

Abstract
The fluoroacetate-producing bacterium Streptomyces cattleya has evolved a fluoroacetyl-CoA thioesterase (FlK) that exhibits a remarkably high level of discrimination for its cognate substrate compared to the cellularly abundant analogue acetyl-CoA, which differs only by the absence of the fluorine substitution. A major determinant of FlK specificity derives from its ability to take advantage of the unique properties of fluorine to enhance the reaction rate, allowing fluorine discrimination under physiological conditions where both substrates are likely to be present at saturating concentrations. Using a combination of pH–rate profiles, pre-steady-state kinetic experiments, and Taft analysis of wild-type and mutant FlKs with a set of substrate analogues, we explore the role of fluorine in controlling the enzyme acylation and deacylation steps. Further analysis of chiral (R)- and (S)-[2H1]fluoroacetyl-CoA substrates demonstrates that a kinetic isotope effect (1.7 ± 0.2) is observed for only the (R)-2H1 isomer, indicating that deacylation requires recognition of the prochiral fluoromethyl group to position the α-carbon for proton abstraction. Taken together, the selectivity for the fluoroacetyl-CoA substrate appears to rely not only on the enhanced polarization provided by the electronegative fluorine substitution but also on molecular recognition of fluorine in both formation and breakdown of the acyl-enzyme intermediate to control active site reactivity. These studies provide insights into the basis of fluorine selectivity in a naturally occurring enzyme–substrate pair, with implications for drug design and the development of fluorine-selective biocatalysts.
Fluoroacetate is a highly toxic natural product found in plants in Australia, Brazil, and Africa.1,2 Its mechanism of toxicity has been attributed to its cellular activation to fluoroacetyl-coenzyme A (CoA) and further metabolism to fluorocitrate, a mechanism-based inhibitor of the aconitase enzyme of the tricarboxylic acid cycle.3,4 Despite its prevalence in plants, only one genetic host of the fluoroacetate pathway, the soil bacterium Streptomyces cattleya, has been characterized to date.5,6 As one mechanism of biological resistance, S. cattleya has evolved a fluoroacetyl-CoA thioesterase (FlK), which can reverse the activation of fluoroacetate.7,8 Remarkably, this enzyme exhibits a 106-fold preference for its cognate substrate, fluoroacetyl-CoA, over acetyl-CoA, an abundant central metabolite and cellular competitor that differs only in the absence of the fluorine substitution.9 On the basis of its ability to exploit the unique properties of fluorine to achieve substrate specificity, FlK represents an ideal model system in which to query molecular recognition of fluorine and its influence on enzymatic reactivity in a naturally evolved protein–ligand pair.
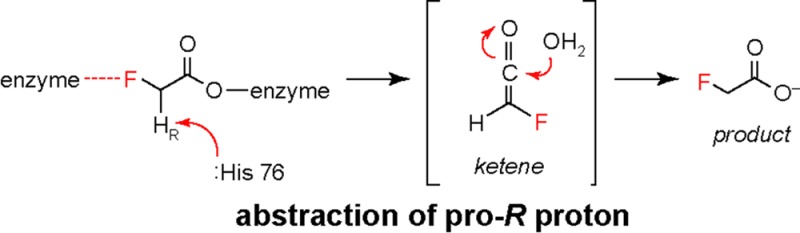
We have previously shown that the hydrolytic mechanism of FlK involves a minimum of three kinetic steps: (i) formation of the enzyme–substrate complex (KD = k–1/k1), (ii) acylation of FlK to form an acyl-anhydride intermediate on Glu 50 (k2), and (iii) FlK deacylation (k3) or breakdown of this intermediate to yield the carboxylic acid product (Scheme 1).10 A large component of fluoroacetyl-CoA (R = F) specificity (104-fold) is based on a change in the rate-limiting step from formation of the acyl-enzyme intermediate for fluoroacetyl-CoA (ii; k2 = kcat) to its breakdown for acetyl-CoA (R = H) (iii; k3 = kcat). Despite similarities to canonical hot dog-fold thioesterases,11 our previous mechanistic studies indicate that the chemical basis for the acceleration of FlK-catalyzed deacylation for the fluorinated substrate is utilization of an unusual hydrolytic mechanism initiated by Cα deprotonation of the acyl-enzyme intermediate.10 The resultant enolate can then break down through a putative ketene intermediate to give fluoroacetate.
Scheme 1. Minimal Kinetic Mechanism for FlK-Catalyzed Acyl-CoA Hydrolysis (fluoroacetyl-CoA, R = F; acetyl-CoA, R = H).

The differences in chemical mechanism between fluoroacetyl-CoA and acetyl-CoA hydrolysis provide a means for FlK to kinetically discriminate between two substrates that are both present at saturating concentrations under physiological conditions based on the high intracellular concentration of acetyl-CoA (2 mM) and the low KM for fluoroacetyl-CoA (8 μM).10 However, it is interesting to note that this kinetic discrimination occurs after the first committed step in the reaction mechanism, which is enzyme acylation (k2), and that the specificity constant kcat/KM depends only on the binding and acylation steps of the reaction (Scheme 1).12 We therefore set out to further explore the function of the active site triad involved in enzyme acylation, consisting of His 76, Glu 50, and Thr 42. We first provide additional evidence supporting the formation of an acyl-enzyme intermediate during the hydrolysis of fluoroacetyl-CoA, and we show that the rate of formation of this intermediate does not depend on the key specificity-determining residue, His 76, which is involved in Cα deprotonation. Instead, the primary determinant of specificity of Glu 50 acylation appears to be based on the inductively electron-withdrawing effect of the α-substituent. We also explore the role of Thr 42 with respect to acylation and deacylation and conclude that its main function in catalysis is to anchor the hydrogen bonding network between Glu 50 and His 76.
In addition to investigating enzyme acylation, we also continue to examine the role of fluorine in controlling the chemical mechanism of enzyme deacylation. Although the fluoroacetyl-CoA α-protons are expected to be more acidic than those of acetyl-CoA, their lower pKa is insufficient to explain why the Cα deprotonation mechanism for deacylation is inaccessible to other α-substituted acyl-CoAs. In particular, acetoacetyl-CoA is predicted to have an α-proton pKa lower than that of fluoroacetyl-CoA based on its ability to form a resonance-stabilized enolate, but free energy relationship analysis of its FlK-catalyzed hydrolysis suggests that it cannot access the Cα deprotonation pathway.10 Using substrates with stereogenic centers at the substrate α-carbon, we demonstrate that polarization is not the sole determinant of reaction through the Cα deprotonation pathway. In this regard, FlK utilizes chiral recognition of fluoroacetyl-CoA’s prochiral fluoromethyl group to position the pro-R proton for preferential abstraction by His 76, indicating that specificity is based not only on the unique reactivity of fluorinated compounds but also potentially on fluorine molecular recognition. Taken together, these results show that fluorine impacts both the acylation and deacylation steps of the FlK reaction mechanism by substrate activation as well as fluorine-specific interactions in the active site.
Materials and Methods
Commercial Materials
Acetyl-coenzyme A sodium salt, coenzyme A hydrate, coenzyme A trilithium salt, anhydrous N,N-dimethylformamide (DMF), 2-(N-morpholino)ethanesulfonic acid (MES), trifluoroacetic acid (TFA), oxalyl chloride (2 M in dichloromethane), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), methyl (S)-(−)-lactate, methyl (R)-(+)-lactate, methanesulfonyl chloride, magnesium chloride hexahydrate, 2-bis(2-hydroxyethyl)amino-2-(hydroxymethyl)-1,3-propanediol (Bis-Tris), 1,3-bis[tris(hydroxymethyl)methylamino]propane (Bis-Tris propane), and tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) were purchased from Sigma-Aldrich (St. Louis, MO). Monosodium phosphate monohydrate, disodium phosphate heptahydrate, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), hexanes, ethyl acetate, methanol, triethylamine, adenosine 5′-triphosphate trisodium salt, and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were purchased from Fisher Scientific (Pittsburgh, PA). 2-Fluoropropionic acid was purchased from Oakwood Products (West Columbia, SC).
