

Abstract
The histidine kinase, CheA, couples environmental stimuli to changes in bacterial swimming behavior, converting a sensory signal to a chemical signal in the cytosol via autophosphorylation. The kinase activity is regulated in the platform of chemotaxis signaling complexes formed by CheW, chemoreceptors, and the regulatory domain of CheA. Our previous computational and mutational studies have revealed that two interdomain linkers play important roles in CheA’s enzymatic activity. Of the two linkers, one that connects the dimerization and ATP binding domains is essential for both basal autophosphorylation and activation of the kinase. However, the mechanistic role of this linker remains unclear, given that it is far from the autophosphorylation reaction center (the ATP binding site). Here we investigate how this interdomain linker is coupled to CheA’s enzymatic activity. Using modern nuclear magnetic resonance (NMR) techniques, we find that by interacting with the catalytic domain, the interdomain linker initiates long-range structural and dynamic changes directed toward the catalytic center of the autophosphorylation reaction. Subsequent biochemical assays define the functional relevance of these NMR-based observations. These findings extend our understanding of the chemotaxis signal transduction pathway.
Histidine kinases (HKs) are common components used in signal transduction pathways that regulate diverse responses in bacteria and fungi, including nutrient acquisition, metabolism, virulence, adaptation to the environment, and cell development.1 Their absence in mammalian organisms makes them potential targets for novel antibiotics and fungicides.2−4 Essential for the infection by some prokaryotic pathogens,5−8 bacterial chemotaxis9,10 uses a HK, CheA, to convert the sensory information detected by transmembrane chemoreceptors to the form of a chemical signal (phosphoryl group) in the cytosol. The ligand occupancy of the periplasmic domain of the chemoreceptors modulates the autophosphorylation activity of CheA. CheY, a response regulator, receives the phosphoryl group from CheA. Phosphorylated CheY subsequently interacts with flagellar motors, resulting in the reversal of the rotation of flagella and cell tumbling. Because the net direction of the movement of the bacterium depends upon the switching of the flagellar rotation in response to attractant or repellent gradients, CheA acts as an input–output element that couples sensory information to the swimming behavior of a bacterium.
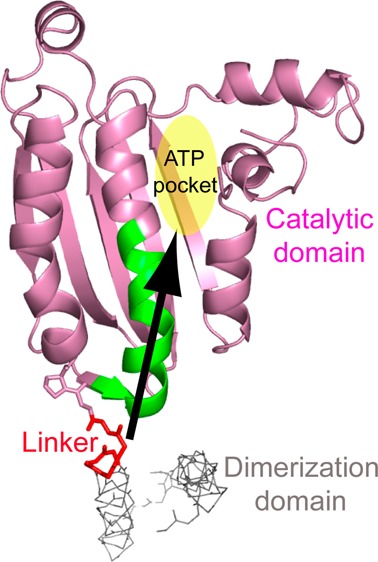
CheA is a dimeric protein with five domains. These include the histidine-containing phosphotransfer domain (P1), the CheY binding domain (P2), the dimerization domain (P3), the ATP binding/catalytic domain (P4), and the regulatory domain (P5) (Figure S1 of the Supporting Information). The P4 domain of one subunit utilizes ATP to transphosphorylate a conserved histidine residue in the P1 domain of the other subunit.11 The P2 domain binds CheY and facilitates the subsequent phosphotransfer from the P1 domain to CheY. Free CheA undergoes basal autophosphorylation at a slow rate. The P5 domain plays no role in basal autophosphorylation but is essential for the regulation of CheA autophosphorylation. The interactions of the P5 domain with chemoreceptors and a small adapter protein, CheW, result in dramatic changes in kinase activity: the rate of autophosphorylation of CheA increases several hundred-fold by the apo or repellent-bound (“kinase-on” state) receptors, while the attractant-bound (“kinase-off” state) receptors suppress CheA autophosphorylation.12−14 Thus, environmental stimuli appear to shift an equilibrium between the kinase-on and kinase-off states in chemoreceptor patches, thereby allosterically modulating net kinase activity and flagellar rotation.10 The P3–P5 domains, connected by two short interdomain linkers, form the kinase core that is able to phosphorylate an isolated P1 domain and supports chemotaxis in vivo when supplied with the P1 domain in trans.15
To elucidate the mechanism by which CheA is regulated in the CheA–CheW–receptor ternary complex, significant progress has been made recently in structural studies of these proteins and their interactions.16−28 These studies depict the architecture of the signaling complex, the platform where CheA is regulated (a structural model of the signaling complex is shown in Figure S2 of the Supporting Information). More recently, our computational and mutational studies have revealed that two interdomain linkers of CheA play important roles in CheA’s enzymatic activity. The P3–P4 linker that connects the dimerization and ATP binding domains is essential for both basal autophosphorylation and activation of the kinase.29 Because the autophosphorylation reaction takes place at the ATP binding site (the autophosphorylation active site) that is ∼25 Å from the P3–P4 linker, the mechanistic role of the linker in affecting the catalytic site is not clear. Here, we investigate this interdomain linker and find that the P3–P4 linker is able to initiate long-range structural and dynamic changes toward the catalytic center of the P4 domain, and the P4 residues that are affected by this linker are crucial for the basal autophosphorylation and activation of CheA.
