

Abstract
Pancreatic cancer has a dismal 5 year survival rate of 5.5% that has not been improved over the past 25 years despite an enormous amount of effort. Thus, there is an urgent need to identify truly novel yet druggable protein targets for drug discovery. The human protein DnaJ homologue subfamily A member 1 (DNAJA1) was previously shown to be downregulated 5-fold in pancreatic cancer cells and has been targeted as a biomarker for pancreatic cancer, but little is known about the specific biological function for DNAJA1 or the other members of the DnaJ family encoded in the human genome. Our results suggest the overexpression of DNAJA1 suppresses the stress response capabilities of the oncogenic transcription factor, c-Jun, and results in the diminution of cell survival. DNAJA1 likely activates a DnaK protein by forming a complex that suppresses the JNK pathway, the hyperphosphorylation of c-Jun, and the anti-apoptosis state found in pancreatic cancer cells. A high-quality nuclear magnetic resonance solution structure of the J-domain of DNAJA1 combined with a bioinformatics analysis and a ligand affinity screen identifies a potential DnaK binding site, which is also predicted to overlap with an inhibitory binding site, suggesting DNAJA1 activity is highly regulated.
Despite the decline of cancer-related mortality in the past decade, effective approaches to early diagnosis and treatment of pancreatic cancer remain elusive. Although it accounts for only 3% (43000 new cases every year) of all cancers, pancreatic cancer is the fourth leading cause of cancer death in the United States (37000 deaths annually) and has the highest mortality rate of any cancer.1,2 Those with an operable early stage of the disease have a 5 year survival rate of ∼20%.2,3 Unfortunately, 80% of all pancreatic cancer diagnoses indicate an advanced stage of the disease that is beyond the point of surgery.1,3,4 Inoperable forms of pancreatic cancer have a 5 year survival rate of only 3%. The difficulty in detecting or diagnosing pancreatic cancer has several causes: the early stages of pancreatic cancer do not typically exhibit symptoms; the symptoms that do occur are often similar to those of other illnesses; and the location of the pancreas behind other organs can hinder detection.1
Most patients with advanced pancreatic cancer are treated with chemotherapy based on gemcitabine, which is a cytotoxic nucleoside drug that primarily inhibits DNA synthesis.5 However, this treatment is only mildly effective for patients with an advanced stage of pancreatic cancer and provides an only 5.91 month increase in the median survival rate.6 Also, gemcitabine-resistant forms of pancreatic cancer and acquired resistance during treatment are common problems.7 Correspondingly, there have been numerous attempts to combine gemcitabine with other cytotoxic agents, such as 5-fluorouracil or capecitabine. However, these approaches have been mostly unsuccessful.8 It is apparent that a cytotoxic approach to treating pancreatic cancer is not an effective therapy. Therefore, identifying novel, but druggable, protein targets for the treatment of pancreatic cancer and improving the quality of life for patients are essential needs.
The DnaJ proteins, also known as heat shock protein 40 (Hsp40 or Hsc40), are proteins originally identified in Escherichia coli that act as cochaperones to the molecular chaperone DnaK (Hsp70), which is responsible for several cellular processes such as rescuing misfolded proteins, folding polypeptide chains, transport of polypeptides through membranes, assembly and disassembly of protein complexes, and control of regulatory proteins.9−11 DnaJ primarily facilitates the hydrolysis of ATP from DnaK that is necessary for the chaperone activity of DnaK.11−13 In general, J-domain proteins modulate protein assembly, disassembly, and translocation.14
Human protein DnaJ subfamily A member 1 (DNAJA1) has been shown to associate on its own with unfolded polypeptide chains and prevent their aggregation,15 to regulate androgen receptor signaling and spermatogenosis in mice,16 and to contribute to the resistance of glioblastomas to radiotherapy.17 DNAJA1 has also been targeted as a biomarker for pancreatic cancer to evaluate the effects of farnesyl protein transferase inhibitors18,19 and has been shown to be downregulated 5-fold in a genomics analysis of pancreatic cancer cells relative to normal healthy cells and cells undergoing pancreatitis.20 Additionally, DNAJA1 appears to be involved in importing proteins into the mitochondria.21,22 Of note, the mitochondrial pathway to apoptosis protects against cancer and requires importing apoptotic factors into the mitochondrial membrane.23−26 Additionally, DNAJA1 is an interesting target based on its association with DnaK, which is expressed abundantly in various tumors and can potentially promote tumorigenesis by inhibiting cell death.27−32 However, there have been no studies of whether the conserved J-domain of DnaJ alone has any role in cancer biology independent of DnaK.10
Thus, DNAJA1 was selected for further analysis as a potentially interesting therapeutic target for the treatment of pancreatic cancer. The potential importance of DNAJA1 to pancreatic cancer was demonstrated by stress response cell-based assays using cell lines overexpressing DNAJA1. DNAJA1 was shown to regulate the hyperphosphorylation of c-Jun and cell survivability under stress. The resulting nuclear magnetic resonance (NMR) structure of the J-domain of DNAJA1 (DNAJA1-JD) combined with a detailed bioinformatics analysis predicted the location of overlapping activating and inhibitory protein binding sites. A subsequent NMR–ligand affinity screen provided further experimental support for the existence of the predicted inhibitory protein binding sites and also demonstrated that the J-domain of DNAJA1 is a potential druggable target. In total, our results suggest DNAJA1 activity is highly regulated and that DNAJA1 is involved in a stress response signaling pathway that suppresses the anti-apoptosis state found in pancreatic cancer cells.
Experimental Procedures
Effect of DNAJA1 Overexpression on Cellular Stress Modulation in Pancreatic Cancer Cells
MiaPaCa2 cells were obtained from the American Type Culture Collection (Rockville, MD). Cells were cultured as previously described.33 Briefly, MiaPaCa2 cells were maintained in Dulbecco’s modified Eagle’s medium (Life Technologies, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS), nonessential amino acids, sodium pyruvate, and penicillin/streptomycin in a 37 °C incubator with 5% CO2. To stably express the full-length His-tagged DNAJA1 construct, retroviral transductions were conducted essentially as described previously.34 The DNAJA1 construct was purchased from the Arizona State University Biodesign Institute (http://dnasu.org/DNASU/Home.jsp).