Protein Expression and Purification
FlK and FlK-T42S were expressed and purified as described previously.9 Acetate kinase from Escherichia coli and phosphotransacetylase from S. cattleya were cloned, expressed, and purified as described previously.8
Synthesis of Substrates
Fluoroacetyl-CoA, chloroacetyl-CoA, and bromoacetyl-CoA were synthesized as described previously.10 Tetrahydrofuran (THF) used in chemical synthesis was dried using a VAC Solvent Purifier System (Vacuum Atmospheres Co., Amesbury, MA). Acyl-CoAs were purified by high-performance liquid chromatography (HPLC) using an Agilent Eclipse XDB-C18 column (9.4 mm × 250 mm, 5 μm) connected to an Agilent 1200 binary pump and an Agilent G1315D diode-array detector, which was used to monitor coenzyme A absorbance at 260 nm. Following sample loading, the column was washed with 0.2% aqueous TFA (3 mL/min) until the absorbance at 260 nm returned to baseline. A linear gradient from 0 to 100% methanol over 90 min (3 mL/min) with 0.2% aqueous TFA as the aqueous mobile phase was then applied. Fractions were collected using an Agilent 1260 fraction collector and then assayed for the desired acyl-CoAs by liquid chromatography and mass spectrometry using an Agilent 1290 binary pump coupled to an Agilent 6130 single-quadrupole electrospray ionization mass spectrometer. The fractions were then analyzed using a Phenomenex Kinetex C18 column (4.6 mm × 30 mm, 2.6 μm) with a linear gradient from 0 to 100% acetonitrile over 2 min (0.7 mL/min) using 0.1% formic acid as the aqueous mobile phase. Fractions containing the desired compound were pooled and lyophilized. Acyl-CoAs were then dissolved in water, quantified by their absorbance at 260 nm, and stored at −80 °C until they were used further. High-resolution mass spectra were acquired at the QB3/Chemistry Mass Spectrometry Facility at the University of California (Berkeley, CA). Nuclear magnetic resonance (NMR) spectra were recorded at the College of Chemistry NMR Facility at the University of California (Berkeley, CA) or at the Central California 900 MHz NMR Facility (Berkeley, CA). One-dimensional spectra were recorded on Bruker AV-600 or AVQ-400 NMR spectrometers at 298 K. Chemical shifts are expressed in parts per million (δ) downfield from tetramethylsilane (1H, 2H, and 13C) or trichlorofluoromethane (19F) and are referenced to the solvent signal using standard CoA numbering (Table S1 of the Supporting Information). Heteronuclear multiple-bond correlation (HMBC) experiments were performed on a Bruker AV-500 NMR spectrometer at 298 K.
(RS)-2-Fluoropropionyl-CoA
2-Fluoropropionic acid (92 mg, 1 mmol) was placed in an oven-dried round-bottom flask equipped with a stir bar and a reflux condenser. The flask was placed under nitrogen pressure, and dry THF (2 mL), DMF (100 μL), and oxalyl chloride (2 M in dichloromethane, 500 μL) were added by syringe. The reaction mixture was heated to 65 °C and stirred for 3–4 h. After the mixture had been cooled, a portion of the reaction mixture (300 μL) was added to a stirred, ice-cooled solution of coenzyme A hydrate (50 mg, ∼0.06 mmol) and triethylamine (41.8 μL, 0.3 mmol) in anhydrous DMF (1 mL). After 1 min, the reaction was quenched by addition of water (20 mL) and the mixture lyophilized. The lyophilizate was dissolved in water (1 mL) and purified by reverse-phase HPLC (15 μmol, 25%): 1H NMR (600 MHz, D2O, 25 °C) δ 8.56 (s, 1H, H8), 8.38 (s, 1H, H2), 6.15 (d, J = 6 Hz, 1H, H1′), 5.12 (dq, JHH = 7.2 Hz, JHF = 49.5 Hz, 1H, CHF), 4.84 (m, 2H, H2′ and H3′), 4.56 (s, 1H, H4′), 4.24 (d, J = 15 Hz, 2H, H5′), 3.97 (s, 1H, H3″), 3.85 (m, 1H, H1″), 3.61 (m, 1H, H1″), 3.41 (t, J = 6.6 Hz, 2H, H5″), 3.31 (t, J = 6.6 Hz, 2H, H8″), 3.00 (t, J = 6 Hz, 2H, H9″), 2.39 (t, J = 6.6 Hz, 2H, H6″), 1.46 (dd, JHH = 6.6 Hz, JHF = 24.9 Hz, 3H, CH3), 0.90 (s, 3H, H10″), 0.78 (s, 3H, H11″); 13C NMR (150.9 MHz, D2O, 25 °C) δ 203.2, 203.0 (FCHCO), 174.6 (C7″), 173.9 (C4″), 149.8 (C6), 144.7 (C2), 142.7 (C4), 142.4 (C8), 118.5 (C5), 87.9 (C1′), 87.5, 86.8 (FCHCO), 86.8 (C4′), 74.4 (C2′), 74.3 (C3″), 74.2 (C3′), 73.7 (C1″), 65.1 (C5′), 38.3 (C8″), 36.9 (C2″) 35.3 (C5″), 35.2 (C6″), 27.1 (C9″), 20.7 (CH3), 18.3 (C10″), 17.9 (C11″); 19F NMR (564.7 MHz, D2O, 25 °C) δ −181.2 (sextet, J = 26.4 Hz); HR-ESI-MS calcd (M – H+) m/z 840.1247, found (M – H+) m/z 840.1236.
(S)- and (R)-2-Fluoropropionyl-CoA
Sodium (S)-2-fluoropropionate and sodium (R)-2-fluoropropionate were synthesized from methyl (R)-(−)-lactate and methyl (S)-(+)-lactate using literature methods.13 2-Fluoropropionyl-CoAs were then synthesized from the corresponding carboxylic acids (1 mmol scale) as described for (RS)-2-fluoropropionyl-CoA (10 μmol, 17%).
(S)-2-Fluoropropionyl-CoA (Figure S1 of the Supporting Information):
1H NMR (600 MHz, D2O, 25 °C) δ 8.54 (s, 1H, H8), 8.33 (s, 1H, H2), 6.10 (d, J = 6 Hz, 1H, H1′), 5.08 (dq, JHH = 7.2 Hz, JHF = 49.5 Hz, 1H, CHF), 4.87 (m, 2H, H2′ and H3′), 4.52 (s, 1H, H4′), 4.21 (d, J = 15 Hz, 2H, H5′), 3.93 (s, 1H, H3″), 3.81 (m, 1H, H1″), 3.58 (m, 1H, H1″), 3.37 (t, J = 6.6 Hz, 2H, H5″), 3.27 (t, J = 6.6 Hz, 2H, H8″), 2.97 (t, J = 6 Hz, 2H, H9″), 2.35 (t, J = 6.6 Hz, 2H, H6″), 1.41 (dd, JHH = 6.6 Hz, JHF = 24.9 Hz, 3H, CH3), 0.86 (s, 3H, H10″), 0.75 (s, 3H, H11″); 13C NMR (150.9 MHz, D2O, 25 °C) δ 203.1, 202.9 (FCHCO), 174.6 (C7″), 173.9 (C4″), 149.6 (C6), 145.3 (C2), 143.9 (C4), 141.6 (C8), 118.3 (C5), 87.9 (C1′), 87.8, 86.8 (FCHCO), 86.8 (C4′), 74.8 (C2′), 74.6 (C3″), 74.4 (C3′), 73.7 (C1″), 65.1 (C5′), 38.2 (C8″), 37.5 (C2″), 36.0 (C5″), 35.3 (C6″), 27.9 (C9″), 21.1 (CH3), 20.1 (C10″), 17.6 (C11″); 19F NMR (564.7 MHz, D2O, 25 °C) δ −181.3 (sextet, J = 26.4 Hz); HR-ESI-MS calcd (M – H+) m/z 840.1247, found (M – H+) m/z 840.1229.
(R)-2-Fluoropropionyl-CoA (Figure S2 of the Supporting Information):
1H NMR (600 MHz, D2O, 25 °C) δ 8.60 (s, 1H, H8), 8.39 (s, 1H, H2), 6.17 (d, J = 6 Hz, 1H, H1′), 5.14 (dq, JHH = 7.2 Hz, JHF = 49.5 Hz, 1H, CHF), 4.87 (m, 2H, H2′ and H3′), 4.57 (s, 1H, H4′), 4.25 (d, J = 15 Hz, 2H, H5′), 3.98 (s, 1H, H3″), 3.85 (m, 1H, H1″), 3.62 (m, 1H, H1″), 3.42 (t, J = 6.6 Hz, 2H, H5″), 3.32 (t, J = 6.6 Hz, 2H, H8″), 3.02 (t, J = 6 Hz, 2H, H9″), 2.40 (t, J = 6.6 Hz, 2H, H6″), 1.47 (dd, JHH = 6.6 Hz, JHF = 24.9 Hz, 3H, CH3), 0.90 (s, 3H, H10″), 0.79 (s, 3H, H11″); 13C NMR (150.9 MHz, D2O, 25 °C) δ 203.2, 203.0 (FCHCO), 174.7 (C7″), 174.0 (C4″), 149.8 (C6), 148.5 (C2), 144.7 (C4), 142.5 (C8), 118.5 (C5), 93.5, 92.3 (FCHCO), 87.5 (C1′), 83.9 (C4′), 74.4 (C2′), 74.2 (C3″), 73.7 (C3′), 72.2 (C1″), 65.1 (C5′), 38.34 (C8″), 38.28 (C2″), 35.3 (C5″), 35.2 (C6″), 27.1 (C9″), 20.7 (CH3), 18.3 (C10″), 17.9 (C11″); 19F NMR (564.7 MHz, D2O, 25 °C) δ −181.2 (sextet, J = 26.4 Hz); HR-ESI-MS calcd (M – H+) m/z 840.1247, found (M – 2H+) m/z 840.1236.
