

Abstract

Type II NADH-quinone oxidoreductase (NDH-2) catalyzes the transfer electrons from NADH to the quinone pool and plays an essential role in the oxidative phosphorylation system of Mycobacterium tuberculosis (Mtb). The absence of NDH-2 in the mammalian mitochondrial electron transport chain makes this enzyme an attractive target for antibiotic development. To fully establish the kinetic properties of this enzyme, we studied the interaction of Mtb NDH-2 with substrates, NADH, and various quinone analogues and their products in both membrane and soluble environments. These studies, and comparative analyses of the kinetics with thio-NAD+ and quinone electron acceptors, provided evidence that Mtb NDH-2 catalyzes the transfer electrons from NADH to quinone substrates by a nonclassical, two-site ping-pong kinetic mechanism whereby substrate quinones bind to a site that is distinct from the NADH-binding site. Furthermore, the effects of quinols on Mtb NDH-2 catalytic activity demonstrate the presence of two binding sites for quinone ligands, one favoring the reduced form and the other favoring the oxidized form.
There is an increasing need for the development of antibiotics against new physiological targets to combat the emergence of bacteria resistant to current front-line drugs. TMC207 (Bedaquiline), a potent and specific inhibitor of mycobacterial ATP synthase, is a new antibiotic that is approved for use in the treatment of multi-drug resistant (MDR) tuberculosis (TB).1 This success has validated the oxidative phosphorylation system (OxPhos) of Mycobacterium tuberculosis (Mtb) as a potential target for the development of new antibiotics. The OxPhos pathway is composed of the respiratory chain (electron transport chain), which produces the proton motive force that drives ATP synthase. In mycobacteria, electrons are introduced into the electron transport chain primarily via type II NADH-quinone (Q) oxidoreductase (Mtb NDH-2), a membrane-bound enzyme composed of a single polypeptide chain of 45 kDa and a single FAD cofactor that mediates electron transfer. NDH-2 catalyzes the oxidation of NADH to NAD+ with concomitant reduction of menaquinone to menaquinol.2 There is no NDH-2 counterpart in vertebrate mitochondria, which utilize a much larger, complex oxidoreductase, type I NADH-quinone oxidoreductase (Complex I or NDH-1), to initiate electron transport. Thus, Mtb NDH-2 is an attractive target for the development of selective antimycobacterial agents, and a detailed understanding of NDH-2 kinetics will be valuable to guide the development process.
Steady-state kinetic studies from our laboratory showed that Mtb NDH-2 catalyzes the transfer electrons from NADH to quinone by a ping-pong mechanism.3 Similar studies of NDH-2 counterparts in Saccharomyces cerevisiae (Ndi1)4,5 and Yarrowia lipolytica(6) also concluded that electron transfer occurs by a ping-pong mechanism. More recently, however, a mechanism proceeding through a ternary complex has been proposed for Ndi1. This mechanism was based on the appearance of a broad spectral peak at 740 nm characteristic of a charge transfer complex between NAD+ and FADH2 that underwent an increase in intensity upon addition of electron acceptors,7 and quinol inhibition kinetics suggesting separate binding sites for NADH and quinone.7 It was suggested that the ping-pong kinetics seen previously might be an artifact of enzyme solubilization by detergents and/or activity measurements using small, soluble quinone substrates that can interact at the NADH-binding site.7
Recently, two independent laboratories have published atomic-resolution structures of Ndi1.8,9 Both structures revealed globally similar protein folds but different modes of interaction of the FAD with quinone substrates. In the structure published by Iwata et al.,8 sites for NAD+ and quinone substrates appeared to overlap on the same face of the isoalloxazine ring of the FAD cofactor, suggesting that donation of electrons from NADH and abstraction of electrons by quinone occur on the same face of FAD. The suggestion of overlapping substrate-binding sites is difficult to reconcile with the formation of a ternary complex but is consistent with a classical (one-site) ping-pong mechanism (Scheme 1; see Experimental Procedures). In the structure published by Feng et al.,9 NADH and quinone substrates were observed to bind at sites located on opposite faces of the isoalloxazine ring of the FAD cofactor. Structures containing bound substrates showed the nicotinamide ring of the NADH substrate bound proximal to the re face of the isoalloxazine ring, whereas the sites for the two quinone ligands were adjacent to the si face of the flavin. Independent binding sites for the electron donor and acceptor would favor a nonclassical (two-site) ping-pong mechanism (Scheme 2).
Scheme 1.
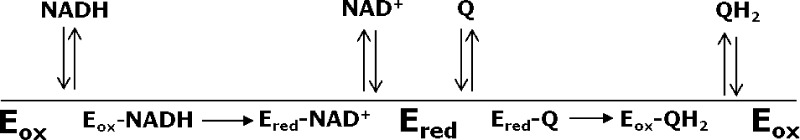
Scheme 2.

In this study, we characterized the kinetics of various quinone substrates and reaction products on the NADH-Q reductase activity of Mtb NDH-2 in both soluble and membrane environments and defined the kinetics of NADH-thio-NAD+ transhydrogenase activity catalyzed by Mtb NDH-2. By comparing the kinetic properties of the two activities, we showed that Mtb NDH-2 catalyzes the transhydrogenase reaction through a one-site ping-pong mechanism and the physiologically relevant NADH-Q reductase reaction through a two-site ping-pong mechanism. Furthermore, the data revealed the existence of two quinone-binding sites with widely different preferences for oxidized and reduced quinones. The two-site ping-pong mechanism and two quinone sites elucidated in this kinetic study are more consistent with the Ndi1 crystal structure of Feng et al.9 than with that of Iwata et al.8
Experimental Procedures
Materials
NADH, NAD+, quinone substrates (UQ0, UQ1, UQ2, and menadione), HEPES buffer, and cosolvents glycerol and DMSO were from Sigma-Aldrich (St. Louis, MO). Thio-NAD+ was from Cell Sciences (Canton, MA), and Big CHAP was from Affimetrix (Santa Clara, CA). The TALON metal affinity column was from Clontech (Mountain View, CA), and the Superose 6 column was from GE Healthcare Life Sciences (Pittsburgh, PA).
Recombinant Mtb NDH-2
NDH-2 was purified according to the method of Yano et al.,3 with the following modifications. Mtb NDH-2 was overexpressed in Mycobacterium smegmatis mc2 4157 with a T7-based expression vector (pYUB derivative).10M. smegmatis was aerobically grown in an autoinduction medium11 in the presence of 20 μg/mL kanamycin and 50 μg/mL hygromycin at 37 °C for 3 days, and the cells were harvested by centrifugation at 5000g for 10 min. Membrane fractions were prepared according to the method of Yano et al.3 and were detergent-solubilized in buffer containing 2% (w/v) Big CHAP. The solubilized membrane fraction was passed over an immobilized metal affinity column and the bound NDH-2 was eluted with an imidazole solution containing 0.25% Big CHAP and was concentrated by ultrafiltration. The concentrated material was further purified by size exclusion chromatography on a Superose 6 HPLC column equilibrated with a solution of 50 mM HEPES/K+ (pH 7.0), 300 mM KCl, 2 mM MgCl2, 0.25% Big CHAP, and 20% (v/v) glycerol. NDH-2 fractions were combined and concentrated by ultrafiltration to 2–3 mg/mL. The final preparation was stored in liquid nitrogen until it was used.
