

Abstract
Antibodies isolated from human donors are increasingly being developed for anti-infective therapeutics. These antibodies undergo affinity maturation in vivo, minimizing the need for engineering of therapeutic leads for affinity. However, the affinities required for some therapeutic applications may be higher than the affinities of the leads obtained, requiring further affinity maturation in vitro. To improve the neutralization potency of natural human antibody MSL-109 targeting human cytomegalovirus (CMV), we affinity matured the antibody against the gH/gL glycoprotein complex. A phage display library where most of the six complementary-determining regions (CDRs) were allowed to vary in only one amino acid residue at a time was used to scan for mutations that improve binding affinity. A T55R mutation and multiple mutations in position 53 of the heavy chain were identified that, when present individually or in combination, resulted in higher apparent affinities to gH/gL and improved CMV neutralization potency of Fab fragments expressed in bacterial cells. Three of these mutations in position 53 introduced glycosylation sites in heavy chain CDR 2 (CDR H2) that impaired binding of antibodies expressed in mammalian cells. One high affinity (KD < 10 pM) variant was identified that combined the D53N and T55R mutations while avoiding glycosylation of CDR H2. However, all the amino acid substitutions identified by phage display that improved binding affinity without introducing glycosylation sites required between two and four simultaneous nucleotide mutations to avoid glycosylation. These results indicate that the natural human antibody MSL-109 is close to a local affinity optimum. We show that affinity maturation by phage display can be used to identify and bypass barriers to in vivo affinity maturation of antibodies imposed by glycosylation and codon usage. These constraints may be relatively prevalent in human antibodies due to the codon usage and the amino acid sequence encoded by the natural human repertoire.
Keywords: affinity, maturation, mutagenesis, glycosylation, codon, cytomegalovirus, antibody, neutralization
Introduction
Natural human antibodies, derived from donors or transgenic mice, and humanized antibodies are the major source of therapeutic antibodies.1 The advantage of these antibodies over antibodies obtained from phage or yeast or mammalian display libraries is that affinity maturation occurs in vivo through the process of somatic hypermutation followed by clonal selection. This usually obviates the need for in vitro affinity maturation, shortening therapeutic antibody development timelines. However, the affinities of antibodies obtained from in vivo sources for certain targets or epitopes may be insufficient for some therapeutic applications. Factors such as the number of donors or immunized animals, immunization protocols, and the number of clones screened, can be controlled in antibody discovery campaigns and contribute to the probability of obtaining leads with high affinities. Other less controllable factors include the identity of the antigen used for immunization, the sequence repertoire, which determines initial shape and chemical complementarity to antigen epitopes, and the natural immune mechanisms of antibody selection.
A factor that imposes a limit on the affinity of natural antibodies is the kinetic selection of clones.2,3 Theoretical considerations on the kinetics of antigen binding and recycling time of B cell receptors suggest that the immune system cannot differentiate clones with equilibrium dissociation constants (KD) below 10−10 M at physiological temperatures.4 This is supported by the finding that B cell receptor-dependent antigen presentation depends on affinity for KD values above but not below 10−10 M.5 Although antibodies with higher affinities could be produced by clonal selection, these would be events without a selective advantage over clones with affinity constants around that threshold. Analysis of antibodies obtained from immunized volunteers indicates that the immune system appears to select clones with a maximum mean affinity constant of about 10−9 M at physiological temperature, with a log-normal distribution of affinity constants around that average.3 These studies found that sequence diversification continues to occur after each boost in these immunized individuals, but without a significant increase in the number of somatic mutations per clone or a decrease of mean maximal affinity constants. In addition, despite continuing sequence diversification, no correlation between the number of somatic mutations and affinity constants can be observed in the antibody panels analyzed. This limit to in vivo affinity maturation would allow affinities suitable for protective immunity in the context of polyclonal antibody responses in conjunction with other adaptive immune responses, but not necessarily for passive immunization with a monoclonal antibody as required for clinical applications.
The immune repertoire and the mechanism of somatic mutation and clonal selection can limit affinity by constraining both the binding affinity of the clones induced in the primary response and the possible mutation path for affinity maturation. In one of the studies described above, IgG from B cell clones without somatic mutations were found to vary in affinity by a few orders of magnitude.2 Regardless of the initial affinity of the germline sequence antibodies, there is an apparent limit of 100-fold improvement of affinity constants in somatically mutated variants. The repertoire may impose such limits based on the overall structures of the antibodies generated and the configuration of the genetic code, which should impose a constraint on mutations likely to be generated based on codon usage. In addition, stepwise selection of a limited set of mutants with one or a few mutations each can lead clonal lineages to local optima from which sequences with further affinity improvements cannot be easily accessed by single point mutations.6 Finally, the number of mutants that need to be sampled to achieve higher affinities increases with affinity.6,7 This may result in clones with suboptimal affinities if clone population size or time is insufficient to generate the necessary number of mutants to find one with improved affinity.
