

Abstract
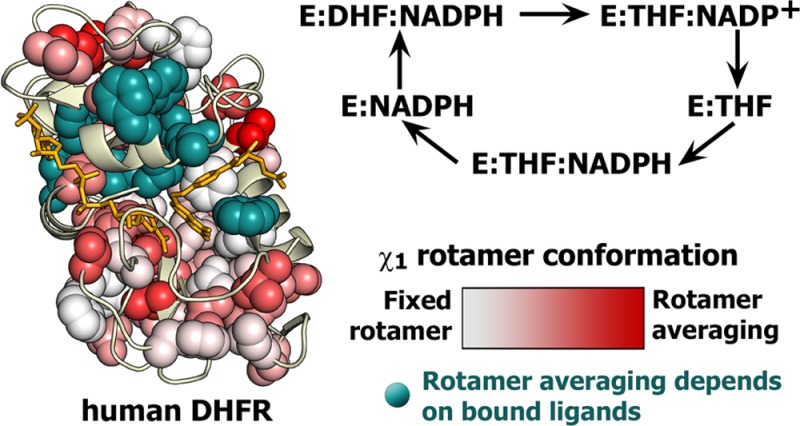
The three-dimensional structures of the dihydrofolate reductase enzymes from Escherichia coli (ecDHFR or ecE) and Homo sapiens (hDHFR or hE) are very similar, despite a rather low level of sequence identity. Whereas the active site loops of ecDHFR undergo major conformational rearrangements during progression through the reaction cycle, hDHFR remains fixed in a closed loop conformation in all of its catalytic intermediates. To elucidate the structural and dynamic differences between the human and E. coli enzymes, we conducted a comprehensive analysis of side chain flexibility and dynamics in complexes of hDHFR that represent intermediates in the major catalytic cycle. Nuclear magnetic resonance relaxation dispersion experiments show that, in marked contrast to the functionally important motions that feature prominently in the catalytic intermediates of ecDHFR, millisecond time scale fluctuations cannot be detected for hDHFR side chains. Ligand flux in hDHFR is thought to be mediated by conformational changes between a hinge-open state when the substrate/product-binding pocket is vacant and a hinge-closed state when this pocket is occupied. Comparison of X-ray structures of hinge-open and hinge-closed states shows that helix αF changes position by sliding between the two states. Analysis of χ1 rotamer populations derived from measurements of 3JCγCO and 3JCγN couplings indicates that many of the side chains that contact helix αF exhibit rotamer averaging that may facilitate the conformational change. The χ1 rotamer adopted by the Phe31 side chain depends upon whether the active site contains the substrate or product. In the holoenzyme (the binary complex of hDHFR with reduced nicotinamide adenine dinucleotide phosphate), a combination of hinge opening and a change in the Phe31 χ1 rotamer opens the active site to facilitate entry of the substrate. Overall, the data suggest that, unlike ecDHFR, hDHFR requires minimal backbone conformational rearrangement as it proceeds through its enzymatic cycle, but that ligand flux is brokered by more subtle conformational changes that depend on the side chain motions of critical residues.
Escherichia coli dihydrofolate reductase (ecDHFR or ecE) has long been a paradigm for the study of the relationship between enzyme dynamics and function.1−13 Interestingly, its human counterpart (hDHFR or hE) achieves the same catalytic function, with arguably better efficiency,14 by utilizing quite different dynamic behavior.15 hDHFR and ecDHFR share the same major catalytic cycle (Figure 1). The flexible Met20 loop of ecDHFR samples distinct closed and occluded conformations as ecDHFR proceeds through its enzymatic cycle: the ground states of the ecE:NADPH complex and the ecE:FOL:NADP+ complex (a model for the Michaelis complex, ecE:DHF:NADPH) have a closed Met20 loop, whereas the product complexes have an occluded Met20 loop.7,16 The equivalent “Met20 loop” regiona of hDHFR has an LPWPP sequence instead of a MPWN sequence in ecDHFR and is locked in a closed conformation throughout the catalytic cycle.15 Ligand binding in hDHFR occurs through a hinge opening motion of the adenosine subdomain relative to the loop subdomain (Figure 2), rather than from the conformational sampling of the Met20 loop as in ecDHFR. The hinge motion in hDHFR is likely mediated by hinge loops longer than those present in ecDHFR: in hDHFR, hinge 1 is defined as residues Thr39–Leu49 and hinge 2 as His127–Leu131, compared to Thr36–Pro39 and Pro105–Gln108 in ecDHFR, respectively.15 Substrate complexes of hDHFR adopt the hinge-closed conformation, where the active site is tightly packed, thereby favoring the hydride transfer reaction. 15N R2 relaxation dispersion experiments reveal conformational fluctuations on a millisecond time scale in all of the intermediates of the major enzymatic cycle of ecDHFR.6,8 In contrast, millisecond time scale dynamics are not observed in the corresponding complexes of hDHFR, but motions on a faster microsecond time scale are observed in the hE:FOL:NADP+ complex.15
Figure 1.
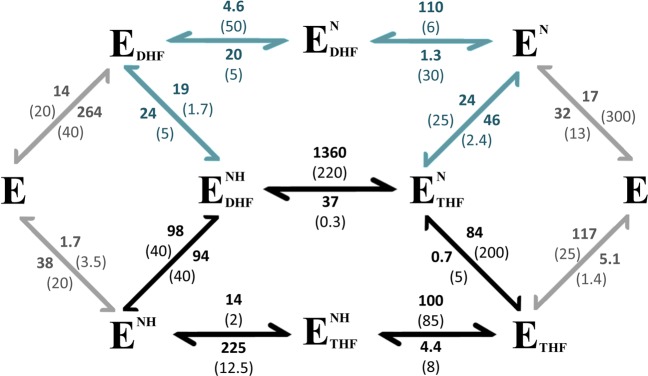
Full kinetic schemes for ecDHFR and hDHFR. hDHFR rates are shown in bold, and ecDHFR rates are shown in parentheses. Black arrows indicate the major catalytic cycle for both ecDHFR and hDHFR. The minor “upper” cycle, shown by blue arrows, is also used by hDHFR because the release of THF from the product complex is much faster than for ecDHFR. Approximately 35% of the product is channeled through the upper branch by human DHFR. Gray arrows show kinetics involving the apoprotein, E, which do not constitute a significant part of the enzymatic cycle for either protein under cellular conditions. ecDHFR kinetic parameters (25 °C, pH 7) were taken from ref (39), and hDHFR parameters (20 °C, pH 7.65) were taken from ref (14). First- and second-order rates are given in units of s–1 and μM–1 s–1, respectively.
Figure 2.

(A) Structure of human DHFR. The Met20 loop (residues 12–27, light blue) remains closed for all hDHFR complexes. The FG loop (residues 139–159) and GH loop (residues 172–175) are colored green and navy, respectively. The adenosine-binding subdomain (residues 40–129) is colored pink for the hE:FOL:NADP+ complex [Protein Data Bank (PDB) entry 4M6K] and dark red for the hE:NADPH complex (PDB entry 4M6J). The structures are aligned on the loop subdomain to illustrate the motion of the subdomains between the hinge-closed (pink) and hinge-opened (dark red) conformations. The Cα atoms of hinge 1 and hinge 2 are shown as small spheres. The NADP/H coloring is consistent with the adenosine domain: dark red for NADPH and pink for NADP+. Folate is colored yellow. (B) View rotated 90° relative to that in panel A. In the hinge-open conformation (hE:NADPH), helix αF slides toward the active site, relative to the hinge-closed conformation (in the hE:FOL:NADP+, hE:THF:NADP+, and hE:THF complexes). Except for the difference in the ligand, the X-ray structures of the hE:FOL (PDB entry 1DHF)47 and hE:ddTHF:NADP+ (PDB entry 4M6L) complexes are highly similar to that of the hE:FOL:NADP+ complex. Structures of the ternary complexes and the hE:NADPH complex were from ref (15).
We have recently demonstrated the exquisite sensitivity of 3JCγCO and 3JCγN couplings to the average χ1 dihedral angle conformation and rotamer averaging of side chains in the complexes of ecDHFR that constitute intermediates in the enzymatic cycle.12 Here, we extend this analysis to hDHFR, with the time scale-independent side chain χ1 conformational information for γ-methyl and aromatic residues obtained from 3JCγCO and 3JCγN coupling measurements. These coupling-derived rotamer data are augmented by rotamer population estimates based on 13Cmethyl chemical shifts for leucine residues.
