


Abstract
Poly(ADP-ribose) polymerases (PARP) attach poly(ADP-ribose) (PAR) chains to various proteins including themselves and chromatin. Topoisomerase I (Top1) regulates DNA supercoiling and is the target of camptothecin and indenoisoquinoline anticancer drugs, as it forms Top1 cleavage complexes (Top1cc) that are trapped by the drugs. Endogenous and carcinogenic DNA lesions can also trap Top1cc. Tyrosyl-DNA phosphodiesterase 1 (TDP1), a key repair enzyme for trapped Top1cc, hydrolyzes the phosphodiester bond between the DNA 3′-end and the Top1 tyrosyl moiety. Alternative repair pathways for Top1cc involve endonuclease cleavage. However, it is unknown what determines the choice between TDP1 and the endonuclease repair pathways. Here we show that PARP1 plays a critical role in this process. By generating TDP1 and PARP1 double-knockout lymphoma chicken DT40 cells, we demonstrate that TDP1 and PARP1 are epistatic for the repair of Top1cc. The N-terminal domain of TDP1 directly binds the C-terminal domain of PARP1, and TDP1 is PARylated by PARP1. PARylation stabilizes TDP1 together with SUMOylation of TDP1. TDP1 PARylation enhances its recruitment to DNA damage sites without interfering with TDP1 catalytic activity. TDP1–PARP1 complexes, in turn recruit X-ray repair cross-complementing protein 1 (XRCC1). This work identifies PARP1 as a key component driving the repair of trapped Top1cc by TDP1.
INTRODUCTION
Topoisomerase I (Top1) is essential in higher eukaryotes, as it relaxes positive DNA supercoiling in advance of replication forks and transcription complexes as well as negative supercoiling behind such complexes (1). Supercoiling relaxation requires the production of transient Top1 cleavage complexes (Top1cc), which are Top1-linked DNA single-strand breaks (SSBs) (2,3). Top1cc catalytic intermediates can be converted into irreversible Top1–DNA cleavage complexes by colliding replication and transcription complexes. These DNA lesions trigger cell death and account for the antitumor activity of camptothecin (CPT) and its clinical derivatives irinotecan and topotecan after the drugs selectively trap Top1cc (3).
A key enzyme for the repair of Top1cc is tyrosyl-DNA phosphodiesterase 1 (TDP1) (4–9). TDP1 hydrolyzes the phosphodiester bond between the Top1 tyrosyl moiety and the DNA 3′-end (10,11). The ability of TDP1 to resolve 3′-phosphotyrosyl linkages is consistent with its role in protecting cells against Top1-induced DNA lesions. TDP1 is conserved in all eukaryotes and present in both the nucleus and mitochondria of human, mouse, chicken and the trypanosome cells (6,12–15). A homozygous mutation of TDP1 causes spinocerebellar ataxia with axonal neuropathy 1 (SCAN1), an autosomal recessive neurodegenerative syndrome (16). Cells from SCAN1 patients or TDP1 knockout mice are hypersensitive to CPT and accumulate elevated Top1-associated DNA breaks in response to CPT (7,9,14,17–20). Top1-linked DNA SSBs can be subsequently transformed into double-strand breaks (DSB) following collision with the replication and transcription machineries (21–23). Top1cc induce the phosphorylation of TDP1 at serine 81 by the protein kinases ataxia-telangiectasia-mutated kinase (ATM) and DNA-dependent protein kinase (DNA-PK), which stabilizes cellular TDP1 and promotes cell survival (6,24). TDP1 is also endogenously SUMOylated on lysine 111, which enhances its recruitment to DNA damage sites and the repair of Top1-induced SSB (20).
Poly(ADP-ribose) polymerase-1 (PARP1) is an ubiquitous chromatin-associated enzyme that binds to DNA base damages and strand breaks, and catalyzes the nicotinamide adenine dinucleotide (NAD+)-dependent addition of ADP-ribose polymers (PAR) onto itself and chromatin proteins including Top1, XRCC1, Ligase III and histones (25–28). Protein modifications by PARP1 play a crucial role in DNA damage response by controlling the cellular localization and biological activities of DNA repair complexes and by remodeling chromatin (25,29–31). PARP1 interacts with several proteins involved in SSB repair, base excision repair and DSB repair (31). PARP1 has been also implicated in the alternative or backup pathway for nonhomologous end joining repair (6,32,33). PARP1 inhibition triggers the activation of ATM (34).
The involvement of PARP1 in the repair of Top1cc stems from several observations: (i) PARP1-deficient cells are hypersensitive to CPT (23,35); (ii) PAR accumulates in CPT-treated cells (36–38); and (iii) PARP inhibitors enhance the activity of CPT and its clinical derivatives (topotecan and irinotecan) by inhibiting the repair of Top1-induced DNA lesions (23,36–38), by inhibiting the release of Top1 from stalled replication complexes (27,39,40) and by inhibiting the restart of replication forks reversed by Top1cc (8). However, the molecular mechanisms by which PARP1 acts in the repair of Top1-induced DNA damage have not been fully elucidated.
PARP1 knockout cells have less TDP1 activity (23) and the clinical PARP inhibitor ABT-888 (veliparib) fails to sensitize TDP1-deficient cells to Top1 inhibitors (36,37). TDP1 is one of several redundant pathways involved in the repair of Top1-mediated damage in yeast and human cells. Yeast cells are sensitized to CPT when both TDP1 and structure-specific endonucleases are inactivated. One such endonuclease is Rad1-Rad10 (41), an ortholog of the human XPF-ERCC1, which has recently been shown to be involved in Top1cc repair in parallel with TDP1 and PARP1 (36). The XPF-related nuclease, Mus81, is also involved in the repair of Top1 lesions in yeast and human cells (41,42). Lastly, the MRN complex (Mre11/Rad50/Nbs1) has been suggested as a supplementary pathway for the repair of Top1-mediated DNA damage (16,43,44). However, what determines the choice between the TDP1 and the endonuclease pathways has not been elucidated.
In the present study, we provide biochemical, cellular, pharmacological and genetic evidence demonstrating the physical and functional coupling of PARP1 and TDP1 for Top1cc repair, and their critical function with XRCC1, suggesting that PARP1 is the primary determinant for Top1cc repair by TDP1.
MATERIALS AND METHODS
Drug, antibodies and expression constructs
The Drug Synthesis and Chemistry Branch, Division of Cancer Treatment, National Cancer Institute (NCI), National Institutes of Health (NIH) provided CPT and Veliparib (ABT-888). Cycloheximide (CHX) was purchased from Sigma (St Louis, MO, USA). Rabbit polyclonal TDP1 (Ab4166) and XRCC1 (Ab1838, mouse monoclonal) were from Abcam (Cambridge, MA, USA). Mouse polyclonal anti-human TDP1 (cat no. H00055775-A01) antibody was from AbNova. Mouse monoclonal anti-Flag (M2) and anti-actin (ACTN05) antibodies were from Sigma (St Louis, MO, USA) and NeoMarkers (Fremont, CA, USA), respectively. The anti-PAR polymer mouse monoclonal (10H) and rabbit polyclonal antibodies were from Trevigen (Gaithersburg, MD, USA). Mouse monoclonal anti-GST antibodies was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal PARP1 antibody and secondary antibodies: Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG or anti-mouse IgG were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Human Flag-TDP1 fusion construct were generated using mammalian expression vectors pCMV-Tag2 (Stratagene, La Jolla, CA, USA) as described previously (6). The Flag-tagged N-terminal (1–185 aa) TDP1 truncated construct was generated by polymerase chain reaction amplification and full-length TDP1 as template and cloned in mammalian expression vectors pCMV-Tag2 (Stratagene, La Jolla, CA, USA). The green fluorescent protein (GFP)- tagged TDP1 construct was kindly provided by Dr. Fritz Boege (Institute of Clinical Chemistry and Laboratory Diagnostics, Heinrich-Heine-University, Medical School, Düsseldorf, Germany). The point mutations FLAG-TDP1K111R and GFP-TDP1K111R were constructed using the ‘QuickChange’ protocol (Stratagene, La Jolla, CA, USA). All polymerase chain reaction–generated constructs were confirmed by DNA sequencing.