(R)- or (S)-1-(Methoxycarbonyl)ethylmethanesulfonate
Methyl lactate (1.0 g, 9.6 mmol) was dissolved in toluene before triethylamine (1.2 g, 1.6 mL, 11.5 mmol) was added. The reaction mixture was cooled in an ice/water bath, and methanesulfonyl chloride (1.2 g, 0.82 mL, 10.6 mmol) was added dropwise over 15 min. The reaction mixture was stirred on ice for 1 h, warmed to room temperature, and filtered. The filtrate was concentrated under vacuum to yield a light pink oil, which was purified on a silica column using an 80:20 hexanes/ethyl acetate mixture as the mobile phase to give (S)- or (R)-1-(methoxycarbonyl)ethylmethanesulfonate as a clear oil (1.43 g, 84%).
(R)-1-(Methoxycarbonyl)ethylmethanesulfonate:
1H NMR (600 MHz, CHCl3, 25 °C) δ 5.11 (q, J = 6.6 Hz, 1H, CH), 3.78 (s, 3H, CH3O), 3.13 (s, 3H, CH3S), 1.59 (d, J = 7.2 Hz, CH3CH); 13C NMR (150.9 MHz, CHCl3, 25 °C) δ 170.1 (COOMe), 74.3 (CH), 52.9 (OCH3), 39.2 (SCH3), 18.5 (CHCH3); HR-ESI-MS calcd (M + Na+) m/z 205.0141, found (M + Na+) m/z 205.0143.
(S)-1-(Methoxycarbonyl)ethylmethanesulfonate:
1H NMR (600 MHz, CHCl3, 25 °C) δ 5.10 (q, J = 7.2 Hz, 1H, CH), 3.77 (s, 3H, CH3O), 3.12 (s, 3H, CH3S), 1.58 (d, J = 7.2 Hz, CH3CH); 13C NMR (150.9 MHz, CHCl3, 25 °C) δ 170.1 (COOMe), 74.3 (CH), 52.9 (OCH3), 39.2 (SCH3), 18.5 (CHCH3); HR-ESI-MS calcd (M + Na+) m/z 205.0141, found (M + Na+) m/z 205.0143.
Methyl (S)- or (R)-2-Fluoropropionate
Formamide (3 mL) was placed in a two-neck round-bottom flask equipped with a stir bar and heated to 60 °C. Potassium fluoride (1.2 g, 22 mmol) was dissolved in the formamide while the mixture was being stirred. A short-path distillation head equipped with a thermometer and a receiving flask cooled with a dry ice/acetone bath was then attached. (S)- or (R)-1-(methoxycarbonyl)ethylmethanesulfonate (1 g, 5.5 mmol) was added slowly. The reaction mixture was then placed under vacuum, and the product was continuously distilled during the course of the reaction to yield methyl (R)- or (S)-2-fluoropropionate as a clear oil (200 mg, 35%).
Methyl (S)-2-fluoropropionate:
1H NMR (600 MHz, CHCl3, 25 °C) δ 5.02 (dq, JHH = 7.2 Hz, JHF = 48.3 Hz, 1H, CH), 3.80 (s, 3H, CH3O), 1.58 (dd, JHH = 6.6 Hz, JHF = 30.6 Hz); 13C NMR (150.9 MHz, CHCl3, 25 °C) δ 171.2, 171.1 (COOMe), 86.5, 85.3 (CH), 52.6 (OCH3), 18.6, 18.4 (CHCH3); 19F NMR (564.6 MHz, CHCl3, 25 °C) δ −185.6 (sextet, J = 24 Hz).
Methyl (R)-2-fluoropropionate:
1H NMR (600 MHz, CHCl3, 25 °C) δ 5.03 (dq, JHH = 7.2 Hz, JHF = 48.6 Hz, 1H, CH), 3.80 (s, 3H, CH3O), 1.59 (dd, JHH = 6.6 Hz, JHF = 30.6 Hz); 13C NMR (150.9 MHz, CHCl3, 25 °C) δ 171.2, 171.0 (COOMe), 86.4, 85.3 (CH), 52.5 (OCH3), 18.5, 18.4 (CHCH3); 19F NMR (564.6 MHz, CHCl3, 25 °C) δ −185.6 (sextet, J = 24 Hz).
Sodium (S)- or (R)-2-Fluoropropionate
Methyl (R)- or (S)-2-fluoropropionate (100 mg, 0.6 mmol) was dissolved in water, and sodium hydroxide (0.6 mmol from a 10 M stock solution) was added. The reaction mixture was stirred at room temperature for 1 h, at which time it was judged to be ∼80% complete based on 19F NMR. The solution was flash-frozen and lyophilized to yield sodium (R)- or (S)-2-fluoropropionate as a white solid (56 mg, 82%).
Sodium (S)-2-fluoropropionate:
1H NMR (400 MHz, D2O, 25 °C) δ 4.91 (dq, JHH = 7.2 Hz, JHF = 51.6 Hz, 1H, CH), 1.49 (dd, JHH = 6.8 Hz, JHF = 17.4 Hz, 3H, CHCH3); 13C NMR (100.6 MHz, D2O, 25 °C) δ 179.2, 179.0 (COONa), 88.9, 87.7 (CH), 18.3, 18.2 (CH3); 19F NMR (375.6 MHz, D2O, 25 °C) δ −174.6 (sextet, J = 26.4 Hz).
Sodium (R)-2-fluoropropionate:
1H NMR (400 MHz, D2O, 25 °C) δ 4.91 (dq, JHH = 6.8 Hz, JHF = 48 Hz, 1H, CH), 1.48 (dd, JHH = 6.8 Hz, JHF = 18 Hz, 3H, CHCH3); 13C NMR (100.6 MHz, D2O, 25 °C) δ 179.3, 179.1 (COONa), 89.2, 87.4 (CH), 18.2, 17.9 (CH3); 19F NMR (375.6 MHz, D2O, 25 °C) δ −172.8 (sextet, J = 28 Hz).
(S)- and (R)-[2H1]Fluoroacetyl-CoA
Sodium (S)-[2H1]fluoroacetate and sodium (R)-[2H1]fluoroacetate were synthesized as described previously.14 (S)- and (R)-[2H1]fluoroacetyl-CoA were synthesized from the corresponding sodium [2H1]fluoroacetates enzymatically using acetate kinase (AckA) from E. coli and phosphotransacetylase (PTA) from S. cattleya. Reaction mixtures containing Tris-HCl (pH 7.5, 100 mM), MgCl2 (5 mM), TCEP (2.5 mM), ATP (50 mM), coenzyme A (40 mM), sodium [2H1]fluoroacetate (50 mM), AckA (10 μM), and PTA (5 μM) in a total volume of 1 mL were incubated at 37 °C for 2 h. [2H1]Fluoroacetyl-CoAs were then purified by reverse-phase HPLC. Fractions containing the desired compounds were pooled and lyophilized to yield (S)- or (R)-[2H1]fluoroacetyl-CoA (6 μmol, 15%).
