

Abstract
Soluble guanylate cyclase (sGC) plays a central role in the cardiovascular system and is a drug target for the treatment of pulmonary hypertension. While the three-dimensional structure of sGC is unknown, studies suggest that binding of the regulatory domain to the catalytic domain maintains sGC in an autoinhibited basal state. The activation signal, binding of NO to heme, is thought to be transmitted via the regulatory and dimerization domains to the cyclase domain and unleashes the full catalytic potential of sGC. Consequently, isolated catalytic domains should show catalytic turnover comparable to that of activated sGC. Using X-ray crystallography, activity measurements, and native mass spectrometry, we show unambiguously that human isolated catalytic domains are much less active than basal sGC, while still forming heterodimers. We identified key structural elements regulating the dimer interface and propose a novel role for residues located in an interfacial flap and a hydrogen bond network as key modulators of the orientation of the catalytic subunits. We demonstrate that even in the absence of the regulatory domain, additional sGC domains are required to guide the appropriate conformation of the catalytic subunits associated with high activity. Our data support a novel regulatory mechanism whereby sGC activity is tuned by distinct domain interactions that either promote or inhibit catalytic activity. These results further our understanding of heterodimerization and activation of sGC and open additional drug discovery routes for targeting the NO–sGC–cGMP pathway via the design of small molecules that promote a productive conformation of the catalytic subunits or disrupt inhibitory domain interactions.
The enzyme soluble guanylate cyclase (sGC) plays a key role in the cardiovascular system and is a validated drug target for the treatment of cardiovascular diseases.1−5 It catalyzes the formation of the cardioprotective signaling molecule cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP).6 Nitric oxide (NO) binds to the N-terminal regulatory domain, thereby inducing the transition from basal to activated sGC, resulting in an increased level of cGMP production. Under conditions of oxidative stress, decreased NO bioavailability7,8 and increased levels of oxidation of sGC both lead to impaired sGC activation and decreased levels of cGMP production.1 Additional physiological regulators of sGC are being discovered, such as the thrombospondin-1/CD47 signaling pathway,9 that are capable of further decreasing sGC activity.10,11 Restoring healthy levels of cGMP in the diseased state thus requires alternative ways to activate sGC. Understanding the molecular events controlling sGC activity may lead to additional classes of cGMP-modulating therapeutics. However, despite great progress in the field, sGC regulation is largely enigmatic. With this goal in mind, we aim to characterize structural changes that occur at the catalytic center during the activation process.
The sGC enzyme is a heterodimer of α and β subunits12 sharing a similar modular organization: an HNOX regulatory domain, an HNOXA domain and a coiled-coil (CC) domain involved in dimerization, and a catalytic guanylate cyclase (GC) domain, from the N- to C-terminus. Crystal structures of independent sGC domains and homologues have been determined,13−20 and recent low-resolution electron microscopy data suggest how these domains might assemble in the full-length enzyme.21 However, the high-resolution three-dimensional structure of full-length sGC is still missing. Both long-range domain–domain interactions and local short-range conformational changes were proposed to account for sGC activation. Several studies point to the inhibitory interaction of the βHNOX domain with the cyclase domain.22−25 Furthermore, the αHNOX and αHNOXA domains were shown to be located near the βHNOX domain to keep it in an inhibited conformation that is released upon NO or YC-1 binding.26,27 From these results, a collective model in which the N-terminal regulatory domains autoinhibit sGC activity was developed. If this model is correct, then isolated cyclase domains should display high levels of activity.
Here we combined X-ray crystallography, activity measurements, and native mass spectrometry analysis of the wild-type human heterodimeric catalytic domains of sGC (hereafter termed αβGC) to characterize the structural features that modulate the orientation of the catalytic subunits leading to sGC activity and to propose a novel model for sGC regulation.
Experimental Procedures
Materials
All chemicals were obtained from Sigma-Aldrich and purification columns from GE Healthcare unless otherwise indicated.
Mutagenesis, Expression, and Purification of the αβGC Heterodimer, αGC, and βGC
Entry clones for human α3GC GUCY1A3 (amino acids 466–690, GenBank accession number JX420281) with an N-terminally His-tagged thioredoxin tag in a pNH-TrxT vector and mutant β1GC GUCY1B3 (amino acids 407–619, GenBank accession number JX420282) with a C-terminally His-tagged Flag tag in a pNIC-CTHF vector were kind gifts of Dr. Allerston (Structural Genomics Consortium). The βGC G476C/C541S double mutant was changed back to the wild type using standard site-directed mutagenesis (Stratagene) with the following primer pairs: forward primer (5′-cgttactgcctgttcggc-3′) and reverse primer (5′-gccgaacaggcagtaacg-3′) for C476G and forward primer (5′-cgaaactgttggcgataagtatatga-3′) and reverse primer (5′-tcatatacttatcgccaacagtttcg-3′) for S541C.
We also designed a shorter construct for αGC, named αGC661 (residues 466–661) by inserting a stop codon at position 662 by site-directed mutagenesis with the following primer pairs: forward primer (5′-gatgcgtatcagtagtgaaccaactcaaaa-3′) and reverse primer (5′-ttttgagttggttcactactgatacgcatc-3′). All mutations were confirmed by DNA sequencing (Genewiz).
Each catalytic subunit was independently co-expressed with the GroEL-ES chaperone system from a pACYC-derived plasmid (Takara Inc.) in Escherichia coli BL21(DE3) cells (Life Technologies). Cells were cultured overnight at 37 °C. Protein expression was induced with 1 mM IPTG and l-arabinose (2 g/L) when OD600 reached 1. Cells were grown for 20 h at 15 °C, pelleted, and frozen at −80 °C until they were used. To purify the αβGC heterodimer, cell pellets from each subunit were mixed and lysed in buffer A [50 mM sodium phosphate (pH 7.4), 0.5 M NaCl, 30 mM imidazole, 0.1% (v/v) Tween 20, 0.1 mg/mL DNaseI, 0.5 mg/mL lysozyme, and protease inhibitor cocktail (Roche)] via sonication. The first chromatography step was a nickel HisTrap affinity column, and αGC and βGC co-eluted with a 15 to 100% gradient of buffer B [50 mM sodium phosphate (pH 7.4), 0.5 M NaCl, and 0.3 M imidazole]. Both proteins were cleaved overnight [in 20 mM Tris-HCl (pH 8.0), 0.15 M NaCl, and 5% glycerol] using a His-tagged tobacco etch virus (TEV) protease yielding αGC(466–690) and βGC(407–626). For βGC, the seven extra C-terminal amino acids are part of the linker for TEV cleavage of the C-terminal His-Flag tag. A second Ni affinity step was performed to remove the cleaved tags and the TEV protease. The proteins were further purified using a HiTrap Q HP anion exchange column and Superdex 200 or Superdex 75 gel filtration in a final buffer consisting of 20 mM Hepes (pH 7.5) and 0.15 M NaCl. The protein was aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C. The same protocol was used to purify all catalytic constructs. The final yields are 3.5 mg of αGC, 0.75 mg of βGC, 0.25–1 mg of copurified αβGC, 0.8 mg of αGC661, and 0.5–0.8 mg of copurified αGC661βGC per liter of cell culture.
Crystallization, Data Collection, and Refinement
Crystals of wild-type αβGC catalytic domains were grown at 20 °C using a vapor diffusion sitting drop setup containing 0.2 μL of 14 mg/mL αβGC with 0.2 μL of a reservoir solution composed of 20% (w/v) PEG3350, 50 mM HEPES (pH 7.0), and 1% Tryptone. The drops were set up using an ArtRobbins Phoenix crystallization robot. Before data collection, the crystal was cryo-protected in mother liquor supplemented with 20% (v/v) ethylene glycol and flash-frozen in the nitrogen cryo-stream. X-ray diffraction data were collected on beamline 8.3.1 at the Advanced Light Source at Lawrence Berkeley National Laboratory. Data processing and reduction were conducted with MOSFLM.28 Phasing by molecular replacement was conducted with PHASER29 using the mutant αβGC structure [Protein Data Bank (PDB) entry 3UVJ] as a starting model. The final model was obtained with iterative cycles of refinement with PHENIX30 and rebuilding with COOT.31 The final model and structure factors were deposited in the PDB (entry 4NI2).
Activity Assay
The cyclase activity reaction was performed using ∼5 μM full-length sGC (based on the absorbance at 431 nm) and ∼10 μM catalytic constructs (based on the absorbance at 280 nm). The reaction assay was conducted in 40 mM HEPES (pH 7.4), 0.5 mM dithiothreitol, 0.3 mM 3-isobutyl-1-methylxanthine, 1 mM GTP, and 3 mM MgCl2 or MnCl2. The reaction mixture was incubated at 15 °C for 20 min and the reaction stopped with the addition of EDTA (final concentration of 10 mM). The reaction mixture was stored at −80 °C until cGMP was quantified using a cGMP enzyme immunoassay kit (R&D) following the manufacturer’s protocol. Activity was measured in duplicate, and the experiments were repeated three times to ensure reproducibility.
Multiangle Light Scattering
The KW 403 gel filtration column was equilibrated with buffer A [50 mM Tris-HCl (pH 8.0), 0.15 M NaCl, 5% glycerol, 1 mM MgCl2, and 1 mM TCEP]. Bovine serum albumin was used as a reference. We injected 50 μL of αβGC at 30 μM.
