

Abstract
Viral entry targets with therapeutic neutralizing potential are subject to multiple escape mechanisms, including antigenic drift, immune dominance of functionally irrelevant epitopes, and subtle variations in host cell mechanisms. A surprising finding of recent years is that potent neutralizing antibodies to viral epitopes independent of strain exist, but are poorly represented across the diverse human population. Identifying these antibodies and understanding the biology mediating the specific immune response is thus difficult. An effective strategy for meeting this challenge is to incorporate multiplexed antigen screening into a high throughput survey of the memory B cell repertoire from immune individuals. We used this approach to discover suites of cross-clade antibodies directed to conformational epitopes in the stalk region of the influenza A hemagglutinin (HA) protein and to select high-affinity anti-peptide antibodies to the glycoprotein B (gB) of human cytomegalovirus. In each case, our screens revealed a restricted VH and VL germline usage, including published and previously unidentified gene families. The in vivo evolution of paratope specificity with optimal neutralizing activity was understandable after correlating biological activities with kinetic binding and epitope recognition. Iterative feedback between antigen probe design based on structure and function information with high throughput multiplexed screening demonstrated a generally applicable strategy for efficient identification of safe, native, finely tuned antibodies with the potential for high genetic barriers to viral escape.
Keywords: monoclonal antibodies, human antibodies, neutralizing antibodies, broadly protective antibodies, immunoglobulin germline, viral epitopes, fusion, influenza, cytomegalovirus
Introduction
Advances in the ex vivo culture, stimulation and cloning of antibody producing B cells from immune blood donors has vastly expanded the possible repertoire of human antibody therapeutics, whose importance was recognized at the outset of human antibody cloning by hybridoma methods.1 For example, accessing the functional successes of in vivo humoral immune system defenses, which have evolved side-by-side with dynamic infectious agents, has allowed the cloning of broadly neutralizing antibodies to complex infectious diseases using a variety of approaches.2-7 A fascinating trend is the discovery of specific Ig germline usage among unrelated and geographically disperse individuals against specific viral antigens.3,8 A parental germline sequence has not generally been anti-viral, but rather provides the best possible scaffold for the development of an affinity-matured, efficacious monoclonal antibody (mAb). Co-crystal structures of antigen and antibody have demonstrated a structural basis for this trend.3,8 This knowledge, however, does not make it any less formidable to clone the optimal mAb from an individual’s polyclonal response, particularly in the context of active viral selection toward immune evasion. It is also likely that the history of exposure to disease, vaccines and allergens will provide certain individuals with better antibody reservoirs than others. Moreover, viruses can also cripple the innate immune response as part of their strategy for survival, adding additional variability to the population response to infection.9
An appreciation of the complexity and diversity of antibody responses in the human population and the resulting rarity of broadly protective memory B cell clones led to the development of a number of human antibody cloning technologies.10,11 Herein, we employed a multiplexed screening process to enable an in-depth characterization of the specificity of naturally occurring antibodies secreted from single memory B cells. Deeming multiplexing a critical component to discovering anti-viral antibodies with cross-clade activity, we counteracted the associated rapid drop in hit frequency with high throughput and miniaturized assay technologies.12
We multiplexed the highly variable influenza A hemagglutinin (HA) fusion protein for antibody discovery using recombinant protein derived from different viral clades and years. Previous studies had shown this target and mechanism to be a good alternative to neuramidase inhibitors for therapy of influenza infections.13 Without a priori knowledge of the best neutralizing epitope, we postulated that some hits would be functional neutralizing mAbs if they bound critical regions conserved among HA subtypes, since conservation of a site in a rapidly mutating virus presumably reflects a critical function. In this way, we discovered antibodies to discontinuous epitopes conserved over many years of influenza A evolution. The biological activity of the subcloned and recombinantly produced mAbs provided direct support for the screening hypothesis.
The functional potency of an antibody can be driven by both affinity and fine epitope specificity; therefore, once a therapeutic epitope has been defined, it is valuable to find B cell clones with the optimal corresponding paratope. To this end, we applied a multiplexed, “affinity metric,” process to identify and selectively clone high-affinity human cytomegalovirus (HCMV) antibodies to known functional epitopes from serum antigen positive human blood donors.12 Specifically, we directed our efforts to AD-2, site I, a rare and poorly immunogenic neutralizing epitope of the abundant HCMV viral surface protein gB.14 Human mAb to site I of AD-2 have been demonstrated to potently cross-neutralize HCMV entry into a broad range of host cell types.7 The use of an optimal, high- affinity antibody in HCMV is of particular importance because low affinity antibodies may be non-neutralizing7 and exacerbate disease by allowing antibody-bound virus access to cells via the neonatal Fc receptor (FcRn).15 Our approach identified a panel of naturally occurring human IgG antibodies that bind equally to both low and high density AD-2 coated particles, thereby selecting for high-affinity antibodies directed to AD-2 based on single arm (Fab) binding events. The subsequent testing of functional potency by cross-neutralization allowed the selection of the optimal therapeutic epitope-paratope fit from in vivo complementary-determining region (CDR) diversification of the known restricted Ig germline usage.8,14
Results
Multiplexed screening strategy for the discovery of optimal antibodies
Building on the previously described CellSpot™ methods,12 human antibodies to viral antigens were discovered starting from frozen vials containing 1 × 106 peripheral blood mononuclear cells (PBMCs) prepared from single blood donors under informed consent. Cells were plated on feeder cells in a master plate in the presence of mitogens and cytokines that stimulate memory B cell proliferation and antibody secretion. An aliquot of the cells was then transferred to a replica plate for use in a destructive, multiplexed phenotyping process to identify the rare wells containing antigen positive (Ag+) B cells meeting the multiple selection criteria.
First, the cells were lightly spun down to the bottom of a well coated with a goat anti-human Fc capture antibody specific for the IgG class, and allowed to incubate for 3 h to capture secreted antibody on the plate in the immediate vicinity of cell (secreted antibody footprint is ~150 µm in diameter). The cells were subsequently lysed, the plate washed, and the expressed antibody footprint of an individual B cell detected by binding up to five distinguishable antigen coated fluorescent beads (300 nm diameter). High throughput digital microscopy and image processing provided a quantitative readout of the number of each bead type bound by the secreted IgG in a given footprint. Once a desired B cell antibody footprint was identified, siblings still growing in the corresponding well of the master plate were recovered by limiting dilution dispersal into 384-well plates and detected using a lower resolution, but non-destructive, imaging process (i.e., with no cell lysis). Recovered single Ag+ B cells were lysed and the mRNA for the antibody cloned by single cell RT-PCR using mixtures of degenerate primers covering all the human VH and VL genes. For each antibody, 2–8 siblings of a single B cell clone were recovered from the master well. An important feature of the process is the correlation between sibling cell phenotype and genotype. In the primary screen (in the replicate plates), the ability to detect replicates reduces both the false negative and false positive rates. In the limiting dilution cloning step, it provides multiple independent opportunities to clone the VH/VL genes, improving the success rate of the single cell cDNA cloning step.
Human blood donors vary widely in their frequency of high-quality neutralizing antibodies for a particular pathogen. Therefore, we used the CellSpot™ discovery platform first in a survey mode to identify the best blood donors, reserving the more labor-intensive cloning step for use on the most promising blood samples. The incorporation of secondary screening criteria into the primary screen required cloning of relatively few mAbs (~25), to identify a lead mAb.
Influenza A antibody discovery
We aimed to identify antibodies to the HA protein of a wide range of influenza A viruses. Influenza A is categorized into 16 different clades in two families designated as group 1 and 2 and further HA variation occurs among strains within a particular clade. The highest sequence conservation of HA molecules across clades is in the fusion domain (color-coded sub-regions of the fusion domain are shown in Table 1). Challenging the HA sequence conservation, however, is a lack of corresponding structural conservation, particularly between group 1 and 2. In Figure 1, positions in the native trimer of the β strand (blue), stalk (green) and N-terminal region (orange) surrounding the fusion peptide (yellow) are seen to be markedly distinct in group 1 vs. group 2 trimers.