Methods
Protein Purification and Sample Preparation
Human DHFR was expressed and purified as described previously.17 Briefly, hDHFR was expressed in BL21(DE3)(DNAY) cells in M9 minimal medium with 2 mM folate at 37 °C. Different isotope labeling patterns were used for assignment, dynamics, and rotamer experiments. Samples uniformly labeled with 1H and 15N were produced using [15N]ammonium chloride (0.5 g/L) and [15N]ammonium sulfate (0.5 g/L). All 3J coupling experiments were performed with samples uniformly labeled with 1H, 13C, and 15N and grown in 100% H2O, [U-13C]glucose (3 g/L), and 15N salts. A sample uniformly labeled with 13C and 15N and labeled with 1H or 2H (with a CHD2 dominant methyl isotopomer) was prepared by growth in 100% D2O medium. Samples for [13C]methyl CPMG relaxation dispersion experiments were prepared using 100% H2O, 15N salts, and [1-13C]glucose to achieve the labeling pattern described by Lundström et al.18 Samples for the stereochemical assignment of leucine and valine methyls were produced by the method of Neri et al.19 using 10% [13C]glucose (0.4 g/L) and 90% [12C]glucose (3.6 g/L). Cultures (1 L) were grown to an OD600 of ∼0.9 and induced with 1 mM IPTG at 15 °C. Cells were harvested after ∼24 h, and pellets were frozen until they were purified. Cells were lysed in the presence of folate. hDHFR was eluted from a Q-Sepharose column with weak salt (<100 mM NaCl) and further purified by high-performance liquid chromatography purification, taking care to avoid an impurity that elutes at a slightly higher percentage of acetonitrile. Complexes were prepared by unfolding lyophilized protein in 8 M urea containing 10 mM DTT, followed by rapid dilution into Tris buffer (pH 8.5) containing the desired ligands. After concentration of the solution, exchange into the final degassed buffer was performed in an argon-equilibrated glovebox because of the highly oxygen-sensitive nature of THF and NADPH solutions. NMR buffer contained a 10% D2O/90% H2O mixture or 100% D2O, as applicable, with 50 mM KPi, 50 mM KCl, 1 mM DTT, and 1 mM EDTA (pH 6.5) for all samples, except for the hE:NADPH complex, which was prepared at pH 8.0 for increased stability. Final sample concentrations ranged from ∼300 μM for binary complexes to 1 mM for ternary complexes. The complexes were formed by addition of a 6-fold excess of cofactor (NADP+ or NADPH) and a 10-fold excess of substrate or product (FOL or THF), as applicable. NMR samples were further degassed on a vacuum line, overlaid with argon, and flame-sealed in an amberized tube. Under these conditions, the hE:FOL:NADP+ sample is very stable, the hE:THF:NADP+ and hE:THF samples are stable for up to 1 month, the hE:NADPH sample is stable for ∼1 week, and the hE:THF:NADPH sample is extremely unstable, with spectra changing in a matter of hours. The quality of the ligands in the final samples was ascertained by comparison of one-dimensional proton spectra of the samples to reference ligand spectra. High-purity (6S)-THF was purchased from Schircks Laboratories, and high-purity NADPH and NADP+ were purchased from Sigma.
Methyl Side Chain Assignments
Backbone assignments (HN, N, Cα, and partial Cβ) for all hDHFR complexes were made using triple-resonance methods, as reported previously.15 Side chain assignments for the hE:FOL:NADP+ complex were determined at 300 K utilizing standard two-dimensional 15N and 13C HSQC experiments and the following three-dimensional NMR experiments, with the labeling scheme, the percent D2O in the NMR buffer, and 1H spectrometer frequency given in parentheses: HCCH-TOCSY and HCCH-COSY20 ([U-1H,13C,15N] in 100% D2O, 600 MHz), 120 ms mixing time 13C NOESY21 ([U-1H,13C,15N] in 100% D2O, 900 MHz), 15N TOCSY ([U-1H,15N] in 10% D2O, 600 MHz), and CHD2-selective TOCSY22 and HNCACB23 ([U-13C,15N], 1H/2H in 10% D2O, 800 MHz). Stereochemical assignments for the hE:FOL:NADP+, hE:THF, and hE:NADPH complexes were obtained from 10% 13C- and 15N-labeled protein using the CHD2-selective 13C HSQC experiment described by Otten et al.22 in which Leu-δ2 and Val-γ2 resonances have the same phase as Met-ε resonances. Methyl assignments obtained for the hE:FOL:NADP+ complex were used as a starting point for assignments of 13C methyl resonances in the hE:THF and hE:NADPH complexes, which were finalized using 13C NOESY and 15N TOCSY experiments for each complex.
13C Methyl R2 Relaxation Dispersion Experiments
Relaxation dispersion experiments report on millisecond to microsecond time scale exchange processes. Methyl 13C CPMG relaxation dispersion experiments were performed on a [1-13C,15N]hE:FOL:NADP+ sample at multiple temperatures (280, 300, and 310 K) using a Bruker DRX 800 MHz spectrometer and the pulse schemes described by Skrynnikov et al.24 and Lundström et al.25 Using [1-13C]glucose as the sole carbon source leads to isolated 13C-enriched methyls for most methyl side chains; however, the presence of 13C–13Cmethyl labeling for Thr-γ2 and Ile-δ1 methyls can interfere with accurate spin relaxation measurements due to the one-bond C–C coupling.18 Experiments were performed using a total relaxation period (TCPMG) of 40 ms and refocusing delays (1/τcp) of 100, 200, 400*, 600, 1000, 1400*, 1800, and 1900 s–1, where asterisks denote experiments completed in duplicate. Dispersion curves were fit using GLOVE.26
Measurement of 3J Couplings and Determination of χ1 Rotamer Populations
The 3JCγCO and 3JCγN coupling constants describe the average χ1 orientation of the side chain with respect to the backbone. There are established NMR experiments for measuring 3JCγCO and 3JCγN coupling constants for Ile, Thr, and Val γ-methyls27,28 and for aromatic residues.29 These rely on 13C HSQC and 15N HSQC difference spectra, respectively. A reference spectrum has maximal intensity as coupling is not evolved, and a second spectrum, where the coupling of interest is allowed to evolve, results in peak intensities that are attenuated as a function of that coupling. For the methyl-containing residues, the couplings are calculated from the relationship (Ia – Ib)/Ia = 2 sin2(πJCγXT), where Ia is the reference spectrum intensity, Ib is the intensity for either the 3JCγCO or 3JCγN experiment, and the delay T is 28.6 ms. Similarly, for aromatic residues, the couplings are calculated from the relationship Ib/Ia = cos(2πJCγXT), where Ia is the reference spectrum intensity and Ib is the intensity for the amide of residue i + 1 in the 3JCγCO experiment or for residue i in the 3JCγN experiment. The delay T was 25 ms for 3JCγCO experiments and 50 ms for 3JCγN experiments. All 3J measurements were taken in triplicate or better to reduce the contribution of spectral noise to rotamer calculations.
The measured coupling constants allow for the calculation of χ1 rotamer populations assuming a three-site jump model according to the following equations:12,30
| 1 |
| 2 |
| 3 |
where 3Jmeas,CγN and 3Jmeas,CγCO are the experimentally measured coupling constants; p–60, p60, and p180 are the populations of the respective χ1 rotamer states; and Jt, Jg, and Jh for CγCO and CγN are the expected coupling values for the fully populated 180°, 60°, and −60° χ1 rotamers, respectively (Table 1 and Figure S1 of the Supporting Information). Populations were fit by minimizing the squared difference between measured and calculated 3JCγN and 3JCγCO couplings for each residue: ∑(1/σJmeas)2(Jcalc – Jmeas)2, where σ is the standard deviation based on three or more measurements of 3J. Populations for Val residues represent a simultaneous fit of γ1 and γ2 couplings. The extent of rotamer averaging is related to the major rotamer population by the relationship pmajor ≡ max[p60, p180, p–60], where more rotamer averaging is associated with a smaller pmajor value. Population ranges were determined using 3J ± σ. Note that small values of 3J are prone to larger errors (by percent) when the primary contribution to error is from noise in the NMR spectra. Unique rotamer averaging is present when the 3J values of a residue in one or more complexes have nonoverlapping error ranges.
