

Abstract
Copper amine oxidases are a family of enzymes with quinone cofactors that oxidize primary amines to aldehydes. The native mechanism proceeds via an iminoquinone intermediate that promotes high selectivity for reactions with primary amines, thereby constraining the scope of potential biomimetic synthetic applications. Here, we report a novel bioinspired quinone catalyst system, consisting of 1,10-phenanthroline-5,6-dione/ZnI2, that bypasses these constraints via an abiological pathway involving a hemiaminal intermediate. Efficient aerobic dehydrogenation of non-native secondary amine substrates, including pharmaceutically relevant nitrogen heterocycles, is demonstrated. The ZnI2 cocatalyst activates the quinone toward amine oxidation and provides a source of iodide, which plays an important redox-mediator role to promote aerobic catalytic turnover. These findings provide a valuable foundation for broader development of aerobic oxidation reactions employing quinone-based catalysts.
Introduction
Enzymatic transformations have provided the inspiration for numerous advances in synthetic chemistry and catalysis. In connection with widespread interest in the development of aerobic oxidation reactions, numerous researchers have turned to metalloenzymes as a starting point for development of small-molecule transition-metal catalysts. Organic cofactors are also common in naturally occurring oxidases and oxygenases, but these have been less extensively developed for use in synthetic applications. Copper amine oxidases promote aerobic oxidation of primary amines to aldehydes in nature (Figure 1).1 Copper is present in the enzyme, but substrate oxidation is promoted exclusively by a quinone cofactor in the active site. The mechanism of the reaction was the subject of considerable historical debate and focused on two possible pathways: 2, 3 a “transamination” pathway involving the formation and oxidation of an iminoquinone intermediate (Figure 1A), and an “addition-elimination” pathway involving substrate oxidation via a hemiaminal intermediate (Figure 1B). Extensive mechanistic studies of the enzyme and model systems by Klinman, Sayre and others convincingly demonstrated that the reaction proceeds via the transamination pathway.4,5
Figure 1.

Mechanism of aerobic amine oxidation mediated by copper amine oxidase enzymes. (A) “Transamination” mechanism involving covalent imine intermediates. (B) “Addition-elimination” mechanism of amine oxidation, involving a hemiaminal intermediate.
Recently, several groups have begun to explore quinone-based catalysts6–9 as alternatives to metal-based catalysts for amine dehydrogenation.10–12 Use of quinones Q16 and Q27 (Scheme 1) enables efficient and selective production of homo- and heterocoupled imines under mild reaction conditions (Scheme 1). These catalysts show exquisite selectivity for primary amines, similar to the native enzymes. Secondary amines are not compatible with the transamination mechanism, and they often serve as inhibitors via formation of irreversible covalent adducts.13,14
Scheme 1.
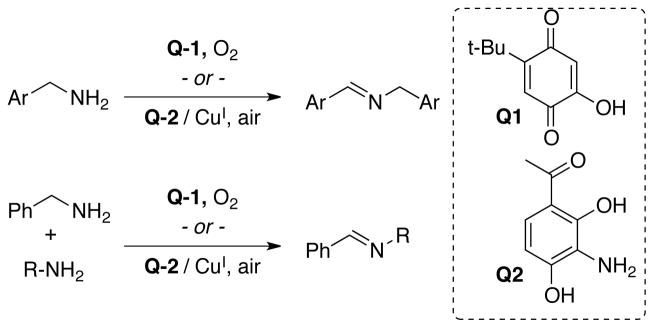
Biomimetic pre-catalysts Q1 and Q2 and their synthetic application to oxidative homo- and cross-coupling of primary amines.
