

Abstract
A component of the shikimate biosynthetic pathway, dehydroquinate dehydratase (DHQD) catalyzes the dehydration of 3-dehydroquniate (DHQ) to 3-dehydroshikimate. In the type I DHQD reaction mechanism a lysine forms a Schiff base intermediate with DHQ. The Schiff base acts as an electron sink to facilitate the catalytic dehydration. To address the mechanism of Schiff base formation, we determined structures of the Salmonella enterica wild-type DHQD in complex with the substrate analogue quinate and the product analogue shikimate. In addition, we determined the structure of the K170M mutant (Lys170 being the Schiff base forming residue) in complex with quinate. Combined with nuclear magnetic resonance and isothermal titration calorimetry data that revealed altered binding of the analogue to the K170M mutant, these structures suggest a model of Schiff base formation characterized by the dynamic interplay of opposing forces acting on either side of the substrate. On the side distant from the substrate 3-carbonyl group, closure of the enzyme’s β8−α8 loop is proposed to guide DHQ into the proximity of the Schiff base-forming Lys170. On the 3-carbonyl side of the substrate, Lys170 sterically alters the position of DHQ’s reactive ketone, aligning it at an angle conducive for nucleophilic attack. This study of a type I DHQD reveals the interplay between the enzyme and substrate required for the correct orientation of a functional group constrained within a cyclic substrate.
The shikimate pathway has been recognized as an attractive antibiotic target due to the fact that it is essential in bacteria but absent in humans.1 The product of this seven-step pathway is chorismate, a precursor required for the synthesis of aromatic amino acids and other important metabolites. Step 3 of the shikimate pathway is catalyzed by dehydroquinate dehydratase (DHQD), an enzyme that converts 3-dehydroquinate (DHQ) to 3-dehydroshikimate (DHS) (Figure 1). Two nonhomologous and mechanistically dissimilar DHQD types with discrete phylogenetic coverage have been characterized.2 Members of the greater class I aldolase superfamily, type I DHQDs establish a Schiff base with the substrate that acts as an electron sink to promote the catalytic dehydration.3−5 By contrast, type II DHQDs employ an unrelated noncovalent mechanism that proceeds via an enolate intermediate.6,7
Figure 1.

Type I DHQD reaction intermediates and analogues. The DHQD-catalyzed reaction is depicted centrally. Quinate and shikimate, boxed on the sides, biosynthetically relate to DHQ and DHS, respectively. Schiff base formation occurs between the ε-amino group of Lys170 and C3 of the substrate. The atoms that are a part of Schiff base formation are colored red.
The focus of this work is the type I DHQD from the human pathogen Salmonella enterica. Our interest in this enzyme is twofold: (1) to improve our understanding of the mechanism used by this enzyme to convert DHQ to DHS and (2) to ultimately exploit this improved understanding to inform the development of DHQD inhibitors. Such inhibitors could act as a new class of antibiotics.
The greater class I aldolase superfamily consists of a group of enzymes that share a TIM barrel (α/β)8 fold and an active site lysine that forms a covalent Schiff base with the reaction substrate.8 Within this superfamily, formation of the catalytic Schiff base is initiated by nucleophilic attack of the active site lysine Nε atom on the electrophilic carbonyl carbon of the substrate, generating a carbinolamine intermediate that gives way to the Schiff base (Figure 1).
On the basis of accepted nucleophilic approach principles, it can be deduced that significant angular rotation changes must occur between the substrate–protein interactions over the course of Schiff base formation.9 The reasoning behind this argument is as follows: if, as predicted by physical chemistry principles, the nucleophilic approach of the Schiff base-forming lysine Nε atom relative to the plane of a substrate double bond (carbonyl) follows an obtuse trajectory but ultimately the same Nε atom is contained within the plane of that double bond (in the Schiff base), then over the course of bond formation >90° of angular rotation (the Nε atom toward the substrate and/or the substrate back toward the Nε atom) must occur.9
To ascertain the precise form this angular rotation takes in a representative enzyme, in previous work we analyzed the individual steps that occur during Schiff base formation for the enzyme transaldolase.9 These studies revealed that bond formation is associated with significant conformational change that is predominantly confined to the substrate, specifically to the bonds adjoining the substrate carbonyl, with little change in the conformation of active site residues. A comparison to related enzymes suggests that this form of substrate conformational change likely represents a common feature of Schiff base formation in enzymes acting on linear ketone substrates.9
However, the Schiff base-forming mechanism observed in transaldolase cannot be extended to type I DHQDs. Unlike transaldolase and the majority of other characterized Schiff base-forming enzymes, the DHQD substrate carbonyl is a carbocyclic ring constituent. This unique property constrains the allowed dihedral change in the carbonyl adjoining bonds, the defining feature associated with transaldolase Schiff base formation, and means that Schiff base formation must qualitatively differ from the process for enzymes that act upon linear substrates.