Table 1. Predicted Values for 3J Couplings at χ1 Rotamer Angles of 180°, 60°, and −60°a.
|
3JCγN (Hz) |
3JCγCO (Hz) |
|||||
|---|---|---|---|---|---|---|
| residue type | Jt | Jg | Jh | Jt | Jg | Jh |
| His | 2.73 | 0.50 | 0.50 | 1.09 | 1.09 | 4.97 |
| Phe, Trp, Tyr | 2.62 | 0.38 | 0.38 | 0.55 | 0.55 | 4.43 |
| Ile | 0.95 | 0.31 | 2.01 | 0.79 | 3.83 | 0.45 |
| Thr | 0.27 | 0.73 | 1.65 | 1.18 | 3.35 | 0.18 |
| Val Cγ1 | 2.00 | 0.47 | 0.68 | 0.45 | 0.79 | 3.83 |
| Val Cγ2 | 0.31 | 2.01 | 0.95 | 3.83 | 0.45 | 0.79 |
Expected 3J values for fully populated staggered rotamers. All values are given with respect to the χ1 angle. Values for the aromatic residues are derived from fits of Karplus curves to the DHFR 3J data vs the χ1 angle as reported previously.12 Values for the γ-methyl residues are from the parametrizations of Chou et al.48 Plots of each Karplus curve are shown in Figure S1 of the Supporting Information.
While the interpretation of these 3J coupling values in terms of a three-site rotamer hopping model is generally justified, there are clear cases in which the predicted minor rotamer is not sterically feasible, as was observed also for ecDHFR.12 This is especially evident for some well-packed aromatic residues. In such cases, it is more appropriate to interpret the 3J couplings in terms of local motions within a rotamer well, where these side chains undergo local averaging about the χ1 angle, or in terms of a relatively fixed but skewed rotamer (i.e., with a noncanonical χ1). Case and co-workers have demonstrated that local χ1 averaging can lead to a broadening of the expected Karplus curves such that the maximal values are reduced and the minimal values are increased.31 Consideration of these issues for individual residues can provide insights into local conformational changes between complexes for residues that can clearly not accommodate the otherwise predicted minor rotamers; pmajor remains a proxy for these types of in-well rotamer averaging because it is a measure of the deviation of the 3J couplings from the expected values for the fully populated staggered rotamers.
Chemical Shift-Derived Rotamer Populations
There has been recent success in correlating 13C methyl chemical shifts with valine χ1 and leucine and isoleucine χ2 rotamer populations.32,33 The correlation between the chemical shift and dihedral angle has been largely attributed to a γ-substituent effect, the influence of an atom in the gauche position three bonds away, based upon early studies of small molecule 13C chemical shifts.34,35 For leucines, a simple relationship between the chemical shifts of the methyls has been reported
| 4 |
where ptrans is the population of the χ2 = 180° rotamer. Despite the nine possible χ1, χ2 pairs for leucine, only two of these are significantly sampled in structures in the Protein Data Bank (PDB): (180°, 60°) and (−60°, 180°).36 Therefore, knowledge of the χ2 rotamer is largely predictive of the χ1 rotamer conformation. This approach was used to determine Leu χ1, χ2 rotamer populations in hDHFR based on the 13Cmethyl chemical shift assignments for the hE:FOL:NADP+, hE:THF, and hE:NADPH complexes. To account for contributions of neighboring aromatic rings to the 13C chemical shift, ring current corrections were calculated using Shifts-4.3.37
Results
Resonance Assignments
Methyl side chain assignments for several intermediates of the hDHFR major enzymatic cycle are summarized in Table S1 of the Supporting Information. The methyl and backbone assignments for all complexes have been deposited in the BioMagResBank. The 15N HSQC and 13C HSQC spectra of the hE:THF:NADP+ and hE:FOL:NADP+ complexes are virtually indistinguishable, whereas the spectrum of the hE:THF:NADPH complex shows extensive broadening for resonances of residues near the active site. Complete assignments could not be obtained for this complex. There are substantial chemical shift changes in the hE:NADPH and hE:THF binary complexes for both backbone and side chain atoms relative to the ternary complexes. The average chemical shift differences from the ternary hE:FOL:NADP+ Michaelis model complex are plotted on the structures of the hE:NADPH and hE:THF complexes in Figure 3.
Figure 3.
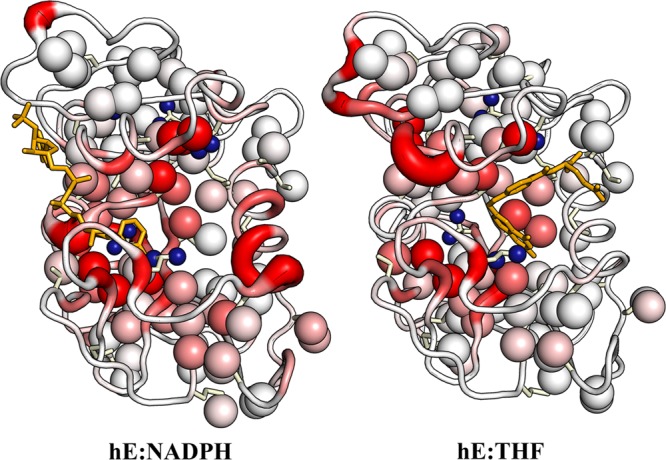
Backbone and methyl side chain chemical shift differences between the binary complexes (hE:NADPH, left; hE:THF, right) and the ternary hE:FOL:NADP+ complex. The backbone color and ribbon diameter report on the average amide and Cα chemical shift differences: ΔδNHC = [ΔδH2 + (ΔδN/5)2 + (2ΔδCα/5)2]1/2. The backbone is colored by a gradient from white, indicating minimal chemical shift difference, to red for ΔδNHC values of >1.0 ppm. Ile, Thr, Val, and Leu side chains are shown as sticks, with the methyl groups shown as spheres. The methyl groups are colored with a white to red gradient according to the average methyl chemical shift difference: ΔδCH3 = [ΔδHmethyl2 + (ΔδCmethyl/3)2]1/2, where red indicates a ΔδCH3 of >0.4 ppm. Leu residues with unassigned or ambiguous methyl chemical shifts are shown as small blue spheres.
Millisecond Time Scale Methyl Dynamics
We have shown previously using 15N CPMG R2 relaxation dispersion experiments that there are no detectable millisecond time scale backbone fluctuations in the hE:FOL:NADP+, hE:THF:NADP+, hE:THF, and hE:NADPH complexes.15 To determine whether hDHFR side chains experience microsecond to millisecond time scale conformational fluctuations, such as those observed for ecDHFR, 13Cmethyl CPMG relaxation dispersion experiments were performed on the hE:FOL:NADP+ complex at temperatures ranging from 280 to 310 K. A representative set of methyl relaxation dispersion data is shown in Figure S2 of the Supporting Information. While some residues show hints of dispersion at 280 K, e.g., Ile114-Cδ and Ile151-Cγ2, none of the methyl-containing residues show clearly defined 13Cmethyl dispersion curves. This is contrary to what was observed for ecDHFR, which exhibits exchange on the microsecond to millisecond time scale for most methyls,38 but is consistent with 15N CPMG experiments for hDHFR.15 On the basis of these results, and given that the hE:FOL:NADP+ complex exhibits a single set of resonances in HSQC spectra, any conformational exchange processes must be occurring on a time scale faster than the scale that can be detected by CPMG relaxation dispersion experiments and/or involve very small changes in chemical shifts.
3JCγCO and 3JCγN Couplings and Side Chain Rotamer Populations
χ1 rotamer populations for the complexes corresponding to the intermediates in the major enzymatic cycle of hDHFR have been determined from 3JCγCO and 3JCγN coupling constants for Ile, Thr, Val, and aromatic residues. Populations were determined by minimizing the difference between 3Jmeas and 3Jcalc as described in Methods. The rotamer populations at each residue are summarized graphically in Figures S3 and S4 of the Supporting Information. χ2 rotamer populations for Leu residues were estimated from 13C methyl chemical shift values according to eq 4. The χ2 rotamer populations for the Leu residues imply a similar population of the corresponding χ1 rotamer because Leu rotamer pairs are predominantly (χ1, χ2) = (180°, 60°) or (−60°, 180°).36 The 3J coupling constants for methyl and aromatic residues are shown in Figures 4 and 5, respectively, with complete tables of measured couplings and rotamer populations for each complex given in Tables S2–S11 of the Supporting Information. The 13C chemical shifts values and calculated χ2 rotamer populations for Leu residues are listed in Table S12 of the Supporting Information, and the χ2 180° populations are shown in Figure 6.