NADH-Quinone (Q) Reductase and NADH-tNAD+ Transhydrogenase Activity Assays
Activity measurements were typically performed at room temperature in 0.5 or 1.0 cm cuvettes using a Beckman 640 spectrophotometer. NADH oxidation was measured by absorbance at 340 nm; tNAD+ reduction was measured at 398 nm. Absorbance measurements were taken at 1.0–3.0 s intervals, and initial activities were determined from linear-least-squares fitting of progress curves over 0.5–3.0 min periods where <10% of the substrates were consumed. The reaction mixture in NADH-Q reductase assays consisted of 0.1 M HEPES/Na+ (pH 7.0) (NADH-Q buffer), substrates, and 2–5 nM enzyme, and in transhydrogenase assays, it consisted of 50 mM HEPES/K+ (pH 7.0), 2 mM MgCl2 (transhydrogenase buffer), substrates, and 10–50 nM NDH-2. Unless otherwise indicated, reactions were initiated by addition of purified recombinant NDH-2 or membrane preparations from cells overexpressing NDH-2. The dilution of the purified enzyme or membrane to start reactions was >100-fold. This dilution virtually removed any influence of the detergent on purified enzyme activity or substrate solubility. In studies with less soluble substrates (UQ1, UQ2, and menadione), 10% (v/v) DMSO was included in the assay buffer. Removal of detergent and addition of DMSO did not result in a loss of activity during the assay period, which were usually followed until NADH consumption was complete (10–20 min).
To obtain kcat or turnover numbers (TN), the change in absorbance was converted into NADH consumed using an extinction coefficient of 6.22 cm–1 mM–1 for NADH (ε340) or 11.9 mM–1 cm–1 for tNADH (ε398)12 and divided by NDH-2 concentration. The stock concentration of purified NDH-2 was determined by FAD measurement. The amount of membrane-bound NDH-2 was estimated assuming it had the same specific activity as purified NDH-2. Under assay conditions of 0.3 mM NADH, 2.0 mM UQ0, and HEPES buffer (pH 7.0), 1.0 nM NDH-2 produced a change in Abs340 of 0.1 min–1.
Assays in the Presence of Reaction Products
Most studies that included reaction products were assayed as just described except for the addition of NAD+ or quinols. NAD+ stock solutions were made in water or 50 mM HEPES-KOH buffer (pH 7.0) containing 2 mM MgCl2. Ubiquinols (Q0H2, Q1H2, and Q2H2) were prepared from commercially available ubiquinone substrates by reduction with sodium borohydride. Briefly, quinones were dissolved in 50 mM HCl/DMSO at 20–100 mM and reduced with a stoichiometric excess of sodium borohydride at room temperature, followed by the addition of 12 N HCl to quench unreacted sodium borohydride. The quinol solution was neutralized with HEPES buffer and confirmed by the loss of visible absorption. Studies to determine the effect of NAD+ on NADH-Q reductase activity were performed in a TECAN M-1000 plate reader by monitoring the change in absorbance at 340 nm. Assays contained 50 mM HEPES (pH 7.0) with 10% glycerol and were initiated with NADH. Initial velocities were determined by linear regression of progress curves.
Kinetic Mechanisms, Equations, and Data Analysis
Kinetic studies were analyzed as described by Cook and Cleland.13 Scheme 1 depicts a classical (one-site) mechanism, and Scheme 2 depicts a nonclassical (two-site) ping-pong mechanism.13 Both mechanisms proceed through two half-reactions: (1) transfer of a hydride from NADH to FAD in the oxidized enzyme (Eox), producing a reduced, modified enzyme (Ered) and the product NAD+, and (2) transfer of electrons from FADH2 in the reduced enzyme to quinone, generating the quinol product and regenerating the oxidized enzyme. Arrows denoting the progress of the chemical reactions point in one direction because both the overall reaction and each half-reaction are highly exergonic and thus effectively irreversible. In accord with the crystal structures discussed in the introductory section, in the one-site ping-pong mechanism both NADH oxidation and quinone reduction occur on the same face of the isoalloxazine ring. Consequently, the binding of each substrate is mutually exclusive because of the overlap between binding sites. This overlap permits competitive inhibition between substrates as well as between each reaction product with the substrate from the other half-reaction. In the case of the two-site mechanism, oxidation of NADH and reduction of quinone occur on different faces of the isoalloxazine ring, permitting separate binding sites for each substrate denoted by E1 and E2 in Scheme 2. The curved arrow connecting the two sites is intended to convey the delivery of an electron to one face of the isoalloxazine ring and the transfer of an electron from the other face. In this mechanism, competitive inhibition can occur between only the substrate and product of the same half-reaction.
The steady-state rate equation describing ping-pong kinetics is the same for both classical and nonclassical mechanisms (eq 1). A ternary complex mechanism differs from a ping-pong mechanism in that both substrates have to bind before products are generated or released. As a result, there is no modified enzyme formed (Ered in Schemes 1 and 2). The rate equation for a ternary complex mechanism is the same as eq 1 except that the factor KiaNADHKMQ is added to the denominator, where KiaNADH is the dissociation constant for the binding of NADH to the enzyme.
| 1 |
Cook and Cleland13 point out that a ping-pong mechanism can be distinguished from ordered or random mechanisms proceeding through a ternary complex in studies where initial velocities are measured as a function of the concentration of both substrates varied at a constant ratio. In the case of a ping-pong mechanism, a double-reciprocal plot of data will be linear as depicted by eq 2, where [Q] = α[NADH], but in the case of a ternary complex mechanism, it will be parabolic because of the added factor in the denominator, as described by eq 3.
| 2 |
| 3 |
To determine the kinetic parameters for Mtb NDH-2 catalysis, a series of velocity (v) versus substrate concentration curves were obtained with one substrate varied (NADH or Q) and the other held constant. The resulting plots, determined at two or three different concentrations of the nonvaried substrate, were globally fit using eq 4, which is a rearranged version of eq 1. The constant term, KMNADH/Vmax, and the apparent Vmax are the fitted parameters; the apparent Vmax is the factor multiplying [NADH] in the denominator. Vmax for the reaction was estimated from a secondary plot of 1/Vmax apparent versus 1/[Q]. KM for NADH was estimated using Vmax and the constant KMNADH/Vmax obtained from the global fit. KM for Q was determined using Vmax and the slope of the secondary plot. The rate equation with Q as the varied substrate and NADH as the fixed substrate has the same form as eq 4, except the positions in the equation for NADH and Q are reversed. To account for the apparent competitive inhibition between substrates in the transhydrogenase reaction when tNAD+ was the varied substrate and NADH was the fixed substrate, KMNADH in the apparent Vmax term was modified by the factor 1 + [tNAD+]/KsitNAD+. Global fitting was by nonlinear least-squares regression analysis using Igor Pro (Wavemetrics).
| 4 |
Reaction irreversibility precludes reversible product inhibition; therefore, inhibition kinetics by NAD+ were assumed to be due to the formation of dead end complexes competing with substrate binding. Inhibition was initially analyzed using the Michaelis–Menten rate equation to determine the type of inhibition. Depending on the outcome of these analyses, the data were then fit to eq 5 to determine relevant Ki values modifying the KM of each substrate where [S]V and [S]C are the concentrations of the variable and constant substrates, respectively, and the modifying inhibition factors β1 and β2 are 1 + [NAD+]/Ki′ and 1 + [NAD+]/Ki″, respectively. To obtain values for inhibition constants Ki′ and Ki″, fits used KM, Vmax, and Ksi values obtained from other analyses.
