

Abstract
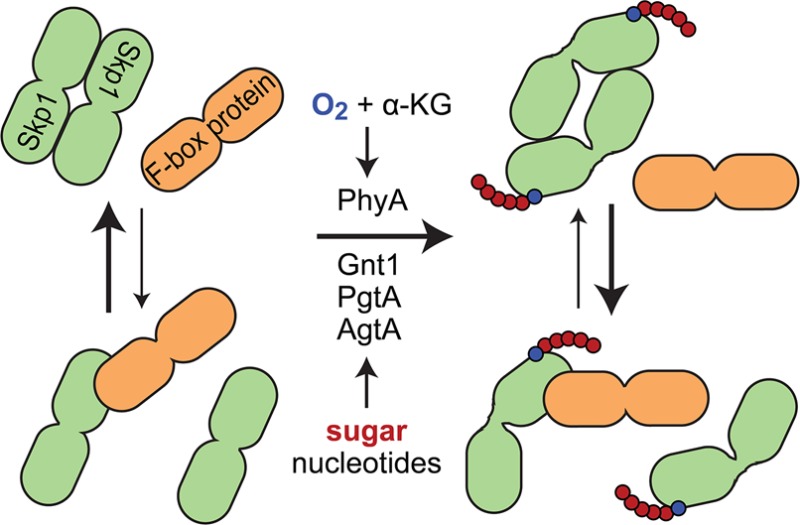
In the social amoeba Dictyostelium, Skp1 is hydroxylated on proline 143 and further modified by three cytosolic glycosyltransferases to yield an O-linked pentasaccharide that contributes to O2 regulation of development. Skp1 is an adapter in the Skp1/cullin1/F-box protein family of E3 ubiquitin ligases that targets specific proteins for polyubiquitination and subsequent proteasomal degradation. To investigate the biochemical consequences of glycosylation, untagged full-length Skp1 and several of its posttranslationally modified isoforms were expressed and purified to near homogeneity using recombinant and in vitro strategies. Interaction studies with the soluble mammalian F-box protein Fbs1/Fbg1/OCP1 revealed preferential binding to the glycosylated isoforms of Skp1. This difference correlated with the increased α-helical and decreased β-sheet content of glycosylated Skp1s based on circular dichroism and increased folding order based on small-angle X-ray scattering. A comparison of the molecular envelopes of fully glycosylated Skp1 and the apoprotein indicated that both isoforms exist as an antiparallel dimer that is more compact and extended in the glycosylated state. Analytical gel filtration and chemical cross-linking studies showed a growing tendency of less modified isoforms to dimerize. Considering that regions of free Skp1 are intrinsically disordered and Skp1 can adopt distinct folds when bound to F-box proteins, we propose that glycosylation, which occurs adjacent to the F-box binding site, influences the spectrum of energetically similar conformations that vary inversely in their propensity to dock with Fbs1 or another Skp1. Glycosylation may thus influence Skp1 function by modulating F-box protein binding in cells.
Skp1 from the social amoeba Dictyostelium is subject to posttranslational modification in the form of prolyl hydroxylation at Pro143 and subsequent glycosylation resulting in, ultimately, assembly of a defined linear pentasaccharide (see Figure 1A).1 Genes for each of the enzymes catalyzing these reactions have been cloned and disrupted, and characterization of the mutants has revealed a role for the modification pathway in O2 regulation of starvation-induced development.2−4 Recent studies have indicated that the Skp1 modification pathway has an O2-sensing role in the regulation of proliferation of the intracellular parasite Toxoplasma gondii.5
Figure 1.

Skp1 isoforms. (A) Skp1 is modified stepwise by the indicated enzymes in Dictyostelium. PhyA is a prolyl 4-trans-hydroxylase, Gnt1 an α-GlcNAc-transferase, PgtA a dual-function β3-galactosyltransferase/α2-fucosyltransferase, and AgtA a dual-function α-galactosyltransferase. The primary structure of the linear Gal-Gal-Fuc-Gal-GalcNAc pentasaccharide appended to Pro143 of Skp1 is represented below the pathway schematic, and the full glycan is abbreviated as GGFGGn. (B) Coomassie blue-stained SDS–PAGE gel of purified Skp1 isoforms. Note the reduced mobility of higher glycoforms. (C) Western blots using isoform-specific antibodies. (D) MALDI-TOF MS analysis of Skp1 isoforms. Expected and observed average m/z values and m/z differences relative to the panel above are indicated. The peak corresponding to a matrix adduct is indicated in the third panel.
A major function of Skp1 is as an adapter in the Skp1/cullin1/F-box protein (SCF) family of E3 ubiquitin ligase complexes, linking the Cul1 scaffold protein to an exchangeable F-box protein (FBP) that targets substrate proteins, or the FBP itself, for polyubiquitination.6 The covalent attachment of ubiquitin and subsequent assembly of a Lys48-linked polyubiquitin chain is a signal for proteasomal degradation.7 The SCF complex is diversified by the existence of multiple FBPs, which range in number from ∼20 in yeast to 70 in humans and 800 in some plants,8 with each FBP potentially recognizing multiple targets for polyubiquitination.8 Some substrates are activated by phosphorylation or glycosylation. The SCF E3 ligases themselves are also regulated by neddylation of cullin-1,9 which influences docking of the Skp1/FBP subcomplex with cullin-1, but what is missing is a mechanism specific to the SCF class of cullin ring ligases. Selective regulation may involve its unique subunit Skp1, which is subject to prolyl hydroxylation, glycosylation, and phosphorylation in different organisms and is represented by a gene family in others.1,10 Indeed, the interaction of Skp1 with some FBPs appears to be regulated in cells.11,12 Though the Skp1–F-box interface is hydrophobic and characterized by a high affinity, the potential for equilibrative exchange is suggested by in vitro studies13 and the observation that some FBPs form distinct complexes with Skp1 and other proteins in cells.14
Structural studies of Skp1 in complex with six different FBPs reveal a common mode of interaction that involves the 70 C-terminal amino acids of Skp1 with so-called core and variable subregions.6,13 Pro143 is located at the start of a stretch that in some cases folds into an α-helix that contributes to the variable component of the interface13 but can exhibit alternative secondary structure15 and is thus positioned to influence the conformation of Skp1 and its interaction with F-box domains. However, the conformation of native Skp1 is not known because of its recalcitrance to crystallization. Solution studies indicate that the FBP Fbs1 (also known as Fbg1 or OCP1), which is unusual in its solubility in the absence of Skp1, confers conformational order to unstructured regions of Skp1 upon binding.16 This effect is reminiscent of the Skp1 homologue elongin C that, based on nuclear magnetic resonance (NMR) studies, exhibits dynamic flexibility that is stabilized by the binding of a von Hippel–Lindau peptide or elongin A.17,18
These observations led to our hypothesis that the glycosylation of Skp1 has an impact on interactions with FBPs because of intrinsic effects on its conformation. To address this, we first developed methods for preparing milligram quantities of homogeneous glycoforms, using a combination strategy of co-expressing untagged Skp1 in Escherichia coli with its early modification enzymes and in vitro extension of the monosaccharide with recombinant enzymes. While all Skp1 isoforms were competent to bind Fbs1, glycosylated glycoforms bound preferentially. This correlated with an increased level of secondary structure and order as indicated by CD and SAXS experiments and with a decreased level of homodimerization. We suggest that the conformational changes in Skp1 induced by glycosylation are responsible for enhanced binding to Fbs1 and propose that this represents a novel form of specific regulation of E3SCFUb ligases and contributes to the role of Skp1 glycosylation in O2 sensing in cells.