Figure 4.

3JCγN and 3JCγCO values for Ile, Thr, and Val residues of the hE:FOL:NADP+ (squares), hE:THF:NADP+ (circles), hE:NADPH (diamonds), and hE:THF (triangles) complexes. Blue, green, and red bars show the expected 3J values for fully populated 180°, 60°, and −60° χ1 rotamers, respectively; shaded areas represent the variation in 3J values over a ±5° span about the staggered rotamer, according to the relevant Karplus curves (Figure S1 of the Supporting Information). The average coupling over all complexes is shown as an X, except for residues with different rotamer populations in one or more complexes, which are labeled with an asterisk. Complexes are excluded from the average if the error in the coupling constant is >1 Hz. The standard deviation of 3Javg is shown as a dark gray vertical bar.
Figure 5.
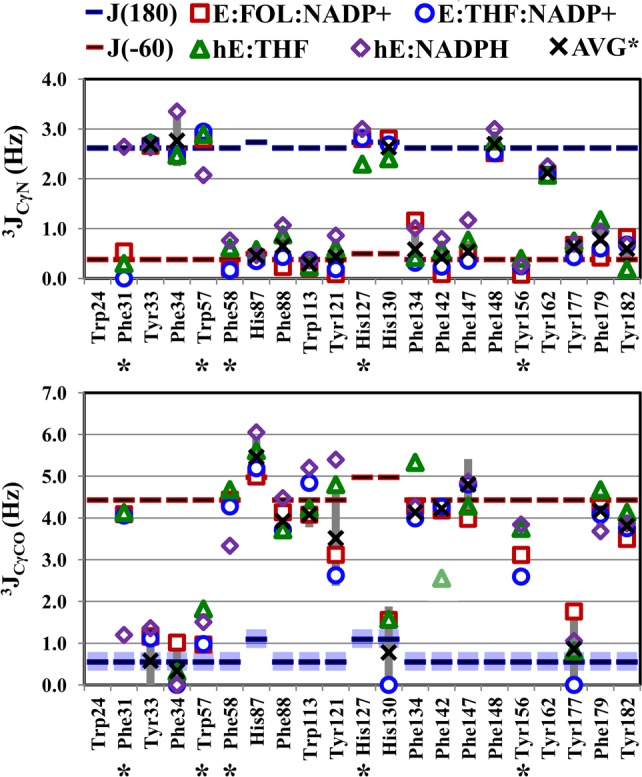
3JCγN and 3JCγCO values for aromatic residues of the hE:FOL:NADP+ (squares), hE:THF:NADP+ (circles), hE:NADPH (diamonds), and hE:THF (triangles) complexes. Symbols are as described in the legend of Figure 4. The 3JCγCO value for Phe142 of the hE:THF complex is a lower bound because of resonance overlap. The average coupling over all complexes is shown as an X, except for residues with different rotamer populations in one or more complexes, which are labeled with an asterisk. Complexes are excluded from the average if the error in the coupling constant is >1 Hz. The standard deviation of 3Javg is shown as a dark gray vertical bar.
Figure 6.
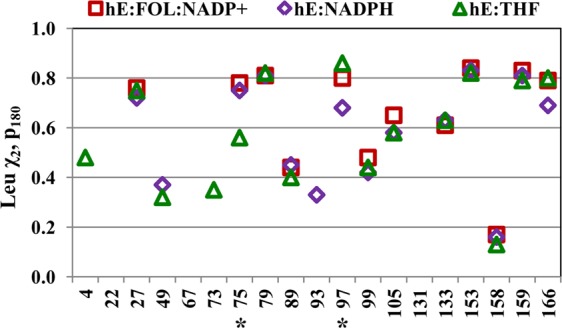
Population of the χ2 = 180° rotamer for Leu residues, determined from the 13Cmethyl chemical shifts. The hE:FOL:NADP+ and hE:THF:NADP+ complexes have essentially identical chemical shifts and therefore similar rotamer populations. The Leu side chains are assumed to populate only the χ2 = 180° or 60° rotamer (eq 4). Asterisks denote residues with a unique average χ2 rotamer conformation in one or more hDHFR complexes.
As was the case for ecDHFR, many residues exhibit the same rotameric averaging in all complexes of hDHFR. These are shown as spheres in Figure 7, colored according to pmajor with a white (pmajor = 1) to red (pmajor < 0.5) gradient. Residues that exhibit rotamer populations that differ in one or more of the hDHFR complexes are colored teal. These “variable population” residues cluster largely in the active site and in regions proximal to the ligand binding pockets. Some residues beyond these zones also exhibit variations in rotamer averaging: of particular note are three residues (Tyr156, His127, and Leu97) proximal to helix αF, which slides ∼2.5 Å between the hinge-closed and hinge-open states of the protein. Rotamer populations determined using the three-site staggered rotamer assumption are indicated for all residues, but as mentioned in Methods, this interpretation may not be appropriate for some well-packed aromatic residues, for which the 3J couplings are better interpreted as reporting on local motions about the major χ1 rotamer instead of rotamer hopping.12
Figure 7.

Average χ1 rotamer conformation for the complexes of hDHFR shown on the hE:FOL:NADP+ structure. Ile, Thr, Val, Leu, and aromatic side chains are shown as spheres, when the pmajor could be determined. Each quadrant (clockwise from the top left) is rotated 90° into the page. Residues with a unique rotamer conformation in one or more complexes of hDHFR are colored and labeled in teal. Residues showing the same average χ1 rotamer conformation in each hDHFR complex are labeled in red and colored with a white (pmajor = 1) to red (pmajor < 0.5) gradient.
Discussion
As Dunbrack and co-workers have shown, the backbone dihedral angles are predictive of the primary χ1 rotamer sampled by side chains.36 Indeed, the major rotamers for the hDHFR complexes in solution are consistent with the favored rotamer based on the X-ray backbone dihedrals for that complex (Figure S5 of the Supporting Information). 3J couplings and chemical shift values can provide information about the distributions of rotamer populations sampled in solution, in terms of both staggered rotamer hopping and in-well rotamer averaging. Analysis of these rotamer distributions in the intermediate complexes of the hDHFR enzymatic cycle provides insights into the structural variation hDHFR undergoes during catalysis.
Comparison of Rotamer Averaging in the hDHFR and ecDHFR Michaelis Model Complexes
Rotamer populations were previously determined for the complexes representing the intermediates in the E. coli DHFR catalytic cycle.12 Many residues in the active sites of hDHFR and ecDHFR are conserved. These are colored green in Figure 8A if the side chains are identical or cyan if the residue is conserved (Leu, Ile, or Val in both ecDHFR and hDHFR, or aromatic in both). A comparison of the 3J couplings for the identical methyl and aromatic residues indicates that the average χ1 conformation is essentially the same in the human and E. coli enzymes (Figure 8B). The residues showing the greatest deviation are Val50 and Tyr121 (Val40 and Tyr100 in ecDHFR), which are sensitive to the bound ligands.
Figure 8.