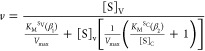 |
5 |
Quinol inhibition of the transhydrogenase and NADH-Q reactions did not appear to be competitive and therefore was evaluated using a relationship assuming binding to a single discrete site as described by eq 6, where FA is fractional activity and f is the fraction of activity remaining at an infinite quinol concentration.
| 6 |
Results
Kinetics of NADH-Q Reductase Activity
To examine the validity of the proposed ping-pong mechanism for Mtb NDH-2, we evaluated its kinetic properties with a series of quinone substrates (UQ0, UQ1, UQ2, and menadione) using both detergent-solubilized and membrane-bound enzyme preparations. The latter membrane preparations were isolated from bacteria in which recombinant Mtb NDH-2 was overexpressed. These membranes contained 50–100-fold more NDH-2 activity than wild-type membranes based on NADH reductase activity in the presence of exogenous UQ0.
As pointed out by Cook and Cleland,13 an ordered or random sequential mechanism can be distinguished from a ping-pong mechanism (classical or non-classical) by the dependence of activity on the concentration of both substrates varied together at a fixed ratio (eq 2, Experimental Procedures). In Figure 1A, the NADH-Q reductase activity of purified Mtb NDH-2 and Mtb NDH-2 overexpressed membranes was measured at varying concentrations of NADH and UQ0 set to a fixed concentration ratio of [UQ0] = 6.25[NADH]. As discussed in Experimental Procedures, the linearity of the double-reciprocal plots for both soluble and membrane-bound NDH-2 preparations strongly supports a ping-pong mechanism.
Figure 1.

Kinetic analysis of NADH-UQ0 reduction by membrane-bound and purified (soluble) Mtb NDH-2. (A) Double-reciprocal plots of the initial velocity vs substrate concentration where both substrates were varied while maintaining a constant ratio of [UQ0] = 6.25[NADH]. The line through the data is the fit of soluble Mtb data to eq 2. Error bars show the range from duplicate experiments. (B) Analysis of NADH-UQ0 kinetics with soluble NDH-2. Initial velocities were determined with NADH as the varied substrate and three different concentrations of UQ0 as the fixed substrate. Lines through the data were determined by global fitting of the three data sets to eq 4. Insets represent secondary plots used to determine Vmax for the reaction and KM values. (C) Plots are similar to those in panel B but were obtained using membranes isolated from M. smegmatis overexpressing recombinant Mtb NDH-2.
KM values for NADH and UQ0 were determined from velocity versus substrate concentration data, as shown in panels B and C of Figure 1. Initial velocities were determined as a function of the concentration of a varied substrate, NADH, at three fixed concentrations of the second substrate, UQ0. The data were globally fit using eq 4 to obtain the constant parameter KMNADH/Vmax, which is characteristic of a ping–pong mechanism, and the apparent Vmax at each of the constant substrate concentrations. Soluble and membrane-bound enzyme data were fit well by eq 4 (solid lines through data). Insets are secondary plots used to obtain KM and kcat values as described in Experimental Procedures. The kinetic parameters are listed in Table 1 along with values obtained with UQ0 as the varied substrate. The KM value for NADH with soluble NDH-2 (average of 0.13 mM) is somewhat lower than the KM for membrane-bound NDH-2 (average of 0.28 mM). The KM for UQ0 varied between 0.23 and 0.64 mM with both free and membrane-bound enzymes. The kcat for the NADH-UQ0 reductase reaction ranged between 330 and 580 s–1. These results indicate that detergent solubilization of NDH-2 from the membrane does not significantly alter electron transfer kinetics.
Table 1. Kinetic Parameters for NADH Oxidation Catalyzed by NDH-2 from Soluble and Overexpressed Membrane Preparations with Various Electron Acceptorsa.
| [NADH] varied | [quinone] varied | ratio plot | |
|---|---|---|---|
| NDH-2 Overexpressed Membranes with UQ0 | |||
| KMNADH (mM) | 0.24 ± 0.07b | 0.32 ± 0.15 | – |
| KMUQ0 (mM) | 0.64 ± 0.20 | 0.23 ± 0.10 | – |
| kcat (s–1) | 580.0 ± 170 | 570.0 ± 200 | 330 ± 15 |
| Soluble NDH-2 with UQ0 | |||
| KMNADH (mM) | 0.14 ± 0.02 | 0.12 ± 0.2 | – |
| KMUQ0 (mM) | 0.58 ± 0.11 | 0.35 ± 0.5 | – |
| kcat (s–1) | 480.0 ± 60 | 425.0 ± 35 | 390 ± 70 |
| Soluble NDH-2 with UQ1 | |||
| KMNADH (mM) | ndc | ndc | 0.13d |
| KMUQ1 (mM) | ndc | ndc | 0.050 ± 0.02e |
| kcat (s–1) | ndc | ndc | 500.0 ± 50 |
| Soluble NDH-2 with UQ2 | |||
| KMNADH (mM) | 0.12 ± 0.2 | ndc | 0.12 |
| KMUQ2 (mM) | <0.02 | ndc | 0.051 ± 0.05 |
| kcat (s–1) | 640.0 ± 40 | ndc | 1150.0 ± 350 |
| Soluble NDH-2 with Menadione | |||
| KMNADH (mM) | ndc | 0.23 ± 0.01 | 0.23 |
| KMmenadione (mM) | ndc | 0.10 ± 0.01 | 0.04 ± 0.02 |
| kcat (s–1) | ndc | 423.0 ± 70 | 400.0 ± 25 |
Kinetic constants determined by global fitting using eq 4 in Experimental Procedures.
Errors are standard errors from fits or propagation errors calculated from fitted values when division of errors was necessary.
Not determined.
The KM value is the average for NADH with UQ0 and UQ2.
KM calculated according to eq 2 assuming KM for NADH where KMUQ1 = α(slope × Vmax – KMNADH).
To provide additional support for a ping-pong mechanism and to evaluate whether apparent ping-pong kinetic behavior might be an artifact of the use of the soluble quinone substrate UQ0, the NADH-Q reductase reaction was assayed with quinone substrates having one or two isoprenoid units at position 5 of the benzoquinone ring (UQ1 and UQ2), and with menadione, which shares the same naphthoquinone headgroup as the physiological substrate in Mtb membranes, menaquinone-9. Because all of these quinones are less soluble than UQ0 in water, 10% DMSO was included in the assay buffer to increase the solubility of each analogue and ensure a broader concentration range for kinetic measurements. Addition of DMSO reduced NADH-UQ0 activity by approximately 10%.
Soluble NDH-2 reduced UQ1, UQ2, and menadione more efficiently than UQ0. At a constant substrate concentration ratio, double-reciprocal plots of NADH-Q reductase activity versus NADH were linear for the three quinone analogues, indicating the general applicability of the ping-pong mechanism (Figure 2A). Kinetic parameters for UQ2 and menadione were determined from velocity versus substrate concentration plots using eq 4. The KM value for UQ1 was estimated from the plots in Figure 2A as described in the footnotes of Table 1. While KM values for NADH were relatively constant with all quinones (∼0.15 mM), the KM values for quinones decreased approximately 10-fold relative to those for UQ0 with each added isoprenoid unit. A double-reciprocal plot for UQ2 in Figure 2B shows the virtual overlap of velocity versus NADH concentration plots at UQ2 concentrations ranging from 0.02 to 0.2 mM, indicating that NDH-2 was saturated with UQ2 at all concentrations analyzed and that the KM for UQ2 is <0.02 mM.
