

Abstract
There are five distinct core structures in the lipopolysaccharides of Escherichia coli and at least two in Salmonella isolates, which vary principally in the outer core oligosaccharide. Six outer core glycosyltransferases, E. coli K-12 WaaG, WaaB, and WaaO and Salmonella typhimurium WaaI, WaaJ, and WaaK, were cloned, overexpressed, and purified. A novel substrate for WaaG was isolated from ΔwaaG E. coli overexpressing the lipid A phosphatase lpxE and the lipid A late acyltransferase lpxM. The action of lpxE and lpxM in the ΔwaaG background yielded heptose2-1-dephospho Kdo2-lipid A, a 1-dephosphorylated hexa-acylated lipid A with the inner core sugars that is easily isolated by organic extraction. Using this structurally defined acceptor and commercially available sugar nucleotides, each outer core glycosyltransferases was assayed in vitro. We show that WaaG and WaaB add a glucose and galactose sequentially to heptose2-1-dephospho Kdo2-lipid A. E. coli K-12 WaaO and S. typhimurium WaaI add a galactose to the WaaG/WaaB product but can also add a galactose to the WaaG product directly without the branched core sugar added by WaaB. Both WaaI and WaaO require divalent metal ions for optimal activity; however, WaaO, unlike WaaI, can add several glucose residues to its lipid acceptor. Using the product of WaaG, WaaB, and WaaI, we show that S. typhimurium WaaJ and WaaK transfer a glucose and N-acetylglucosamine, respectively, to yield the full outer core. This is the first demonstration of the in vitro assembly of the outer core of the lipopolysaccharide using defined lipid A-oligosaccharide acceptors and sugar donors.
Lipopolysaccharide (LPS) is the major constituent of the outer leaflet of the outer membrane of Gram-negative bacteria and essential for the integrity of the outer membrane, making it a potential target for the development of novel therapeutics. The general features of many LPS structures have been characterized; it is composed of lipid A (the hydrophobic membrane anchor), the core region (a nonrepeating oligosaccharide), and O-antigen (a distal, repeating oligosaccharide).1−3
The core oligosaccharide (OS) can be further divided into two structurally distinct regions: the inner (lipid A proximal) and outer core OS. The inner core typically contains residues of 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) and l-glycero-d-manno-heptose4 and is less variable among Gram-negative bacteria. The outer core OS, in contrast, varies considerably and helps define five distinct core structures in Escherichia coli, termed K-12 and R1–R4, and at least two core structures in Salmonella isolates.5,6 Nevertheless, the overall outer core OS structure shows conserved structural themes as shown in the simplified outer core structures of E. coli K-12 and Salmonella typhimurium in Figure 1. Both have three connected hexose residues on the main chain, and the first and third hexose residues are branched with other sugars. In all of the E. coli and Salmonella outer core OS, HexI, the first hexose on the main chain, is a glucose residue.2 WaaG has been identified as the UDP-glucose:(heptosyl) lipopolysaccharide α-1,3-glucosyltransferase in E. coli K-12 and S. typhimurium.7,8 In E. coli K-12, HexII and HexIII are both glucose residues, and in S. typhimurium, HexII is a galactose residue and HexIII is a glucose residue (Figure 1). WaaO [UDP-glucose:(glucosyl) lipopolysaccharide α-1,3-galactosyltransferase] from E. coli K-12, WaaI [UDP-galactose:(glucosyl) lipopolysaccharide α-1,3-galactosyltransferase] from S. typhimurium, and WaaJ [UDP-glucose:(galactosyl) lipopolysaccharide α-1,2-glucosyltransferase] are responsible for the transfer of these hexose residues.1,2,9,10
Figure 1.

E. coli K-12 and S. typhimurium LPS outer core oligosaccharides (OS’s). The enzymes proposed to catalyze each glycosylation are shown. The linkages among the sugars are indicated;1 all are α linkages except sugars added by WaaU from E. coli K12, which are β linkages. The dashed line attached to HepII indicates where the rest of the inner core and lipid A is attached. The location at which the O-antigen is attached to the outer core OS is indicated.
In both E. coli K-12 and S. typhimurium, branched sugars are added to the outer core OS. In both organisms, galactose is added to HexI in an α-1→6 linkage by WaaB, the UDP-galactose:(glucosyl) lipopolysaccharide α-1,6-galactosyltransferase.5 HexIII is appeneded with an α-1→2-linked N-acetylglucosamine residue in S. typhimurium by WaaK, the UDP-N-acetyl glucosamine:(glucosyl) lipopolysaccharide α-1,6-N-acetyl glucosamine transferase. E. coli K-12 differs at this position; it is partially substituted at the C-6 position with a β-d-GlcNAc-(1→7)-l-α-d-Hep disaccharide, both of which are proposed to be added by WaaU.1,2
The presence of an intact inner and outer core OS is critical for cell function in both E. coli and S. typhimurium. The outer membranes of E. coli cells possessing a mutated waaG have a decreased level of phosphorylation of the inner core heptose, leading to decreased membrane stability; this phosphorylation increases membrane stability by mediating cross-linking with divalent cations.9 In Salmonella enterica serovar Typhi, the outer core terminal glucose residue (Glc II) is required for the entry of the bacteria into epithelial cells.11 In addition, the inner and outer core OS links lipid A and the O-antigen, both of which are potent modulators of innate immunity.1,12
Detailed analysis of the outer core OS glycosyltransferases of E. coli and S. typhimurium has been limited by the lack of biochemical data derived from purified enzymes and structurally defined acceptors and structural characterization of in vitro reaction products. Previously, only WaaJ from E. coli R3 has been characterized in vitro using pure enzyme and substrates.13,14 Previous work on the biochemical characterization of the other outer core OS glycosyltransferases has relied on in vivo complementation of chromosomal insertion mutations. In these studies, the LPS was characterized using polyacrylamide gel electrophoresis, not by directly assessing the required lipid acceptors and sugar donors required.15,16 In this work, we generated an outer core OS acceptor that is easily isolated by extraction with organic solvents and has a defined structure. The overexpression of the lipid A late acyltransferase lpxM(17) and lipid A 1-phosphatase18,19 in the ΔwaaG E. coli strain yields a lipid A OS, with decreased hydrophilicity due to removal of the 1-phosphate (by lpxE), a decreased level of phosphorylation of the inner core heptoses (due to ΔwaaG), and a higher proportion of secondary acyl chains (due to lpxM). Using this lipid acceptor and commercially available sugar nucleotides, we report the in vitro characterization of six outer core glycosyltransferases, E. coli WaaG, WaaB, and WaaO and S. typhimurium WaaI, WaaJ, and WaaK, and demonstrate the assembly of the entire S. typhimurium outer core OS in vitro.
Materials and Methods
Materials
Reagent grade chloroform, methanol, and silica gel 60 thin-layer chromatography (TLC) plates (layer thickness of 150–200 μm) were obtained from EMD Chemicals Inc. (Gibbstown, NJ). [32P]Pi, UDP-[U-14C]glucose, UDP-[U-14C]galactose, and GDP-[U-14C]mannose were from PerkinElmer Life and Analytical Sciences, Inc. (Waltham, MA). Yeast extract, agar, and tryptone were from BD Biosciences. Sodium chloride and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were from VWR International (West Chester, PA). Triton X-100 was from Thermo Fisher Scientific (Waltham, MA). Isopropyl 1-thio-β-d-galactopyranoside (IPTG) was from Invitrogen Corp. (Carlsbad, CA). UDP-glucose, UDP-galactose, UDP-glucuronic acid, UDP-galacuronic acid, ADP-glucose, GDP-glucose, UDP-N-acetylglucosamine, and ADP-mannose were purchased from Sigma-Aldrich. The P-10 desalting column was from GE. All other chemicals were reagent grade and were purchased from either Sigma-Aldrich (St. Louis, MO) or Mallinckrodt Baker, Inc. (Phillipsburg, NJ).