The function of quinone cofactors in nature is not limited to primary amine oxidation. For example, pyrroloquinoline quinone (PQQ)-dependent alcohol dehydrogenases (Figure 2) mediate alcohol oxidation via a mechanism that involves a hemiacetal intermediate, resembling the addition-elimination mechanism in Figure 1B.15–17 Identification of new quinone-based catalysts that operate via an addition-elimination mechanism could significantly enhance the synthetic scope of such oxidation reactions. Kobayashi proposed the involvement of hemiaminal intermediates in diverse amine oxidation reactions that use Pt/Ir nanoclusters and 4-tert-butylcatechol as cocatalysts.8 Here, we expand this concept by showing that 1,10-phenanthroline-5,6-dione (phd) (Figure 2) is an effective catalyst for secondary amine oxidation. Fundamental studies provide direct evidence for the addition-elimination pathway, including spectroscopic characterization of the hemiaminal intermediate. We further show that coordination of the distal nitrogen atoms to Zn2+ enhances phd amine oxidation activity, and that a catalyst system composed of phd/ZnI2 promotes efficient aerobic oxidation of a variety of secondary amines and nitrogen heterocycles.
Figure 2.
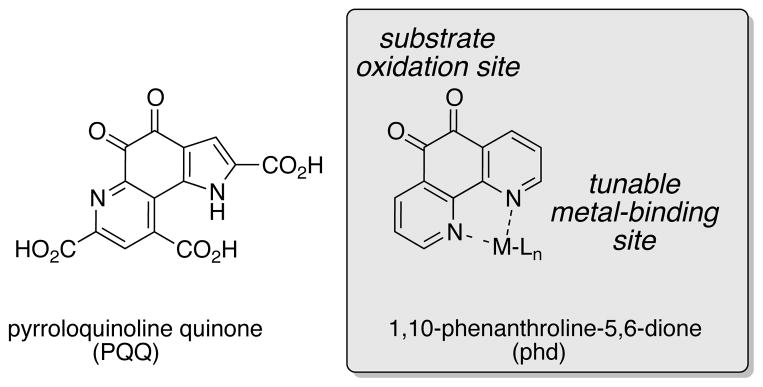
Pyrroloquinoline quinone (PQQ), and phd: a modular, bifunctional catalyst for amine oxidation. The o-quinone moiety is responsible for substrate oxidation, while remote metal-binding sites can be used to tune the quinone reactivity.
Results and Discussion
Stoichiometric secondary amine oxidation and characterization of a hemiaminal adduct
In an effort to expand upon our earlier studies of quinone-mediated amine oxidation,6 we were drawn to the structure of 1,10-phenanthroline-5,6-dione (phd) as a potential catalyst because of its bifunctional character associated with the o-quinone and distal chelating nitrogen atoms (Figure 2). Independently, the coordination chemistry of phd with a number of different metals has been investigated.18–20 We speculated that these two features could be combined to achieve unique amine oxidation reactivity.21 The dehydrogenation of cyclic secondary amines was targeted because these substrates have been ineffective with traditional amine oxidase mimics. For example, Bruice and coworkers have studied isomeric phenanthroline-derived o-quinones as models of the cofactor pyrroloquinoline quinone (PQQ), and they observed stoichiometric oxidation of various amines, including the secondary amine morpholine. No catalytic reactivity was observed, however, and the reaction with morpholine led to formation of an irreversible covalent adduct.14,22 Catalytic dehydrogenation of secondary amines is also an important target because the unsaturated heterocyclic products are prevalent in pharmaceuticals and other biologically active molecules.23
Our initial studies probed the stoichiometric reaction of 1,2,3,4-tetrahydroisoquinoline 1 with phd in MeCN. The reaction proceeded quantitatively at room temperature within 18 h to afford 3,4-dihydroisoquinoline 2 and 1,10-phenanthroline-5,6-diol (phd-H2) as a yellow-green precipitate (eq 1). The effectiveness of this reaction was better than expected in light of Bruice’s precedent showing that secondary amines could form irreversible covalent adducts with phd analogs.14
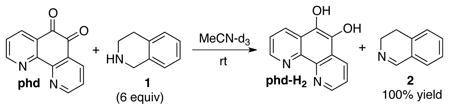 |
(1) |
The reaction of 1 with phd was monitored by 1H NMR spectroscopy to determine whether intermediates could be observed. Upon addition of 6 equiv of 1 in MeCN-d3 at room temperature, the characteristic phd resonances disappeared with concomitant formation of new broad peaks. Variable temperature studies demonstrated that these broad peaks were associated with an equilibrium exchange process occurring on the NMR time scale. The broad peaks resolved at lower temperature (Figure 3 and S1) to reveal the presence of a new species, and the chemical exchange process was sufficiently slow at −40 °C to enable full characterization of this intermediate by NMR spectroscopy.24 It was identified as hemiaminal A (Figure 3A) on the basis of 1H-13C and 1H-15N gHSQC and gHMBC as well as 1D NOESY data (see Supporting Information, Figures S2–S6).