Figure 2.

Kinetics for reduction of various quinones by soluble NDH-2. Assays were conducted in NADH-Q buffer containing 10% (v/v) DMSO. (A) Double-reciprocal plots of velocity vs substrate concentration data obtained with NADH and quinone concentrations maintained at a constant ratio. Ratios were [UQ1] and [UQ2] = 0.28[NADH] and [menadione] = 0.68[NADH]. To minimize spontaneous reoxidation, the assay with menadione was performed under anaerobic conditions by addition to the assay of an oxygen scavenging reaction mixture consisting of 0.2 μg/mL glucose oxidase and 1.0 mM glucose. (B) Double-reciprocal plots describing UQ2 reduction kinetics by soluble NDH-2.
Unlike KM values for quinones, kcat values were similar in all cases, including when menadione was the electron acceptor. Naphthoquinone has a lower midpoint redox potential than benzoquinone (−70 to −110 mV vs 70–110 mV), demonstrating that kcat values were not simply a function of the midpoint redox potential of each quinone. This observation indicates that the transfer of an electron from FADH2 to quinone substrates does not contribute appreciably to the rate-limiting step of the reaction and that the transfer of a hydride from NADH to FAD may be the rate-limiting in the NADH-Q reductase reaction. It should be noted that all quinone substrates exhibited exclusively two-electron reduction reactions, as determined by the stoichiometry of NADH consumption relative to quinone concentration under assay conditions where NADH was in excess.
The decreased KM values for quinones with longer isoprenoid side chains indicate that hydrophobicity is an important determinant of quinone binding affinity, and that the quinone-binding site can accommodate at least two isoprenoid units. Importantly, we did not observe any evidence of substrate inhibition of NADH oxidation by any of the quinone substrates examined, including UQ2 for which the KM is at least 10-fold lower than that of NADH (see Figure 2B). The absence of detectable substrate inhibition is consistent with a nonclassical ping-pong mechanism (Scheme 2).
Characterization of NADH-tNAD+ Transhydrogenase Activity
Product inhibition studies are routinely employed to distinguish between classical and nonclassical ping-pong kinetic mechanisms. However, in the case of NADH-Q reduction, product inhibition is not feasible because the highly exergonic nature of both half-reactions renders each effectively irreversible. Under these conditions, products (NAD+ and QH2) can function only as dead-end inhibitors. To discriminate between classical and nonclassical ping-pong mechanisms, we turned to a comparison of the NADH-Q oxidoreductase and transhydrogenase activities of NDH-2. As a member of the flavoprotein disulfide reductase family, Mtb NDH-2 is capable of catalyzing the transfer of a hydride from NADH via FAD to various NAD+ analogues in vitro.(14) The midpoint redox potential for the tNAD+/tNADH couple (Em,7 = −270 mV) is slightly higher than that for the NAD+/NADH couple (Em,7 = −320 mV), making tNAD+ reduction favorable. Given that the electron acceptor for the transhydrogenase reaction [thio-NAD+ (tNAD+)] resembles NADH, we predicted that the transhydrogenase reaction should proceed through a classical (one-site) ping-pong mechanism, with both the donor and the acceptor interacting with NDH-2 at the NADH site. Markedly different properties of the transhydrogenase and NADH-Q reductase activities of NDH-2 toward substrates and reaction products acting as dead-end inhibitors might therefore provide evidence for discriminating between classical and nonclassical ping-pong mechanisms.
Characterization of the NDH-2 kinetics using tNAD+ as the electron acceptor is shown in Figure 3. Assays monitored tNAD+ reduction by an increase in absorbance at 398 nm, a wavelength at which tNADH exhibits significant absorbance and NADH has virtually no absorbance. Shown in Figure 3A is a double-reciprocal plot of velocity versus substrate data obtained at a fixed concentration ratio of NADH = tNAD+. The kcat value obtained from the y-intercept was 0.275 s–1, which is ∼1000-fold lower than the kcat observed with quinone acceptors. The linear relationship observed is consistent with a ping-pong mechanism. The dependence of velocity on tNAD+ concentration with NADH fixed at two different concentrations is shown in Figure 3B. The curve at high NADH concentrations reached a plateau indicative of saturation, whereas the curve at the lower NADH concentration trended downward at the higher tNAD+ concentrations. The downward curvature is indicative of competition between the two substrates, which is consistent with a one-site ping-pong mechanism.
Figure 3.

NADH-tNAD+ transhydrogenase reaction catalyzed by soluble Mtb NDH-2. Initial velocity data converted to turnover numbers (TNs) as described in Experimental Procedures. (A) Double-reciprocal plots of velocity vs substrate concentration data where substrates were maintained at a fixed ratio of [NADH] = [tNAD+]. The reaction mixture contained 40 nM NDH-2. (B) Initial velocity vs substrate concentration plots to obtain kinetics of the transhydrogenase reaction. The reaction mixture contained 48 nM NDH-2, varying concentrations of tNAD+, and 200 μM NADH (○) or 25 μM NADH (●). Solid lines through the data were produced from global fits to eq 4 modified with a term to account for substrate inhibition.
To obtain the KM values for the reaction, the data were fit to eq 4 with an added term to account for substrate inhibition of NADH by tNAD+ (see Experimental Procedures). The Vmax term in eq 4 was assigned the kcat value determined from the intercept in Figure 3A (0.25 s–1), reducing the fitted parameters to three values. KM values for NADH and tNAD+ were 0.01 and 0.02 mM, respectively, and the substrate inhibition constant (KsitNAD+) for tNAD+ was 0.90 mM (values listed in Table 2). The kcat for the transhydrogenase reaction is roughly 2000-fold lower than that observed for NADH-Q reductase activity (∼500 s–1), suggesting that the rate-determining step may have changed from the first to the second half-reaction, i.e., transfer of a hydride from FADH2 to tNAD+ instead of transfer of a hydride from NADH to FAD. A change in the rate-determining step would be consistent with the marked decrease in the KM for NADH (see the Supporting Information). The presence of substrate inhibition alters eq 2, making it no longer linear. Curvature was not evident in the double-reciprocal plot of Figure 3A because the magnitude of Ksi was 50-fold higher than the KM for NADH, thereby minimizing its influence on the reaction velocities in this analysis.
Table 2. Kinetic Parameters Determined for the NADH-tNAD+ Transhydrogenase Reaction Catalyzed by Mtb NDH-2 and Inhibition Constants Produced by Addition of NAD+ to Each Reaction Mixturea.
| NADH-UQ0 reductase reaction | NADH-tNAD+ transhydrogenase reaction | |
|---|---|---|
| KMNADH (mM) | 0.13–0.15b | 0.01 ± 0.001c |
| KMtNAD+ (mM) | – | 0.02 ± 0.003c |
| KitNAD+ (mM) | 1.0 ± 0.13d | – |
| KsitNAD+ (mM) | – | 0.90 ± 0.35c |
| Ki′NAD+ (mM) | 0.54, 1.37e | 1.4 ± 0.34 |
| Ki″NAD+ (mM) | – | 0.03–0.05f |
Reaction steps represented by constants are noted on Schemes 3–5 in Figures 4 and 5. Values were obtained from global fits using eq 4 or 5 as described in Experimental Procedures. Analyses of NAD+ inhibitory activity were performed with each substrate varied (see Figure 4); hence, two values are reported for these constants.
Data taken from Table 1.