Cell lysates were prepared by scraping cells (80–90% confluent) into lysis buffer [50 mM Tris-HCl (pH 7.5), 0.15 M NaCl, 1% (v/v) Triton X-100, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 5 mM EDTA, and 1 mM phenylmethanesulfonyl fluoride]. Lysates were incubated, on ice, for 30 min and centrifuged at 4 °C for 15 min at 13000 rpm to remove cell debris. Supernatants were transferred to fresh tubes, and the protein content was determined using the Bradford protein assay reagent (Bio-Rad, Hercules, CA) with various concentrations of bovine serum albumin as standards. Cell lysates were stored at −20 or −80 °C. Cell lysate proteins were resolved on 10 or 12% Tris-glycine denaturing polyacrylamide gels in 1× sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) buffer (1 g/L SDS, 3 g/L Tris base, and 14.4 g/L glycine). Western blotting was performed as previously described.35,36 The membranes were probed with primary antibodies against phosphoS63-c-Jun [2361 (Cell Signaling Technology, Danvers, MA)], total c-Jun [9165 (Cell Signaling Technology)], the His tag [2365 (Cell Signaling Technology)], and tubulin [E7 (Developmental Studies Hybridoma Bank, Iowa City, IA)]. The primary antibody treatments were performed overnight at 4 °C at 1:1000 dilutions.
Cell survival was evaluated by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay as described elsewhere.37 Cells were plated in triplicate in 96-well plates at a density of 5000 cells/well and incubated at 37 °C. Twelve hours later, cells were treated with anisomysin (10 μg/mL) or solvent control [dimethyl sulfoxide (DMSO)] and incubated for 24 h. At the end point, the culture medium was removed and 20 μL of the MTT solution [5 mg/mL (Sigma-Aldrich, St. Louis, MO)] was added per well, followed by a 2 h incubation. MTT was removed, and 200 μL of DMSO was added to each well to dissolve formazan. The formazan optical density was determined by utilizing a microplate reader (FLUOstar Omega, BMG LABTECH, Cary, NC) at a wavelength of 540 nm.
Solution Structure of the DNAJA1 J-Domain
The full-length human DNAJA1 protein (397 amino acids) has been targeted by the Northeast Structural Genomics Consortium [NESG (http://www.nesg.org)] for structural elucidation as target HR3099 (UniProt entry P31689) (Figure 1).
Figure 1.

Protein sequence of DNAJA1 (UniProt entry P31689). The red residues are the 67 amino acids of the J-domain (DNAJA1-JD).
Samples of approximately 100% 15N-enriched, 5% 13C-enriched, and uniformly 13C- and 15N-enriched DNAJA1-JD [77 amino acids with 10 non-native residues, MGHHHHHHSH, at the N-terminus for purification (NESG entry HR3099K)] were produced using the standard protocols of the NESG.38,39 The protein construct containing the sequence for DNAJA1-JD was transformed into BL21(DE3)+Magic cells. The soluble fraction of the lysed cells was collected and purified with a Ni+ affinity column (HisTrap HP 5 mL column, GE Healthcare, Pittsburgh, PA) and a gel filtration column (HiLoad 26/60 Superdex 75 prep grade, GE Healthcare). The sample identity and isotopic enrichment were validated by matrix-assisted laser desorption ionization time-of-flight mass spectrometry, and the homogeneity (>97%) was assessed by SDS–PAGE. The NMR protein sample was stored in a sealed Shigemi tube with 20 mM 2-(4-morpholino)ethanesulfonic acid [MES (Sigma-Aldrich)] buffer [pH 6.5 (uncorrected)] with 10% D2O (Sigma-Aldrich), 0.02% NaN3 (Sigma-Aldrich), 10 mM dithiothreitol [DTT (Sigma-Aldrich)], 5 mM CaCl2 (Sigma-Aldrich), 100 mM NaCl (Sigma-Aldrich), and 50 μM 4,4-dimethyl-4-silapentane-1-sulfonic acid [DSS (Sigma-Aldrich)].
NMR experiments used for the protein backbone and side-chain assignments of DNAJA1-JD were collected at 298 K on a 600 MHz Bruker Avance spectrometer equipped with a 5 mm TXI probe. The backbone and side-chain assignments were completed using the standard triple-resonance approach40−42 of the following NMR experiments: two-dimensional (2D) 1H–15N HSQC, 2D 1H–13C HSQC, and three-dimensional (3D) HNCO; HN(CA)CO; HNCA; HN(CO)CA; CBCA(CO)NH; CBCANH; HNHA; HBHA(CO)NH; CC(CO)NH; HCC(CO)NH; HCCH-COSY; and HCCH-TOCSY. 15N-edited NOESY-HSQC and 13C-edited NOESY-HSQC experiments (mixing times of 120 ms) were conducted on a 500 MHz Bruker Avance DRX spectrometer equipped with a triple-resonance, Z-axis gradient cryoprobe to identify nuclear Overhauser effects (NOEs). Amide hydrogen exchange rates were also evaluated on the 500 MHz Bruker Avance DRX spectrometer using the (CLEANEX-PM)-FHSQC experiment.
The NMR experimental data were processed using NMRPipe43 and evaluated in CCPNMR Analysis (http://www.ccpn.ac.uk).44 An initial model of DNAJA1-JD was generated from the protein backbone resonances using CS-ROSETTA on the WeNMR GRID-enabled web portal (http://www.enmr.eu/webportal).45−47
The initial model of DNAJA1-JD was refined with XPLOR-NIH version 2.3148 using the following experimental restraints: 1070 NOE distance restraints, 50 H-bond distance and angle restraints, 38 3JNHα coupling constant restraints, 127 13Cα/13Cβ chemical shift restraints, and 116 dihedral angle restraints predicted from TALOS+.49 All peptide bonds were constrained to be planar and trans. A total of 400 structures were calculated, with the 20 lowest-energy structures being subjected to explicit water refinement based on the RECOORD protocols.50 An average DNAJA1-JD structure was calculated on the basis of the average atom coordinates of the 20 water-refined structures and subsequently minimized using the same explicit water refinement described above.
The resulting structures were evaluated using the RPF webserver (http://nmr.cabm.rutgers.edu/rpf/)51 and PSVS software suite (http://psvs-1_5-dev.nesg.org/),52 which includes Verify3D,53 ProsaII,54 PROCHECK,55 and Molprobity.56 The three-dimensional structures of the proteins are represented here using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (http://www.cgl.ucsf.edu/chimera/).57 ClustalW58 was used (with default settings) to align the sequences of DNAJA1-JD with the J-domains of four homologous proteins: E. coli DnaJ J-domain [Protein Data Bank (PDB) entry 1XBL],59Homo sapiens Hsp40 (HDJ-1) J-domain (PDB entry 1HDJ),60H. sapiens HSJ1a (PDB entry 2LGW),61 and H. sapiens DnaJ subfamily C member 12 (PDB entry 2CTQ). Electrostatic surface potentials of the protein were calculated using Delphi.62 The identification of evolutionarily conserved residues using both sequence and structure was performed using the ConSurf server with default settings.63
Identification of a Ligand Binding Site on the DNAJA1 J-Domain
To experimentally determine a potential functional epitope or functionally relevant ligand binding site, we used a high-throughput NMR ligand affinity screen using a function-based compound library.64,65 The one-dimensional (1D) 1H line broadening ligand-based screen and 2D 1H–15N HSQC protein-based screen follow the same procedure outlined elsewhere66,67 using 10 and 30 μM DNAJA1-JD (5% 13C-labeled and 100% 15N-labeled), respectively. The concentrations for the compounds were 100 μM in the 1D 1H line broadening screen and 400 μM in the 2D 1H–15N HSQC screen.