Table 1. Alignment of HA protein surrounding stalk region and epitopes of antibodies.
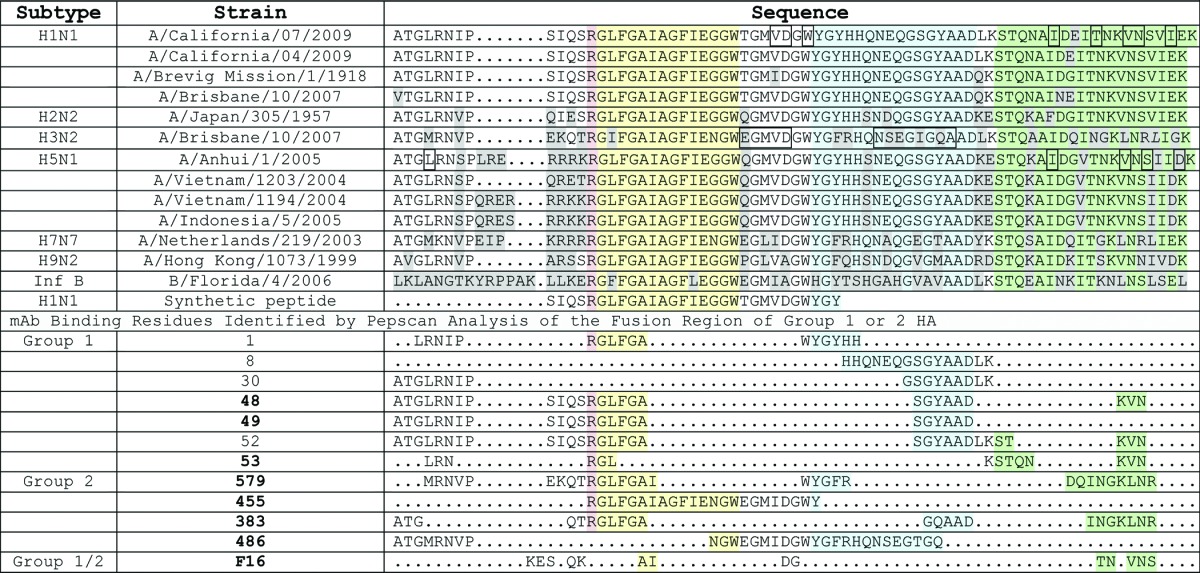
Residues are highlighted according to conserved HA structural domains (see Fig. 1) surrounding the stalk domain(in green). The Arginine colored red is conserved among HA proteins and is cleaved by extracellular proteases to trigger the fusion conformation. In sequence order: a loop region (yellow)is next to an anti-parallel β-sheet (blue),followed by the alphα-helical stalk region (green). Anti-HA antibodies (neutralizers in bold) with Pepscan analysis data are shown in alignment. Boxed residues are key contacts in the F10 antibody (H1N1), CR6261 (H5N1) or CR8020 (H3N2).

Figure 1. Structures of influenza A group 1 (A) and group 2 (B) hemagglutinin. Published crystal structures for H1N1 HA (pdb 3AL4) and H3N2 HA (pdb 1MQM) were compared by simplifying the trimers into “pretty” monomer formats revealing the tertiary positions of the sequential N-terminal HA1 region (orange), pre-fusion (light blue) and fusion region (yellow; with arginine 329 shown in red) loops, anti-parallel β sheet (royal blue) and finally the α-helical stalk region (green). Shown just below the pretty models are the corresponding 3D crystal structures with the discontinuous epitopes of the lead group 1 (mAb 53) and group 2 (mAb 579) antibodies, determined by PepScan methods. The identified epitope regions are colored according the fusion region domains as described above.
Pan-influenza antibody discovery was initially attempted using beads conjugated with a synthetic HA fusion peptide (as shown in the yellow highlighted region of the HA alignment, Table 1), but this did not work because of excessive non-specific binding. Instead, we successfully multiplexed our antibody discovery using monomers of five HA clades with pandemic potential: H1, H5, and H9 (group 1), H3 and H7 (group 2); together, the probes used span > 10 y of viral evolution. This combination of beads was used first in survey mode to screen over 100 blood donors, of which 28 were selected for B cell cloning. This approach eliminated clade restricted antibodies and found 22 high-affinity antibodies with cross-reactivity to HA from multiple group 1 clades. Seven of these cross-reactive antibodies were found to bind to the fusion region of HA with different kinetic profiles, and three of these showed potent neutralizing activity (Fig. 2). Data showing binding of these mAb to peptides spanning the fusion region of H1N1 as well as their clade cross-reactive, HA binding profiles are provided in supplementary data (Fig. S1 and S2).

Figure 2. Kinetic binding and neutralizing activity of a panel of antibodies cross-reactive to the fusion region of group 1 HA. (A) Comparison of the binding kinetics of mAb 1, 8, 30, 48, 49, 52 or 53 (captured on an anti-human Fc sensors), to 200nM of HA monomer of the H1N1 A/CA/04/2009 strain. (B) Comparative fusion peptide binding (gray bars) as a percent relative to a rabbit pAb directed to and purified by the fusion peptide; and neutralizing activity of antibodies (colored bars), at a constant IgG concentration of 5 µg/ml in MDCK cells infected with the H1N1 A/CA/04/2009 virus. Plaques were counted in duplicate wells and expressed as a percent of plaques in an uninhibited infection. A comparison of the breadth and potency of HA coverage for selected neutralizing group 1 antibodies can be found in Table 4 and Figure S2.
Our best group 1 high-affinity neutralizing antibody, mAb 53, (Table 2), did not neutralize group 2 viruses nor did it bind to group 2 HA in trimer form (Table 3 kinetic binding; Table 4 neutralization), although it did bind to H7 HA monomers. These data suggest a greater conservation of epitope sequence than of structure. Having learned this lesson in our group 1 screen, we only used trimeric H3 and H7 for a subsequent group 2 directed screen, including strains spanning > 40 y of viral evolution. From 38 mAbs cloned from 7 donors, we discovered mAb 579, a group 2 specific, broadly neutralizing antibody with high-affinity binding for the HA trimeric state (Table 3 kinetic binding and Table 4 neutralization). Five antibodies (mAbs 699–723) closely related in sequence to mAb 579, (Table 2), from the same donor did not cross-neutralize (Table 4). Notably, the mAbs we cloned that bind to both group 1 and 2 HA were either restricted in neutralizing activity by strain or non-neutralizing (e.g., mAb 486, Fig. 3).
Table 2. (A) Comparative VH regions of human mAb for influenza A groups 1 and 2. (B) Comparative VL regions of human mAb for influenza A groups 1 and 2.