Affinity maturation by in vitro molecular engineering can potentially overcome these barriers to in vivo affinity maturation by imposing more stringent selection than can be achieved in vivo, by decoupling the mutations that can be generated from the natural codon usage and by allowing combinatorial mutations to access regions of the sequence space that are not easily accessible to the immune system. The design of affinity maturation libraries and the selection systems used have a significant effect on the possible constraints to affinity improvement that can be successfully overcome.8 For instance, the design of libraries with each amino acid position mutated individually would be expected to only improve the affinity of clones that have reached a kinetic limit for selection or that cannot easily escape a local optimum due to the constraint of codon usage. Libraries allowing combinatorial amino acid substitutions and variable domain shuffling would be expected to access wider regions of the sequence space, including simultaneous mutations whose combined, synergistic effect on affinity may not be identified with libraries with single amino acid mutations. However, in the context of therapeutic antibody engineering, this approach has the potential to introduce hitchhiking mutations with neutral effects on affinity, but detrimental effects on off-target binding, pharmacokinetic, physicochemical or manufacturing properties of the antibody. This issue can be minimized if the role of individual and combined mutations on affinity is subsequently determined in order to define the mutations needed for affinity improvement without introducing off-target binding,9 a potentially laborious task.
We affinity-matured MSL-109, a natural antibody against the gH glycoprotein of human cytomegalovirus (CMV) derived from a human donor, to enhance its neutralization potency. Using Fab fragment phage display libraries with single amino acid substitutions in each complementary-determining region (CDR), we identified a region in heavy chain CDR 2 (CDR H2) that was somatically mutated in vivo into a sequence resulting in suboptimal binding affinity of MSL-109. This region is constrained from readily evolving to higher affinity variants by introduction of glycosylation sites in CDR H2 deleterious for binding and by codon usage restrictions. Mutations that in combination overcome these limits were identified and used to create a very high affinity variant with improved neutralization of a diverse panel of CMV strains.
Results
Selection of phage libraries identifies at least two CDR H2 positions with mutations favorable for binding
Two libraries were constructed randomizing CDR positions in the heavy and light chains, respectively. All three CDRs were randomized in each library but each CDR had a maximum of 1 mutation in each clone except CDR H3, which could have two simultaneous mutations in Kabat positions 100l (Gly or Ala) and 100 min (Leu, Phe, Ile or Val). Except for these two positions, each position was randomized with an NNK codon (IUPAC code) encoding any amino acid or an amber stop. The combined heavy and light chain library designs had a potential diversity of 1.1 × 107 unique full-length clones without stop or cysteine codons and an expected distribution of about 0.02%, 1.1%, 17% and 82% of clones with 0, 1, 2, and 3 mutations, respectively. A minor fraction of heavy chain clones was expected to have 4 mutations due to positions 100l and 100m being randomized together. Sequencing of 96 clones from each library confirmed the randomization of each position (not shown), although not all amino acid mutations could be observed in every position due to the limited sampling depth. About 22% and 35% of the light and heavy chain libraries had full-length randomized clones, enough to cover all the potential diversity of the design with the 1010 independent clones generated even with moderate incorporation biases in oligonucleotide synthesis and library construction.
Both libraries were selected by binding to biotinylated CMV gH/gL glycoprotein complex from strain VR1814 followed by competition with excess MSL-109 IgG. After two rounds of selection random clones were sequenced and amino acid mutations scored (Fig. 1). Most sites in each CDR had low frequency mutations, although in a few positions the combined number of mutations could reach about 33% of all sequences. The frequency of mutations in each CDR after three rounds of selection was relatively high, ranging from 57% in CDR H3 to 95% in CDR H2. The selective pressure and number of rounds of selection used appears to reveal not only the most favored mutations, but possibly also lower tier mutations, despite the limited sequencing depth with concomitant noise. No sequences with stop codons were observed in the sequenced clones, indicating efficient enrichment for binders during selection.

Figure 1. Prevalence of mutations in MSL-109 after two rounds of selection of phage by binding to biotinylated CMV strain VR1814 gH/gL followed by competition with 100 nM of MSL-109 IgG. A total of 92 and 88 clones were sequenced for the heavy and light chains respectively. CDR positions with no mutations identified in any of the sequenced clones are omitted for clarity. Mutations occurring in at least 15% of all clones are highlighted in black. MSL-109 CDR positions with somatic mutations are highlighted, with the original germline residues shown in parentheses. Only unambiguous somatic mutations in CDR H3 are indicated in gray.