Experimental Procedures
Expression and Purification of Unmodified and Hydroxylated Skp1 in E. coli
Unmodified Skp1A (without peptide tags) was expressed and purified as previously described.19 For the expression of HO-Skp1, plasmid pET19b-Skp1A-PhyA was constructed by excision of the full-length PhyA cDNA containing an N-terminal His6 tag from pET15TEVi-PhyA,20 using HindIII and BglII, and ligation into the HindIII and BamHI sites of pET19b-Skp1A,20 generating a dual-expression plasmid termed pET19b-Skp1A-PhyA. pET19b-Skp1A-PhyA (AmpR) was transformed into E. coli Gold BL21 Competent Cells. At an OD600 of 0.5–0.6 in LB-ampicillin, Skp1A and His6PhyA co-expression was induced by addition of 1 mM IPTG at 22 °C. After 16 h, cells were recovered in 20 mM Tris-HCl (pH 8.0) and resuspended in cell lysis buffer [100 mM Tris-HCl (pH 8.2), 5 mM benzamidine, 0.5 μg/mL pepstatin A, 5 μg/mL aprotinin, 5 μg/mL leupeptin, 0.5 mM PMSF, and 1 mg/mL lysozyme] on ice using a Dounce tissue grinder. Cells were lysed using a French pressure cell and supplemented with 10 μg/mL DNase I and 50 μg/mL RNase A (final concentrations). The lysate was centrifuged at 100000g and 4 °C for 1 h. The supernatant was purified by successive chromatography over DEAE-Sepharose Fast Flow, Phenyl-Sepharose Fast Flow (Hi-Sub), Q-Sepharose High Performance, and Superdex 200 HiLoad 16/60 columns under nondenaturing conditions as described previously.20 Essentially, quantitative hydroxylation of Skp1A was confirmed by Western blotting with pAb UOK87 and pAb UOK85, which selectively recognize Skp1 and HO-Skp1,3 respectively, and detection of unmodified and hydroxylated peptides, after digestion with endo Lys-C, by MALDI-TOF-TOF MS using α-cyanocinnamic acid as the matrix.3
Expression and Purification of Gn-Skp1 in E. coli
Expression plasmid pACYCDuet-DdDpGnt1 was constructed by excision of the chimeric DdDpGnt1 coding sequence (a chimeric Gnt1 constructed from sequences of two closely related Dictyostelium species, D. discoideum and D. purpureum) from pDEST527-DdDpGnt119 using HindIII and MluI, followed by ligation into the first MCS of pACYCDuet-1 (EMD Millipore) using the same restriction sites. pACYCDuet-DdDpGnt1 (CmR) and pET19b-Skp1A-PhyA (AmpR) were cotransformed into E. coli Gold BL21 Competent Cells. Expression and purification were performed as described for HO-Skp1. Glycosylation of Skp1A was confirmed by Western blotting with pAb UOK85 and mAb 1C9, which selectively recognize HO-Skp1 and Gn-Skp1,4 respectively, and by direct analysis of the highly purified protein by MALDI-TOF-TOF MS.
Preparation and Purification of FGGn-Skp1
The full-length cDNA for the dual-function glycosyltransferase PgtA21 was amplified from pTYCBD-FT22 by polymerase chain reaction using primers 5′-GCAGATCTATGAATGATTCACCAATAATAAGTGTAGTT-3′ and 5′-CGTGATCATTAAGAAATAAAAAGTTCACCAATATGAACAC-3′ and ligated into pCR4-TOPO (Invitrogen). The pgtA cDNA was excised by digestion with BglII and SpeI (underlined in primer sequences) and ligated into a similarly digested extrachromosomal pDM320 vector23 (gift of Y. Abu-Kwaik), designed to introduce a FLAG tag at the N-terminus of PgtA (sequence). The pDM320-FLAG-PgtA vector was electroporated into the D. discoideum strain Ax3.24 Transformed cells were selected under 20 μg/mL G418 without being cloned and grown to a density of 1 × 108 cells/mL. An S100 cytosolic fraction was prepared from 1 L of culture medium, and FLAG-PgtA was purified on a 90 mL DEAE-Sepharose Fast Flow anion exchange column as described previously.25 Eluted DEAE fractions containing FLAG-PgtA, as determined by Western blotting with anti-FLAG M2 Ab, were applied to a column containing 250 μL of M2-EZ view beads (Sigma) equilibrated in 50 mM HEPES (pH 7.4), 15% (v/v) glycerol, 150 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 5 μg/mL aprotinin, and 5 μg/mL leupeptin, washed under gravity flow with equilibration buffer, and eluted with 300 μL aliquots of 0.1 mg/mL 3×-FLAG peptide in equilibration buffer.
Gn-Skp1 (4 mg) was incubated with FLAG-PgtA (5% of the yield from the preparation described above) in 50 mM HEPES-NaOH (pH 7.4), 100 mM NaCl, 15 mM MgCl2, 2 mM MnCl2, 2 mM DTT, 0.1% (v/v) Tween 20, 2 mM ATP, 3 mM NaF, 0.004 unit/mL calf intestinal phosphatase, 15 μM GDP-Fuc, and 15 μM UDP-Gal for 18 h at 22 °C. Reaction progress was monitored by the disappearance of binding by mAb 1C9 (recognizes Gn-Skp1 but not GGn-Skp1 or FGGn-Skp1) in a dot-blot protocol.
The reaction mixture was extensively dialyzed against 50 mM Tris-HCl (pH 7.4), 0.1 mM EDTA, and 1 mM DTT and loaded onto a 1 mL Q-Sepharose HiTrap column (GE Healthcare) equilibrated in the same buffer. PgtA-modified Skp1 was eluted using a gradient from 0 to 300 mM NaCl in the same buffer. Formation of FGGn-Skp1 was verified by MALDI-TOF MS using DHB matrix and SDS–PAGE analysis after staining with Coomassie Brilliant Blue.
Preparation and Purification of GGFGGn-Skp1
His6AgtA was prepared in E. coli and purified on a 1 mL Ni2+-Sepharose HiTrap column as described previously.26 Fractions containing His6AgtA were identified using an anti-His mAb (Novagen) in a dot-blot protocol and immediately chromatographed at 4 °C on a Superdex 200 HiLoad size-exclusion column in 50 mM Tris-HCl (pH 8.0), 0.2 mM EDTA, and 5 mM β-mercaptoethanol.
FGGn-Skp1 was incubated at 22 °C with His6AgtA in 50 mM HEPES-NaOH (pH 7.2), 50 mM NaCl, 2 mM MnCl2, 5 mM DTT, and 20 μM UDP-Gal. After 24 h, the mixture was dialyzed for 6 h at 4 °C in the same buffer lacking UDP-Gal, and the reaction was continued for an additional 24 h after the mixture had been supplemented with additional equal amounts of His6AgtA and UDP-Gal. Reaction progress was monitored by MALDI-TOF MS. After >95% modification, Skp1 was purified from His6AgtA on a Superdex 200 HiLoad column at 22 °C in 50 mM HEPES-NaOH (pH 7.2), 50 mM NaCl, 2 mM MnCl2, and 1 mM DTT. Essentially complete formation of the expected product was confirmed by MALDI-TOF MS analysis of intact Skp1 and after endo Lys-C digestion as described above.
Ni2+-Sepharose Pull-Down Assay
His6Fbs1 was expressed and purified as previously described.16 Skp1 isoforms (0.1 μM) were preincubated with 1 mg/mL bovine serum albumin in the presence or absence of His6Fbs1 (0.3 μM) for 1.5 h at 4 °C in pull-down buffer containing 20 mM imidazole, 50 mM HEPES-NaOH (pH 7.4), 250 mM NaCl, 1 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 10 μg/mL aprotinin, and 10 μg/mL leupeptin. Ten microliters was used to resuspend 10 μL of packed Ni2+-Sepharose HP beads (GE Healthcare). After rotation for 1.5 h at 4 °C, beads were collected by centrifugation at 2000g for 1 min and washed three times with 150 μL of pull-down buffer, exchanging tubes after the first wash. Beads were then boiled in SDS–PAGE sample buffer for 3 min and analyzed by SDS–PAGE and Western blotting using an anti-His mAb (Novagen) or the pan-specific Skp1 mAb 4E1, and Alexa-680-conjugated rabbit anti-mouse IgG. Fluorescence was detected using an Odyssey infrared scanner (Li-Cor), and images were densitometrically analyzed using ImageJ (version 1.47, National Institutes of Health, Bethesda, MD), at intensity levels that were linearly related to input protein levels.
Analytical Gel Filtration Chromatography
Skp1 (and isoforms) at 2 μM with or without His6Fbs1 at a limiting concentration of 1.5 μM was preincubated for 1 h at 4 °C in a total reaction volume of 100 μL of column buffer containing 20 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, and 1 mM DTT. Of the 100 μL reaction mixture, 50 μL was injected onto a Superdex 200 PC 3.2/30 column using Pharmacia SMARTSystem high-performance liquid chromatography at a flow rate of 80 μL/min in column buffer. Fractions of 50 μL were collected and analyzed by SDS–PAGE, Western blotting, and densitometry as described above.