Fitted parameters obtained from a global fit of data in Figure 3B to a modified eq 3, assuming the kcat value (0.25 s–1) for the transhydrogenase reaction determined from data in Figure 3A.
Value obtained from progress curves in Figure 5.
Fitted parameter obtained from data in panels C and D of Figure 4.
Fitted parameters obtained from data in panels A and B of Figure 4.
NAD+ Inhibition of Mtb NDH-2 Transhydrogenase and NADH-Q Reductase Activities
The kinetics of NAD+ inhibition of transhydrogenase and NADH-UQ0 reductase activities are characterized in Figure 4. In panels A and B of Figure 4, inhibition of transhydrogenase activity was measured with NADH and tNAD+ as the varied substrates, respectively. Data shown are presented in double-reciprocal form with velocities converted to turnover numbers (TNs); velocity versus substrate concentration plots were used in global fits to obtain inhibition constants. Hyperbolic plots of this data are presented in the Supporting Information. With NADH as the varied substrate, a marked decrease in apparent kcat values was evident, whereas a decrease in KM was seen with tNAD+ as the varied substrate. Given the structural similarity of NAD+ with each substrate, the inhibition constants fitted using eq 5 were assumed to modify the KM of each substrate. KM values of each substrate and the Ksi value for substrate inhibition were assumed from the studies in Figure 3 (listed in Table 2), so that the only fitted constants in eq 5 were inhibition constants for NAD+. The inhibition kinetics with NADH (Figure 4A) as the varied substrate were best described by noncompetitive (mixed) inhibition, with a weak effect on the KM for NADH (Ki′NAD+ = 1.4 ± 0.34 mM) and a much stronger effect on kcat (Ki″NAD+ = 0.03–0.05 mM). With tNAD+ as the varied substrate (Figure 4B), the inhibition was best described as being competitive with a strong effect on the KM for tNAD+ (Ki″NAD+ = 0.03 mM). The results are fully consistent with a classical (one-site) ping-pong mechanism (Scheme 3 in Figure 4), wherein NAD+ is a competitive inhibitor of both the electron donor and acceptor. Substrate inhibition between tNAD+ and NADH is added in Scheme 3. The kinetics in Figure 4B appear to be competitive rather than noncompetitive as expected because the relatively high NADH concentrations used in the study minimized NAD+/NADH competition, virtually eliminating an effect on the apparent kcat.
Figure 4.

Kinetics of NAD+ inhibition for NADH-tNAD+ transhydrogenase and NADH-UQ0 reductase activities. Data are presented in double-reciprocal plot form. Lines through data were generated using kinetic and inhibition constants from global fits of the original velocity vs substrate data with assumptions as described in the text. (A) Inhibition of transhydrogenase activity by NAD+ with NADH as the varied substrate. Reaction mixtures contained 25 nM NDH-2 and 1.0 mM tNAD+ as the fixed substrate with 0.0 mM NAD+ (○), 1.0 mM NAD+ (●), or 2.0 mM NAD+ (△). Fitting of data to obtain Ki values assumed noncompetitive inhibition. (B) Inhibition of transhydrogenase activity by NAD+ with tNAD+ as the varied substrate. Reaction mixtures contained 25 nM NDH-2 and 0.2 mM NADH with 0.0 mM NAD+ (○), 0.2 mM NAD+ (●), or 1.0 mM NAD+ (△). Data were fit assuming competitive inhibition. (C) NAD+ inhibition of NADH-UQ0 activity with NADH as the variable substrate and UQ0 at a fixed concentration. Reaction mixtures contained 5.0 nM NDH-2 and 2.0 mM UQ0 with 0.0 mM NAD+ (○), 0.5 mM NAD+ (●), or 1.5 mM NAD+ (△). Data were fit globally assuming uncompetitive inhibition. (D) Inhibition of NAD+ on NADH-UQ0 reductase activity with UQ0 as a variable substrate and NADH at a fixed concentration. Reaction mixtures contained 5.0 nM NDH-2, buffer, and 0.125 mM NADH with 0.0 mM NAD+ (○), 0.5 mM NAD+ (●), or 1.5 mM NAD+ (△). Data were fit assuming competitive inhibition.
A parallel study with UQ0 replacing tNAD+ as the electron acceptor is shown in panels C and D of Figure 4. The NAD+ inhibition pattern observed is markedly different from that describing the transhydrogenase reaction. Fitting of the data using either the Michaelis–Menten rate law or eq 5 with the appropriate substitutions found competitive inhibition by NAD+ when NADH was the varied substrate (Figure 4C) and uncompetitive inhibition when UQ0 was the varied substrate (Figure 4D). This inhibition pattern indicates that the only kinetic constant affected by NAD+ was the KM for NADH. The inhibition constant (Ki′NAD+) obtained in both studies using eq 5 varied between 0.5 and 1.3 mM. Both values (average of 0.9 mM) are comparable to the corresponding constant for NAD+ competitive inhibition of NADH in the transhydrogenase reaction (Table 2).
The singular effect of NAD+ on the KM for NADH in the NADH-Q reductase reaction could be consistent with either a one-site or a two-site ping-pong mechanism. However, to be consistent with the one-site mechanism, NAD+ would have to be a very poor competitive inhibitor of UQ0 so that inhibition would go undetected in our measurements. This does not appear to be the case because NAD+ was a relatively strong inhibitor of tNAD+ exhibiting a Ki of 0.03–0.05 mM, which is approximately 10-fold lower than the KM of UQ0. Therefore, the absence of competition between NAD+ and UQ0 for the reduced enzyme strongly supports a two-site ping-pong mechanism (Scheme 4 in Figure 4) where NAD+ and UQ0 interact at discrete, independent sites.
NDH-2 Kinetics When Electron Acceptors UQ0 and tNAD+ Are Present in the Same Reaction Mixture
Kinetic studies with electron acceptors, tNAD+ and UQ0, in the same reaction mixture were performed to determine if there was any competition between the two acceptors. NADH consumption monitored in the presence of UQ0 alone or with both UQ0 and tNAD+ as electron acceptors is shown in Figure 5. The UQ0 concentration in both data sets was 4-fold higher than its KM value, and the tNAD+ concentration in the data set with the electron acceptor mixture was 100-fold higher than its KM value. The concentration of tNAD+ was at enzyme saturation levels to maximize the potential effect of its low KM on the reaction (KM values differ by >30-fold), and the concentration of UQ0 was somewhat greater than its KM to distinguish its higher rate of reduction from that of tNAD+ (kcat values differ by >103-fold). The curve with UQ0 alone reflects the high activity observed with this electron acceptor. A similar study with tNAD+ alone at a concentration of 100KM would have a maximal turnover number of <1, which was therefore not graphed. When both electron acceptors were present in the reaction mixture, the activity was reduced only slightly and in a manner consistent with competitive inhibition as determined using the Michaelis–Menten rate equation or eq 5 to fit the data (Scheme 4).
Figure 5.

Effect of tNAD+ and UQ0 on NADH-UQ0 oxidoreductase activity with NADH the varied substrate. The reaction mixture contained 5.0 nM NDH-2, 0.2 mM NADH, and 2.0 mM UQ0 in the absence (○) or presence (●) of 1.0 mM tNAD+. Data were fit globally using eq 4 assuming competitive inhibition.