The NMR spectra for the ligand affinity screens were recorded on a 500 MHz Bruker Avance DRX spectrometer equipped with a triple-resonance, Z-axis gradient cryoprobe with a Bruker autotune and match (ATM) and BACS-120 sample changer. All 1D 1H NMR spectra were processed with the ACD/NMR Processor (ACD/Laboratories), and 2D 1H–15N HSQC spectra were processed with NMRPipe43 and visualized in CCPNMR Analysis.44
Chemical shift perturbations (CSPs) between the free and ligand-bound 2D 1H–15N HSQC spectra of DNAJA1-JD were used to define the consensus binding site. The CSPs were calculated using a common weighting approach68 based on changes in 15N and 1H chemical shifts that occur upon ligand binding. AutoDock version 4.2.369−71 with the AutoDockTools version 1.5.471,72 graphical interface was used to calculate 120 protein–ligand costructures, which were filtered using AutoDockFilter73 to identify the costructures that best agree with the experimental CSPs.
Results and Discussion
Effect of DNAJA1 Overexpression on Cellular Stress Modulation in Pancreatic Cancer Cells
DNAJA1 is related to the cellular response to stress, a common event in the tumor cells due to fluctuations in the nutrient and oxygen levels in the tumor microenvironment or due to therapy, and can govern the response of the tumor to chemo- and radiotherapies. Hence, we performed cell-based functional assays of c-Jun phosphorylation to identify DNAJA1-mediated activation of the stress response pathway in cancer cells. c-Jun is a member of the JNK signaling pathway, and its transcriptional activity and expression are primarily regulated by the phosphorylation of two serines, Ser63 and Ser73.74−77 c-Jun regulates a range of cellular processes, including apoptosis, tumorigenesis, and cell proliferation, which includes protecting cells from stress-induced cell death.75,77 This makes c-Jun both a positive and negative regulator of cell death. However, in several cancers, c-Jun has been shown to inhibit apoptosis, leading to the uncontrolled growth typical of cancer.78−80
Human MiaPaCa2 cells stably expressing full-length DNAJA1 or the vector control were subjected to a 1 h incubation with anisomycin (370 nM), a protein synthesis inhibitor, or UV treatment at 20 or 50 J/m2 doses to mimic stress. Activation of the stress-induced JNK pathway was measured by evaluating the downstream phosphorylation of c-Jun 20 min after treatment. The results indicate that the overexpression of DNAJA1 (i.e., in cell line MiaPaCa2-DNAJA1) diminishes the level of anisomycin and UV-induced c-Jun phosphorylation at residue Ser63 (Figure 2A). Please note the doubling of the c-Jun bands in Figure 2A is a result of different phosphorylation states of c-Jun and, correspondingly, different gel migration rates. Cell survival was also evaluated by performing MTT assays 24 h after treatment with anisomycin. The results indicate that DNAJA1 expression decreased the level of cell survival under conditions of anisomycin treatment (Figure 2B).
Figure 2.

(A) Expression of full-length DNAJA1 (UniProt entry P31689) in MiaPaCa2 pancreatic cancer cells suppresses the activation of c-Jun in response to anisomycin (370 nM) or UV treatment (20 or 50 J/m2 doses). Twenty minutes after being treated, cells were subjected to lysis, and the levels of phospho-c-Jun (S63) and total c-Jun were evaluated by Western blotting. Expression levels of exogenously expressed His-tagged DNAJA1 were evaluated by immunoblotting with the anti-His antibody, while immunoblotting with the anti-tubulin antibody was performed as a loading control. (B) Expression of full-length DNAJA1 in MiaPaCa2 pancreatic cancer cells decreases the level of survival in response to anisomycin treatment-induced stress. The level of cell survival was measured by the MTT assay 24 h after treatment (**p < 0.01; ***p < 0.001).
Expression of the DNAJA1 protein in MiaPaCa2 pancreatic cancer cell lines results in the cells being more susceptible to the stress-induced (UV-induced DNA damage or anisomycin-induced inhibition of protein synthesis) diminution of the level of cell survival (Figure 2). DNAJA1 expression also suppresses the phosphorylation-mediated activation of the oncogenic transcription factor, c-Jun, which is often overexpressed and hyperphosphorylated in cancer81−83 and has an essential role in pancreatic cancer.84,85
Interestingly, heat shock proteins have also been shown to be regulators of apoptosis, where DnaK (Hsp70) suppresses JNK activity.86−88 Thus, our preliminary results suggest DNAJA1 potentially stimulates the DnaK suppression of a JNK-induced anti-apoptotic signaling pathway by forming a complex with DnaK. This hypothesis is consistent with the 5-fold downregulation of DNAJA1 in pancreatic cancer cells, the resulting suppression of c-Jun phosphorylation, and the corresponding susceptibility to stress-induced apoptosis by expressing DNAJA1. Obviously, protection from stress-induced apoptosis by downregulating DNAJA1 would be beneficial because cancer cells, by definition, exist in a stressful environment. To further investigate this role of DNAJA1 in pancreatic cancer, we determined an NMR solution structure.
Solution Structure of the DNAJA1 J-Domain
The backbone resonance assignments were determined for 63 (98.4%) of the assignable 64 amino acid residues (excluding the 10 non-native residues to the N-terminal side of the His tag, and the three Pro residues). The one amino acid for which the amide could not be assigned, Met11, is the first residue following the His tag. The side-chain assignments were completed using a combination of CC(CO)NH, HCC(CO)NH, HCCH-COSY, and HCCH-TOCSY 3D NMR experiments. The backbone and side-chain assignments, not including the 10-residue His tag, were nearly complete with 63 of 67 N, 63 of 64 HN, 67 of 67 Cα, 70 of 70 Hα, 63 of 64 Cβ, 113 of 115 Cβ, 43 of 62 Cγ, 66 of 74 Hγ, 26 of 48 Cδ, 40 of 54 Hδ, 11 of 21 Cε, 14 of 34 Hε, 0 of 9 Cζ, and 0 of 1 Hζ atoms assigned.
Using the backbone resonance assignments, a model of DNAJA1-JD was generated using CS-ROSETTA, which utilizes chemical shifts to select protein fragments from the PDB followed by Monte Carlo assembly and relaxation by Rosetta.46,47 This tool has been shown to be effective in predicting protein structures for small proteins (≤16 kDa). The model generated from CS-ROSETTA exhibited the same secondary structure as most DnaJ proteins and agreed with the secondary structure predicted from TALOS+. All available backbone and side-chain chemical shift assignments have been deposited into the Biological Magnetic Resonance Data Bank [BMRB (http://www.brmb.wisc.edu)] as entry 19163.