To understand Schiff base formation in type I DHQDs, we previously attempted to capture the DHQD Michaelis complex. Our strategy was to analyze the structure of the inactive Schiff base-forming lysine to methionine mutant (K170M) cocrystallized with DHQ.10 The resulting complex revealed that DHQ initially docks in an orientation similar to its Schiff base-bound state. This was a surprising result, because this binding mode is inconsistent with nucleophilic approach by Lys170 following an obtuse trajectory. On the basis of this observation, we reasoned that formation of a bond between the Lys170 ε-amino group and the C3 atom of DHQ (i.e., Schiff base bond) must either follow a higher-energy pathway with the substrate binding in its observed orientation or, alternatively, follow a lower-energy pathway with the substrate binding in a different orientation at which the obtuse approach angle can be accessed.10
Complicating the analysis of DHQD Schiff base formation is the question of whether binding of substrate to the K170M mutant accurately reflects the wild-type Michaelis state. At issue is the position of the ε-amino group of Lys170, which when modeled into the K170M structure could sterically clash with the substrate carbonyl oxygen. To address this concern, we sought an alternative strategy for probing the Michaelis state.
Quinate and shikimate are one biosynthetic step removed from the DHQD substrate and product, respectively. Differing from DHQ and DHS only in containing a hydroxyl rather than a carbonyl at the reactive 3-position (Figure 1), quinate and shikimate should bind noncovalently to DHQD. With the objective of clarifying the mechanism of Schiff base formation, we characterized the binding of quinate and shikimate to the wild-type and K170M mutant DHQD using ITC, NMR, and X-ray crystallography. The data presented here reveal three distinct binding modes of the reaction analogues and demonstrate enhanced affinity in the K170M mutant. Incorporating these findings with previous data, we propose that the dynamic interplay of residues on either side of the active site acts to correctly position the substrate for nucleophilic attack.
Experimental Procedures
Protein Expression and Purification
As previously described, wild-type and K170M constructs were expressed in Escherichia coli strain BL21(DE3) in the pMCSG7 vector.10−12 After being inoculated with an overnight culture, cells were grown for 4 h at 37 °C. Then, the temperature was reduced to 25 °C and protein overexpression induced by the addition of isopropyl 1-thio-d-galactopyranoside to a final concentration of 0.5 mM. Cells were grown overnight before being harvested by centrifugation, resuspended in a buffer containing 10 mM Tris (pH 8.3), 500 mM NaCl, 10% glycerol, and 5 mM β-mercaptoethanol, and lysed by sonication. Protein was purified by Ni-NTA affinity chromatography and step-eluted with 0.5 M imidazole. To remove the expression tag, purified DHQD was incubated overnight at 4 °C with hexahistidine-tagged TEV protease and repurified by Ni-NTA chromatography.
Protein Crystallization and Collection of X-ray Data
Sitting drop crystallization experiments were performed at room temperature using a 1:1 ratio of DHQD (7.5 mg/mL) to reservoir. Wild-type crystals were incubated for ∼15 min in 5 M sodium quinate or 2 M sodium shikimate solutions before being frozen. The K170M mutant crystal was soaked for ∼15 min in a mother liquor, consisting of reservoir solution supplemented with 5 mM quinate. The wild-type crystals were harvested from a condition containing 0.01 M nickel chloride, 0.1 M Tris (pH 8.5), and 20% poly(ethylene glycol) monomethyl ether 2000. The K170M mutant crystal was harvested from a condition containing 0.1 MIB buffer (pH 4) and 25% poly(ethylene glycol) 1500. Crystals were frozen in liquid nitrogen, and diffraction data were collected at 100 K at the Life Sciences Collaborative Access Team at the Advance Photon Source (Argonne, IL).
Crystal Structure Determination and Refinement
Data were processed using HKL-3000 for indexing, integration, and scaling.13 Structures were determined by molecular replacement in Phaser, using the unliganded DHQD structure [Protein Data Bank (PDB) entry 3L2I] as a search model.14 Structures were iteratively refined with Refmac15 after working models were displayed in Coot16 and manually adjusted on the basis of electron density maps. All structure figures were prepared using PyMOL Molecular Graphics System, version 1.3 (Schrödinger, LLC).
Isothermal Titration Calorimetry (ITC)
ITC experiments were performed using the MicroCal ITC200 instrument (GE Healthcare) with the jacket temperature set at 25 °C. Wild-type and K170M DHQD samples were prepared by dialysis into a buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, and 5 mM 2-mercaptoethanol. Titrations of 10 mM quinate or shikimate dissolved in the same buffer were performed with either buffer or 200 μM wild-type or K170M DHQD. In each case, injections from a 40 μL syringe rotating at 500 rpm were spaced at 2 min intervals. An initial 0.2 μL injection, which was subsequently removed during data analysis, was followed by 17 injections of 2.0 μL each. For K170M DHQD, binding parameters (Kd, ΔH, and −TΔS) were obtained by fitting the resultant data to a single-site binding model using Origin 7 after subtracting heats of dilution into buffer. The Kd was calculated as the inverse of the Ka.