Only two sites had mutations occurring in at least 15% of clones, both in CDR H2 (Fig. 1). Heavy chain residues 53 and 55 were mutated from Asp to Ser and Thr to Arg in 15% and 20% of the clones, respectively. Position 53 not only had a high frequency of Ser mutations, but also had about 50% of clones with mutations to other residues, with a combined 66% of clones mutated in this position. The mutations in position 53 did not show any obvious pattern of allowed residues, which included small, large, charged, hydrophobic and polar residues. Due to library design constraints, mutations in positions 53 and 55 were not found in combination with each other. However, mutations in each of these two sites were often combined with mutations in other CDRs.
Residues in positions 53 and 55 selected by phage display improve affinity of MSL-109 for gH/gL
The selection and screening strategy we used assumed that the frequency of each mutation correlates with affinity improvements. Therefore, we selected the two most frequent mutations, heavy chain T55R and D53S, for further characterization. Other mutations in heavy chain position 53 were also characterized because these had the highest combined frequency for any site despite the fact that position 53 mutations were in direct competition with the dominant D53S and T55R mutations during selection, whereas mutations in other CDRs were not. This suggests a strong selection against the wild-type residue in position 53, with a preference for Ser. Clones with individual and combined mutations in positions 53 and 55 were tested in a phage IC50 assay to determine affinity improvements (Fig. 2A). The clone with the T55R mutation bound to gH/gL with a 3-fold higher apparent affinity than the parental clone. The clones with the D53T, D53N and D53S mutations individually or in combination with the T55R mutation bound to antigen with 5-fold or higher apparent affinity relative to the parental clone. Clones with mutations in position 53 only or in combination with the T55R mutation were expressed as Fab fragments in Escherichia coli and used in neutralization assays with CMV strain VR1814. All tested mutant Fab fragments neutralized CMV more potently than the parental MSL-109 Fab fragment (Fig. 2B). Additivity of mutations in positions 53 and 55 varied for different residues in position 53. Whereas the D53L and D53F single mutants neutralized CMV slightly better than the D53N single mutant, combination with the T55R mutation resulted in a larger additive effect with the D53N mutation than with the D53L or D53F mutations (Fig. 2B). Surface plasmon resonance (SPR) experiments confirmed that the D53N/T55R double mutant Fab fragment bound to gH/gL with an estimated affinity at least 10-fold higher than the wild-type MSL-109 Fab fragment (Table 1).
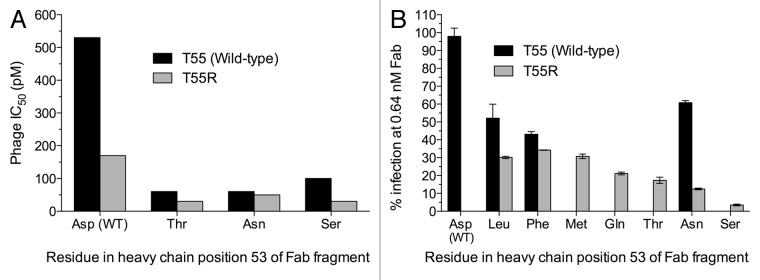
Figure 2. Apparent affinity (A) and neutralization potency (B) of Fab fragments expressed in E. coli. CMV strain VR1814 used as source of antigen and virus for binding and neutralization assays. The single mutants T55R, D53M, D53Q, D53T and D53S were not tested in neutralization assays. Only the neutralization data with Fab at concentration of 0.64 nM of the 5-fold dilution series between 0.128 and 400 nM is shown. Average and range of duplicate neutralization assays are shown.
Table 1. Binding kinetics of Fab fragments with different residues in heavy chain positions 52, 53 and 55 to CMV strain VR1814 gH/gL.
| Variant | Sequencea | kon (M−1s−1)b | koff (s−1)b | KD (pM) |
|---|---|---|---|---|
| MSL-109 | NSDST | 5.2 × 106 | 5.2 × 10−4 | 100 |
| D53N/T55R | NSNSR | 1.0 × 107 | 6.0 × 10−5 | < 10c |
| Germline (IGHV3–21) | SSSSS | 4.6 × 106 | 2.6 × 10−4 | 57 |
| Asp-53 only | SSDSS | 2.9 × 106 | 4.6 × 10−4 | 160 |
| Thr-55 only | SSSST | 2.3 × 106 | 7.7 × 10−4 | 330 |
aSequence between heavy chain Kabat positions 52 and 55 shown. The second residue is position 52a. b Binding kinetics measured at 37 °C. c Upper-limit of estimated KD shown due to kon and koff values close to or above the detection limit.