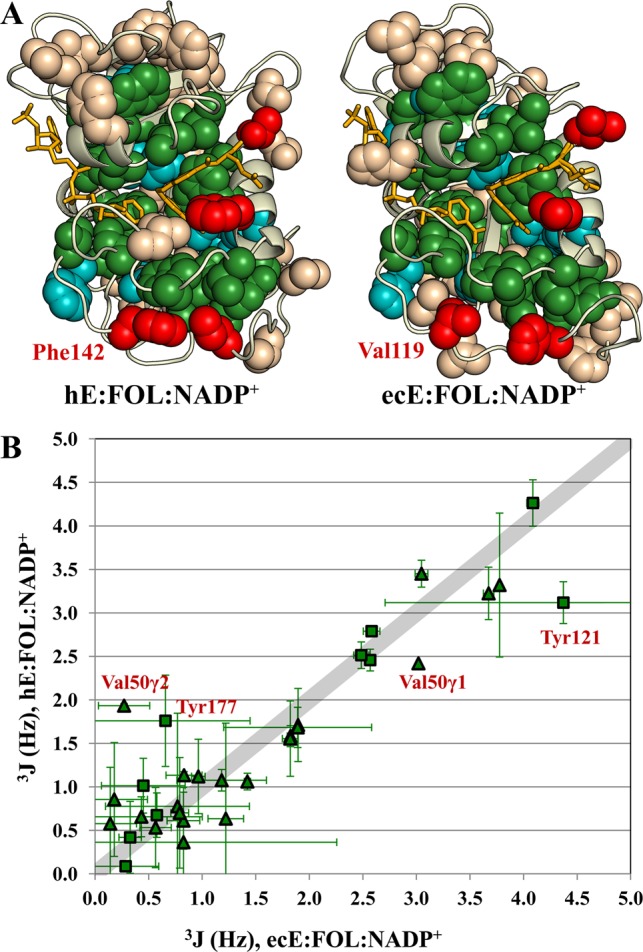
Identical residues in human and E. coli DHFR show remarkably similar average χ1 rotamer conformations. (A) Spheres are shown for Ile, Thr, Val, Leu, and aromatic side chains on the structures of the human and E. coli E:FOL:NADP+ complexes. Identical residues are colored green. Conserved residues (Leu, Val, and Ile in both species or aromatic in both species) are colored cyan. Methyl (aromatic) residues that are aromatic (methyl) residues in the other species are colored red. (B) 3JCγCO and 3JCγN couplings for the identical residues (colored green in panel A) are plotted for the ecE:FOL:NADP+ complex vs the hE:FOL:NADP+ complex. Triangles indicate methyl residues; squares indicate aromatic residues. Val50 and Tyr121, which deviate the most from the line with a slope of 1 (broad gray line), are sensitive to the bound ligands in both human and E. coli DHFR.
Differences in rotameric averaging are observed for those residues that are not conserved between hDHFR and ecDHFR (termed nonequivalent residues). In situations where an aromatic residue has been substituted for a methyl residue (red residues in Figure 8A), the aromatic residue shows a decrease in rotamer averaging relative to the methyl residue. An example of this is a residue at the interface of the Met20 and FG loops, Phe142 in hDHFR, which is replaced by Val119 in ecDHFR. Val119 displays a very high level of rotamer averaging in the occluded, but not the closed, complexes of ecDHFR. In hDHFR, which has a closed Met20 loop throughout the catalytic cycle, Phe142 is predominantly in the χ1 = −60° rotamer conformation in all complexes. The nonequivalent Ile, Thr, and Val residues of hDHFR show an enhanced degree of rotameric averaging compared to that of the residues of ecDHFR, with an average pmajor of 0.68 ± 0.18 in hDHFR versus a value of 0.90 ± 0.07 in ecDHFR for the E:FOL:NADP+ complex (see Figure S6 of the Supporting Information for histograms of the pmajor distributions for hDHFR and ecDHFR E:FOL:NADP+ complexes). Many of the residues with enhanced rotamer averaging in hDHFR are located in loop regions that represent insertions into the core secondary structure shared by the human and E. coli enzymes. This is particularly apparent in the hDHFR hinge regions, where increased rotamer averaging of small side chains along domain interfaces may facilitate the quasi-rigid body movements seen in the hinge-open versus hinge-closed states.
Rotamer Averaging in the hDHFR Catalytic Intermediates
Residues showing variable rotamer averaging in one or more complexes of hDHFR (teal residues in Figure 7) are located predominantly in the adenosine binding subdomain (top half of each structure). Unlike ecDHFR, which exhibits variable rotamer averaging for numerous residues in the loop subdomain,12 hDHFR, in which the Met20 loop remains closed throughout the catalytic cycle, shows similar levels of rotamer averaging in the loop subdomain for all complexes (Figure 7). The major exception is Phe31 on helix αC, which has a different primary rotamer in the hE:NADPH complex and in the substrate/analogue-bound complexes. Many of the residues with variable rotamer averaging in the hDHFR complexes appear to be sensitive to variations in the position of helix αF, which is adjacent to hinge 2 (Figure 2). In contrast, the methyl side chains in and around the hinge 1 region show similar, elevated levels of rotamer averaging in each of the hDHFR complexes.
The hE:FOL:NADP+ Complex Compared to the hE:THF:NADP+ Complex
The 15N HSQC and 13C HSQC spectra for the hE:THF:NADP+ and hE:FOL:NADP+ complexes are virtually identical, indicating that the bound FOL and THF interact in very similar manner with the protein backbone and side chains in the ternary complexes (Figure S7 of the Supporting Information). This is in marked contrast to the case for E. coli DHFR, where substantial changes in chemical shift are observed between FOL and THF complexes due to different interactions between the protein and the pterin ring; the protonated N8 atom of the pterin ring in THF (and DHF) forms a hydrogen bond with the carbonyl of Ile5, an interaction that cannot be made by FOL.7,11 It is interesting that these differences between the human and E. coli enzymes are paralleled in the binding affinities. FOL, DHF, and THF all bind hDHFR with similar affinities.14 In contrast, ecDHFR binds FOL, DHF, and THF incrementally more tightly, with ∼1 order of magnitude difference in affinity between each successive ligand.39
Methyl 13C chemical shifts are very sensitive to rotamer populations.32,40−42 The 13C HSQC spectra of the hE:FOL:NADP+ and hE:THF:NADP+ complexes are very similar, indicating that the distributions of rotamers for these complexes are virtually indistinguishable. This is also confirmed by comparison of 3JCγCO and 3JCγN coupling constants. On the basis of the 3J couplings, Val8 is an apparent exception; however, the large uncertainties in the small 3J values for this residue make the rotamer analysis unreliable, and methyl 13C chemical sifts suggest that the rotamer population is similar (predominantly χ1 = 60°) in all complexes.
Average χ1 Conformation in the hE:NADPH Complex
As shown in Figure 3, many backbone and side chain resonances of the hE:NADPH complex, generally associated with residues in the substrate binding pocket and the cofactor binding site, exhibit different chemical shifts than in the ternary complexes. The changes in the average χ1 conformation also span these regions. Unlike the ecE:NADPH complex, which shows a widespread increase in χ1 rotameric averaging throughout the complex compared to the ecE:FOL:NADP+ complex, several residues in the hE:NADPH complex show decreased rotamer averaging relative to that of the ternary complexes. This is the case for Tyr156, which has an average χ1 conformation that depends on the conformation of helix αF, which will be discussed in detail below. Val50 also shows decreased rotamer averaging in the hE:NADPH complex compared to that of each of the other complexes. This is likely to be a consequence of the hinge-open conformation of the hE:NADPH complex compared to the hinge-closed conformation of each of the other complexes. Phe31 adopts a unique χ1 conformation in the hE:NADPH complex compared to the conformations in the other complexes, as will be discussed below. Several residues do show increased rotamer averaging in the hE:NADPH complex compared to the rotamer averaging in the other hDHFR complexes. The reduced 3JCγN and 3JCγCO coupling constants of Trp57 and Phe58, respectively, in the hE:NADPH complex compared to those of the hinge-closed complexes (Tables S7–S11 of the Supporting Information) suggest an increase in in-well χ1 rotamer averaging of these residues. Ile114 also shows an increase in rotamer averaging. Ile114 and Val115 are analogous to Val93 and Ile94, respectively, of ecDHFR. In ecDHFR, Ile94, with its side chain oriented toward the substrate, was found to be especially sensitive to the nature of the bound ligand in the substrate binding pocket.12 Val115, which is oriented toward the substrate in hDHFR, does not appear to have the sensitivity to substrate that the larger Ile side chain in ecDHFR does. Instead, Ile114 of hDHFR faces away from the substrate binding pocket and toward helix αF and shows elevated rotamer averaging in the binary complexes. It is not clear if this is a consequence of changes in the position of helix αF, which appears to drive the rotamer averaging in numerous other residues, or if it is an effect propagated from the substrate binding site.