Nuclear Magnetic Resonance (NMR)
NMR experiments were performed on a Bruker 900 MHz AVANCE spectrometer equipped with a cryogenic triple-resonance probe. Experimental conditions were 10 μM DHQD and 500 μM quinate or shikimate in 20 mM PO4 (pH 7.4) and 100 mM NaCl in 90% 1H2O and 10% 2H2O at 25 °C in 3 mm NMR tubes. The WaterLOGSY experiments were performed as previously described.17 Water was selectively saturated using a 2 ms square-shaped pulse with a mixing time of 2 s and a relaxation delay of 2.5 s. Spectra were processed by NMRPipe with a 5 Hz line broadening function and analyzed by NMRDraw.18
Results
Wild-Type DHQD–Quinate and −Shikimate Complexes
Wild-type DHQD was cocrystallized with 0.5 M quinate or shikimate. However, no discernible electron density for the compounds was observed at the DHQD active site. Consequently, we resorted to soaking crystals in 5 M quinate or 2 M shikimate prior to data collection. Employing this soaking regime, structures determined to 1.78 and 1.50 Å resolution (Table 1) revealed clear active site electron density for quinate and shikimate, respectively.
Table 1. Data Collection and Refinement Statisticsa.
| WT–quinate | WT–shikimate | K170M–quinate | |
|---|---|---|---|
| PDB entry | 4GUI | 4GUJ | 4IUO |
| Data Collection | |||
| space group | P21 | P21 | P212121 |
| unit cell dimensions | a = 48.67 Å | a = 48.62 Å | a = 36.91 Å |
| b = 74.43 Å | b = 74.97 Å | b = 72.80 Å | |
| c = 63.13 Å | c = 63.04 Å | c = 170.91 Å | |
| α = 90.00° | α = 90.00° | α = 90.00° | |
| β = 100.57° | β = 100.49° | β = 90.00° | |
| γ = 90.00° | γ = 90.00° | γ = 90.00° | |
| resolution range (Å) | 30.00–1.78 (1.81–1.78) | 30.00–1.50 (1.53–1.50) | 28.51–1.80 (1.85–1.80) |
| completeness (%) | 97.2 (99.3) | 96.8 (95.0) | 99.2 (95.3) |
| redundancy | 3.0 (2.3) | 3.9 (3.9) | 5.3 (4.2) |
| ⟨I/σ(I)⟩ | 17.5 (2.4) | 25.5 (3.8) | 16.4 (2.8) |
| Rmerge (%) | 5.6 (31.4) | 4.5 (36.7) | 11.0 (55.0) |
| Refinement | |||
| resolution range (Å) | 28.66–1.78 (1.83–1.78) | 28.66–1.50 (1.54–1.50) | 28.51–1.80 (1.85–1.80) |
| no. of reflections | 41302 (3071) | 68926 (4983) | 43418 (3058) |
| Rwork/Rfreeb | 15.9/18.8 | 15.5/18.2 | 17.8/20.8 |
| no. of atoms | |||
| protein | 3619 | 3501 | 3583 |
| water | 239 | 420 | 435 |
| shikimate or quinate | 26 | 24 | 26 |
| average B factor (Å2) | |||
| protein | 33.8 | 20.5 | 18.9 |
| water | 40.3 | 30.8 | 27.8 |
| shikimate or quinate | 37.6 | 19.8 | 16.2 |
| root-mean-square deviation | |||
| bond lengths (Å) | 0.008 | 0.009 | 0.007 |
| bond angles (deg) | 1.35 | 1.42 | 1.26 |
| Ramachandran analysis (%) | |||
| favored regions | 98.0 | 98.7 | 97.8 |
| allowed regions | 100 | 100 | 100 |
| disallowed regions | 0 | 0 | 0 |
Data for the highest-resolution shell are given in parentheses.
Definition of Rwork and Rfree: R = ∑hkl||Fobs| – |Fcalc||/∑hkl|Fobs|, where hkl are the reflection indices used in the refinement for Rwork and the 5% not used in the refinement for Rfree. Fobs and Fcalc are structure factors deduced from measured intensities and calculated from the model, respectively.
As in previously described S. enterica DHQD structures, the crystallographic asymmetric units of the shikimate and quinate complexes each contain the physiological homodimer (Figure 2a). Interestingly, in both the quinate and shikimate complexes, the two protomers in the asymmetric unit, here termed protomer A and protomer B, exhibit disparate conformational behaviors.
Figure 2.