Figure 3.

Observation of hemiaminal intermediate A by variable temperature 1H NMR.
NMR titration studies of phd with 1 in MeCN-d3 (Figure S7) were used to establish the equilibrium constant for hemiaminal formation: K = 0.10 mM−1 at −40 °C. Exchange spectroscopy (EXSY) experiments were carried out with 6 equiv of 1 and revealed exchange between 1 and the hemiaminal, and between the hemiaminal and free phd (Figures S8 and S9).
Zn2+-promoted amine oxidation and characterization of Zn-phd complexes
The prospect that metal ions could promote phd-mediated amine oxidation was tested by adding various quantities of Zn(OTf)2 to the reaction mixture. The most significant rate enhancement was observed with 0.5 equiv of Zn(OTf)2 (i.e., phd/Zn2+ = 2:1), which led to an 11-fold increase in the initial rate of the oxidation of 1 by phd (Figure 4). Formation of large quantities of precipitate, presumably corresponding to a Zn2+/phd-H2 coordination polymer, slowed the reaction after approx. 40–50% conversion under these conditions.
Figure 4.

Rates for the stoichiometric reaction of 1 with phd at −10 °C in acetonitrile with and without 0.5 equiv Zn(OTf)2. Reaction conditions: [phd] = 19 mM (0.019 mmol), [1] = 114 mM (0.114 mmol), [Zn(OTf)2] = 9.5 M (0.095 mmol), MeCN (1 mL), −10 °C.
NMR titration studies of Zn(OTf)2 and phd in MeCN-d3 revealed sequential formation of three discrete species in solution, corresponding to [Zn(phd)3]2+, [Zn(phd)2]2+ and [Zn(phd)]2+ (Figures 5 and S10). 1H-15N HMBC experiments reveal that the phd 15N resonances shift from 313 ppm to 251 ppm in the presence of Zn(OTf)2 (Figures S11 and S12), consistent with coordination of the pyridyl nitrogen atoms to Zn. X-ray quality crystals of a [Zn(phd)2]2+ species were obtained from a 2:1 mixture of phd/Zn(OTf)2 in MeCN, confirming phd coordination to Zn (Figure 6).
Figure 5.

1H NMR titration and speciation plot at different Zn(OTf)2/phd ratios. Lines do not represent fits, but are included to guide the eye.
Figure 6.
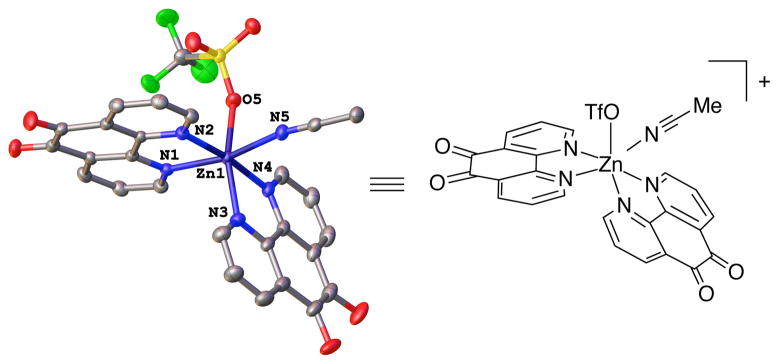
X-ray crystal structure of [Zn(phd)2(MeCN)(OTf)]+ shown with 50% probability ellipsoids. All H atoms and disorder are omitted for clarity (see Supporting Information for details).