Mass Spectrometry (MS)
The protein samples were buffer exchanged into 0.1 M ammonium acetate (AA) buffer (pH 7.4) using either size-exclusion chromatography spin columns (Bio-Rad) or Amicon Ultra-4 centrifugal filter units (EMD Millipore). Subsequently, the concentrations of the buffer-exchanged proteins were calculated by measuring the A280, and a solution of 0.1 M triethylammonium acetate (TEAA) was added to the protein samples in a 1:4 (TEAA:AA) ratio to produce charge-reduced catalytic dimers, unless otherwise stated. In the KD analysis of the catalytic dimers, various protein concentrations were obtained by serial dilution starting from the stock solution of the buffer-exchanged protein sample. For all other mass spectrometry analyses, the concentration of catalytic dimers used was 10 μM, unless otherwise stated.
Nano-electrospray ionization mass spectrometry (nano-ESI/MS) analysis was conducted by utilizing a modified Quadrupole Ion Mobility Time of Flight (Q-IM-TOF) instrument (Synapt G2, Waters Corp., Manchester, U.K.) with a customized surface-induced dissociation (SID) device installed before the IM chamber as previously described.32 All experiments were conducted using a nanoelectrospray source, using a capillary voltage of 1.0–1.5 kV and a cone voltage of 50–75 V. No heating was applied to the cone. Nano-ESI glass capillary and surface preparation procedures can be found elsewhere.33 The following instrumental conditions were used: 5 mbar for the source/backing pressure, 2 mbar for the nitrogen gas pressure in the IM cell, rate of 120 mL/min for the flow of gas into the helium cell, ∼6 × 10–7 mbar in the TOF analyzer, and a wave velocity and a height of 300 ms–1 and 20 V, respectively, for IM experiments. Nano-ESI/MS is a gentle method of ionization in which salts and solvent are often retained, giving m/z values higher than those calculated from sequence, especially for oligomers. This problem is sometimes eliminated in nano-ESI–MS/MS measurements of fragments, if the MS/MS approach unfolds the monomers causing a loss of salts, solvents, and ligands.
Results
Overall Structure of Wild-Type Human Heterodimeric αβGC and Comparison with Mutant Heterodimeric αβGC
To identify the structural determinants for sGC catalytic activity, we determined the X-ray structure of the wild-type heterodimeric αβGC catalytic domains of human sGC in the apo form at 1.9 Å resolution (Figure 1). The final model shows good statistics with Rwork and Rfree values of 0.159 and 0.198, respectively, and 98.2% of the residues in the allowed regions of the Ramachandran plot (Table 1). The model comprises one heterodimer per asymmetric unit and contains αGC residues 471–662, βGC residues 411–608, 342 water molecules, and six ethylene glycol molecules. Residues at the N- and C-termini of αGC (466–470 and 663–690, respectively) and βGC (407–410 and 609–626, respectively) were not visible in the electron density and were not included in the final model. To rule out the possibility that these proteins may be proteolyzed or degraded and to unambiguously determine the masses of the purified proteins, we performed nano-ESI/MS analysis on the αGC and βGC proteins purified individually or copurified (Table 2, Supporting Information, and Figure S1 of the Supporting Information). We confirmed that we crystallized heterodimeric αβGC with a truncated αGC(466–662) subunit.
Figure 1.

Overall structure of heterodimeric wild-type αβGC catalytic domains that resembles the Chinese yin-yang symbol with both subunits arranged in a head-to-tail conformation. (A and B) Ventral face of the heterodimer as a cartoon (A) and a solvent accessible surface representation (B). The deep extended substrate groove (shown by the oval in panel B) bisects the ventral face, which contains the C-termini of both subunits (denoted with C). The αGC subunit is colored blue, and the βGC subunit is colored orange. The substrate binding regions of αGC (residues 523–534) and βGC (residues 470–480) adopt an extended conformation in the absence of substrate and metals (arrows). (C and D) The dorsal face of the heterodimer as a cartoon (C) and a solvent accessible surface representation (D) is flatter than the ventral face and contains the N-termini of both subunits (denoted with N).
Table 1. X-ray Data Collection and Refinement Statistics.
| Data Collectiond | |
| space group | P212121 |
| wavelength (Å) | 1.116 |
| resolution (Å) | 69.7–1.9 (2.0–1.9) |
| unit cell parameters (Å) | a = 49.5, b = 55.8, c = 139.4 |
| no. of measurements | 240390 (31623) |
| no. of unique reflections | 31270 (4438) |
| redundancy | 7.7 (7.1) |
| completeness (%) | 99.8 (98.7) |
| ⟨I/σ(I)⟩ | 14.2 (2.6) |
| Rmerge (%)a | 8.6 (68) |
| Refinementd | |
| resolution range (Å) | 69.7–1.9 (1.96–1.90) |
| no. of protein atoms | 2964 |
| no. of water atoms | 342 |
| no. of heteroatoms | 30 |
| rmsd of bond lengths (Å) | 0.012 |
| rmsd of bond angles (deg) | 1.3 |
| Rwork (%)b | 15.9 (23.8) |
| Rfree (%)c | 19.8 (26.8) |
| Ramachandran plot (%) | |
| favored | 98.2 |
| allowed | 0.8 |
| generous | 0.0 |
| disallowed | 0.0 |
Rmerge = ∑h∑i|I(h,i) — ⟨I(h)⟩|/∑h∑iI(h,i), where I(h,i) is the intensity of the ith observation of reflection h and ⟨I(h)⟩ is the average intensity of redundant measurements of reflection h.
Rwork = ∑h||Fobs| – |Fcalc||/∑h|Fobs|.
Rfree = ∑h||Fobs| – |Fcalc||/∑h|Fobs| for 5% of the reserved reflections.
Values in parentheses apply to the highest-resolution shell.
Table 2. Comparison of Calculated and Experimental Molecular Masses for the Different sGC Constructs Determined by Nano-ESI/MS and Nano-ESI/MS/MS.
| construct | calculated mass (Da)a | calculated mass (Da)b | experimental mass (Da) |
|---|---|---|---|
| αGC(466–690) (monomer) | 24671.2 | 24687.5 | 24740.8 ± 17.9 (10 μM)e |
| αGC(466–661) (monomer)c | 21621.7 | 21636.1 | 21642.9 ± 18.3 (10 μM)e |
| αGC(466–690) (dimer) | 49342.4 | 49375 | 49489 ± 20.3 (10 μM)e |
| αGC(466–662) (dimer)d | 43499.6 | 43528.6 | 43500.1 ± 55.4 (10 μM) |
| αGC(466–661) (dimer) | 43243.4 | 43272.2 | 43403.1 ± 5.5 (10 μM) |
| βGC(407–626) (monomer) | 24769.2 | 24785.2 | 24820.5 ± 47.8 (10 μM)e |
| βGC(407–626) (dimer) | 49538.4 | 49570.4 | 49636.7 ± 36.6 (10 μM) |
| βGC(407–626) (tetramer) | 99076.8 | 99140.8 | 99929.9 ± 68.7 (10 μM) |
| αβGC (heterodimer) |
49440.4 | 49472.7 | 49660.2 ± 38.7 (10 μM) |
| αGC661βGC (heterodimer)c | 46390.9 | 46421.3 | 46449.5 ± 20.2 (10 μM) |
Monoisotopic masses were calculated from amino acid sequences with PeptideMass66 from the Expasy Web site.
Average masses were calculated from amino acid sequences with PeptideMass66 from the Expasy Web site.
Species obtained by introducing a stop codon at position 662 in the αGC(466–690) construct.
Species obtained by cleavage of αGC(466–690) at the N- and C-terminal TEV cleavage sites.
Measured mass obtained from nano-ESI/MS/MS at a SID voltage of 100 V.
The structure of heterodimeric αβGC resembles the Chinese yin-yang symbol with the two subunits arranged in a head-to-tail conformation (Figures 1A,C). The catalytic domains were crystallized without metal or substrate. Accordingly, the structure reveals an inactive heterodimer conformation compared to that of active adenylate cyclase.34 This is evidenced both by the relative orientation of the two subunits leading to an open active site and by the extended loop conformation of the core regions containing the catalytic residues (Figure 1A).
We superimposed our structure on that of the mutant heterodimeric human αβGC containing an engineered disulfide bridge (αGC C595–C476 βGC) at the dimer interface.20 Aside from the mutations (G476C and C541A), the two structures are very similar, with an rmsd of 0.52 Å for 397 amino acids, but present subtle differences at the dimer interface (Movie S1 of the Supporting Information) and in surface-exposed regions (a detailed description of similarities and differences is presented in the Supporting Information). As a result, the αGC subunit and the βGC subunit present a slightly different orientation relative to each other in our structure. Overall, the αGC and βGC subunits are rotated by ∼3° in the structure of the wild-type catalytic domains compared to the mutant structure. While these differences may seem subtle, similar conformational changes are observed in related adenylate cyclase between the inactive and active structures, in which one of the subunit rotates by 7° and secondary structure elements shift by 1–2 Å.34 This example illustrates how small structural changes at the dimer interface in this protein family have profound effects on catalytic activity. In comparison to the structure of the mutant catalytic domains of sGC, our structure reveals an αGC−βGC interface with high plasticity that is necessary for sGC activity. Regardless, our structure is the closest to that of the catalytic domains in full-length sGC, as it is heterodimeric and devoid of mutations. As such, it represents an excellent starting model for the docking of small molecules that either inhibit or activate sGC.