(S)-[2H1]Fluoroacetyl-CoA:
1H NMR (600 MHz, D2O, 25 °C) δ 8.62 (s, 1H, H8), 8.41 (s, 1H, H2), 6.19 (d, J = 6 Hz, 1H, H1′), 4.98 (d, J = 46 Hz, 1H, CH2F), 4.87 (m, 2H, H2′ and H3′), 4.58 (s, 1H, H4′), 4.26 (d, J = 15 Hz, 2H, H5′), 3.99 (s, 1H, H3″), 3.85 (m, 1H, H1″), 3.62 (m, 1H, H1″), 3.43 (t, J = 6.6 Hz, 2H, H5″), 3.35 (t, J = 6.6 Hz, 2H, H8″), 3.07 (t, J = 6.6 Hz, 2H, H9″), 2.42 (t, J = 6.6 Hz, 2H, H6″), 0.92 (s, 3H, H10″), 0.80 (s, 3H, H11″); 13C NMR (225 MHz, D2O, 25 °C) δ 202.8, 202.7 (FCH2CO), 177.5 (C7″), 176.8 (C4″), 152.7 (C6), 151.3 (C2), 147.3 (C4), 145.3 (C8), 121.4 (C5), 90.5 (C1′), 86.9, 86.1 (FCH2CO), 83.0 (C4′), 77.0 (C2′), 76.9 (C3″), 76.6 (C3′), 76.5 (C1″), 67.8 (C5′), 45.0 (C8″), 41.1 (C2″), 38.1 (C5″), 38.0 (C6″), 29.7 (C9″), 23.5 (C10″), 21.0 (C11″); 19F NMR (564.7 MHz, D2O, 25 °C) δ −226.6 (dt, JHF = 49.2 Hz, JDF = 7.8 Hz); HR-ESI-MS calcd (M – H+) m/z 827.1154, found (M – H+) m/z 827.1151.
(R)-[2H1]Fluoroacetyl-CoA:
1H NMR (600 MHz, D2O, 25 °C) δ 8.64 (s, 1H, H8), 8.42 (s, 1H, H2), 6.20 (d, J = 6 Hz, 1H, H1′), 4.99 (d, J = 46 Hz, 1H, CH2F), 4.87 (m, 2H, H2′ and H3′), 4.59 (s, 1H, H4′), 4.26 (d, J = 13 Hz, 2H, H5′), 4.00 (s, 1H, H3″), 3.87 (m, 1H, H1″), 3.62 (m, 1H, H1″), 3.44 (t, J = 6 Hz, 2H, H5″), 3.36 (t, J = 5.4 Hz, 2H, H8″), 3.08 (t, J = 6 Hz, 2H, H9″), 2.43 (t, J = 6.6 Hz, 2H, H6″), 0.92 (s, 3H, H10″), 0.81 (s, 3H, H11″); 13C NMR (225 MHz, D2O, 25 °C) δ 202.8, 202.7 (FCH2CO), 177.5 (C7″), 176.8 (C4″), 152.7 (C6), 151.3 (C2), 147.4 (C4), 145.3 (C8), 121.4 (C5), 90.2 (C1′), 86.8, 86.4 (FCH2CO), 83.0 (C4′), 76.9 (C2′), 76.7 (C3″), 76.6 (C3′), 76.5 (C1″), 67.8 (C5′), 45.0 (C8″), 41.3 (C2″), 38.1 (C5″), 38.0 (C6″), 29.7 (C9″), 23.6 (C10″), 20.9 (C11″); 19F NMR (564.7 MHz, D2O, 25 °C) δ −226.6 (dt, JHF = 48.6 Hz, JDF = 7.2 Hz); HR-ESI-MS calcd (M – H+) m/z 827.1154, found (M – H+) m/z 827.1146.
Pre-Steady-State Kinetic Analysis
Pre-steady-state kinetic experiments were performed using rapid chemical quench followed by HPLC separation of coenzyme A from unhydrolyzed acyl-CoA. FlK [50 or 150 μM in 20 mM Tris-HCl (pH 7.6) and 50 mM NaCl] or FlK-T42S [40 μM in 20 mM Tris-HCl (pH 7.6) and 50 mM NaCl] was mixed with substrate diluted in water [chloroacetyl-CoA and [2H1]fluoroacetyl-CoAs, 500 μM; cyanoacetyl-CoA and fluoroacetyl-CoA, 750 μM; bromoacetyl-CoA and (R)-2-fluoropropionyl-CoA, 1 mM; (S)-2-fluoropropionyl-CoA, 5 mM; acetyl-CoA, 6 mM] using a Chemical Quench Flow Model RQF-3 (KinTek). The reaction was stopped at various times by mixing with 50% TFA to achieve a final concentration of 17% TFA. Quenched samples were analyzed by HPLC on an Agilent Eclipse XDB-C18 (3.5 μm, 3.0 mm × 150 mm) column using a linear gradient from 0 to 100% methanol over 15 min (0.6 mL/min) with 50 mM sodium phosphate and 0.1% TFA (pH 4.5) as the mobile phase. Conversion percentages were calculated on the basis of the 260 nm peak areas for acyl-CoA substrate and free CoA product. Plots of coenzyme A formed versus time were fit to the burst-phase equation:
where [E0] is the enzyme concentration, k2 is the burst-phase (acylation) rate constant, and k3 is the steady-state (deacylation) rate constant.15 This equation is a reduced form of the general burst equation:
where kcat = (k2k3)/(k2 + k3), the burst height (β) = k22/(k2 + k3)2, and the rate of burst formation (λ) = k2 + k3, which results from making the assumption that k2 ≫ k3.15 In cases where this assumption was invalid, only kcat is reported. In cases that could not be fit with a burst phase, they were fit as pseudo-first-order reactions according to the equation [CoA] = k[FlK]t + [CoA]0, where the y-intercept, [CoA]0, is the concentration of CoA released before the first data point, k is the rate constant, and t is time. For fluoroacetyl-CoA and [2H1]fluoroacetyl-CoAs, a sum-of-squares F test was used to determine whether the data were best fit with a line or with the burst-phase equation. The null hypothesis was a linear fit, and the alternative hypothesis was a burst-phase fit with the burst amplitude fixed at 25 μM. The p value threshold was set at 0.01, meaning that if the F test gave a p value of >0.01, there was no compelling reason to reject the null hypothesis, whereas if the p value was <0.01, the null hypothesis was rejected in favor of a burst-phase fit. The F test gave p = 0.0309 for [1H2]fluoroacetyl-CoA, p = 0.0866 for (S)-[2H1]fluoroacetyl-CoA, and p = 0.0022 for (R)-[2H1]fluoroacetyl-CoA. Therefore, data for [1H2]fluoroacetyl-CoA and (S)-[2H1]fluoroacetyl-CoA were fit with a line, and data for (R)-[2H1]fluoroacetyl-CoA were fit with the burst-phase equation.
For pre-steady-state kinetic experiments at pH 6, FlK was exchanged into 100 mM Bis-Tris (pH 6.0) prior to the experiment using a MicroBiospin 6 column (Bio-Rad) that had been pre-equilibrated with the appropriate buffer. FlK was then quantified by absorbance and diluted to 50 μM with the same buffer prior to use in rapid quench experiments.
Taft Free Energy Relationship Analysis of FlK Acylation Rates
Taft plots were constructed by plotting the log of the acylation rate constant (log k2) versus the Taft polar substituent constant σ* for the α-substituent of each acyl-CoA tested.16 Acylation rate constants and the corresponding error bars were derived from nonlinear curve fitting of triplicate pre-steady-state kinetic data sets. Taft plots were fit to the equation log k = ρ*σ* + c, where ρ* is the polar sensitivity factor and c is a constant.
Steady-State pH–Rate Profiles
Steady-state kinetic experiments for determining the pH–rate profile of FlK were conducted on a Beckman Coulter DU800 spectrophotometer by continuously monitoring the absorbance of the acyl-CoA thioester bond at 232 nm (ε = 4500 M–1 cm–1) in a 0.2 cm path length quartz cuvette at 25 °C. Reaction mixtures contained the appropriate buffer [100 mM; Bis-Tris propane (pH 6.3–7), Tris-HCl (pH 7–9), or Bis-Tris propane (pH 9)], fluoroacetyl-CoA (10, 25, 50, or 100 μM), and FlK (1 nM) or FlK-H76A (1 μM). Each rate was measured in triplicate. Experiments at saturation (kcat conditions) were repeated with similar results in a series of sulfonate buffers [MES (pH 6–7), HEPES (pH 7–8), and TAPS (pH 7.5–9)] to ensure the observed activity resulted from changes in pH and not buffer composition. Our ability to measure FlK-catalyzed hydrolysis rates below pH 6 was limited by protein instability, while our ability to measure enzymatic rates above pH 9 was limited by rapid nonenzymatic substrate hydrolysis. Kinetic parameters (kcat and KM) were determined by fitting the initial rate data to the equation V0 = (Vmax[S])/(KM + [S]), where V0 is the initial rate and [S] is the substrate concentration, using Origin version 6.0 (OriginLab Corp., Northampton, MA). The pH–rate profile for wild-type FlK in which decreases in activity were observed at both low and high pH was fit to the equation
where kobs is the observed kcat at a given pH, kmax is the pH-independent rate constant, pKa1 is the pKa of the group that ionizes at basic pH, and pKa2 is the pKa of the group that ionizes at acidic pH. A comparison between the pH–rate profile for wild-type FlK and pH dependence studies of the FlK-H76A mutant led to the conclusion that the mutant lacks an ionization at acidic pH.