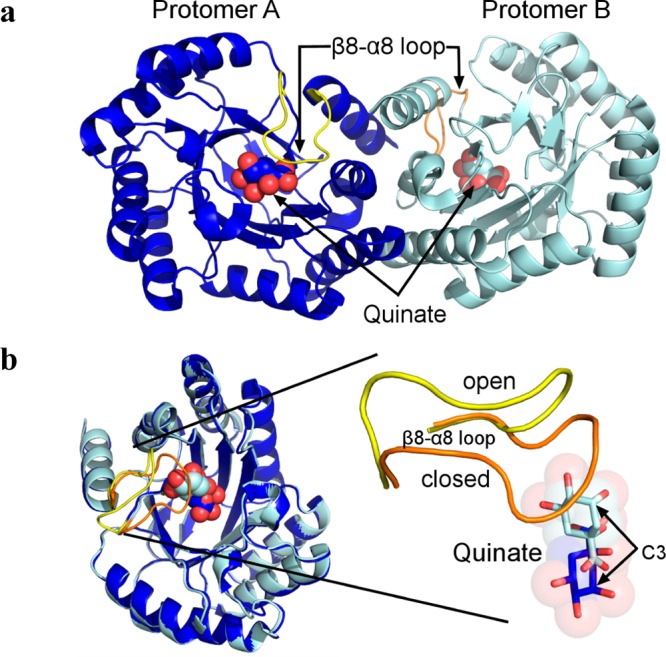
Distinct conformational behavior within the physiological DHQD homodimer present in the crystallographic asymmetric unit. (a) The DHQD homodimer present in the quinate asymmetric unit is depicted in cartoon representation. Quinate molecules are shown as spheres, and the β8−α8 loops are colored yellow and orange. (b) Superposition of the two protomers highlights the high degree of overall structural similarity but pronounced differences in the β8−α8 loop and quinate conformations.
The first difference between protomers A and B concerns the conformation adopted by the Val228-to-Gln236 loop that connects β-strand 8 to α-helix 8 (β8−α8 loop). The β8−α8 loop has previously been observed open and partially disordered in the unliganded state but closed over the active site and hydrogen bonding with DHQ in the K170M mutant and Schiff base reaction intermediate structures in the wild-type enzyme.10,11 In protomer A of both the quinate and shikimate complex structures, the β8−α8 loop adopts the closed conformational state (colored yellow in Figure 2b). By contrast, in protomer B of these complexes the loop adopts the open conformational state (colored orange in Figure 2b).
The second difference between protomers A and B concerns the mode of ligand binding. Correlating with the difference in β8−α8 loop conformation, quinate and shikimate exhibit distinct binding modes within the two protomers (Figure 2b).
Binding of Quinate and Shikimate to Protomer A of Wild-Type DHQD
Quinate (Figure 3a) and shikimate (Figure 3b) assume similar binding modes within protomer A of their respective complexes. With the closed β8−α8 loop, shikimate and quinate are positioned within the enclosed active site like the covalent reaction intermediate observed in a previous wild-type complex with DHQ and noncovalently bound DHQ in the K170M–DHQ complex (Figure 3c).10
Figure 3.

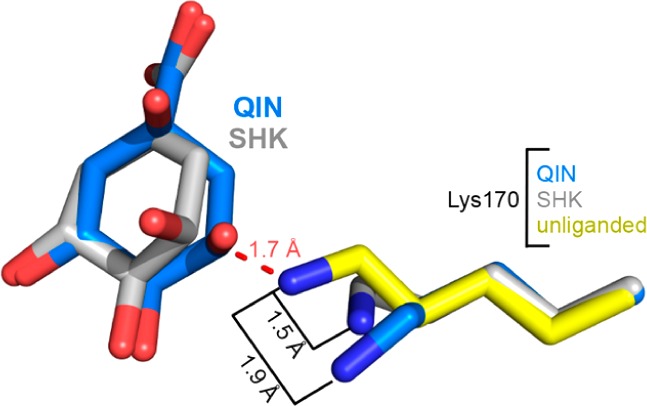
Forms of protomer A of the quinate and shikimate complexes. (a) Stick model of the quinate complex active site (protomer A). (b) Stick model of the shikimate complex active site (protomer A). The Fo – Fc map was calculated with ligand omitted and is contoured at 3σ. (c) Superposition of forms of protomer A of quinate (blue) and shikimate (gray) to the K170M–DHQ complex (pink, PDB entry 3NNT) illustrates the similar mode of ligand binding and closed loop conformational states. (d) Superposition of forms of protomer A of quinate (rmsd = 0.31 Å over 196 Cα atoms) and shikimate (rmsd = 0.20 Å over 196 Cα atoms) to the unliganded structure (yellow, PDB entry 3L2I). The retraction of Lys170 from its unliganded state conformation prevents a clash (dashed red line) with quinate or shikimate.
Establishing essentially the same interactions that have been observed for DHQ in the K170M–DHQ complex, the presence of Lys170 represents the principal feature differentiating the wild-type quinate and shikimate complex structures from the mutant complex. Within both quinate and shikimate structures, the Lys170 side chain has retracted, with its Nε atom displaced 1.5–1.9 Å from its extended unliganded state position (Figure 3d). This retraction of Lys170 is rationalized by a superposition with the unliganded structure, which reveals that the unliganded state Lys170 conformation would otherwise sterically clash with the quinate or shikimate 3-hydroxyl (dashed red line in Figure 3d). This indicates that the change in the Lys170 conformation, from fully extended to retracted, is necessitated by the proximity of the bound ligand.