Amino Acid Sequence Conservation Suggests Docking Sites for Other sGC Domains and Key Interfaces for Activation of sGC
To identify conserved regions in the catalytic heterodimer, we performed multiple-sequence alignments for both the αGC and βGC subunits (Supporting Information) and mapped the sequence identity percentage of each residue onto the αβGC structure (Figure 2). The regions with the highest degrees of sequence conservation are located in three regions of the heterodimer: (i) the substrate channel on the ventral face of the αβGC heterodimer (94% identical sequence), (ii) the C-terminal subdomain of αGC present on the ventral face of the heterodimer (78% identical sequence), and (iii) the dorsal face of the heterodimer (94% identical sequence). While the substrate channel is expected to be mainly invariant, the high level of sequence conservation of the αGC C-terminal domain (residues 616–662) is surprising and suggests that this domain may play an important role in the assembly and/or regulation of sGC. This hypothesis is supported by recent studies showing that the βHNOX–HNOXA domains directly interact with the C-terminal domain of αGC23,35 and could modulate the conformation of the active site. Interestingly, mutation of solvent-exposed residue αGC Arg624 in that domain to Ala leads to a dramatic increase in full-length sGC activity.36 It is tempting to speculate that this residue plays a key role in domain–domain interaction with the N-terminal regulatory domains. Importantly, this region of the catalytic subunit could be targeted for the rational design of small molecules or protein therapeutics disrupting the inhibitory interactions between the regulatory and catalytic domains.
Figure 2.
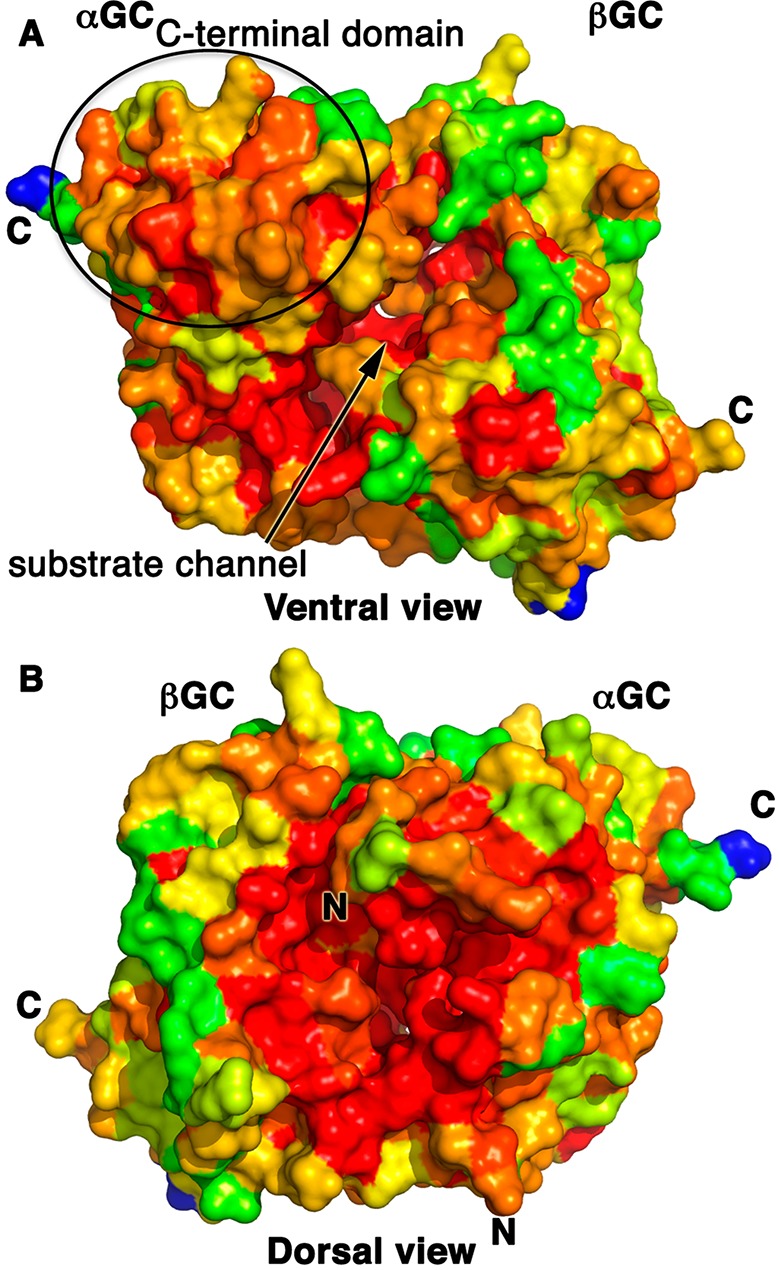
Map of the high levels of sequence conservation on the ventral face substrate channel and the αGC C-terminal domain and the heterodimer cleft on the dorsal face of the αβGC structure. We aligned >20 sequences of eukaryotic αGC and βGC domains (Supporting Information) and mapped the level of amino acid sequence conservation onto the αβGC crystal structure, colored from blue (0% identical) to red (100% identical). C-Termini of both subunits are marked with C, and N-termini are marked with N. (A) The highly conserved substrate channel on the ventral face is marked with an arrow. The C-terminal subdomain of αGC is moderately to strongly conserved (orange to red) as indicated by an oval. (B) The dimer interface region close to the N-termini of both subunits on the dorsal face is largely invariant.
Second, the highly conserved region of the dorsal face of αβGC serves as the point of contact with the preceding αβCC dimerization domain, where it could modulate the interface between the αGC and βGC subunits. There is no crystal structure for heterodimeric αβCC, but recent studies unambiguously demonstrated a parallel orientation of the CC domains in sGC.21,26 In this arrangement, both C-termini of αβCC would be able to connect with the N-termini of the αβGC heterodimer separated by ∼28 Å on the dorsal face of the heterodimer and located in the region with the highest level of sequence conservation. This position of αβCC is consistent with recent mass spectrometry23 and FRET data.25 This model is also coherent with the CC domain acting as a platform on which all the sGC domains assemble in full-length sGC.26 The fact that the entire region surrounding the N-termini of the αβGC heterodimer is highly conserved in the sGC family points to more than simply an anchoring area for the preceding CC domain. Instead, we propose that the interface between the cyclase and the CC domains is essential for catalysis and that the CC promotes an optimal conformation of the catalytic subunits for activity.
Structural Determinants for Heterodimerization of the Catalytic Subunits
Our crystallization trials with copurified wild-type αβGC protein yielded mostly homodimeric β1β2GC crystals (98%) and some heterodimeric αβGC crystals (2%). Others also noted difficulties in obtaining heterodimer crystals of the wild-type catalytic domains and engineered a non-natural disulfide bridge at the interface to favor heterodimerization.20 This suggests that β1β2GC has a stronger propensity to crystallize and/or that the β1β2GC interface is stronger. To address the first point, we performed native mass spectrometry and crystal packing analysis (Supporting Information) and showed that, in our case, TEV cleavage at the N-terminal and C-terminal sites in αGC yielded a minor population of αGC662βGC heterodimers that were subsequently crystallized, thus explaining our low success in crystallizing αβGC heterodimers (Supporting Information). Second, to identify the structural determinants promoting heterodimerization of the catalytic subunits, we compared the heterodimeric αβGC structure with the homodimeric β1β2GC structure (PDB entry 2WZ1) and analyzed both dimeric interfaces (Supporting Information). The structural superposition shows that the β2GC subunit of the homodimer and the βGC subunit of the heterodimer overlay closely, while the β1GC subunit of the homodimer and the αGC subunit of the heterodimer present distinct orientations (Figure 3A). In the heterodimer, αGC is rotated ∼13° compared to β1GC in the homodimer, resulting in a different dimer interface. Both the heterodimeric and homodimeric interfaces contain a large number of hydrophobic interactions stabilizing the dimer core formed by both subunits and lining the substrate-binding site (Table S1 of the Supporting Information and Figure 3C,D). In addition, both interfaces contain several polar interactions between surface flexible structural elements (Figure 3E,F).
Figure 3.

Structural determinants of catalytic domain dimerization. (A and B) Homodimeric β1β2GC (PDB entry 2WZ1) is colored light green (β1GC) and dark green (β2GC). The αβGC heterodimer is colored blue (αGC) and orange (βGC). The ventral view (A) shows that βGC and β2GC subunits superimpose well, while αGC adopts a conformation different from that of β1GC in the heterodimer and homodimer, respectively. The dorsal view (B) shows the double flap-wrap conformation of the homodimer flaps (light and dark green cartoon). In the heterodimer depicted as a semitransparent solvent accessible surface, only the βGC flap (orange cartoon) wraps on the αGC subunit while the αGC flap (blue cartoon) is flipped out. (C and D) The core of the heterodimeric interface is formed by numerous hydrophobic interactions between residues from the βGC subunit (orange) and the αGC subunit (blue), depicted as sticks. (E and F) Polar interactions (hydrogen bonds and salt bridges) also contribute to the heterodimeric interface. Residues from βGC (orange) and αGC (blue) participating in these interactions are shown as sticks.