The solution structure of DNAJA1-JD was calculated using 1120 distance restraints, 116 dihedral restraints, 127 Cα/Cβ chemical shift restraints, and 38 3JNHα coupling constant restraints. The restraints used during the structure calculation are summarized in Table 1. XPLOR-NIH was used to calculate 400 structures, and the 20 lowest-energy structures were selected for further refinement in water using the RECOORD protocol implemented in XPLOR-NIH. The resulting ensemble and average structures (Figure 3A,B) agreed well with the NMR data, where the experimental restraints had low root-mean-square deviations (rmsds) (Table 1). The water-refined average structure had no NOE violations greater than 0.5 Å or dihedral violations greater than 5°. The water-refined ensemble of 20 structures had a backbone rmsd of 0.709 ± 0.119 Å from the unrefined average coordinates. This result improves to 0.399 ± 0.086 Å when only the residues involved in the more stable secondary structure elements are evaluated, which indicates the consistency of the structure calculation using the experimental restraints (Table 2). Comparing the original CS-ROSETTA homology model to the final water-refined average structure showed backbone rmsds of 1.803 Å (full protein) and 0.889 Å (secondary structure).
Table 1. Structure Calculation Statisticsa.
| ⟨SA⟩ | (SA)r | |
|---|---|---|
| rmsd for distance restraints (experimental) (Å) | ||
| all (1120) | 0.083 ± 0.001 | 0.082 |
| inter-residue sequential (|i – j| = 1) (269) | 0.082 ± 0.003 | 0.086 |
| inter-residue short-range (1 < |i – j| < 5) (221) | 0.078 ± 0.005 | 0.073 |
| inter-residue long-range (|i – j| ≥ 5) (80) | 0.108 ± 0.008 | 0.098 |
| intraresidue (500) | 0.084 ± 0.002 | 0.085 |
| H-bonds (50) | 0.038 ± 0.008 | 0.029 |
| rmsd for dihedral angle restraints (deg) (116) | 0.027 ± 0.044 | 0.00 |
| rmsd for Cα and Cβ shifts restraints (ppm) (127) | 0.873 ± 0.036 | 0.907 |
| rmsd for 3JNHα restraints (Hz) (38) | 0.536 ± 0.038 | 0.540 |
| rmsd (covalent geometry) | ||
| bonds (Å) | 0.007 ± 0.000 | 0.007 |
| angles (deg) | 0.649 ± 0.015 | 0.647 |
| impropers (deg) | 0.794 ± 0.037 | 0.778 |
| energy (kcal/mol) | ||
| total | –2620.08 ± 70.18 | –2834.43 |
| bond | 37.44 ± 2.35 | 36.16 |
| angle | 94.07 ± 4.69 | 95.65 |
| dihedral | 0.02 ± 0.04 | 0.00 |
| impropers | 33.44 ± 2.77 | 31.56 |
| van der Waals | –262.40 ± 10.76 | –273.46 |
| NOE | 230.11 ± 7.09 | 226.01 |
| 3JNHα | 10.99 ± 1.60 | 11.08 |
| Cα and Cβ shifts | 49.16 ± 4.30 | 52.92 |
⟨SA⟩ represents the final 20 water-refined simulated annealing structures. (SA)r represents the water-refined average structure of all 20 water-refined structures.
Figure 3.
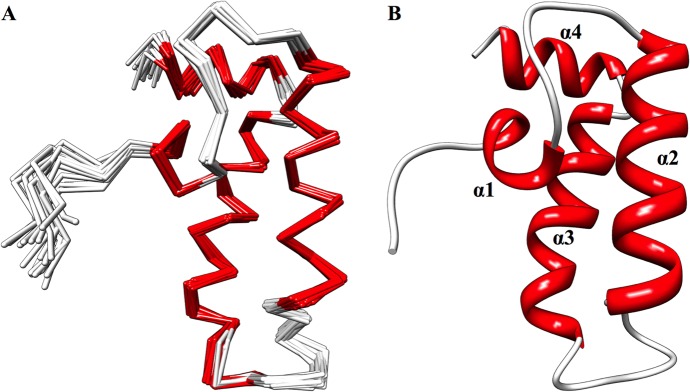
(A) Overlay of the backbone trace of the 20 lowest-energy, water-refined structures. (B) Ribbon representation of the average structure generated from the average atomic coordinates of the 20 lowest-energy, water-refined structures, followed by water refinement of the average structure. Both structures are colored according to secondary structure: red for α-helix and white for loop.
Table 2. Atomic Root-Mean-Square Differencesa.
| full
protein (residues 11–77) |
secondary
structure |
|||
|---|---|---|---|---|
| backbone atoms | all heavy atoms | backbone atoms | all heavy atoms | |
| ⟨SA⟩ vs SA | 0.709 ± 0.119 | 1.417 ± 0.110 | 0.399 ± 0.086 | 1.232 ± 0.134 |
| ⟨SA⟩ vs (SA)r | 0.888 ± 0.136 | 1.739 ± 0.139 | 0.639 ± 0.155 | 1.605 ± 0.131 |
| (SA)r vs SA | 0.649 | 1.217 | 0.523 | 1.183 |
⟨SA⟩ represents the final 20 water-refined simulated annealing structures. SA represents the average structure of all 20 water-refined structures. (SA)r represents the water-refined average structure of all 20 water-refined structures.
The RPF webserver and PSVS software suite were used to verify the quality of the ensemble and average structures (Table 3). Few unreasonable atom clashes were identified by the Molprobity module, and our DNAJA1-JD NMR structure has a very good Z-score (−1.59) compared to the average Z-score for NMR structures in the PDB (−10.74).52 This is actually comparable to the Z-scores of medium-resolution X-ray crystal structures in the PDB (−1.39).52 An evaluation of the probable dihedral angles expected for each residue using PROCHECK also shows excellent Z-scores (1.65 for ϕ and ψ dihedrals and 1.48 for all dihedrals). Additionally, 98.5% of the residues have backbone dihedral angles in the most favored regions of Ramachandran space; only one residue, Val12, has backbone ϕ and ψ values in the more generously allowed region. The discriminating power (DP-score), Recall, Precision, and F-measure scores provide global measures of the goodness of fit between the DNAJA1-JD NMR structure and the NOESY peak list, where a DP-score of >0.75 and an F-measure of >0.8 are consistent with high-quality structures. The DP-score and F-measure calculated for the DNAJA1-JD NMR structure are 0.852 and 0.733, respectively. Overall, the results indicate that the final ensemble and average models for DNAJA1-JD are good-quality structures with few to no unreasonable structural features. The coordinates of the water-refined ensemble have been deposited in the PDB as entry 2M6Y.