Average χ1 Conformation in the hE:THF Complex
As in the hE:NADPH complex, several residues in the hE:THF complex are sensitive to the conformation of helix αF, discussed below. Unlike the hE:NADPH complex, most of the chemical shift differences in the hE:THF complex relative to the ternary complexes are associated with residues clustered near the vacant cofactor binding site (Figure 3). The absence of NADP/H leads to an increase in rotamer averaging for Leu75. Ile60, Ile114, and Val115 also show increased rotamer averaging relative to the ternary complexes. Ile114 shows elevated rotamer averaging that is similar to that of the hE:NADPH complex. In the hE:THF complex, the methyl groups of Ile60 and Val115 show significant chemical shift changes relative to those of the hE:FOL:NADP+ complex, which is consistent with a change in rotamer averaging. Increased averaging of these residues, which are both near the bound substrate, may be a consequence of their position along the trajectory taken by helix αF as it slides from one position to the other.
Phe31 Is a Substrate and Product Gatekeeper
It has been suggested on the basis of X-ray structures and molecular dynamics simulations that Phe31, located on helix αC following the Met20 loop, functions as a gatekeeper residue that controls access to the active site.15,43,44 In solution, the 3J coupling constants show that the Phe31 side chain is fully in the χ1 = −60° rotamer when the substrate or product is bound and fully occupies the χ1 = 180° rotamer in the hE:NADPH complex, where the substrate/product binding pocket is empty (Figure 9). The combination of hinge opening and rotation of Phe31 into the χ1 = 180° rotamer in the hE:NADPH holoenzyme opens the active site to facilitate entry of the substrate. However, once bound, the p-aminobenzoylglutamate (pABG) moiety of the substrate would clash sterically with the phenyl ring,15,43 triggering the conformational transition to the −60° rotameric state of Phe31. In the −60° rotamer, the phenyl ring is packed tightly against the pterin ring of the substrate and may contribute to transition state stabilization, because the hydride transfer rate of Phe31 mutants is significantly decreased.45 Indeed, when the equivalent residue at this site in ecDHFR, Leu28, is mutated to phenylalanine, the hydride transfer rate is greatly enhanced, emphasizing the importance of this residue in active site organization.46 Product release requires the reverse transition, in which the conformation of the Phe31 side chain changes from the χ1 = −60° rotamer to the χ1 = 180° rotamer of the holoenzyme. It is notable that the energetics are such that Phe31 fully populates the −60° or 180° rotamer in the various catalytic intermediates, and we see no evidence of rotamer averaging in any of the complexes at the level of uncertainty of our measurements.
Figure 9.
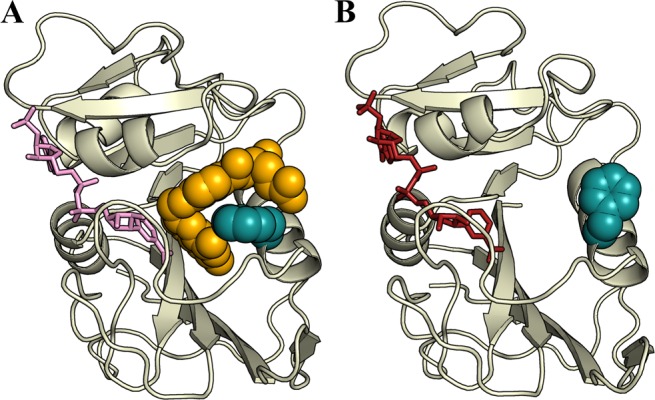
Phe31 acts as a gatekeeper for the substrate and product. (A) Phe31 (blue spheres) is fully in the χ1 = −60° rotamer when THF or FOL (yellow spheres) is bound, packing tightly against the substrate and pseudosubstrate. (B) In the hE:NADPH complex, with the vacant substrate binding pocket, Phe31 is fully in the χ1 = 180° rotamer, with the side chain packing against helix αC, leaving the substrate pocket accessible.
Role of Side Chain Disorder in the Movement of Helix αF
The primary conformational change that hDHFR undergoes as it progresses through the catalytic cycle is between a hinge-closed state, in complexes where the substrate or product is bound, and a hinge-open state, formed when the substrate or product binding pocket is vacant, i.e., in the E:NADPH complex. One of the structural consequences of this is a change in the location of helix αF, which is adjacent to hinge 2. In undergoing the transition from the hinge-open state to the hinge-closed state, helix αF slides ∼2.5 Å toward the active site (Figure 2B).15 In the hinge-closed state, helix αF is restrained by a pair of hydrogen bonds involving the side chains of His127 and Tyr156. Tyr156, which is on the β-sheet of the loop subdomain, forms a hydrogen bond from its hydroxyl group to the backbone CO group of Met125 in the hinge-closed complexes but not in the hinge-open complexes, because the shift in helix αF in the hinge-open conformation increases the distance between these atoms from 2.6 to 4.3 Å (Figure 10A,B). In the hE:THF:NADP+ and hE:FOL:NADP+ complexes, the 3JCγCO value is reduced relative to those of the binary complexes, suggesting that Tyr156 in the ternary complexes adopts a more skewed χ1 rotamer to form the hydrogen bond or that the side chain undergoes more in-well rotamer averaging while tethered to the Met125 backbone.
Figure 10.

Role of rotamer averaging in conformational changes associated with hinge motions. (A) Location of helix αF in the X-ray structure of the hE:FOL:NADP+ complex (PDB entry 4M6K).15 Helix αF is stabilized in its hinge-closed conformation by hydrogen bonds, shown by the black dashed lines. (B) X-ray structure of the hE:NADPH complex (PDB entry 4M6J), showing movement of helix αF toward the active site and breaking of the restraining hydrogen bonds upon opening of the hinges.15 Leu97 undergoes increased rotamer averaging in the hE:NADPH complex. Helix αF is colored red in panels A and B, and side chains are shown as sticks. NADP/H is colored cyan and atom colors and folate yellow and atom colors. (C) Superposition of the structures of the hE:NADPH (green) and hE:FOL:NADP+ (pink) complexes, showing conformational changes associated with sliding of helix αF and at the C-terminus of helix αE. Methyl-containing side chains (in PDB entry 4M6J) that undergo rotamer averaging in the hE:NADPH complex are shown as spheres: red for side chains on or in contact with helix αF, blue for side chains that pack against the C-terminal region of helix αE, pink for side chains in hinge 1, and gray for all other methyl-containing side chains with rotamer averaging. Leu133 also exhibits rotamer averaging in the crystal structure of the hE:NADPH complex. The NADPH in PDB entry 4M6J is colored cyan and atom colors; the folate and NADP+ ligands have been omitted from the structure of the hE:FOL:NADP+ complex for the sake of clarity.
His127 is located in hinge 2, at the C-terminus of helix αF. In the hinge-closed conformation, His127 Nε2 is close enough to Asp94 COδ to form a hydrogen bond, which cannot be formed in the hinge-open hE:NADPH complex (Figure 10A,B). The 3JCγN coupling constants for His127 in the ternary complexes and the hE:NADPH complex indicate a fully populated χ1 = 180° rotamer, in accord with the X-ray structures. The 3JCγN value is decreased in the E:THF complex, consistent with the skewed rotamer (χ1 = 209°) observed in the crystal structure of the E:FOL complex (PDB entry 1DHF). Leu97 is close in space to His127. In the hinge-closed complexes, the Leu97 Cδ1 methyl is next to the face of the His127 aromatic ring (Figure 10A) and is subject to a ring current shift; in the hinge-open hE:NADPH complex, Leu97 Cδ1 moves away from the face of the histidine ring (Figure 10B). The 13Cmethyl chemical shifts of Leu97, corrected for the ring current contributions from His127, indicate increased χ2 rotamer averaging of the Leu97 side chain in the hE:NADPH complex compared to that of either of the ternary complexes or the hE:THF complex (Table S12 of the Supporting Information). In each of the hDHFR complexes, the space is available to Leu97 to sample multiple rotamer conformations and the backbone dihedrals would support either rotamer (Figure S5 of the Supporting Information), but the chemical shifts suggest a very low level of rotamer averaging for the hinge-closed complexes. The χ2 = 60° conformation would cause the hydrophobic methyl groups to be substantially solvent-exposed, such that the better packed χ2 = 180° rotamer is substantially preferred. This suggests that in the hE:NADPH hinge-open state that the χ2 = 60° rotamer of Leu97 is more protected from solvent than in the hinge-closed complexes, primarily as a consequence of the changed conformation of the adjacent αF helix.