Steady-State Kinetic Analysis of 2-Fluoropropionyl-CoAs
Steady-state rates of FlK-catalyzed 2-fluoropropionyl-CoA hydrolysis were measured by monitoring the increase in absorbance at 412 nm from the reaction of 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) with enzymatically produced coenzyme A in a Beckman Coulter DU800 spectrophotometer. Assays (100 μL) were performed at 25 °C in Tris-HCl (pH 7.6, 100 mM) containing DTNB (0.5 mM), the appropriate 2-fluoropropionyl-CoA, and FlK (5 nM). Each rate was measured in triplicate. Absorbance values were converted to coenzyme A concentration using a standard curve. Kinetic parameters (kcat and KM) were determined by fitting the initial rate data to the equation V0 = (Vmax[S])/(KM + [S]), where V0 is the initial rate and [S] is the substrate concentration, using Origin version 6.0.
Results and Discussion
Probing the Role of the Catalytic Residues in Acylation Specificity
Because acylation of Glu 50 represents the committed step in FlK-catalyzed hydrolysis, we were interested in further exploring the contribution of the other catalytic residues, His 76 and Thr 42, to this step (Figure 1A). Our previous pre-steady-state kinetic analyses indicated that the acylation rate constant differs by at least 2 orders of magnitude between the fluoroacetyl-CoA and acetyl-CoA substrates and that mutation of His 76 to Ala led to a loss of specificity in both the acylation and deacylation steps.10 While the defect in deacylation can be attributed to the loss of the key base required for the Cα deprotonation pathway, the 10-fold reduction in the acylation rate constant for the H76A mutant is more difficult to interpret because of the participation of the His 76 side chain in a hydrogen bond network involving Thr 42 and Glu 50 that has been shown to be important for their positioning (Figure S3 of the Supporting Information).9,17 However, active roles for His 76 in determining substrate specificity for enzyme acylation beyond simple structural function are also possible. One possibility is that His 76 could be involved in the deprotonation of the Glu 50 nucleophile to promote nucleophilic attack on the fluoroacetyl-CoA carbonyl group. Another potential although unlikely role could involve deprotonation of the fluoroacetyl-CoA α-carbon by His 76 to initiate an E1Cb-like (elimination–addition) mechanism to generate a ketene, which could then be trapped by Glu 50 to form the acyl-enzyme intermediate in a manner analogous to its breakdown. We therefore set out to clarify whether general base catalysis involving His 76 is essential for controlling the observed specificity in acylation.
Figure 1.

Steady-state and pre-steady-state kinetic analysis to investigate the role of the FlK catalytic residues in acylation specificity. (A) The catalytic triad of FlK consists of Thr 42, Glu 50, and His 76 (PDB entry 3P2Q). (B) pH–rate profile for wild-type FlK. (C) pH–rate profile for FlK-H76A. Values are reported as means ± the standard deviation (n = 3). (D) Pre-steady-state kinetics of FlK-catalyzed fluoroacetyl-CoA hydrolysis of fluoroacetyl-CoA at pH 6. (E) Pre-steady-state kinetics of FlK-T42S-catalyzed acetyl-CoA hydrolysis. (F) Pre-steady-state kinetics of FlK-T42S-catalyzed fluoroacetyl-CoA hydrolysis. Dotted lines indicate 1 equiv of enzyme.
We initiated these studies by examining FlK acylation under conditions in which His 76 is protonated and therefore unable to participate in general base catalysis, which should minimize structural perturbations in the active site compared to those occurring during mutagenesis. To determine appropriate conditions, we first constructed a pH–rate profile of FlK and observed an ionization event at acidic pH (Figure 1B). The apparent absence of this ionization in the pH–rate profile of the FlK-H76A mutant is consistent with its assignment to His 76 (Figure 1C). Although a second ionization event at basic pH was also observed, it is not clearly attributable to any of the residues known to be involved in catalysis. The existence of a similar ionization in the H76A mutant (Figure 1C), as well as in other hot dog-fold thioesterases,18 suggests that the origin of this deprotonation event is more complex and not directly related to His 76. We then performed pre-steady-state kinetic studies of FlK at pH 6, where His 76 should be predominantly protonated based on pH dependence studies. Under these conditions, we observed a burst phase of product formation consistent with enzyme acylation followed by a slower steady-state rate, which we interpret to be caused by the inaccessibility of the Cα deprotonation pathway in the absence of general base catalysis by His 76 (Figure 1D). The observed burst-phase rate constant (270 ± 120 s–1) demonstrates the kinetic competence of the protonated His 76 in forming the acyl-enzyme intermediate. In comparison, no burst phase is observed in FlK at pH 7.6 and fluoroacetyl-CoA is turned over at a steady-state rate of 220–270 s–1.9,10 These results demonstrate that general base catalysis involving His 76 is not required for acylation activity or fluoroacetylation specificity.
Because His 76 appears to play a structural role in FlK acylation, we turned our attention to exploring the contribution of Thr 42 to Glu 50 acylation. Indeed, steady-state kinetic studies have shown that the FlK-T42S mutant retains only ∼200-fold selectivity for fluoroacetyl-CoA over acetyl-CoA, an effect that is partially attributable to a 30-fold decrease in the KM for acetyl-CoA.9 Using pre-steady-state measurements, we determined that the acylation rate constant (k2) for the T42S mutant for acetyl-CoA was within error of the rate constant measured for wild-type FlK while the deacylation rate constant (k3) was reduced by 6-fold (Figure 1E). Kinetic modeling suggests that the observed decrease in KM for acetyl-CoA in FlK-T42S can be explained by this defect in the deacylation rate in the absence of any changes in substrate affinity (Figure S4 of the Supporting Information). For fluoroacetyl-CoA, the T42S mutation leads to a 3-fold decrease in acylation rate (k2) and a 25-fold decrease in deacylation rate (k3) (Figure 1F). From the different behavior of the two substrates with respect to acylation, it seems as if Thr 42 may make a small contribution to fluoroacetyl-CoA acylation specificity. Crystallographic studies have shown that Thr 42 is important in orienting Glu 50 and His 76 for reactivity. In the crystal structure of the T42S mutant, the Glu 50 side chain is rotated by 90°, both Ser 42 and His 76 populate two rotamers, and the hydrogen bonding network among the catalytic residues is disrupted (Figure S3 of the Supporting Information).17 The observed acylation defect is therefore likely to be related to structural effects that result in misorientation of the catalytic residues. Furthermore, the lack of an acylation defect for acetyl-CoA in both the H76A and T42S mutants suggests that the catalytic site is not optimized for utilization of this substrate.
Determining the Impact of Substrate Polarization on the Acylation Rate
On the basis of our studies, the catalytic residues, His 76 and Thr 42, appear to play a limited role in controlling the specificity of Glu 50 acylation. We therefore set out to probe the participation of the substrate itself in determining acylation specificity. On the basis of the electron-withdrawing nature of the fluorine atom, the fluoroacetyl-CoA carbonyl group is expected to be more activated toward nucleophilic attack than acetyl-CoA. We have previously compared the rates of chemical hydrolysis of these two substrates and showed that the rate of fluoroacetyl-CoA hydrolysis is at least 10-fold higher than the rate for acetyl-CoA.10 This ratio increases with an increasing pH up to 100-fold, which is similar to the observed magnitude of discrimination between fluoroacetyl-CoA and acetyl-CoA in the acylation step. To probe the role of carbonyl inductive polarization in acylation specificity, we examined the pre-steady-state kinetics of FlK-catalyzed hydrolysis of a series of acyl-CoAs with different functional groups at the α-position to access a wide range of values of the Taft polar substituent constant (σ*) to define the impact on this particular step.16
The rate of acylation (k2) for each substrate was measured using pre-steady-state kinetic experiments. For acetyl-CoA, acetoacetyl-CoA (33 ± 3 μM), bromoacetyl-CoA (30 ± 3 μM), and chloroacetyl-CoA (28 ± 3 μM), a burst phase of product formation corresponding to 1 equiv of enzyme (25 μM) was observed, followed by a slower steady-state rate identical to the previously measured steady-state rate (Figure 2A–C), indicating that hydrolysis of these substrates is likely to proceed through the same acyl-enzyme intermediate that was trapped for acetyl-CoA (Scheme 1). For FlK-catalyzed cyanoacetyl-CoA hydrolysis, plots of CoA formed versus time could not be fit with an equation describing burst-phase kinetic behavior and were instead best fit by a simple linear fit (Figure 2D). However, the dead time of the rapid quench instrument is 2 ms, precluding the measurement of rate constants approaching or exceeding 500 s–1. To distinguish between the absence of a burst phase and the presence of a burst phase that is over before the first quenched time point, we examined the amount of CoA formed after 2 ms. In the absence of a burst phase, the CoA concentration would be given by [CoA] = kcat[FlK]t, corresponding to a line with a y-intercept of zero. On the other hand, if a burst phase had occurred before the first quenched time point, the CoA concentration would be given by [CoA] = kcat[FlK]t + [FlK], corresponding to a line with a y-intercept of [FlK]. Linear fitting of the data gave a y-intercept of 77 ± 7 μM, within error of the FlK concentration of 75 μM used in the assay (Figure 2D). We therefore conclude that cyanoacetyl-CoA is also hydrolyzed through a mechanism involving an acyl-enzyme intermediate, although we could not include this substrate in our free energy relationship analysis because the acylation rate could not be accurately measured.