Table 3. Structural Evaluationa.
| ⟨SA⟩ | (SA)r | |
|---|---|---|
| PSVS Z-scoreb | ||
| Verify3D | –0.80 | –0.16 |
| ProsaII (−ve) | 2.40 | 2.52 |
| Procheck (ϕ and ψ) | 1.65 | 1.46 |
| Procheck (all) | 1.48 | 1.54 |
| MolProbity clash score | –1.59 | –1.56 |
| RPF scoresc | ||
| Recall | 0.968 ± 0.001 | 0.965 |
| Precision | 0.762 ± 0.006 | 0.737 |
| F-measure | 0.852 ± 0.004 | 0.836 |
| DP-score | 0.733 ± 0.020 | 0.656 |
| Ramachandran spaced (%) | ||
| most favored regions | 98.5 | 98.5 |
| allowed regions | 1.5 | 1.5 |
| disallowed regions | 0.0 | 0.0 |
⟨SA⟩ represents the final 20 water-refined simulated annealing structures. (SA)r represents the water-refined average structure of all 20 water-refined structures.
Calculated with PSVS (http://psvs-1_5-dev.nesg.org/). More positive scores are better.
Calculated with RPF (http://nmr.cabm.rutgers.edu/rpf/). A DP-score of >0.75 and an F-measure of >0.8 are consistent with high-quality structures.
Calculated with the Molprobity module in PSVS.
The secondary structure and fold for DNAJA1-JD are similar to those of other J-domains found in DnaJ homologues. In effect, our DNAJA1-JD structure adopts the characteristic J-domain found in most species. The structure consists of four α-helices: residues 17–21 (α1), 29–42 (α2), 52–65 (α3), and 68–75 (α4) (Figure 3B). The loop between helices α2 and α3 (residues 43–51) contains the highly conserved His-Pro-Asp (HPD) motif (residues 44–46).
The structures of 28 DnaJ proteins in various organisms have been determined. Most of these proteins (16 structures) are from humans, with the majority belonging to DnaJ subfamily B (six) and DnaJ subfamily C (eight). The tertiary structure of DNAJA1-JD was compared to a few representative structures: E. coli DnaJ J-domain (PDB entry 1XBL), human DnaJ homologue subfamily B member 1 J-domain (PDB entry 1HDJ), human DnaJ homologue subfamily B member 2 J-domain (PDB entry 2LGW), and human DnaJ homologue subfamily C member 12 J-domain (PDB entry 2CTQ). All four proteins have essentially the same tertiary structure as DNAJA1-JD with PDBeFold Z-scores of 5.148 (2.08 Å rmsd), 6.872 (1.65 Å rmsd), 4.541 (2.24 Å rmsd), and 6.904 (1.37 Å rmsd), respectively (Figure 4A–D). The different DnaJ homologue subfamilies do not appear to exhibit a significant difference in structure, especially because the best matched structure to DNAJA1 belongs to DnaJ homologue subfamily C. Thus, the structural and functional difference between these subfamilies likely occurs in the other DnaJ domains.
Figure 4.

Overlay of the ribbon structure for DNAJA1-JD (red) with (A) the E. coli DnaJ J-domain (PDB entry 1XBL), (B) the H. sapiens DnaJ homologue subfamily B member 1 J-domain (PDB entry 1HDJ), (C) H. sapiens DnaJ homologue subfamily B member 2 (PDB entry 2LGW), and (D) H. sapiens DnaJ homologue subfamily C member 12 (PDB entry 2CTQ). (E) ClustalW comparison of DNAJA1-JD (HR3099K) with PDB entries 1HDJ (blue), 1XBL (green), 2LGW (yellow), and 2CTQ (cyan). The highly conserved HPD sequence is outlined with a black box. The residues that make up helix α2 are outlined with a red box.
The human DNAJA1-JD sequence is also very well conserved in J-domains from other organisms, with 56 proteins having ≥49% identical sequences. The sequences of representative DnaJ proteins 1XBL, 1HDJ, 2LGW, and 2CTQ have 51, 56, 47, and 32% identical sequences, respectively (Figure 4E). As expected, all five proteins have the highly conserved HPD motif. Additionally, helix α2 is highly conserved except for protein 2CTQ. This is interesting and potentially functionally relevant because helix α2 tends to be positively charged and represents a possible binding site for DnaK (Hsp70).
Identification of Potential Protein Binding Sites on the DNAJA1 J-Domain
One of the primary functions for DnaJ is to stimulate the ATPase activity of DnaK. The primary function of the J-domain of DnaJ is to bind to the ATPase domain on DnaK. As previously mentioned, the main feature of J-domain proteins is a highly conserved HPD motif, which may indicate its importance in binding to DnaK. Thus, identifying a ligand binding site that overlaps with this proposed DnaK binding site would provide further validation of the location and existence of a potential DnaK binding site. Critically, a specific DnaK or DnaK-like protein that binds DNAJA1 has not been identified. It is simply hypothesized to exist on the basis of analogy with other DnaJ proteins.11
The PDB contains only one example of a J-domain in complex with DnaK. This example (PDB entry 2QWN) is a crystal structure of the bovine auxilin (DnaJ homologue subfamily C) J-domain chemically cross-linked with bovine DnaK at the conserved HPD motif.9,89 A sequence alignment of the residues between the bovine auxilin J-domain and DNAJA1-JD allows for a prediction of the proposed DnaJ–DnaK interaction site (Figure 5A). This proposed binding site includes the highly conserved HPD motif. However, there is some contention about whether the cross-linked complex is biologically relevant or whether auxilin accurately represents most interactions of DnaJ with DnaK.89−91
Figure 5.

(A) Transparent surface and ribbon representation of DNAJA1-JD highlighting another proposed DnaK binding site based upon the bovine auxilin–bovine Hsp70 complex (PDB entry 2QWN) (colored red and green; conserved HPD motif). (B) Transparent surface and ribbon representation of DNAJA1-JD (rotated ∼90°) with the proposed DnaK binding site based upon NMR titration data (colored blue). (C) Transparent surface and ribbon of DNAJA1-JD (rotated ∼90°) with the proposed inhibition site based upon the TIM14–TIM16 complex (colored purple). (D) Sequence of DNAJA1-JD with the proposed interaction sites indicated: DnaK binding site from titrations (blue circle), DnaK inhibition site (purple triangles), DnaK binding site from the cross-linked auxilin–Hsp70 complex (red stars), and the highly conserved HPD motif (green box). The conserved HPD motif and helix α2, which is potentially an important component of the DnaJ–DnaK interaction site and the TIM16-like inhibitory binding site, are labeled.