Binding of Quinate and Shikimate to Protomer B of Wild-Type DHQD
The ligand electron density is not as well-resolved within the respective forms of protomer B but is nonetheless of sufficient quality to allow quinate (not shown) and shikimate (Figure 4a) to be confidently modeled with partial occupancy. Unlike the similar mode of quinate and shikimate binding exhibited in protomer A, dissimilar binding modes are exhibited between protomer B of the quinate and shikimate complexes (not shown). This distinction persists despite nearly identical open β8−α8 loop conformational states in the two protomers.
Figure 4.

Protomer B of the shikimate complex. (a) Stick model of the protomer B active site of the quinate complex. The Fo – Fc electron density map was calculated and contoured as described in the legend of Figure 3. (b) Superposition of protomers A (light gray) and B (dark gray) of the shikimate complex. The difference in shikimate position (arrow 1) tracks with the conformational change of Arg213 (arrow 2).
In protomer B of the quinate complex, the carbocyclic quinate ring is flipped relative to its protomer A conformation (Figure 2b). As a result of this flipped orientation, the quinate carboxylic moiety interacts with Lys170 and the C3 atom that corresponds to the reactive atom in DHQ is located ∼7.5 Å from the Lys170 Nε atom (not shown). This mode of binding is similar to the mode of DHS binding observed in the previously described E86A mutant DHS complex.12 As the functional implications of this presumably nonproductive binding mode have been thoroughly discussed in a recent publication12 and do not pertain to the mechanism of Schiff base formation, we will leave discussion of quinate complex protomer B here.
In contrast to the quinate protomer B, shikimate in protomer B (light gray in Figure 4b) retains its protomer A orientation (dark gray in Figure 4b). However, the carbocyclic shikimate ring has shifted from its protomer A conformation (arrow 1 in Figure 4b). Like in protomer A, the shikimate 1-carboxyl group forms a bidentate salt bridge with Arg213. The difference in the protomer B shikimate position is related to the distinct conformation of Arg213 (arrow 2 in Figure 4b). Arg213 has previously been observed to undergo a coordinated conformational change upon β8−α8 loop closure.11 The Arg213 guanidinium group points away from Lys170 in its open loop conformation but swivels to point toward Lys170 following loop closure (Figure 4b).11 Tracking with the open loop conformation of Arg213, shikimate is shifted away from Lys170 and its protomer A position in protomer B (Figure 4b).
Binding of Quinate to the K170M Variant
To obtain the wild-type quinate and shikimate complex structures, it was necessary to soak crystals in molar concentrations of these compounds. By contrast, 2 mM substrate DHQ was sufficient to obtain the previously described K170M–DHQ complex.10 To determine if this difference in required ligand concentration resulted from the K170M mutation, diffraction data were collected from a preformed unliganded K170M crystal soaked in 5 mM quinate.
Indeed, a 1.80 Å crystal structure (Table 1) revealed that, in contrast to wild-type DHQD, a low millimolar quinate concentration was sufficient to generate unambiguous ligand electron density at the K170M mutant active site (Figure 5a). Within both of the protomers in the asymmetric unit, quinate adopts the closed loop binding mode observed in wild-type protomer A (Figure 5b). We could also discern quinate at the K170M active site following a 0.5 mM soak, albeit with partial occupancy (not shown). Thus, despite representing a 1000-fold reduction from the concentration that failed to generate a wild-type complex, 0.5 mM quinate proved to be sufficient to produce unambiguous quinate electron density in the K170M variant.
Figure 5.
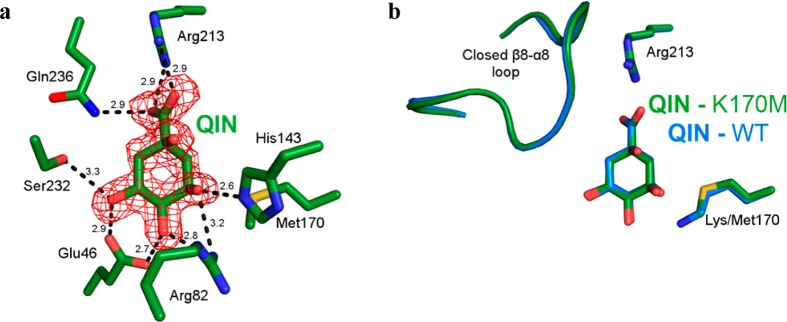
K170M–quinate complex. (a) Stick model of the K170M mutant–quinate complex active site (protomer A). Density map calculated and contoured as in Figure 2. (b) Overlay of the K170M mutant–quinate (green) and wild-type–quinate (blue) complexes.
Next, we sought to confirm the different ligand concentrations required for successful crystal soaks were indicative of differing binding affinities to the two DHQD variants. For the K170M variant, Kd values of 224 μM for quinate and 862 μM for shikimate were measured by ITC (Figure 6a,b). By contrast, titrations of quinate and shikimate into the wild-type enzyme provided no indication of binding (Figure 6a,b). We further probed quinate and shikimate binding using WaterLOGSY NMR, a technique previously shown to be useful for detecting binding of small molecules to large protein complexes.17,19 In this experiment, a positive sign of the quinate peaks indicates binding to K170M DHQD (bottom spectrum in Figure 6c,d). On the other hand, a negative sign of the quinate and shikimate peaks indicates little or no binding to wild-type DHQD (top spectrum in Figure 6c,d).