Cell culture, treatment and transfections
Cell cultures were maintained at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum (Life Technologies, Rockville, MD, USA). The colon carcinoma cell line (HCT116) was obtained from the Developmental Therapeutics Program (NCI, NIH), and the isogenic HeLa cells stably transfected with PARP1-shRNA or control-shRNA were cultured in the presence of 125 µg/ml hygromycin B (Sigma, St Louis, MO, USA) as described previously (45). Chicken lymphoma DT40 cells were provided by Dr. Shunichi Takeda, Kyoto University (14,46). DT40 cells were cultured at 37°C with 5% CO2 in RPMI-1640 medium supplemented with 1% chicken serum (Invitrogen, Carlsbad, CA, USA), 10−5 M β-mercaptoethanol, penicillin, streptomycin and 10% fetal calf serum. For the CHX experiments, cells (60–70% confluent) were grown in six-well plates and CHX was added to a final concentration of 100 µg/ml. Plasmid DNAs were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol.
Generation of TDP1−/−; PARP1−/− double-knockout DT40 cells
TDP1−/− and PARP1−/− DT40 cells have been described previously (14,46,47). To generate the double-knockout TDP1−/−;PARP1−/− cells the PARP1 knockout constructs carrying blasticidin and l-histidinol–resistant genes were sequentially transfected in TDP1−/− cells (46). Gene disruption was confirmed by Southern blotting and Western blotting analyses.
In vitro PARylation assays
The in vitro PARylation assay was performed as described (48). Briefly, purified human PARP1 (hPARP1, 1 µg) alone or in the presence of varying concentration (1.5–3 µg) of purified human TDP1 (hTDP1) were incubated with 200 ng of calf thymus DNA previously treated with DNase I in 100 μl solution containing 100 mM Tris–HCl, pH 8.0, 10 mM MgCl2, 1 mM dithiothreitol and 800 nM [α-32P] NAD+ (100 nCi/nmol). After incubation at 25°C for 20 min, reactions were stopped with ice-cold acetone (80% v/v) and incubated for 30 min at −20°C. Insoluble material was pelleted by centrifugation for 20 min at 4°C, washed once with 100% acetone, once with water-saturated ether and dried. Pellets were resolubilized in 40 μl of 1× Laemmli buffer and analyzed on 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Gels were stained with Coomassie Blue, dried and exposed to PhosphorImager screens (GE Healthcare, UK).
Ni2+ will be the power of Ni NTA-agarose co-immobilization binding assay
In vitro Ni2+-NTA-Agarose pull-down assays were performed as described (49). Briefly, protein complexes of hexahistidine-tagged purified hTDP1 (1 µg) were mixed with purified hPARP1 (1 µg) with pre-equilibrated Ni2+-NTA agarose beads (Qiagen Inc, Santa Clarita, CA, USA) in 100 μl of pull-down buffer [50 mM potassium phosphate, pH 7.5, 0.5 mM dithiothreitol, 1 mM EDTA, with protease inhibitors (Complete, Roche Diagnostics, Indianapolis, IN, USA)] in the presence or absence of calf thymus DNA previously treated with DNase I (400 ng) with or without NAD+ (400 µM). After incubation at 25°C for 30 min, protein complexes were further incubated at 4°C for 1 h with gentle shaking. The beads were pelleted by centrifugation and washed twice with 500 μl of pull-down buffer containing 50 mM imidazole so that all of the unbound proteins were removed. The protein samples were eluted from the beads with 100 μl of pull-down buffer containing 300 mM imidazole and were analyzed by 8% SDS-PAGE and detected by Western blotting with indicated antibodies.
Cell extracts, immunoblotting and immunoprecipitation
Preparation of whole cell extracts, immunoprecipitation and immunoblotting were carried out as described (6). Briefly, cells were lysed in 10 mM Tris–HCl (pH 8), 150 mM NaCl, 0.1% SDS, 1% NP40, 0.5% Na-deoxycholate supplemented with protease inhibitors (Complete, Roche Diagnostics, Indianapolis, IN, USA) and phosphatase inhibitors (Phosphatase Inhibitor Cocktail 1, Sigma). After thorough mixing and incubation at 4°C for 2 h, lysates were then centrifuged at 12 000g at 4°C for 20 min. Supernatants were collected, aliquoted and stored at −80°C.
For immunoprecipitation, cells were lysed in 50 mM Tris–HCl, pH 7.4, 300 mM NaCl, 0.4% NP40, 10 mM MgCl2, 0.5 mM dithiothreitol with protease and phosphatase inhibitors. Cell lysates were pretreated with Benzonase® nuclease 10 U/µl (Sigma Aldrich, St Louis, MO, USA) as indicated. Supernatants were obtained by centrifugation at 15 000g at 4°C for 20 min and precleared with 50 µl of protein A/G-PLUS-Agarose beads (Santa Cruz, CA, USA). Three to five milligrams of precleared lysate was incubated overnight at 4°C with the indicated antibodies (2–5 µg/ml) and 50 µl of protein A/G-PLUS-Agarose beads. Isolated immunocomplexes were recovered by centrifugation, washed three times with lysis buffer and were subjected to 8% Tris-glycine gels (Invitrogen). Immunoblotting was done by standard procedures, and immunoreactivity was detected by Electrochemiluminescence (ECL) reaction (Amersham). Densitometry analyses of immunoblots were performed using Image J software (NIH).
Immunocytochemistry and confocal microscopy
Immunofluorescence staining and confocal microscopy were performed as described (6). Briefly, cells were grown and drug treated on chamber slides (Nalge Nunc International, Naperville, IL, USA). Following 10-min fixation with 4% paraformaldehyde at room temperature, primary antibodies against PAR, PARP1 and XRCC1 were detected with anti-mouse or anti-rabbit IgG secondary antibodies labeled with Alexa 488/568 (Invitrogen). Cells were mounted in anti-fade solution with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) and examined using a laser scanning confocal microscope (Zeiss LSM510) with a 63× oil objective. Images were collected and processed using the Zeiss AIM software and sized in Adobe Photoshop 7.0.
Oligonucleotides and preparation of DNA substrates
The N14Y oligonucleotide (5′-GATCTAAAAGACTTY-3′), which contains a 3′-phosphotyrosine (Y) and the 14-mer DNA substrate containing the internally labeled quencher (TAMRA) and a fluorophore (6-FAM) attached to the 3′-DNA end [5′-GATC(TAMRA-T)AAAAGACTT-FAM] were synthesized by Midland Certified Reagents Company (Midland, TX, USA). The N14Y oligonucleotide was 5′-end labeled using T4 polynucleotide kinase and [γ-32P] ATP. Unincorporated radioactive nucleotides were removed using a mini Quick Spin Oligo column (Roche Diagnostics, Indianapolis, IN, USA) after inactivation of the kinase by heating for 5 min at 95°C.