These findings are best explained by Scheme 5, which shows the transhydrogenase reaction proceeding by a one-site ping-pong mechanism and the NADH-UQ0 reductase reaction proceeding in parallel through a two-site ping-pong mechanism. Because electron acceptors tNAD+ and UQ0 bind to different sites in the two-site mechanism, they are not competitive with each other for interaction with the reduced form of the enzyme. If they were, tNAD+ would have prevented UQ0 from binding given its concentration was at virtual saturation (100KM). Instead, the reaction rate appeared mostly to reflect the difference in rate constants governing abstraction of electrons by each acceptor from reduced FAD. Because the half-reaction for UQ0 reduction is much more efficient than that for tNAD+ reduction, the electron flux likely proceeds exclusively through UQ0 and the two-site pathway (the dashed line in Scheme 5 signifies the absence of electron flux in the tNAD+ half-reaction). The rapid rate of UQ0 reduction maintains the enzyme predominately in the oxidized form, a form to which tNAD+ binds weakly as a competitive inhibitor of NADH (KitNAD+ in Table 2). Via substitution of the appropriate values into eq 5, a Ki value in the range of 1.0 mM was obtained for the competitive inhibition, which is similar to that obtained for substrate inhibition in Figure 3.
This kinetic study was repeated monitoring tNADH production instead of NADH oxidation. In accord with the interpretation mentioned above, the presence of UQ0 in the assay inhibited tNAD+ reduction. These studies further demonstrate that tNAD+ and UQ0 do not bind to the same site and that the transhydrogenase reaction proceeds through a one-site ping-pong mechanism, whereas NADH-UQ0 reductase activity proceeds through a two-site ping-pong mechanism.
Effects of Quinols on Mtb NDH-2 Catalyzing Transhydrogenase and NADH-UQ Reductase Activities
To further substantiate the two-site ping-pong mechanism and explore the properties of the quinone-binding site, inhibition of NDH-2 transhydrogenase and NADH-Q reductase activities by quinol products was evaluated. Because of the irreversible reaction, quinols, like NAD+, can act as only dead end inhibitors. Given the above evidence establishing different binding sites for NADH/tNAD+ and UQ0, we were surprised to find that reduced forms of UQ0, UQ1, and UQ2 inhibited the transhydrogenase reaction (Figure 6A). The inhibition was not complete and was observed despite virtually saturating concentrations of NADH and tNAD+ being used. The maximal level of inhibition of transhydrogenase activity was ∼60% for all three quinols. Apparent Ki values roughly estimated by fitting the data in Figure 6A to a model assuming a single discrete binding site (eq 6, Experimental Procedures) were ∼0.4 mM for UQ0H2, 0.08 mM for UQ1H2, and 0.003 mM for UQ2H2. The decrease in Ki values with an increasing isoprenoid chain length mirrors KM values for the parent quinone substrates in the NADH-Q reductase reaction (Table 1). In the presence of 0.05 mM UQ2H2, which should be near saturating according to the Ki reported above, the KM values for NADH and tNAD+ were unaffected while kcat decreased by 60% (data not shown), further indicating that the inhibitory effects of quinol ligands on the transhydrogenase reaction are not mediated by binding to the NADH/tNAD+ substrate-binding site.
Figure 6.

Effect of quinols on NDH-2 transhydrogenase and NADH-Q reductase activities. (A) Effect of quinols on the transhydrogenase activity of soluble NDH-2. Reaction mixtures contained 20 nM NDH-2, transhyrogenase buffer, 0.2 mM NADH, 0.2 mM tNAD+, and varying concentrations of UQ0H2 (○), UQ1H2 (●), or UQ2H2 (△). Fractional activities were plotted as a function of quinone concentration, and plots were fit with eq 6. (B) Effects of UQH2 on the NADH-Q reductase activity of soluble NDH-2. Reaction mixtures contained 0.3 mM NADH, 0.025–0.05 mM UQ, 1–5 nM NDH-2, and varying concentrations of UQH2. Activities relative to those of control reaction mixtures that contained no reduced quinone were plotted as a function of UQH2 concentration. Activation was fit with a modification of eq 6. (C) Kinetic studies characterizing UQ1H2 activation of membrane-bound NADH-UQ0 reductase activity. Reaction mixtures contained 0.1 μL of recombinant membrane, NADH-Q buffer, and 0.3 M NADH in the absence (○) or presence (●) of 0.1 M Q1H2. Data were fit to the Michaelis–Menten rate equation (—) to obtain kinetic constant values.
The effects of quinols on NADH-Q reductase activity also showed incomplete inhibition, except for the activation of UQ0 activity by UQ1H2 (Figure 6B). To maximize the possibility of demonstrating competitive inhibition by quinol products, NADH-Q activity was measured at subsaturating concentrations of substrate quinones. Modest, partial inhibition was again observed for most combinations of quinone and quinol ligands, plateauing in the range of 35–40% inhibition. Estimates of inhibition constants were made as described above. UQ2H2 exhibited relatively strong inhibition of activity when UQ0 and UQ1 (Ki = 0.003–0.006 mM) were the electron acceptors. UQ1H2 also inhibited activity when UQ1 was the electron acceptor, but ∼10-fold more weakly than UQ2H2 (Ki ≈ 0.070 mM). In contrast to the general inhibitory activity of most quinols, UQ1H2 activated NADH oxidase activity 2-fold when UQ0 was the electron acceptor. Although the inhibition constants mirror the KM values observed with quinone substrates, the observation that quinol binding is not competitive with either NADH or quinone substrates suggests that quinol inhibition results from binding to a unique site that affects both NADH-Q and transhydrogenase activities.
To demonstrate that activation and inhibition were not artifacts of enzyme solubilization and purification, parallel experiments were repeated with membrane-bound recombinant NDH-2 under conditions similar to those described in the legend of Figure 1. Consistent with the results obtained with soluble NDH-2, addition of 1.0 mM UQ1H2 to an assay containing 0.05 mM UQ1 as an electron acceptor resulted in 30 ± 10% inhibition, whereas the same quinol added to an assay containing UQ0 as an electron acceptor produced an up to 5-fold increase in activity (data not shown). The dependence of NADH-UQ0 activation on UQ1H2 concentration was hyperbolic, indicative of a discrete site with a binding constant of 0.070 mM. Kinetic analysis of UQ1H2 activation of NADH-UQ0 reductase activity is shown in Figure 6C. The data indicate that activation results from a 5–6-fold decrease in the apparent KM value for UQ0 (from 0.35 ± 0.007 to 0.06 ± 0.02 mM; Vmax values under both conditions were 0.12 ± 0.01 A340 min–1 nM–1). Because the KM for UQ0 is composed of rate constants for association and dissociation of the substrate as well as the chemical steps for both half-reactions (see the Supporting Information), the precise mechanism through which quinol binding decreases the KM of UQ0 is unclear. Nevertheless, in the case of UQ1H2 activation of UQ0, the quinol appears to function as an allosteric activator. The incomplete inhibition of NDH-2 transhydrogenase and quinone reductase activities and apparent allosteric activation of NADH-UQ0 reduction by UQ1H2 provide unequivocal evidence of the existence of at least two binding sites with different affinities for quinone and quinol ligands on Mtb NDH-2.