Bacterial Strains and Molecular Biology Applications
All bacteria were grown in Luria-Bertani broth (LB, 10 g of NaCl, 10 g of bacto-tryptone, and 5 g of yeast extract per liter at 37 °C).20 When required for the selection of plasmids, cells were grown in the presence of 100 μg/mL ampicillin, 30 μg/mL chloramphenicol, or 30 μg/mL kanamycin. Plasmids were prepared using the Qiagen mini-prep kit (Qiagen). Restriction endonucleases (New England Biolabs) and T4 ligase (Invitrogen) were used according to the manufacturers’ instructions. Genomic DNA was isolated using the protocol for bacterial cultures in the Easy-DNA kit (Invitrogen). Transformation-competent E. coli cells were prepared by the method of Inoue et al.21 Double-stranded DNA sequencing was performed with an ABI Prism 377 instrument at the Duke University DNA Analysis Facility. Primers were purchased from Integrated DNA Technologies, Inc.
Cloning, Overexpression, and Purification of E. coli K-12 WaaG, WaaB, and WaaO and S. typhimurium WaaI, WaaJ, and WaaK
E. coli K-12 waaG, waaB, and waaO were amplified via PCR from genomic DNA of W3110 using the primers described in Table S1 of the Supporting Information. For each gene, an NdeI site was incorporated at the N-terminus and a BamHI or XhoI site was incorporated at the C-terminus. Each PCR product was digested with NdeI and BamHI or XhoI and ligated into a similarly digested pET-21b or pET-28b expression vector as indicated in Table 1. S. typhimurium waaI, waaJ, and waaK were similarly cloned from genomic DNA of S. typhimurium into the pET-28b expression vector.
Table 1. Bacterial Strains and Plasmids.
| strain or plasmid | description | source or reference |
|---|---|---|
| strains | ||
| S. typhimurium | ||
| LT2 | wild type | Salmonella Genetic Stock Center (University of Calgary, Calgary, AB) |
| E. coli | ||
| W3110 | wild type, F-λ | E. coli Genetic Center (Yale University, New Haven, CT) |
| BL21(DE3) | F–ompT hsdSB(rB–, mB–) gal dcm (DE3) | Novagen |
| CWG303 | E. coli F470 (waaG::Gen) | gift from C. Whitfield9 |
| JQ2 | W3110 (waaG:: Gen) pWFnlpxE, pBAD-lpxM | |
| plasmids | ||
| pWSK29 | low-copy number vector, lac promoter, AmpR | (38) |
| pWFnlpxE | pWSK29 harboring Francicella novicida lpxE | (19) |
| pBAD33 | medium-copy number vector, CamR | (39) |
| pBAD-lpxM | pBAD33 harboring lpxM | gift from D. Sixa |
| pECG28 | pET28b harboring E. coli K-12 waaG | |
| pECB21 | pET21b harboring E. coli K-12 waaB | |
| pECI21 | pET21b harboring E. coli K-12 waaO | |
| pSTI28 | pET28b harboring S. typhimurium waaI | |
| pSTJ28 | pET28b harboring S. typhimurium waaJ | |
| pSTK28 | pET28b harboring S. typhimurium waaK |
lpxM was subcloned out of the pET vector and into the pBAD vector using the SacI and XbaI sites, yielding a fragment that included the pET-21a(+) ribosome binding site.
Each plasmid was transformed into BL21(DE3) for overexpression of the respective protein. LB (750 mL) containing the appropriate antibiotics was inoculated from a 50 mL overnight culture to a final A600 of 0.02 and grown to an A600 of 0.7 at 37 °C. Protein expression was induced by the addition of IPTG (final concentration of 1 mM). For JQ2, arabinose (0.2%) was also added for the induction. The cultures were grown for an additional 4 h and then harvested at 4 °C by centrifugation at 4000g for 20 min. The cell pellet was washed with phosphate-buffered saline (PBS) [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 (pH 7.5)], and the cells were harvested by centrifugation. This and all subsequent steps were conducted at 4 °C. Cell debris and unbroken cells were removed by centrifugation at 27000g for 30 min. The final cell pellet was resuspended in 30 mL of buffer A [50 mM Hepes (pH 7.5) containing 300 mM KCl] with 0.5% Triton X-100 and disrupted by being passed through a French pressure cell at 18000 psi.
Each of the overexpressed proteins was purified using Ni-NTA chromatography. The membrane-free cytosol (derived from a 750 mL culture of E. coli overexpressing an outer core glycosyltransferase) was loaded onto 1 mL of Ni-NTA resin at 4 °C and washed with 30 column volumes of buffer A with 30 mM imidazole and 1 column volume of buffer A with 120 mM imidazole. The enzyme was eluted with buffer A with 500 mM imidazole. The imidazole was removed from the enzyme preparation using a P10 desalting column. Purified, desalted enzymes were stored at −80 °C until they were needed.
Preparation of Radiolabeled Substrates
A 10 mL culture of G5622 medium containing 100 μg/mL ampicillin, 30 μg/mL chloramphenicol, 1 mM IPTG, and 0.02% arabinose was inoculated with 100 μL of an overnight culture of JQ2 (Table 1). The culture was incubated at 37 °C while being shaken at 225 rpm until A600 reached 0.5. Cells were collected by centrifugation at 4 °C and 2000g for 20 min and then resuspended in 10 mL of G56 medium without phosphate. The cell suspension was centrifuged again, the supernatant discarded, and the cell pellet resuspended in 10 mL of fresh G56 medium without phosphate. [32P]Pi (1 mCi) was added, and the cells were grown at 37 °C for 4 h. The radiolabeled cells were pelleted by centrifugation in a clinical centrifuge for 15 min. The cell pellet was resuspended in 3.8 mL of a neutral single-phase Bligh–Dyer extraction mixture [1:2:0.8 (v/v/v) CHCl3/CH3OH/PBS]23 and incubated at room temperature for 30 min. The cell debris was pelleted by centrifugation as described above. The supernatant was transferred to a fresh tube, and 1 mL of CHCl3 and 1 mL of PBS were added to generate a two-phase neutral Bligh–Dyer extraction mixture [2:2:1.8 (v/v/v) CHCl3/CH3OH/PBS].23 After the mixture had been vortexed, the phases were resolved by centrifugation for 15 min as described above. The lower phase was transferred to a fresh glass tube. The remaining upper phase was extracted a second time by the addition of the fresh, pre-equilibrated neutral lower phase. The two lower phases were pooled and dried under a stream of N2. The dried lipid films were redissolved in a CHCl3/CH3OH mixture [4:1 (v/v)] and spotted onto a 10 cm × 20 cm TLC plate. The TLC plate was developed in the CHCl3/CH3OH/CH3COOH/H2O solvent system [25:15:4:4 (v/v/v/v)]. The plate was dried under a cold air stream and exposed to X-ray film for 30 s to locate the [32P]heptose2-1-dephosphorylated-KDO2-lipid A (Hep2-1-deP-KLA). The region of the silica plate containing the product was dampened with H2O, removed by scraping, transferred to a thick-walled glass tube, and resuspended in 4 mL of a neutral single-phase Bligh–Dyer mixture. The suspension was vigorously mixed and subjected to sonic irradiation in a sonicator bath for 30 s. The silica particles were pelleted by centrifugation as in a clinical centrifuge for 10 min. The supernatant containing the 32P-labeled Hep2-1-deP-KLA was removed and passed through a 4 mL glass-wool column to remove the remaining silica particles. The solution was converted to a neutral two-phase Bligh–Dyer mixture by the addition of CHCl3 and PBS. The phases were separated in a clinical centrifuge as described above. The lower phase was transferred to new glass tubes. The upper phases was extracted a second time by the addition of 1.8 mL of fresh, pre-equilibrated neutral lower phase. The lower phases were pooled and dried under a stream of N2. The [32P]Hep2-1-deP-KLA was resuspended in buffer B [25 mM Tris-HCl (pH 7.8) containing 1 mM EDTA, 1 mM EGTA, and 0.1% Triton X-100] and then stored at −20 °C. Typically, the yield of [32P]Hep2-1-deP-KLA recovered was ∼0.6% of the total input 32[P]Pi.