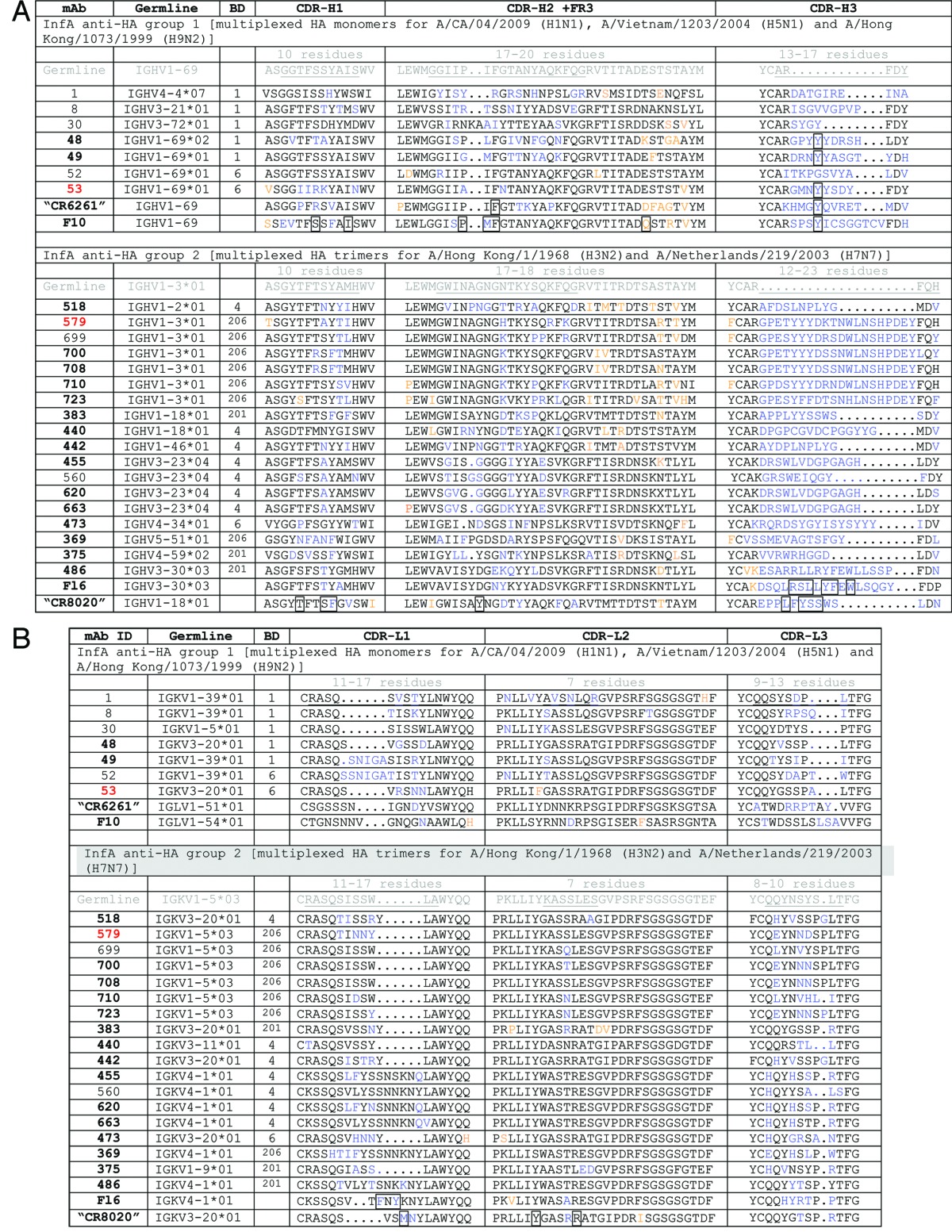
Table 3. Kinetic binding of optimal influenza A antibodies.
–, no measureable activity
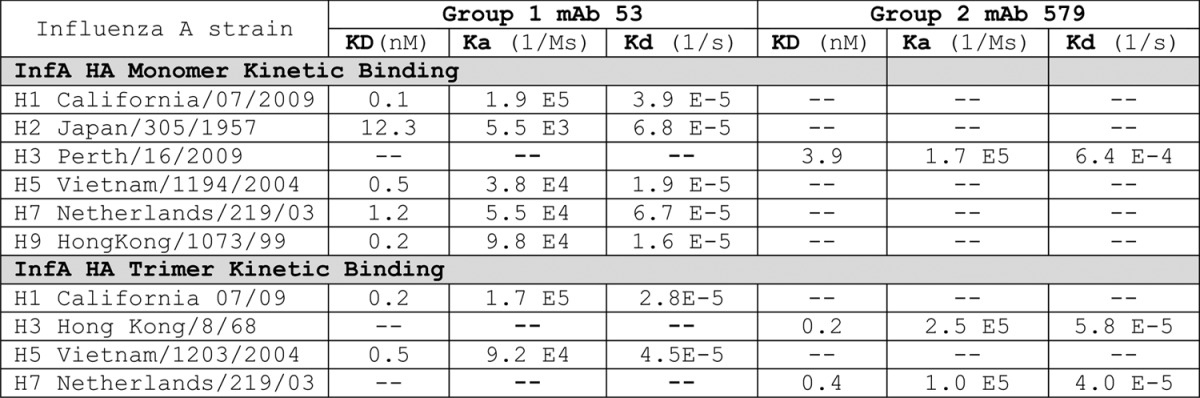
Table 4. Neutralization IC50(μg/ml)of influenza antibodies in MDCK plaque formation assay.
–, no measureable activity; blank not done; ^ only neutralizing if antibody allowed to bind virus prior to activation with trypsin. Group 1 mAb 48 and 49 did not bind to the H2 strain. All of the other antibodies listed in this table showed high affinity binding to the breadth of group 1 or group 2 HA proteins, respectively, corresponding to the viruses tested. Only mAb 486 showed high affinity binding to both group 1 and 2 HA proteins in monomeric and trimeric forms. †All wild bird viruses kindly provided by Dr David Stallknecht (UGA). ‡Reverse genetics virus containing modified H5 and N1 of A/Vietnam/1203/2004 and approved for non-BSL3 containment. Gift of Dr Ruben Donis (CDC).

Figure 3. Kinetic binding and limited neutralizing activity of group 1 and group 2 HA cross-reactive mAb 486. (A) Comparison of the binding kinetics of mAb 486 after capture on an anti-human Fc sensors, to 200 nM of HA trimers representative of group 1 (H1 California 07/09 and H5 Vietnam/1203/2004) and group 2 (H3 Hong Kong/8/68 and H7 Netherlands/219/03). (B) Plaque formation neutralization activity of mAb 486, rabbit pAb directed to the fusion peptide, or mAb 579 of H1 A/CA/04/09 or H3 A/Perth/16/09 infected MDCK cells, with (black) or without (blue) prior trypsin activation of viruses.
Human cytomegalovirus (HCMV) antibody discovery
For this screen, we targeted the known neutralizing linear site I, AD-2 epitope of glycoprotein B (gB). Blood donor sera were first screened by a direct ELISA against the AD-2 peptide and a recombinant gB protein (data not shown). To create an affinity metric, two populations of AD-2 linked beads were prepared: one with a high density of antigen and one with a low density. A third population of beads conjugated with the recombinant gB was used to select for specificity and native binding of antibodies to the AD-2 epitope. To aid in the development of optimal antigens and conditions for bead coupling methods, we used the previously discovered mAb 4A2, cloned in-house, that we wished to improve upon 10-fold. Using a bead-based ELISA method, we determined that the low and high density coated AD-2 beads could be detected by 4A2 with EC50 values separated by 10-fold (Fig. 4A). Upon mutliplexing both AD-2 bead types and gB beads, the low density AD-2 beads showed a reduction in 4A2 binding EC50 and saturation, consistent with competition from the high density AD-2 bead population. And, as expected, the high density bead binding by 4A2 was unchanged in the multiplex format (Fig. 4A). These optimized beads were then tested in CellSpot™ mode using the affinity metric approach, and shown to be effective in differentiating high (mAb 310) and low (mAb 300) affinity antibodies secreted by memory B cells (Fig. 4B and C). Using this 3-plex approach we identified one blood donor with an extensive natural reservoir of in vivo affinity matured anti-AD2 antibodies with functional viral neutralizing activity. Thirty closely related, high-affinity antibodies were cloned from ~2 million memory B cells screened from this blood donor. Selected mAb sequences are shown in Table 5 and affinities and neutralization potencies are shown in Table 6.

Figure 4. Development of a multiplexed strategy for discovery of high-affinity antibodies to the neutralizing site I, AD-2 region of the gB protein on human CMV. (A) A bead-based ELISA was used to track the optimization of low and high density AD-2 beads with a 10-fold binding difference to a benchmark reagent, mAb 4A2. As desired, binding of 4A2 to the low density beads was reduced when multiplexed with high density and gB beads, whereas high density beads bound 4A2 equally in single or multiplexed formats. With these bead properties we expected to be able to screen and differentiate mAb at least 10-fold improved in AD-2 binding over 4A2. (B) Digital fluorescent microscopy was used to count and quantify antibody binding to low affinity beads (high density, 20% AD-2 coating in 1% BSA), high-affinity beads (low density, 1% AD-2 coating in 1% BSA) and gB beads. Data was transformed to a bar graph format and an “affinity metric” value was assigned. A high-affinity antibody would bind equally, without bias, to both densities of AD-2 coated beads and have a calculated “affinity metric” of 1. Low-affinity antibodies would preferentially bind the high density AD-2 coated beads, and demonstrate affinity metric values < 1. Antibodies that cannot bind the peptide in the context of the native gB protein will show no signal on the gB-ECD coated beads. (C) Kinetic AD-2 binding sensorgrams for the high- (affinity metric = 0.87) and low- (affinity metric = 0.15) affinity monoclonal antibodies corresponding to these CellSpot™ phenotypes.
