standard pathogen-free conditions. The experiments were conducted in accordance with the guidelines of the National Cancer Institute Animal Care and Use Committee. MEFs were prepared from 13.5-d embryos derived from crosses between homozygous parents of either Wt or Ppm1d−/− genotype. Following removal of the head and the red organs, the torso was rinsed with phosphate-buffered saline (PBS), minced, and digested with 0.5 ml of 0.05% trypsin-EDTA. Trypsin was inactivated by addition of Dulbecco modified Eagle medium (Invitrogen) containing 10% fetal bovine serum (Atlanta Biologicals), 50 units/ml penicillin (Invitrogen), and 50 µg/ml streptomycin (Gibco Life Technologies), and then the suspension was placed into a 100-mm dish and incubated at 37 °C with 5% CO2 for 2 d. At approximately 90% confluency, cells that grew from tissue fragments were harvested and used for experiments. These cells were considered as passage 0. Cells (5 × 105) that were derived from a single embryo were cultured in 5% CO2 plus 3% or 20% O2 by subculturing in a 75-cm2 flask every 3 d, and cell numbers were determined at each passage. Oxygen concentrations in a CO2 incubator were adjusted by an oxygen sensor and a regulated nitrogen source.
Kinase inhibitor
The specific inhibitor for ATM kinase activity, KU55933 (Tocris Biosciences), was prepared as a 10 mM stock solution in DMSO.
Apoptosis assays
Cellular apoptosis of MEFs was assessed by using the Annexin-V-FITC Apoptosis Detection Kit (BD PharMingen) according to the manufacturer’s instructions. In order to exclude necrotic cells from the analysis, MEFs were also treated with propidium iodide (PI) without permeabilization. Double-positive cells of both PI and annexin-V are considered necrotic and were excluded from analysis.87,88
Senescence-associated β-galactosidase (SA-β-Gal)
Fluorecence based assays of SA-β-Gal activity were performed by using FACSCalibur (Becton Dickinson) as described previously.27 Wt and Ppm1d−/− MEFs at Passage 3 in 3% or 20% O2 conditions were pretreated with 100 nM bafilomycin A1 (Sigma-Aldrich) followed by 1.5 h incubation with 33 µM dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG) (Invitrogen). The percentage of SA-β-Gal-positive cells was determined on a 2-parameter display of C12-fluorescein fluorescence (FL1) vs. FSC, and the data were analyzed with CELLQuest software (Becton Dickinson). Wt MEFs at passage 0 were used as a negative control to establish the quadrant gate.
Cell cycle analysis
Subconfluent cultures at passage 2 cultured in 3% or 20% O2 were labeled with 100 µM BrdU (Sigma-Aldrich) for 1 h at 37 °C. Trypsinized cells were fixed in 70% cold ethanol and stored at −20 °C overnight. The cells were treated with 2 N HCL for 20 min at room temperature, suspended in 0.1 M Borate Buffer (pH 8.4), incubated with Alexa Fluor 488-conjugated anti-BrdU antibody (B35139, Invitrogen Life Technologies), diluted 1:20 with PBS containing 0.5% Tween 20 and 1% BSA for 1 h at room temperature in the dark, and finally incubated with a 50 µg/ml propidium iodide (PI) solution containing 0.1 mg/ml RNaseA (Roche Diagnostics GmbH) for 20 min at room temperature in the dark. The cells were washed with PBS between each step. Samples were analyzed by 2-dimensional (2D) flow cytometry to detect both BrdU incorporation and DNA content by using FACSCalibur (Becton Dickinson). Cytometric analysis was performed with CELLQuest software (Becton Dickinson).