Circular Dichroism
Skp1 and its various isoforms were purified essentially to homogeneity and extensively dialyzed simultaneously against CD buffer [20 mM sodium phosphate (pH 7.4), 50 mM NaCl, 1 mM MgCl2, 0.1 mM EDTA, and 1 mM TCEP]. Prior to analysis, samples were filtered through a Corning Costar Spin-X 0.22 μm cellulose acetate microcentrifuge tube filter. Far-UV (ultraviolet) CD spectra were recorded for each protein at 0.2 mg/mL in 210 μL of CD buffer in a N2-purged 1.0 mm quartz cuvette on a Jasco J-715 spectropolarimeter equipped with a Jasco PTC-348WI Peltier temperature controller operated at a 1.0 nm bandwidth in 1 nm steps from 200 to 260 nm. Three spectra were summed for the final readout. For secondary structure prediction, the Jasco mdeg data files were converted into molar ellipticity per residue and analyzed using three predictors (SELCON3, CDSSTR, and CONTINLL) in CDPro (http://lamar.colostate.edu/∼sreeram/CDPro/main.html) using reference file no. 4 for soluble, nonmembrane proteins. Thermal denaturation experiments were performed by monitoring the ellipticity at 220 nm from 20 to 95 °C, and vice versa, at a rate of 2 °C/min. Thermal denaturation data were fit to a two-state Boltzmann sigmoidal model using GraphPad Prism (GraphPad Software, La Jolla, CA) to obtain the reported thermal unfolding midpoint temperatures (Tm).
CD data have been deposited at the Protein Circular Dichroism Data Bank (PCDDB) located at http://pcddb.cryst.bbk.ac.uk. The sample codes are as follows: unmodified DdSkp1A, CD0004330000; HO-Skp1, CD0004331000; Gn-Skp1, CD0004332000; FGGn-Skp1, CD0004333000; GGFGGn-Skp1, CD0004334000.
Small-Angle X-ray Scattering
Frozen, unmodified, and fully glycosylated Skp1 samples were thawed and extensively dialyzed against SAXS buffer [50 mM HEPES-NaOH (pH 7.4), 50 mM NaCl, 2 mM MnCl2, 1 mM TCEP, and 3% (v/v) glycerol]. Samples were immediately shipped to Stanford Synchrotron Radiation Lightsource (SSRL) beamline 4-227 on ice packs and, just prior to the collection of SAXS data, were centrifuged through a Corning Costar Spin-X 0.22 μm cellulose acetate microcentrifuge tube filter. Scattering data were collected over a concentration range of 1.2–9.4 mg/mL for unmodified Skp1 and 1.5–3.0 mg/mL for fully modified Skp1 in 30 μL volumes at 10 °C in oscillating quartz cuvettes with a Rayonix MX255-HE detector. Samples were loaded with a custom-built liquid handling robot.28 The X-ray wavelength was 1.1263 Å. The sample to detector distance was set to 1700 mm. Twelve 1 s exposures were recorded for each sample. SAXS data were collected from buffer samples between each protein sample. SAXS curves [I(q) vs q, where I(q) is the scattering intensity, q = 4π sin θ/λ, 2θ is the scattering angle, and λ is the X-ray wavelength] were generated from image files using SAXSPipe (SSRL). The scattering curves from successive images were compared to check for radiation damage before being averaged together. Averaging and buffer subtraction were performed using PRIMUS.29 Guinier analysis in PRIMUS was used to screen for concentration-dependent effects on the radius of gyration (Rg); the SAXS curves from concentrations showing linearity in the Guinier regions were averaged, and Kratky and P(r) function plots were generated using GNOM(30,31) to determine the maximal particle distance, Dmax. Molecular weight estimation using the experimental SAXS curves was performed using the “SAXS MoW”32 online web tool (http://www.if.sc.usp.br/∼saxs/saxsmow.html) and SCÅTTER version 1.7i (http://www.bioisis.net/scatter) for comparison. For each protein, 10 independent ab initio molecular envelopes were generated using DAMMIF(33) in “slow” mode imposing P2 symmetry using the scattering data from the highest-concentration sample. The 10 models were aligned, averaged, and filtered by a cutoff volume (the averaged volume of the models) using DAMAVER.34 Figures were prepared using PyMOL.35 SAXS data have been deposited at http://www.bioisis.net. Reference codes are SKP1UP (unmodified DdSkp1A) and SKP1FP (Dd-GGFGGn-Skp1A).
Glycan Modeling
The Skp1 glycan conformation was modeled using the GLYCAM Carbohydrate Builder at http://glycam.ccrc.uga.edu.36 The predicted Galα1,2Galα1,3Fucα1,2Galβ1,3GlcNAcα- structure was used.1
Chemical Cross-Linking
Skp1 preparations at 0.2, 2, or 3 μM were mixed with the nonspecific chemical cross-linking reagent disuccinimidyl suberate [DSS (Thermo Scientific/Pierce)] at either 0.1 or 1 mM in cross-linking buffer [20 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, and 0.1 mM EDTA] in the presence or absence of 5 mM MgCl2. Reaction mixtures were incubated at room temperature for 5 min and reactions quenched by the addition of 1 M Tris-HCl (pH 7.4) to a final concentration of 50 mM. Samples were analyzed by gel filtration as described above or SDS–PAGE followed by staining with Coomassie Brilliant Blue or Western blotting using the pan-specific Skp1 mAb 4E1 and Alexa-680-conjugated rabbit anti-mouse IgG. Fluorescence was quantitated as described above.
Results
Preparation of Skp1 Isoforms
Previous studies on Skp1 isoforms have been constrained by the availability of native material that can be isolated from Dictyostelium, so recombinant methods were developed to generate adequate quantities for biophysical and biochemical studies. Native Dictyostelium Skp1A is expressed well in E. coli(19) and was purified to near homogeneity under nondenaturing conditions based on DEAE-Sepharose, phenyl Sepharose, Q-Sepharose, and gel filtration chromatographies. Epitope tags were not employed because of evidence of interference with Skp1 modifications in cells.37 HO-Skp1 was prepared by co-expression of DdPhyA from a dual-expression plasmid with Skp1 in E. coli and purified in the same way. Gn-Skp1 was similarly prepared from an E. coli strain harboring a second plasmid encoding DdGnt1. Because DdPgtA was not expressed well as a soluble active enzyme in E. coli, FLAG-PgtA was transiently overexpressed in Dictyostelium, partially purified by DEAE ion exchange chromatography and anti-FLAG affinity chromatography, and used to quantitatively convert Gn-Skp1 to FGGn-Skp1 in vitro. FGGn-Skp1 was repurified, and assembly of the final pentasaccharide isoform was achieved by incubation with purified Dd-His6AgtA prepared from E. coli. Examples of preparations used for this study are shown using SDS–PAGE gels stained for total protein (Figure 1B). A decreasing mobility corresponds to higher glycosylation states. The modification status of Skp1 was confirmed by Western blot analysis using isoform-specific antibodies (Figure 1C) and MALDI-TOF MS of the intact protein (Figure 1D). MALDI-TOF MS analysis of proteolytic fragments generated by endo Lys-C confirmed that the modifications occurred on Pro143 as expected (data not shown).
The FBP Fbs1 Preferentially Binds Glycosylated Skp1s
To test the functionality of the recombinantly expressed Skp1 isoforms, we prepared the naturally soluble mammalian FBP His6Fbs1, also known as Fbg1 and OCP1, from E. coli as an expected Skp1 binding partner. Previous biophysical studies showed that Fbs1 and mammalian Skp1 form a high-affinity 1:1 complex.16 Pull-down studies with Ni2+ beads showed that each Skp1 isoform was fully competent at 0.1 μM to bind Fbs1 in a 3-fold concentration excess, indicative of normal function (Figure 2A). This interaction was analyzed in 20 mM imidazole and high salt (250 mM) to prevent intrinsic binding of Skp1 to the beads, potentially mediated by an internal His-rich region, R(59)HHHQHP.
Figure 2.