Table 5. (A) Comparative VH regions of human mAb for human cytomegalovirus. (B) Comparative VL regions of human mAb for human cytomegalovirus.
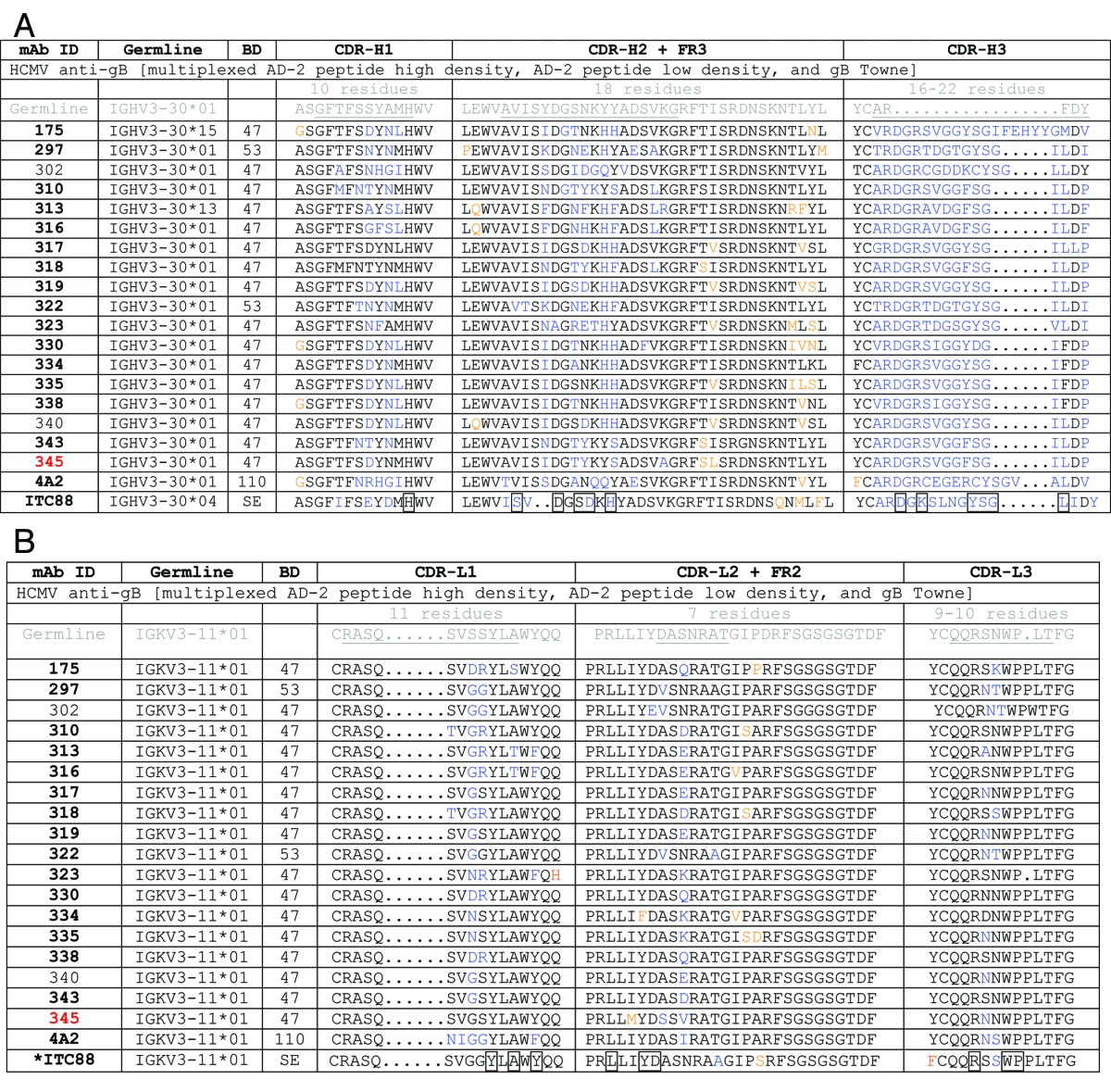
BD, blood donor; bold mAb number, neutralizing activity; underlined text, CDR regions; blue text, changes from CDR germline; orange text, changes from Framework germline; boxed residues, part of antigen contact determined by X-ray crystal structure analysis; *boxed residues for ITC88 are based on homology to mAb 8F9 binding AD-2S1 peptide (69ETIYNTTLKY78)
Table 6. Kinetic binding and neutralization activities of human cytomegalovirus antibodies.
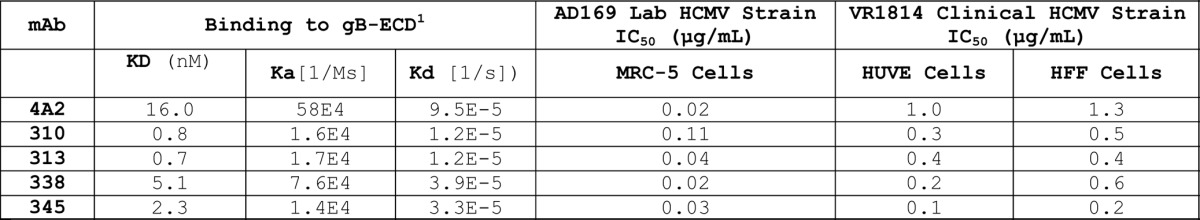
1 The recombinant gB was purchased from Sino Biological, and is a 130–140kDa, glycosylated, C-terminal His-tagged, disulfide bonded linkage of the ECD and cytoplasmic domains (with the transmembrane region deleted). Functional ELISA binding to CD209-Fc suggests this gB is folded in an active state, but it is not a well characterized product. Therefore KD values should be interpreted for the purposes of internal comparisons only.
Sequence diversity of human mAbs obtained from selective screening
The bead based CellSpot™ screening process uses a well-characterized commercial anti-human Fc pAb directed specifically to the IgG class, and therefore has an intrinsic bias toward the high-affinity IgG class of mAbs in the memory B cell repertoire. This affinity selection was further reinforced by an intentional reduction of the avidity potential of the beads. Accordingly, nearly all of the cloned mAbs have undergone significant somatic mutation.
Influenza A antibody sequences
The group 1 antibodies were derived from multiple germlines (Table 2), and although all demonstrated binding to the fusion region, only a subset were neutralizing (Fig. 2B). Overlap with the fusion region was determined by PepScan (Fig. S1) and by measuring synthetic fusion peptide binding in an ELISA assay (Fig. 2B). The individual antibody binding kinetics as seen in the sensorgrams of Figure 2A were linked to the sub-epitope (see section below), not the antibody germline. However, all of the neutralizing mAbs we identified used the VH1–69 family and shared key features of previously cloned group 1 antibodies, including CR626116 and F1017 (CDR-H2 F54 and CDR-H3 Y98).
The lack of neutralizing activity in high-affinity, VH1–69, mAb 52, demonstrates that neutralizing potency is an unpredictable attribute that develops during the affinity maturation path from the Ig germline (i.e., compare sensorgrams of VH1–69 mAbs 48, 49, 52 and 53, in Figure 2A). A single G98Y mutation in CDR-H3 was not sufficient to confer neutralizing activity to mAb 52 (data not shown). The VH1–69 germline gene was highly mutated in all CDRs, including length diversity in CDR-H3 (Table 2A). Mutations in the supporting VH framework 3 regions were also observed. While the previously described CR6261 and F10 are both lambda chain antibodies, we cloned only kappa antibodies (our RT-PCR used multiplexed kappa and lambda primers that had been validated to clone from mixtures of both chains). The germline usage of the light chain (LC) in our set was more diverse, with length differences and sequence mutation occurring in both CDR-L1 and L3 (Table 2B). Recombinant swapping of LC was tolerated in the limited antibodies we tested (e.g., we made recombinant mAb 53 VH with VL from mAb 48, 49 or 52), but the optimal cross-reactive binding properties came from native VH and VL pairs (data not shown).