Radiolabeled Hep2-1-deP-KLA was used to prepare [32P]Gal-Glc-Hep2-1-deP-KLA using WaaG and WaaB. WaaG (0.2 mg/mL) was incubated with 3 μCi of [32P]Hep2-1-deP-KLA and 5 mM UDP-Glc in the presence of 50 mM Hepes (pH 7.5) and 0.1% Triton X-100 for 2 h at 25 °C in a total volume of 100 μL. The reaction mixture was converted to a one-phase neutral Bligh–Dyer mixture by the addition of CHCl3 and CH3OH. After a brief centrifugation, the single-phase mixture was transferred to a fresh tube and converted to a two-phase neutral Bligh–Dyer mixture by the addition of CHCl3 and PBS. After centrifugation to resolve the phases, the upper phase was washed as described above and the two lower phases were combined. After being dried under a stream of N2, the lipid films were dissolved in 50 μL of buffer B. WaaB (0.2 mg/mL) and 5 mM UDP-Gal were added to the extracted WaaG product, producing a final volume of 100 μL, and incubated at 25 °C for 3 h. The reaction mixture was spotted onto a 10 cm × 20 cm TLC plate and developed in a CHCl3/CH3OH/CH3COOH/H2O system [25:15:4:4 (v/v/v/v)]. The plate was dried under a cold air stream and exposed to X-ray film for 3 min to locate the [32P]Gal-Glc-Hep2-1-deP-KLA. The region of product was scraped, extracted, and purified as described above for the purification of [32P]Hep2-1-deP-KLA, with a yield of ∼10% of the starting lipid.
Large-Scale Preparation of Hep2-1-deP-KLA
JQ2 cells were grown under conditions similar to those described above for the expression of the outer core enzymes. A 3 L culture of JQ2 was grown to an A600 of 1.8 and harvested by centrifugation at 4000g for 10 min at 4 °C. The cell pellet was washed once with 40 mL of PBS and the centrifugation repeated. The final cell pellet was resuspended in 40 mL of PBS, and 50 mL of CHCl3 and 100 mL of CH3OH were added to generate a neutral single-phase Bligh–Dyer mixture. The extraction mixture was incubated for 1 h at room temperature. The cell debris was removed by centrifugation at 4000g for 15 min at room temperature. The supernatant was transferred to a clean 250 mL Teflon bottle and converted to a two-phase neutral Bligh–Dyer system by the addition of 50 mL of CHCl3 and 50 mL of PBS. The phases were resolved by centrifugation as described above. The upper phase was removed and re-extracted with 90 mL of the fresh, pre-equilibrated neutral lower phase. The lower phases were pooled and dried by rotary evaporation. Next, the dried lipid film was redissolved in 25 mL of a CHCl3/CH3OH/H2O mixture [2:3:1 (v/v/v)] and applied to a 5 mL diethylaminoethyl (DEAE) cellulose (acetate form) column24 equilibrated with the same solvent mixture. The column was washed with 10 column volumes of the CHCl3/CH3OH/H2O mixture [2:3:1 (v/v/v)]. The bound phospholipids and LPS were eluted using 25 mL steps of the CHCl3/CH3OH/CH3COONH4 mixture [2:3:1 (v/v/v)], with successive CH3COONH4 concentrations of 60, 120, 240, and 480 mM in the aqueous component. For each elution step, 5 mL fractions were collected. The fractions containing Hep2-1-deP-KLA were identified by spotting 10 μL portions of each fraction onto a TLC plate. The plate was developed in a CHCl3/CH3OH/CH3COOH/H2O system [25:15:4:4 (v/v/v/v)]. Lipids were visualized by spraying the plate with 10% sulfuric acid in ethanol and charring on a hot plate. All fractions containing Hep2-1-deP-KLA were pooled and the appropriate amounts of CHCl3 and H2O added to generate a 2:2:1.8 (v/v/v) CHCl3/CH3OH/aqueous mixture. The lower phase was dried under a stream of N2. The collected lipid was redissolved in a CHCl3/CH3OH mixture [4:1 (v/v)] and spotted along the bottom of a 10 cm × 20 cm TLC plate. A 10 μL sample was separately spotted onto the same TLC plate as a control to identify the location of the Hep2-1-deP-KLA. After the plate had been developed in the CHCl3/CH3OH/CH3COOH/H2O solvent system [25:15:4:4 (v/v/v/v)], the plate was dried under cold air, and the portion of the plate containing the control was charred. The region of the TLC plate containing the Hep2-1-deP-KLA was scraped, extracted, and purified as described above. Typically, 3 mg of Hep2-1-deP-KLA was isolated from 3 L of JQ2 grown to an A600 of 1.8.
The concentration of Hep2-1-deP-KLA was determined as follows. UDP-[14C]Glc (1 mM) and different concentrations of Hep2-1-deP-KLA (from 0.01 to 0.05 mM) were reacted with 0.2 mg/mL WaaG for 30 min as described above until all of the Hep2-1-deP-KLA had been completely consumed. Assays with [32P]Hep2-1-deP-KLA showed that WaaG can convert most of the Hep2-1-deP-KLA to Glc-Hep2-1-deP-KLA at high UDP-Glc concentrations (Figure 3). By following the incorporation of [14C]Glc into [14C]Glc-Hep2-1-deP-KLA, we could determine the concentration of the unlabeled Hep2-1-deP-KLA.
Figure 3.

Glycosylations catalyzed by E. coli WaaG and WaaB. [32P]Hep2-1-deP-KLA (marked A) was incubated with WaaG and/or WaaB (0.01 mg/mL each) for the times indicated as described in Materials and Methods. Both UDP-Glc (1 mM) and UDP-Gal (1 mM) were included in the reaction mixture. B marks the location of the WaaG product, Glc-Hep2-1-deP-KLA. C marks the location of the WaaB product, Gal-Glc-Hep2-1-deP-KLA.
In Vitro Assay Conditions
The standard reaction mixtures contained purified glycosyltransferase(s) (0.1 mg/mL), 50 mM Hepes (pH 7.5), 0.1% Triton X-100, 10 μM [32P]Hep2-1-deP-KLA, and 1 mM UDP-hexose donor(s). Reactions were initiated by the addition of enzyme and mixtures incubated at 25 °C for the times indicated. For coupling reactions of WaaG, WaaB, and WaaO from E. coli K-12, WaaG (0.01 mg/mL) was reacted with UDP-Glc for 15 min using the standard assay conditions described above (final volume of 50 μL). Next, WaaB (0.1 mg/mL) and 1 mM UDP-Gal were added to the reaction mixture (final volume of 52 μL) and incubated at 25 °C for 30 min. WaaO (0.1 mg/mL) and 1 mM MgCl2 were added to the reaction solution after WaaB reacted at 25 °C for 30 min (final volume of 54 μL).
For reactions of E. coli K-12 WaaO and S. typhimurium WaaI with Gal-Glc-Hep2-1-deP-KLA, the product of the reaction of WaaG and WaaB, only the [32P]Gal-Glc-Hep2-1-deP-KLA product (final concentration in the nanomolar range) was included as the LPS acceptor along with the appropriate UDP-hexoses and 1 mM MgCl2. The preparation of [32P]Gal-Glc-Hep2-1-deP-KLA is described above.
Reactions were stopped by spotting 3–5 μL portions of the reaction mixtures onto a TLC plate. After being dried in a stream of cold air, plates were developed in the CHCl3/CH3OH/CH3COOH/H2O solvent [25:15:4:4 (v/v/v/v)]. The amount of product formed was calculated from the percent conversion of radioactive substrate to product, quantified using a Molecular Dynamics PhosphoImager.
Data Fitting
To determine the kinetic parameters for outer core enzymes, one substrate, either a sugar donor or an acceptor, was varied from approximately 5-fold below to 5-fold above the predicted Km while the other substrate was held constant 5-fold above the predicted Km. The data were fit to the Michaelis–Menten equation using Kaleidagraph (Synergy Software, Reading, PA).