Materials and Methods
Protein Preparation
Genes encoding P4 (residues 356–540), P4l (residues 346–540), and P3P4 (residues 290–540) of CheA from Thermotoga maritima were subcloned into vector pET22b (Novagen), and the expressed proteins with C-terminal His6 tags were purified using Ni-NTA affinity chromatography (Qiagen). Purified proteins were dialyzed against the buffer containing 50 mM Na2HPO4 (pH7.4) and 20 mM NaCl and concentrated to a final concentration of 0.5–1.5 mM. Strain RP309830 was used to express Escherichia coli proteins. CheA,31 CheW,32 CheY,33 and Tsr-containing membranes12 were isolated and purified following published protocols. All site-directed mutants were generated by QuickChange mutagenesis (Stratagene).
NMR Spectroscopy
NMR data for backbone assignment as well as 1H–15N TROSY-HSQC spectra were collected at 50 °C on a Varian 600 MHz or Bruker 800 MHz spectrometer. Sequential assignments of the P4l resonances were accomplished with a 2H-, 15N-, and 13C-labeled sample using TROSY versions of the triple-resonance HNCACB and HN(CO)CACB experiments.34 Except for some regions [the P3–P4 linker and the ATP lid (residues 490–510)] that appeared to be exchange-broadened, a total of 142 of 154 non-proline backbone residues of the P4 domain were successfully assigned. TROSY versions of the HNCA and HN(CO)CA spectra35 of 2H-, 15N-, and 13C-labeled P3P4 mutants were recorded for assignments. The assignments of the wild-type P3P4 and P4 constructs were previously reported.36
CPMG Relaxation Dispersion Experiments
Relaxation dispersion (CPMG) experiments were performed at 50 °C using an ∼1.5 mM U-[15N,2H], ILV 13CH3 sample on 500 and 800 MHz Varian Inova spectrometers equipped with standard room-temperature triple-resonance probes. 1H CPMG experiments were performed using the relaxation-compensated CPMG pulse sequence37,38 with a constant-time CPMG evolution delay39 of 15 ms, and the 15N CPMG experiments were performed using a TROSY CPMG sequence40,41 with a constant-time relaxation delay of 30 ms. Data were collected at ∼15 different CPMG frequencies in an interleaved manner. Errors in the relaxation rates were estimated from repeat data points. The data were processed using NMRPipe42 and visualized using SPARKY.43 Peak intensities were quantified using FuDA. The CPMG relaxation dispersion data were fit to a two-state model using CATIA (http://pound.med.utoronto.ca/software.html).
ATPase Assay
The basal autophosphorylation rates of CheA variants were measured as previously described.25,44 An Amersham Ultrospec3300 spectrophotometer was used to monitor NADH oxidation at room temperature. The ATP hydrolysis rate was determined by calculating the slope of a linear decay in NADH absorbance at 340 nm using the extinction coefficient (6220 M–1 cm–1) of NADH.
CheA Activation Assay
The activation of CheA by Tsr-containing membranes was quantified according to the published protocols.12,25
Nucleotide Binding Assay
The binding of ATP analogue TNP-ATP to CheA enhanced the fluorescence at 541 nm when the nucleotide was excited at 410 nm. The dissociation constants between CheA variants and the ATP analogue were measured at 25 °C on a Varian Cary Eclipse fluorometer as previously reported by Stewart et al.45 The buffer used in the measurements contained 50 mM Tris-HCl (pH 7.5), 100 mM KCl, and 10 mM MgCl2.
Results
To specifically investigate the role of the P3–P4 linker (residues 351–355), we performed NMR experiments on two P4 constructs from CheA of T. maritima: P4 (residues 356–540) without the P3–P4 linker and P4l (residues 346–540) with the linker residues. Most of our atomic-resolution understanding of bacterial chemotaxis has used proteins from the hyperthermophile T. maritima whose chemotaxis system has components similar to those of mesophilic bacteria.46 The primary structure of the P4 domain of T. maritima CheA is 51% identical and 73% similar to its E. coli counterpart. Figure 1a shows two superimposed 1H–15N TROSY-HSQC (transverse relaxation-optimized and heteronuclear single-quantum correlation spectroscopy)47 spectra taken from these two constructs. The addition of 10 native residues at the N-terminal P4 domain causes some significant changes in the NMR spectrum. The largest chemical shift changes are observed in the resonances of residues D392–T394. These residues are located at the interface between the P3 and P4 domains in the crystal structure of the dimeric P3P4P5 kinase core (Figure 1b). This observation argues that the interaction between the P3–P4 linker and the P4 domain seen in the crystal structure exists in solution rather than being a crystal packing artifact (ref (16), and see below). Interestingly, some other resonances in the NMR spectra show smaller, but significant, chemical shift changes. When mapped onto the structure of the P4 domain, these changes appear to propagate from the linker–P4 interface through the middle helix of P4’s three major α-helices toward the catalytic center of the P4 domain (Figure 1b). The structural difference in this region is not seen when comparing the crystal structures of the P4 construct lacking the native linker and the linker-containing P3P4P5 construct (Figure S4 of the Supporting Information).