Immunocytochemistry
Immunocytochemistry for detection of γH2AX was performed as previously described89 with slight modifications. Wt and Ppm1d−/− MEFs at passage 3 in 3% or 20% O2 conditions were grown on 4-well chamber slides for 3 d prior to immunostaining. After washing with PBS, the slides were fixed with freshly prepared 2% paraformaldehyde in PBS for 20 min and then fixed with 70% cold ethanol at −20 °C for 20 min. The slides were then washed with PBS for 15 min and blocked with 5% BSA in PBS containing 0.5% Tween-20 and 0.1% Triton X-100 (PBS-TT) for 1 h. After a 5 min wash with PBS, the slides were incubated for 2 h with the mouse monoclonal anti-γH2AX antibody (Ser139) (#05–636, EMD Millipore) (dilution 1:500 in 1% BSA in PBS-TT) followed by 1 h incubation with the goat anti-mouse Alexa Fluor 488-conjugated IgG (A-11001, Invitrogen Life Technologies) (dilution 1:500 in 1% BSA in PBS-TT). After 15 min wash and 30 min incubation at 37 °C with 0.1 mg/ml RNase A (Roche Diagnostics GmbH) in PBS, the slides were mounted with mounting medium containing propidium iodide (PI) (Vectashield) and sealed with nail polish. Laser-scanning confocal microscopy was performed with a Nikon PCM 2000 (Nikon Inc). The foci were visually counted in at least 200 cells per group. Immunocytochemistry for phospho-ATM (p-ATM) was performed as well by using anti-p-ATM antibody (#05-740, EMD Millipore) (dilution 1:500 in 1% BSA in PBS-TT).
Western blot analysis
Whole-cell lysates were prepared by homogenizing cell pellets in TNE buffer (10 mM TRIS-HCl pH 7.8, 1 mM EDTA, 150 mM NaCl, 1% NP-40, 50 mM NaF) supplemented with a protease inhibitor cocktail (Roche Diagnostics) and phosphatase inhibitor cocktails (Sigma-Aldrich). The protein concentration in the lysate was determined by using the BCA protein assay reagent (Thermo Scientific). Forty micrograms of protein were separated by SDS-PAGE and electrophoretically transferred onto a polyvinylidene difluoride membrane (EMD Millipore). The blots were probed with the following primary antibodies overnight at 4 °C: Cell Signaling Technology: anti-phospho-p53 (pSer15, #9284), anti-phospho-ATR (pSer428, #2853), anti-ATR (#2790), anti-phospho-p38 MAPK (pThr180/pTyr182, #9216), anti-p38 MAPK (#9212); Santa Cruz Biotechnology: anti-p53 (sc-1311), anti-p21 (sc-397G), anti-p16 (sc-1207); EMD Millipore: anti-PCNA (NA03), anti-γH2AX (pSer139, #05–636), anti-phospho-ATM (pSer1981, #05-740), anti-ATM (#07-1286); BD Biosciences: anti-pRb (G3-245), anti-Wip1 (custom mouse monoclonal antibody); Sigma-Aldrich: anti-β-actin (A5441); Abcam: anti-p19 (ab80). The protein bands were visualized by using horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence reagents (PerkinElmer Inc).
Immunoprecipitation
Lysates containing 500 µg of nuclear protein were immunoprecipitated with Protein G-coupled magnetic beads (Invitrogen Life Technologies) conjugated to Chk1 antibody (sc-8408, Santa Cruz Biotechnology). Briefly, cell lysates were incubated with anti-Chk1 antibody-Protein G-conjugated magnetic beads in a rotating mixer overnight at 4 °C. After incubation, the tubes were placed on a magnetic rack, the supernatant was carefully aspirated, and the pellet was washed 3 times with wash buffer (PBS containing 0.02% Tween-20). The magnetic beads were suspended in 20 µl of 1 × LDS sample buffer (Invitrogen Life Technologies) without reducing agents and boiled for 10 min at 70 °C. Eighteen microliters of the eluted samples were analyzed by western blotting to detect phospho-Chk1 protein expression. The blotted PVDF membrane was blocked in SuperBlock Blocking buffer (Thermo Scientific) in TBS containing 0.05% Tween-20 for 1 h at room temperature. After washing, the membrane was incubated with anti-phospho-Chk1 antibody (pSer345) (#2348, Cell Signaling Technology) overnight at 4 °C with gentle shaking. The membrane was washed and incubated with Clean-Blot IP Detection Reagent (Thermo Scientific) for 1 h at room temperature. Specific bands were detected by ECL Plus (Thermo Scientific).