Figure 2.

Pre-steady-state kinetic and Taft analysis of a series of acyl-CoAs. The dashed line indicates 1 equiv of enzyme: (A) acetoacetyl-CoA (R = Ac), (B) bromoacetyl-CoA (R = Br), (C) chloroacetyl-CoA (R = Cl), and (D) cyanoacetyl-CoA (R = CN). (E) Taft plot for the acylation step of FlK-catalyzed acyl-CoA hydrolysis. Acylation rate constants (k2) are derived from nonlinear curve fitting of pre-steady-state kinetic time courses. Values are reported as means ± the standard deviation (n = 3). The linear fit gave a ρ* value of 1.7 ± 0.2 (R2 = 0.986).
A plot of log k2 versus σ* for the series of acyl-CoAs revealed a linear relationship (Figure 2E), indicating that acylation of all of the substrates proceeds through a common mechanism and transition state, which is in contrast to what is observed when kcat is plotted similarly.16 On the basis of this linear relationship, specificity in FlK acylation appears to be based on inductive carbonyl polarization rather than a change in mechanism, rate-limiting step, or transition-state structure induced by the fluorine substituent. The slope of the line, ρ*, is the polar sensitivity factor that compares the reaction under study to the reference reaction, which is methyl ester hydrolysis.16 A slope of >1 shows that the reaction under study is more sensitive to substituents than the reference reaction, while a slope of <1 shows that it is less sensitive. The Taft plot for acylation gave a ρ* value of 1.7 ± 0.2, indicating that FlK acylation is more sensitive to the influence of substituents that alter induced carbonyl polarization than the chemical hydrolysis of methyl esters on which the Taft scale is based. This sensitivity may be attributable to the inductive polarization of thioesters being higher than that of esters, or to additional activation of the carbonyl group on the enzyme. In combination with the mutagenesis studies discussed above, our Taft analysis of FlK substrates suggests that specificity in the rate of enzyme acylation is determined mainly by the intrinsic polarization of the fluoroacetyl-CoA thioester compared to the acetyl-CoA thioester rather than by features of the enzyme that specifically accelerate reaction with the fluorinated substrate.
Investigating the Role of Fluorine Molecular Recognition in FlK-Catalyzed Thioester Hydrolysis
Although our studies suggest that the main selectivity determinant for FlK acylation is the intrinsic reactivity of the fluoroacetyl group, this finding does not exclude the possibility that the C–F bond is recognized in the enzyme active site. If so, interactions between FlK and the fluoroacetyl-CoA substrate could reduce free rotation of the fluoromethyl group such that each of the two prochiral α-protons would occupy a different position in three-dimensional space. In this case, we might expect that FlK could exhibit a kinetic preference for substrates with an available pro-R or pro-S proton. To test this hypothesis, we prepared (RS)-2-fluoropropionyl-CoA, a substrate in which one hydrogen at the α-carbon of a fluoroacetyl-CoA has been replaced by a methyl group (Figure 3A). Upon incubation of this substrate with FlK, we observed two steady-state kinetic phases (Figure 3B and Figure S5 of the Supporting Information), suggesting that the two stereoisomers may be hydrolyzed at different rates specified by their relative values of kcat/KM. A plot of the fast and slow rates as a function of substrate concentration revealed that both phases exhibit saturation kinetic behavior (Figure 3C). Although the true kcat and KM for the two individual stereoisomers cannot be extracted from this data, we hypothesized that these two phases might correspond to different rates of hydrolysis for the different stereoisomers.
Figure 3.

Kinetic analysis of 2-fluoropropionyl-CoA hydrolysis. (A) 2-Fluoropropionyl-CoA stereoisomer structures. (B) Time course for hydrolysis of 100 μM (RS)-2-fluoropropionyl-CoA. (C) Observed rate constants (kobs) for the slow phases and fast phases plotted over a range of substrate concentrations. (D) Steady-state kinetic analysis of (S)-2-fluoropropionyl-CoA. Values are reported as means ± the standard deviation (n = 3). (E) Steady-state kinetic analysis of (R)-2-fluoropropionyl-CoA. Values are reported as means ± the standard deviation (n = 3). (F) Pre-steady-state kinetic analysis of FlK-catalyzed hydrolysis of (S)-2-fluoropropionyl-CoA. (G) Pre-steady-state kinetic analysis of FlK-catalyzed hydrolysis of (R)-2-fluoropropionyl-CoA. (H) Analysis of FlK acylation by 2-fluoropropionyl-CoA in terms of the Felkin–Ahn model. The steric and electronic preferences of the substrate in combination with the structural constraints of the enzyme may explain the defect in acylation rate for (S)-2-fluoropropionyl-CoA compared to (R)-2-fluoropropionyl-CoA.
To test this idea, we prepared the individual (S)- and (R)-2-fluoropropionyl-CoA stereoisomers (Figure 3A) from the corresponding chiral methyl lactates and measured the steady-state kinetic parameters for each stereoisomer (Figure 3D,E). Indeed, we observed that each stereoisomer was hydrolyzed at a different rate. Interestingly, when these substrates are added to the steady-state Taft plot for log kcat versus σ*,10 the S isomer falls more on the same line as fluoroacetyl-CoA and chloroacetyl-CoA while the R isomer appears to behave more like the less inductively polarized substrates (Figure S6 of the Supporting Information). This result can be interpreted to indicate that there is a change in mechanism or rate-limiting step between the S and R isomers and that hydrolysis of the S isomer proceeds through the same kinetic and chemical mechanism as fluoroacetyl-CoA, while hydrolysis of the R isomer proceeds through the same kinetic and chemical mechanism as acetyl-CoA. However, we cannot rule out the possibility that a rate-limiting rearrangement of the R isomer acyl-enzyme intermediate produces differing kinetic behavior but allows this substrate to access the same chemical mechanism for hydrolysis as the S isomer. Nonetheless, the kinetic data indicate that fluorine-based polarization of the substrate alone is insufficient to dictate the reaction pathway through which thioester hydrolysis proceeds.
In addition to the difference in kcat, the two stereoisomers diverge with regard to their KM values, with the R isomer exhibiting a KM lower than that measured for the S isomer. This observation could be explained by either preferential binding of the R isomer, faster acylation by the R isomer, or slower deacylation of the R isomer. To determine the basis of the observed KM difference, we examined the pre-steady-state kinetic behavior of each isomer under saturating conditions (Figure 3F,G). For (S)-2-fluoropropionyl-CoA, we observed single-phase kinetics with a rate constant equal to kcat for the S isomer, which indicates that acylation is rate-limiting for this substrate as it is for the native substrate (Figure 3F). In contrast, we observed a burst of 1 equiv of CoA formed with (R)-2-fluoropropionyl-CoA, followed by a slower steady-state rate with a rate constant matching the kcat of the R isomer (Figure 3G). We interpret this kinetic behavior to demonstrate that rate-liming step for the R isomer has changed to breakdown of the acyl-enzyme intermediate (k3 = kcat). Taken together, the results of the pre-steady-state kinetic experiments are also consistent with the behavior of the isomers with regard to the linear free energy plot (Figure S6 of the Supporting Information). Additional kinetic modeling using these rate constants shows that the differences in the rates of chemical catalysis between the two substrates are sufficient to explain their differing steady-state kinetics without the need to invoke differential binding affinities (Figure S7 of the Supporting Information).