Natural somatic mutations in MSL-109 heavy chain positions 52, 53 and 55 are not required for high affinity binding
An unexpected result was the relatively strong selection for the D53S mutation and the higher neutralization potency of the D53S/T55R mutant than any other double mutant Fab fragment. The D53S substitution is a reversion of an in vivo somatic mutation back to the germline residue (Fig. 1). In addition, although Thr-55 is the product of an in vivo somatic mutation, our results indicate that an Arg rather than Thr is the optimal residue in position 55 for affinity. Thus, residues 53 and 55 of MSL-109 seem to be suboptimal for affinity. We tested whether reversion of the CDR H2 sequence between positions 52 and 55 to the corresponding germline residues (Fig. 1) would significantly reduce the affinity for CMV VR1814 gH/gL. We included residue 52 in our analysis because a germline Ser residue in position 53 would create a potential glycosylation site at Asn-52. Surprisingly, the Fab fragment with the germline sequence from residue 52 to 55 bound to antigen with an affinity comparable or superior to MSL-109 or variants with the Asp-53 or Thr-55 somatic mutations individually (Table 1). Collectively, the results indicate that the MSL-109 somatic mutations in positions 53 and 55 individually, and positions 52, 53 and 55 combined, have a neutral to deleterious effect on binding affinity relative to the corresponding germline residues.
The D53S and D53N single mutations result in glycosylation of heavy chain positions 52 and 53 and impair antigen binding
The presence of somatic mutations in the CDR H2 sequence between residues 52 and 55 that are neutral or deleterious in the non-glycosylated, E. coli-expressed context suggests that glycosylation on positions 52 and 53 was a critical factor during in vivo selection of the clonal lineage leading to MSL-109. Two of the best mutants identified, D53S/T55R and D53T/T55R, introduce a potential glycosylation site on Asn-52. Another improved variant with the D53N mutation would introduce a potential glycosylation site on position 53 if not combined with the T55R mutation. We tested whether these potential N-linked glycosylation sites are glycosylated in IgG expressed in CHO and human 293 cells. We analyzed the mass of heavy chains without deglycosylation by mass spectrometry (Table 2). As expected, the masses of the heavy chains of variants without the potential glycosylation sites in positions 52 or 53 were all between 51,747 to 52,125 Da (Fig. S1), consistent with the mass of the respective native IgG1 heavy chains with only one N-linked glycan moiety. In contrast, variants with the potential glycosylation sites had heavy chains with higher masses than expected for heavy chains with only one glycosylated site, indicating that these variants were glycosylated on both CDR H2 and the Fc region. An exception was the D53S/T55R mutant, which had heavy chain masses compatible with two distinct subpopulations, either with one or two glycosylated sites, when expressed in either 293 or CHO cells (Fig. S1). The added masses suggest the addition of a complex type carbohydrate moiety to Asn-53 and a smaller high-mannose glycan moiety or a mixture or different glycan types to Asn-52 in IgG expressed in both 293 and CHO cells.
Table 2. Summary of VH region glycosylation of antibody IgG expressed in 293 and CHO cells determined by mass spectrometry.
| VH glycosylation in IgG expressed in: | |||
|---|---|---|---|
| Variant | Sequencea | 293 cells | CHO cells |
| MSL-109 | NSDST | No | NDb |
| D53S | NSSST | Yes | Yes |
| D53N | NSNST | Yes | Yes |
| T55R | NSDSR | No | ND |
| D53S/T55R | NSSSR | Partial | Partial |
| D53N/T55R | NSNSR | No | ND |
aSequence between heavy chain Kabat positions 52 and 55 shown. Potential glycosylation sites are underlined. Mutations from MSL-109 are highlighted. b Not determined.
We determined whether glycosylation of residues 52 or 53 impairs antigen binding in ELISA. Two variants without potential glycosylation sites bound to gH/gL with a better apparent affinity than MSL-109 IgG (Fig. 3A). In contrast, the IgG of two variants that are apparently fully glycosylated in CDR H2, D53S and D53N, bind to gH/gL with an EC50 5 or 25 times higher than MSL-109, suggesting that glycosylation on CDR H2 positions 52 or 53 impairs antigen binding. The D53S/T55R variant glycosylated in CDR H2 in a fraction of the molecules when expressed in 293 cells bound to gH/gL at levels comparable with MSL-109. We tested whether this variant, when expressed in CHO cells, could neutralize virus at levels comparable to the non-glycosylated IgG expressed in E. coli. Non-glycosylated, E. coli-expressed, D53S/T55R double mutant IgG and Fab fragments neutralized CMV strain VR1814 more potently than E. coli-expressed MSL-109 IgG (Fig. 3B). In contrast, the D53S/T55R double mutant IgG expressed in CHO cells neutralized virus with a 5-fold higher IC50 than the glycosylated MSL-109 IgG. These results indicate that glycosylation of CDR H2 on position 52 and 53 impairs antigen binding.