Skp1 binds guinea pig Fbs1. (A) Ni2+-Sepharose pull-down assay using 0.3 μM His6Fbs1 and purified Skp1 isoforms (0.1 μM), labeled according to the substitution status of Pro143. Data derive from densitometric analysis of Western blots. Average values ± SEM from two independent trials are shown. (B) His6Fbs1 (1.5 μM) and a pool of three Skp1 isoforms (Skp1, HO-Skp1, and Gn-Skp1, 2 μM each) were fractionated on a Superdex 200 gel filtration column separately or after preincubation. Elution was monitored by A280, which favors detection of Fbs1 because of its higher extinction coefficient. Elution positions for bovine serum albumin (66000) and carbonic anhydrase (29000) are indicated. (C) Western blots of the fractions using the pan-specific anti-Skp1 mAb 4E1 or the isoform-specific antibodies shown in Figure 1C. (D) Densitometric analysis of the data depicted in panel C. (E) Similar gel filtration experiment with His6Fbs1 and a mixture of Skp1 and GGFGGn-Skp1 monitored by SDS–PAGE and silver staining. As indicated by the double bar, GGFGGn-Skp1 migrates more slowly than Skp1 during SDS–PAGE. (F) Densitometric analysis of the data depicted in panel E. Also included are data, denoted with an asterisk, from probing a parallel Western blot for unmodified Skp1 using pAb UOK87. Results similar to those in panels C and E were obtained in independent experiments.
To probe for possible differential binding in a more physiological buffer, a pool of three Skp1 isoforms (unmodified, HO-Skp1, and Gn-Skp1, 2 μM each) was preincubated in the absence or presence of a limiting concentration of Fbs1 and subjected to gel filtration in column buffer [20 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, and 1 mM DTT]. As shown in Figure 2B, His6Fbs1 eluted as a nearly symmetrical peak corresponding approximately to its monomeric Mr value of 35000, with a shoulder potentially corresponding to the previously reported homodimer.16 Skp1 (unmodified Mr = 18710) eluted as a symmetrical peak between that of Fbs1 and the Mr = 29000 globular standard, which is likely due to partial dimerization, a high axial ratio, and a partially unfolded state, leading to an increased apparent volume, as discussed below. Western blot analysis of the fractions using isoform-specific antibodies and densitometry showed that Skp1 isoforms eluted in fractions 12 and 13 (Figure 2C,D), with unmodified Skp1 showing a slightly enhanced presence in fraction 12 versus fraction 13, consistent with a stronger tendency to dimerize (see below). Preincubation of Fbs1 and the Skp1 pool resulted in an earlier eluting peak that depleted the Fbs1 and Skp1 peaks, consistent with formation of a stable complex. The peak eluted after the Mr = 66000 standard, consistent with a 1:1 stoichiometry as previously reported for mammalian Skp1,16 contained His6Fbs1 based on Western blotting (not shown), and was followed by a trailing shoulder. Western blot analysis and densitometry showed that of the three Skp1 isoforms, Gn-Skp1 was most heavily represented in peak fraction 10, whereas HO-Skp1 was most heavily represented in fraction 11 and unmodified Skp1 in fraction 12 (Figure 2E,F). Thus, Gn-Skp1 preferentially associated in this competitive setting in which the Fbs1 concentration was limiting.
To extend the analysis, Fbs1 (1.5 μM) and a mixture of unmodified and fully glycosylated GGFGGn-Skp1 (2.5 μM each) were analyzed similarly using silver staining because specific probes are not available for the longer glycoforms (Figure 2E). As in the experiment depicted in Figure 2C, the Skp1 mixture alone eluted in fractions 12 and 13 and emerged earlier in the presence of Fbs1. On the basis of densitometric analysis (Figure 2F), the peak elution position of GGFGGn-Skp1 shifted to fraction 11 whereas that of unmodified Skp1 remained in fraction 12, and proportionately more GGFGGn-Skp1 than unmodified Skp1 emerged in fraction 10. The elution profile of unmodified Skp1 was confirmed by Western blotting using pAb UOK87 (Figure 2F). Although it was not feasible to differentiate GGFGGn-Skp1 and Gn-Skp1 in the same run, comparison of elution positions between runs (Figure 2D,F) suggests that Fbs1 preferentially associates with Gn-Skp1. Thus, a hierarchy of Fbs1 association was observed: Gn-Skp1 > GGFGGn-Skp1 ≥ HO-Skp1 > Skp1.
Glycosylation Alters the Secondary Structure Composition
To investigate the basis for preferential binding of glycosylated isoforms of Skp1 to Fbs1, their conformations were assessed using circular dichroism (CD) spectroscopy (Figure 3A). When compared at the same concentration in the same phosphate-based CD buffer, all isoforms exhibited differential far-UV absorption from 200 to 245 nm, with discrete minima at 208–222 nm indicative of β-strand and α-helical secondary structure, consistent with previous studies of guinea pig and Dictyostelium Skp1s.16,19 Whereas Skp1 and HO-Skp1 exhibited indistinguishable spectra, Gn-Skp1 showed increased negative ellipticity indicative of a substantially increased level of secondary structure under these buffer conditions that correlated with an increased level of binding to Fbs1. This difference is unlikely to be contributed directly by the sugar moiety given the typically low mean residue ellipticity of glycans.38,39 FGGn-Skp1 was indistinguishable from Gn-Skp1, whereas fully modified GGFGGn-Skp1 exhibited intermediate values also correlated with an increased level of Fbs1 binding. Secondary structure deconvolution analysis indicated that addition of α-GlcNAc results in greater α-helical content, at the partial expense of β-sheet content (Figure 3B). To a lesser extent, this trend was maintained by the pentasaccharide-modified glycoform, which correlated with slightly less apparent Fbs1 binding potential. In comparison, a previous study reported a higher α-helical content of unmodified Dictyostelium Skp119 when the analysis was conducted in a no-salt Tris buffer [10 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 0.1 mM EDTA, and 1 mM DTT], in comparison to the more physiological and CD-compatible buffer [20 mM sodium phosphate (pH 7.4), 50 mM NaCl, 1 mM MgCl2, 0.1 mM EDTA, and 1 mM TCEP] employed for the present comparison. Conformational sensitivity to ionic strength is known for other proteins (e.g., ref (40)).
Figure 3.
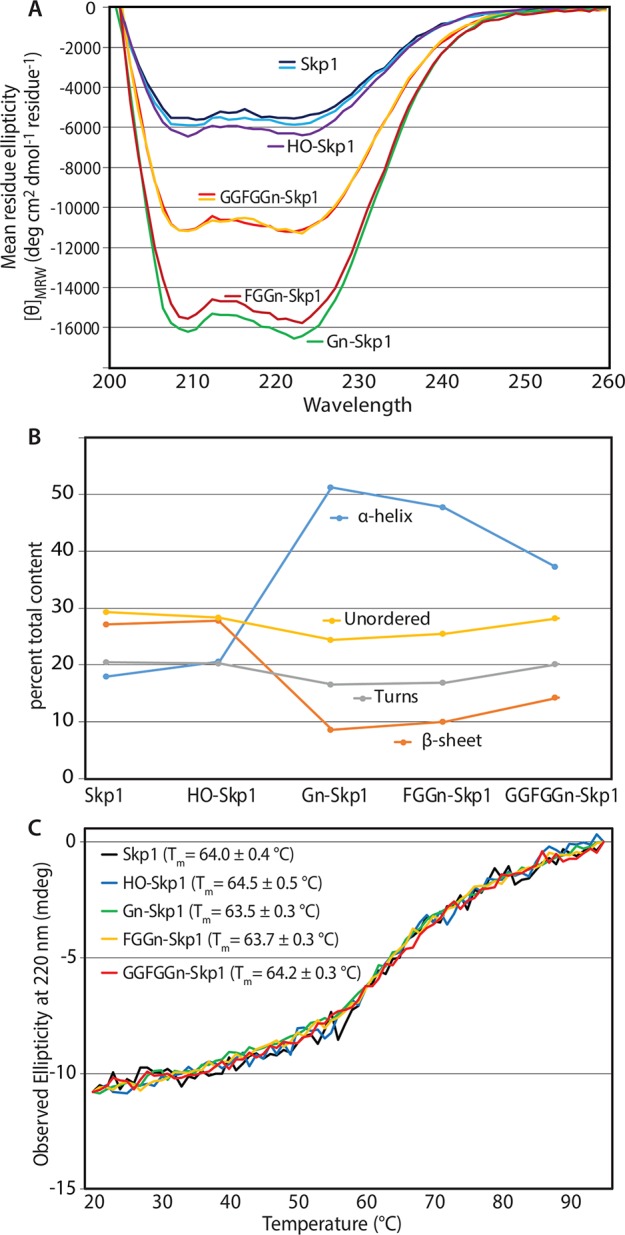
Circular dichroism spectroscopy of Skp1 isoforms. (A) CD spectra of Skp1 isoforms (10 μM) in CD buffer. Replicates of samples subjected to additional freeze–thaw cycle are included. Replicate studies on independent protein preparations qualitatively confirmed these differences (data not shown). (B) Secondary structure analysis using three predictors (SELCON3, CDSSTR, and CONTINLL) agreed within 7%. The averages of estimates for α-helix, β-sheet, β-turn, and disordered content are plotted as a function of Skp1 modification status. (C) Thermal denaturation of Skp1 isoforms monitored by ellipticity at 220 nm. Data were proportionately scaled to similar initial and terminal values to facilitate comparison. Apparent Tm values based on the inflection between 50 and 80 °C are listed.