Electrospray Ionization Mass Spectrometry
Mass spectra were acquired on an ABI QSTAR XL tandem quadrupole time-of-flight mass spectrometer (ABI/MDS-Sciex, Toronto, ON) equipped with an electrospray ionization (ESI) source. Spectra were acquired in the negative ion mode and typically were the accumulation of 60 scans collected from m/z 200 to 2000. Typically, lipids were dissolved in 200 μL of a CHCl3/CH3OH mixture [2:1 (v/v)], supplemented with 1% piperidine, and immediately infused into the ion source at a rate of 5–10 μL/min. The negative ion ESI was conducted at −4200 V.18 The acquisition and analysis of data were performed using Analyst QS (ABI/MDS-Sciex).
Results
Preparation of the E. coli WaaG Substrate, Hep2-1-deP-KLA
An LPS lacking an outer core OS accumulates in CW303, an E. coli strain with the gentamycin gene inserted into the waaG gene.10 The lipid A 1-phosphatase lpxE(19) and the lipid A late acyl transferase lpxM(17) were introduced into CW303 to generate strain JQ2 and promote the accumulation of a modified LPS that contains a fully hexa-acylated lipid A portion that is dephosphorylated at the 1-position and the inner core sugars, two Kdo and two heptose residues (Hep2-1-deP-KLA). The accumulating Hep2-1-deP-KLA (Figure 2) was purified as described in Materials and Methods, and the structure was confirmed using negative ion electrospray ionization mass spectrometry (ESI-MS) (Figure 4A). The major ions at m/z 1269.73 and 846.15 correspond to the [M – 2H]2– and [M – 3H]3– ions of Hep2-1-deP-KLA, respectively (expected values of m/z 1269.7409 and 846.1582, respectively).
Figure 2.

Structure of Hep2-1-deP-KLA. The structure of the lipid A OS purified from JQ2 is shown.
Figure 4.

Negative ion ESI-MS of the lipid A OS’s, Hep2-1-deP-KLA, Glc-Hep2-1-deP-KLA, and Gal-Glc-Hep2-1-deP-KLA. Panel A shows the mass spectrum from m/z 800 to 1600 for the Hep2-1-deP-KLA isolated from JQ2. The [M – 2H]2– ion at m/z 1269.73 and the [M – 3H]3– ion at m/z 846.15 correspond by mass to Hep2-1-deP-KLA (expected values of m/z 1269.7409 and 846.1582, respectively). Panel B shows the mass spectrum from m/z 800 to 1600 for the product isolated from the reaction catalyzed by WaaG using Hep2-1-deP-KLA and UDP-Glc as substrates. The [M – 2H]2– ion at m/z 1351.34 and the [M – 3H]3– ion at m/z correspond by mass to the expected WaaG product, Glc-Hep2-1-deP-KLA (expected values of m/z 1351.2690 and 900.1758, respectively). Panel C shows the mass spectrum from m/z 800 to 1600 for the product of the WaaG and WaaB coupled reaction. The product, Gal-Glc-Hep2-1-deP-KLA, is detected as an [M – 2H]2– ion at m/z 1431.78 (expected value of m/z 1431.7937). Unreacted substrate, Hep2-1-deP-KLA, is also detected as an [M – 2H]2– ion at m/z 1269.73.
WaaG Transfers the Outer Core HexI
When purified E. coli WaaG was incubated in an in vitro reaction mixture with UDP-Glc as the sugar donor and [32P]Hep2-1-deP-KLA (A, Figure 3) as the lipid acceptor, a more slowly migrating product is formed (B, Figure 3), consistent with the addition of a glucose to the Hep2-1-deP-KLA to form Glc-Hep2-1-deP-KLA. This product was purified and analyzed by negative ion ESI-MS (Figure 4B). The two predominant ions at m/z 1351.34 and 900.23 correspond to the [M – 2H]2– and [M – 3H]3– ions expected for Glc-Hep2-1-deP-KLA, respectively (expected values of m/z 1351.2690 and 900.1758, respectively).
Using this in vitro reaction, WaaG was further characterized. The Glc-Hep2-1-deP-KLA product was formed in a time-dependent manner (Figure 3). No Glc-Hep2-1-deP-KLA was formed when other sugar donors, such as UDP-galactose, UDP-glucuronic acid, UDP-galacuronic acid, GDP-mannose, ADP-glucose, and GDP-glucose, were used in place of UDP-Glc (Figure S1A,B of the Supporting Information). The apparent Km values for UDP-Glc and Hep2-1-deP-KLA are 0.162 ± 0.028 mM and 3.85 ± 1.80 μM, respectively, and the kcat is 0.5 s–1, similar to the values reported previously when WaaG was characterized in crude extracts.7,8
The Triton X-100 dependence of the WaaG-catalyzed formation of Glc-Hep2-1-deP-KLA was investigated (Figure S2 of the Supporting Information). The activity of WaaG increased as the Triton X-100 concentration was increased from 0 to 0.2%; higher concentrations of detergent were inhibitory. On the basis of these results, 0.1% Triton X-100 was used in subsequent in vitro assay reactions of WaaG.
The WaaG activity is dramatically decreased by inclusion of Mg2+ in the in vitro assay reaction mixture (Figure S3 of the Supporting Information). Less than 20% of WaaG activity is left when 0.8 mM Mg2+ is added to the assay. WaaG was similarly inhibited by other divalent metal ions such as Mn2+, Ca2+, Zn2+, Co2+, Ni2+, and Cu2+ (Figure S4 of the Supporting Information).
WaaG can hydrolyze the sugar donor, UDP-Glc, in the absence of a lipid acceptor as shown in Figure 5. When 14C-labeled UDP-Glc is incubated with WaaG, glucose is released in a WaaG-dependent reaction (in Figure 5, compare lanes 1 and 2). The Km and kcat values for this hydrolysis reaction are 3.6 ± 0.8 mM and 0.014 s–1, respectively. WaaG was not able to catalyze the release of the 4-epimer of glucose, galactose, from UDP-Gal (Figure S5 of the Supporting Information).
Figure 5.

WaaG hydrolyzes UDP-Glc in the absence of a lipid acceptor. UDP-[U-14C]Glc was incubated in the presence (lane 1) or absence (lane 2) of WaaG (0.25 mg/mL) and then displayed using TLC as described in Materials and Methods. [U-14C]Glc was spotted as a control in lane 3 to indicate the migration of free glucose.
WaaB Catalyzes the Transfer of Galactose to the E. coli K-12 Outer Core
Using the product of WaaG, Glc-Hep2-1-deP-KLA, the ability of E. coli WaaB to catalyze the transfer of galactose from UDP-Gal to the outer core glucose was assessed. When purified WaaB was coupled in vitro with the WaaG reaction, a more slowly migrating product was formed (product C, Figure 3), consistent with the addition of galactose to Glc-Hep2-1-deP-KLA. This product, Gal-Glc-Hep2-1-deP-KLA, was isolated and analyzed using negative ion ESI-MS (Figure 4C). The ion at m/z 1431.78 is consistent with the [M – 2H]2– ion of Gal-Glc-Hep2-1-deP-KLA (expected value of m/z 1431.7937). No product was formed when Hep2-1-deP-KLA was used as the sugar acceptor. This strongly indicates that the galactose added by WaaB depends only on the glucose added by WaaG. In addition, no activity was observed for other sugar nucleotides, such as UDP-glucose, UDP-glucuronic acid, UDP-galacuronic acid, GDP-mannose, ADP-glucose, and GDP-glucose (Figure S5A,B of the Supporting Information). As with WaaG, in the absence of a lipid acceptor, WaaB can hydrolyze its sugar donor, UDP-galactose, to UDP and galactose but was not able to hydrolyze UDP-Glc (Figure S5A,B of the Supporting Information).