Figure 3. Binding and neutralization of MSL-109 variants with glycosylation in CDR H2. (A) ELISA with IgG expressed in 293 cells. Legends indicate sequence of region between heavy chain residues 52 and 55 of each clone and mutant name. Underlined residues are mutated relative to MSL-109. Asn residues shown in red indicate potential N-linked glycosylation sites. Green and red lines indicate binding of glycosylated and glycosylated MSL-109 variants, respectively. (B) Virus neutralization assay with D53S mutant Fab and IgG expressed in E. coli and CHO cells. Legends indicate mutated and potential glycosylation sites.
The D53N/T55R double mutant antibody neutralizes CMV strains from different groups
One possible reason for the presence of somatic mutations in MSL-109 with seemingly neutral and deleterious effects on affinity is that phage selection simply changed residues that were originally optimized in vivo for binding to gH/gL from an unidentified CMV strain to residues that favor binding to gH/gL from strain VR1814. That is, instead of affinity maturation, phage selection may have simply shifted strain specificity. We tested whether the mutations identified by phage display improve neutralization of divergent CMV strains as a proxy for binding affinity. We selected four additional strains that included members from both gH protein sequence branches (Fig. S2). The D53N/T55R double mutant IgG neutralized all tested strains more potently than the wild-type MSL-109 IgG with an average reduction in neutralization IC50 of about 5-fold. This indicates that the mutations identified by phage display replace neutral or deleterious somatic mutations with residues that improve binding affinity for the gH/gL protein of divergent CMV strains (Table 3).
Table 3. Potency of neutralization of diverse human cytomegalovirus strains by parental and mutant MSL-109 antibodies.
| Neutralization IC50 (pM) by IgG variant: | |||
|---|---|---|---|
| CMV strain | MSL-109 | D53N/T55R | Fold improvement |
| VR1814 | 4,850 | 730 | 6.6 |
| Brown | 2,560 | 570 | 4.5 |
| Dement | 350 | 60 | 5.8 |
| Cano | 950 | 190 | 5.0 |
| TR | 1,810 | 490 | 3.7 |
The D53N/T55R double mutation does not alter off-target binding of parental MSL-109
One possible reason for MSL-109 not having the mutations identified by phage display is that in vivo affinity maturation may select against non-specific binding to endogenous antigens, whereas in phage display there is no such selection. This is particularly relevant for therapeutic antibodies as non-specific binding can potentially lead to faster than expected clearance and reduced in vivo potency.9 We tested five double mutants and the single-site T55R mutant for potential off-target binding using a baculovirus (BV) particle non-specific ELISA that correlates with in vivo antibody clearance in humans and cynomolgus monkeys.10 All variants tested had low binding in the non-specific ELISA similar to the parental MSL-109 and negative control antibodies (Fig. S3), indicating that phage-selected mutations do not significantly alter off-target binding.
Discussion
Here, we used phage display libraries and CDR single-site mutagenesis to identify mutations in MSL-109 that favor binding to CMV gH/gL. Our mutation scan procedure identified amino acid substitutions at positions in CDR H2 that can be combined to produce an antibody with high affinity to CMV gH/gL and increased neutralization potency without an apparent increase in off-target binding. Although our mutational procedure did not allow for combinatorial effects between the heavy and light chains or different mutations within the same CDR, it did enable optimization through combinations of individual mutations in different CDRs of the same polypeptide chain. Nonetheless, our top variant came from combining two substitutions in CDR H2. Randomization schemes allowing combination of mutated residues in the same CDR could also lead to identification of this or possibly other combinations of mutations with synergistic effects that do not introduce glycosylation sites. However, this requires larger library sizes to include as many mutation combinations as possible in the library, which can be limiting in eukaryotic display systems and even in phage display, and may not lead to mutants that are detectably better than the D53N/T55R mutant, given that the estimated affinity of this mutant is beyond the limit of detection of most SPR systems (KD < 10 pM). One limitation of the procedure described here is that lower frequency mutations are found in most sites in the selected clones, as shown in Figure 1. The relatively low sequencing depth we used allowed us to determine the significance only of higher frequency mutations, such as heavy chain D53S and T55R, or sites with an unusually low frequency of the wild-type residue, such as heavy chain position 53, directly from the sequencing data. The other lower frequency mutations we found in other sites could be neutral for affinity, and therefore simply noise, or could add to binding affinity, which can only be determined by follow-up experiments or by deep sequencing of libraries and selected clones to identify mutations with minor contributions to affinity with confidence.11