The majority of antibodies we cloned to group 2 HA possessed some neutralizing activity. Among 38 neutralizers, 15 different VH and VL germlines were represented, standing in strong contrast to the exclusive use of VH1–69 in the group 1 set (Table 2A). However, as already observed in the group 1 mAb families, there was poor correlation between the sequence, breadth of binding and breadth of neutralizing activity. For example, four closely related mAbs from a single donor and sharing germline origins (mAb 455, 560, 620 and 663, Table 2) showed high-affinity binding to three H3N2 strains of HA that did not correlate with the corresponding potency of neutralization (Fig. S3). As with group 1 HA, we cloned several group 2 HA antibodies that are closely related to those previously described in the literature, including CR802018 and F1619 (Table 2). Although mAb 486 (closely related to mAb F16) had limited neutralizing activity (Table 4; Fig. 3), it was interesting because of its unusually broad binding at high-affinity, including both group 1 and 2 HA antigen trimer forms (Fig. 3). This mAb was able to bind all tested HA antigens in ELISA and FACS formats as well (not shown). We therefore screened this particular blood donor’s PBMC repertoire extensively, only to clone the identical mAb three times, with no variants of it found. Our binding and biological activity evaluations led to the selection of mAb 579 as the lead neutralizing clinical candidate. This mAb is derived from an entirely different germline than previously reported for broadly neutralizing antibodies directed to group 2 HA.
Human cytomegalovirus antibody sequences
High-affinity antibodies directed to the linear N-terminal AD-2 (site I) epitope region of gB solely used the restricted germlines VH3–30 and VK3–11. This restriction of both VH and VL germline usage toward AD-2 was also observed in three published studies from different countries8 and in three different blood donors we identified as well. We concentrated our cloning efforts on one donor found to have a rich natural memory B cell reservoir of in vivo affinity matured anti-AD2 antibodies. This individual showed mutated residues in both the VH and VL CDR regions, particularly in CDR-H2 and CDR-H3 (Table 5). There was no diversity of CDR length and there was a higher percentage of germline residues retained in comparison to the influenza HA antibodies. In this case, perhaps because the epitope was a well-defined linear motif, there was a direct correlation between binding affinity and neutralizing activity. This correlation, as well as a requirement for avidity, was reported in an earlier phage display technology study using combinatorial synthetic libraries for CDR randomization of mAb AE11F.20 In addition, our subsequent studies (see below) on the cloned diversity of this directed CDR evolution revealed a functional advantage to particular fine epitope specificity.
Characteristics and selection of optimal neutralizing antibodies
A unifying characteristic of the best neutralizing antibodies in each of our examples was their high-affinity to a single functionally and structurally conserved epitope. That is, nothing in our selection process for the influenza example would have precluded discovery of a high-affinity, strain-independent antibody to an epitope outside the fusion region. A restricted germline origin for the path to neutralization was apparent in both of the influenza fusion HA protein targets, and for the HCMV gB target. Notably, the affinity maturation process often developed contacts to regions surrounding the target epitope that were detrimental to virus neutralization. That is, neutralization potency was not a steadily improving parameter along the affinity maturation path.
Influenza A hemagglutinin
Epitope mapping was performed using the Pepscan, Inc. technology, which involved synthesizing an array of ∼6000 conformationally constrained peptides spanning the HA fusion region. This led to the identification of the crucial neutralizing sub-epitope criteria among our mAbs and an understanding of the functional hotspots (Table 1). By the PepScan profile, both the group 1 and group 2 lead mAbs were directed to more than one sub-region of the HA fusion region that would be discontinuous across two protomers of the naturally-occurring trimer. Also, optimal virus neutralization in both cases included antibody binding to the alphα-helical stalk region.
Our non-neutralizing, group 1, VH1–69 mAb 52 appeared to have energetically-favored binding in the β sheet of the fusion region (Fig. S1), which may abrogate neutralization. The detrimental effects of binding to the group 1 HA β sheet is also suggested by the optimal activity of mAb 53 that excludes this region, and the non-neutralizing activity of mAb 8 that binds well to a peptide derived from the β sheet region (PepScan data Fig. S1; Table 1). In contrast, binding to the group 2 HA β sheet does appear to facilitate neutralization, supported by the binding by mAb 579 (Table 1) and CR802018 to this region. A fine specificity of the mAb 579 epitope is evidenced by its superior neutralizing activity over closely related clones from the same donor (Table 2 and 4).
Our weakly neutralizing group 1/2 cross-reactive binding mAb 486, related in antibody sequence to F16,19 bound to the fusion region by capturing the β sheet and not the stalk region (PepScan data in Table 1). H1N1 viral plaque formation was only inhibited by mAb 486 if it was allowed to bind the virus before activation with trypsin, suggesting it cannot inhibit group 1 HA in its active fusion conformation. Neutralization of H3N2 was weak either with or without trypsin pre-treatment (Fig. 3). By contrast, the group 1 and 2 leads, mAb 53 and 579, were able to neutralize virus if added before or after HA activation (Fig. Three mAb579, mAb 53 data not shown), and bound irrespective of the antigen N-glycosylation state (data not shown), ideal characteristics for in vivo activity.
The AD-2, site I, region of human cytomegalovirus glycoprotein B was known to be highly conserved and susceptible to neutralizing antibody attack. We compared a series of high-affinity antibodies directed to the AD-2 epitope, cloned from memory B cells of human donors with no active infection, to identify critical functional properties of an optimal mAb. First, all high-affinity antibodies directed to AD-2 were able to neutralize MRC-5 cell infection by the AD169 lab strain of HCMV at sub-nanomolar concentrations. Potencies could not be well-differentiated by this plaque microneutralization assay format, possibly due to the general attenuation of the AD169 lab strain and the fact that these virus preparations contain a low percentage of non-infectious particles relative to clinical HCMV strains. In contrast, the measurement of antibody neutralizing activity with HUVEC cells infected by the VR1814 HCMV strain (thought to be closer to a primary clinical isolate) was useful for potency ranking with 10-fold difference in EC50 observed between the 4A2 benchmark mAb and our closely related high affinity antibodies (Table 6). To further select a lead mAb, we looked for equal potency of antibody neutralization of VR1814 infection on HUVEC and HFF cells (Table 6). Perhaps because we had already selected for high-affinity mAb, binding kinetics to the AD-2 peptide (Fig. 5A) or gB protein (Table 6) were not useful in ranking our mAbs. We could not report ka/kd values for the kinetic binding of mAbs to the AD-2 peptide because it could only be evaluated successfully upon ligand immobilization (i.e., by capture of biotinylated N- or C-terminal peptide on streptavidin sensors), which gave values in the pM range, below the accuracy limits of the instrumentation (Forte Octet). Competitive ELISA binding assays showed varying levels of AD-2 peptide competition for gB binding. AD-2 sensorgrams of these antibodies showed fast on- and slow off-rates, and binding are kinetics are therefore not thought to contribute or correlate to the results of the 30 min, competitive gB ELISA format. This suggests that some antibodies, such as mAbs 310, 313, and 338, possess additional, energetically favored, and presumably undesirable, contacts in gB outside the site I, AD-2 region (Fig. 5B).

Figure 5. Binding of selected high-affinity human CMV antibodies to the neutralizing AD-2 epitope alone and in the context of glycoprotein B. (A) Sensorgrams of 100 nM IgG (mAb 310, 313, 338, 345 and 4A2) binding to biotinylated AD-2 peptide captured on streptavidin probes. (B) Competitive binding ELISA using a constant amount of the same panel of mAb IgG pre-incubated with increasing amounts of AD-2 peptide for 30 min prior to binding plate-coated gB. Concentrations of IgG were normalized among the antibodies to give an OD = 1 when binding 0.5 µg/ml gB for 1 h in the absence of any competing peptide. High-affinity antibodies cloned from memory B cells of naturally infected humans with HCMV, such as mAbs 310, 313 and 338, demonstrate additional contacts with gB outside of the AD-2 region causing them to bind gB more strongly in the presence of competing peptide.