Despite the resemblance between the two dimeric interfaces, one major difference between the two structures is the conformation of the β-hairpins of residues 532–539 in β1GC and 587–593 in αGC. We will herein refer to these hairpins as “flaps”. In the symmetrical homodimer, both flaps—one contributed by each βGC subunit—participate in the dimer interface by wrapping securely onto the α2 helix on the partner subunit, in a “double flap-wrap” conformation (Figure 3B). Residues from both flaps contribute one salt bridge and ten of the thirteen polar interactions that stabilize the homodimer. In the heterodimer, residues from the β1GC flap contribute three of the nine hydrogen bonds that stabilize the dimer (Table S1 of the Supporting Information), while the α1GC flap is swung away and makes no interaction with the βGC subunit (Figure 3B). This suggests a different role of the flaps in the homodimer and the heterodimer. We further performed a structural comparison of all guanylate cyclase and adenylate cyclase catalytic domain structures (Supporting Information) and proposed that these surface-exposed structural flaps not only are involved in regulating the relative orientations of the subunits in various dimers but also may be important in modulating interactions with other domains or proteins. For sGC, our results indicate a key role of the interfacial flaps in stabilizing different dimer interfaces and suggest that the flaps may also be important for the proper orientation of the catalytic subunits in full-length sGC (see below).
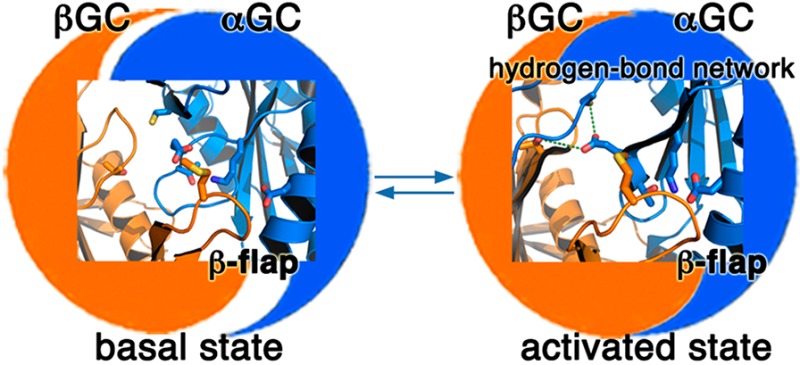
Residues in the Interfacial Flap and Hydrogen Bond Network Play Key Roles in Modulating the Dimer Interface
To begin to understand the mechanisms involved in modulating sGC activity, we mapped sGC residues that have been shown to significantly impact activity and activation on our structure of the inactive heterodimer (Figures 4A,B) and our model for the active heterodimer (Figures 4C,D). These residues fall in two areas of the dimer interface. (a) The first includes residues located in the flap or in the region interacting with the flap on the partner subunit. In full-length sGC, the Met537Asn mutation in βGC leads to increased constitutive activity and a very strong response to NO and/or YC-1 activators.37 Met537 is located on the βGC flap, and its position is not expected to change drastically between the inactive and active conformation. However, reorientation of αGC in the active heterodimer leads to movement of its α2 helix and β3 strand closer to the βGC flap and places Met537 closer to hydrogen-bonding residues αGC Lys524 and Thr527 located on the β3 strand (Figures 4B,D). Thus, the Met537Asn mutation would enhance interactions between the βGC flap and αGC by providing extra hydrogen bond contacts between the two subunits. This hypothesis is strongly supported by mutagenesis studies showing that two mutations abolishing hydrogen bonds between αGC and the βGC flap (αGC Lys524Ala and αGC Asp514Ala) in full-length sGC impair dimerization and activity.36 Similar mutations introduced into adenylate cyclase yielded similar phenotypes. The C2a Lys1014Asn mutation (analogous to the βGC Met537Asn mutation) increased activity and affinity between the two subunits in the absence of forskolin or Gsα.38 In addition, the C1a Asp424Ala mutation (analogous to the αGC Asp514Ala mutation) severely impaired activity and activation.36 (b) Other residues modulating sGC activity are located in the core of the heterodimer at the interface between both subunits. The αGC residues Glu526 and Cys595 are located in interfacial regions that would undergo major conformational changes in the active form (Figure 4B). On the basis of the structure of active AC,34 we hypothesize that αGC Glu526, αGC Cys595, and βGC Thr474 form an interfacial hydrogen bond network (equivalent to the C1a Lys436–C1a Asp505–C2a Thr939 adenylate cyclase triad), as these residues are also in the proximity of each other in the modeled catalytic domain active structure (within 3.5 Å of each other). This is strongly supported by studies showing that mutations that abrogate the ability to form hydrogen bonds at this interface severely impair sGC activity (αGC Cys595Tyr, αGC Cys595Asp, αGC Glu526Ala, and αGC Cys595Asp/Glu526Lys)36,37 and adenylate cyclase activity (C1a Lys436Ala and C1a Asp505Ala).38,39 In contrast, the αGC Cys595Ser mutation dramatically increases sGC basal activity.40 The Cys to Ser substitution is often described as “silent”. However, the two amino acids have distinct properties because of differences in size and polarity between oxygen and sulfur. Thus, it is possible that the Cys595Ser mutation would enhance the ability of this residue to form hydrogen bonds.41 While we cannot rule out the possibility that the Cys595Ser mutation may prevent Cys595 oxidation and subsequent enzyme inhibition, it is more likely that Cys595 plays a key role in the communication between the two subunits. Previous studies proposed that Cys595 may participate in binding of the sGC stimulator YC-1 and modulate YC-1/NO activation.37 However, several groups have unambiguously ruled out the possibility of binding of YC-1 to the catalytic domains.27,42−49 Instead, we propose that both the interfacial flap and the hydrogen bond network enhance interactions between the two catalytic subunits and guide an optimal conformation of the active center necessary for activity.
Figure 4.

Interfacial mutations in adenylate cyclase and sGC that modulate enzyme activity underscore the key role of the heterodimeric interface for catalysis. We mapped AC and sGC mutations on the structures of the inactive αβGC heterodimer (A and B; PDB entry 4NI2) and the modeled active structure of αβGC (C and D). The model for the active conformation was generated with SWISSMODEL67 by using active adenylate cyclase (PDB entry 1CJU) as a template. Residues that affect sGC or AC activity map to two regions of the αβGC heterodimer: (i) the interfacial hydrogen bond network among sGC residues αGC Cys595, αGC Glu526, and βGC Thr 474 (indicated by dashed green lines) and (ii) the sGC flap region (βGC Met537) or the region interacting with the flap in the partner subunit (αGC Asp514). Panels B and D are close-up views of panels A and C, respectively.
Copurified αβGC Catalytic Domains Assemble as a Mixture of Monomers, Homodimers, and Heterodimers
To confirm our structural prediction that the homodimeric β1β2GC interface is stronger than the heterodimeric αβGC interface and the ααGC interface, we used size-exclusion chromatography coupled to multiangle light scattering (SEC-MALS) and nano-ESI/MS (Figure S1 of the Supporting Information). Both the SEC (Figure S2A of the Supporting Information) and SEC-MALS profiles (Figure S2C of the Supporting Information) showed subtle deviations from perfectly symmetric peaks at higher elution volumes, suggesting slight heterogeneity in the sample composition. The molecular weight determined by SEC-MALS ranged from 96 to 106% of the theoretical molecular weight for the αβGC heterodimer. The subtle tail of the elution peak at high elution volumes contained more αGC than βGC as determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). This suggested that the αβGC sample is a mixture of different oligomeric species in solution. However, we were unable to distinguish the different species in solution by SEC, MALS, or even mass spectrometry. Our result is not surprising given our overwhelming success in crystallizing ββGC homodimers over αβGC heterodimers and has important implications when working with truncated sGC domains (see below).
Design of the Shorter αGC661βGC Protein Allows Precise Quantification of Heterodimers in Solution
We showed above that the copurified αβGC protein is heterogeneous both in the length of the αGC subunit and in oligomer composition and that we were unable to quantify the different species in solution. This result is problematic for two reasons. First, structural mechanistic studies of the αβGC catalytic domains in the presence of metals, GTP, or GTP analogues require a reproducible homogeneous heterodimeric sample, and second, specific activity calculations require precise quantification of the αβGC heterodimers in solution, as only heterodimers are catalytically active.
Mass spectrometry and crystal packing analysis suggested that we had crystallized the heterodimeric αβGC catalytic domains with a truncated αGC(466–662) subunit (Supporting Information). To increase the likelihood of obtaining high-quality crystals of heterodimeric αβGC, we designed a shorter αGC construct. The XtalPred and DISOPRED2 web servers50 predicted the region of residues 662–690 of αGC to be disordered. Therefore, we designed the shorter αGC661 construct that encompasses residues 466–661 after TEV cleavage of the N-terminal tag. The size-exclusion profile of copurified αGC661βGC showed two overlapping peaks (Figure S2B of the Supporting Information) identified as homodimers and heterodimers by SDS–PAGE.
The calculated molecular masses for αGC661 and βGC differ by ∼3 kDa (Table 2), allowing us to discriminate all species present in the αGC661βGC sample using mass spectrometry. First, we performed nano-ESI/MS for each subunit purified independently. Like that of αGC (Figure S1A of the Supporting Information), the spectrum for αGC661 showed that the protein was predominantly monomeric (mass of 21642.9 ± 18.3 Da) with a small amount of homodimers (mass of 43403.1 ± 5.5 Da) in a ratio of 79:21, as determined by AUC analysis (Figure 5A). As seen previously, the spectrum for βGC and AUC analysis showed that the monomer:dimer:tetramer ratio was 8:75:17 (Figure 5B). These results confirmed our prediction based on the crystal structure and modeling, and our size-exclusion experiments, and showed that the KD for homodimerization of αGC661 was higher than that of βGC.