Catalytic aerobic oxidation of secondary amines
The oxidation of tetrahydroisoquinoline 1 to the dihydroisoquinoline 2 was then tested with catalytic quantities of phd (5 mol %) and different Zn2+ sources (2.5 mol %) under 1 atm of O2 (Table 1). Negligible catalytic turnover was observed in the oxidation of 1 by phd in the absence of Zn2+ ions (7% yield), and little improvement was achieved by including Zn(OTf)2, Zn(OAc)2, ZnCl2, or ZnBr2 (Table 1, entries 1–5). Use of ZnI2, however, resulted in significant catalytic turnover (55% yield; entry 6). The yield further improved upon adding catalytic quantities of a Brønsted acid (75% yield with 15 mol % pyridinium p-toluenesulfonic acid, PPTS; entry 7). Control experiments showed that no amine oxidation occurred in the absence of phd under these conditions (entry 8), and removal of the ZnI2 leads to only stoichiometric oxidation (7%; entry 9). Replacement of phd with 1,10-phenanthroline also results in no substrate oxidation (entry 10).
Table 1.
Optimization of the reaction conditions for the catalytic aerobic oxidation of tetrahydroisoquinoline, 1.a

| ||||
|---|---|---|---|---|
| Entry | quinone | additive | additive | % yield 2 |
| 1 | 5% phd | ---- | --- | 7 |
| 2 | 5% phd | 2.5% ZnOTf2 | --- | 7 |
| 3 | 5% phd | 2.5% ZnOAc2 | --- | 7 |
| 4 | 5% phd | 2.5% ZnCl2 | --- | 7 |
| 5 | 5% phd | 2.5% ZnBr2 | --- | 10 |
| 6 | 5% phd | 2.5% ZnI2 | --- | 55 |
| 7 | 5% phd | 2.5% ZnI2 | 15% PPTS | 75 |
|
| ||||
| 8 | --- | 2.5% ZnI2 | 15% PPTS | NR |
| 9 | 5% phd | --- | 15% PPTS | 7 |
| 10 | 5% phen | 2.5% ZnI2 | 15% PPTS | NR |
Reaction conditions: 1,2,3,4-tetrahydroisoquinoline (0.130 mmol), MeCN (0.5 mL), O2 atmosphere, 24 h. PPTS, Pyridinium p-toluenesulfonic acid. Yields determined by 1H NMR spectroscopy.
Further studies of the oxidation of 1 to 2, as well as the oxidation of dibenzylamine 3 to N-benzylidene benzylamine 4, revealed that both Zn2+ and iodide are important to the success of the catalytic reactions. Oxidation of 3 under the optimized reaction conditions resulted in an 80% yield of 4 (Table 2, entry 1). Upon replacement of ZnI2 with tetrabutylammonium iodide (Bu4NI), significant catalytic activity was retained in the oxidation of 1, but only stoichiometric reactivity was observed in the oxidation of 3 (entry 2). A similar observation was made when ZnI2 was replaced with molecular iodine (entry 3). Use of catalytic I2 in the absence of phd led to negligible reactivity, even in the reaction of 1 (entry 4).
Table 2.
Beneficial effect of Zn2+ and iodide on catalytic aerobic amine oxidation.a
| |||||
|---|---|---|---|---|---|
| Entry | quinone | additive | additive % yield 2 | % yield 4 | |
| 1 | 5% phd | 2.5% ZnI2 | 15% PPTS | 75 | 80 |
| 2 | 5% phd | 5% Bu4NI | 15% PPTS | 61 | 6 |
| 3 | 5% phd | 2.5% I2 | 15% PPTS | 52 | 8 |
| 4 | --- | 2.5% I2 | 15% PPTS | 2 | 3 |
Reaction conditions: amine (0.130 mmol), MeCN (0.5 mL), O2 atmosphere, 24 h. PPTS, Pyridinium p-toluenesulfonic acid. Yields determined by 1H NMR spectroscopy.