Energetic Analyses
To examine the thermodynamic consequences of these differences, thermal denaturation experiments were conducted in CD buffer by monitoring ellipticity at 220 nm. As shown in Figure 3C, Skp1 (black trace) exhibited a gradual loss of secondary structure upon being heated from 22 to 95 °C, with a modest inflection at 64 °C indicative of cooperative partial loss of secondary structure, consistent with previous studies.16,19 Indistinguishable results were obtained for the other isoforms, which were scaled to facilitate comparison in Figure 3C. As shown previously for Skp1,16,19 temperature-dependent unfolding is largely reversible for all isoforms (data not shown). Thus, the similar thermal instabilities indicate similar energetics of folding, suggesting that glycosylation influences secondary structure in an energy neutral way that may allow facile interconversion between α-helix- and β-sheet-rich conformations.
Glycosylation Increases the Folding Order and Alters the Overall Shape
Small-angle X-ray scattering (SAXS) experiments were performed on unmodified and fully glycosylated Skp1 in solution to interpret the impact of secondary structure changes and glycosylation on Skp1 structure and conformation. Studies were conducted in SAXS buffer [50 mM HEPES-NaOH (pH 7.4), 50 mM NaCl, 2 mM MnCl2, 1 mM TCEP, and 3% glycerol], which had an ionic strength (12% increase) similar to that of CD buffer but employed a different buffer type. Other experimental parameters are listed in Table 1. To address potential concentration-dependent effects, SAXS exposures were conducted on a series of Skp1 dilutions. The scattering curves showed no evidence of aggregates and interparticle effects at very low q values. The Guinier analysis of the SAXS curves41 revealed linearity in the low-q regions (where qRg < 1.3), indicating that samples were monodisperse and free from aggregation (Figure 4A). The parallel plots indicate no change in the radius of gyration (Rg) with a change in concentration. The Guinier plots also showed that the Rg of Skp1 was 11.4% larger when it was fully modified (Figure 4A and Table 1). The experimental SAXS data shown in the Guinier plots were averaged (Figure 4B) and then transformed into Kratky plots [q2I(q) vs q]. The Kratky plots showed that both proteins exhibited typical globular behavior at 60–460 μM, with a bell-shaped curve at low q values (Figure 4C). However, unmodified Skp1 displayed increasing values of q2I(q) at higher q values, indicating a larger unfolded component.42 This interpretation was confirmed by analysis of the Porod–Debye plot of the scattering data (Figure 4D), where the Porod–Debye plateau of GGFGGn-Skp1 implies a homogeneous conformation and the continuing rise for unmodified Skp1 indicates flexibility.42 Pairwise distance distribution functions [P(r)] of the averaged SAXS curves were calculated (Figure 4E), and inverse Fourier transformations showed excellent agreement with the experimental data (Figure 4B, red traces). The P(r) plots revealed an increased maximal particle size (Dmax) for fully glycosylated Skp1, independent of concentration (Figure 4C and Table 1). Three independent methods of Mr estimation for unmodified Skp1 were in close agreement and yielded an average value of 39300, similar to the expected Mr (37174) of a Skp1 dimer (Table 1). The calculations for GGFGGn-Skp1 yielded an average increment of 4200, which contrasts with the expected difference of 1704 (852 × 2). Thus, increased Rg, Dmax, and Mr values were consistent with the covalent addition of the pentasaccharide; the Skp1s were dimeric, and unmodified Skp1 was partially disorganized.
Table 1. Data Collection and Scattering-Derived Parameters.
| unmodified Skp1 | GGFGGn-Skp1 | |
|---|---|---|
| Data Collection Parameters | ||
| beamline | SSRL beamline 4-2 | |
| beam geometry | 0.3 mm × 0.3 mm, pinhole camera | |
| detector | Rayonix MX255-HE | |
| wavelength (Å) | 1.1263 | |
| q range (Å–1) | 0.03–0.30 | |
| exposure time (s) | 12 × 1 | |
| temperature (K) | 283 | |
| concentration range (mg/mL) | 1.2–9.4 | 1.5–3.0 |
| Structural Parametersa | ||
| I(0) (real space) | 7438 ± 3 | 3191 ± 6 |
| Rg (Å) (real space) | 25.3 ± 0.1 | 27.2 ± 0.1 |
| I(0) (Guinier) | 7130 ± 10 | 3158 ± 7 |
| Rg (Å) (Guinier) | 23.9 ± 1.1 | 26.6 ± 0.6 |
| Dmax (Å) | 79 | 91 |
| Porod volume estimate (Å3) | 64693 | 76000 |
| DAMMIF volume (Å3)b | 87753 | 98971 |
| mean NSDc of 10 DAMMIF models | 0.750 ± 0.080 | 0.728 ± 0.059 |
| Molecular Mass Determinationa | ||
| calculated monomeric Mr from sequence | 18587 | 19481 |
| molecular mass Mr estimate | ||
| from SAXS MoW | 38800 | 46400 |
| from SCÅTTER (real space) | 38800 | 40200 |
| from SCÅTTER (Guinier) | 40300 | 43900 |
| Software | ||
| primary data reduction | SAXSPipe (SSRL) | |
| data processing | PRIMUS, GNOM, SCÅTTER, SAXS MoW | |
| ab initio analysis | DAMMIF | |
| validation and averaging | DAMAVER | |
| three-dimensional graphics representations | PyMOL | |
Reported for averaged data over a concentration range.
Reported for the highest-concentration data.
Normalized spatial discrepancy.
Figure 4.

SAXS analysis of Skp1 and GGFGGn-Skp1. Skp1 and GGFGGn-Skp1 were analyzed at concentrations of 60–460 μM in SAXS buffer. (A) Guinier analysis at different Skp1 concentrations, where q = 4π sin θ/λ (2θ is the scattering angle and λ the X-ray wavelength). (B) Scattering intensity plots averaged from the concentrations shown in panel A, for unmodified Skp1 (black) and GGFGGn-Skp1 (blue). Fourier transformations of the scattering data are shown as the P(r) functions in panel E; the red traces represent the inverse Fourier transformations of the P(r) functions. (C) Kratky plots of Skp1 were generated from the averaged data shown in panel B. (D) Porod–Debye representation of the data shown in panel B. (E) Pairwise distance distribution functions of the averaged data.
SAXS ab initio molecular envelopes were generated for each isoform using DAMMIF, imposing P2 symmetry according to evidence of dimerization (Figure 5A). Because the CD data suggested a change in the secondary structure content of Skp1, we decided that it was not appropriate to use multiphase bead modeling by using the scattering data simultaneously from Skp1 and GGFGGn-Skp1. The envelope for unmodified Skp1 easily accommodates two copies of the Skp1 structure present in complexes with F-box proteins, with additional volume that might represent conformational dynamics. In agreement with the greater Rg and Dmax from the raw scattering data, the molecular envelope of GGFGGn-Skp1 presents an elongated structure (Figure 5B), most consistent with localization of the C-terminal glycans at or near the opposite ends of an antiparallel dimer (Figure 5C). The calculated increase in volume of 12.7% for GGFGGn-Skp1 relative to that of Skp1 (Table 1) is in close agreement with the increase in Rg and apparent Mr, but ∼2.5-fold greater than the actual Mr increment of 4.6%. This discrepancy likely reflects a combination of contributions from the increased volume that can be attributed to glycan chain flexibility43 and the decreased volume of the polypeptide core because of increased folding order (Figure 4C,E).
Figure 5.