TDP1 activity assays
TDP1 gel-based assays were performed as described (13,50). Briefly, TDP1 and varying ratio of PARP1 recombinant proteins were premixed in the presence of NAD+ (400 µM) and incubated at 25°C for 30 min in 50 mM Tris–HCl, pH 8.0, 10 µM MgCl2, 100 mM NaCl, 100 ng/µl bovine serum albumin, and DNase I–treated calf thymus DNA (17 ng/µl). At the beginning of each experiment, 1 volume of TDP1–PARP1 solution was incubated with 3 volumes of 32P-labeled N14Y substrate at 0°C in ice-cold buffer containing 1× phosphate buffered saline, pH 7.4, 80 mM KCl, 0.01% Tween-20 to achieve a final concentration of 1 nM N14Y substrate and 5 nM TDP1. At the indicated time points, 10 µl of aliquot was withdrawn and mixed with equal volume of formamide loading dye [96% (v/v) formamide, 10 mM EDTA, 1% (w/v) xylene cyanol and 1% (w/v) bromophenol blue] to stop the reactions. Samples were analyzed by 16% denaturing PAGE gels, dried and exposed on PhosphorImager screens. Imaging and quantification were done using a Typhoon 8600 and ImageQuant software (GE Healthcare, UK). The resulting kinetic curves were fit to a single-exponential function, yielding the rate constant k (s−1). The obtained k values for different samples were then normalized and compared with each other.
For fluorescence resonance energy transfer (FRET)-based assays, TDP1 and varying ratio of PARP1 recombinant proteins were premixed in the presence of NAD+ (400 µM) and incubated at 25°C for 30 min. At the beginning of each experiment, 1 volume of TDP1–PARP1 solution was mixed with 1 volume of FRET substrate [5′-GATC(TAMRA-T)AAAAGACTT-3′FAM] (51) in buffer containing 50 mM Tris–HCl (pH 7.5), 80 mM KCl, 2 mM EDTA, 1 mM Dithiothreitol (DTT), DNase I–treated calf thymus DNA (17 ng/µl) and 40 µg/ml bovine serum albumin to achieve a final concentration of 50 nM substrate and 50 nM TDP1. The changes in fluorescein signal were monitored in real time at 520 nm on a SpectraMax Germini XS microplate reader from Molecular Devices (Sunnyvale, CA, USA). The resulting fluorescence spectra were fit to a single-exponential function, yielding the rate constant k (s−1).
Cytotoxicity assays
To measure the sensitivity of cells to CPT, cells from different genotypes were continuously exposed to various concentrations of the CPT. Two hundred cells were seeded into 384-well white plate (#6007680 Perkin Elmer Life Sciences, Waltham, MA, USA) with 40 µl of medium per well. Plates were incubated at 37°C for 72 h. Cell survival was determined using the ATPlite 1-step kit (PerkinElmer). Briefly, 40 µl of ATPlite solution was added to each well. After 5 min, luminescence was measured by Envision 2104 Multilabel Reader (PerkinElmer).
Live cell microscopy and photobleaching experiments
Live cell imaging, micro-irradiation and photobleaching experiments were carried out as described (6,52) with a Zeiss LSM510 confocal laser-scanning microscope equipped with a 365 nm UV laser and 63×/1.4 NA oil objective (Carl Zeiss MicroImaging, Inc.). Fluorophores were excited using a 488/514 nm Ar-laser line. The microscope was equipped with a heated environmental chamber set to 37°C. FRAP analyses were carried out with living HCT116 cells grown on chamber cover glass (Nalge Nunc International, Naperville, IL, USA). Cells were transfected with GFP fusion proteins and mounted on an incubation chamber filled with medium 24 h after transfection. Cells were pretreated with PARP inhibitor (ABT888/5 µM for 2 h) as indicated.
For FRAP analysis, a subnuclear spot was bleached for 300 ms at highest intensity of an argon laser line (488 nm for GFP) adapted to the fluorescent protein of interest. DNA damage was carried out with a UV laser set to 50% transmission. For imaging, the laser power was attenuated to 0.1% of the bleach intensity. Subsequently, the recovery of fluorescence in the spot was monitored at intervals of 5 s for ∼130 s. Relative fluorescence intensities of the bleached region were corrected for background. To display the FRAP curves, the fluorescence signal measured in a region of interest (ROI) was individually normalized to the pre-bleach signal in the ROI according to the following equation: ROI = (It − Ibg)/(Io − Ibg) × 100, where Io is the intensity in the ROI during pre-bleaching, It is the intensity in the ROI at time point t and Ibg is the background signal determined in a region outside of the cell nucleus.
RESULTS
PARP1 and TDP1 are epistatic for the repair of Top1cc
To determine the functional relationship between PARP1 and TDP1, we tested the impact of the PARP inhibitor (veliparib, ABT-888) on TDP1 knockout lymphoma chicken DT40 cells deficient in 3′-tyrosyl-DNA phosphodiesterase activity and hypersensitive to CPT (14). Cell survival assays (Figure 1A) showed supra-additive effect in wild-type cells treated with CPT and PARP inhibitor (ABT-888) (veliparib), which is consistent with experiments performed in murine and human cancer cells (36–38). Notably, ABT-888 failed to enhance the cytotoxicity of CPT in the TDP1−/− DT40 cells (Figure 1A), suggesting that TDP1 and PARP are in the same pathway for the repair of Top1cc.
Figure 1.

PARP1 and TDP1 are epistatic for the repair of Top1cc. (A) Survival of wild-type or TDP1−/− DT40 cells treated with CPT continuously for 72 h in the absence or presence of ABT-888. Cell viability was determined by ATPlite® assays. Error bars represent standard deviation (n ≥ 3). (B) Western blotting of the indicated whole-cell lysates. Blots were probed with anti-PARP1 and anti-actin antibodies. (C) Survival curves of DT40 cells of the indicated genotypes treated with CPT. (D) Survival curves of PARP1−/− DT40 cells stably complemented with human PARP1 (PARP1−/−; hPARP1) or human PARP2 (PARP1−/−; hPARP2) (46,47) cells treated with CPT. Cell viability was determined by ATPlite® assays. Error bars represent SD (n ≥ 3); where not visible, error bars are smaller than the symbols.
To further establish the functional genetic relationship between TDP1 and PARP1, we generated PARP1–TDP1 double-knockout DT40 cells (referred to as TDP1−/−;PARP1−/− cells). PARP1 disruption in the TDP1−/−;PARP1−/− cells was confirmed by western blotting (Figure 1B). The double-knockout TDP1−/−;PARP1−/− cells were hypersensitive to CPT, similar to the TDP1−/− or PARP1−/− single-mutant cells (Figure 1C). Recent studies show that CtIP S322/−/− mutant is deficient in interacting with BRCA1 and confers hypersensitivity to CPT (44,53,54). The double mutant TDP1−/−;CtIP S332/−/− DT40 cells (14) showed greater than additive sensitivity to CPT compared with TDP1−/− or PARP1−/− single-mutant or TDP1−/−; PARP1−/− double-mutant cells (Figure 1C), indicating that CtIP and TDP1 are in different pathways. This contrasts with PARP1 and TDP1, which we found to be epistatic for Top1cc repair (Figure 1C).
Next, we took advantage of the fact that DT40 cells only express PARP1 and not PARP2 (46,47), and compared the CPT sensitivity of PARP1−/− DT40 cells stably complemented either with human PARP1 (PARP1−/−; hPARP1) or PARP2 (PARP1−/−; hPARP2) (46,47). PARP2 failed to rescue the CPT sensitivity under condition where PARP1 did (Figure 1D), indicating that PARP1 is the preferential regulator of TDP1.
The N-terminus domain of TDP1 binds the C-terminal domain of PARP1
To test whether PARP1 directly interacts with TDP1, we performed binding assays with purified recombinant PARP1 and TDP1. Figure 2A shows that His-tag-TDP1 pulls down PARP1 both in the absence and presence of PARylating activity induced by DNA plus NAD+ (Figure 2A, Lanes E1, E2 and E3). PARP1 alone was not retained by the Ni2+-NTA-agarose beads either in the presence or absence of DNA plus NAD+ (Figure 2A, Lanes E4 and 5), indicating the direct binding between TDP1 and PARP1.
Figure 2.