Figure 1.
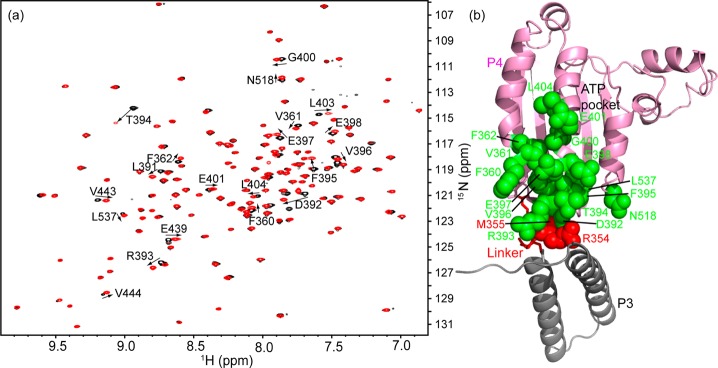
Comparison of the 1H–15N TROSY-HSQC spectra of P4 and P4l. (a) Overlay of the 1H–15N TROSY-HSQC spectra of P4 (black) and P4l (red). Resonances showing significant chemical shift changes upon the addition of the linker are assigned and numbered, and the direction of the peak movement is indicated with an arrow. The assignments of the rest of the resonances are shown in Figure S3 of the Supporting Information. (b) These residues (green) are mapped onto the structure of P3P4P5 (Protein Data Bank entry 1B3Q). The P5 domain has been omitted. The location of the gray P3 domain is shown, but this domain was not included in the constructs from which the spectra in panel A were taken.
The relaxation dispersion (CPMG) experiments we performed on the P4l construct showed that for several amide protons (residues 360, 390–398, 400, 401, 403, and 439) and a few amide nitrogens (residues 391–395, 517, and 518), the transverse relaxation rate (R2,eff) depended on the frequency (νCPMG) at which refocusing π pulses were applied during the relaxation delay (Figure 2 and Figure S5 of the Supporting Information), indicating that there is conformational exchange on the microsecond to millisecond time scale in the protein. Consistently, all of these residues (residues 360, 390–398, 400, 401, 403, 439, 517, and 518) are either in the helix or near it, and all the data could be fit to a single two-state process (reduced χ2 = 1.2) with an exchange rate constant kex of ∼1250 ± 100 s–1. The population of the minor state could not be defined because exchange is fast compared to the change in the chemical shift of the nuclei between the ground and excited states.48 In addition to recording CPMG relaxation dispersion profiles of the P4l construct, we have also recorded data sets using the P4 construct that did not contain the linker. Notably, both 1H and 15N relaxation dispersion profiles were significantly reduced in the P4 construct, as illustrated in Figures S6 and S7 of the Supporting Information. These results indicate that the deletion of the linker strongly damped relaxation in the residues that were microsecond to millisecond dynamic in the linker-containing P4l construct. Thus, the P3–P4 linker affects the dynamics of the residues of the P4 domain.
Figure 2.
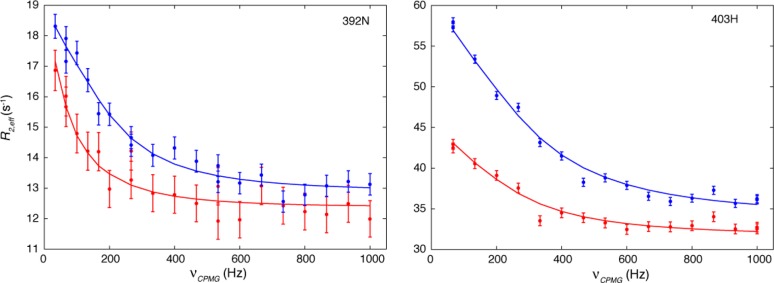
Relaxation dispersion curves show that there is millisecond time scale conformational exchange in P4l. 15N CPMG data for the amide nitrogen of residue 392 are shown in the left panel, and the 1H CPMG data for the amide proton of residue 403 are shown in the right panel. Data recorded at 500 and 800 MHz are shown as red and blue dots, respectively, and the solid lines are the best fits to a global two-state process.