Our analysis indicates that the somatic mutations introduced during in vivo affinity maturation in the CDR H2 region have a neutral to slightly deleterious effect on binding affinity in the context of the final MSL-109 sequence. There were two types of apparent constraints on affinity maturation of MSL-109, one created by introduction of potential glycosylation sites and another by codon usage. The glycosylation constraints were created both by the germline sequence and by somatic mutations introduced during in vivo affinity maturation whereas the codon usage restrictions were caused solely by one of the somatic mutations. The S52N somatic mutation constrains position 53 from mutating to Ser or Thr due to the creation of a glycosylation site that impairs binding. In addition, Asp-53 cannot mutate to Asn because the presence of a Thr in position 55 creates a glycosylation site. The Thr-55 is also a product of in vivo somatic mutation, but the germline Ser-55 should also preclude position 53 from mutating to Asn due to glycosylation. Consequently, introducing Asn, Ser or Thr in position 53 requires a preceding or simultaneous mutation of Asn-52 or Thr-55. Codon usage constraints occur in both codons 53 and 55. In position 53, other alternatives are possible, including Leu, Phe, Met, Gln and Thr (Fig. 1B). All these amino acid mutations require two simultaneous nucleotide changes from the Asp codon in MSL-109 or an intermediate Ala, Val, Tyr or His amino acid to be selected. Of these, only Ala and, to a lesser extent, Val were present in position 53 in the selected clones (Fig. 1). A codon usage constraint was also introduced by mutation of the germline Ser-55 to Thr. Changing this Thr to an Arg codon requires two or three simultaneous nucleotide transversions, an intermediate reversion to germline Ser or silent mutation to a different Thr codon that would have no selective advantage. Notably, the germline Ser-55 encoded by an AGC codon could have mutated to an Arg codon by a single C to A/G or A to C transversion. Instead, in the lineage leading to MSL-109 position 55 was mutated to an ACC Thr codon, making a subsequent mutation to Arg at that site less likely. The reason for this outcome, either chance or selection against Arg in position 55 in vivo, is not clear. It is also not known if other clones derived from the same naïve B cell as MSL-109 had maturation pathways that led to the Asn/Ser/Thr-53 and Arg-55 configuration identified by phage display. It should be noted that generation of the S53D somatic mutation in vivo should also have been constrained by an intermediate, glycosylated, S53N or a possible suboptimal S53G mutation. Alternatively, the S53D somatic mutation could have arisen from a two-nucleotide mutation in codon 53, bypassing either of these intermediates. Although both codon usage and glycosylation could constrain affinity maturation of MSL-109 in vivo, an additional barrier to in vivo affinity maturation of MSL-109 may be simply the fact that this clone may already have reached the affinity ceiling for effective clonal selection in vivo.
The limit imposed by glycosylation on affinity maturation may be a relatively common situation in natural human antibodies. Analysis of the codon usage of the most used human variable region germlines indicates that about 13% of CDR codons cannot mutate to Asn, Ser or Thr by 1 bp mutations without a previous or simultaneous change of other residues due to the creation of potential glycosylation sites (Fig. S4). Single-site saturation scans in conjunction with bacterial-based phage display can identify affinity-enhancing mutations that cannot be selected in vivo or in eukaryotic cell systems and indicate ways to use those mutations without introducing glycosylation sites deleterious to binding.
Materials and Methods
Construction and selection of phage-displayed Fab fragment libraries
A previously described phagemid vector12 was modified by standard molecular biology techniques to construct a phagemid designed to display MSL-109 Fab fragments on the surface of M13 bacteriophage as a fusion with the N-terminus of a fragment of the gene-3 minor coat protein. An amber stop was introduced before the g3 sequence to allow expression of Fab fragments directly from phagemid clones. The phagemid was used as the template to construct phage-displayed libraries containing 1010 unique members as described.13,14 Phage displaying Fab fragments were produced as previously described.14 Binding clones were selected by incubating the phage display libraries with 1 and 0.1 nM biotinylated gH/gL in successive rounds of selection, each round with a post-binding competition step with 100 nM MSL-109 IgG for 1 h at room temperature to select against lower affinity clones. Bound clones were captured on ELISA plates coated with neutravidin or streptavidin, washed with PBS and eluted in 10 mM HCl for 10 min at room temperature. The eluted phage was neutralized with 1/10 volume of 1 M Tris pH 8.0 and used to infect E. coli XL1-Blue (Agilent Technologies, Cat. 200249) for amplification for the next round of selection. Clones from the second round of selection were sequenced to determine mutation frequencies. The apparent affinity of phage clones with favored mutations was estimated by a competition phage ELISA as described.15 The IC50 was calculated by nonlinear regression using the GraphPad Prism version 5 software.
Expression of IgG in mammalian cells and purification
Heavy chain variable regions from selected phage clones were subcloned into a human IgG1 heavy chain mammalian expression vector.16 The heavy chain expression vectors were co-transfected into CHO or 293T cells with a mammalian expression vector expressing MSL-109 light chain at a 1:1 ratio using JetPEI (Polyplus Transfection, Cat. 101–10N) and cultured for 5 (293T) or 14 d (CHO). Human IgG1 was purified from culture supernatants by protein A affinity chromatography and analyzed by gel filtration chromatography.