E. coli K-12 LPS Outer Core HexII Is Transferred by WaaO
In E. coli K-12 LPS, the outer core HexII is a glucose residue, which is transferred by WaaO.1 When purified E. coli WaaO is coupled to the reaction of WaaG and WaaB, several more slowly migrating products were detected (Figure 6). When 0.1 mM Mg2+ was included in the assay, low levels of only one product, presumably Glc-[Gal]-Glc-Hep2-1-deP-KLA ([Gal] indicates the branched galactose added to the 6-position of the glucose as shown in Figure 1), was detected (D, Figure 6). As the concentration of Mg2+ is increased, the number of products increased (D*, Figure 6). These glucosylated products (Glcn-[Gal]-Glc-Hep2-1-deP-KLA) were detected only when Mg2+ was included in the reaction mixture (Figure 6), indicating that WaaO activity is dependent on divalent metal ions. Taken together, these data strongly suggest that WaaO is capable of adding multiple sugars to the outer core OS. As seen in Figure 7, the formation of these multiglucosylated products, Glcn-[Gal]-Glc-Hep2-1-deP-KLA, increases with time in the presence of WaaO and 1 mM Mg2+.
Figure 6.

Mg2+ concentration affects the activity of E. coli K-12 WaaO. Hep2-1-deP-KLA (labeled A) was reacted with WaaG (0.01 mg/mL) for 15 min to first produce Glc-Hep2-1-deP-KLA (labeled B). WaaB (0.1 mg/mL) was then added and incubated for an additional 135 min to produce Gal-Glc-Hep2-1-deP-KLA (labeled C). WaaO was added (0.1 mg/mL) in the presence of various concentrations of Mg2+ as indicated after WaaB reacted as indicated. When Mg2+ was excluded, no WaaO-dependent glycosylation was detected. However, when Mg2+ was included, a product with migration consistent with glucose addition was detected. At a low Mg2+ concentration (0.1 mM), one product is detected (labeled D), but as the Mg2+ concentration is increased, multiple products (labeled D*) are detected, consistent with the addition of multiple glucoses to Gal-Glc-Hep2-1-deP-KLA.
Figure 7.

Time dependence of the E. coli K-12 WaaO reaction. Hep2-1-deP-KLA (labeled A) was reacted with WaaG (0.01 mg/mL) and WaaB (0.1 mg/mL) for 285 min as described in the legend of Figure 6. E. coli K-12 WaaO (0.1 mg/mL) and 1 mM MgCl2 were added, and a portion of the reaction mixture was spotted onto a TLC plate at the times indicated. WaaO catalyzes the formation of a single glucose addition (labeled D) and at increased times additional glucoses (labeled D*).
To determine if the galactose added by WaaB was necessary for the glucosylation catalyzed by WaaO, we coupled the WaaO reaction to only the WaaG reaction. In the presence of WaaO, multiple products whose migration is consistent with glucosylation of the WaaG product Glc-Hep2-1-deP-KLA to Glc2-Hep2-1-deP-KLA [product CO (Figure 8)] and Glcn-Hep2-1-deP-KLA [CO* (Figure 8)] are formed. The glucosylation of Glc-Hep2-1-deP-KLA by WaaO was similarly dependent on Mg2+ (data not shown); multiple glucosylated products were observed only in the presence of Mg2+. These results confirm the divalent cation dependence of WaaO and indicate that WaaO does not require the branched galactose added by WaaB. These data also strongly suggest that UDP-Glc is the sugar donor for the multiple sugar additions catalyzed by WaaO (Figure 8) because there is no UDP-galactose present in this reaction mixture.
Figure 8.

E. coli K-12 WaaO does not require the galactose added by WaaB. Hep2-1-deP-KLA (labeled A) was reacted with WaaG (0.01 mg/mL) for 270 min to produce Glc-Hep2-1-deP-KLA (labeled B). WaaO (0.1 mg/mL) and 1 mM MgCl2 were added to the WaaG reaction mixture and incubated for the times indicated. Initially, the product, Glc2-Hep2-1-deP-KLA (labeled CO), is formed. The products labeled CO* indicate the addition of multiple sugars by WaaI.
To exclude the possibility that the products observed in the presence of WaaO might be due to the coupling enzyme WaaG or WaaB, Gal-Glc-Hep2-1-deP-KLA, the product of WaaG and WaaB, was isolated (C, Figure 9) and used directly as the substrate for WaaO. As shown in Figure 9, multiple products consistent with the addition of a single glucose (product D, Figure 9) but also multiple glucoses (D*, Figure 9) were observed, indicating that WaaO alone is responsible for the formation of those lipid A OS’s.
Figure 9.

E. coli K-12 WaaO glycosylates Gal-Glc-Hep2-1-deP-KLA, the product of WaaG and WaaB. Purified Gal-Glc-Hep2-1-deP-KLA (labeled C), produced from Hep2-1-deP-KLA by WaaG and WaaB as described in Materials and Methods, was reacted with WaaO in the presence of 1 mM MgCl2 and UDP-Glc to verify that the products attributed to WaaO were not due to WaaG or WaaB. Products consistent with the addition of one glucose (labeled D) and then additional glucoses (labeled D*) are formed.
Like WaaB and WaaG, WaaO can hydrolyze its sugar donor, UDP-Glc, but not UDP-Gal (Figure S5A,B of the Supporting Information). The Km for UDP-Glc in this hydrolysis reaction is 1.95 ± 0.4 mM, and the kcat is 0.013 s–1.
S. typhimurium LPS Outer Core HexII Is Transferred by WaaI
The LPS outer core structure of S. typhimurium is similar to that of E. coli K-12; both HexI and the branched substitution for HexI are the same (Figure 1). HexII, added by WaaO in E. coli and WaaI in S. typhimurium, differs between the two organisms; in S. typhimurium, a galactose is added instead of glucose. This suggests the S. typhimurium WaaI will preferentially add a galactose to Gal-Glc-Hep2-1-deP-KLA, the product of the coupled reaction of E. coli K-12 WaaG and WaaB. To test this, S. typhimurium WaaI was purified and reacted with Gal-Glc-Hep2-1-deP-KLA in the presence of UDP-galactose or UDP-glucose. Similar to E. coli WaaO, S. typhimurium WaaI catalyzed the formation of a product consistent with the addition of a sugar to Gal-Glc-Hep2-1-deP-KLA (Ds, Figure 10). The glycosylation occurred only in the presence of UPD-galactose, not UDP-glucose, consistent with the predicted specificity for the HexII glycosyltransferase based on the outer core OS structure.
Figure 10.

S. typhimurium WaaI uses UDP-Gal but not UDP-Glc. S. typhimurium WaaI (0.1 mg/mL) was incubated with Gal-Glc-Hep2-1-deP-KLA (labeled C), in the presence of UDP-Gal or UDP-Glc and 1 mM MgCl2 as indicated. The major product, Gal-[Gal]-Glc-Hep2-1-deP-KLA, is indicated by Ds. No product was formed when UDP-Glc was used as the sugar donor.
Like E. coli WaaO, S. typhimurium WaaI also requires magnesium ions for optimal activity. When the S. typhimurium WaaI was reacted with Glc-Hep2-1-deP-KLA through coupling only to the WaaG reaction, a faint product consistent with Gal*-Glc-Hep2-1-deP-KLA (the asterisk indicates this Gal is added in a linkage different from that of the Gal added by WaaB) is observed, suggesting that WaaI does not require the branched α1→6 galactose added by WaaB (Figure S6 of the Supporting Information).
The S. typhimurium LPS Outer Core HexIII Is Transferred by WaaJ
S. typhimurium WaaJ is proposed to add the outer core HexIII. To test this, purified S. typhimurium WaaJ was coupled to the reaction of S. typhimurium WaaI with Gal-Glc-Hep2-1-deP-KLA in the presence of UDP-Gal or UDP-Glc (Figure 11). S. typhimurium WaaJ shows some promiscuous activity when UDP-Gal was used as the only sugar donor; however, the activity is greatly increased when UDP-Glc is added as the sugar donor. The enhanced formation of what is presumed to be Glc-Gal-[Gal]-Glc-Hep2-1-deP-KLA (Es in Figure 11, last two lanes) is consistent with the structure of the S. typhimurium LPS outer core,2 though WaaJ likely has some ability to catalyze the addition of HexIII using UDP-Gal as the sugar donor.