Overall, a correlation of potent near-clinical strain virus neutralization, on both endothelial and epithelial cells, with high-affinity binding, and a fine specificity to the AD-2 peptide as demonstrated by competitive binding with gB, provided the optimal characteristics for the selection of lead HCMV antibody, mAb 345. The fact that it was difficult to differentiate greatly between the high-affinity mAbs lends support to the effectiveness of the affinity metric CellSpot™ discovery approach on a functionally defined epitope.
Discussion
By combining a multiplexed high throughput screening technology with single memory B cell antibody cloning from immune human PBMC, we captured a diverse set of cross-reactive, functional human antibodies that reveal common features in the in vivo response to complex viral targets. We plan to detail the potent, strain cross-reactive, in vivo neutralization properties of the influenza- and HCMV-targeted mAbs, in the future.
The work presented here is focused on the discovery approach and analysis of the optimal genetic and biophysical properties of the molecules and has three clear conclusions. First, although most individuals have antibody responses to infectious agents, the epitopes corresponding to optimal neutralizing antibodies are not consistently immunogenic. This means that a large proportion of the biological response must rely on post-binding effector function activities (e.g., phagocytosis, complement-dependent cytotoxicity, and antibody-dependent cell-mediated cytotoxicity), which are not always sufficient or appropriately directed to prevent disease. For example, during HCMV infections of pregnant women, the binding of weak-affinity maternal antibodies to the virus may actually facilitate viral transport to the fetus via the neonatal FcRn, causing subsequent infection and harm upon dissociation of the mAb.15 In the case of influenza viruses, their well-known rapid antigenic drift and potential for crossing host species barriers increase the risk for a pandemic outbreak with high mortality.13 In each case, our multiplexed method for the discovery of high-affinity, clade and strain independent antibodies neutralizing viral entry, promises a substantial advance in the on-going co-evolutionary battle against viruses.
Second, despite the diversification of exposed antigens during viral co-evolution with the host immune system, the regions targeted by optimal neutralizing antibodies apparently serve a vital function, resulting in high sequence conservation (i.e., mutations in these sites must compromise the viral fitness). In addition to the influenza and HCMV examples described here, such conserved epitopes have also been described for the respiratory syncytial virus, hepatitis C virus, and human immunodeficiency virus.4-6 However, we also provide evidence that the optimal neutralizing antibody features may drift on large antigens, such as HA, as repeated exposures recruit contacts with additional binding sites surrounding the core epitope, which increases binding affinity but is detrimental to the neutralizing activity. We observed this disconnect between affinity and neutralization among closely related antibodies of the same germline origin, cloned from single blood donors, in the group 1 and 2 fusion region of HA. These disconnects appeared to be caused by subtle CDR evolution recruiting energetically favored contacts detrimental to neutralization (e.g., in the β-sheet of the group 1 HA fusion region). The combined effects of antibody and viral evolution would serve to increase the chances for viral escape and clinical sequela. Crystal structure analysis was beyond the scope of our studies, and antibody epitopes may be larger and more complex than what we identified by Pepscan methods. Understanding the fine details of the epitope structure and paratope of the antibodies, as well as that of closely related antibodies with reduced function, may provide additional insights. In the case of HCMV antibodies directed to site I of the AD-2 region of gB, all high-affinity antibodies possessed neutralizing properties despite evidence of significant CDR evolution. This lends support to the idea that once a conserved, limited, fine epitope is understood, and the immune response is driven to the appropriate target functional affinity, the repercussions of epitope and paratope drift are minimized.
Third, to realize the clinical utility of these research advances, therapeutic mAbs need to meet additional criteria, including ease of manufacturing (e.g., stability and low aggregation) and lack of off-target reactivity that might cause toxicity. A notable advantage of the multiplexed, single B cell screening platform we used is that it yields enough promising candidates, isolated directly from human blood donors, that these secondary considerations, if a problem, can be applied to select the lead candidate. The tools described here for sensitive and high throughput surveying of blood donors and subsequent cloning of rare native antibodies thus facilitate the identification of the best possible antibodies for clinical development. While direct administration of the optimal mAbs offers a quicker route to clinical utility than vaccine development, the longer lasting effects of vaccines, available at lower cost to a larger population and geographical area, makes that class of intervention important for the longer-term. The suite of mAbs described here should be useful for directing the design of optimal vaccine antigens. That is, immunogens that do not elicit antibodies cross-blocked by or with similar biophysical, epitope and functional properties to the mAbs we have discovered are less likely to be broadly effective. An understanding of such mAb panels can be used to engineer the best immunogens to direct the in vivo antibody evolution toward the minimal and optimal conserved neutralizing contacts while avoiding the less conserved or non-neutralizing immunogenic hotspots. This knowledge-based approach will provide a broadly protective B cell memory for the long-term over the course of repeated natural exposures and offset resistance arising from epitope and paratope evolution.
Materials and Methods
Blood donor PBMC preparation and memory B cell culture
Blood was collected, with heparin as an anticoagulant, from donors at the Stanford University blood bank. An aliquot of plasma was collected by centrifuging 5 ml of blood at 1000 rpm for 10 min. The remaining blood was diluted 1:1 at room temperature into cell culture tested phosphate buffered saline (PBS), without Ca+2 or Mg+2. PBMCs were isolated by carefully layering the diluted blood (20 ml/tube) over an equal volume of 1.077 g/ml Histopaque media (Sigma-Aldrich) in 50 ml Leucosep tubes (Greiner Bio-One Inc.). After 2 washes with RPMI, centrifuging at 200 x g for 10 min, cell pellets were resuspended at 1 × 107 cells/vial in 10% DMSO/90% heat-inactivated FBS for cryopreservation. PBMC were thawed in a 37 °C water bath and immediately diluted in 10 ml of DMEM, 20% FBS, 1x PSG (100 I.U. Penicillin, 100 µg streptomycin, 0.292 mg L-glutamine/ ml) in a 15 ml conical tube and centrifuged 1200 rpm for 5 min. Cells were re-suspended and added at a density of 20 000 per U-bottom 96-well of an irradiated MRC-5 (ATCC®, 55-X™) feeder plate (prepared 24 h prior), in B cell stimulation media (DMEM, 1x PSG, 10% FBS-HI ultralow IgG, 1x ITS (all from Life Technologies), 2 µg/ml CpG, 10 ng/ml IL-10, 1 ng/ml IL-2, and 5 ng/ml IL-6 (all from R&D Systems). This created the “master plate” in which memory B cells were stimulated to replicate (2–3 rounds) and secrete antibody, and constitutes the source plate from which antibody profiling and cloning was performed.
CellSpot™ phenotyping
To survey the antibody repertoire of a donor master plate (typically performed at day 5), 25% of the master plate cells were replica-plated into 96-well V-bottom plates and washed 2 times with cell media (DMEM, 1xPSG, 10% low IgG HI-FBS). Cells were transferred in a volume of 100 μl to black flat-bottom plates (Greiner 96-well clear bottom, high protein binding plates) coated with 2 µg/ml goat anti-human Fc IgG (Jackson ImmunoResearch) and blocked with 3% BSA/PBS. In order to gauge the number of antibody secreting cells per well, a 20 μl sample of 8 representative wells for each plate is serially diluted and 100 μl added to anti-human IgG coated plates. After 5 h at 37 °C, 5% CO2 plates were washed in cell lysis buffer (water/0.2% polysorbate-20) followed by PBS. Multihued particles of 5 distinguishable types were used in these experiments. Particle functionalization and bead-based ELISA characterizations were performed as described6 and stored at 0.1% solids by volume. One hundred μl of a 1:500 dilution of particles (which mixtures of particles depended on the screen) in 0.1 M citrate, 2% BSA, 0.2% polysorbate-20, pH 6, was added to the plates and allowed to incubate overnight, in the dark at ambient temperature. After washing in PBS, 0.2% polysorbate-20 the plates were fixed with 4% paraformaldehyde/PBS, washed with de-ionized distilled water, followed by 200-proof ethanol and then dried at 37 °C before imaging. Quantitative imaging was performed with a custom built, automated Leica fluorescent microscope and in-house analysis software. In each CellSpot™ the quantity of each particle type was reported and used to grade cross-reactivity or affinity.