The unique beneficial effect of iodide counterions raised the possibility of a catalytic redox role for iodide, and use of a starch-iodine test provides evidence for the formation of I3− in the absence of substrate under the standard reaction conditions. On the basis of this result, at least two reasonable mechanisms could be considered for the amine oxidation reactions (Scheme 2). Mechanism A involves phd-mediated amine oxidation, similar to the stoichiometric reactivity shown in eq 1 and Figure 4. Catalytic turnover involves an iodide/triiodide cycle25 that mediates aerobic reoxidation of phd-H2 (Scheme 2A). Mechanism B reflects literature precedents for stoichiometric oxidation of certain amines by molecular iodine,26 and the catalytic cycle involves I2/I3−-promoted amine oxidation coupled to a phd-based redox cycle that mediates aerobic reoxidation of iodide (Scheme 2B). The latter pathway resembles metal-catalyzed oxidation reactions in which quinones have been used to facilitate aerobic oxidation of the reduced catalyst.27
Scheme 2.

Two proposed reaction mechanisms.
Kinetic isotope effect experiments were carried out in order to distinguish between these possibilities (Scheme 3). The reaction of 1-d1 was subjected to three different reaction conditions, including those employing (a) stoichiometric phd as the oxidant under anaerobic conditions, (b) stoichiometric iodine as the oxidant under anaerobic conditions, and (c) the optimized catalytic conditions with 5 mol % phd/2.5 mol % ZnI2 under aerobic conditions. Comparison of the KIEs from these experiments showed that the KIE obtained under catalytic conditions matched that obtained with stoichiometric phd (KIE = 6.4 in both cases), and differed from the KIE observed with I2 as the oxidant (KIE = 3.8). These results provide strong support for the quinone-mediated amine oxidation pathway associated with Mechanism A in Scheme 2A.
Scheme 3.

Intramolecular competition kinetic isotope effect experiments.
Substrate scope and synthetic applications
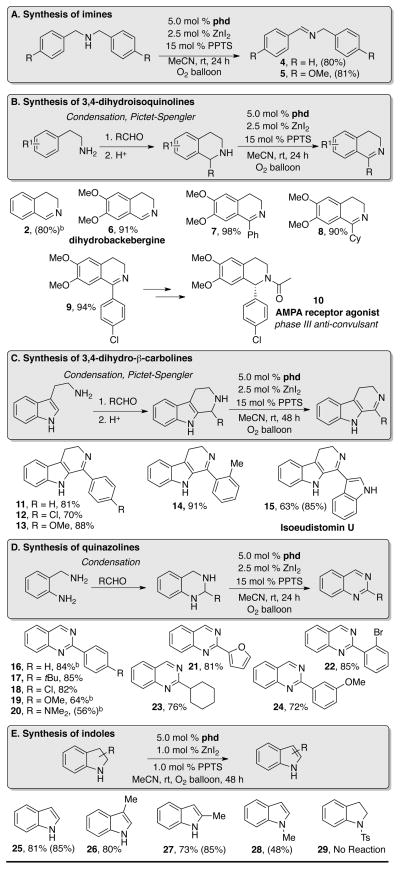
The optimized catalytic conditions were tested with a number of different substrates (Table 3), ranging from simple dibenzyl amines (A) to various nitrogen heterocycles, such as tetrahydroisoquinolines (B), tetrahydro-β-carbolines (C), and tetrahydroquinazolines (D), as well as indolines (E). Substrate classes (B)–(D) are particular appealing because the saturated heterocycles may be accessed readily via simple condensation and Pictet-Spengler reactions (Table 3B–3D).
Table 3.
Substrate scope and synthetic applications.a
|
Reaction conditions: substrate amine (1.0 mmol), phd (0.05 mmol), ZnI2 (0.025 mmol), PPTS (0.15 mmol), MeCN (4.0 mL), O2 balloon, 24–48 h.
48 h reaction time. Yields are reported as isolated yields, numbers in parenthesis indicate NMR yields.