Figure 11.

S. typhimurium WaaJ preferentially uses UDP-Glc but not UDP-Gal. Gal-Glc-Hep2-1-deP-KLA (labeled C) and UDP-Gal were reacted with S. typhimurium WaaI (0.1 mg/mL) to produce Gal-[Gal]-Glc-Hep2-1-deP-KLA (labeled Ds). After 90 min, S. thyphimurium WaaJ was added and incubated for the times indicated in the presence of UDP-Gal or UDP-Glc. The product labeled Es is proposed to be Glc-Gal-[Gal]-Glc-Hep2-1-deP-KLA.
The S. typhimurium LPS Outer Core HexIII Substitution with UDP-GlcNAc Is Catalyzed by WaaK
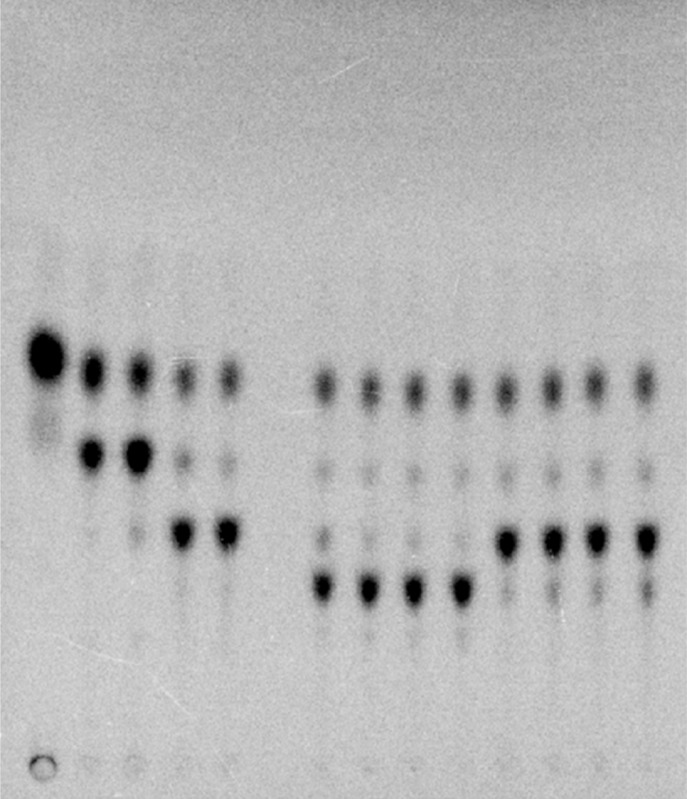
The ability of S. typhimurium WaaK to add a GlcNAc residue to Glc-Gal-[Gal]-Glc-Hep2-1-deP-KLA was tested. Gal-Glc-Hep2-1-deP-KLA, the product of E. coli WaaG and WaaB, was isolated using preparative TLC. S. typhimurium WaaI and WaaJ were reacted with Gal-Glc-Hep2-1-deP-KLA in the presence of UDP-Glc and UDP-Gal to form Gal-[Gal]-Glc-Hep2-1-deP-KLA (Figure 12, product Ds) and then Glc-Gal-[Gal]-Glc-Hep2-1-deP-KLA (Figure 12, product Es). When S. typhimurium WaaK was added to the reaction mixture in the absence of UDP-GlcNAc, a faint band was observed, suggesting WaaK has some activity using UDP-Glc or UDP-Gal as a sugar donor. However, when UDP-GlcNAc is added, WaaK activity is dramatically enhanced, strongly suggesting that UDP-GlcNAc is a preferred sugar donor. The formation of product Fs, GlcNAc-Glc-Gal-[Gal]-Glc-Hep2-1-deP-KLA, is dependent on WaaK. The formation of this product demonstrates, for the first time, the in vitro assembly of the entire S. typhimurium outer core OS using purified proteins and defined substrates.
Figure 12.

In vitro synthesis of the entire S. typhimurium outer core OS. Gal-Glc-Hep2-1-deP-KLA (labeled C) was reacted with S. typhimurium WaaI (0.1 mg/mL) for 30 min to generate Gal-[Gal]-Glc-Hep2-1-deP-KLA (labeled Ds). Next, WaaJ (0.1 mg/mL) was added to generate Glc-Gal-[Gal]-Glc-Hep2-1-deP-KLA (labeled Es). S. typhimurium WaaK (0.1 mg/mL) was added in the presence or absence of 1 mM UDP-GlcNAc and incubated for the times indicated. Only in the presence of UDP-GlcNAc was the product labeled Fs formed. This product represents the in vitro formation of the entire outer core on a defined lipid acceptor.
Discussion
This work relates several leaps forward for the biochemical characterization of the enzymes involved in outer core OS synthesis in Gram-negative bacteria. First, we have developed a method for the production of a lipid A OS to use as an acceptor for the enzymes involved in outer core OS assembly. The LPS isolated from CWG303, an E. coli strain deficient in waaG, possesses only the inner core sugars and decreased the level of phosphorylation of the inner core heptoses.9 However, the major lipid A OS in CWG303 is still too hydrophilic to be easily purified using standard lipid extraction protocols. The incorporation of lpxE, the Francisella novocida lipid A 1-phosphatase,19 into a strain lacking waaG decreases the hydrophilicity of the major LPS species. Furthermore, the incorporation of lpxM, the lipid A late acyltransferase,25 increases the proportion of hexa-acylated lipid A. All of these modifications result in a high yield of a lipid A OS, Hep2-1-deP-KLA (Figure 2), that is structurally defined, can be isolated via organic extraction, and can be radiolabeled efficiently with 32P.
Detailed analysis of outer core glycosyltransferases has been limited by the purity of the LPS acceptor and the assay method.19 Usually, assays depended on following the incorporation of [14C]glucose or [14C]galactose with scintillation counting into the LPS acceptor. The availability of Hep2-1-deP-KLA allows us to follow the addition of the outer core sugars in vitro using TLC analysis. These reactions can be followed either by labeling the lipid A OS acceptor with 32P or by using 14C-labeled sugar donors. The increased sensitivity afforded by the use of radiolabeled substrates allows us to fully define the biochemical properties of the outer core glycosyltransferases by detecting expected and unexpected products.
We purified three E. coli and three S. typhimurium outer core glycosyltransferases previously shown through numerous genetic and in vivo experiments to be involved in outer core biosynthesis.2,5,13,15 Each protein was efficiently overexpressed in E. coli and purified to homogeneity using Ni affinity chromatography.
Combining the purified enzymes with a defined lipid acceptor and sugar donors, we characterized the biochemical activity of each of these enzymes from E. coli and S. typhimurium. We have demonstrated by TLC and ESI-MS that WaaG can add a glucose residue to its LPS acceptor, Hep2-1-deP-KLA (Figures 3 and 4). While E. coli WaaG is inhibited by Mg2+ (Figure S2 of the Supporting Information), E. coli WaaO can add multiple sugars under similar conditions (Figure 6). Because the physiological concentration of Mg2+ in E. coli could be in the millimolar range, these in vitro activities may be physiologically relevant.10,16
WaaB has not been characterized previously in pure enzyme form in vitro. The obvious activity of WaaB is observed onlywhen UDP-Gal is used as the sugar donor for addition to the WaaG product, Glc-Hep2-1-deP-KLA (Figure S5 of the Supporting Information). ESI-MS (Figure 4) of the WaaB in vitro product is consistent with the addition of a galactose residue to Glc-Hep2-1-deP-KLA. The role of WaaB as the second enzyme to act in the LPS outer core assembly in E. coli K-1210,16 is supported by these in vitro results. Furthermore, both HexII glycosyltransferases, E. coli K-12 WaaO and S. typhimurium WaaI, have much lower activity toward Glc-Hep2-1-deP-KLA than toward Gal-Glc-Hep2-1-deP-KLA, the product of WaaG and WaaB, suggesting that the addition of HexII occurs after HexI substitution by WaaB in vivo (compare Figures 8 and 9 for E. coli WaaO and Figure S6 of the Supporting Information and Figure 10 for S. typhimurium WaaI).