Flow cytometric quantification of γH2AX
Wt and Ppm1d−/− MEFs at Passage 2 in 3% or 20% O2 conditions were collected by trypsinization, fixed with 1% formaldehyde for 8 min at 4 °C, and then fixed with 70% cold ethanol for 2 h at −20 °C. After washing with PBS-T (PBS containing 0.5% Triton X-100), cells were blocked with 5% BSA in PBS-T for 30 min at room temperature. Cells were washed with PBS-T containing 1% BSA and incubated with anti-γH2AX antibody (pSer139) (#05–636, EMD Millipore) diluted 1:250 in PBS-T containing 1% BSA for 2 h at room temperature. Cells were washed and suspended in Alexa Fluor 488-conjugated anti-mouse IgG antibody (A-11001, Invitrogen Life Technologies) diluted 1:250 in PBS-T containing 1% BSA for 1 h in the dark. Cells were then washed and suspended in 50 µg/ml PI containing 100 µg/ml RNaseA (Roche Diagnostics) for 30 min at 4 °C. Fluorescent intensity of Alexa Fluor 488 (γH2AX) and propidium iodide (PI) (DNA content) were analyzed by using a FACSCalibur (Becton Dickinson) and CELLQuest software (Becton Dickinson).
Detection of intracellular ROS and 8-oxodG levels
Intracellular ROS levels of either Wt or Ppm1d−/− MEFs at passage 2 in 3% or 20% O2 conditions were determined by using H2DCFDA (Invitrogen Life Technologies) as described previously90 with FACSCalibur (Becton Dickinson). The data were analyzed with CELLQuest software (Becton Dickinson) and the mean fluorescent intensity was used to quantify the response. The levels of 8-oxodG were detected by immunocytochemistry according to the manufacture’s protocol (Trevigen) with slight modifications. Briefly, MEFs growing on poly-L-lysine (Sigma-Aldrich) coated coverslip were fixed in absolute cold methanol, and then fixed in cold acetone at −20 °C for 15 min each. Cells were incubated with 100 µg/mL RNaseA (Roche Diagnostics) at 37 °C for 1 h. After blocking with 5% normal goat serum, cells were incubated with anti-8-oxodG antibody (clone 2E2, Trevigen) diluted 1:2000 at 37 °C overnight followed by Alexa Fluor 488-conjugated anti-mouse IgG antibody (#A-11001, Invitrogen Life Technologies) diluted 1:500 at room temperature for 1 h in the dark. Coverslips were mounted on glass slides using mounting medium containing PI (Vectashield) and viewed under a fluorescence microscope (Nikon PCM 2000, Nikon Inc). The fluorescence intensities of at least 100 cells was quantified by using Adobe Photoshop (Adobe Systems Inc) as described.91
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Christophe Redon from National Cancer Institute for his helpful comments and suggestions.
Funding
This research was supported by the Intramural Research Program of the Center for Canter Research, National Cancer Institute, National Institutes of Health.