A previous NMR analysis revealed chemical shift perturbations (CSPs) in 2D 1H–15N HSQC experiments when the E. coli DnaJ J-domain is bound to E. coli DnaK.92 The majority of the perturbed residues in the E. coli DnaJ J-domain occurred along helix α2. An alignment of the E. coli DnaJ J-domain and human DNAJA1-JD sequences allowed us to map those same perturbed residues onto the human DNAJA1-JD sequence and structure (Figure 5B). This process identifies a possible DnaK binding site on DNAJA1-JD that includes helix α2 instead of the HPD motif predicted from the DnaJ–DnaK X-ray structure. Helix α2 is an intriguing location for a potential protein binding site because it presents a positively charged surface. Coincidentally, the proposed DnaJ binding site on DnaK has a negatively charged surface, which further supports the identification of helix α2 as a binding site for DnaK. Additionally, mutations of residues in helix α2 inhibit the DnaJ–DnaK interaction, further implicating the importance of helix α2 in binding DnaK.90,92
Another potential protein interaction site on DNAJA1-JD may be linked to the inhibition of DnaJ activity. TIM14 is a related DnaJ protein that is essential for the transport of proteins across the outer membrane of mitochondria by stimulating ATPase activity of mitochondrial Hsp70.93 Any mutation in the HPD motif of TIM14 effectively inhibits its activity, which indicates the importance of the HPD motif in the function of DnaJ proteins. Importantly, TIM14 activity is inhibited when TIM14 is bound to TIM16. TIM16 is another J-domain-like protein, but it lacks the HPD motif. The location of the TIM14–TIM16 interaction site partially overlaps with the proposed DnaK binding site that includes residues in helix α2.94 A sequence alignment of TIM14 with DNAJA1-JD indicates an essentially identical overlap (Figure 5C), where four of the 15 residues that make up the proposed DnaK binding site are also part of the potential inhibition site (Figure 5D). Thus, the binding of a TIM16-like protein to DNAJA1 would sterically interfere with DnaK binding because of this partial overlap in protein binding sites. Again, a TIM16-like protein that binds DNAJA1 has not been identified. Thus, the DnaK binding site is located in the highly conserved HPD motif or helix α2, where a potential inhibitory binding site also involves helix α2. Our ligand binding assay may provide some further insight regarding these potential protein binding sites on DNAJA1-JD.
Identification of a Ligand Binding Site on the DNAJA1 J-Domain
A high-throughput NMR ligand affinity screen66,67 was performed with a function-based compound library.64,65 The library contains ∼460 biologically active compounds consisting of known drugs, protein inhibitors, metabolites, substrates, carbohydrates, fatty acids, hormones, and cofactors. The 1D 1H line broadening screen identified 27 possible binders. A 2D 1H–15N HSQC screen identified only seven compounds that induced chemical shift perturbations (CSPs) upon binding with DNAJA1-JD: O-phospho-l-serine, pyridoxal 5′-phosphate, Bay 11-7082, β-NADPH, ureidosuccinic acid, 2-aminophenol, and d-glucosamine. The seven compounds induced CSPs to the same set of residues, inferring a consistent and unique ligand binding site. Most of the compounds are small, and five of the compounds have a phosphate group or a carboxylic acid group, both of which are likely to be deprotonated at pH 7.0, leaving negatively charged molecules. Importantly, the chemical library contained other compounds with a phosphate or carboxylic acid groups that did not bind DNAJA1-JD, eliminating the likelihood of a nonspecific binding interaction driven only by charge interactions. No other common chemical motif or scaffold is apparent.
Of the seven compounds, O-phospho-l-serine had the greatest number of CSPs (nine) in the 2D 1H–15N HSQC screen (Figure 6A). Using the size of O-phospho-l-serine and our CSP-Consensus program, every residue with an observed CSP was determined to be part of a consensus ligand binding site. A DNAJA1-JD–O-phospho-l-serine costructure was then determined on the basis of these CSPs using AutoDock and AutoDockFilter, which identifies the docked pose that best matches the chemical shift perturbation data.73 The costructure selected by AutoDockFilter (Figure 6B) had an AutoDock binding energy of −2.66 kcal/mol, which fits with the average AutoDock binding energy of −2.54 ± 0.40 kcal/mol for all of the docked poses. The costructure has nine residues with any atom within 6 Å of the docked ligand: Ala40, Leu41, Lys42, Tyr43, His44, Lys47, Asn48, Glu51, and Lys54 (Figure 6C). Six of these residues were perturbed in the 2D 1H–15N HSQC spectrum (Figure 6A). However, there may be some differences between the predicted costructure and the actual complex in solution because of local structural rearrangements upon ligand binding. This is likely to occur because AutoDock used a static model of DNAJA1-JD.
Figure 6.

(A) Overlay of 2D 1H–15N HSQC spectra of free DNAJA1-JD (black) and DNAJA1-JD with O-phospho-l-serine (red). (B) Transparent surface representation and ribbon diagram of DNAJA1-JD bound with O-phospho-l-serine, with the residues showing a chemical shift perturbation upon binding of O-phospho-l-serine colored blue and the one residue, Glu51, that shows the greatest chemical shift perturbation with every binding ligand colored red. (C) Expanded view of O-phospho-l-serine bound to a ribbon diagram of DNAJA1-JD, where residues with side chains directed toward the ligand are displayed.
On the basis of the protein–ligand costructure, the binding site coincides with the predicted inhibition site based on the TIM14–TIM16 complex (Figure 7A). The O-phospho-l-serine binding site is also consistent with the locations of chemical shift perturbations observed for the other compounds shown to bind in the FAST-NMR assay.66,67 Surprisingly, this ligand binding site is not particularly well conserved (Figure 7B). This fact would benefit a drug discovery effort that targets the proposed DNAJA1–TIM16-like complex. While the J-domain is a common fold, the predicted DNAJA1–TIM16-like interaction site appears to be sequence specific, which means drug selectivity can be achieved. Because most of the binding ligands have negatively charged groups, it seems likely that the compounds have an electrostatic interaction with the positively charged region on helix α2 (Figure 7C). Additionally, most of the protein surface, including the binding site, consists of hydrophilic residues, indicating that hydrophobic interactions are unlikely to be energetically favorable. These properties may explain why most of the ligands shown to bind to DNAJA1-JD were small and negatively charged. Nevertheless, these results provide further support for the existence of an inhibitory protein binding site on DNAJA1-JD and the importance of helix α2 to the function of DNAJA1-JD. It is important to note that the ligands shown to bind DNAJA1-JD have a low binding affinity (KD > 100 to 50 μM), are unlikely to inhibit protein–protein interactions, and have not been shown to inhibit DNAJA1-JD protein binding or to be biologically active in pancreatic cancer cells. Instead, they are simple chemical probes of DNAJA1-JD functional epitopes and provide experimental evidence that supports the identification of the predicted inhibitory protein binding site on DNAJA1-JD.
Figure 7.