Chemical shift changes with different magnitudes and directions are also seen from the same residues of the P3P4 (residues 290–540) construct,36 when we compare the spectra of the P3P4, P4l, and P4 constructs (Figure 3). This observation suggests that the behavior of the interdomain linker and its effects on the P4 domain are influenced by the presence of the P3 domain. Bilwes et al.17 reported that a linker-containing P4 construct (P4 residues 350–540) had very low kinase activity when compared with a dimeric P3P4P5 construct. The structural differences revealed by NMR may account for such a dramatic difference in enzymatic activity. In addition, chemical mapping studies have shown that some P3 residues are important for kinase autophosphorylation, signaling complex assembly, and CheA activation.18 These results suggest that the state of the P3 domain may affect the conformation of the linker, thereby affecting CheA activity.
Figure 3.
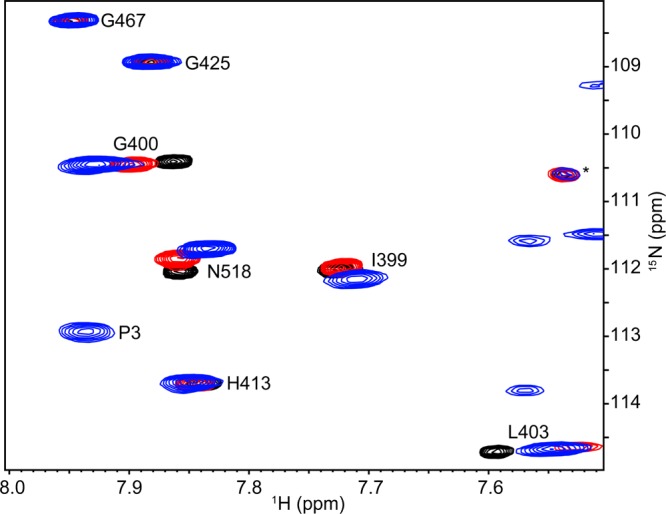
Differences in P4 amide NMR resonances in three constructs. Shown is a representative region of the 1H–15N TROSY-HSQC spectrum of P3P4 (blue) superimposed with the spectra of P4 (black) and P4l (red).
To further assess the structural and dynamic effects of the linker on the P4 domain, we introduced single-amino acid substitutions (R354D and M355A) into the linker of the T. maritima P3P4 construct. The corresponding E. coli CheA mutants (R325D and M326A) displayed different decreases in rates of basal autosphosphorylation (reduced by 95 and 58%, respectively) and activation (reduced by 99 and 91%, respectively) of E. coli CheA.29 The 1H–15N TROSY-HSQC spectrum (Figure S8 of the Supporting Information) shows that the replacement of Arg354 with Asp dramatically changes the dynamics of the same resonances of the P4 domain as well as some resonances of the P3 domain, resulting in missing peaks and significant line broadening. The interdomain linker is in two different conformations in the crystal structure of the P3P4P5 dimer. In one conformation, the interaction between the linker and the P4 domain is a salt bridge formed by the conserved Arg354–Asp392 pair, while the salt bridge is broken in the other conformation (ref (16) and Figure S9 of the Supporting Information). Moreover, despite different linker conformations, the Arg-Asp pairing is seen in all the structures of our previous MD simulations (Figure S10 of the Supporting Information and ref (29)). Thus, we suggest that breakage of the Arg354-Asp392 pair by mutation might give rise to more states that coexist in solution, resulting in the observed resonance broadening. The spectrum of the other mutant, M355A, of P3P4 (Figure S11 of the Supporting Information) also shows that changes in the linker produce small but significant chemical shift changes to the region close to the catalytic center of the P4 domain.
In an attempt to clarify the significance of the NMR data, we mutated some P4 residues whose resonances were perturbed by the P3–P4 linker and tested their activities in E. coli CheA both alone and when activated by assembly into complexes with E. coli transmembrane receptor Tsr and E. coli CheW. Figure 4a shows that single-amino acid substitutions at the linker–P4 interface (residues 363 and 364 of E. coli CheA, corresponding to residues 392 and 393, respectively, of T. maritima CheA) dramatically impair the basal autophosphorylation activity of CheA. Furthermore, we chose to mutate several residues situated along the middle helix of P4’s three major α-helices (residues 368, 369, 372, and 375 of E. coli CheA, corresponding to residues 397, 398, 401, and 404, respectively, of T. maritima CheA). In the crystal structures of T. maritima CheA, these residues are exposed and do not interact with other residues. Thus, these single-amino acid substitutions seem unlikely to cause dramatic changes in the structure. However, these mutants also exhibit severely decreased basal autophosphorylation activity (Figure 4a). In addition, we notice that a cysteine mutant (I388C) of Salmonella typhimurium CheA (corresponding to residue 400 of T. maritima CheA and residue 371 of E. coli CheA) from previous work in the Falke laboratory falls into the same region and showed an ∼70% reduction in basal autophosphorylation activity.49 In the kinase activation assay, these mutants also show impaired activation by the E. coli transmembrane chemoreceptor Tsr (Figure 4b), indicating that these residues are important for CheA activation, as well. Moreover, panels a and b of Figure 4 show that kinase activation appears parallel to kinase basal activity as these amino acid replacements disrupt CheA autophosphorylation activity to relatively the same extent in the basal and activated states. These results echo our previous observation that the P3–P4 linker is related to both basal activity and activation of CheA.29
Figure 4.