Glossary
Abbreviations:
- 8-oxodG
8-hydroxy-2′-deoxyguanosine
- ATM
ataxia telangiectasia-mutated
- ATR
ataxia telangiectasia and Rad3-related
- BrdU
bromodeoxyuridine
- C12FDG
dodecanoylaminofluorescein di-β-D-galactopyranoside
- Chk1
checkpoint kinase 1
- CPT
camptothecin
- DDR
DNA damage response
- DMSO
dimethyl sulfoxide
- DNA-PK
DNA-dependent protein kinase
- DSBs
double-stranded breaks
- FSC
forward scatter
- γH2AX
phosphorylated form of the histone 2A variant H2AX
- H2DCFDA
2′, 7′-dichlorodihydrofluorescein diacetate
- MEFs
mouse embryonic fibroblasts
- PBS
phosphate-buffered saline
- PI
propidium iodide
- PIKK
phosphatidylinositol 3-kinase-related kinase
- Ppm1d
Protein phosphatase Mg2+/Mn2+ dependent 1D
- ROS
reactive oxygen species
- SA-β-Gal
senescence-associated β-galactosidase
- SSBs
single-strand breaks
- SSC
side scatter
- Wip1
wild-type p53-induced phosphatase 1
- Wt
wild-type
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27920
References
- 1.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 2.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 3.Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, Vande Woude GF, O’Connor PM, Appella E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94:6048–53. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cha H, Lowe JM, Li H, Lee JS, Belova GI, Bulavin DV, Fornace AJ., Jr. Wip1 directly dephosphorylates gamma-H2AX and attenuates the DNA damage response. Cancer Res. 2010;70:4112–22. doi: 10.1158/0008-5472.CAN-09-4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–74. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macůrek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010;29:2281–91. doi: 10.1038/onc.2009.501. [DOI] [PubMed] [Google Scholar]
- 7.Moon SH, Lin L, Zhang X, Nguyen TA, Darlington Y, Waldman AS, Lu X, Donehower LA. Wild-type p53-induced phosphatase 1 dephosphorylates histone variant gamma-H2AX and suppresses DNA double strand break repair. J Biol Chem. 2010;285:12935–47. doi: 10.1074/jbc.M109.071696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–64. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–5. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 11.Castellino RC, De Bortoli M, Lu X, Moon SH, Nguyen TA, Shepard MA, Rao PH, Donehower LA, Kim JY. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol. 2008;86:245–56. doi: 10.1007/s11060-007-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuku T, Semba S, Yutori H, Yokozaki H. Increased wild-type p53-induced phosphatase 1 (Wip1 or PPM1D) expression correlated with downregulation of checkpoint kinase 2 in human gastric carcinoma. Pathol Int. 2007;57:566–71. doi: 10.1111/j.1440-1827.2007.02140.x. [DOI] [PubMed] [Google Scholar]
- 13.Loukopoulos P, Shibata T, Katoh H, Kokubu A, Sakamoto M, Yamazaki K, Kosuge T, Kanai Y, Hosoda F, Imoto I, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007;98:392–400. doi: 10.1111/j.1349-7006.2007.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito-Ohara F, Imoto I, Inoue J, Hosoi H, Nakagawara A, Sugimoto T, Inazawa J. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003;63:1876–83. [PubMed] [Google Scholar]
- 15.Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 16.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr., Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 17.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, Appella E, Fornace AJ., Jr. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 18.Nannenga B, Lu X, Dumble M, Van Maanen M, Nguyen TA, Sutton R, Kumar TR, Donehower LA. Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol Carcinog. 2006;45:594–604. doi: 10.1002/mc.20195. [DOI] [PubMed] [Google Scholar]
- 19.Choi J, Nannenga B, Demidov ON, Bulavin DV, Cooney A, Brayton C, Zhang Y, Mbawuike IN, Bradley A, Appella E, et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol Cell Biol. 2002;22:1094–105. doi: 10.1128/MCB.22.4.1094-1105.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–76. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 22.Itahana K, Campisi J, Dimri GP. Mechanisms of cellular senescence in human and mouse cells. Biogerontology. 2004;5:1–10. doi: 10.1023/B:BGEN.0000017682.96395.10. [DOI] [PubMed] [Google Scholar]
- 23.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–15. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 24.Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–7. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Micco R, Cicalese A, Fumagalli M, Dobreva M, Verrecchia A, Pelicci PG, di Fagagna Fd. DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle. 2008;7:3601–6. doi: 10.4161/cc.7.22.7152. [DOI] [PubMed] [Google Scholar]