Substituted tetrahydroisoquinolines were smoothly converted to 3,4-dihydroisoquinolines under these conditions (Table 3B). Electron-donating groups improved reaction yields and diminished reaction times. 6,7-Dimethoxytetrahydroisoquinoline was oxidized to dihydrobackebergine 6 in 91% isolated yield and proceeds more rapidly than the parent substrate 1. Aryl and alkyl substitution at the 1-position is well-tolerated: 1-phenyl- and 1-cyclohexyl-substituted 6,7-dimethoxy-3,4-dihydroisoquinolines were isolated in excellent yields (98% and 90% yields of 7 and 8, respectively). The 3,4-dihydroisoquinoline products of these reactions have been widely used as precursors to chiral tetrahydroisoquinolines, which are widely represented in natural products and pharmaceuticals. For example, 1-(4-chlorophenyl)-3,4-dihydro-6,7-dimethoxyisoquinoline 9, which is obtained in 94% yield under our aerobic oxidation conditions, is an intermediate to the phase III antiepileptic AMPA receptor agonist 10.28
Substituted tetrahydro-β-carbolines are readily converted to 3,4-dihydro-β-carbolines using the same optimized conditions (Table 3C). Aryl substitution in the 1-position is again tolerated, with yields slightly improved for substrates containing electron-rich substituents (13, R = OMe, 88% yield), relative to those containing electron-deficient substituents (11, R = Cl, 70% yield). o-Methyl substitution is also well-tolerated (14, 91% yield), and the natural product isoeudistomin U (15) was obtained in 63% isolated yield (85% NMR yield).
Quinazolines were formed in good yields from tetrahydroquinazolines (Table 3D). Unlike other substrate classes, wherein electron-donating substituents improved yields, quinazoline products containing electron-withdrawing substitution at the 2-position were better substrates. Ring-chain tautomerism in 2-substituted tetrahydroquinazolines could occur, and the improved yields of electron-deficient quinazolines may reflect the stabilization of the ring tautomer in the respective tetrahydroquinazoline substrates.
With slight modification to the reaction conditions (5 mol % phd, 1.0 mol % ZnI2, and 1.0 mol % PPTS) indoline could be converted to indole 25 (Table 3E) in 81% isolated yield. 3-Methyl, and 2-methyl indolines were also oxidized to the corresponding indoles in good yields (80% and 73% isolated yields, 26 and 27 respectively). Even the tertiary amine substrate, N-methylindoline, afforded the indole product 28 in 48% yield (Table 3E); however, the electron-deficient N-tosylindoline 29 was not oxidized under these conditions. Bruice has previously demonstrated stoichiometric oxidation of tertiary amines with phenanthroline-derived o-quinones, and he proposed a mechanism analogous to the addition-elimination mechanism in Figure 1B, involving an ammonium-hemiaminal intermediate.14 Further studies are ongoing to develop improved quinone-based catalysts for tertiary amine oxidation.
Conclusion
In conclusion, we have identified a new strategy for aerobic oxidation of secondary amines, employing 1,10-phenanthroline-5,6-diones as a bifunctional o-quinone catalyst. The success of these reactions can be traced to the non-biomimetic reaction mechanism, which involves an addition-elimination pathway, rather than the transamination pathway employed by copper amine oxidase enzymes and many quinone model systems. Direct spectroscopic evidence was obtained for the hemiaminal intermediate. The bifunctional character of the phd catalyst was exploited in the use of Zn2+ to promote amine oxidation, and iodide was fortuitously discovered to promote aerobic catalytic turnover. Control experiments and mechanistic studies reveal that iodide plays a critical redox role in mediating aerobic reoxidation of the reduced quinone catalyst. Collectively, these results provide a foundation for broader exploration of quinones and related redox-active organic catalysts in selective aerobic oxidation reactions.
Supplementary Material
Acknowledgments
We would like to thank Mr. Paul White for helpful discussions and assistance with NMR studies, and Brian S. Dolinar and Dr. Ilia A. Guzei for X-ray crystallographic determination. We are grateful for financial support from the NIH (R01-GM100143). Analytical instrumentation was partially funded by the National Science Foundation (CHE-1048642, CHE-9208463, CHE-0342998, CHE-9974839, CHE-9304546) and National Institutes of Health (NIH 1 S10 RR13866-01)
Footnotes
Supporting Information. Full experimental procedures and characterization data for all products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For selected reviews, see: Klinman JP. J Biol Chem. 1996;271:27189. doi: 10.1074/jbc.271.44.27189.Mure M. Acc Chem Res. 2004;37:131. doi: 10.1021/ar9703342.