Figure 5.

Nano-ESI/MS and nano-ESI/MS/MS (SID) reveal different oligomeric species present in αGC661, βGC, and αGC661βGC samples. (A–C) Nano-ESI/MS spectra were obtained by spraying a 10 μM protein sample in 0.1 M NH4OAc (pH 7.4). (A) αGC661 exists mostly as a monomer. (B) βGC exists predominantly as a dimer, with a small proportion of monomers and tetramers also present. (C) αGC661βGC is present in approximately equal amounts of monomer and dimer. The inset represents the spectrum for αGC661βGC (10 μM) in 80 mM NH4OAc and 20 mM TEAA (pH 7.4). The major species present was the αGC661βGC heterodimer. αGC661 and βGC monomers and ααGC661 and ββGC homodimers were also present. (D–F) Samples (10 μM) in 80 mM NH4OAc and 20 mM TEAA were sprayed, and the +11 precursor ion was chosen for MS/MS analysis at an SID voltage of 100 V. (D) αGC661 and (E) βGC dimers dissociate to give αGC661 and βGC monomers, respectively. (F) Dissociation of αGC661βGC results in an equal population of αGC661 and βGC monomers.
Second, we performed nano-ESI/MS for copurified αGC661βGC (Figure 5C). The complex spectrum showed a mixture of αGC661 monomers, βGC monomers, and αGC661βGC heterodimers, with an experimental mass for the heterodimer of 46449.5 ± 20.2 Da, which is close to the calculated value. After the addition of TEAA to reduce the overall charge of the complex and better separate the different charged species (Figure 5C, inset), the spectrum showed a mixture of αGC661 monomers, βGC monomers, ααGC661 homodimers, ββGC homodimers, and αGC661βGC heterodimers. We further used SID to confirm the identity of the different peaks (Figure 5D–F). These results unambiguously confirmed that the copurified αGC661βGC protein is a complex mixture of different oligomeric species.
To estimate the relative abundance of each species, we measured nano-ESI/MS spectra for αGC661, βGC, and αGC661βGC proteins at various protein concentrations. For αGC661, we observed only monomers and dimers. Thus, the KD for αGC661 homodimerization was defined as the concentration at which the concentrations of the ααGC661 dimer and the αGC661 monomer are equal and was determined to be 30 μM (Figure S3A of the Supporting Information). For βGC, we observed several species in solution, including monomers, dimers, and tetramers, complicating the determination of KD. Furthermore, the ionization efficiency of the βGC protein was significantly reduced at low concentrations. As a result, we can only estimate that the KD for the ββGC dimer is <2 μM (Figure S3B of the Supporting Information). We repeated this analysis for various concentrations of αGC661βGC (Figure 6). First, we noted that at high concentrations, the relative abundance of all αGC species is almost equal to that of all βGC species. However, at low concentrations, the fraction of αGC is much higher than that of βGC. This may be due to the decreased ionization efficiency of βGC at low concentrations, as mentioned above. Therefore, we postulated that the KD for the αGC661βGC heterodimer can be determined from the point at which the relative abundances of αGC and αGC661βGC are equal, which corresponds to a KD of ∼6.9 μM. Finally, mass spectrometry analysis showed that mixing independently purified αGC661 and βGC subunits yielded less αGC661βGC heterodimer than purifying them together (data not shown).
Figure 6.
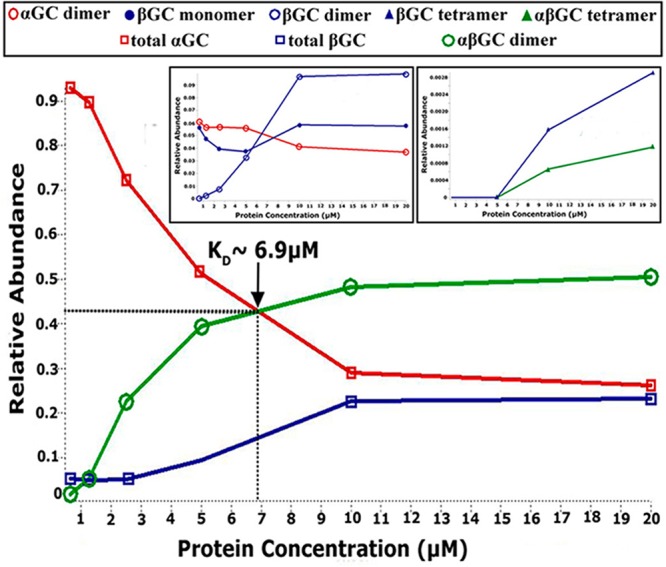
Determination of KD for αGC661βGC heterodimers by nano-ESI/MS. Spectra for αGC661βGC were obtained by spraying the protein sample in 80 mM NH4OAc and 20 mM TEAA (pH 7.4). The concentration of αGC661βGC was varied by performing serial dilutions from an initial 50 μM stock solution over a range of 0.6–20 μM. The main panel shows the changes in the relative abundance of αGC661βGC, total αGC661, and total βGC as a function of protein concentration. The insets show the relative abundance of the minor species in the αGC661βGC sample as the concentration changes. For all experiments, the abundance of a particular species was determined by extracting its intensity from the ion mobility mobilogram that is produced with the mass spectrum, and the area under the curve (AUC) was determined using Origin.
Overall, the use of the truncated αGC661 construct allowed us to determine the KD for the αGC661βGC heterodimer and quantify the amount of heterodimers present in solution at any given concentration to calculate the true specific activity normalized per mole of heterodimer. We firmly established that the monomer–dimer dissociation constants for the catalytic domains increase as follows: ββGC < αβGC ≪ ααGC. This is in contrast to earlier studies suggesting that the KD of ββGC or ααGC homodimers was much higher than the KD of the αβGC heterodimer.22 These conflicting results may be due to distinct techniques used to characterize oligomeric assemblies. As opposed to low-resolution size-exclusion chromatography used previously,22 we used native mass spectrometry coupled to SID, which affords high accuracy in the stoichiometry of noncovalent complexes while preserving a nativelike quaternary structure.51 Importantly, our results support and extend a recent study showing that homodimers and heterodimers of the catalytic domains are both present in solution.20
Our results have important implications for activity assay measurements with truncated sGC constructs commonly used by various laboratories. (i) A protein concentration well above the KD should be chosen to ensure adequate heterodimer formation in the reaction mixture, and (ii) the true specific activity should be calculated on the basis of the heterodimer concentration only, as monomers and homodimers display no activity but contribute to the total protein concentration. While these requirements may not apply to wild-type full-length sGC, which mostly forms heterodimers in vitro, these results are critical for accurate activity assay measurements with mutant full-length sGC that show impaired dimerization.24,36,52−54
Finally, our mass spectrometry approach to determine dimerization dissociation constants can be applied to truncated sGC constructs, encompassing various domains to precisely determine the contribution of each domain to heterodimerization of full-length sGC. Dimerization determinants were shown to be predominantly contained in the HNOXA and coiled-coil (CC) domains,24,42,53−56 with some contribution from the catalytic domains.22,57 However, X-ray structures of homodimers were determined for HNOXA and CC domains,16,57 suggesting that, under these conditions, both domains can also homodimerize with KD values that were estimated to be in the micromolar range.16,57 While a major role for the CC domain was proposed,57 the precise determinants favoring sGC heterodimerization remain ambiguous and warrant further studies.
Isolated Catalytic Domains Display Low Activity Despite Forming Heterodimers
Because we crystallized the apo αβGC heterodimer, we wanted to confirm that the isolated catalytic domains were nonetheless active. Others have shown that the regulatory N-terminal βHNOX–HNOXA domains inhibit the activity of the catalytic domains.22,23 Thus, in the absence of the regulatory domains, the isolated αβGC domains should be as active as activated full-length sGC. We measured cGMP formation with our purified αβGC and αGC661βGC proteins, and human full-length sGC that was overexpressed in baculovirus (kind gift from E. Martin, The University of Texas Health Science Center at Houston, Houston, TX). We used αGC protein purified independently as a negative control.
The results presented above show that the copurified αβGC protein is a heterogeneous mixture of monomers, homodimers, and heterodimers. Because heterodimeric αβGC is the only catalytically active species, the true specific activity should be calculated by normalizing to the heterodimer concentration. This has proven to be difficult with wild-type αβGC protein (see above). The design of the truncated αGC661βGC construct has allowed us to calculate the ratio of heterodimers at different total protein concentrations. First, we showed that αGC661βGC [1.2 + 0.4 or – 0.3 fmol of cGMP min–1 (pmol of enzyme)−1] and untruncated αβGC catalytic domains [1.8 + 0.7 or – 0.5 fmol of cGMP min–1 (pmol of enzyme)−1] display comparable activity. Thus, the remainder of the analysis is based on αGC661βGC for which we can calculate the true specific activity by normalizing to heterodimer concentration based on mass spectrometry data. For full-length sGC, mass spectrometry analysis showed that the protein is 100% heterodimeric at the concentration used for the cGMP reaction (data not shown). Importantly, copurified αGC661βGC displayed only a fraction (0.01%) of the specific activity of basal full-length sGC in the presence of Mg2+ (Table 3). In the presence of Mn2+, the activities of αGC661βGC and full-length sGC increased (779- and 2-fold, respectively), but αGC661βGC still displayed <6% of the activity of full-length sGC. These results suggest that the isolated catalytic domains are not catalytically competent compared to basal full-length sGC, despite their ability to heterodimerize. Our structural studies of αβGC catalytic domains corroborate this result, as we crystallized the heterodimer in an inactive conformation. In the related adenylate cyclase, binding of Gsα, forskolin, and ATP induces conformational changes leading to a closed and active catalytic center.34 A similar mechanism is likely for sGC, whereby the catalytic center alternates between inactive and active conformations via structural rearrangements.