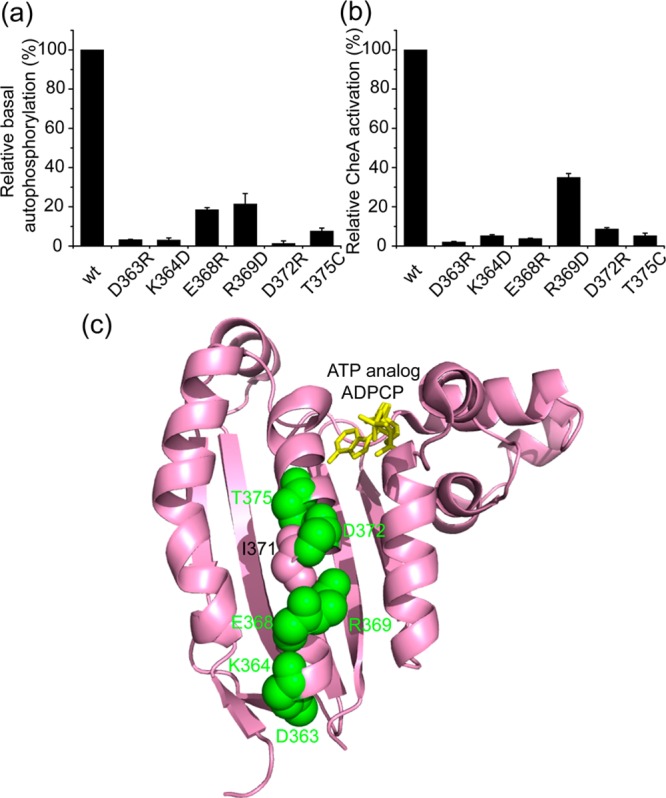
Mutational analyses of the residues affected by the linker in E. coli CheA. (a) ATPase assays (standard deviation; n = 3) for the basal autophosphorylation activities. (b) CheA activation assays (standard deviation; n = 3) for the activation abilities. (c) Mapping the mutation sites onto the structure of the P4 domain.
Crystal structures of the P4 domain in complex with ATP analogues show that the P3–P4 linker and the influenced residues are not involved in nucleotide binding (ref (17) and Figure 4c). Our NMR results also show that the linker does not perturb the chemical shifts of the residues responsible for nucleotide binding (residues 405, 408, 409, 412, 413, 449, 453, 531, etc.) (Figure S3 of the Supporting Information). In addition, we tested the affinity of some of these mutants for the ATP analogue TNP-ATP45 and found that these amino acid replacements did not significantly alter the dissociation constants for the analogue (Table S1 of the Supporting Information). Therefore, these residues may be involved in interactions with the P1 domain and/or activation for ATP hydrolysis.
Discussion
In the previous work from our laboratory,36 some differences in the chemical shifts of the P4 residues were observed when the spectra of the P4 and P3P4 constructs were compared, but the significance of this observation and the physical interaction underlying these chemical shift differences were not clear then. In light of our recent results describing the importance of the interdomain linker,29 we focus on the P3–P4 linker and establish the structural and functional connection between the linker and the catalytic center of the P4 domain. The results from our current NMR and biochemical work indicate that the interdomain linker is able to propagate long-range structural and dynamic changes through the middle helix of the P4 domain to the catalytic site of the P4 domain.
Prior NMR36 and mutational studies18,49 have characterized the interaction between the P1 and P4 domains and identified a group of P4 residues that are important for the binding of the P1 domain to the P4 domain. These residues include some of the residues influenced by the P3–P4 linker. Thus, it seems that the role of the linker and the perturbed P4 residues might be related to the P1–P4 interaction. However, we find that it is imprudent to make such an assignment. Some NMR chemical shifts of the P4 residues perturbed by the P1 interaction might result from unproductive collisions as both disulfide cross-linking49 and pulsed dipolar ESR22 experiments have revealed that the P1 domain is in the vicinity of the P4 domain of the other subunit, rapidly and nonspecifically colliding with the P4 domain. Also, a lack of kinetic data and structural details about the mechanism of CheA autophosphorylation prevents one from distinguishing the involvement of these residues in P1 binding from the catalytic events that lead to autophosphorylation. Recent mutational studies of the P1 domain of CheA50 support a P1–P4 complex model derived from computational docking and molecular dynamics simulations.51 In this model, P1 interacts with some P4 residues surrounding the ATP binding site. More work on the P4 domain may assign the involvement of different groups of P4 residues to either function.