Discussion
Comparison of the kinetics for Mtb NDH-2 catalysis of the NADH-Q and transhydrogenase reactions allowed us to conclude that the NADH-Q reductase activity follows a nonclassical, two-site ping-pong kinetic mechanism as has been reported for a number of other oxidoreductase systems.15−17 The establishment of this two-site mechanism differs from the previously reported ternary complex mechanism proposed for the yeast NDH-2 analogue, Ndi1, and supports the recent Ndi1 crystal structure of Feng et al.,9 showing separate binding sites for NADH and quinone on opposite faces of the isoalloxazine ring of the FAD cofactor. In addition, studies with quinol products revealed a second quinone-binding site more suited to binding quinol than quinone. Occupation of this second site produced either partial inhibition or activation depending on the specific quinone/quinol pair. It is unclear if these two sites reflect the two sites found in the Ndi1 crystal structure of Feng et al.,9 both of which were predicted to play a role in catalysis.
Discrimination between a one-site and a two-site ping-pong mechanism was based on considerable evidence. (1) Kinetic studies with UQ2 as the electron acceptor found no evidence of competition with NADH, even though its KM was at least 10-fold lower than that for NADH (Figure 2B). The absence of substrate competition under these conditions is consistent with independent electron donor- and acceptor-binding sites. In contrast, the transhydrogenase reaction, which was predicted to follow a one-site ping-pong mechanism, displayed substrate inhibition between NADH and tNAD+; KM values for NADH and tNAD+ were approximately equal in this reaction (Figure 3B). (2) NAD+ was a potent competitive inhibitor of tNAD+ in the transhydrogenase reaction, consistent with overlap in donor- and acceptor-binding sites. In contrast, NAD+ inhibited NADH-UQ0 activity only weakly, despite UQ0 having a KM 30-fold higher than that of tNAD+ (Figure 4C,D). The lack of appreciable competition between NAD+ and UQ0 strongly indicates that donor and acceptor substrates bind to different sites, consistent with a two-site ping-pong mechanism. (3) Finally, in a reaction mixture containing both UQ0 and tNAD+, initial NADH oxidation rates reflected the high kcat of the NADH-UQ0 reductase reaction instead of the low kcat of the transhydrogenase reaction (Figure 5). On the basis of a KM for tNAD+ 30-fold lower than that for UQ0, the concentration of tNAD+ in the reaction mixture should have prevented UQ0 from binding to reduced NDH-2 if both electron acceptors were interacting at the same site.
The relatively low Ki of NAD+ for reduced NDH-2 compared to the KM of NADH (differing by 3-fold) for the oxidized NDH-2 might appear to be counterproductive to overall reaction efficiency, especially in the bacterium where the steady-state NAD+/NADH ratio usually favors NAD+. However, as discussed above, in the two-site ping-pong mechanism, quinone and NAD+ are not competitive for the reduced enzyme, and therefore, NAD+ will not impede the reduction of quinone. Furthermore, quinone reduction is fast compared to FAD reduction by NADH, the likely rate-determining step (Table 1). The greater efficiency of quinone reduction will maintain the enzyme largely in the oxidized state to which NAD+ binds weakly (Ki ∼5–7-fold greater than KM for NADH). Our kinetic data also suggest a potential protective role for the tight binding of NAD+ to reduced NDH-2. Tight binding of NAD+ may limit the reactivity of reduced FAD toward soluble oxidants such as molecular oxygen, thereby suppressing the formation of toxic reactive oxygen species. Physiologically, a two-site ping-pong mechanism is ideal for catalyzing the transfer of electrons from a charged, cytoplasmic electron donor (NADH) to a hydrophobic electron acceptor that is buried in a membrane (menaquinone).
Recently, Yang et al.7 proposed a ternary complex mechanism for Ndi1. Their data were heavily based on the appearance of a broad 740 nm spectral peak characteristic of a charge transfer complex. The peak was barely detectable upon addition of NADH but intensified upon addition of an electron acceptor. The need for both substrates to maximize the 740 nm signal was interpreted to indicate that the charge transfer complex was a ternary complex between the enzyme and both substrates and that the oxidation–reduction process was concerted and proceeded through this ternary complex. In support of a ternary complex, Yang et al.7 report inhibition kinetics with a quinol consistent with separate binding sites for quinone and NADH. Although this group initially reported a ping-pong mechanism for Ndi1, they recently attributed this result to an artifact resulting from enzyme solubilization and/or the use of small, soluble quinone substrates capable of entering the NADH substrate site.5 There is, however, an alternative explanation for their data. First, as shown here, the presence of independent binding sites is consistent with a two-site ping-pong mechanism. Second, the kinetics of the 740 nm peak intensification appeared to be too slow (∼5 min with O2 or UQ6) to be consistent with steady-state kinetics. We suggest that the slow intensification may be related to the accumulation of NAD+ during the reaction, especially if the interaction of NAD+ with the reduced form of Ndi1 is as strong as described for NDH-2 in this study. Broad absorption peaks at 740 nm between reduced flavoprotein (FADH2, acting as the charge donor) and NAD+ (acting as the charge acceptor) have been described for flavoproteins exhibiting ping-pong kinetics, e.g., glutathione reductase and lipoyl dehydrogenase.18
When Ndi1 was crystallized in the presence of UQ4, electron density for 2 equiv of quinone ligands (designated UQI and UQII) was observed on the si face of the FAD isoalloxazine ring, which opens to the face of Ndi1 that associates with the membrane.9 The authors further supported this observation through an EPR study, in which two distinct semiquinone species were observed. This structural evidence is consistent with studies of Ndi1, indicating weak and strong quinone-binding sites.5 On the basis of their structural and EPR studies, Feng et al.9 proposed a model whereby 2 equiv of quinone participates in the transfer of an electron from FADH2.
In our studies, addition of quinols (UQ1H2 and UQ2H2) to the NADH-Q and transhydrogenase reaction mixtures produced partial inhibition (maxima of 30 and 60 μM, respectively) in most cases. Thus, they were not competitive inhibitors of NADH, tNAD+, or quinones and therefore not interacting at the substrate-binding site(s). The order of inhibitor effectiveness for the various quinols was the same for the transhydrogenase and NADH-Q reactions, consistent with a single quinol-binding site capable of affecting both reactions. Perhaps the best evidence for a unique quinol site is the observation that UQ0 reduction was activated in the presence of UQ1H2 (Figure 6B). Activation resulted from a 5.5-fold reduction in the KM for UQ0 (Figure 6C), suggesting an allosteric interaction with NDH-2. A binding constant of 0.07 mM was measured for the activation, which is similar to that observed for Q1H2 inhibition of transhydrogenase and NADH-Q reactions. Thus, we believe that both inhibition and activation of NDH-2 reductase activity are mediated through a single quinol-binding site. How this site affects the transhydrogenase reaction remains to be determined. Feng et al.9 do not provide information about quinol inhibition for Ndi1 activity, making it difficult to compare their structural results with our kinetic results. However, Yang et al.7 report competitive inhibition of Ndi1 catalytic activity by Q1H2. This would suggest the Mtb quinol-binding site described in our study is not reflective of either of two quinone sites described in the crystal structure of Feng et al.9
This study establishes that Mtb NDH-2 catalyzes quinone reduction through a two-site ping-pong mechanism, which is distinct from the sequential, ternary complex mechanism proposed for yeast homologue Ndil.7 Our studies also provide evidence of a binding site that favors quinols over quinones. In other studies, we showed that a drug, trifluoperazine,19 inhibits NDH-2 in a noncompetitive manner, and the drug clofazimine is a substrate of NDH-2, its reduction leading to ROS production.20 The identification of a unique binding site that favors quinols and can influence activity might help explain the relatively selective interaction of mycobacteria with these drugs and aid in the design of inhibitory agents that target enzyme regulation. Work to fully understand the function and utility of the quinol/quinone-binding sites is underway.