CellSpot™ cloning
Siblings of B cells identified in the CellSpot™ phenotyping process were isolated from the master plate. First, the total number antibody secreting cells in a given master plate well was calculated from the number of Fc spots (i.e., detected with a mixture of anti-kappa/anti-lambda particles). Single B cells were subsequently cloned by limiting dilution in the presence of antigen specific particles prepared in phenol red-free DMEM/10% low IgG media. In the absence of washing or fixing, wells containing antigen-specific B cells were identified by imaging, all media was carefully removed from the well, and the cell was lysed with 10 μl 1x Qiagen One-Step RT-PCR buffer, 0.5% NP-40, 16 units RNasin Plus (Promega) in RNase-free water (Ambion), and stored at -70 °C. The lysates were divided in half and RT-PCR performed separately for the VH (IgG 1, 2, 3, and 4) and VL (primers including kappa and lambda) regions using the Qiagen One step RT-PCR Kit supplemented with 16 units/reaction of RNasin plus. Degenerate Ig primers21,22 were ordered from Integrated DNA Technologies, Inc., prepared as 100 µM equimolar mixed stocks and used at 1.2 µM. RT-PCR included 60 min 50 °C, 15 min 95 °C, 2 cycles 94 °C 30 s, 54 °C 30 s, 72 °C 30 s, 25 cycles 94 °C 30 s, 55 °C 30 s, 72 °C 30 s and 72 °C 8 min. RT-PCR was followed by a semi-nested PCR (i.e., same forward primers and nested reverse primers containing restriction sites for cloning into the pTT5 expression vector (licensed from the National Research Council, Canada). Aliquots of the PCR were sent to Sequetech Inc. for sequencing using the same nested PCR reverse primers. Sequence analysis was done using the online Fab analysis program at www.vbase2.org.
Viral antigens for screens
Influenza A screens were done with recombinant HA protein purchased in monomer form from Sino Biological Inc. (human 293 cell derived H1, H2, H3, H5, H7, and H9) or trimer form from eEnzyme LLC (human 293 cell derived H1 and H3) and Protein Sciences Corp (Baculovirus derived H5 and H7). A fusion peptide (FP) based on the H1N1 sequence (KNVPSIQSRGLFGAIAGFIEGGWTGMVDGWYGY) and a corresponding affinity purified rabbit anti-FP pAb was generated at Genemed Synthesis Inc. Cytomegalovirus screens were done with AD-2 peptide (SHRANETIYNTTLKYGD) biotinylated on either the C- or N- termini combined with a biotin spacer peptide (Genemed Synthesis Inc.). Recombinant glycoprotein B (gB, Towne strain) was purchased from Sino Biological Inc.
HCMV Screening and Competitive ELISA
Patient serum was pre-screened for AD-2 reactivity by binding at three targeted titers (1/50, 1/250, and 1/1250) on high protein binding ELISA plates coated with 2 μg/ml AD-2 and blocked with BSA. The highest titer binding patient serum was chosen for memory B cell surveys and screening from PBMC.
For comparisons of recombinant antibody binding, plates were coated with 0.5 μg/ml gB overnight in PBS. The next day, plates were blocked with 3% BSA/PBS and then incubated for 1 h with mAb titrated from 5 μg/ml in 0.5% BSA/PBST using 1/3 dilutions. After washing 3 × PBST, secondary anti-human kappa-HRP antibody was added for 45 min., plates were washed 3 x PBST and developed with TMB substrate. The concentration of each antibody giving an OD450 of 1 was used in the subsequent competitive ELISA. The selected constant concentration of antibody was pre-incubated for 30 min with a titration of AD-2 peptide in 0.5%BSA/PBST before adding to the gB coated/blocked plate. The remaining steps were done with the same timings and washes as the direct ELISA.
Kinetic binding assays
The kinetic binding and affinity of recombinant antibodies were determined on the ForteBio Octet model QK using HBST (10 mM HEPES, pH 7.2, 150 mM NaCl, 0.005% Tween-20). Human antibody capture was performed with new ForteBio anti- hIgG Fc capture (AHC) biosensors for each data point without any regeneration. IgG was loaded for 900 s from a solution of 2 μg/ml (to give a signal of 2 units), baseline was equilibrated for 120 s, and then analyte (HA or gB) associated for 900 s followed by 900 s disassociation. Biotinylated ligands were captured with new ForteBio streptavidin SA biosensors. N-terminally biotinylated AD-2 peptide was loaded from a 5 nM solution for 900 s (to give a signal of 0.2 units), baseline was equilibrated for 120 s, and then analyte (IgG) associated for 900 s followed by 900 s disassociation.
Virus neutralization assays
The influenza A neutralizing activity of the mAbs was titrated to determine EC50 concentrations in a 72 h plaque-formation assay on MDCK cells (ATCC, CCL-34). The day before the assay, one 12-well plate per test antibody was seeded with 2 × 105 MDCK/well in MEM/ 5% FBS/1% L-glutamine. Pre-titered virus stocks (prepared in Dr. Tripp’s lab23) were rapidly thawed and diluted in MEM/1% L-glutamine and 1 μg/ml TPCK trypsin (Worthington), vortexed and kept on ice. Antibody was added to the virus from 0–20 μg/ml, vortexed and incubated at 37 °C for 1 h. Cell monolayers were washed 3 times with PBS and 100 μl MEM/1% L-glutamine and 1 μg/ml TPCK trypsin added to each well before adding 200 μl of the pre-incubated virus/antibody preparation. After 2 h at 37 °C the supernatant was aspirated, the monolayer was washed once with PBS and a 0.5 ml volume of 1.2% Avicel RC-581 (FMC Bio-polymer Inc.) overlay containing 1 μg/ml TPCK trypsin in MEM, 20mM HEPES, 2mM L-glutamine, 0.075% NaHCO3, and 100 units/ml penicillin G/ 100 µg/ml Streptomycin/0.25 µg/ml amphotericin B, was applied for 72 h at 37 °C/5% CO2. The plaques were visualized by fixing and staining with Crystal Violet. First, the Avicel overlay was aspirated and monolayers gently washed with PBS. Monolayers were then fixed by applying 1 ml of ice cold 80%/20% methanol/acetone for 10 min. Stock solutions of 0.13% (w/v) crystal violet were prepared in deionized water containing 5% methanol and 11% formaldehyde. Working stocks of crystal violet were made by diluting the stock solution 1:2 in PBS. After monolayer fixation, enough crystal violet working solution was added to a well to just cover the cells and allowed to stain for 10 to 20 min. Wells were rinse in distilled deionized water and the average number of plaques counted from duplicate wells, expressed as a percentage of plaques in uninhibited (100% infected) control wells.