Figure 6.

Binding of quinate and shikimate to wild-type and K170M DHQD. (a) Data from ITC experiments in which 10 mM quinate was injected into 200 μM wild-type (red) or K170M (black) DHQD are shown. The top panel shows the direct heat generated per injection, while the bottom panel shows the integrated and normalized heat data with a curve fit to the K170M DHQD data. (b) Data from ITC experiments in which 10 mM shikimate was injected into 200 μM wild-type (red) or K170M (black) DHQD. The experimental setup and presentation are the same as in panel a. (c) WaterLOGSY NMR spectrum of quinate in the presence of wild-type DHQD (top) or K170M DHQD (bottom). Red arrows denote the resonances corresponding to those of quinate. The negative sign of the quinate peaks in the wild-type spectrum indicates little or no binding. The positive sign of the quinate peaks in the K170M spectrum indicates binding. (d) WaterLOGSY NMR spectrum of shikimate in the presence of wild-type DHQD (top) or K170M DHQD (bottom). Red arrows denote the resonances corresponding to those of shikimate. The negative sign of the shikimate peaks in the wild-type spectrum indicates little or no binding. The positive sign of the shikimate peaks in the K170M spectrum indicates binding.
Discussion
Significance of Quinate and Shikimate Binding Modes Observed in Protomer A
While quinate and shikimate complexes were pursued with the objective of clarifying the noncovalent basis of substrate and product binding, it may be more instructive to view protomer A binding (Figure 3) in the context of analyzing the carbinolamine intermediates. In the forward direction of the reaction, the carbinolamine #1 intermediate precedes Schiff base formation, whereas the carbinolamine #2 intermediate follows Schiff base hydrolysis (Figure 1). The quinate and shikimate 3-hydroxyl contains the sp3 geometry and stereochemistry that defines these intermediate states (Figure 1). Quinate, in particular, is positioned very much like the Schiff base-bound dehydroquinate, with C3 of quinate shifted 0.3 Å from C3 of the Lys170-bound dehydroquinate. It thus stands to reason that quinate should form interactions characteristic of the carbinolamine intermediates.
Interestingly, the quinate 3-hydroxyl interacts with His143 and Arg82 (Figure 3a). Recent computational studies of Schiff base formation and hydrolysis predicted such an interaction, as well as a key role for these residues in coordinating the attacking water in Schiff base hydrolysis.20 Thus, this experimentally observed interaction combined with recent computational predictions makes an emerging case for an important role for Arg82 in DHQD catalysis.
A Proposal for the Mechanism of Schiff Base Formation in Type I DHQDs
The first insight into Schiff base formation in type I DHQDs came from the structure of the K170M mutant in complex with DHQ.10 In that structure, DHQ assumes an orientation similar to that of the Schiff base-bound intermediate states within the wild-type enzyme. Notably, such a DHQ orientation vis-à-vis Lys170 is inconsistent with the required nucleophilic approach trajectory for Schiff base formation. This observation combined with the enhanced affinity of the K170M variant for substrate and product reaction analogues makes a compelling case that the K170M–DHQ complex does not accurately represent the Michaelis state.
We are aware that the high ligand concentration necessary to obtain the shikimate and quinate complexes raises questions about the functional relevance of the binding modes observed in these structures. Nevertheless, the binding mode observed in protomer B of the shikimate structure is intriguing. Previous structural and kinetic data make it clear that the β8−α8 loop closure and the accompanying Arg213 conformational change fundamentally relate to substrate binding.11 In protomer A of the wild-type DHQD complex with shikimate, the β8−α8 loop is open and Arg213 adopts its unliganded conformation (Figure 7a,b). Because Arg213 engages the shikimate carboxyl group in a fashion similar to that in closed β8−α8 loop complexes, it is easy to envision how loop closure and the accompanying Arg213 conformational change would cause the substrate to shift toward Lys170 (Figure 4b). Importantly, as these conformational changes (of the β8−α8 loop and Arg213) push the substrate deeper into the active site, some rearrangement must be necessary to avoid a steric clash between Lys170 and substrate carbonyl (Figure 7c,d) and differentiate this state from the one observed in the K170M–DHQ complex structure (Figure 7, box).
Figure 7.