Direct interaction between the N-terminus of TDP1 and the C-terminus of PARP1. (A) His6-tagged hTDP1 pulls down recombinant human PARP1 (hPARP1). L1, L2 and L3: Loaded samples following 1-h incubation at 4°C (reaction conditions are indicated at the bottom). W1, W2 and W3: excess unbound proteins after washing the Ni2+-NTA-agarose beads with 50 mM imidazole. E1, E2 and E3: bound proteins eluted with 300 mM imidazole. Right panel: control reactions showing that hPARP1 alone (L4) or in the presence of DNA plus NAD+ (L5) does not bind the Ni2+-NTA-agarose beads and is recovered in the flow through (Ft). W4, W5 and E4, E5 are washed and eluted fractions with 50 and 300 mM imidazole, respectively. PARP1 and TDP1 were detected by Western blotting after 8% SDS-PAGE. (B) Schematic representation of Flag-tagged full-length hTDP1 and truncated hTDP1 (residues 1–185) constructs. The regulatory NTD and the catalytic domain are indicated with different colors. (C) PARP1 binds the NTD of TDP1. Flag-tagged TDP1 constructs were expressed in HCT116 cells. TDP1 was immunoprecipitated using anti-Flag antibody and the immune complexes were probed with anti-PARP1 antibody. Blots were subsequently stripped and probed with anti-Flag antibody. Lower panel shows PARP1 input corresponding to one-tenth of the lysate. Control immunoprecipitation with anti-IgG demonstrates the specificity of the reactions. (D) Schematic representation of GST-fused full-length hPARP1 and hPARP1 truncated domains corresponding to the DNA binding domain (DBD) (residues 1–371), the BRCT (C-terminal domain of a breast cancer susceptibility protein; residues 384–524) and the C-terminal domain (CTD) harboring the catalytic site (residues 525–1014). (E) TDP1 binds the CTD of PARP1. The GST-tagged full-length and truncated domains of PARP1 were expressed in HCT116 cells. PARP1 and its truncated domains were immunoprecipitated using anti-GST antibody, and the immune complexes were detected by western blotting after 4–20% SDS-PAGE with anti-TDP1 antibody. Lower panel shows TDP1 input corresponding to one-tenth of the lysates.
To determine the interacting domains between PARP1 and TDP1, we performed pull-down experiments in cells transfected with the FLAG-TDP1 and Glutathione S-transferase (GST)-tagged PARP constructs shown in Figure 2B and D. The FLAG-tagged-N-terminal domain of TDP1 (1–185 aa) was sufficient to pull-down endogenous PARP1 (Figure 2C), indicating that the catalytic domain of TDP1 is not required for the interaction of TDP1 with PARP1. To identify the PARP1 domain(s) interacting with TDP1, we used the GST-tagged fragments of PARP1 corresponding to different domains of PARP1 (55) (Figure 2D). In these cellular experiments, GST-tagged full-length PARP1 pulled down endogenous TDP1 (Figure 2E), confirming the association between the two proteins shown by in vitro binding assays (Figure 1A). GST-pull-down experiments with truncated PARP1 constructs showed that TDP1 bound the C-terminal domain of PARP1, within amino acids 524–1014 of PARP1 (Figure 2C). We also observed a weak binding of TDP1 with the BRCT domain of PARP1 (384–524 aa) (Figure 2E), an important domain for protein–protein interactions (55). Thus, the N-terminal domain (NTD) of TDP1 binds with the C-terminal end of PARP1.
TDP1 is PARylated but not inactivated by PARP1
PARP1 itself is the main PAR acceptor (90% of PAR is found on PARP1) (48). Because PARP1 also PARylates other chromatin-associated proteins (25,56), we tested whether TDP1 could be PARylated by PARP1. We incubated purified PARP1 activated by DNase I–treated DNA and [32P]-NAD+ in the presence of increasing concentration of purified TDP1 and looked for radiolabeled PARylated protein products by SDS-PAGE and autoradiography. As expected, PARP1 was auto-PARylated (Figure 3A, Lane 1). TDP1 was also radiolabeled as indicated by the appearance of a labeled band with a molecular weight corresponding to TDP1, the intensity of which increased with increasing TDP1 (Figure 3A, compare Lanes 2 and 3). These experiments demonstrate that TDP1 not only binds to PARP1 but is also a substrate of PARP1.
Figure 3.

TDP1 is PARylated but not inactivated by PARP1. (A) ADP-ribosylation of TDP1. Purified hPARP1 (1 µg, Lanes 1–3) was incubated without (Lane 1) or with 1.5 µg (Lane 2) or 3 µg (Lane 3) purified hTDP1 in the presence of [32P]-NAD+ and DNase I–treated DNA for 20 min at 25°C. Samples were analyzed by 10% SDS-PAGE. Autoradiography of the Coomassie Blue–stained gel shown at left. Protein molecular weight markers (kDa) are indicated at left. (B) Schematic representation of the TDP1 biochemical assays using a single-stranded oligopeptide 14Y. 32P-radiolabeling (*) was at the 5′-terminus of the oligopeptide. TDP1 converts 14Y to an oligonucleotide with 3′-phosphate, 14P (14). (C) Representative gels showing TDP1 catalytic activity (5 nM) in the presence of PARP1 in PARylation conditions (5–20 nM, increasing concentrations as indicated). Reactions were performed at 0°C for the indicated times. (D) Schematic representation of the FRET-based TDP1 assay using a 14-mer single-stranded substrate with an internally labeled quencher (TAMRA) and a fluorophore (fluorescein) attached to the 3′-DNA-end. Hydrolysis of the 3′-phosphodiester bond by TDP1 releases the free fluorescein, which was measured in real time at 520 nm. (E) TDP1 activity measured by FRET-based assays plotted as a function of PARP1/TDP1 ratio.
Next we tested the biochemical impact of TDP1 PARylation. Using gel-based assays with recombinant proteins (13,57), we measured TDP1 activity as the conversion of a 3′-tyrosyl-DNA nucleopeptide substrate (14-Y) to a product with a 3′-phosphate (14-P) (11,14,19) (Figure 3B). Addition of PARP1 and NAD+ had no impact on TDP1 activity (Figure 3C). These results were confirmed in FRET-based TDP1 assays using a 14-mer oligonucleotide with an internally labeled quencher (TAMRA) and a fluorophore (fluorescein) attached to the 3′-end of the DNA substrate (Figure 3D). Cleavage of the 3′-fluorescein by TDP1 abolishes FRET, giving rise to fluorescence that can be detected in real time. Addition of PARP1 under PARylation conditions had no detectable impact on TDP1 catalytic activity (Figure 3E). Together, these results demonstrate that, contrary to other enzymes [including Top1 (28)], TDP1 remains active despite its PARylation.
PARylation and SUMOylation both recruit TDP1 to DNA damage sites
To test whether PAR polymers affect TDP1 accumulation at DNA damage sites, we generated DNA damage using UV laser micro-irradiation and measured the recruitment of GFP-tagged wild-type TDP1 (GFP-TDP1WT) under live cells microscopy by photobleaching (FRAP) analysis (6,52,58). We previously used a similar approach to show that TDP1 is recruited at laser-induced DNA damage sites, depending on its phosphorylation at Ser81 by ATM and DNA-PK (6). Recruitment kinetics of GFP-TDP1WT in the presence of ABT-888 (Figure 4A and B) was determined under conditions that blocked PARylation (Figure 5) (47). The fluorescence recovery of TDP1 (GFP-TDP1WT) was significantly reduced (43–56% versus 80–95%) in the presence of ABT-888 (Figure 4A and B), demonstrating that PARylation promotes TDP1 recruitment to DNA damage sites.
Figure 4.