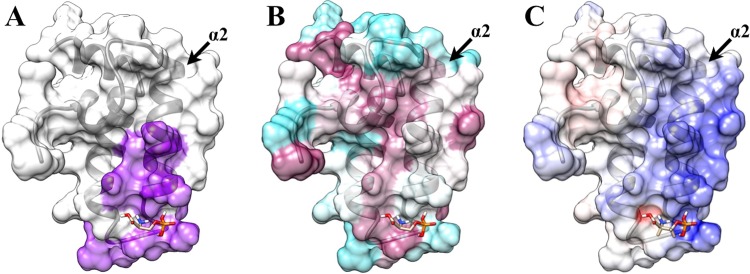
Transparent surface representation and ribbon diagram of DNAJA1-JD bound with O-phospho-l-serine with (A) the proposed inhibition site based on the TIM14–TIM16 interaction (purple), (B) the highly conserved (magenta) and poorly conserved (cyan) residues from Consurf, and (C) the positively charged surface (blue) and negatively charged surface (red) from Delphi. Helix α2, which is potentially an important component of the DnaJ–DnaK interaction site and the TIM16-like inhibitory binding site, is labeled.
Conclusions
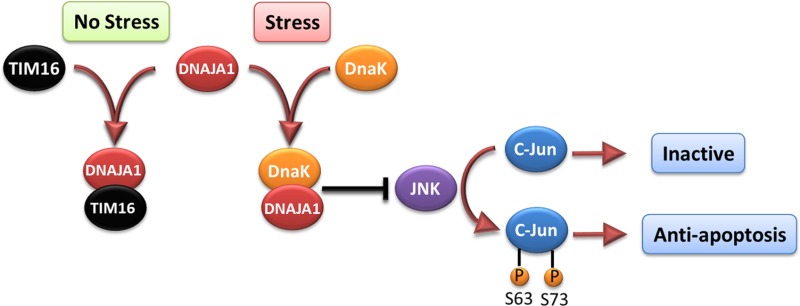
Cell-based functional assays showed that the overexpression of DNAJA1 suppresses the stress response capabilities of the oncogenic transcription factor, c-Jun, which is often overexpressed and hyperphosphorylated in cancer cells. c-Jun is part of the JNK signaling pathway, and its phosphorylation state can promote apoptosis or cell proliferation. DnaK has previously been shown to suppress the JNK pathway, which inhibits the hyperphosphorylated, anti-apoptosis state found in pancreatic cancer cells. The DNAJA1-JD NMR structure combined with a bioinformatics analysis and a ligand affinity screen identified a potential DnaK-like binding site and TIM16-like inhibitory binding site. Interestingly, both of these binding sites involve conserved helix α2 and suggest DNAJA1 activity is highly regulated. The NMR ligand affinity screen provided further experimental support for the existence of the TIM16-like inhibitory binding site. On the basis of these observations, we propose a stress response pathway involving DNAJA1 (Figure 8). In the absence of stress, DNAJA1 would be inactivated by forming an inhibitory complex with a TIM16-like protein. In the presence of environmental stress, DNAJA1 would activate a DnaK protein by forming a complex that suppresses the JNK pathway, the hyperphosphorylation of c-Jun, and the anti-apoptosis state. Conversely, the downregulation of DNAJA1 in pancreatic cancer cells likely lowers the activity of DnaK even under conditions of stress, which allows for the hyperphosphorylation of c-Jun and the cell proliferation that is a hallmark of cancer. Thus, the overlapping protein binding interfaces on DNAJA1-JD may be interesting targets for future drug discovery efforts related to pancreatic cancer. Specifically, a drug that disrupts the predicted DNAJA1–TIM16-like inhibitory complex may activate DnaK even in the absence of a stress response. Thus, a constitutively active DnaK may lead to a cascade of events resulting in cell death through the inactivation of JNK and a decrease in the level of c-Jun phosphorylation. Because the overexpression of DNAJA1 reduces pancreatic cancer cell survivability under stress, the J-domain of DNAJA1 itself may be a valuable biological target for treating pancreatic cancer as part of a combination therapy.
Figure 8.
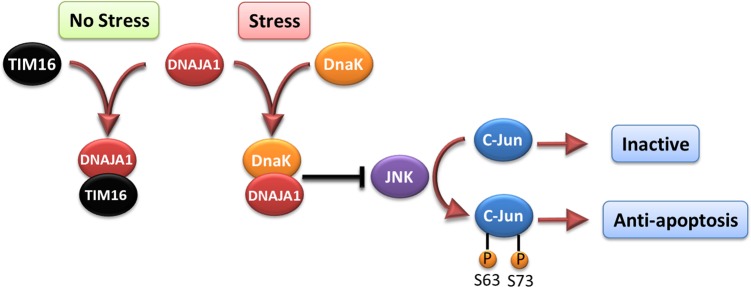
Illustration of the proposed role of DNAJA1 and DnaK on the JNK pathway and c-Jun phosphorylation. The activation of DNAJA1 through the interaction with DnaK appears to suppress the JNK pathway, thus keeping c-Jun in the inactive state. However, inhibiting binding of DNAJA1 to DnaK, as TIM16 does, would allow for the hyperphosphorylation of c-Jun, which has an anti-apoptotic effect.
Acknowledgments
The pET expression plasmid for DNAJ, HR3099K-1-67-14.1, is available through the PSI Materials Repository (http://psimr.asu.edu/) at the Arizona State University Biodesign Institute.
Glossary
Abbreviations
- 2D
two-dimensional
- 3D
three-dimensional
- ATM
auto tune and match
- CSPs
chemical shift perturbations
- DNAJA1
human protein DnaJ homologue subfamily A member 1
- DNAJA1-JD
J-domain of DNAJA1
- DSS
4,4-dimethyl-4-silapentane-1-sulfonic acid
- HPD
His-Pro-Asp
- HSQC
heteronuclear single-quantum coherence
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NESG
Northeast Structural Genomics.
The authors declare no competing financial interest.
This work was supported in part by funds from National Institute of Allergy and Infectious Diseases Grant R21 AI081154 (to R.P.), SPORE Career Development Award P50 CA127297 (to P.K.S.), National Cancer Institute (NCI) Grant R01 CA163649 (to P.K.S. and R.P.), American Association for Cancer Research (AACR)-Pancreatic Cancer Action Network (PanCAN) Career Development Award 30-20-25-SING (to P.K.S.), NCI Pancreatic Tumor Microenvironment Research Network Grant U54 CA163120 (to P.K.S.), Gretchen Swanson Center for Nutrition (GSCN) Cancer Prevention and Control Nutrition Seed Grant 15618 (to P.K.S.), and Grant U54 GM094597 from the Protein Structure Initiative of the National Institutes of Health (to G.T.M.). The research was performed in facilities renovated with support from the National Institutes of Health (Grant RR015468-01).