As expected, WaaI from S. typhimurium can add a galactose residue to the E. coli K-12 WaaG, WaaB product, Gal-Glc-Hep2-1-deP-KLA (Figure 10). It can also add a second galactose residue to its first product, though the activity is low; after reaction for 30 min, a faint second product band can be observed (Figure 10). Building off of the major WaaI product, the S. typhimurium WaaJ adds a glucose residue (Figure 11), and S. typhimurium WaaK adds a GlcNAc residue to yield a fully assembled S. typhimurium LPS outer core (Figure 12). This defined, in vitro-assembled S. typhimurium LPS outer core on a defined lipid A anchor can serve as a substrate for investigating the ligation of the O-antigen.
Previously, it has been reported that only one hexose residue was observed in the outer core of the majority of LPS isolated from E. coli K-12 waaB and Δgal mutants, but both mutants still produce minor LPS bands that migrate as though they have additional sugars added to the core.16 The authors proposed that E. coli K-12 WaaO cannot work unless the branched galactose added by WaaB is present and that the HexIII glycosyltransferase (WaaR in E. coli K-12) might mistake the GlcI added by WaaG for GlcII added by WaaO and inefficiently add a terminal glucose.16 Our in vitro data for S. typhimurium WaaI do not support this proposal. However, WaaI can add, with a low efficiency, glucose to Glc-Hep2-1-deP-KLA (Figure 10, slight spot that appears after 30 min). This suggests that the more glycosylated LPS species detected in the waaB and Δgal mutants are due to WaaI adding glucose to Glc-Hep2-1-deP-KLA. This is also consistent with previous observations with an S. typhimurium waaB mutant.26 Despite lacking the branched galactose, additional core sugars were detected in a fraction of the isolated LPS molecules. Our work clearly shows that the branched galactose added by WaaB is not absolutely necessary for the action of other enzymes involved in outer core biosynthesis, consistent with the LPS structures observed in waaB mutants.
WaaB and WaaG are members of glycosyltransferase carbohydrate-active enzyme (CAZy) family 4,27−29 characterized by a GT-B fold as well as retention of configuration. WaaO and WaaR from E. coli K12 and WaaJ and WaaR from S. typhimurium, however, belong to CAZy family 8,27−29 typified by LgtC being a retaining glycosyltransferase of CAZy family 8 that has been well characterized enzymatically and structurally.30,31 These glycosyltranferases are characterized by the GT-A fold, a DXD motif that is implicated in coordination of catalytically important divalent metal ions involved in sugar binding, and by retention of configuration at the anomeric carbon of the donor sugar.4,14 One LgtC mechanism invokes an aspartate fairly distant (∼9 Å) from the sugar binding site31 and structural dynamics.32 Like LgtC, E. coli K-12 WaaO is capable of specifically hydrolyzing their uridine diphosphate sugar donors in the absence of a lipid acceptor, perhaps suggesting a similar reliance on dynamic structural changes for catalysis. No obvious hydrolysis activity was observed for S. typhimurium outer core enzymes, indicating that sugar nucleotide hydrolysis is not necessarily a shared characteristic of GT-A fold glycosyltransfereases. The comparison of these similar, yet catalytically distinct, glycosyltransferases may shed further light on the mechanism of this class of enzymes.
WaaO from E. coli K-12 was originally proposed to be a nonprocessive enzyme.33 However, we have shown that, in vitro, WaaO can add several sugars (at least three) to Gal-Glc-Hep2-1-deP-KLA and Glc-Hep2-1-deP-KLA, especially in the presence of MgCl2. Similar results have been observed previously; Shibayama et al.33 reported that overexpression of E. coli K-12 waaO in the waaO-inactivated E. coli K-12 C600 strain revealed a minor slow-migrating band besides the prominent band of LPS in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE), both of which could be the WaaO product, and the minor band may be due to the overexpression of waaO adding an additional glucose residue due to overexpression of waaI.33 In addition, Schnaitman et al.10 reported a doublet band on the SDS–PAGE gel for LPS of E. coli K-12 lacking waaR. Given our observation that WaaO can add multiple glucoses, the additional sugars observed in vivo may be due to the processive activity of WaaO and deserve further investigation. S. typhimurium WaaI, which, like E. coli WaaO, also transfers a HexII to Gal-Glc-Hep2-1-deP-KLA, does not add multiple sugars (Figure 10). As E. coli WaaO and S. typhimurium WaaI are quite similar (∼56% identical and ∼75% similar),34 further comparisons will yield insights into the mechanism by which E. coli WaaO adds multiple sugars to Gal-Glc-Hep2-1-deP-KLA.
The synthesis of these defined lipid A OS’s opens the possibility for careful characterization of the glycosyltransferase mechanisms of these enzymes as well as glycosyltransferases from other Gram-negative bacteria. The synthesis of the entire S. typhimurium OS on a defined lipid anchor opens the possibility for careful biochemical characterization of enzymes involved in O-antigen ligation.
Further experiments might focus on creating novel lipid A OS’s by using outer core glycosyltransferases from other Gram-negative bacteria that have different sugar nucleotide specificities. Both Hep2-Kdo2-lipid A and the various in vitro-synthesized lipid A OS’s we report here may be useful clinically as vaccine adjuvants.35 4′-Monophosphoryl-lipid A, prepared by chemical hydrolysis of LPS,36 is routinely used as a vaccine adjuvant; Salmonella with 1-dephosphorylated LPS were attenuated for virulence, yet their ability to elicit an immune response was not diminished.37 Here we demonstrate the ability to produce 1-dephosphorylated lipid A OS’s that can be tested for their ability to modulate the immune response.
Glossary
Abbreviations
- LPS
lipopolysaccharide
- OS
oligosaccharide
- l,d-Hep
l-glycero-d-manno-heptose
- KO
knockout
- ESI-MS
electrospray ionization mass spectrometry
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- TLC
thin-layer chromatography
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- UDP-Glc
uridine diphosphate glucose
- UDP-Gal
uridine diphosphate galactose
- Hep2-1-deP-KLA
heptose2-1-dephospho-Kdo2-lipid A (see Table S2 of the Supporting Information for full abbreviations of other lipid A OS’s)
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- Hex
hexose.
Supporting Information Available
A list of oligonucleotides used in cloning (Table S1), abbreviations used for lipid A OS’s (Table S2), WaaG sugar-nucleotide dependence (Figure S1), Triton X-100 dependence (Figure S2), divalent cation dependence (Figures S3 and S4), WaaB sugar dependence (Figure S5), and WaaI UDP-Gal dependence (Figure S6). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ J.Q.: General Atomics, San Diego, CA 92121.