Table 3. Guanylate Cyclase Activitya.
| αGC661βGC |
basal
full-length sGC |
αGC | |||
|---|---|---|---|---|---|
| Mg2+ | Mn2+ | Mg2+ | Mn2+ | Mg2+ | |
| specific activity [fmol of cGMP min–1 (pmol of enzyme)−1] | 1.2 | 899.9 | 10075.5 | 20578.5 | 0.5 |
| upper bound errorb | 0.4 | 259.8 | 3163.7 | 5819.0 | 0.3 |
| lower bound errorb | 0.3 | 200.7 | 2300.2 | 4433.3 | 0.2 |
| relative abundance of the heterodimer (%)c | 82.5 | 82.5 | 100 | 100 | NAd |
| adjusted specific activity [fmol of cGMP min–1 (pmol of heterodimer)−1] | 1.4 | 1090.8 | 10075.5 | 20578.5 | NAd |
| upper bound errorb | 0.5 | 314.9 | 3163.7 | 5819.0 | NAd |
| lower bound errorb | 0.4 | 243.2 | 2300.2 | 4433.3 | NAd |
| activity normalized to full-length sGC (%) | 0.01 | 5.3 | 100 | 100 | NAd |
| x-fold increase in specific activity with Mn2+ | 1 | 779 | 1 | 2 | NAd |
The reaction was performed as described in Experimental Procedures.
Error boundaries describe the 95% confidence interval.
Determined by mass spectrometry.
Not applicable.
We propose that other sGC domains (including the coiled coil) will guide conformational changes leading to an optimal alignment of the active site residues. This model is supported by recent data showing that an αβHNOXA-CC-GC construct displays activity comparable to that of full-length sGC,58 and by our results showing that substitution of Mg2+ with Mn2+ increases the basal activity of full-length sGC and αGC661βGC (Table 3). We propose that Mn2+ ions allow catalysis in both proteins without the need for the catalytic domains to undergo the transition to an optimal conformation. While Mg2+ requires a very specific coordination geometry,59 Mn2+ has been shown to be less stringent.60 In DNA polymerases, Mn2+ easily replaces Mg2+ but it also allows reactions to occur with mutated catalytic residues and decreased substrate specificity because of its higher tolerance for suboptimal metal coordination.61 Similarly, for full-length sGC and isolated αβGC catalytic domains, Mn2+ may favor a suboptimal conformation of the catalytic domains. This hypothesis is supported by studies showing that NO and/or YC-1 fails to fully activate sGC in the presence of Mn2+,40,55,62,63 suggesting that the enzyme is locked in an intermediate catalytic state. In conclusion, our activity measurements strongly support a role for other sGC domains in promoting a competent conformation of the active center.
Discussion
The combined structural, biophysical, and mass spectrometry analyses presented here clarify widely accepted ideas regarding heterodimerization and activity of the isolated catalytic subunits of sGC and further provide the basis for understanding the regulation of sGC catalytic activity. We propose a novel role for interfacial structural elements in modulating the conformation of the active site for optimal activity. Finally, our results allow us to propose a comprehensive regulatory model in which distinct domain–domain interactions in sGC prevent or guide an optimal conformation of the sGC catalytic center associated with high activity.
Overall Structure of αβGC Catalytic Domains and Comparison with the Mutant Structure
To determine whether conformational changes are required for catalytic activity, we determined the X-ray structure of the αβGC catalytic domains from human sGC. We successfully obtained crystals of the wild-type catalytic domains, without the need for an engineered disulfide.20 In our case, cleavage of the 28 C-terminal amino acids of αGC yielded a small amount of truncated αGC662βGC catalytic domains that formed favorable crystal contacts, unlike the untruncated catalytic domains. The structural comparison of the mutant and wild-type αβGC catalytic domains reveals that the two structures are very similar but exhibit subtle differences at the dimer interface. Both our structure and that of the mutant αβGC catalytic domains were obtained in the absence of metal and nucleotide and show inactive conformations of the active center. This is evidenced by the extended loop conformation of the substrate binding regions at the core of the dimer interface, and the drastically different orientation of the αGC subunit relative to the βGC subunit, compared to the structure of active adenylate cyclase catalytic domains.34 Potent sGC inhibitors have recently been described.64 These nucleotide analogues show Ki values in the low nanomolar range and were predicted by docking to bind to an active conformation of the catalytic domains. We are currently exploring the use of these molecules to favor the crystallization of the active heterodimeric catalytic domains of sGC.
Residues at the Heterodimeric Interface Guide an Optimal Conformation of the Active Center
Our structure allowed us to propose a key role for structural elements of the catalytic domains in guiding an optimal conformation of the active center for activity. Our mass spectrometry studies, yielding a relatively weak heterodimerization affinity, and our structural comparisons strongly suggest that the dimer interface is flexible for achieving structural transitions from an inactive open form to an activated closed form, which has been observed for adenylate cyclase.34 On the basis of our structural studies and mutagenesis data,36,37,40 we propose that both the interfacial flap and the hydrogen bond network enhance interactions between the two catalytic subunits and guide an optimal conformation of the active center necessary for activity. Studies are now underway to determine whether these activating mutations can facilitate structural studies of the sGC catalytic domains in the active state.
The Cyclase Domains Require Other sGC Domains for Optimal Activity
Our activity assay results demonstrate that even in the absence of the regulatory domains, additional sGC domain interactions are required to guide the appropriate conformation of the catalytic subunits associated with high activity. We propose that sGC activity is modulated by domain–domain interactions that allow the catalytic domains to undergo the transition from an inactive to an active conformation according to the following model: in the basal state, catalytic domains are constrained in a suboptimal conformation via inhibitory interactions with sGC domains, including the N-terminal βHNOX (and HNOXA) domain,22,23 which is itself maintained in an inhibited conformation by the αHNOX and αHNOXA domains.27 Binding of NO and/or activators releases these inhibitory interactions and induces conformational changes transmitted to the catalytic domains via the coiled-coil domain25 to yield the activated state (Figure 7). How the conformational transitions of these domains are orchestrated in sGC is still unknown. However, recent electron microscopy and hydrogen–deuterium exchange mass spectrometry suggest that full-length sGC presents multiple conformations with several interdomain pivot points allowing conformational changes to be transmitted from the regulatory domains to the catalytic domains.21,65 The observed “closing” of the catalytic interface upon NO binding65 corroborates our proposal that other sGC domains, including the coiled coil, will guide conformational changes in the catalytic domains, leading to an optimal alignment of the active site residues for full sGC activity.
Figure 7.
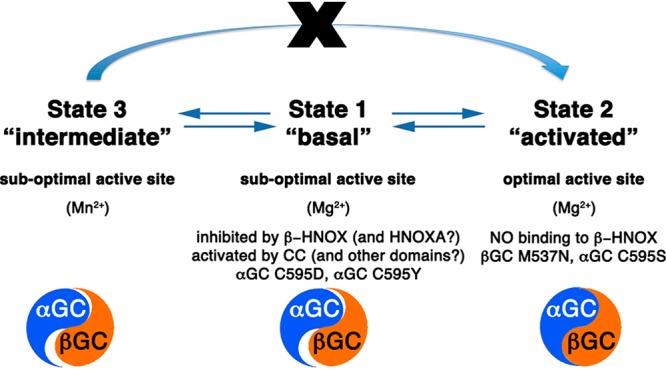
Model for domain–domain interactions that influence the conformation of the heterodimeric GC interface to modulate the activity of sGC. In state 1 (“basal”), competing domain–domain interactions yield sGC with low activity. While some sGC domains (including the coiled coil) promote conformational changes of the catalytic subunits, the regulatory βHNOX domain (and possibly the HNOXA domain) inhibits activity via direct binding to αGC. Mutations in the αGC subunit (Cys595Asp and Cys595Tyr) also prevent conformational changes in the active site. In state 2 (“activated”), binding of NO to βHNOX removes the inhibition and allows further conformational changes of the catalytic domains to yield fully active sGC. Mutations in catalytic subunits (βGC Met537Asn and αGC Cys595Ser) also yield an “activated” phenotype. In state 3 (“intermediate”), Mn2+ allows catalysis with a nonoptimal conformation of the catalytic domains. However, the enzyme is now locked in an intermediate state and cannot be further activated by NO and/or activators. The catalytic domains (represented as a blue and orange yin-yang) are in an inactive conformation (poor alignment) in states 1 and 3, and an active conformation in state 2 (perfect alignment of yin and yang).