- 2.Mure M, Mills SA, Klinman JP. Biochem. 2002;41:9269. doi: 10.1021/bi020246b. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ohshiro Y, Itoh S. Bioorg Chem. 1991;19:169. [Google Scholar]; (b) Rodriguez EJ, Bruice TC. J Am Chem Soc. 1989;111:7947. [Google Scholar]
- 4.(a) Mure M, Klinman JP. J Am Chem Soc. 1995;117:8698. [Google Scholar]; (b) Mure M, Klinman JP. J Am Chem Soc. 1995;117:8707. [Google Scholar]
- 5.(a) Lee Y, Sayre LM. J Am Chem Soc. 1995;117:11823. [Google Scholar]; (b) Lee Y, Sayre LM. J Am Chem Soc. 1995;117:3096. [Google Scholar]
- 6.Wendlandt AE, Stahl SS. Org Lett. 2012;14:2850. doi: 10.1021/ol301095j. [DOI] [PubMed] [Google Scholar]
- 7.Largeron M, Fleury MB. Angew Chem Int Ed. 2012;51:5409. doi: 10.1002/anie.201200587. [DOI] [PubMed] [Google Scholar]
- 8.(a) Yuan H, Yoo WJ, Miyamura H, Kobayashi S. J Am Chem Soc. 2012;134:13970. doi: 10.1021/ja306934b. [DOI] [PubMed] [Google Scholar]; (b) Yuan H, Yoo WJ, Miyamura H, Kobayashi S. Adv Synth Catal. 2012;354:2899. [Google Scholar]
- 9.Largeron M, Fleury MB. Science. 2013;339:43. doi: 10.1126/science.1232220. [DOI] [PubMed] [Google Scholar]
- 10.For selected reviews highlighting different approaches to amine oxidation, esp employing molecular oxygen as the terminal oxidation, see: Li CJ. Acc Chem Res. 2009;42:335. doi: 10.1021/ar800164n.Murahashi SI. Angew Chem Int Ed. 1995;34:2443.Largeron M. Eur J Org Chem. 2013:5225.Schümperli MT, Hammond C, Hermans I. ACS Catalysis. 2012;2:1108.
- 11.For alternative approaches to aerobic, organocatalytic C-N bond dehydrogenation, see: Chen S, Hossain MS, Foss FW. ACS Sustainable Chem Eng. 2013;1:1045.Fang X, Liu YC, Li C. J Org Chem. 2007;72:8608. doi: 10.1021/jo701796n.Liu L, Wang Z, Fu X, Yan CH. Org Lett. 2012;14:5692. doi: 10.1021/ol302708r.Srogl J, Voltrova S. Org Lett. 2009;11:843. doi: 10.1021/ol802715c.
- 12.For selected examples of aerobic, transition-metal catalyzed amine oxidation, see: Sonobe T, Oisaki K, Kanai M. Chem Sci. 2012;3:3249.Samec JSM, Éll AH, Bäckvall JE. Chem – Eur J. 2005;11:2327. doi: 10.1002/chem.200401082.Yamaguchi K, Mizuno N. Angew Chem Int Ed. 2003;42:1480. doi: 10.1002/anie.200250779.Lang X, Ji H, Chen C, Ma W, Zhao J. Angew Chem Int Ed. 2011;50:3934. doi: 10.1002/anie.201007056.Wang JR, Fu Y, Zhang BB, Cui X, Liu L, Guo QX. Tetrahedron Lett. 2006;47:8293.Hu Z, Kerton FM. Org Biomol Chem. 2012;10:1618. doi: 10.1039/c2ob06670j.Zhang E, Tian H, Xu S, Yu X, Xu Q. Org Lett. 2013;15:2704. doi: 10.1021/ol4010118.Patil RD, Adimurthy S. Adv Synth Catal. 2011;353:1695.
- 13.(a) Lee Y, Ling KQ, Lu X, Silverman RB, Shepard EM, Dooley DM, Sayre LM. J Am Chem Soc. 2002;124:12135. doi: 10.1021/ja0205434. [DOI] [PubMed] [Google Scholar]; (b) Lee Y, Huang H, Sayre LM. J Am Chem Soc. 1996;118:7241. [Google Scholar]; (c) Zhang Y, Ran C, Zhou G, Sayre LM. Bioorg Med Chem. 2007;15:1868. doi: 10.1016/j.bmc.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 14.Eckert TS, Bruice TC. J Am Chem Soc. 1983;105:4431. [Google Scholar]
- 15.For leading reference, see: Anthony C, Ghosh M, Blake CC. Biochem J. 1994;304:665. doi: 10.1042/bj3040665.