Colleagues in the field have argued that signal transmission along the chemotaxis receptors involves a coupled series of frozen–dynamic transitions from the ligand binding sites, through the HAMP domains, methylation region, and CheA/CheW interaction regions.10,52−54 The structural model of the chemotaxis signaling complex derived from EM images and X-ray structures (Figure S2 of the Supporting Information) describes a scenario in which two trimers of receptor dimers manipulate one CheA dimer with the help of two CheW molecules. In this context, it is possible that the dynamic events in the receptors change the conformation and/or dynamics of the P3–P4 linkers and further affect the kinase active site via the middle helix of the P4 domain.
Glossary
Abbreviations
- NMR
nuclear magnetic resonance
- TROSY
transverse relaxation-optimized spectroscopy
- HSQC
heteronuclear single-quantum correlation spectroscopy
- CPMG
Carr–Purcell–Meiboom–Gill
- HK
histidine kinase
- Ni-NTA
nickel-nitrilotriacetic acid, nickel-charged resin
- Tris
2-amino-2-(hydroxymethyl)propane-1,3-diol
- ESR
electron spin resonance
- TNP-ATP
2′(3′)-O-(trinitrophenyl)adenosine 5′-triphosphate
- HAMP
histidine kinases, adenyl cyclases, methyl-accepting proteins, and phosphatases.
Supporting Information Available
Structural models of CheA (Figure S1) and the chemotaxis signaling complex (Figure S2), additional NMR spectra and assignments (Figures S3, S8, and S11), structural alignment of the P3P4P5 construct against the isolated P4 domain (Figure S4), additional CPMG relaxation dispersion curves (Figure S5), differences in microsecond to millisecond dynamics between the P4 and P4l constructs (Figures S6 and S7), the Arg354-Asp392 pair in the crystal structure and simulation structures of the P3P4P5 construct (Figures S9 and S10), and dissociation constants of CheA variants for TNP-ATP (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by National Institutes of Health Grant GM59544 (to F.W.D.) and National Science Foundation (NSF) Grant MCB-1158577 (to J.-E.S.). Simulations were performed using the XSEDE resources (Grant TG-MCA05S027) supported by the NSF (Grant OCI-1053575). We also acknowledge support from the Center for Scientific Computing from the CNSI, MRL: an MRSEC (DMR-1121053) and NSF CNS-0960316.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Krell T.; Lacal J.; Busch A.; Silva–Jimenez H.; Guazzaroni M.; Ramos J. L. (2010) Bacterial sensor kinases: Diversity in the recognition of environmental signals. Annu. Rev. Microbiol. 64, 539–559. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Okada A.; Gotoh Y.; Utsumi R. (2008) Inhibitors targeting two-component signal transduction. Adv. Exp. Med. Biol. 631, 229–236. [DOI] [PubMed] [Google Scholar]
- Barrett J. F.; Hoch J. A. (1998) Two-component signal transduction as a target for microbial anti-infective therapy. Antimicrob. Agents Chemother. 42, 1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosu M.; Begari E. (2010) Bacterial protein kinase inhibitors. Drug Dev. Res. 71, 168–187. [Google Scholar]
- Josenhans C.; Suerbaum S. (2002) The role of motility as a virulence factor in bacteria. Int. J. Med. Microbiol. 291, 605–614. [DOI] [PubMed] [Google Scholar]
- Lertsethtakarn P.; Ottemann K. M.; Hendrixson D. R. (2011) Motility and chemotaxis in Campylobacter and Helicobacter. Annu. Rev. Microbiol. 65, 389–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler S. M.; Camilli A. (2005) Going against the grain: Chemotaxis and infection in Vibrio cholerae. Nat. Rev. Microbiol. 3, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux R.; Shi W. (2004) Chemotaxis-guided movements in bacteria. Crit. Rev. Oral Biol. Med. 15, 207–220. [DOI] [PubMed] [Google Scholar]
- Wadhams G. H.; Armitage J. P. (2004) Making sense of it all: Bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 5, 1024–1037. [DOI] [PubMed] [Google Scholar]
- Hazelbauer G. L.; Falke J. J.; Parkinson J. S. (2008) Bacterial chemoreceptors: High-performance signaling in networked arrays. Trends Biochem. Sci. 33, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R. V.; Bourret R. B.; Simon M. I. (1993) Intermolecular complementation of the kinase activity of CheA. Mol. Microbiol. 8, 435–441. [DOI] [PubMed] [Google Scholar]
- Borkovich K. A.; Kaplan N.; Hess J. F.; Simon M. I. (1989) Transmembrane signal transduction in bacterial chemotaxis involves ligand-dependent activation of phosphate group transfer. Proc. Natl. Acad. Sci. U.S.A. 86, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegner J. A.; Graham D. R.; Roth A. F.; Dahlquist F. W. (1992) Assembly of an MCP receptor, CheW, and kinase CheA complex in the bacterial chemotaxis signal transduction pathway. Cell 70, 975–982. [DOI] [PubMed] [Google Scholar]