Expression of IgG in E. coli and purification
MSL109 and MSL109 IgG variants were cloned into an E. coli expression vector and expressed in E. coli strain 64B4 as previously described.17 IgG was purified from culture supernatants by protein A affinity chromatography and analyzed by gel filtration chromatography.
Expression of Fab fragments in E. coli and purification
E. coli 62A7 non-suppressor cells were transformed with selected phagemid clones, inoculated in C.R.A.P. phosphate-limiting media17 containing 50 μg/mL carbenicillin and grown for 24 h at 30 °C with shaking. Fabs were purified from culture supernatants by protein A affinity chromatography. The eluted Fabs were dialyzed against PBS and analyzed by size-exclusion chromatography.
Fab fragment affinity determinations by BIAcore
To determine the binding affinity of Fab fragments by single-cycle kinetics, SPR measurement with a BIAcoreTM T100 instrument was used. Briefly, a series S sensor chip CM5 (GE Healthcare, Cat. BR100530) was activated with EDC and NHS reagents according to the supplier’s instructions, and streptavidin (Thermo Scientific, Cat. 21122) was coupled to achieve approximately 2000 response units (RU), followed by blocking un-reacted groups with 1 M ethanolamine. For kinetics measurements, biotinylated CMV gH/gL was first injected at a 10 µl/min flow rate to capture approximately 25 RU on 3 different flow cells (FC), except for FC1 (reference), and then 5-fold serial dilutions of Fab fragment in HBS-P buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 0.005% surfactant P20) from low (1.6 nM) to high (1 µM) were injected (flow rate: 30 µl/min) one after the other in the same cycle with no regeneration between injections. The sensorgram was recorded and subject to reference and buffer subtraction before evaluating by using BIAcoreTM T100 Evaluation Software (version 2.0). Association rates (kon) and dissociation rates (koff) were calculated using a simple one-to-one Langmuir binding model. The equilibrium dissociation constant (KD) was calculated as the ratio koff/kon.
Cloning, expression and purification of CMV soluble proteins
Purified glycoproteins in this study are from HCMV strain VR1814. For gH/gL expression, gH/gL residues 27 to 718 of gH and 41 to 278 of gL were each cloned into a modified pAcGP67A vector (PharMingen, Cat. 554756), resulting in the addition of three N-terminal residues (AGS) and a C-terminal His-tag. For protein production T.ni insect cells were grown to a density of 2 million cells/ml and co-infected with recombinant gH and gL stocks. Cell supernatants were harvested 72 h post-infection and protein was bound to a Ni affinity chromatography column and eluted with 50 mM HEPES pH 7.5, 150 mM NaCl and 500 mM Imidazole. Protein was and further purified on a S200 gel filtration column (GE Healthcare) in 25 mM HEPES pH 7.5, 150 mM NaCl buffer (gH/gL). The protein was concentrated to 2–10 mg/ml.
Mass spectrometry
Approximately 2 µg IgG sample was reduced with 100 mM dithiothreitol at 37 °C for 20 min. Polypeptide chains were separated with an Agilent PLRP-S reversed phase column and analyzed on an Agilent 6224 TOF LC-MS instrument. Intact masses were obtained by Maximum Entropy deconvolution using the MassHunter software (Qualitative Analysis B.03.01).
Phylogenetic analysis of the gH protein of CMV strains
The following strains were obtained from Dr Jay Nelson (University of Oregon Health and Science University, OHSU): Adinis, Brown, Cano, Davis, Dement, Grunden, Harris, Keone, Lysistrata, NewRock, Phoebe, Powers, Salvo, Schmoe, Simpson, and Watkins. The following strains were obtained from Dr Sunwen Chou (OHSU): C079, C323, C327, C336, C352, C353, and C359. VR1814 was obtained from Dr Maria Grazia Revello (Servizio di Virologia).18 Cells infected by each strain were lysed and DNA was extracted using the DNA blood/tissue extraction kit (Qiagen, Cat 69581). Primers were designed to conserve sequences according to an alignment of AD169, FIX, TB40E, Toledo, and Towne sequences available in GenBank (National Center for Biotechnology Information [NCBI]) database). The gH gene was amplified from each clinical strain from the start codon through base 2196, just short of the stop codon. The polymerase chain reaction (PCR) products were sequenced using dye-terminator reactions. Additional gH sequences from CMV strains from the United States, Japan, England, and Europe were obtained from the NCBI database. Glycoprotein sequences without the signal peptides were aligned using ClustalW2 and clustered into a tree.
Cells, viral strains, and antibodies
Fibroblast MRC-5 cells obtained from American Type Culture Collection (ATCC) were grown, using ATCC’s suggested conditions. Human cytomegalovirus strains VR1814, Brown, Dement, Cano and TR were grown in human HUVEC cells and virus in supernatant was concentrated by ultracentrifugation, resuspended in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal calf serum, and frozen at -80 °C.