The research was supported by National Institutes of Health Grant GM-51310 to C.R.H.R.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Raetz C. R. H.; Whitfield C. (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs D. E.; Yethon J. A.; Whitfield C. (1998) Molecular basis for structural diversity in the core regions of the lipopolysaccharides of Escherichia coli and Salmonella enterica. Mol. Microbiol. 30, 221–232. [DOI] [PubMed] [Google Scholar]
- Raetz C. R. H. (1996) Bacterial lipopolysaccharides: A remarkable family of bioactive macroamphiphiles. In Escherichia coli and Salmonella. Cellular and Molecular Biology, American Society for Microbiology, Washington, DC. [Google Scholar]
- Raetz C. R. H. (1990) Biochemistry of endotoxins. Annu. Rev. Biochem. 59, 129–170. [DOI] [PubMed] [Google Scholar]
- Kaniuk N.; Monteiro M.; Parker C.; Whitfield C. (2002) Molecular diversity of the genetic loci responsible for lipopolysaccharide core oligosaccharide assembly within the genus Salmonella. Mol. Microbiol. 46, 1305–1318. [DOI] [PubMed] [Google Scholar]
- Amor K.; Heinrichs D. E.; Frirdich E.; Ziebell K.; Johnson R. P.; Whitfield C. (2000) Distribution of core oligosaccharide types in lipopolysaccharides from Escherichia coli. Infect. Immun. 68, 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeger E. S.; Rothfield L. I. (1978) Cloning of genes for bacterial glycosyltransferase I. Selection of hybrid plasmids carrying genes for two glucosyltransferase. J. Biol. Chem. 254, 804–810. [PubMed] [Google Scholar]
- Müller E.; Hinckley A.; Rothfield L. (1972) Studies of phospholipids-requiring bacterial enzymes. III purification and properties of uridine diphosphate glucose: Lipopolysaccharide glucosyltransferase I. J. Biol. Chem. 247, 2614–2622. [PubMed] [Google Scholar]
- Yethon J. A.; Vinogradov E.; Perry M. B.; Whitfield C. (2000) Mutation of the lipopolysaccharide core glycosyltransferase encoded by waaG destabilizes the outer membrane of Escherichia coli by interfering with core phosphorylation. J. Bacteriol. 182, 5620–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardel E.; Parker C. T.; Schnaitman C. A. (1992) The structure of the rfaB, rfaI, rfaJ, and rfaS gene of Escherichia coli K-12 and their roles in the assembly of the lipopolysaccharide core. J. Bacteriol. 174, 4736–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare A.; Bittner M.; Carter J.; Alvarez S.; Zaldívar M.; Bravo D.; Valvano M. A.; Contreras I. (2006) The outer core lipopolysaccharide of Salmonella enterica serovar Typhi is required for bacterial entry into epithelial cells. Infect. Immun. 74, 1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A.; He X.; Smirnova I.; Liu M. Y.; Huffel C. V.; Du X.; Birdwell D.; Alejos E.; Silva M.; Galanos C.; Freudenberg M.; Ricciardi-Castagnoli P.; Layton B.; Beutler B. (1998) Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Leipold M. D.; Kaniuk N. A.; Whitfield C. (2007) The C-terminal domain of the Escherichia coli WaaJ glycosyltransferase is important for catalytic activity and membrane association. J. Biol. Chem. 282, 1257–1264. [DOI] [PubMed] [Google Scholar]
- Leipold M. D.; Vinogradov E.; Whitfield C. (2007) Glycosyltransferases involved in biosynthesis of the outer core region of Escherichia coli lipopolysaccharides exhibit broader substrate specificities than is predicted from lipopolysaccharide structures. J. Biol. Chem. 282, 26786–26792. [DOI] [PubMed] [Google Scholar]
- Austin E. A.; Graves J. F.; Hite L. A.; Parker C. T.; Schnaitman C. A. (1990) Genetic analysis of lipopolysaccharide core biosynthesis by Escherichia coli K-12: Insertion mutagenesis of the rfa locus. J. Bacteriol. 172, 5312–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaitman C. A.; Klena J. D. (1993) Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol. Rev. 57, 655–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz T.; Zhou Z.; Raetz C. R. (1997) Function of the Escherichia coli msbB gene, a multicopy suppressor of htrB knockouts, in the acylation of lipid A. Acylation by MsbB follows laurate incorporation by HtrB. J. Biol. Chem. 272, 10353–10360. [DOI] [PubMed] [Google Scholar]
- Ingram B. O.; Sohlenkamp C.; Geiger O.; Raetz C. R. (2010) Altered lipid A structures and polymyxin hypersensitivity of Rhizobium etli mutants lacking the LpxE and LpxF phosphatases. Biochim. Biophys. Acta 1801, 593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Karbarz M. J.; McGrath S. C.; Cotter R. J.; Raetz C. R. H. (2004) MsbA transporter dependent lipid A 1-dephosphorylation on the periplasmic surface of the inner membrane: Topography of Francisella novicida LpxE expressed in Escherichia coli. J. Biol. Chem. 279, 49470–49478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. R. (1972) Experiments in Molecular Genetics, Cold Spring Harbor Laboratory Press, Plainview, NY. [Google Scholar]
- Inoue H.; Nojima H.; Okayama H. (1990) High efficiency transformation of Escherichia coli with plasmids. Gene 96, 23–28. [DOI] [PubMed] [Google Scholar]
- Radika K.; Raetz C. R. H. (1988) Purification and properties of lipid A disaccharide synthase of Escherichia coli. J. Biol. Chem. 263, 14859–14867. [PubMed] [Google Scholar]
- Bligh E. G.; Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Kanipes M. I.; Lin S.; Cotter R. J.; Raetz C. R. H. (2001) Ca2+-induced phosphoethanolamine transfer to the outer 3-deoxy-d-manno-octulosonic acid moiety of Escherichia coli lipopolysaccharide. J. Biol. Chem. 276, 1156–1163. [DOI] [PubMed] [Google Scholar]
- Raetz C. R. H.; Reynolds C. M.; Trent M. S.; Bishop R. E. (2007) Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollin R.; Creeger E. S.; Rothfield L. I.; Stocker B. A. D.; Lindberg A. A. (1983) Salmonella typhimurium mutants defective in UDP-d-galactose:lipopolysaccharide α-1,6-d-galactosyltransferase. Structural, immunochemical, and enzymologic studies of rfaB mutants. J. Biol. Chem. 258, 3769–3774. [PubMed] [Google Scholar]
- Campbell J. A.; Davies G. J.; Bulone V.; Henrissat B. (1997) A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 326, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V.; Golaconda Ramulu H.; Drula E.; Coutinho P. M.; Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P. M.; Deleury E.; Davies G. D.; Henrissat B. (2003) An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328, 307–317. [DOI] [PubMed] [Google Scholar]
- Persson K.; Ly H. D.; Dieckelmann M.; Wakarchuk W. W.; Withers S. G.; Strynadka N. C. (2001) Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat. Struct. Biol. 8, 166–175. [DOI] [PubMed] [Google Scholar]
- Lairson L. L.; Chiu C. P. C.; Ly H. D.; He S.; Wakarchuk W. W.; Strynadka N. C.; Withers S. G. (2004) Intermediate trapping on a mutant retaining α-galactosyltransferase identifies an unexpected aspartate residue. J. Biol. Chem. 279, 28339–28344. [DOI] [PubMed] [Google Scholar]
- Chan P. H. W.; Cheung A. H.; Okon M.; Chen H.; Withers S. G.; McIntosh L. P. (2013) Investigating the structural dynamics of α-1,4-galactosyltransferase C from Neisseria meningitidis by nuclear magnetic resonance spectroscopy. Biochemistry 52, 320–332. [DOI] [PubMed] [Google Scholar]
- Shibayama K.; Ohsuka S.; Tanaka T.; Arakawa Y.; Ohta M. (1998) Conserved structural regions involved in the catalytic mechanism of Escherichia coli K-12 WaaO (RfaI). J. Bacteriol. 180, 5313–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Reed S. G.; Bertholet S.; Coler R. N.; Friede M. (2009) New horizons in adjuvants for vaccine development. Trends Immunol. 30, 23–32. [DOI] [PubMed] [Google Scholar]
- Garçon N.; Chomez P.; Van Mechelen M. (2007) GlaxoSmithKline adjuvant systems in vaccines: Concepts, achievements and perspectives. Expert Rev. Vaccines 6, 723–739. [DOI] [PubMed] [Google Scholar]
- Kong Q.; Six D. A.; Roland K. L.; Liu Q.; Gu L.; Reynolds C. M.; Wang X.; Raetz C. R. H.; Curtiss R. III (2011) Salmonella synthesizing 1-dephosphorylated lipopolysaccharide exhibits low endotoxic activity while retaining its immunogenicity. J. Immunol. 187, 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. F.; Kushner S. R. (1991) Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100, 195–199. [PubMed] [Google Scholar]
- Guzman L.; Belin D.; Carson M. J.; Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose P(BAD) promoter. J. Bacteriol. 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.