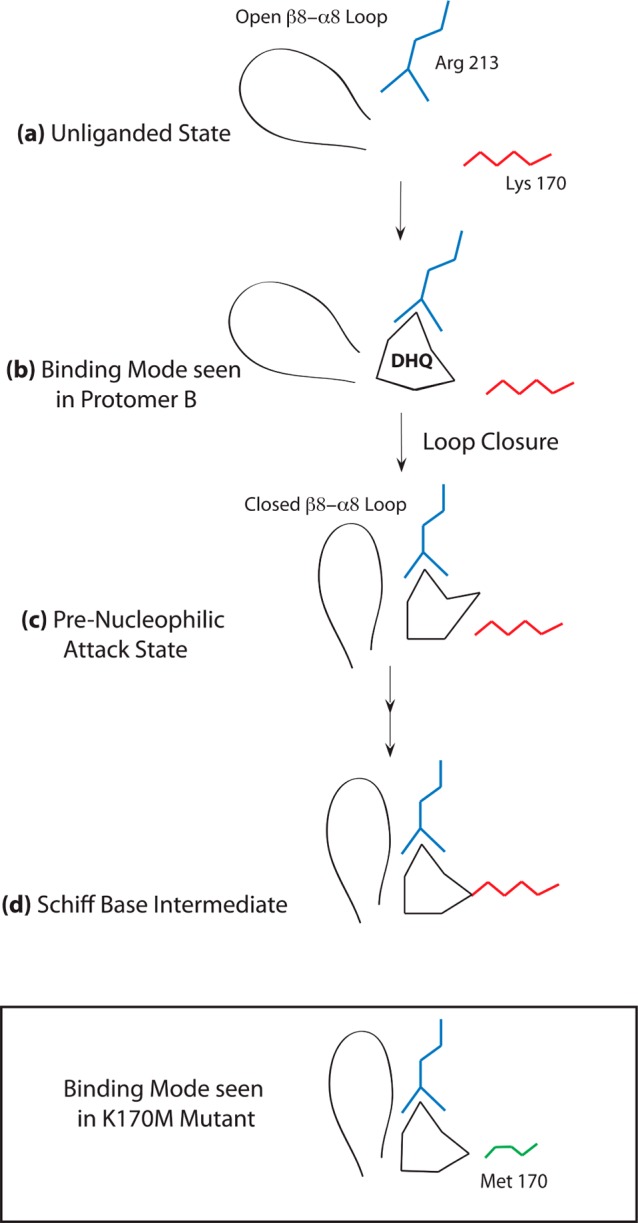
Proposed mechanism of Schiff base formation. (a) DHQD adopts an open loop conformation in the absence of ligand. (b) DHQ binds to the open loop conformational state, as observed in protomer B of the shikimate complex. (c) Closure of the loop requires Arg213 and DHQ to shift deeper into the active site. The presence of Lys170 in the wild-type enzyme sterically disallows the K170M binding mode (black box), forcing the DHQ 3-carbonyl to pivot away from Lys170 and generating a prenucleophilic attack state like that described by Pan et al.21 (d) The reactive carbonyl is positioned at a stereoelectronically reasonable nucleophilic approach angle in the prenucleophilic attack state that allows reaction with Lys170 to generate the Schiff base intermediate.
In protomer A of the quinate and shikimate complexes, such a steric clash is avoided by the retraction of the Lys170 side chain (Figure 3d). However, the high concentration of ligand necessary to generate these structures as compared to the lower concentration required for the K170M mutant suggests that this retraction is energetically unfavorable. An alternative mechanism by which the steric clash could be resolved would involve the substrate, or a portion of its carbocyclic ring, pivoting away from Lys170. In addition to preventing a clash, such a rearrangement has the potential advantage of exposing the carbonyl to an energetically favorable nucleophilic approach angle and thus promoting Schiff base formation.
The notion that the reactive portion of the DHQ ring might pivot away from Lys170 falls in line with recent docking/molecular dynamic experiments, which, performed using the closed β8−α8 loop conformational state as a starting model, identified a stable binding mode for DHQ, similar to its position in the K170M structure but differing in that the 3-carbonyl has rotated away from Lys170.21 When this model was subjected to quantum chemical calculations, small movements of the substrate and/or Lys170 side chain positioned the reactive components at a reasonable nucleophilic approach angle.21 Taken together with the computational findings, the new structural data suggest that, following the initial binding event, β8−α8 loop closure helps guide the substrate into the active site. Here, the DHQ 3-carbonyl pivots away from the K170M binding mode to prevent a steric clash and to allow nucleophilic approach to follow a favorable trajectory (Figure 7).
In summary, the most striking feature of this model of Schiff base formation concerns the interplay of residues on either side of the substrate in promoting Schiff base formation. On the external side of the active site cavity, the β8−α8 loop critically interacts with the substrate, effectively pushing it into sufficient proximity of Lys170 for covalent bond formation to occur. The direct contribution of the β8−α8 loop to the catalytic rate is confirmed by studies of loop mutant Q236A, which reveal a 30-fold decrease in kcat.11 On the other side of the substrate, as evidenced by structural revelations demonstrating perturbed substrate binding to the K170M mutant, Lys170 sterically orients the carbonyl, presumably positioning it at a favorable nucleophilic approach angle. In this way, the β8−α8 loop and Lys170 act in concert, pressing the substrate from opposing sides, drawing it in and positioning it for nucleophilic attack. Importantly, Lys170 causes DHQ to rotate away from the Schiff base-forming residue, exposing it to a favorable nucleophilic approach by the lysine amino group, a reaction that results in Schiff base formation. These results reveal the various substrate binding modes that must occur during the catalytic cycle, and it is the mimicking of these binding modes that can be used by inhibitors to prevent the dehydration reaction.