- 16.(a) Itoh S, Kawakami H, Fukuzumi S. J Am Chem Soc. 1997;119:439. [Google Scholar]; (b) Itoh S, Kawakami H, Fukuzumi S. Biochem. 1998;37:6562. doi: 10.1021/bi9800092. [DOI] [PubMed] [Google Scholar]
- 17.Itoh S, Mure M, Ogino M, Ohshiro Y. J Org Chem. 1991;56:6857. [Google Scholar]
- 18.For leading reference, see: Goss CA, Abruna HD. Inorg Chem. 1985;24:4263.
- 19.Yuasa J, Suenobu T, Fukuzumi S. ChemPhysChem. 2006;7:942. doi: 10.1002/cphc.200500640. [DOI] [PubMed] [Google Scholar]
- 20.Lei Y, Anson FC. J Am Chem Soc. 1995;117:9849. [Google Scholar]; (b) Lei Y, Shi C, Anson FC. Inorg Chem. 1996;35:3044. [Google Scholar]
- 21.For an example of Lewis acid-promoted stoichiometric amine oxidation by TTQ model quinones, see: Itoh S, Taniguchi M, Takada N, Nagatomo S, Kitagawa T, Fukuzumi S. J Am Chem Soc. 2000;122:12087.
- 22.Eckert TS, Bruice TC, Gainor JA, Weinreb SM. Proc Nat Acad Sci. 1982;79:2533. doi: 10.1073/pnas.79.8.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For selected reviews, see: Kochanowska-Karamyan AJ, Hamann MT. Chem Rev. 2010;110:4489. doi: 10.1021/cr900211p.Cao R, Peng W, Wang Z, Xu A. Curr Med Chem. 2007;14:479. doi: 10.2174/092986707779940998.Bentley KW. Nat Prod Rep. 2006;23:444. doi: 10.1039/b509523a.Kawasaki T, Higuchi K. Nat Prod Rep. 2005;22:761. doi: 10.1039/b502162f.Somei M, Yamada F. Nat Prod Rep. 2004;21:278. doi: 10.1039/b212257j.Scott JD, Williams RM. Chem Rev. 2002;102:1669. doi: 10.1021/cr010212u.Allen JRF, Holmstedt BR. Phytochemistry. 1980;19:1573.
- 24.At −40 °C, the oxidation of 1 is sufficiently slow to permit full characterization of the hemiaminal. At higher temperatures, formation of the oxidation product 2 can be detected in the NMR spectrum (cf. the singlet at 8.3 ppm, and multiplets at 7.3 ppm in Figures 3 and S1).
- 25.(a) Dhineshkumar J, Lamani M, Alagiri K, Prabhu KR. Org Lett. 2013;15:1092. doi: 10.1021/ol4001153. [DOI] [PubMed] [Google Scholar]; (b) Huang H, Ji X, Wu W, Jiang H. Adv Synth Catal. 2013;355:170. [Google Scholar]
- 26.For examples of iodine-mediated oxidation of amines, see: Goosen A, McCleland CW, Sipamla AM. J Chem Res (S) 1995:18.Iida S, Togo H. Tetrahedron. 2007;63:8274.Leonard NJ, Leubner GW. J Am Chem Soc. 1949;71:3408.Domínguez E, Lete E. J Heterocyclic Chem. 1984;21:525.Sotomayor N, Domínguez E, Lete E. Tetrahedron. 1995;51:12721.
- 27.Piera J, Bäckvall JE. Angew Chem Int Ed. 2008;47:3506. doi: 10.1002/anie.200700604. [DOI] [PubMed] [Google Scholar]
- 28.Wu Z, Perez M, Scalone M, Ayad T, Ratovelomanana-Vidal V. Angew Chem Int Ed. 2013;52:4925. doi: 10.1002/anie.201301134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.