- 26.Ivanovic Z. Hypoxia or in situ normoxia: The stem cell paradigm. J Cell Physiol. 2009;219:271–5. doi: 10.1002/jcp.21690. [DOI] [PubMed] [Google Scholar]
- 27.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 28.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell. 2007;12:342–54. doi: 10.1016/j.ccr.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 30.Barzilai A, Yamamoto K. DNA damage responses to oxidative stress. DNA Repair (Amst) 2004;3:1109–15. doi: 10.1016/j.dnarep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Lou Z, Chen J. Cellular senescence and DNA repair. Exp Cell Res. 2006;312:2641–6. doi: 10.1016/j.yexcr.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 32.Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–81. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–4. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 34.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 35.Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene. 1993;8:2457–67. [PubMed] [Google Scholar]
- 36.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 37.Chen QM, Bartholomew JC, Campisi J, Acosta M, Reagan JD, Ames BN. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem J. 1998;332:43–50. doi: 10.1042/bj3320043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ames BN. Oxygen radicals and 8-hydroxyguanine in DNA. Jpn J Cancer Res. 1991;82:1460–1. [PubMed] [Google Scholar]
- 39.Shigenaga MK, Ames BN. Assays for 8-hydroxy-2′-deoxyguanosine: a biomarker of in vivo oxidative DNA damage. Free Radic Biol Med. 1991;10:211–6. doi: 10.1016/0891-5849(91)90078-H. [DOI] [PubMed] [Google Scholar]
- 40.Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu ZX, Ferrans VJ, Howard BH, Finkel T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–40. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 41.Ferguson DO, Alt FW. DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene. 2001;20:5572–9. doi: 10.1038/sj.onc.1204767. [DOI] [PubMed] [Google Scholar]
- 42.Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008;22:297–302. doi: 10.1101/gad.452308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 44.Chen JH, Ozanne SE, Hales CN. Heterogeneity in premature senescence by oxidative stress correlates with differential DNA damage during the cell cycle. DNA Repair (Amst) 2005;4:1140–8. doi: 10.1016/j.dnarep.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–28. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–89. doi: 10.1016/S0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 47.Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene. 2006;25:4381–8. doi: 10.1038/sj.onc.1209466. [DOI] [PubMed] [Google Scholar]
- 48.Nghiem P, Park PK, Kim Y, Vaziri C, Schreiber SL. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc Natl Acad Sci U S A. 2001;98:9092–7. doi: 10.1073/pnas.161281798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25:1764–74. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zachos G, Rainey MD, Gillespie DA. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 2003;22:713–23. doi: 10.1093/emboj/cdg060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 52.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 55.Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, Nussenzweig A. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443:222–5. doi: 10.1038/nature05112. [DOI] [PubMed] [Google Scholar]
- 56.So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009;187:977–90. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 58.Shiloh Y, Tabor E, Becker Y. In vitro phenotype of ataxia-telangiectasia (AT) fibroblast strains: clues to the nature of the “AT DNA lesion” and the molecular defect in AT. Kroc Found Ser. 1985;19:111–21. [PubMed] [Google Scholar]
- 59.Riballo E, Kühne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–24. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 60.Halliwell B. Oxidative stress in cell culture: an under-appreciated problem? FEBS Lett. 2003;540:3–6. doi: 10.1016/S0014-5793(03)00235-7. [DOI] [PubMed] [Google Scholar]
- 61.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 62.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–94. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakamura AJ, Redon CE, Bonner WM, Sedelnikova OA. Telomere-dependent and telomere-independent origins of endogenous DNA damage in tumor cells. Aging (Albany NY) 2009;1:212–8. doi: 10.18632/aging.100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A. 2003;100:12871–6. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Macurek L, Benada J, Müllers E, Halim VA, Krejčíková K, Burdová K, Pecháčková S, Hodný Z, Lindqvist A, Medema RH, et al. Downregulation of Wip1 phosphatase modulates the cellular threshold of DNA damage signaling in mitosis. Cell Cycle. 