Glossary
Abbreviations
- DHQ
dehydroquinate
- DHQD
dehydroquinate dehydratase
- DHS
dehydroshikimate
- ITC
isothermal titration calorimetry
- NMR
nuclear magnetic resonance.
The authors declare no competing financial interest.
The Center for Structural Genomics of Infectious Diseases has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contracts HHSN272200700058C and HHSN272201200026C (to W.F.A.). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). This work used resources at the Northwestern University Keck Biophysics Facility, supported by Grant NCI-CCSG-P30-CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center.
Funding Statement
National Institutes of Health, United States
References
- Bentley R. (1990) The shikimate pathway: A metabolic tree with many branches. Crit. Rev. Biochem. Mol. Biol. 25, 307–384. [DOI] [PubMed] [Google Scholar]
- Kleanthous C.; Deka R.; Davis K.; Kelly S. M.; Cooper A.; Harding S. E.; Price N. C.; Hawkins A. R.; Coggins J. R. (1992) A comparison of the enzymological and biophysical properties of two distinct classes of dehydroquinase enzymes. Biochem. J. 282(Part 3), 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J. R.; Alworth W. L.; Nugent M. J. (1974) Mechanism of Dehydroquinase Catalyzed Dehydration. 1. Formation of a Schiff-Base Intermediate. J. Am. Chem. Soc. 96, 1617–1618. [Google Scholar]
- Gourley D. G.; Shrive A. K.; Polikarpov I.; Krell T.; Coggins J. R.; Hawkins A. R.; Isaacs N. W.; Sawyer L. (1999) The two types of 3-dehydroquinase have distinct structures but catalyze the same overall reaction. Nat. Struct. Biol. 6, 521–525. [DOI] [PubMed] [Google Scholar]
- Hanson K. R.; Rose I. A. (1963) Absolute Stereochemical Course of Citric Acid Biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 50, 981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. M.; Gonzalez-Bello C.; Kleanthous C.; Hawkins A. R.; Coggins J. R.; Abell C. (1996) Evidence from kinetic isotope studies for an enolate intermediate in the mechanism of type II dehydroquinases. Biochem. J. 319(2), 333–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker E. J.; Bello C. G.; Coggins J. R.; Hawkins A. R.; Abell C. (2000) Mechanistic studies on type I and type II dehydroquinase with (6R)- and (6S)-6-fluoro-3-dehydroquinic acids. Bioorg. Med. Chem. Lett. 10, 231–234. [DOI] [PubMed] [Google Scholar]
- Choi K. H.; Lai V.; Foster C. E.; Morris A. J.; Tolan D. R.; Allen K. N. (2006) New superfamily members identified for Schiff-base enzymes based on verification of catalytically essential residues. Biochemistry 45, 8546–8555. [DOI] [PubMed] [Google Scholar]
- Light S. H.; Minasov G.; Duban M.-E.; Anderson W. F. (2014) Adherence to Bürgi-Dunitz stereochemical principles requires significant structural rearrangements in Schiff base formation: Insights from transaldolase complexes. Acta Crystallogr. D D70, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light S. H.; Minasov G.; Shuvalova L.; Duban M. E.; Caffrey M.; Anderson W. F.; Lavie A. (2011) Insights into the mechanism of type I dehydroquinate dehydratases from structures of reaction intermediates. J. Biol. Chem. 286, 3531–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light S. H.; Minasov G.; Shuvalova L.; Peterson S. N.; Caffrey M.; Anderson W. F.; Lavie A. (2011) A conserved surface loop in type I dehydroquinate dehydratases positions an active site arginine and functions in substrate binding. Biochemistry 50, 2357–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light S. H.; Anderson W. F.; Lavie A. (2013) Reassessing the type I dehydroquinate dehydratase catalytic triad: Kinetic and structural studies of Glu86 mutants. Protein Sci. 22, 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276(Part A), 307–326. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Storoni L. C.; Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D61, 458–464. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Lebedev A.; Wilson K. S.; Dodson E. J. (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D55, 247–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Antanasijevic A.; Cheng H.; Wardrop D. J.; Rong L.; Caffrey M. (2013) Inhibition of influenza h7 hemagglutinin-mediated entry. PLoS One 8, e76363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A Multidimensional Spectral Processing System Based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Fogliatto G.; Stewart A.; Veronesi M.; Stockman B. (2001) WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 21, 349–359. [DOI] [PubMed] [Google Scholar]
- Yao Y.; Li Z. S. (2012) New insights into the mechanism of the Schiff base hydrolysis catalyzed by type I dehydroquinate dehydratase from S. enterica: A theoretical study. Org. Biomol. Chem. 10, 7037–7044. [DOI] [PubMed] [Google Scholar]
- Pan Q.; Yao Y.; Li Z. S. (2012) New insights into the mechanism of the Schiff base formation catalyzed by type I dehydroquinate dehydratase from S. enterica. Theor. Chem. Acc. 131. [DOI] [PubMed] [Google Scholar]