- Bourret R. B.; Davagnino J.; Simon M. I. (1993) The carboxy-terminal portion of the CheA kinase mediates regulation of autophosphorylation by transducer and CheW. J. Bacteriol. 175, 2097–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzón A.; Parkinson J. S. (1996) Chemotactic signaling by the P1 phosphorylation domain liberated from the CheA histidine kinase of Escherichia coli. J. Bacteriol. 178, 6752–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilwes A. M.; Alex L. A.; Crane B. R.; Simon M. I. (1999) Structure of CheA, a signal-transducing histidine kinase. Cell 96, 131–141. [DOI] [PubMed] [Google Scholar]
- Bilwes A. M.; Quezada C. M.; Croal L. R.; Crane B. R.; Simon M. I. (2001) Nucleotide binding by the histidine kinase CheA. Nat. Struct. Biol. 8, 353–360. [DOI] [PubMed] [Google Scholar]
- Miller A. S.; Kohout S. C.; Gilman K. A.; Falke J. J. (2006) CheA kinase of bacterial chemotaxis: Chemical mapping of four essential docking sites. Biochemistry 45, 8699–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis N. R.; Wolanin P. M.; Stock J. B.; Derosier D. J.; Thomas D. R. (2004) Three-dimensional structure and organization of a receptor/signaling complex. Proc. Natl. Acad. Sci. U.S.A. 101, 17480–17485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Parkinson J. S. (2006) Cysteine-scanning analysis of the chemoreceptor-coupling domain of the Escherichia coli chemotaxis signaling kinase CheA. J. Bacteriol. 188, 4321–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. Y.; Borbat P. P.; Gonzalez–Bonet G.; Bhatnagar J.; Pollard A. M.; Freed J. H.; Bilwes A. M.; Crane B. R. (2006) Reconstruction of the chemotaxis receptor-kinase assembly. Nat. Struct. Mol. Biol. 13, 400–407. [DOI] [PubMed] [Google Scholar]
- Bhatnagar J.; Borbat P. P.; Pollard A. M.; Bilwes A. M.; Freed J. H.; Crane B. R. (2010) Structure of the ternary complex formed by a chemotaxis receptor signaling domain, the CheA histidine kinase, and the coupling protein CheW as determined by pulsed dipolar ESR spectroscopy. Biochemistry 49, 3824–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Li X.; Bilwes A. M.; Hughes K. T.; Jensen G. J.; Crane B. R. (2012) Bacterial chemoreceptor arrays are hexagonally packed trimers of receptor dimers networked by rings of kinase and coupling proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 3766–3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu A.; Wang X.; Zhou H.; Dahlquist F. W. (2012) The receptor-CheW binding interface in bacterial chemotaxis. J. Mol. Biol. 415, 759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Vu A.; Lee K.; Dahlquist F. W. (2012) CheA-receptor interaction sites in bacterial chemotaxis. J. Mol. Biol. 422, 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Hu B.; Morado D. R.; Jani S.; Manson M. D.; Margolin W. (2012) Molecular architecture of chemoreceptor arrays revealed by cryoelectron tomography of Escherichia coli minicells. Proc. Natl. Acad. Sci. U.S.A. 109, E1481–E1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Fleetwod A. D.; Bayas C.; Bilwes A. M.; Ortega D. R.; Falke J. J.; Zhulin I. B.; Crane B. R. (2013) The 3.2 Å Resolution Structure of a Receptor:CheA:CheW Signaling Complex Defines Overlapping Binding Sites and Key Residue Interactions within Bacterial Chemosensory Arrays. Biochemistry 52, 3852–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasta K. N.; Ulliman C. J.; Slivka P. F.; Crane B. R.; Falke J. J. (2013) Defining a Key Receptor-CheA Kinase Contact and Elucidating Its Function in the Membrane-Bound Bacterial Chemosensory Array: A Disulfide Mapping and TAM-IDS Study. Biochemistry 52, 3866–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Wu C.; Vu A.; Shea J.-E.; Dahlquist F. W. (2012) Computational and experimental analyses reveal the essential roles of interdomain linkers in the biological function of chemotaxis histidine kinase CheA. J. Am. Chem. Soc. 134, 16107–16110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames P.; Parkinson J. S. (1994) Constitutively signaling fragments of Tsr, the Escherichia coli serine chemoreceptor. J. Bacteriol. 176, 6340–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock A.; Chen T.; Welsh D.; Stock J. (1988) CheA protein, a central regulator of bacterial chemotaxis, belongs to a family of proteins that control gene expression in response to changing environmental conditions. Proc. Natl. Acad. Sci. U.S.A. 85, 1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegner J. A.; Dahlquist F. W. (1991) Signal transduction in bacteria: CheW forms a reversible complex with the protein kinase CheA. Proc. Natl. Acad. Sci. U.S.A. 88, 750–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry D. F.; Roth A. F.; Rupert P. B.; Dahlquist F. W.; Moy F. J.; Domaille P. J.; Matsumura P. (1994) Signal transduction in chemotaxis. A propagating conformation change upon phosphorylation of CheY. J. Biol. Chem. 269, 26358–26362. [PubMed] [Google Scholar]