Because the only significant difference between the S and R isomers is expected to be the orientation of the α-substituents, the differences in the rate constants of the acylation (2-fold) and deacylation (4-fold) steps between the two stereoisomers suggest that the fluorine atom is specifically recognized by the enzyme and that the α-carbon does not freely rotate. Indeed, the difference observed in the deacylation rate for each isomer can be explained by a requirement for the α-proton to be accessible to His 76 for the substrate to access the faster Cα deprotonation pathway.10 The orientation of the substrate via the fluorine atom could result in the methyl group occupying the space where an α-proton would normally be poised for abstraction by His 76, which could lead to a change in the mechanism from Cα deprotonation to addition of water followed by elimination of the Glu 50 leaving group. On the basis of the kinetic data, we hypothesize the His 76 preferentially abstracts the pro-R proton, leading to a compromised deacylation rate when the methyl group occupies this position instead. However, the basis for the difference between the acylation rates is more difficult to explain. Considering an addition–elimination mechanism for enzyme acylation, the observed difference in the acylation rates is likely due to the increased steric bulk of the additional methyl substituent, which kinetic data suggest is positioned unfavorably for acylation in the S isomer. One possible source of this kinetic selectivity is that recognition of the fluorine substituent by FlK could force nucleophilic attack by Glu 50 to occur along a trajectory that is disfavored based on the Felkin–Anh model for carbonyl addition (Figure 3H).19,20
Stereochemical Course of the FlK Reaction
To directly test the stereochemical course of acylation and deacylation without the need for the enzyme to accommodate an additional α-substituent in its active site, we prepared (S)-[2H1]fluoroacetyl-CoA and (R)-[2H1]fluoroacetyl-CoA from the corresponding carboxylic acids.14 Our previous kinetic studies with [2H2]fluoroacetyl-CoA showed that there is a kinetic isotope effect (KIE) of 2.4 ± 0.1 on the deacylation step of the reaction mechanism, consistent with abstraction of an α-proton from the substrate in this step.10 We therefore predict that if the enzyme utilizes chiral recognition of the prochiral fluoromethyl group in the deacylation step, one of the [2H1]fluoroacetyl-CoA stereoisomers will have its 2H positioned for proton abstraction and will exhibit a primary KIE, while the other stereoisomer will show a much smaller secondary KIE, which may be negligible compared to the error in the rates. In contrast, based on the addition–elimination mechanism that we have proposed for the acylation step, we would expect both substrates to exhibit a secondary inverse KIE on acylation, which may be too small to detect by direct comparison of rates.
To interrogate both the acylation and deacylation steps using the chiral deuterated substrates, we examined the pre-steady-state kinetics of their FlK-catalyzed hydrolysis (Figure 4). Similar to the unlabeled fluoroacetyl-CoA, (S)-[2H1]fluoroacetyl-CoA exhibited single-phase pre-steady-state kinetic behavior with the observed kcat within error of that for the undeuterated substrate. While this result does not conclusively demonstrate the presence of an inverse secondary KIE, it not inconsistent considering the propagated error. In contrast, (R)-[2H1]fluoroacetyl-CoA exhibited burst-phase kinetic behavior. While the acylation rate constant for this substrate was within error of the kcat observed for the undeuterated substrate, a kinetic isotope effect (KIE) of 1.7 ± 0.2 was observed on the steady-state rate, consistent with the previously reported KIE of 2.4 observed with the doubly deuterated [2H2]fluoroacetyl-CoA substrate. This result provides further evidence that while the acylation rate is influenced mainly by polarization, consistent with an addition–elimination-type mechanism, deacylation requires specific recognition of the prochiral fluoromethyl group to position the α-carbon for proton abstraction catalyzed by His 76, which preferentially abstracts the pro-R proton. Notably, the mechanistically related hot dog-fold dehydratases, which utilize a mechanism initiated by Cα deprotonation to catalyze elimination of water from 3-hydroxyacyl-CoAs, also conduct stereoselective deprotonation.21,22 While the pro-2S proton is removed by dehydratases, it has the same relative stereochemistry as the pro-2R proton in fluoroacetyl-CoA because the S designation is derived from a change in substituent priorities based on the Cahn–Ingold–Prelog rules. While the proton abstraction occurs on an acyl-enzyme intermediate in FlK instead of on the substrate itself, the commonality in mechanism between the hot dog-fold dehydratases and FlK is consistent with their surprisingly close evolutionary relationship,10 as well as the structural similarity of their active sites.9,23
Figure 4.
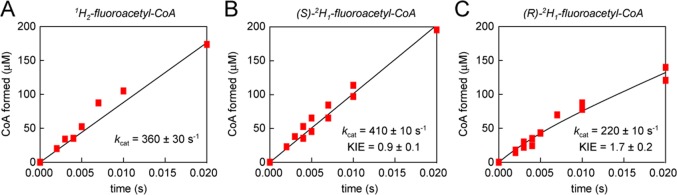
Pre-steady-state kinetic analysis of monodeuterated fluoroacetyl-CoAs. FlK-catalyzed hydrolysis of (A) undeuterated fluoroacetyl-CoA (left) is compared to hydrolysis of (B) (S)-[2H1]fluoroacetyl-CoA (center) and (C) (R)-[2H1]fluoroacetyl-CoA (right).
Conclusions
Our data suggest a chemical, kinetic, and structural mechanism for the selectivity of FlK for fluoroacetyl-CoA (Scheme 2). Following substrate binding, fluoroacetyl-CoA reacts with Glu 50 to acylate this side chain. Taft free energy relationship analysis of the acylation step (Figure 2E) indicates that the observed enhancement of 2 orders of magnitude in the rate of acylation for fluoroacetyl-CoA compared to that of acetyl-CoA is provided by the intrinsic inductive polarization of the fluoroacetyl-CoA carbonyl group. Kinetic studies of (R)- and (S)-2-fluoropropionyl-CoA demonstrated that although these substrates differ only by the orientation of their α-substituents, their rates of acylation differ by 2-fold. Given the differences in acylation rate between the two 2-fluoropropionyl-CoA stereoisomers, we propose that the C–F bond is specifically recognized by FlK, allowing it to distinguish the two enantiotopic faces of the carbonyl group during the acylation step.
Scheme 2. Model for the Influence of Fluorine Recognition on the Course of the FlK-Catalyzed Reaction.
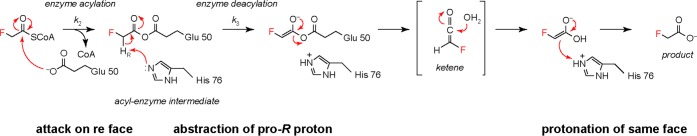
On the basis of the kinetic analysis of substrates with a stereogenic center at the α-carbon, we propose that Glu 50 attacks the fluoroacetyl-CoA thioester bond from the re face. The pro-R proton of the resultant intermediate is then removed by His 76. Following hydration of the putative intermediate, the carboxylic acid enolate is reprotonated on the same face from which the proton was removed.