Acknowledgments
We thank Daniel Gentry, Andrew Ramsey, Trevor Selwood, and Jennifer Yano for helpful discussions.
Glossary
Abbreviations
- Mtb
M. tuberculosis
- NDH-2
type II NADH-quinone oxidoreductase
- Complex I or NDH-1
type I NADH-quinone oxidoreductase
- MDR
multi-drug resistant
- Ndi1
S. cerevisiae NADH dehydrogenase
- NAD+
nicotinamide adenine dinucleotide, oxidized form
- NADH
nicotinamide adenine dinucleotide, reduced form
- FAD
flavin adenine dinucleotide, oxidized form
- FADH2
flavin adenine dinucleotide, reduced form
- TN
turnover number
- UQ
ubiquinone, oxidized form
- UQH2
ubiquinone, reduced form
- tNAD+
thio-NAD+, oxidized form
- tNADH
thio-NAD+, reduced form
- DMSO
dimethyl sulfoxide.
Supporting Information Available
Kinetics of NAD+ inhibition for NADH-tNAD+ transhydrogenase and NADH-UQ0 reductase activities that are the same as those depicted in Figure 4 except that data are presented as s–v plots and the data were globally fit using the same parameters used in Figure 4 (Figure S1), microscopic steps for the NADH-Q reductase and transhydrogenase reactions (Scheme S1), and kinetic parameters for the NADH-Q reductase and transhydrogenase reactions depicted in Scheme S1 (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
T.Y., M.R., K.K.A., N.M.S., and C.P.S. performed the experiments, and T.Y., N.M.S., H.R., and C.P.S. wrote the manuscript. All authors have given approval to the final version of the manuscript.
This work supported by National Institutes of Health Grant R01 AI068942 (to H.R.) and the Global Alliance for TB Drug Development.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Andries K.; Verhasselt P.; Guillemont J.; Gohlmann H. W.; Neefs J. M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E.; Williams P.; de Chaffoy D.; Huitric E.; Hoffner S.; Cambau E.; Truffot-Pernot C.; Lounis N.; Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227. [DOI] [PubMed] [Google Scholar]
- Teh J. S.; Yano T.; Rubin H. (2007) Type II NADH:menaquinone oxidoreductase of Mycobacterium tuberculosis. Infect. Disord.: Drug Targets 7, 169–181. [DOI] [PubMed] [Google Scholar]
- Yano T.; Li L. S.; Weinstein E.; Teh J. S.; Rubin H. (2006) Steady-state kinetics and inhibitory action of antitubercular phenothiazines on Mycobacterium tuberculosis type-II NADH-menaquinone oxidoreductase (NDH-2). J. Biol. Chem. 281, 11456–11463. [DOI] [PubMed] [Google Scholar]
- Velazquez I.; Pardo J. P. (2001) Kinetic characterization of the rotenone-insensitive internal NADH:ubiquinone oxidoreductase of mitochondria from Saccharomyces cerevisiae. Arch. Biochem. Biophys. 389, 7–14. [DOI] [PubMed] [Google Scholar]
- Yamashita T.; Nakamaru-Ogiso E.; Miyoshi H.; Matsuno-Yagi A.; Yagi T. (2007) Roles of bound quinone in the single subunit NADH-quinone oxidoreductase (Ndi1) from Saccharomyces cerevisiae. J. Biol. Chem. 282, 6012–6020. [DOI] [PubMed] [Google Scholar]
- Eschemann A.; Galkin A.; Oettmeier W.; Brandt U.; Kerscher S. (2005) HDQ (1-hydroxy-2-dodecyl-4(1H)quinolone), a high affinity inhibitor for mitochondrial alternative NADH dehydrogenase: Evidence for a ping-pong mechanism. J. Biol. Chem. 280, 3138–3142. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Yamashita T.; Nakamaru-Ogiso E.; Hashimoto T.; Murai M.; Igarashi J.; Miyoshi H.; Mori N.; Matsuno-Yagi A.; Yagi T.; Kosaka H. (2011) Reaction mechanism of single subunit NADH-ubiquinone oxidoreductase (Ndi1) from Saccharomyces cerevisiae: Evidence for a ternary complex mechanism. J. Biol. Chem. 286, 9287–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M.; Lee Y.; Yamashita T.; Yagi T.; Iwata S.; Cameron A. D.; Maher M. J. (2012) The structure of the yeast NADH dehydrogenase (Ndi1) reveals overlapping binding sites for water- and lipid-soluble substrates. Proc. Natl. Acad. Sci. U.S.A. 109, 15247–15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Li W.; Li J.; Wang J.; Ge J.; Xu D.; Liu Y.; Wu K.; Zeng Q.; Wu J. W.; Tian C.; Zhou B.; Yang M. (2012) Structural insight into the type-II mitochondrial NADH dehydrogenases. Nature 491, 478–482. [DOI] [PubMed] [Google Scholar]
- Wang F.; Jain P.; Gulten G.; Liu Z.; Feng Y.; Ganesula K.; Motiwala A. S.; Ioerger T. R.; Alland D.; Vilcheze C.; Jacobs W. R. Jr.; Sacchettini J. C. (2010) Mycobacterium tuberculosis dihydrofolate reductase is not a target relevant to the antitubercular activity of isoniazid. Antimicrob. Agents Chemother. 54, 3776–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 41(8), 207–234. [DOI] [PubMed] [Google Scholar]
- Stein A. M.; Lee J. K.; Anderson C. D.; Anderson B. M. (1963) The Thionicotinamide Analogs of DPN and TPN. I. Preparation and Analysis. Biochemistry 2, 1015–1017. [DOI] [PubMed] [Google Scholar]
- Cook P. F., and Cleland W. W. (2007) Enzyme Kinetics and Mechanism, Garland Science, New York. [Google Scholar]
- Rydström J., Persson B., and Carlenor E. (1987) Transhydrogenases linked to pyridine nucleotides. In Pyridine nucleotide coenzymes: Chemical, biochemical and medical aspects, part B (Dolphin D., Aramovic O., and Poulson R., Eds.) pp 433–461, John Wiley & Sons, New York. [Google Scholar]
- Northrop D. B. (1969) Transcarboxylase VI: Kinetic analysis of the reaction mechanism. J. Biol. Chem. 244, 5808–5819. [PubMed] [Google Scholar]
- Coughlin M. P.; Rajagopala K. V. (1980) The Kinetic Mechanism of Xanthine Dehydroigenase and Related Enzymes. J. Biochem. 105, 81–84. [DOI] [PubMed] [Google Scholar]
- Moxley M. A.; Tanner J. J.; Becker D. F. (2011) Steady-state kinetic mechanim of the proline:ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli. Arch. Biochem. Biophys. 516, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey V.; Ghisla S. (1974) Role of charge-transfer interactions in flavoprotein catalysis. Ann. N.Y. Acad. Sci. 227, 446–465. [Google Scholar]
- Weinstein E. A.; Yano T.; Li L. S.; Avarbock D.; Avarbock A.; Helm D.; McColm A. A.; Duncan K.; Lonsdale J. T.; Rubin H. (2005) Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc. Natl. Acad. Sci. U.S.A. 102, 4548–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T.; Kassovska-Bratinova S.; Teh J. S.; Winkler J.; Sullivan K.; Isaacs A.; Schechter N. M.; Rubin H. (2011) Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: A pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 286, 10276–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.