2013;12:251–62. doi: 10.4161/cc.23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lowe J, Cha H, Lee MO, Mazur SJ, Appella E, Fornace AJ., Jr. Regulation of the Wip1 phosphatase and its effects on the stress response. Front Biosci (Landmark Ed) 2012;17:1480–98. doi: 10.2741/3999. [Landmark Ed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008;27:123–35. doi: 10.1007/s10555-008-9127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freeman AK, Monteiro AN. Phosphatases in the cellular response to DNA damage. Cell Commun Signal. 2010;8:27. doi: 10.1186/1478-811X-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shimada M, Nakanishi M. Response to DNA damage: why do we need to focus on protein phosphatases? Front Oncol. 2013;3:8. doi: 10.3389/fonc.2013.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 71.Carlessi L, Buscemi G, Fontanella E, Delia D. A protein phosphatase feedback mechanism regulates the basal phosphorylation of Chk2 kinase in the absence of DNA damage. Biochim Biophys Acta. 2010;1803:1213–23. doi: 10.1016/j.bbamcr.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 72.Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23:4451–61. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Collis SJ, Schwaninger JM, Ntambi AJ, Keller TW, Nelson WG, Dillehay LE, Deweese TL. Evasion of early cellular response mechanisms following low level radiation-induced DNA damage. J Biol Chem. 2004;279:49624–32. doi: 10.1074/jbc.M409600200. [DOI] [PubMed] [Google Scholar]
- 74.Kitagawa R, Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb Symp Quant Biol. 2005;70:99–109. doi: 10.1101/sqb.2005.70.002. [DOI] [PubMed] [Google Scholar]
- 75.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–6. doi: 10.1158/0008-5472.CAN-03-3207. [DOI] [PubMed] [Google Scholar]
- 76.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 77.Savic V, Yin B, Maas NL, Bredemeyer AL, Carpenter AC, Helmink BA, Yang-Iott KS, Sleckman BP, Bassing CH. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol Cell. 2009;34:298–310. doi: 10.1016/j.molcel.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suzuki M, Suzuki K, Kodama S, Yamashita S, Watanabe M. Persistent amplification of DNA damage signal involved in replicative senescence of normal human diploid fibroblasts. Oxid Med Cell Longev. 2012;2012:310534. doi: 10.1155/2012/310534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Crescenzi E, Raia Z, Pacifico F, Mellone S, Moscato F, Palumbo G, Leonardi A. Down-regulation of wild-type p53-induced phosphatase 1 (Wip1) plays a critical role in regulating several p53-dependent functions in premature senescent tumor cells. J Biol Chem. 2013;288:16212–24. doi: 10.1074/jbc.M112.435149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 81.Yasaei H, Gilham E, Pickles JC, Roberts TP, O’Donovan M, Newbold RF. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene. 2013;32:171–9. doi: 10.1038/onc.2012.45. [DOI] [PubMed] [Google Scholar]
- 82.Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science. 2008;320:1507–10. doi: 10.1126/science.1159051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle 2009; 8:4112-8. [DOI] [PMC free article] [PubMed]
- 84.Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003;299:1355–9. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- 85.Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–23. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- 86.Schito ML, Demidov ON, Saito S, Ashwell JD, Appella E. Wip1 phosphatase-deficient mice exhibit defective T cell maturation due to sustained p53 activation. J Immunol. 2006;176:4818–25. doi: 10.4049/jimmunol.176.8.4818. [DOI] [PubMed] [Google Scholar]
- 87.Albanese C, D’Amico M, Reutens AT, Fu M, Watanabe G, Lee RJ, Kitsis RN, Henglein B, Avantaggiati M, Somasundaram K, et al. Activation of the cyclin D1 gene by the E1A-associated protein p300 through AP-1 inhibits cellular apoptosis. J Biol Chem. 1999;274:34186–95. doi: 10.1074/jbc.274.48.34186. [DOI] [PubMed] [Google Scholar]
- 88.Logue SE, Elgendy M, Martin SJ. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat Protoc. 2009;4:1383–95. doi: 10.1038/nprot.2009.143. [DOI] [PubMed] [Google Scholar]
- 89.Redon CE, Nakamura AJ, Gouliaeva K, Rahman A, Blakely WF, Bonner WM. The use of gamma-H2AX as a biodosimeter for total-body radiation exposure in non-human primates. PLoS One. 2010;5:e15544. doi: 10.1371/journal.pone.0015544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–13. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Polytarchou C, Pfau R, Hatziapostolou M, Tsichlis PN. The JmjC domain histone demethylase Ndy1 regulates redox homeostasis and protects cells from oxidative stress. Mol Cell Biol. 2008;28:7451–64. doi: 10.1128/MCB.00688-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.