The methyl-containing side chains that undergo rotamer averaging in the hinge-open hE:NADPH complex cluster in functionally important regions (Figure 10C). One such cluster (colored red in Figure 10C) includes Leu75, Leu93, Leu97, Ile114, and Leu133, all of which are in contact with helix αF; Val120 on helix αF also displays rotamer disorder. Dynamic disorder in these side chains may well play a direct functional role by facilitating sliding of helix αF during the hinge-open to hinge-closed transition. A second cluster [Leu49, Leu99, Thr100, Leu105, Val109, and Val112 (colored blue in Figure 10C)] is located at the packing interface between the C-terminal end of helix αE and the β-sheet of the adenosine-binding subdomain, a region that contacts hinge 1 and undergoes structural rearrangement during opening or closing of the hinges. Several side chains in hinge 1 (colored pink) also exhibit rotamer averaging. Thus, it appears that the core of the adenosine-binding subdomain has intrinsic side chain flexibility that may function to “lubricate” the movement of secondary structure elements during hinge transitions. Most of these residues also undergo rotamer averaging in the hinge-closed complexes (hE:FOL:NADP+, hE:THF:NADP+, and hE:THF), showing that the flexibility is an intrinsic property of the adenosine-binding subdomain and is not influenced by the nature of the bound ligands. In contrast to the adenosine-binding domain, relatively few residues in the loop subdomain exhibit side chain disorder. Rotamer averaging is observed for Val135 and Ile151, which pack against Leu133 and may help accommodate its motions.
Conclusions
Conformational and dynamics data for a wide range of time scales have been collected for side chain residues of various complexes of human DHFR that represent intermediates in the catalytic cycle. Consistent with what has been found for the backbone,15 there is no evidence of millisecond time scale motions of the side chains in hDHFR complexes. This occurs either because there are exchange processes that are too fast to detect with CPMG relaxation dispersion experiments or because any exchange processes present do not cause significant changes in the chemical shift. Backbone R1ρ dispersion experiments show microsecond exchange processes in the hE:FOL:NADP+ complex,15 suggesting that similarly fast dynamics may be present for hDHFR side chains.
In contrast to ecDHFR, which undergoes transitions between closed and occluded active site conformations that influence side chain rotamer populations in the various catalytic intermediates,12 hDHFR remains in the closed conformation in all complexes. As a consequence, differences in side chain χ1 and/or χ2 rotamer populations and averaging between the various hDHFR complexes are relatively small and are influenced more by the presence or absence of ligands, rather than the conformational change. For hDHFR, the only substantial structural change is associated with a subdomain rotation that opens the active site cleft and causes helix αF to slide relative to neighboring secondary structural elements; most of the residues that pack against this helix exhibit rotamer disorder in all complexes studied and may lubricate the movement of the helix.
These data paint a picture of hDHFR as an enzyme that requires minimal conformational rearrangement as it proceeds through its enzymatic cycle. The hinge-open or hinge-closed state of the adenosine-binding subdomain and the side chain conformation of Phe31 appear to contribute in important ways to ligand flux and active site packing in human DHFR. However, we have thus far found no evidence of transient sampling of higher-energy conformational substates in the catalytic intermediates of hDHFR that may facilitate progression through the reaction cycle. This is in marked contrast to the E. coli enzyme, in which millisecond time scale fluctuations play a major role in modulating the energy landscape and regulating ligand flux.8 Backbone motions may be present and necessary for hDHFR activity but have not been detected by the experiments thus far performed, which probe only millisecond time scale fluctuations. On the basis of our observation of pervasive backbone motions on the microsecond time scale in human DHFR,15 it seems highly likely that catalytically relevant motions in the human enzyme are much faster than in E. coli DHFR. Characterization of these motions will be the subject of future investigations of human DHFR complexes.
Acknowledgments
We thank Gerard Kroon for his assistance with NMR experiments and Maria Martinez-Yamout and Gira Bhabha for helpful discussion and assistance in sample preparation.
Glossary
Abbreviations
- hDHFR or ecDHFR
human or E. coli dihydrofolate reductase, respectively
- NADP+
nicotinamide adenine dinucleotide phosphate
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- DHF
7,8-dihydrofolate
- THF
(6S)-5,6,7,8-tetrahydrofolate
- FOL
folate
- MTX
methotrexate
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single-quantum correlation.
Supporting Information Available
Tables showing methyl chemical shift assignments and complete rotamer populations and 3JCγCO and 3JCγN coupling constants for each of the complexes and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Chemical shift assignments and values for the 3JCγN and 3JCγCO coupling constants have been deposited in the BioMagResBank as accession numbers 19567 (hE:FOL:NADP+), 19566 (hE:NADPH), 19565 (hE:THF:NADP+), 19564 (hE:THF), and 19563 (hE:FOL).
The authors declare no competing financial interest.
This work was supported by National Institutes of Health Grant GM75995.
Funding Statement
National Institutes of Health, United States
Footnotes
hDHFR does not contain a Met at position 20, but for the sake of being consistent with the ecDHFR terminology, we refer to the region of residues 12–27 of hDHFR as the Met20 loop. Note also that the residue numbering in hDHFR starts from Met0 to be consistent with the numbering in X-ray structures.
Supplementary Material
References
- Falzone C. J.; Wright P. E.; Benkovic S. J. (1994) Dynamics of a flexible loop in dihydrofolate reductase from Escherichia coli and its implication for catalysis. Biochemistry 33, 439–442. [DOI] [PubMed] [Google Scholar]
- Epstein D. M.; Benkovic S. J.; Wright P. E. (1995) Dynamics of the dihydrofolate reductase folate complex: Catalytic sites and regions known to undergo conformational change exhibit diverse dynamical features. Biochemistry 34, 11037–11048. [DOI] [PubMed] [Google Scholar]
- Osborne M. J.; Schnell J.; Benkovic S. J.; Dyson H. J.; Wright P. E. (2001) Backbone dynamics in dihydrofolate reductase complexes: Role of loop flexibility in the catalytic mechanism. Biochemistry 40, 9846–9859. [DOI] [PubMed] [Google Scholar]
- Schnell J. R.; Dyson H. J.; Wright P. E. (2004) Effect of cofactor binding and loop conformation on side chain methyl dynamics in dihydrofolate reductase. Biochemistry 43, 374–383. [DOI] [PubMed] [Google Scholar]
- Schnell J. R.; Dyson H. J.; Wright P. E. (2004) Structure, dynamics and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 33, 119–140. [DOI] [PubMed] [Google Scholar]
- McElheny D.; Schnell J. R.; Lansing J. C.; Dyson H. J.; Wright P. E. (2005) Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. U.S.A. 102, 5032–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitakrishnan R. P.; Zaborowski E.; McElheny D.; Benkovic S. J.; Dyson H. J.; Wright P. E. (2004) Conformational changes in the active site loops of dihydrofolate reductase during the catalytic cycle. Biochemistry 43, 16046–16055. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. (2006) The dynamic energy landscape of dihydrofolate reductase catalysis. Science 313, 1638–1642. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; Dyson H. J.; Wright P. E. (2006) An NMR perspective on enzyme dynamics. Chem. Rev. 106, 3055–3079. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; Dyson H. J.; Wright P. E. (2008) Conformational relaxation following hydride transfer plays a limiting role in dihydrofolate reductase catalysis. Biochemistry 47, 9227–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. (2010) Millisecond timescale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proc. Natl. Acad. Sci. U.S.A. 107, 1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle L. M.; Dyson H. J.; Wright P. E. (2013) Side-chain conformational heterogeneity of intermediates in the Escherichia coli dihydrofolate reductase catalytic cycle. Biochemistry 52, 3464–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; Schnell J. R.; McElheny D.; Bae S. H.; Duggan B. M.; Benkovic S. J.; Dyson H. J.; Wright P. E. (2013) A distal mutation perturbs dynamic amino acid networks in dihydrofolate reductase. Biochemistry 52, 4605–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleman J. R.; Beard W. A.; Delcamp T. J.; Prendergast N. J.; Freisheim J. H.; Blakley R. L. (1990) Unusual transient- and steady-state kinetic behavior is predicted by the kinetic scheme operational for recombinant human dihydrofolate reductase. J. Biol. Chem. 265, 2740–2748. [PubMed] [Google Scholar]