SAXS envelopes. Ab initio molecular envelopes were calculated from data collected from 9.4 mg/mL unmodified Skp1 (A) or 3.0 mg/mL GGFGGn-Skp1 (C), based on the average of 10 iterations using DAMMIF. (B) Overlay of panels A and C. In panels A and C, images of two Skp1 molecules excerpted from the crystal structure of a complex with the FBP Tir1 (Protein Data Bank entry 2P1N)46 are modeled with P2 symmetry within the envelope of Skp1 and GGFGGn-Skp1. The Skp1 pentasaccharide was modeled using GLYCAM, and the reducing terminus is positioned near the Pro143 attachment site. The bottom two rows represent 90° relative rotations as indicated.
Modifications Weaken Skp1 Dimerization
The SAXS analyses indicate that at higher concentrations both isoforms exist as homodimers, in agreement with data for free mammalian Skp1s.17,44,45 We also consistently observed that via denaturing SDS–PAGE, a fraction of unmodified Skp1 larger than that of HO-Skp1 and Gn-Skp1 migrates as a dimer (Figure 6A), suggesting that modifications inhibit dimerization. When the suspected dimer state was stabilized by mild treatment with a nonspecific amine-targeted cross-linker with a 13 Å linker distance, DSS, a greater fraction of the Skp1s migrated as the dimer. The fraction of Skp1 in the dimer was greatest for unmodified Skp1, as confirmed by densitometric analysis (Figure 6C, fourth panel from the top). The difference between Skp1 and HO-Skp1 was also apparent as determined by analytical gel filtration, in which both cross-linked proteins migrated predominantly as dimers, HO-Skp1 predominantly as monomer, and Skp1 at an intermediate position consistent with a mixture of monomer and dimer states (Figure 6B). The unexpectedly small difference in elution volumes is attributed to a conformation of the dimer that is more globular than that of the monomer, which may occupy a larger than expected volume because of a larger axial ratio and partially unfolded conformation based on the SAXS data (Figures 4 and 5). The effect of glycosylation on dimerization was examined for all isoforms at a variety of Skp1 concentrations and ratios of cross-linker to protein. As shown in Figure 6C, an increasing level of glycosylation resulted in a modest but reproducible reduction in the level of dimerization under all conditions, regardless of the average degree of cross-linking achieved. Mg2+ (≤5 mM) did not have a systematic effect on cross-linking. The decreased dimerization potential of glycosylated relative to nonglycosylated Skp1s correlated with a stronger propensity to associate with Fbs1 but did not precisely hold between partially and fully glycosylated Skp1s.
Figure 6.

Analysis of Skp1 dimerization. (A) Skp1 isoform solutions (3 μM) in cross-linking buffer were heated at 100 °C in SDS and 50 mM DTT followed by standard SDS–PAGE and Western blotting using mAb 4E1. Parallel samples were first subjected to DSS cross-linking for 5 min as indicated. (B) Similar samples (3 μM) were subjected to gel filtration on an analytical Superdex 200 column, and elution was monitored at A280. (C) Summary of densitometric measurements of the dimer fraction after DSS cross-linking for 5 min at the indicated concentrations of Skp1s and DSS. SDS–PAGE gels were stained with Coomassie blue (○) or Western blotted with mAb 4E1 (■). Error bars represent the SEM of technical replicates.
Discussion
E. coli was found to be an efficient biosynthetic vessel for producing prolyl 4-hydroxylated and α-GlcNAcylated Skp1 based on enzyme co-expression (Figure 1). Further glycosylation was not amenable to this approach, though, because insufficient functional PgtA could be produced in E. coli. However, PgtA could be efficiently overexpressed in Dictyostelium, which allowed for quantitative production of FGGn-Skp1 in an in vitro reaction. Finally, fully glycosylated Skp1 could be generated by a subsequent in vitro reaction conducted in the presence of AgtA, which was easily expressed in and purified from E. coli. Skp1, which was expressed as its native sequence without epitope tags and in the absence of native binding partners, was purified under nondenaturing conditions to maximize the likelihood of assuming its native conformation(s). Thus, multimilligram quantities of highly purified native Skp1 isoforms with zero, one, three, or five sugars could be evaluated. An isoform with two sugars could in principle be produced by withholding the donor for the third sugar, GDP-Fuc, from the PgtA diglycosyltransferase reaction.21 An isoform with four sugars would be more challenging to produce, as the bifunctional AgtA utilizes the same donor, UDP-Gal, for both additions of sugar. However, the finding that the tetrasaccharide form of Skp1 transiently accumulated in the AgtA reaction opens the possibility of recovering this intermediate for future studies.
Each Skp1 isoform was capable of high-affinity binding to a natively soluble F-box protein, Fbs1, based on pull-down studies (Figure 2A), indicating that the modifications are not required for FBP interaction as previously shown for FbxA.19 However, analytical gel filtration analysis of competitive binding reactions revealed a preference of Fbs1 for binding of glycosylated relative to unmodified Skp1s, with evidence for the following hierarchy: Gn-Skp1 > GGFGGn-Skp1 ≥ HO-Skp1 > Skp1 (Figure 2C–F). Because probes for selectively detecting GGFGGn-Skp1 are not available, it was not feasible to directly compare the binding of fully glycosylated and Gn-Skp1 using the analytical gel filtration method. Pull-down approaches based on the N-terminal His6 tag of Fbs1 were confounded by intrinsic binding to Ni2+ beads, possibly because of a His-rich region in Dictyostelium Skp1 that might also contribute to Skp1 folding.
Because preferential binding of Fbs1 to glycosylated isoforms of Skp1 occurred in binary isolation, it is likely that the sugars either influence the conformation of the F-box binding region of Skp1 or expand the interaction surface that is recognized by Fbs1. However, the latter seemed unlikely as Fbs1 is a mammalian protein and Skp1 is evidently not glycosylated in chordates.1 In favor of the former prediction, CD revealed substantially increased levels of α-helical secondary structure in glycosylated Skp1 isoforms, compared to those of unmodified and hydroxylated Skp1 (Figure 3A,B). The increase came mainly at the expense of apparent β-sheet organization. Thus, glycosylation invoked substantial conformational reorganization of the Skp1 polypeptide, which may be the basis for improved binding to Fbs1. Notably, the higher α-helical content of glycosylated Skp1s is consistent with the value for Skp1s from Arabidopsis, Saccharomyces, and mammals that are complexed with various FBPs (e.g., refs (13), (15), and (46)), suggesting that glycosylation promotes a conformational state of free Skp1 that better mimics that of the final complex and therefore may promote the interaction with Fbs1. The indistinguishable Tm values as inferred from temperature-induced denaturation (Figure 3C) suggest that the conformational differences at 22 °C are not separated by a substantial energy barrier and that both conformational extremes may be accessible by all isoforms, thus explaining the intrinsic ability of each isoform to bind Fbs1 with high affinity.
Previous studies revealed that mammalian Skp1 homodimerizes with a Kd of 1 μM,44 and the SAXS data confirmed the homodimeric status of Dictyostelium Skp1s at 60 μM (Figure 5 and Table 1). Furthermore, gel filtration analysis indicated that at 2 μM, unmodified Skp1 exhibits a strong tendency to homodimerize relative to that of HO-Skp1 unless the latter was stabilized with a nonspecific amine cross-linker (Figure 6B). Analysis of cross-linked Skp1s (0.2–3 μM) by SDS–PAGE indicated that an increasing level of modification resulted in a progressively decreased level of homodimerization irrespective of cross-linker concentration. On the basis of findings that binding of Skp1 to Fbs1 and binding to itself are mutually antagonistic,16 a weakened tendency to homodimerize may contribute to, but does not fully explain, the increased level of binding of glycosylated isoforms of Skp1 to Fbs1.
Further support for conformational differences between unmodified and glycosylated Skp1s emerged from SAXS analysis. While the Kratky plots confirmed a typical globular organization, a substantially higher degree of disorder was observed for unmodified Skp1 relative to GGFGGn-Skp1 (Figure 4C). Substantial disorder for unmodified mammalian Skp1 at a similar salt concentration, but at a lower pH of 6.0 compared to the pH of 7.4 used here, was described on the basis of 1H–15N HSQC NMR studies.16 A comparison of pairwise distance distribution functions and envelopes indicates that glycosylated Skp1 is elongated with a 13% larger volume, greater than the 4.6% Mr increment of the dimer (Figure 4E and Table 1). The increased volume was concentrated at opposing ends of the structure (Figure 5) and may in part be assigned to the glycans whose typically dynamic time-averaged conformations normally occupy more volume than stably folded polypeptides of the same mass.43 However, it is difficult to exclude the possibility that the terminal knobs were occupied by C-terminal peptide regions that were reoriented by internally disposed glycans. Because the predicted overall volume of unmodified Skp1 was smaller than that of GGFGGn-Skp1, the increased volume expected to be contributed by the disordered polypeptide in unmodified Skp1 might be accommodated within voids that are smaller than the SAXS resolution of 20 Å (Figure 5A). Indeed, the modestly slimmer profile of the GGFGGn-Skp1 dimer envelope (Figure 5C) had difficulty in accommodating the homodimer model, excerpted from a crystal structure with the FBP Tir1,46 that easily fit inside the unmodified envelope (Figure 5A). Thus, some reorganization of the Skp1 polypeptide dimer caused by glycosylation was implied. Although the increased disorder of unmodified Skp1 correlated with a reduced α-helical content relative to that of GGFGGn-Skp1, it did not correlate with an increased β-sheet content. These results suggested that increased disorder lies in the organization of secondary structure elements relative to each other rather than to the total secondary structure content.