Conclusions
Our results allow us to propose that novel structural elements, the interfacial β-flap and hydrogen bond network, play a key role in sGC catalytic activity by enhancing interactions between the two catalytic subunits and guiding the active center to an optimal conformation.
This is the first study demonstrating that the catalytic αβGC domains require additional sGC domains for activity, despite their ability to heterodimerize. Overall, collective results provide evidence that other sGC domains modulate the relative orientation of the catalytic subunits and control the proper orientation of key residues in the catalytic domain for full enzyme activity. The fine balance between inhibitory and activating domain–domain interactions is modulated by NO and/or activators. As such, small molecules that influence the orientation of the catalytic subunits have a strong potential to shift this equilibrium. This work provides the basis for a novel model for sGC activation and opens additional drug discovery routes for targeting the NO–cGMP pathway.
Acknowledgments
We thank Susan E. Tsutakawa, Robert P. Rambo, Scott Classen, Thomas Miller, and James Fishbein for critical reading of the manuscript, technical support, and insightful discussions. Part of this work was conducted at the Advanced Light Source (ALS), a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy (DOE), Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by the DOE Office of Biological and Environmental Research. Additional support comes from the National Institute of Health project MINOS (R01GM105404).
Glossary
Abbreviations
- sGC
full-length soluble guanylyl cyclase or soluble guanylate cyclase
- CC
coiled coil
- H-NOX or HNOX
heme-nitric oxide oxygen binding
- HNOXA
HNOX-associated
- GC
guanylate cyclase catalytic domain
- SEC-MALS
size-exclusion chromatography coupled to multiangle light scattering
- SID
surface-induced dissociation
- αβGC
construct containing only the α and β catalytic domains of sGC
- rmsd
root-mean-square deviation.
Supporting Information Available
Movie showing the superimposition of the wild-type and mutant (PDB entry 3UVJ) heterodimeric structures of the sGC catalytic domains (Movie S1), additional results, Table S1, sequence alignment, and Figures S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported in part by grants from the American Heart Association (10SDG2600345 to E.D.G. and 13PRE17000045 to F.S.), the National Institutes of Health (R01GM039345 to J.A.T.), and the National Science Foundation (DBI0923551 to V.H.W.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Voetsch B.; Jin R. C.; Loscalzo J. (2004) Nitric oxide insufficiency and atherothrombosis. Histochem. Cell Biol. 122, 353–367. [DOI] [PubMed] [Google Scholar]
- Ghofrani H.-A.; Galiè N.; Grimminger F.; Grünig E.; Humbert M.; Jing Z.-C.; Keogh A. M.; Langleben D.; Kilama M. O.; Fritsch A.; Neuser D.; Rubin L. J. (2013) Riociguat for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 369, 330–340. [DOI] [PubMed] [Google Scholar]
- Follmann M.; Griebenow N.; Hahn M. G.; Hartung I.; Mais F.-J.; Mittendorf J.; Schäfer M.; Schirok H.; Stasch J.-P.; Stoll F.; Straub A. (2013) The Chemistry and Biology of Soluble Guanylate Cyclase Stimulators and Activators. Angew. Chem., Int. Ed. 52, 9442–9462. [DOI] [PubMed] [Google Scholar]
- Conole D.; Scott L. (2013) Riociguat: First global approval. Drugs 73, 1967–1975. [DOI] [PubMed] [Google Scholar]
- Stasch J.-P., and Evgenov O. V. (2013) Soluble Guanylate Cyclase Stimulators in Pulmonary Hypertension. In Pharmacotherapy of Pulmonary Hypertension (Humbert M., Evgenov O. V., and Stasch J.-P., Eds.) pp 279–313, Springer, Berlin. [DOI] [PubMed] [Google Scholar]
- Moncada S.; Higgs A. (1993) The l-Arginine-Nitric Oxide Pathway. N. Engl. J. Med. 329, 2002–2012. [DOI] [PubMed] [Google Scholar]
- Naseem K. (2005) The role of nitric oxide in cardiovascular diseases. Mol. Aspects Med. 26, 33–65. [DOI] [PubMed] [Google Scholar]
- Thomas D. D.; Ridnour L. A.; Isenberg J. S.; Flores-Santana W.; Switzer C. H.; Donzelli S.; Hussain P.; Vecoli C.; Paolocci N.; Ambs S.; Colton C. A.; Harris C. C.; Roberts D. D.; Wink D. A. (2008) The chemical biology of nitric oxide: Implications in cellular signaling. Free Radical Biol. Med. 45, 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg J. S.; Ridnour L. A.; Perruccio E. M.; Espey M. G.; Wink D. A.; Roberts D. D. (2005) Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 102, 13141–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. W.; Isenberg J. S.; Roberts D. D. (2010) Thrombospondin-1 is an inhibitor of pharmacological activation of soluble guanylate cyclase: TSP-1 inhibits sGC activators. Br. J. Pharmacol. 159, 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan S.; Mazzalupo S.; Boitano S.; Montfort W. R. (2011) Thrombospondin-1 and Angiotensin II Inhibit Soluble Guanylyl Cyclase through an Increase in Intracellular Calcium Concentration. Biochemistry 50, 7787–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisaki Y.; Saheki S.; Nakane M.; Palmieri J. A.; Kuno T.; Chang B. Y.; Waldman S. A.; Murad F. (1986) Soluble guanylate cyclase from rat lung exists as a heterodimer. J. Biol. Chem. 261, 7236–7241. [PubMed] [Google Scholar]
- Pellicena P.; Karow D. S.; Boon E. M.; Marletta M. A.; Kuriyan J. (2004) Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc. Natl. Acad. Sci. U.S.A. 101, 12854–12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nioche P.; Berka V.; Vipond J.; Minton N.; Tsai A. L.; Raman C. S. (2004) Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science 306, 1550–1553. [DOI] [PubMed] [Google Scholar]
- Ma X.; Sayed N.; Beuve A.; van den Akker F. (2007) NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 26, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X.; Sayed N.; Baskaran P.; Beuve A.; van den Akker F. (2008) PAS-mediated dimerization of soluble guanylyl cyclase revealed by signal transduction histidine kinase domain crystal structure. J. Biol. Chem. 283, 1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit R.; Weichsel A.; Montfort W. R. (2013) Crystal structure of the α subunit PAS domain from soluble guanylyl cyclase. Protein Sci. 22, 1439–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A.; Leipelt M.; Russwurm M.; Steegborn C. (2008) Crystal structure of the guanylyl cyclase Cya2. Proc. Natl. Acad. Sci. U.S.A. 105, 15720–15725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winger J. A.; Derbyshire E. R.; Lamers M. H.; Marletta M. A.; Kuriyan J. (2008) The crystal structure of the catalytic domain of a eukaryotic guanylate cyclase. BMC Struct. Biol. 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allerston C. K.; von Delft F.; Gileadi O. (2013) Crystal Structures of the Catalytic Domain of Human Soluble Guanylate Cyclase. PLoS One 8, e57644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M. G.; Underbakke E. S.; Potter C. S.; Carragher B.; Marletta M. A. (2014) Single-particle EM reveals the higher-order domain architecture of soluble guanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 111, 2960–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winger J. A.; Marletta M. A. (2005) Expression and characterization of the catalytic domains of soluble guanylate cyclase: Interaction with the heme domain. Biochemistry 44, 4083–4090. [DOI] [PubMed] [Google Scholar]
- Underbakke E. S.; Iavarone A. T.; Marletta M. A. (2013) Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 110, 6777–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkegel C.; Schmidt P. M.; Atkins D. J.; Hoffmann L. S.; Schmidt H. H.; Schroder H.; Stasch J. P. (2007) Dimerization region of soluble guanylate cyclase characterized by bimolecular fluorescence complementation in vivo. Mol. Pharmacol. 72, 1181–1190. [DOI] [PubMed] [Google Scholar]