Neutralization assay
Neutralization assays were performed as previously described19 but with infected nuclei detected using anti-IE antibody Mab810 (Millipore, Cat. MAB810-500UG) followed by Alexa Fluor 488 conjugated secondary (Invitrogen, Cat. ab150113). Total nuclei were stained with Hoechst (Invitrogen, Cat. H1399) and cells were imaged and counted using the ImageXpress Micro and MetaXpress software (Molecular Devices). Data were graphed using Prism (GraphPad Software), and EC50 values were calculated from best-fit curves using the GraphPad Prism version 5 software.
BV particle ELISA
The non-specific BV particle ELISA was performed as previously described.10
Supplementary Material
Disclosure of Potential Conflicts of Interest
All authors are current or past employees of Genentech.
Acknowledgments
We thank Germaine Fuh for helpful discussions, Pamela Chan and Renuka Sibia with the BV particle ELISA, and Rong Deng for review of the manuscript.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27875
References
- 1.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 2.Poulsen TR, Meijer PJ, Jensen A, Nielsen LS, Andersen PS. Kinetic, affinity, and diversity limits of human polyclonal antibody responses against tetanus toxoid. J Immunol. 2007;179:3841–50. doi: 10.4049/jimmunol.179.6.3841. [DOI] [PubMed] [Google Scholar]
- 3.Poulsen TR, Jensen A, Haurum JS, Andersen PS. Limits for antibody affinity maturation and repertoire diversification in hypervaccinated humans. J Immunol. 2011;187:4229–35. doi: 10.4049/jimmunol.1000928. [DOI] [PubMed] [Google Scholar]
- 4.Foote J, Eisen HN. Kinetic and affinity limits on antibodies produced during immune responses. Proc Natl Acad Sci U S A. 1995;92:1254–6. doi: 10.1073/pnas.92.5.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batista FD, Neuberger MS. Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity. 1998;8:751–9. doi: 10.1016/S1074-7613(00)80580-4. [DOI] [PubMed] [Google Scholar]
- 6.Kauffman SA, Weinberger ED. The NK model of rugged fitness landscapes and its application to maturation of the immune response. J Theor Biol. 1989;141:211–45. doi: 10.1016/S0022-5193(89)80019-0. [DOI] [PubMed] [Google Scholar]
- 7.Macken CA, Perelson AS. Protein evolution on rugged landscapes. Proc Natl Acad Sci U S A. 1989;86:6191–5. doi: 10.1073/pnas.86.16.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voigt CA, Kauffman S, Wang ZG. Rational evolutionary design: the theory of in vitro protein evolution. Adv Protein Chem. 2000;55:79–160. doi: 10.1016/s0065-3233(01)55003-2. [DOI] [PubMed] [Google Scholar]
- 9.Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368:652–65. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 10.Hötzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs. 2012;4:753–60. doi: 10.4161/mabs.22189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–8. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee CV, Liang WC, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol. 2004;340:1073–93. doi: 10.1016/j.jmb.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 13.Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol. 2004;338:299–310. doi: 10.1016/j.jmb.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 14.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods Enzymol. 2000;328:333–63. doi: 10.1016/S0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 15.Bostrom J, Lee CV, Haber L, Fuh G. Improving antibody binding affinity and specificity for therapeutic development. Methods Mol Biol. 2009;525:353–76, xiii. doi: 10.1007/978-1-59745-554-1_19. [xiii.] [DOI] [PubMed] [Google Scholar]
- 16.Eaton DL, Wood WI, Eaton D, Hass PE, Hollingshead P, Wion K, Mather J, Lawn RM, Vehar GA, Gorman C. Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochemistry. 1986;25:8343–7. doi: 10.1021/bi00374a001. [DOI] [PubMed] [Google Scholar]
- 17.Simmons LC, Reilly D, Klimowski L, Raju TS, Meng G, Sims P, Hong K, Shields RL, Damico LA, Rancatore P, et al. Expression of full-length immunoglobulins in Escherichia coli: rapid and efficient production of aglycosylated antibodies. J Immunol Methods. 2002;263:133–47. doi: 10.1016/S0022-1759(02)00036-4. [DOI] [PubMed] [Google Scholar]
- 18.Grazia Revello M, Baldanti F, Percivalle E, Sarasini A, De-Giuli L, Genini E, Lilleri D, Labò N, Gerna G. In vitro selection of human cytomegalovirus variants unable to transfer virus and virus products from infected cells to polymorphonuclear leukocytes and to grow in endothelial cells. J Gen Virol. 2001;82:1429–38. doi: 10.1099/0022-1317-82-6-1429. [DOI] [PubMed] [Google Scholar]
- 19.Abai AM, Smith LR, Wloch MK. Novel microneutralization assay for HCMV using automated data collection and analysis. J Immunol Methods. 2007;322:82–93. doi: 10.1016/j.jim.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.