Pro143 is near but oriented away from the F-box combining region and initiates the C-terminal α-helix that is observed in most crystal structures with FBPs. Regions upstream of Pro143 contribute to the core interface that is sufficient for high-affinity binding in vitro to the FBP Skp2. Additional contacts are contributed by the downstream region, and this so-called variable region of the interface is essential for in vivo function.13 This region can fold as α-helix 8 or as a loop in the complex with Fbs1, which is evidently not an artifact of crystal packing.15 This indicates that Skp1 is conformationally flexible in its complexes with FBPs, which is consistent with the proposed conformational sensitivity to Pro143 modifications. Previous thermodynamic studies have suggested that the Fbs1–mammalian Skp1 interaction does not satisfy conditions of a rigid body interaction, implying that matching conformational changes accompany complex formation,16 which is consistent with NMR data for the Skp1 homologue elongin C as it associates with a peptide from its von Hippel–Lindau protein binding partner.17,18 We speculate that posttranslational modifications of Pro143 modify the ensemble of energetically similar conformations of Skp1 as indicated by the results of the CD and SAXS studies and Fbs1 binding studies.
Although evidence of the conformational effects of glycans on polypeptide folding is generally sparse, there is precedence for the effects of covalently attached sugars on peptide conformation and protein dimerization. Notable examples at the peptide level include effects of single sugars and neutral glycans on mucin domains,47−49 and β-linked GlcNAc on an estrogen receptor peptide.50 In another peptide, O-glycosylation can affect the trans:cis rotamer ratio of nearby Pro residues,51 and at the protein level, N-glycans affect dimerization of the FGF receptor.52 Potentially, modification of Skp1 Pro143 could modify the pucker of its pyrrolidine ring and/or trans:cis isomerization of the Thr–Pro143 peptide bond that could initiate the global conformational reorganization that we propose results in improved Fbs1 binding. In cells, an increased level of binding to FBPs would be expected to promote the activity of the respective E3SCFubiquitin ligases, resulting in timely turnover of target proteins consistent with normal O2-dependent developmental progression.
Acknowledgments
We thank Dr. Augen Pioszak for his advice on multiplasmid co-expression studies in E. coli and Drs. Thomas Weiss, Tsutomu Matsui, and Lester Carter (SSRL beamline 4-2) for assistance with SAXS data collection and initial evaluation.
Glossary
Abbreviations
- CD
circular dichroism
- CRL
cullin ring ligase
- Dd
prefix indicating origination from D. discoideum
- FBP
F-box protein
- SAXS
small-angle X-ray scattering
- GGFGGn-
glycan notation in which G is d-galactose, F is l-fucose, and Gn is d-GlcNAc or N-acetyl-d-glucosamine
- MALDI-TOF MS
matrix-assisted laser desorption ionization mass spectrometry
- SCF
CRL1 subcomplex consisting of Skp1, cullin-1, and an F-box protein
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SEM
standard error of the mean
- Ub
ubiquitin.
The authors declare no competing financial interest.
This work was supported by grants to C.M.W. from the National Institutes of Health (NIH) (R01 GM037539) and the Oklahoma Center for Advancement of Science and Technology (HR10-0181) and to B.H.M.M. from the NIH [R01 AI088011 (Principal Investigator, Blaine Mooers) and P20 GM103504 (Principal Investigator, Ann West)]. Portions of this research were conducted at the Stanford Synchrotron Radiation Lightsource, a Directorate of the Stanford Linear Accelerator Center National Accelerator Laboratory, and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by the National Institute of General Medical Sciences (including Grant P41 GM103393) and the National Center for Research Resources (Grant P41 RR001209). J.T.P. was supported by funding to the University of Oklahoma Health Sciences Center (OUHSC) Summer Undergraduate Research Program (SURE) program from the OUHSC Graduate School and the Provost’s Office.
Funding Statement
National Institutes of Health, United States
References
- West C. M.; Wang Z. A.; van der Wel H. (2010) A cytoplasmic prolyl hydroxylation and glycosylation pathway modifies Skp1 and regulates O2-dependent development in Dictyostelium. Biochim. Biophys. Acta 1800, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West C. M.; van der Wel H.; Wang Z. A. (2007) Prolyl 4-hydroxylase-1 mediates O2 signaling during development of Dictyostelium. Development 134, 3349–3358. [DOI] [PubMed] [Google Scholar]
- Zhang D.; van der Wel H.; Johnson J. M.; West C. M. (2012) Skp1 prolyl 4-hydroxylase of Dictyostelium mediates glycosylation-independent and -dependent responses to O2 without affecting Skp1 stability. J. Biol. Chem. 287, 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. A.; van der Wel H.; Vohra Y.; Buskas T.; Boons G. J.; West C. M. (2009) Role of a cytoplasmic dual-function glycosyltransferase in O2 regulation of development in Dictyostelium. J. Biol. Chem. 284, 28896–28904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Brown K. M.; Wang Z. A.; van der Wel H.; Teygong C.; Zhang D.; Blader I. J.; West C. M. (2012) The Skp1 protein from Toxoplasma is modified by a cytoplasmic prolyl 4-hydroxylase associated with oxygen sensing in the social amoeba Dictyostelium. J. Biol. Chem. 287, 25098–15110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman E. S.; Schulman B. A.; Zheng N. (2010) Structural assembly of cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 20, 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. [DOI] [PubMed] [Google Scholar]
- Skaar J. R.; Pagan J. K.; Pagano M. (2013) Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 14, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies R. J.; Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434. [DOI] [PubMed] [Google Scholar]
- Beltrao P.; Albanese V.; Kenner L. R.; Swaney D. L.; Burlingame A.; Villen J.; Lim W. A.; Fraser J. S.; Frydman J.; Krogan N. J. (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumimoto K.; Muneoka T.; Tsuboi T.; Nakayama K. I. (2013) Substrate binding promotes formation of the Skp1-Cul1-Fbxl3 (SCFFbxl3) complex. J. Biol. Chem. 288, 32766–32776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen J. L.; Flick K.; Papagiannis C. V.; Mathur R.; Tyrrell A.; Ouni I.; Kaake R. M.; Huang L.; Kaiser P. (2012) Signal-induced disassembly of the SCF ubiquitin ligase complex by Cdc48/p97. Mol. Cell 48, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman B. A.; Carrano A. C.; Jeffrey P. D.; Bowen Z.; Kinnucan E. R.; Finnin M. S.; Elledge S. J.; Harper J. W.; Pagano M.; Pavletich N. P. (2000) Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature 408, 381–386. [DOI] [PubMed] [Google Scholar]
- Nelson D. E.; Laman H. (2011) A competitive binding mechanism between Skp1 and exportin 1 (Crm1) controls the localization of a subset of F-box proteins. J. Biol. Chem. 286, 19804–19815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima T.; Yoshida Y.; Kumanomidou T.; Hasegawa Y.; Suzuki A.; Yamane T.; Tanaka K. (2007) Structural basis for the selection of glycosylated substrates by SCF(Fbs1) ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 104, 5777–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A.; Tanner J. J.; Henzl M. T. (2008) Energetics of OCP1-OCP2 complex formation. Biophys. Chem. 134, 64–71. [DOI] [PubMed] [Google Scholar]