- Busker M.; Neidhardt I.; Behrends S. (2013) Nitric Oxide Activation of Guanylate Cyclase Pushes the α1 Signaling Helix and the β1 Heme-binding Domain closer to the Substrate-binding Site. J. Biol. Chem. 289, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz B. G.; Roberts S. A.; Ahmed A.; Breci L.; Li W.; Weichsel A.; Brailey J. L.; Wysocki V. H.; Tama F.; Montfort W. R. (2013) Molecular Model of a Soluble Guanylyl Cyclase Fragment Determined by Small-Angle X-ray Scattering and Chemical Cross-Linking. Biochemistry 52, 1568–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit R.; Fritz B. G.; The J.; Issaian A.; Weichsel A.; David C. L.; Campbell E. V.; Hausrath A. C.; Rassouli-Taylor L.; Garcin E. D.; Gage M. J.; Montfort W. R. (2014) YC-1 binding to the β subunit of soluble guanylyl cyclase overcomes allosteric inhibition by the α subunit. Biochemistry 53, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A. G. W., and Powell H. R. (2007) Processing diffraction data with mosflm. In Evolving Methods for Macromolecular Crystallography (Read R. J., and Sussman J. L., Eds.) pp 41–51, Springer, Dordrecht, The Netherlands. [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. (2002) PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Dagan S.; Wysocki V. (2012) Protein subunits released by surface collisions of noncovalent complexes: Nativelike compact structures revealed by ion mobility mass spectrometry. Angew. Chem., Int. Ed. 51, 4336–4339. [DOI] [PubMed] [Google Scholar]
- Galhena A.; Dagan S.; Jones C.; Beardsley R.; Wysocki V. (2008) Surface-induced dissociation of peptides and protein complexes in a quadrupole/time-of-flight mass spectrometer. Anal. Chem. 80, 1425–1436. [DOI] [PubMed] [Google Scholar]
- Tesmer J. J.; Sunahara R. K.; Johnson R. A.; Gosselin G.; Gilman A. G.; Sprang S. R. (1999) Two-metal-Ion catalysis in adenylyl cyclase. Science 285, 756–60. [DOI] [PubMed] [Google Scholar]
- Haase T.; Haase N.; Kraehling J. R.; Behrends S. (2010) Fluorescent Fusion Proteins of Soluble Guanylyl Cyclase Indicate Proximity of the Heme Nitric Oxide Domain and Catalytic Domain. PLoS One 5, e11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen P. S.; Doolittle L. K.; Garbers D. L. (1994) Dominant negative mutants of nitric oxide-sensitive guanylyl cyclase. J. Biol. Chem. 269, 791–793. [PubMed] [Google Scholar]
- Lamothe M.; Chang F. J.; Balashova N.; Shirokov R.; Beuve A. (2004) Functional characterization of nitric oxide and YC-1 activation of soluble guanylyl cyclase: Structural implication for the YC-1 binding site?. Biochemistry 43, 3039–3048. [DOI] [PubMed] [Google Scholar]
- Hatley M. E. (2000) Isolation and Characterization of Constitutively Active Mutants of Mammalian Adenylyl Cyclase. J. Biol. Chem. 275, 38626–38632. [DOI] [PubMed] [Google Scholar]
- Tang W.-J.; Stanzel M.; Gilman A. G. (1995) Truncation and Alanine-Scanning Mutants of Type I Adenylyl Cyclase. Biochemistry 34, 14563–14572. [DOI] [PubMed] [Google Scholar]
- Friebe A.; Russwurm M.; Mergia E.; Koesling D. (1999) A Point-Mutated Guanylyl Cyclase with Features of the YC-1-Stimulated Enzyme: Implications for the YC-1 Binding Site?. Biochemistry 38, 15253–15257. [DOI] [PubMed] [Google Scholar]
- Gregoret L. M.; Rader S. D.; Fletterick R. J.; Cohen F. E. (1991) Hydrogen bonds involving sulfur atoms in proteins. Proteins: Struct., Funct., Genet. 9, 99–107. [DOI] [PubMed] [Google Scholar]
- Koglin M.; Behrends S. (2004) Native human nitric oxide sensitive guanylyl cyclase: Purification and characterization. Biochem. Pharmacol. 67, 1579–1585. [DOI] [PubMed] [Google Scholar]
- Hu X.; Feng C.; Hazzard J. T.; Tollin G.; Montfort W. R. (2008) Binding of YC-1 or BAY 41-2272 to Soluble Guanylyl Cyclase Induces a Geminate Phase in CO Photolysis. J. Am. Chem. Soc. 130, 15748–15749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Murata L. B.; Weichsel A.; Brailey J. L.; Roberts S. A.; Nighorn A.; Montfort W. R. (2008) Allostery in recombinant soluble guanylyl cyclase from Manduca sexta. J. Biol. Chem. 283, 20968–20977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denninger J. W.; Schelvis J. P.; Brandish P. E.; Zhao Y.; Babcock G. T.; Marletta M. A. (2000) Interaction of soluble guanylate cyclase with YC-1: Kinetic and resonance Raman studies. Biochemistry 39, 4191–4198. [DOI] [PubMed] [Google Scholar]
- Derbyshire E. R.; Fernhoff N. B.; Deng S.; Marletta M. A. (2009) Nucleotide Regulation of Soluble Guanylate Cyclase Substrate Specificity. Biochemistry 48, 7519–7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharina I. G.; Jelen F.; Bogatenkova E. P.; Thomas A.; Martin E.; Murad F. (2008) α1 Soluble Guanylyl Cyclase (sGC) Splice Forms as Potential Regulators of Human sGC Activity. J. Biol. Chem. 283, 15104–15113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo B.-K.; Lamarre I.; Rappaport F.; Nioche P.; Raman C. S.; Martin J.-L.; Negrerie M. (2012) Picosecond to Second Dynamics Reveals a Structural Transition in Clostridium botulinum NO-Sensor Triggered by the Activator BAY-41-2272. ACS Chem. Biol. 7, 2046–2054. [DOI] [PubMed] [Google Scholar]
- Li Z.; Pal B.; Takenaka S.; Tsuyama S.; Kitagawa T. (2005) Resonance Raman Evidence for the Presence of Two Heme Pocket Conformations with Varied Activities in CO-Bound Bovine Soluble Guanylate Cyclase and Their Conversion. Biochemistry 44, 939–946. [DOI] [PubMed] [Google Scholar]
- Slabinski L.; Jaroszewski L.; Rychlewski L.; Wilson I. A.; Lesley S. A.; Godzik A. (2007) XtalPred: A web server for prediction of protein crystallizability. Bioinformatics 23, 3403–3405. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Jones C. M.; Wysocki V. H. (2013) Dissecting the Large Noncovalent Protein Complex GroEL with Surface-Induced Dissociation and Ion Mobility–Mass Spectrometry. Anal. Chem. 85, 8262–8267. [DOI] [PubMed] [Google Scholar]
- Zabel U.; Hausler C.; Weeger M.; Schmidt H. H. (1999) Homodimerization of soluble guanylyl cyclase subunits. Dimerization analysis using a glutathione S-transferase affinity tag. J. Biol. Chem. 274, 18149–18152. [DOI] [PubMed] [Google Scholar]
- Wagner C.; Russwurm M.; Jager R.; Friebe A.; Koesling D. (2005) Dimerization of nitric oxide-sensitive guanylyl cyclase requires the α1 N terminus. J. Biol. Chem. 280, 17687–17693. [DOI] [PubMed] [Google Scholar]
- Shiga T.; Suzuki N. (2005) Amphipathic α-helix mediates the heterodimerization of soluble guanylyl cyclase. Zool. Sci. 22, 735–742. [DOI] [PubMed] [Google Scholar]
- Wedel B.; Harteneck C.; Foerster J.; Friebe A.; Schultz G.; Koesling D. (1995) Functional domains of soluble guanylyl cyclase. J. Biol. Chem. 270, 24871–24875. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Gross S.; Roussos C.; Meurer S.; Muller-Esterl W.; Papapetropoulos A. (2004) Structural and functional characterization of the dimerization region of soluble guanylyl cyclase. J. Biol. Chem. 279, 24935–24943. [DOI] [PubMed] [Google Scholar]
- Ma X.; Beuve A.; van den Akker F. (2010) Crystal structure of the signaling helix coiled-coil domain of the β1 subunit of the soluble guanylyl cyclase. BMC Struct. Biol. 10, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharina I.; Sobolevsky M.; Doursout M.-F.; Gryko D.; Martin E. (2011) Cobinamides Are Novel Coactivators of Nitric Oxide Receptor That Target Soluble Guanylyl Cyclase Catalytic Domain. J. Pharmacol. Exp. Ther. 340, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding M. M. (1999) The geometry of metal-ligand interactions relevant to proteins. Acta Crystallogr. D55, 1432–1443. [DOI] [PubMed] [Google Scholar]
- Yang W.; Lee J. Y.; Nowotny M. (2006) Making and Breaking Nucleic Acids: Two-Mg2+-Ion Catalysis and Substrate Specificity. Mol. Cell 22, 5–13. [DOI] [PubMed] [Google Scholar]
- Tabor S.; Richardson C. C. (1989) Effect of manganese ions on the incorporation of dideoxynucleotides by bacteriophage T7 DNA polymerase and Escherichia coli DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 86, 4076–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenicka M.; Becker M.; Apeler H.; Sirichoke T.; Schroder H.; Gerzer R.; Stasch J. P. (1999) Purified soluble guanylyl cyclase expressed in a baculovirus/Sf9 system: Stimulation by YC-1, nitric oxide, and carbon monoxide. J. Mol. Med. 77, 14–23. [DOI] [PubMed] [Google Scholar]
- Beste K. Y.; Burhenne H.; Kaever V.; Stasch J.-P.; Seifert R. (2012) Nucleotidyl Cyclase Activity of Soluble Guanylyl Cyclase α1β1. Biochemistry 51, 194–204. [DOI] [PubMed] [Google Scholar]
- Dove S.; Danker K. Y.; Stasch J.-P.; Kaever V.; Seifert R. (2014) Structure/Activity Relationships of (M)ANT- and TNP-Nucleotides for Inhibition of Rat Soluble Guanylyl Cyclase α1β1. Mol. Pharmacol. 85, 598–607. [DOI] [PubMed] [Google Scholar]
- Underbakke E. S.; Iavarone A. T.; Chalmers M. J.; Pascal B. D.; Novick S.; Griffin P. R.; Marletta M. A. (2014) Nitric Oxide-Induced Conformational Changes in Soluble Guanylate Cyclase. Structure DOI: 10.1016/j.str.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins M. R.; Lindskog I.; Gasteiger E.; Bairoch A.; Sanchez J.-C.; Hochstrasser D. F.; Appel R. D. (1997) Detailed peptide characterization using PEPTIDEMASS: A World-Wide-Web-accessible tool. Electrophoresis 18, 403–408. [DOI] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. (2006) The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.