TDP1 recruitment to DNA damage sites is determined by PARP1 activity in addition to TDP1 SUMOylation. (A) Representative images showing the recruitment of wild-type TDP1 (GFP-TDP1WT) or SUMOylation mutant TDP1 (GFP-TDP1K111R) transiently expressed in HCT116 cells in response to laser-induced DNA damage. Cells expressing the ectopic proteins were kept untreated or pretreated for 2 h with the PARP inhibitor (ABT-888, 5 µM) and were analyzed by micro-irradiation with live cell microscopy and photobleaching (FRAP analysis). A subnuclear spot indicated by a circle was bleached (BLH) for 300 ms and photographed at regular intervals of 5 s thereafter. Successive images taken for ∼130 s after bleaching illustrate fluorescence return into the bleached areas. (B) Quantitation of FRAP data (n = 5) showing mean curves. Error bars represent the standard error of the mean.
Figure 5.

PARP1 recruits both XRCC1 and TDP1 to Top1-induced damage sites. (A) Endogenous PARP1 co-immunoprecipitates TDP1. HCT116 cells were treated with CPT (10 µM for 2 h) alone or with ABT-888 (5 µM for 2 h). Endogenous PARP1 was immunoprecipitated with anti-PARP1 antibody and immune complexes were blotted with anti-PAR or anti-TDP1 antibodies. Blots were subsequently stripped and probed with anti-PARP1 antibody. Control immunoprecipitation with anti-IgG antibody demonstrates the specificity of the reactions. (B) TDP1 co-immunoprecipitates PARP1. Flag-tagged hTDP1 was ectopically expressed in HCT116 cells. Following CPT treatment without or with ABT-888 (as in A), TDP1 was immunoprecipitated with anti-Flag antibody and immune complexes were probed with anti-PAR, anti-PARP1 or anti-XRCC1 antibodies. Blots were subsequently stripped and probed with anti-TDP1 antibody. Protein molecular weight markers (kDa) are indicated at right. (C) Knocking down PARP1 abrogates the TDP1-XRCC1 interaction. Flag-tagged hTDP1 was ectopically expressed in isogenic HeLa cells stably transfected with PARP1-shRNA or control (Ctr)-shRNA. Following CPT treatment (as in A), ectopic TDP1 was immunoprecipitated using anti-Flag antibody and the immune complexes were blotted with anti-XRCC1 antibodies. (D) TDP1–PARP1 association is not mediated through DNA. Flag-tagged hTDP1 was ectopically expressed in HCT116 cells treated with CPT (as in A). Cell lysates was pretreated with Benzonase® nuclease before co-immunoprecipitation. The immune complexes were probed with anti-PARP1 or anti-XRCC1 antibodies. Blots were subsequently stripped and probed with anti-TDP1 antibody. Migration of protein molecular weight markers (kDa) is indicated at right. Aliquots (10%) of the input show the level of indicated proteins before immunoprecipitation. (E) CPT-induced XRCC1 foci are abrogated by ABT-888. Representative immunofluorescence microscopy image of HCT116 cells treated with CPT (1 µM for 3 h) alone or with ABT-888 (5 µM for 3 h). Focal accumulation of PAR-polymers and XRCC1 are shown in green. (F) Defective XRCC1 focus formation in HeLa cells stably transfected with PARP1-shRNA. Representative immunofluorescence microscopy image of control-shRNA (Ctr) or PARP1-shRNA HeLa cells were treated with CPT (1 µM for 3 h) and subsequently fixed and immunostained for XRCC1 (green) or PARP1 (red). Nuclei are outlined as dashed white line circle.
A recent study showed that TDP1-SUMOylation at lysine 111 also enhances its accumulation at DNA damage sites (20). To characterize the impact of SUMOylation versus PARylation on the recruitment of TDP1 at laser-induced DNA damage, we compared the fluorescence recovery of the SUMOylation mutant TDP1 (GFP-TDP1K111R) with wild-type TDP1 (GFP-TDP1WT) in the presence and absence of PARP inhibitor. In the absence of ABT-888, the fluorescence recovery of the GFP-TDP1K111R was defective (50–60% at 1 min) and reached a maximum intensity (55–70% at 1.5 min) compared with the rapid fluorescence recovery of wild-type TDP1 (75–86% in 1 min with a maximum intensity of 80–95% within 1.5 min; Figure 4A and B). The deficiency in the recruitment of GFP-TDP1K111R at DNA damage sites confirms the results of El-Khamisy and coworkers (20). ABT-888 further reduced the recruitment of GFP-TDP1K111R to the levels of the GFP-TDP1WT + ABT-888 (Figure 4A and B). Moreover, the TDP1K111R mutant retains PARP1 interaction (Supplementary Figure S1). Taken together, these results indicate that SUMOylation and PARylation both promote the recruitment of TDP1 to laser-induced DNA damage sites.
PARP1–TDP1 complexes recruit XRCC1 to Top1cc-induced damage sites
Because TDP1 is component of XRCC1 complexes (6,59,60), we investigated the potential involvement of PARP1 in the formation of TDP1-XRCC1 complexes in cells treated with CPT. Co-immunoprecipitation experiments with endogenous PARP1 (Figure 5A) confirmed the results obtained with recombinant and tagged proteins (see Figures 1 and 2). PARP1 co-immunoprecipitated endogenous TDP1 both in the absence and presence of CPT or PARP inhibitor (Figure 5A). To determine the involvement of XRCC1, we immunoprecipitated ectopic FLAG-TDP1 in cells treated with or without CPT or/and ABT-888. XRCC1 was readily found in the immunoprecipitates in the absence of drug treatment (Figure 5B, second lane). Under these conditions, limited PAR signal was observed with the most prominent band ∼100 kDa, consistent with endogenous PARylation of PARP1. CPT markedly enhanced PARylation measured by western blotting (Figure 5A and B, third lanes, top panels) and by confocal immunofluorescence microscopy (Figure 5D) (36). As expected, ABT-888 abrogated CPT-induced PARylation in the FLAG-TDP1 co-immunoprecipitation experiments (Figure 5B, top panel) as well as in the immunofluorescence experiments (Figure 5D), consistent with the potent activity of ABT-888 as PARP catalytic inhibitor (36,47). The PARP1–TDP1 interactions, both in the case of exogenous FLAG-TDP1 and endogenous TDP1 were not affected by inhibiting PARylation with ABT-888 (Figure 5A and B, fourth lanes). The persistent co-immunoprecipitation of FLAG-TDP1 in the presence of Benzonase® nuclease indicated that the formation of TDP1–PARP1–XRCC1 complexes was not mediated by DNA (Figure 5D). These results, together with the binding experiments (see Figure 2), demonstrate that PARP1 and TDP1 associate in stable complexes independently of DNA and PARP enzymatic activation.
The immunoprecipitation experiments also revealed that ABT-888 inhibited XRCC1 pull-down by FLAG-TDP1 (Figure 5B, fourth lane), indicating that PARylation promotes the binding of TDP1 to XRCC1. Further evidence for PARylation-mediated XRCC1 recruitment to the PARP1–TDP1 complexes was obtained from confocal microscopy experiments (Figure 5E), which showed that ABT-888 abrogated CPT-induced XRCC1 foci. To further test this conclusion, we examined the ability of TDP1 to pull-down XRCC1 in PARP1-knockdown cells. Using isogenic HeLa cells stably transfected with PARP1-shRNA (45), we found that knockdown of PARP1 abrogated the association of FLAG-TDP1 with XRCC1 both in the presence and absence of CPT (Figure 5C). If PARP1–TDP1 complexes are required to recruit XRCC1 to the sites of Top1cc-induced DNA damage, then XRCC1 should also fail to assemble into foci in cells lacking PARP1. Accordingly, PARP1-knockdown cells failed to generate XRCC1 foci after CPT treatment (Figure 5F).