- Botuyan M. V.; Koth C. M.; Mer G.; Chakrabartty A.; Conaway J. W.; Conaway R. C.; Edwards A. M.; Arrowsmith C. H.; Chazin W. J. (1999) Binding of elongin A or a von Hippel-Lindau peptide stabilizes the structure of yeast elongin C. Proc. Natl. Acad. Sci. U.S.A. 96, 9033–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan M. V.; Mer G.; Yi G. S.; Koth C. M.; Case D. A.; Edwards A. M.; Chazin W. J.; Arrowsmith C. H. (2001) Solution structure and dynamics of yeast elongin C in complex with a von Hippel-Lindau peptide. J. Mol. Biol. 312, 177–186. [DOI] [PubMed] [Google Scholar]
- van der Wel H.; Johnson J. M.; Xu Y. C.; Karunaratne C. V.; Wilson K. D.; Vohra Y.; Boons G. J.; Taylor C. M.; Bendiak B.; West C. M. (2011) Requirements for Skp1 processing by cytosolic prolyl 4(trans)-hydroxylase and α-N-acetylglucosaminyltransferase enzymes involved in O2 signaling in Dictyostelium. Biochemistry 50, 1700–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel H.; Ercan A.; West C. M. (2005) The Skp1 prolyl hydroxylase from Dictyostelium is related to the hypoxia-inducible factor-α class of animal prolyl 4-hydroxylases. J. Biol. Chem. 280, 14645–14655. [DOI] [PubMed] [Google Scholar]
- van der Wel H.; Fisher S. Z.; West C. M. (2002) A bifunctional diglycosyltransferase forms the Fucα1,2Galβ1,3-disaccharide on Skp1 in the cytoplasm of Dictyostelium. J. Biol. Chem. 277, 46527–46534. [DOI] [PubMed] [Google Scholar]
- van der Wel H.; Morris H. R.; Panico M.; Paxton T.; North S. J.; Dell A.; Thomson J. M.; West C. M. (2001) A non-Golgi α-1,2-fucosyltransferase that modifies Skp1 in the cytoplasm of Dictyostelium. J. Biol. Chem. 276, 33952–33963. [DOI] [PubMed] [Google Scholar]
- Veltman D. M.; Akar G.; Bosgraaf L.; Van Haastert P. J. M. (2009) A new set of small, extrachromosomal expression vectors for Dictyostelium discoideum. Plasmid 61, 110–118. [DOI] [PubMed] [Google Scholar]
- Sassi S.; Sweetinburgh M.; Erogul J.; Zhang P.; Teng-Umnuay P.; West C. M. (2001) Analysis of Skp1 glycosylation and nuclear enrichment in Dictyostelium. Glycobiology 11, 283–295. [DOI] [PubMed] [Google Scholar]
- West C. M.; ScottWard T.; Tengumnuay P.; vanderWel H.; Kozarov E.; Huynh A. (1996) Purification and characterization of an α-1,2-l-fucosyltransferase, which modifies the cytosolic protein FP21, from the cytosol of Dictyostelium. J. Biol. Chem. 271, 12024–12035. [DOI] [PubMed] [Google Scholar]
- Ercan A.; Panico M.; Sutton-Smith M.; Dell A.; Morris H. R.; Matta K. L.; Gay D. F.; West C. M. (2006) Molecular characterization of a novel UDP-galactose:fucoside α3-galactosyltransferase that modifies Skp1 in the cytoplasm of Dictyostelium. J. Biol. Chem. 281, 12713–12721. [DOI] [PubMed] [Google Scholar]
- Smolsky I. L.; Liu P.; Niebuhr M.; Ito K.; Weiss T. M.; Tsuruta H. (2007) Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J. Appl. Crystallogr. 40, S453–S458. [Google Scholar]
- Martel A.; Liu P.; Weiss T. M.; Niebuhr M.; Tsuruta H. (2012) An integrated high-throughput data acquisition system for biological solution X-ray scattering studies. J. Synchrotron Radiat. 19, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev P. V.; Volkov V. V.; Sokolova A. V.; Koch M. H. J.; Svergun D. I. (2003) PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282. [Google Scholar]
- Semenyuk A. V.; Svergun D. I. (1991) Gnom: A program package for small-angle scattering data-processing. J. Appl. Crystallogr. 24, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503. [Google Scholar]
- Fischer H.; Neto M. D.; Napolitano H. B.; Polikarpov I.; Craievich A. F. (2010) Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43, 101–109. [Google Scholar]
- Franke D.; Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V. V.; Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. (2002) The PyMOL molecular graphics system, DeLano Scientific, San Carlos, CA. [Google Scholar]
- Woods Group (2005–2013) GLYCAM Web, Complex Carbohydrate Research Center, University of Georgia, Athens, GA. [Google Scholar]
- Wang Z. A.; Singh D.; van der Wel H.; West C. M. (2011) Prolyl hydroxylation- and glycosylation-dependent functions of Skp1 in O2-regulated development of Dictyostelium. Dev. Biol. 349, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisman L. S.; Friedman J. H.; Neaga A.; Cobb B. A. (2007) Structure and function relations with a T-cell-activating polysaccharide antigen using circular dichroism. Glycobiology 17, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K.; Namatame H.; Taniguchi M.; Gekko K. (2009) Vacuum-ultraviolet circular dichroism analysis of glycosaminoglycans by synchrotron-radiation spectroscopy. Biosci. Biotechnol. Biochem. 73, 557–561. [DOI] [PubMed] [Google Scholar]
- Godderz L. J.; Rahman N. S.; Risinger G. M.; Arbuckle J. L.; Rodgers K. K. (2003) Self-association and conformational properties of RAG1: Implications for formation of the V(D)J recombinase. Nucleic Acids Res. 31, 2014–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens H. D.; Svergun D. I. (2010) Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 172, 128–141. [DOI] [PubMed] [Google Scholar]
- Rambo R. P.; Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M.; Weinkam P.; Sali A.; Lee K. K. (2013) All-atom ensemble modeling to analyze small-angle X-ray scattering of glycosylated proteins. Structure 21, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzl M. T.; Thalmann I.; Thalmann R. (1998) OCP2 exists as a dimer in the organ of Corti. Hear. Res. 126, 37–46. [DOI] [PubMed] [Google Scholar]
- Ng R. W.; Arooz T.; Yam C. H.; Chan I. W.; Lau A. W.; Poon R. Y. (1998) Characterization of the cullin and F-box protein partner Skp1. FEBS Lett. 438, 183–189. [DOI] [PubMed] [Google Scholar]
- Tan X.; Calderon-Villalobos L. I.; Sharon M.; Zheng C.; Robinson C. V.; Estelle M.; Zheng N. (2007) Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645. [DOI] [PubMed] [Google Scholar]
- Barchi J. J. Jr. (2013) Mucin-type glycopeptide structure in solution: Past, present, and future. Biopolymers 99, 713–723. [DOI] [PubMed] [Google Scholar]
- Borgert A.; Heimburg-Molinaro J.; Song X.; Lasanajak Y.; Ju T.; Liu M.; Thompson P.; Ragupathi G.; Barany G.; Smith D. F.; Cummings R. D.; Live D. (2012) Deciphering structural elements of mucin glycoprotein recognition. ACS Chem. Biol. 7, 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.-g.; Pasternack L.; Huang D.-H.; Koeller K. M.; Lin C.-C.; Seitz O.; Wong C.-H. (1999) Structural study on O-glycopeptides: Glycosylation-induced conformational changes of O-GlcNAc, O-LacNAc, O-sialyl-LacNAc, and O-sialyl-Lewis-X peptides of the mucin domain of MAdCAM-1. J. Am. Chem. Soc. 121, 2409–2417. [Google Scholar]
- Chen Y. X.; Du J. T.; Zhou L. X.; Liu X. H.; Zhao Y. F.; Nakanishi H.; Li Y. M. (2006) Alternative O-GlcNAcylation/O-phosphorylation of Ser16 induce different conformational disturbances to the N terminus of murine estrogen receptor β. Chem. Biol 13, 937–944. [DOI] [PubMed] [Google Scholar]
- Narimatsu Y.; Kubota T.; Furukawa S.; Morii H.; Narimatsu H.; Yamasaki K. (2010) Effect of glycosylation on cis/trans isomerization of prolines in IgA1-hinge peptide. J. Am. Chem. Soc. 132, 5548–5549. [DOI] [PubMed] [Google Scholar]
- Liu Y. C.; Yen H. Y.; Chen C. Y.; Chen C. H.; Cheng P. F.; Juan Y. H.; Chen C. H.; Khoo K. H.; Yu C. J.; Yang P. C.; Hsu T. L.; Wong C. H. (2011) Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. U.S.A. 108, 11332–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]