- Bhabha G.; Ekiert D. C.; Jennewein M.; Zmasek C. M.; Tuttle L. M.; Kroon G.; Dyson H. J.; Godzik A.; Wilson I. A.; Wright P. E. (2013) Divergent evolution of enzyme dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 20, 1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Kraut J. (1997) Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 36, 586–603. [DOI] [PubMed] [Google Scholar]
- Bhabha G.; Tuttle L.; Martinez-Yamout M. A.; Wright P. E. (2011) Identification of endogenous ligands bound to bacterially expressed human and E. coli dihydrofolate reductase by 2D NMR. FEBS Lett. 585, 3528–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundström P.; Teilum K.; Carstensen T.; Bezsonova I.; Wiesner S.; Hansen D. F.; Religa T. L.; Akke M.; Kay L. E. (2007) Fractional 13C enrichment of isolated carbons using [1-13C]- or [2- 13C]-glucose facilitates the accurate measurement of dynamics at backbone Cα and side-chain methyl positions in proteins. J. Biomol. NMR 38, 199–212. [DOI] [PubMed] [Google Scholar]
- Neri D.; Szyperski T.; Otting G.; Senn H.; Wüthrich K. (1989) Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry 28, 7510–7516. [DOI] [PubMed] [Google Scholar]
- Kay L. E.; Xu G.-Y.; Singer A. U.; Muhandiram D. R.; Forman-Kay J. D. (1993) A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson., Ser. B 101, 333–337. [Google Scholar]
- Marion D.; Driscoll P. C.; Kay L. E.; Wingfield P. T.; Bax A.; Gronenborn A. M.; Clore G. M. (1989) Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: Application to Interleukin 1β. Biochemistry 28, 6150–6156. [DOI] [PubMed] [Google Scholar]
- Otten R.; Chu B.; Krewulak K. D.; Vogel H. J.; Mulder F. A. A. (2010) Comprehensive and cost-effective NMR spectroscopy of methyl groups in large proteins. J. Am. Chem. Soc. 132, 2952–2960. [DOI] [PubMed] [Google Scholar]
- Grzesiek S.; Döbeli H.; Gentz R.; Garotta G.; Labhardt A. M.; Bax A. (1992) 1H, 13C, and 15N NMR backbone assignments and secondary structure of human interferon-γ. Biochemistry 31, 8180–8190. [DOI] [PubMed] [Google Scholar]
- Skrynnikov N. R.; Mulder F. A. A.; Hon B.; Dahlquist F. W.; Kay L. E. (2001) Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: Application to methionine residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 123, 4556–4566. [DOI] [PubMed] [Google Scholar]
- Lundström P.; Vallurupalli P.; Religa T. L.; Dahlquist F. W.; Kay L. E. (2007) A single-quantum methyl 13C-relaxation dispersion experiment with improved sensitivity. J. Biomol. NMR 38, 79–88. [DOI] [PubMed] [Google Scholar]
- Sugase K.; Konuma T.; Lansing J. C.; Wright P. E. (2013) Fast and accurate fitting of relaxation dispersion data using the flexible software package GLOVE. J. Biomol. NMR 56, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S.; Vuister G. W.; Bax A. (1993) A simple and sensitive experiment for measurement of JCC couplings between backbone carbonyl and methyl carbons in isotopically enriched proteins. J. Biomol. NMR 3, 487–493. [DOI] [PubMed] [Google Scholar]
- Vuister G. W.; Wang A. C.; Bax A. (1993) Measurement of three-bond nitrogen-carbon J couplings in proteins uniformly enriched in 15N and 13C. J. Am. Chem. Soc. 115, 5334–5335. [Google Scholar]
- Hu J. S.; Grzesiek S.; Bax A. (1997) Two-dimensional NMR methods for determining χ1 angles of aromatic residues in proteins from three-bond JC′Cγ and JNCγ couplings. J. Am. Chem. Soc. 119, 1803–1804. [Google Scholar]
- Hennig M.; Bermel W.; Spencer A.; Dobson C. M.; Smith L. J.; Schwalbe H. (1999) Side-chain conformations in an unfolded protein: χ1 distributions in denatured hen lysozyme determined by heteronuclear 13C, 15N NMR spectroscopy. J. Mol. Biol. 288, 705–723. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Scheurer C.; Brüschweiler R. (2000) Static and dynamic effects on vicinal scalar J couplings in proteins and peptides: A MD/DFT analysis. J. Am. Chem. Soc. 122, 10390–10397. [Google Scholar]
- London R. E.; Wingad B. D.; Mueller G. A. (2008) Dependence of amino acid side chain 13C shifts on dihedral angle: Application to conformational analysis. J. Am. Chem. Soc. 130, 11097–11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder F. A. A. (2009) Leucine side-chain conformation and dynamics in proteins from 13C NMR chemical shifts. ChemBioChem 10, 1477–1479. [DOI] [PubMed] [Google Scholar]
- Grant D. M.; Paul E. G. (1964) Carbon-13 magnetic resonance. II. Chemical shift data for the alkanes. J. Am. Chem. Soc. 86, 2984–2990. [Google Scholar]
- Tonelli A. E. (1978) Calculated γ effects on the 13C-NMR spectra of 3,5,7,9,11,13,15-heptamethylheptadecane stereoisomers and their implications for the conformational characteristics of polypropylene. Macromolecules 11, 565–567. [Google Scholar]
- Shapovalov M. V.; Dunbrack R. L. Jr. (2011) A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19, 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. P.; Case D. A. (2001) Automated prediction of 15N, 13Cα, 13Cβ and 13C′ chemical shifts in proteins using a density functional database. J. Biomol. NMR 21, 321–333. [DOI] [PubMed] [Google Scholar]
- Schnell J. R. (2003) NMR studies of backbone and side-chain dynamics in dihydrofolate reductase, pp 1–200, The Scripps Research Institute, La Jolla, CA. [Google Scholar]
- Fierke C. A.; Johnson K. A.; Benkovic S. J. (1987) Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 26, 4085–4092. [DOI] [PubMed] [Google Scholar]
- Hansen D. F.; Neudecker P.; Kay L. E. (2010) Determination of isoleucine side-chain conformations in ground and excited states of proteins from chemical shifts. J. Am. Chem. Soc. 132, 7589–7591. [DOI] [PubMed] [Google Scholar]
- Hansen D. F.; Neudecker P.; Vallurupalli P.; Mulder F. A. A.; Kay L. E. (2010) Determination of Leu side-chain conformations in excited protein states by NMR relaxation dispersion. J. Am. Chem. Soc. 132, 42–43. [DOI] [PubMed] [Google Scholar]
- Hansen D. F.; Kay L. E. (2011) Determining valine side-chain rotamer conformations in proteins from methyl 13C chemical shifts: Application to the 360 kDa half-proteasome. J. Am. Chem. Soc. 133, 8272–8281. [DOI] [PubMed] [Google Scholar]
- McTigue M. A.; Davies J. F. I.; Kraut J. (1992) Crystal structure of chicken liver dihydrofolate reductase complexed with NADP+ and biopterin. Biochemistry 31, 7264–7273. [DOI] [PubMed] [Google Scholar]
- Shrimpton P.; Mullaney A.; Allemann R. K. (2003) Functional role for Tyr 31 in the catalytic cycle of chicken dihydrofolate reductase. Proteins 51, 216–223. [DOI] [PubMed] [Google Scholar]
- Tsay J.-T.; Appleman J. R.; Beard W. A.; Prendergast N. J.; Delcamp T. J.; Freisheim J. H.; Blakley R. L. (1990) Kinetic investigation of the functional role of phenylalanine-31 of recombinant human dihydrofolate reductase. Biochemistry 29, 6428–6436. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Wagner C. R.; Benkovic S. J. (1994) Nonadditivity of mutational effects at the folate binding site of Escherichia coli dihydrofolate reductase. Biochemistry 33, 11576–11585. [DOI] [PubMed] [Google Scholar]
- Davies J. F. II; Delcamp T. J.; Prendergast N. J.; Ashford V. A.; Freisheim J. H.; Kraut J. (1990) Crystal structures of recombinant human dihydrofolate reductase complexed with folate and 5-deazafolate. Biochemistry 29, 9467–9479. [DOI] [PubMed] [Google Scholar]
- Chou J. J.; Case D. A.; Bax A. (2003) Insights into the mobility of methyl-bearing side chains in proteins from 3JCC and 3JCN couplings. J. Am. Chem. Soc. 125, 8959–8966. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.