To determine whether TDP1 facilitates CPT-induced nuclear XRCC1 foci formation, we tested cells lacking TDP1 (TDP1−/− MEFs cells) by immunofluorescence microscopy. CPT treatment induced nuclear XRCC1 foci (Figure 6A and B) both in TDP1 proficient and deficient cells. However, a significant reduction in the number of XRCC1 foci was observed in the TDP1−/− cells (Figure 6B). This effect was not due to reduced expression of XRCC1 in TDP1−/− cells (Figure 6C, bottom panel). XRCC1 interaction with GST-PARP1 was also reduced in the TDP1−/− cells treated with CPT (Figure 6C, upper panel). Taken together, these results suggest that not only PARP activity but also PARP1–TDP1 complexes facilitate XRCC1 recruitment at Top1cc-induced DNA lesions.
Figure 6.
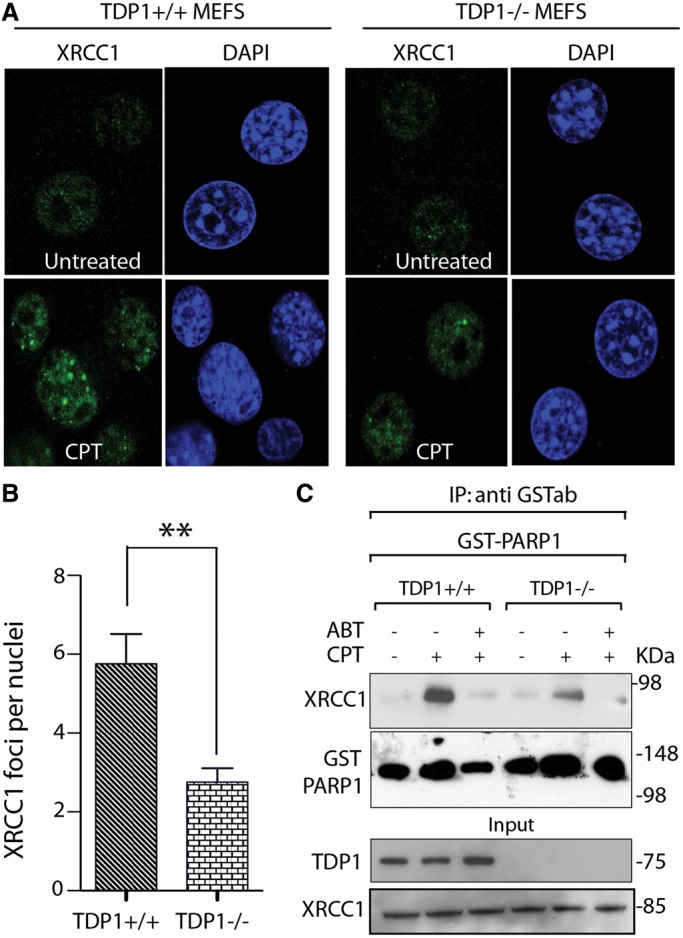
TDP1–PARP1 association stimulates CPT-induced XRCC1 foci formation. (A) Representative immunofluorescence microscopy images showing CPT-induced (1 µM for 3 h) nuclear XRCC1 foci in TDP1+/+ and −/− MEFs cells. XRCC1 foci are in green. Nuclei were stained with DAPI (blue). (B) Quantitation of XRCC1 foci per nuclei after CPT treatment in TDP1+/+ (dark bar) and −/− cells (light gray bar) calculated from 25 to 30 cells per sample (Error bar: mean values ± SEM). Asterisks denote statistically significant difference (**P < 0.001; t-test). (C) TDP1 deficiency impairs the CPT-induced PARP1–XRCC1 interaction. GST-tagged hPARP1 was ectopically expressed in isogenic TDP1+/+ and −/− MEFs cells. Following CPT treatment (5 µM for 2 h) alone or with ABT-888 (5 µM for 2 h), ectopic PARP1 was immunoprecipitated using anti-GST antibody and the immune complexes were blotted with anti-XRCC1 antibody. Blots were subsequently stripped and probed with anti-GST antibody to evaluate PARP1 expression. Aliquots (10%) of the input demonstrate equal protein levels before immunoprecipitation.
PARP1 activity stabilizes TDP1
Because TDP1 has a relatively short half-life in the absence of DNA damage (6,24), we tested whether PARP1 inactivation affected TDP1 stability. Experiments performed with the protein synthesis inhibitor CHX in cells treated with or without CPT in the presence or absence of ABT-888 (6) (Figure 7A and B) showed that the extended half-life of TDP1 in CPT-treated cells (6,24) was abolished by co-treatment with ABT-888. Independent experiments in the PARP1-knockdown cells (Figure 7C and D) confirmed the accelerated disappearance of TDP1 in PARP1-knockdown cells. These data indicate that PARP1 stabilizes TDP1 in response to Top1cc-induced damage.
Figure 7.

PARP1 stabilizes TDP1. (A and B) PARP inhibition by ABT-888 interferes with CPT-induced TDP1 stabilization. HCT116 cells treated with 10 µM CPT and/or ABT-888 (10 µM) were co-treated with CHX for the indicated times. Endogenous TDP1 was detected by western blotting (panel A, representative experiment) and quantified by densitometry analyses after normalization to actin (panel B). Data represent the mean ± standard error of at least three independent experiments. (C and D) PARP1 knockdown (isogenic HeLa cells stably transfected with PARP1-shRNA) reduces TDP1 stabilization by CPT. Cells were treated with CPT (10 µM) in the presence CHX for the indicated times. TDP1 levels were determined by western blotting (panel C, representative experiment) and quantified by densitometry normalized to actin (panel D). Data represents mean ± standard errors. (E) Representative experiment showing the kinetics of TDP1 phosphorylation at Ser81 (pS81-TDP1), γH2AX and the disappearance of total TDP1 in HCT116 cells treated with CPT (1 µM) in the absence or presence of ABT-888 (5 µM) for the indicated times. Protein levels were determined by western blotting.
CPT-induced Top1cc activate ATM, which in turn phosphorylates TDP1 at S81 leading to TDP1 accumulation at Top1cc-induced γH2AX (6). We investigated the impact of PARP inhibitor on TDP1-S81 phosphorylation. As reported (6), CPT treatment increased pS81-TDP1 (Figure 7E). Notably, ABT-888 abrogated the total TDP1 accumulation while increasing pS81-TDP1 levels (Figure 7E). However, ABT-888 markedly increased the accumulation of Top1cc-induced DSBs as measured by γH2AX, indicating PARP inhibition clearly increased CPT-induced DSBs (Figure 7E).
DISCUSSION
The present study reveals new regulatory mechanisms linking TDP1 and PARP1. Using biochemical assays, we show that PARP1 directly binds TDP1, and PARylates TDP1 without blocking its catalytic activity. PARP1 forms molecular complexes with TDP1 independently of DNA damage and PARylation. Yet, PARylation stabilizes TDP1 in response to Top1cc-induced DNA damage and promotes the recruitment of both TDP1 and XRCC1 at Top1cc-induced DNA damage sites. Moreover, genetic evidence indicates that PARP1 and TDP1 are epistatic for the repair of Top1cc, in parallel with the endonuclease pathway (see Figure 8). The epistatic relationship between PARP1 and TDP1 was suggested in an earlier study (36), and has independently been confirmed in a parallel study released while our manuscript was under revision (61). The same study also confirms the importance of TDP1 for the repair of base damage induced by monofunctional alkylating agents (14).
Figure 8.

Schematic representation of PARP1–TDP1 coupling for the repair of Top1–DNA covalent complexes. (1) The C-terminus domain of PARP1 binds the N-terminus regulatory domain of TDP1 (double-headed arrow). The PARP1–TDP1 molecular complex is shown as a black node. (2) PARP coupling with TDP1 stimulates (open arrow) the excision of Top1-DNA covalent complexes by the phosphodiesterase activity of TDP1 (jigsaw line) (3). The parallel pathway for Top1–DNA complex removal involves various endonucleases (4) including XPF-ERCC1, CtIP and Mre11. Graphical symbols for molecular interactions are derived from Kohn’s MIM conventions (68).