One day prior to HCMV infection, MRC-5 (ATCC, CCL-171) cells were seeded at 2 × 104 cells per well in a flat bottom 96-well plate in growth media (DMEM, 10% FCS, 2 mM L-glutamine, 100 units/ml penicillin and 100 µg/ml streptomycin). Test antibodies were diluted in growth media from 0–10 μg/ml and then combined with HCMV AD169 virus (Advanced Biotechnologies Inc., ~1 × 108 PFU/mL stock) and allowed to incubate at ambient temperature. The MOIs was determined by the titer of the viral stock such that 95–100% of the cells stained positive in the absence of any inhibition (i.e., the greatest dilution in which all the cells in a well looked stained). Control human IgG1κ antibody (Southern Biotech) was used as a negative control. After a 1 h pre-incubation, 100 µl of antibody/ virus was added to cells for overnight infection in a 37 °C/5% CO2 incubator. The next day, the media was aspirated and cells were fixed for 30 min with 150 µl/ well of 100% ethanol, followed by a 10 min PBS re-hydration and 20 min blocking in 10% goat serum/PBS. Wells were then incubated with 0.1µg/ml of a mouse mAb directed against the cytomegalovirus early nuclear antigen (1E1, MAB810, Millipore) in blocking buffer. After 1 h, wells were washed once in blocking buffer and incubated for 1 h with goat-anti-mIgG2a-biotin (Jackson ImmunoResearch). After two washes in PBST, a 1:200 dilution of Streptavidin-HRP in PBS was added for 15 min, wells were washed 3 × PBST, and Histomark TrueBlue substrate (KPL) was added for 30 min in PBS (or as soon as nuclei staining was observed in control uninhibited infected wells). Plates were washed with water, rinsed with absolute ethanol and allowed to dry before stained foci were counted in one microscope field, in triplicate wells. Quantitation of foci in test wells were expressed as a percentage of the average counted in uninhibited control wells. Average EC50 values from three independent assays are reported. Assays using primary human foreskin fibroblasts or HUVE cells were performed as previously described, with VR1814 virus stock prepared in Dr Pereira’s lab.24
Epitope mapping
For each antibody, 5808 different Chemically Linked Peptides on Scaffolds (CLIPS) peptides covering linear, conformational and discontinuous epitopes were designed and synthesized as previously reported25 using 75 amino acid fraction of group 1 HA2 (ATGLRNIPSIQSRGLFGAIAGFIEGGWTGMVDGWYGYHHQNEQGSGYAADLKSTQNAIDEITNKVNSVIEKMNTQ from H1N1 A/California/04/2009) or a 105 amino acid discontinuous fraction of group 2 HA1/2 (HHAVPNGTLVKTITDDQIEVTNATELV QSS/ATGMRNVPEKQTRGLFGAIAGFIENGWEGMIDGWYGFRHQNSEGTGQAADLKSTQAAIDQINGKLNRVIEKTNEK from H3N2 A/Hong Kong/68). In general, antibodies did not strongly bind peptides, which allowed for only putative, discontinuous epitope prediction.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
A special thank you to the late Dr William Usinger, whose fostering of key collaborations linking the expertise of academic researchers in the infectious disease area with Trellis’ antibody discovery tools was important to the development of this work. Grant support for this research came from Dr Tripp at the University of Georgia, Dr Pereira at the University of San Francisco and from Trellis Biosciences.
Glossary
Abbreviations:
- mAb
monoclonal antibody
- PBMC
peripheral blood mononuclear cells
- CDR
complementary-determining region
- VH
variable domain of heavy chain
- VL
variable domain of light chain
- Fab
antigen binding fragment
- Fc
fragment crystallizable region
- InfA
influenza A
- HCMV
human cytomegalovirus
- HA
hemagglutinin
- FP
fusion peptide
- gB
glycoprotein B
- AD-2
antigenic determinant 2
- Ig
immunoglobulin
- IgG
immunoglobulin gamma
- Ag+
antigen positive
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27760
References
- 1.Glassy MC, Handley HH, Hagiwara H, Royston I. UC 729-6, a human lymphoblastoid B-cell line useful for generating antibody-secreting human-human hybridomas. Proc Natl Acad Sci U S A. 1983;80:6327–31. doi: 10.1073/pnas.80.20.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ter Meulen J. Monoclonal antibodies in infectious diseases: clinical pipeline in 2011. Infect Dis Clin North Am. 2011;25:789–802. doi: 10.1016/j.idc.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Han T, Marasco WA. Structural basis of influenza virus neutralization. Ann N Y Acad Sci. 2011;1217:178–90. doi: 10.1111/j.1749-6632.2010.05829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pedersen J, Carlsen TH, Prentoe J, Ramirez S, Jensen TB, Forns X, Alter H, Foung SK, Law M, Gottwein J, et al. Neutralization resistance of hepatitis C virus can be overcome by recombinant human monoclonal antibodies. Hepatology. 2013;58:1587–97. doi: 10.1002/hep.26524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammond PW. Accessing the human repertoire for broadly neutralizing HIV antibodies. MAbs. 2010;2:157–64. doi: 10.4161/mabs.2.2.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collarini EJ, Lee FE, Foord O, Park M, Sperinde G, Wu H, Harriman WD, Carroll SF, Ellsworth SL, Anderson LJ, et al. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from B cells of infected patients. J Immunol. 2009;183:6338–45. doi: 10.4049/jimmunol.0901373. [DOI] [PubMed] [Google Scholar]
- 7.Macagno A, Bernasconi NL, Vanzetta F, Dander E, Sarasini A, Revello MG, Gerna G, Sallusto F, Lanzavecchia A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol. 2010;84:1005–13. doi: 10.1128/JVI.01809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomson CA, Bryson S, McLean GR, Creagh AL, Pai EF, Schrader JW. Germline V-genes sculpt the binding site of a family of antibodies neutralizing human cytomegalovirus. EMBO J. 2008;27:2592–602. doi: 10.1038/emboj.2008.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melchjorsen J. Learning from the messengers: innate sensing of viruses and cytokine regulation of immunity - clues for treatments and vaccines. Viruses. 2013;5:470–527. doi: 10.3390/v5020470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang S. Advances in the production of human monoclonal antibodies. Antibody Technology Journal. 2011 doi: 10.2147/ANTI.S20195. [DOI] [Google Scholar]
- 11.Beerli RR, Rader C. Mining human antibody repertoires. MAbs. 2010;2:365–78. doi: 10.4161/mabs.2.4.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harriman WD, Collarini EJ, Sperinde GV, Strandh M, Fatholahi MM, Dutta A, Lee Y, Mettler SE, Keyt BA, Ellsworth SL, et al. Antibody discovery via multiplexed single cell characterization. J Immunol Methods. 2009;341:135–45. doi: 10.1016/j.jim.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mancini N, Solforosi L, Clementi N, De Marco D, Clementi M, Burioni R. A potential role for monoclonal antibodies in prophylactic and therapeutic treatment of influenza. Antiviral Res. 2011;92:15–26. doi: 10.1016/j.antiviral.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Ohlin M, Sundqvist VA, Mach M, Wahren B, Borrebaeck CA. Fine specificity of the human immune response to the major neutralization epitopes expressed on cytomegalovirus gp58/116 (gB), as determined with human monoclonal antibodies. J Virol. 1993;67:703–10. doi: 10.1128/jvi.67.2.703-710.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am J Pathol. 2006;168:1210–26. doi: 10.2353/ajpath.2006.050482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Throsby M, van den Brink E, Jongeneelen M, Poon LL, Alard P, Cornelissen L, Bakker A, Cox F, van Deventer E, Guan Y, et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One. 2008;3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, Santelli E, Stec B, Cadwell G, Ali M, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16:265–73. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science. 2011;333:843–50. doi: 10.1126/science.1204839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–6. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 20.Lantto J, Fletcher JM, Ohlin M. Binding characteristics determine the neutralizing potential of antibody fragments specific for antigenic domain 2 on glycoprotein B of human cytomegalovirus. Virology. 2003;305:201–9. doi: 10.1006/viro.2002.1752. [DOI] [PubMed] [Google Scholar]
- 21.Xu MY, Xu XH, Chen GZ, Deng XL, Li J, Yu XJ, Chen MZ. Production of a human single-chain variable fragment antibody against esophageal carcinoma. World J Gastroenterol. 2004;10:2619–23. doi: 10.3748/wjg.v10.i18.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang ZC, Hu XJ, Yang Q. Generation of high affinity human single-chain antibody against PreS1 of hepatitis B virus from immune phage-display antibody library. Hepatobiliary Pancreat Dis Int. 2004;3:77–81. [PubMed] [Google Scholar]
- 23.Cottey R, Rowe CA, Bender BS. Influenza Virus. Current Protocols in Immunology. 2001. Unit 19.11. [DOI] [PubMed]
- 24.Andreoni M, Faircloth M, Vugler L, Britt WJ. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J Virol Methods. 1989;23:157–67. doi: 10.1016/0166-0934(89)90129-8. [DOI] [PubMed] [Google Scholar]
- 25.Timmerman P, Puijk WC, Meloen RH. Functional reconstruction and synthetic mimicry of a conformational epitope using CLIPS technology. J Mol Recognit. 2007;20:283–99. doi: 10.1002/jmr.846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.