- Salzmann M.; Wider G.; Pervushin K.; Senn H.; Wüthrich K. (1999) TROSY-type Triple-Resonance Experiments for Sequential NMR Assignments of Large Proteins. J. Am. Chem. Soc. 121, 844–848. [Google Scholar]
- Salzmann M.; Pervushin K.; Wider G.; Senn H.; Wüthrich K. (1998) TROSY in triple-resonance experiments: New perspectives for sequential NMR assignment of large proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 13585–13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel D. J.; Zhou H.; Starich M. R.; Byrd R. A.; Dahlquist F. W. (2006) Chemical-shift-perturbation mapping of the phosphotransfer and catalytic domain interaction in the histidine autokinase CheA from Thermotoga maritima. Biochemistry 45, 9509–9517. [DOI] [PubMed] [Google Scholar]
- Loria J. P.; Rance M.; Palmer A. G. (1999) A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J. Am. Chem. Soc. 121, 2331–2332. [Google Scholar]
- Ishima R.; Wingfield P. T.; Stahl S. J.; Kaufman J. D.; Torchia D. A. (1998) Using amide H-1 and N-15 transverse relaxation to detect millisecond time-scale motions in perdeuterated proteins: Application to HIV-1 protease. J. Am. Chem. Soc. 120, 10534–10542. [Google Scholar]
- Mulder F. A. A.; Skrynnikov N. R.; Hon B.; Dahlquist F. W.; Kay L. E. (2001) Measurement of slow (μs-ms) time scale dynamics in protein side chains by 15N relaxation dispersion NMR spectroscopy: Application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 123, 967–975. [DOI] [PubMed] [Google Scholar]
- Loria J. P.; Rance M.; Palmer A. G. (1999) A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J. Biomol. NMR 15, 151–155. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P.; Hansen D. F.; Stollar E.; Meirovitch E.; Kay L. E. (2007) Measurement of bond vector orientations in invisible excited states of proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 18473–18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A Multidimensional Spectral Processing System Based on Unix Pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Kneller D. G. (2000) SPARKY 3, University of California, San Francisco. [Google Scholar]
- Ninfa E. G.; Stock A.; Mowbray S.; Stock J. (1991) Reconstitution of the bacterial chemotaxis signal transduction system from purified components. J. Biol. Chem. 266, 9764–9770. [PubMed] [Google Scholar]
- Stewart R. C.; VanBruggen R.; Ellefson D. D.; Wolfe A. J. (1998) TNP-ATP and TNP-ADP as probes of the nucleotide binding site of CheA, the histidine protein kinase in the chemotaxis signal transduction pathway of Escherichia coli. Biochemistry 37, 12269–12279. [DOI] [PubMed] [Google Scholar]
- Swanson R. V.; Sanna M. G.; Simon M. I. (1996) Thermostable chemotaxis proteins from the hyperthermophilic bacterium Thermotoga maritima. J. Bacteriol. 178, 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin K.; Riek R.; Wider G.; Wüthrich K. (1997) Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A. G.; Kroenke C. D.; Loria J. P. (2001) Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 339, 204–238. [DOI] [PubMed] [Google Scholar]
- Gloor S. L.; Falke J. J. (2009) Thermal domain motions of CheA kinase in solution: Disulfide trapping reveals the motional constraints leading to trans-autophosphorylation. Biochemistry 48, 3631–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama S.; Garzon A.; Parkinson J. S. (2014) Mutational analysis of the P1 phosphorylation domain in Escherichia coli CheA, the signaling kinase for chemotaxis. J. Bacteriol. 196, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Xu Y.; Shen J.; Luo X.; Chen J.; Chen K.; Zhu W.; Jiang H. (2005) Dynamic mechanism for the autophosphorylation of CheA histidine kinase: Molecular dynamics simulations. J. Am. Chem. Soc. 127, 11709–11719. [DOI] [PubMed] [Google Scholar]
- Kim K. K.; Yokota H.; Kim S.-H. (1999) Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature 400, 787–792. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Ames P.; Parkinson J. S. (2009) Mutational analyses of HAMP helices suggest a dynamic bundle model of input-output signalling in chemoreceptors. Mol. Microbiol. 73, 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain K. E.; Gonzalez M. A.; Falke J. J. (2009) Engineered socket study of signaling through a four-helix bundle: Evidence for a yin-yang mechanism in the kinase control module of the aspartate receptor. Biochemistry 48, 9266–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.