DNA repair pathways for Top1cc consist in a molecular network of factors that carry out the detection, excision of the 3′-phosphotyrosyl linkage between Top1 and DNA and subsequent repair of the damage (23,3,4,6). PARP1 is activated by Top1-mediated DNA breaks (35–38), and PARylation at DNA lesions likely recruits DNA repair complexes while releasing PARP1 from DNA to facilitate DNA repair (25,62). Thus, it is plausible that TDP1 detects Top1-mediated DNA damage through PARP1, as PARP1 and TDP1 complexes are readily detectable in cells even in the absence of DNA damage (see Figure 5). Consistent with this conclusion, our data indicate that PARP activation is essential for the recruitment of TDP1 at DNA damage sites (see Figure 4). TDP1 has been associated with XRCC1 repair complexes (59), which also include PARP1, polynucleotide kinase 3′-phosphatase and ligase III (17,23,60). PARylation-dependent XRCC1 recruitment has been established at H2O2 or laser-induced DNA lesions (63,64). Our data further indicate that XRCC1 acts downstream from TDP1–PARP1 in the case of Top1cc-induced DNA damage. PARP1–TDP1 complexes promote the recruitment of XRCC1 at CPT-induced Top1cc (see Figure 6), and stabilize TDP1 in cells treated with CPT (see Figure 7).
The regulation of TDP1 activity involves the NTD of TDP1 (see scheme in Figure 2B), which is phosphorylated at serine 81 by ATM and/or DNA-PK, resulting in stabilization TDP1, enhanced DNA repair (6) and interaction with ligase III (24). In addition, TDP1 SUMOylation at lysine 111 promotes the repair of Top1cc- and radiation-induced DNA lesions, and TDP1 accumulation at sites of DNA damage (20). The present study demonstrates that the NTD of TDP1 (1–185 aa) directly binds the C-terminal catalytic domain of PARP1 (residues 525–1014) (see schemes in Figures 2B and 8). Yet, contrary to other DNA repair enzymes (62) and to Top1 (28), PARP1 binding and PARylation do not block the catalytic activity of TDP1 (see Figure 3), which is consistent with the fact that the NTD fragment of TDP1 (spanning residues 1–148) is not required for phosphodiesterase activity (10). The fact that ABT-888 (veliparib) fails to abrogate PARP1–TDP1 interaction (Figure 5A and B) and that TDP1 directly binds PARP1 (see Figures 2 and 5) indicate that PARP1 and TDP1 associate in stable complexes independently of PARP activation and DNA (Figure 5C). Further studies are warranted to elucidate how the three TDP1 regulatory mechanisms, PARylation, SUMOylation and S81 phosphorylation are interrelated to regulate the repair of Top1-induced DNA lesions.
Finally, the present study provides new insights into the mechanism of action of PARP inhibitors in combination with Top1 inhibitors. PARP1 appears to act as a molecular switch between TDP1 and the endonuclease pathway for the repair of Top1cc. As shown in Figure 8 (36), Top1cc are excised by two main pathways: TDP1 that ‘unhooks’ Top1cc from the 3′-end of the DNA (11,65) and nucleases that cleave Top1cc-containing 3′-flaps including XPF-ERCC1 (36,41), Mus81-Eme1 (42), Mre11-Rad50-Nibrin (66) and CtIP (44,53,54). The fact that the double-mutant TDP1−/−;CtIP S332/−/− shows greater than additive hypersensitivity to CPT (14), whereas the TDP1−/−;PARP1−/− double-mutant cells behave like single mutants (see Figure 1) indicates that PARP1 and TDP1 are epistatic, and therefore the tight coupling of TDP1 and PARP for Top1cc repair (36). This conclusion suggests that the combination of PARP and Top1 inhibitors should be most beneficial in endonuclease-deficient cancer cells, such as ERCC1-deficient (36) or Mre11-deficient tumors (67). Because PARP1 can affect Top1cc by other mechanisms beside TDP1, including destabilization of Top1cc (27,28), release of Top1 stalled replication complexes (39) and restart of replication forks reversed by Top1cc (8,40), and because PARP1 affects many other cellular processes besides Top1 (25), TDP1 inhibitors could potentially serve as more specific alternative to PARP inhibitors for synergistic combination with Top1 inhibitors in cancer chemotherapy.
SUPPLEMENTRY DATA
Supplementary Data are available at NAR Online.
FUNDING
The NCI Intramural Program, Center for Cancer Research, National Cancer Institute, NIH, supported this work (Z01 BC 006161). The BBD team is supported by Wellcome Trust/India alliance intermediate fellowship (Award# IA/I/13/1/500888) and IACS startup fund, Department of science and technology, Government of India. VS team is Equipe Labellisée Ligue contre le Cancer and is supported by CNRS, University of Strasbourg, Ligue contre le Cancer (comités du Bas-Rhin et du Haut-Rhin) and Labex Medalis (11LABX065). Funding for open access charge: The NCI Intramural Program, Center for Cancer Research, National Cancer Institute, NIH.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr. Shunichi Takeda (Kyoto University, Kyoto, Japan) for sharing PARP1−/− and PARP1−/− complemented with human PARP1 and PARP2 DT40 cell lines and support and discussion through the course of these studies.
REFERENCES
- 1.Wang JC. A journey in the world of DNA rings and beyond. Annu. Rev. Biochem. 2009;78:31–54. doi: 10.1146/annurev.biochem.78.030107.090101. [DOI] [PubMed] [Google Scholar]
- 2.Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 3.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dexheimer TS, Antony S, Marchand C, Pommier Y. Tyrosyl-DNA phosphodiesterase as a target for anticancer therapy. Anticancer Agents Med. Chem. 2008;8:381–389. doi: 10.2174/187152008784220357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Khamisy SF, Caldecott KW. DNA single-strand break repair and spinocerebellar ataxia with axonal neuropathy-1. Neuroscience. 2007;145:1260–1266. doi: 10.1016/j.neuroscience.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 6.Das BB, Antony S, Gupta S, Dexheimer TS, Redon CE, Garfield S, Shiloh Y, Pommier Y. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 2009;28:3667–3680. doi: 10.1038/emboj.2009.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007;26:4720–4731. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berti M, Chaudhuri AR, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013;20:347–354. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, Choi K, Bhattacharjee MB, Arimura K, Umehara F, Izumo S, et al. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 2007;26:4732–4743. doi: 10.1038/sj.emboj.7601885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Interthal H, Pouliot JJ, Champoux JJ. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc. Natl Acad. Sci. USA. 2001;98:12009–12014. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–555. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 12.El-Khamisy SF. To live or to die: a matter of processing damaged DNA termini in neurons. EMBO Mol. Med. 2011;3:78–88. doi: 10.1002/emmm.201000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das BB, Dexheimer TS, Maddali K, Pommier Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl Acad. Sci. USA. 2010;107:19790–19795. doi: 10.1073/pnas.1009814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J. Biol. Chem. 2012;287:12848–12857. doi: 10.1074/jbc.M111.333963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banerjee B, Roy A, Sen N, Majumder HK. A tyrosyl DNA phosphodiesterase 1 from kinetoplastid parasite Leishmania donovani (LdTdp1) capable of removing topo I-DNA covalent complexes. Mol. Microbiol. 2010;78:119–137. doi: 10.1111/j.1365-2958.2010.07318.x. [DOI] [PubMed] [Google Scholar]
- 16.Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 2002;32:267–272. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 17.El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 18.Miao ZH, Agama K, Sordet O, Povirk L, Kohn KW, Pommier Y. Hereditary ataxia SCAN1 cells are defective for the repair of transcription-dependent topoisomerase I cleavage complexes. DNA Repair (Amst) 2006;5:1489–1494. doi: 10.1016/j.dnarep.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Interthal H, Chen HJ, Champoux JJ. Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J. Biol. Chem. 2005;280:36518–36528. doi: 10.1074/jbc.M508898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hudson JJ, Chiang SC, Wells OS, Rookyard C, El-Khamisy SF. SUMO modification of the neuroprotective protein TDP1 facilitates chromosomal single-strand break repair. Nat. Commun. 2012;3:733. doi: 10.1038/ncomms1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP. Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci. 2000;922:1–10. doi: 10.1111/j.1749-6632.2000.tb07020.x. [DOI] [PubMed] [Google Scholar]