After FlK acylation, a proton is abstracted by His 76 to generate an enolate that can break down through a proposed ketene intermediate. A 4-fold difference in the rates of deacylation of (S)- and (R)-2-fluoropropionyl-CoA suggested that FlK might utilize chiral recognition of the prochiral fluoromethyl group of fluoroacetyl-CoA in the deacylation step to specifically position one proton for abstraction. Indeed, kinetic studies of (S)- and (R)-[2H1]fluoroacetyl-CoA demonstrated a primary KIE of 1.7 ± 0.2 on only the R stereoisomer, suggesting that the pro-R proton is specifically abstracted. Taken together, our studies of FlK substrates with a stereogenic center at the α-carbon provide evidence that the C–F bond is specifically recognized by FlK to orient the α-carbon for proton abstraction catalyzed by His 76. The previously reported lack of exchange between the fluoroacetyl-CoA α-protons and solvent10 along with the pH–rate profiles of FlK and FlK-H76A (Figure 1B,C) suggests that FlK may utilize a one-base mechanism to achieve this transformation, rather than separate catalytic residues that function in general acid catalysis and general base catalysis. Therefore, following hydration of the putative ketene intermediate, we propose that the resultant enolate is reprotonated by the His 76 imidazolium side chain on the same face from which deprotonation occurred. This mechanistic proposal is similar to the one-base mechanism elucidated for the hot dog-fold dehydratases.21,22
Taken together, our results suggest that molecular recognition of fluorine, rather than simple inductive polarization and enolate stability, is involved in making the Cα deprotonation pathway accessible to the fluorinated substrate. Thus, substrate polarization appears to be necessary, but not sufficient, for rate acceleration. This observation provides a possible explanation for the inability of substrates like acetoacetyl-CoA, which is expected to form a more stable enolate than fluoroacetyl-CoA but is much larger in size, to access the Cα deprotonation pathway. In the absence of a high-resolution structure of the FlK–substrate complex, it is difficult to precisely define the structural determinants of chiral fluoromethyl group recognition. However, previously reported structural and mutagenesis studies of FlK have suggested multiple possible mechanisms through which molecular recognition of the C–F bond may be achieved. Computational docking studies have suggested that the guanidinium group of Arg 120 and the backbone amide of Gly 69 may be involved in C–F recognition through dipolar interactions,9,17 while mutagenesis data suggest that the hydrophobic nature of the FlK active site may contribute to molecular recognition of the C–F bond based on its polar hydrophobicity.9
Although fluorine’s ability to initiate unusual enzymatic reaction pathways involved in mechanism-based inhibition is well-known, FlK represents a rare example in which an enzyme exploits the unique properties of fluorine to enhance its reaction rate and to drive substrate selectivity. The results reported here demonstrate that discrimination of the fluoroacetyl-CoA substrate relies not only on the enhanced inductive polarization afforded by fluorine’s high electronegativity but also on the molecular recognition of fluorine to position the substrate for optimal reactivity. These studies of FlK provide insights into how enzymes evolve to recognize unusual functional groups, with implications for fluorine molecular recognition and the design of mechanism-based inhibitors.
Acknowledgments
We thank Dr. Mark Walker and Benjamin Thuronyi for purification of acetate kinase and phosphotransacetylase. 13C NMR spectra of (S)- and (R)-[2H1]fluoroacetyl-CoA were collected by Dr. Jeffrey Pelton at the Central California 900 MHz NMR Facility (supported by National Institutes of Health Grant P41GM68933).
Glossary
Abbreviations
- CoA
coenzyme A
- KIE
kinetic isotope effect
- PDB
Protein Data Bank.
Supporting Information Available
Additional figures as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ A.M.W.: Department of Pharmaceutical Chemistry, University of California, San Francisco, CA 94158.
This work was funded by generous support from University of California, Berkeley, and National Institutes of Health (NIH) New Innovator Award 1 DP2 OD008696. A.M.W. acknowledges the support of an NIH National Research Service Award Training Grant (1 T32 GMO66698), a National Science Foundation Graduate Research Fellowship, and an Aldo DeBenedictis Fellowship.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- O’Hagan D.; Perry R.; Lock J. M.; Meyer J. J. M.; Dasaradhi L.; Hamilton J. T. G.; Harper D. B. (1993) High levels of monofluoroacetate in Dichapetalum braunii. Phytochemistry 33, 5. [Google Scholar]
- Harper D. B.; O’Hagan D. (1994) The fluorinated natural products. Nat. Prod. Rep. 11, 123–133. [DOI] [PubMed] [Google Scholar]
- Clarke D. D. (1991) Fluoroacetate and fluorocitrate: Mechanism of action. Neurochem. Res. 16, 1055–1058. [DOI] [PubMed] [Google Scholar]
- Lauble H.; Kennedy M. C.; Emptage M. H.; Beinert H.; Stout C. D. (1996) The reaction of fluorocitrate with aconitase and the crystal structure of the enzyme-inhibitor complex. Proc. Natl. Acad. Sci. U.S.A. 93, 13699–13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H.; O’Hagan D.; Schaffrath C. (2004) Fluorometabolite biosynthesis and the fluorinase from Streptomyces cattleya. Nat. Prod. Rep. 21, 773–784. [DOI] [PubMed] [Google Scholar]
- Sanada M.; Miyano T.; Iwadare S.; Williamson J. M.; Arison B. H.; Smith J. L.; Douglas A. W.; Liesch J. M.; Inamine E. (1986) Biosynthesis of fluorothreonine and fluoroacetic acid by the thienamycin producer, Streptomyces cattleya. J. Antibiot. 39, 259–265. [DOI] [PubMed] [Google Scholar]
- Huang F.; Haydock S. F.; Spiteller D.; Mironenko T.; Li T. L.; O’Hagan D.; Leadlay P. F.; Spencer J. B. (2006) The gene cluster for fluorometabolite biosynthesis in Streptomyces cattleya: A thioesterase confers resistance to fluoroacetyl-coenzyme A. Chem. Biol. 13, 475–484. [DOI] [PubMed] [Google Scholar]
- Walker M. C.; Wen M.; Weeks A. M.; Chang M. C. (2012) Temporal and fluoride control of secondary metabolism regulates cellular organofluorine biosynthesis. ACS Chem. Biol. 7, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks A. M.; Coyle S. M.; Jinek M.; Doudna J. A.; Chang M. C. (2010) Structural and biochemical studies of a fluoroacetyl-CoA-specific thioesterase reveal a molecular basis for fluorine selectivity. Biochemistry 49, 9269–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks A. M.; Chang M. C. (2012) Catalytic control of enzymatic fluorine specificity. Proc. Natl. Acad. Sci. U.S.A. 109, 19667–19672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F.; Thoden J. B.; Zhuang Z.; Latham J.; Trujillo M.; Holden H. M.; Dunaway-Mariano D. (2012) The catalytic mechanism of the hotdog-fold enzyme superfamily 4-hydroxybenzoyl-CoA thioesterase from Arthrobacter sp. strain SU. Biochemistry 51, 7000–7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L. (2002) Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4524. [DOI] [PubMed] [Google Scholar]
- Fritz-Langhals E.; Schutz G. (1993) Simple synthesis of optically-active 2-fluoropropanoic acid and analogs of high enantiomeric purity. Tetrahedron Lett. 34, 293–296. [Google Scholar]
- Wadoux R. D. P.; Lin X.; Keddie N. S.; O’Hagan D. (2013) Chiral fluoroacetic acid: Synthesis of (R)- and (S)-[2H1]-fluoroacetate in high enantiopurity. Tetrahedron: Asymmetry 24, 719–723. [Google Scholar]
- Purich D. L. (2002) Covalent enzyme-substrate compounds: Detection and catalytic competence. Methods Enzymol. 354, 1–27. [DOI] [PubMed] [Google Scholar]
- Taft R. W. Jr. (1952) Polar and steric substituent constants for aliphatic and o-benzoate groups from rates of esterification and hydrolysis of esters. J. Am. Chem. Soc. 74, 3120–3128. [Google Scholar]
- Dias M. V.; Huang F.; Chirgadze D. Y.; Tosin M.; Spiteller D.; Dry E. F.; Leadlay P. F.; Spencer J. B.; Blundell T. L. (2010) Structural basis for the activity and substrate specificity of fluoroacetyl-CoA thioesterase FlK. J. Biol. Chem. 285, 22495–22504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F.; Zhuang Z.; Finci L.; Dunaway-Mariano D.; Kniewel R.; Buglino J. A.; Solorzano V.; Wu J.; Lima C. D. (2006) Structure, function, and mechanism of the phenylacetate pathway hot dog-fold thioesterase PaaI. J. Biol. Chem. 281, 11028–11038. [DOI] [PubMed] [Google Scholar]
- Anh N. T.; Eisenstein O.; Lefour J. M.; Tran Huu Dau M. E. (1973) Orbital factors and asymmetric induction. J. Am. Chem. Soc. 95, 6146–6147. [Google Scholar]
- Burgi H. B.; Dunitz J. D.; Lehn M.; Wipff G. (1974) Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 30, 1563–1572. [Google Scholar]
- Labonte J. W.; Townsend C. A. (2013) Active site comparisons and catalytic mechanisms of the hot dog superfamily. Chem. Rev. 113, 2182–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab J. M.; Habib A.; Klassen J. B. (1986) A thorough study of the stereochemical consequences of the hydration/dehydration reaction catalyzed by β-hydroxydecanoyl thioester dehydrase. J. Am. Chem. Soc. 108, 5304–5308. [Google Scholar]
- Hisano T.; Tsuge T.; Fukui T.; Iwata T.; Miki K.; Doi Y. (2003) Crystal structure of the (R)-specific enoyl-CoA hydratase from Aeromonas caviae involved in polyhydroxyalkanoate biosynthesis. J. Biol. Chem. 278, 617–624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.