- 22.Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, Conti C, Nakamura AJ, Das BB, Nicolas E, et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009;10:887–893. doi: 10.1038/embor.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, et al. Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2006;81:179–229. doi: 10.1016/S0079-6603(06)81005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiang SC, Carroll J, El-Khamisy SF. TDP1 serine 81 promotes interaction with DNA ligase IIIalpha and facilitates cell survival following DNA damage. Cell Cycle. 2010;9:588–595. doi: 10.4161/cc.9.3.10598. [DOI] [PubMed] [Google Scholar]
- 25.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell. Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 26.Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010;35:208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Malanga M, Althaus FR. Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and Induces DNA strand break resealing. J. Biol. Chem. 2004;279:5244–5248. doi: 10.1074/jbc.C300437200. [DOI] [PubMed] [Google Scholar]
- 28.Park SY, Cheng YC. Poly(ADP-ribose) polymerase-1 could facilitate the religation of topoisomerase I-linked DNA inhibited by camptothecin. Cancer Res. 2005;65:3894–3902. doi: 10.1158/0008-5472.CAN-04-4014. [DOI] [PubMed] [Google Scholar]
- 29.Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell. 2010;39:736–749. doi: 10.1016/j.molcel.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem Pharmacol. 2012;84:137–146. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 32.Audebert M, Salles B, Calsou P. Effect of double-strand break DNA sequence on the PARP-1 NHEJ pathway. Biochem. Biophys. Res. Commun. 2008;369:982–988. doi: 10.1016/j.bbrc.2007.11.132. [DOI] [PubMed] [Google Scholar]
- 33.Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34:1685–1691. doi: 10.1093/nar/gkl108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatterjee S, Cheng MF, Trivedi D, Petzold SJ, Berger NA. Camptothecin hypersensitivity in poly(adenosine diphosphate-ribose) polymerase-deficient cell lines. Cancer Commun. 1989;1:389–394. doi: 10.3727/095535489820875129. [DOI] [PubMed] [Google Scholar]
- 36.Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011;39:3607–3620. doi: 10.1093/nar/gkq1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel AG, Flatten KS, Schneider PA, Dai NT, McDonald JS, Poirier GG, Kaufmann SH. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J. Biol. Chem. 2012;287:4198–4210. doi: 10.1074/jbc.M111.296475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowman KJ, Newell DR, Calvert AH, Curtin NJ. Differential effects of the poly (ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro. Br. J. Cancer. 2001;84:106–112. doi: 10.1054/bjoc.2000.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugimura K, Takebayashi S, Taguchi H, Takeda S, Okumura K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008;183:1203–1212. doi: 10.1083/jcb.200806068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 41.Vance JR, Wilson TE. Yeast Tdp1 and Rad1-Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc. Natl Acad. Sci. USA. 2002;99: 13669–13674. doi: 10.1073/pnas.202242599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Regairaz M, Zhang YW, Fu H, Agama KK, Tata N, Agrawal S, Aladjem MI, Pommier Y. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol. 2011;195:739–749. doi: 10.1083/jcb.201104003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamilton NK, Maizels N. MRE11 function in response to topoisomerase poisons is independent of its function in double-strand break repair in Saccharomyces cerevisiae. PLoS One. 2010;5:e15387. doi: 10.1371/journal.pone.0015387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartsuiker E, Neale MJ, Carr AM. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell. 2009;33:117–123. doi: 10.1016/j.molcel.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, Hall J, Favaudon V. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008;36:4454–4464. doi: 10.1093/nar/gkn403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006;25:1305–1314. doi: 10.1038/sj.emboj.7601015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Menissier-de Murcia J, de Murcia G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 49.Das BB, Sen N, Dasgupta SB, Ganguly A, Majumder HK. N-terminal region of the large subunit of Leishmania donovani bisubunit topoisomerase I is involved in DNA relaxation and interaction with the smaller subunit. J. Biol. Chem. 2005;280:16335–16344. doi: 10.1074/jbc.M412417200. [DOI] [PubMed] [Google Scholar]
- 50.Dexheimer TS, Stephen AG, Fivash MJ, Fisher RJ, Pommier Y. The DNA binding and 3′-end preferential activity of human tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2010;38:2444–2452. doi: 10.1093/nar/gkp1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang SY, Murai J, Dalla Rosa I, Dexheimer TS, Naumova A, Gmeiner WH, Pommier Y. TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 2013;41:7793–7803. doi: 10.1093/nar/gkt483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mortusewicz O, Leonhardt H. XRCC1 and PCNA are loading platforms with distinct kinetic properties and different capacities to respond to multiple DNA lesions. BMC Mol. Biol. 2007;8:81. doi: 10.1186/1471-2199-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakamura K, Kogame T, Oshiumi H, Shinohara A, Sumitomo Y, Agama K, Pommier Y, Tsutsui KM, Tsutsui K, Hartsuiker E, et al. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 2010;6:e1000828. doi: 10.1371/journal.pgen.1000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meder VS, Boeglin M, de Murcia G, Schreiber V. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J. Cell Sci. 2005;118:211–222. doi: 10.1242/jcs.01606. [DOI] [PubMed] [Google Scholar]
- 56.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Interthal H, Champoux JJ. Effects of DNA and protein size on substrate cleavage by human tyrosyl-DNA phosphodiesterase 1. Biochem. J. 2011;436:559–566. doi: 10.1042/BJ20101841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004;23:2674–2683. doi: 10.1038/sj.emboj.7600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 60.Caldecott KW. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 61.Alagoz M, Wells OS, El-Khamisy SF. TDP1 deficiency sensitizes human cells to base damage via distinct topoisomerase I and PARP mechanisms with potential applications for cancer therapy. Nucleic Acids Res. 2013;42:3089–3103. doi: 10.1093/nar/gkt1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindahl T. Recognition and processing of damaged DNA. J. Cell Sci. Suppl. 1995;19:73–77. doi: 10.1242/jcs.1995.supplement_19.10. [DOI] [PubMed] [Google Scholar]
- 63.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mortusewicz O, Ame JC, Schreiber V, Leonhardt H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007;35:7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pouliot JJ, Robertson CA, Nash HA. Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells. 2001;6:677–687. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 66.Sacho EJ, Maizels N. DNA repair factor MRE11/RAD50 cleaves 3′-phosphotyrosyl bonds and resects DNA to repair damage caused by topoisomerase 1 poisons. J. Biol. Chem. 2011;286:44945–44951. doi: 10.1074/jbc.M111.299347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takemura H, Rao VA, Sordet O, Furuta T, Miao ZH, Meng L, Zhang H, Pommier Y. Defective Mre11-dependent activation of Chk2 by Ataxia telangiectasia mutated in colorectal carcinoma cells in response to replication-dependent DNA double strand breaks. J. Biol. Chem. 2006;281:30814–30823. doi: 10.1074/jbc.M603747200. [DOI] [PubMed] [Google Scholar]
- 68.Kohn KW, Aladjem MI, Weinstein JN, Pommier Y. Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol. Biol. Cell. 2006;17:1–13. doi: 10.1091/mbc.E05-09-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.