Funding Statement
National Institutes of Health, United States
References
- Hidalgo M. (2010) Pancreatic cancer. N. Engl. J. Med. 362, 1605–1617. [DOI] [PubMed] [Google Scholar]
- Siegel R.; Naishadham D.; Jemal A. (2012) Cancer statistics, 2012. Ca-Cancer J. Clin. 62, 10–29. [DOI] [PubMed] [Google Scholar]
- Li D.; Xie K.; Wolff R.; Abbruzzese J. L. (2004) Pancreatic cancer. Lancet 363, 1049–1057. [DOI] [PubMed] [Google Scholar]
- Hidalgo M. (2012) New insights into pancreatic cancer biology. Ann. Oncol. 23(Suppl. 10), x135–x138. [DOI] [PubMed] [Google Scholar]
- Mini E.; Nobili S.; Caciagli B.; Landini I.; Mazzei T. (2006) Cellular pharmacology of gemcitabine. Ann. Oncol. 17(Suppl. 5), v7–v12. [DOI] [PubMed] [Google Scholar]
- Moore M. J., Goldstein D., Hamm J., Figer A., Hecht J. R., Gallinger S., Au H. J., Murawa P., Walde D., Wolff R. A., Campos D., Lim R., Ding K., Clark G., Voskoglou-Nomikos T., Ptasynski M., Parulekar W., and National Cancer Institute of Canada Clinical Trials (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 25, 1960–1966. [DOI] [PubMed] [Google Scholar]
- Sheikh R.; Walsh N.; Clynes M.; O’Connor R.; McDermott R. (2010) Challenges of drug resistance in the management of pancreatic cancer. Expert Rev. Anticancer Ther. 10, 1647–1661. [DOI] [PubMed] [Google Scholar]
- Rivera F.; Lopez-Tarruella S.; Vega-Villegas M. E.; Salcedo M. (2009) Treatment of advanced pancreatic cancer: From gemcitabine single agent to combinations and targeted therapy. Cancer Treat. Rev. 35, 335–339. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Maes E. G.; Taylor A. B.; Wang L.; Hinck A. P.; Lafer E. M.; Sousa R. (2007) Structural basis of J cochaperone binding and regulation of Hsp70. Mol. Cell 28, 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A.; Shevde L. A.; Samant R. S. (2009) Multi-faceted role of HSP40 in cancer. Clin. Exp. Metastasis 26, 559–567. [DOI] [PubMed] [Google Scholar]
- Qiu X. B.; Shao Y. M.; Miao S.; Wang L. (2006) The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 63, 2560–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne B. E.; Li T.; Genevaux P.; Georgopoulos C.; Landry S. J. (2010) The Hsp40 J-domain stimulates Hsp70 when tethered by the client to the ATPase domain. J. Biol. Chem. 285, 21679–21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall D.; Zylicz M.; Georgopoulos C. (1994) The NH2-terminal 108 amino acids of the Escherichia coli DnaJ protein stimulate the ATPase activity of DnaK and are sufficient for lambda replication. J. Biol. Chem. 269, 5446–5451. [PubMed] [Google Scholar]
- Walsh P.; Bursac D.; Law Y. C.; Cyr D.; Lithgow T. (2004) The J-protein family: Modulating protein assembly, disassembly and translocation. EMBO Rep. 5, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada K.; Oike Y. (2010) Multiple molecules of Hsc70 and a dimer of DjA1 independently bind to an unfolded protein. J. Biol. Chem. 285, 16789–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada K.; Yomogida K.; Imai T.; Kiyonari H.; Takeda N.; Kadomatsu T.; Yano M.; Aizawa S.; Mori M. (2005) A type I DnaJ homolog, DjA1, regulates androgen receptor signaling and spermatogenesis. EMBO J. 24, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. C.; Liao Y. P.; Mischel P. S.; Iwamoto K. S.; Cacalano N. A.; McBride W. H. (2006) HDJ-2 as a target for radiosensitization of glioblastoma multiforme cells by the farnesyltransferase inhibitor R115777 and the role of the p53/p21 pathway. Cancer Res. 66, 6756–6762. [DOI] [PubMed] [Google Scholar]
- Chow L. Q.; Eckhardt S. G.; O’Bryant C. L.; Schultz M. K.; Morrow M.; Grolnic S.; Basche M.; Gore L. (2008) A phase I safety, pharmacological, and biological study of the farnesyl protein transferase inhibitor, lonafarnib (SCH 663366), in combination with cisplatin and gemcitabine in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 62, 631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik A.; Eckhardt S. G.; Izbicka E.; Tolcher A. A.; Hammond L. A.; Takimoto C. H.; Schwartz G.; McCreery H.; Goetz A.; Mori M.; Terada K.; Gentner L.; Rybak M. E.; Richards H.; Zhang S.; Rowinsky E. K. (2003) A phase I, pharmacokinetic, and biological study of the farnesyltransferase inhibitor tipifarnib in combination with gemcitabine in patients with advanced malignancies. Clin. Cancer Res. 9, 4761–4771. [PubMed] [Google Scholar]
- Crnogorac-Jurcevic T.; Gangeswaran R.; Bhakta V.; Capurso G.; Lattimore S.; Akada M.; Sunamura M.; Prime W.; Campbell F.; Brentnall T. A.; Costello E.; Neoptolemos J.; Lemoine N. R. (2005) Proteomic analysis of chronic pancreatitis and pancreatic adenocarcinoma. Gastroenterology 129, 1454–1463. [DOI] [PubMed] [Google Scholar]
- Kanazawa M.; Terada K.; Kato S.; Mori M. (1997) HSDJ, a human homolog of DnaJ, is farnesylated and is involved in protein import into mitochondria. J. Biochem. 121, 890–895. [DOI] [PubMed] [Google Scholar]
- Terada K.; Kanazawa M.; Bukau B.; Mori M. (1997) The human DnaJ homologue dj2 facilitates mitochondrial protein import and luciferase refolding. J. Cell Biol. 139, 1089–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F.; Green D. R. (2011) Apoptosis and oncogenesis: Give and take in the BCL-2 family. Curr. Opin. Genet. Dev. 21, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M.; Gogvadze V.; Orrenius S.; Zhivotovsky B. (2007) Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922. [DOI] [PubMed] [Google Scholar]
- Paschen S. A.; Weber A.; Hacker G. (2007) Mitochondrial protein import: A matter of death?. Cell Cycle 6, 2434–2439. [DOI] [PubMed] [Google Scholar]
- Petit E.; Oliver L.; Vallette F. M. (2009) The mitochondrial outer membrane protein import machinery: A new player in apoptosis?. Front. Biosci. 14, 3563–3570. [DOI] [PubMed] [Google Scholar]
- Gurbuxani S.; Bruey J. M.; Fromentin A.; Larmonier N.; Parcellier A.; Jaattela M.; Martin F.; Solary E.; Garrido C. (2001) Selective depletion of inducible HSP70 enhances immunogenicity of rat colon cancer cells. Oncogene 20, 7478–7485. [DOI] [PubMed] [Google Scholar]
- Jaattela M. (1995) Over-expression of hsp70 confers tumorigenicity to mouse fibrosarcoma cells. Int. J. Cancer 60, 689–693. [DOI] [PubMed] [Google Scholar]
- Jaattela M.; Wissing D.; Bauer P. A.; Li G. C. (1992) Major heat shock protein hsp70 protects tumor cells from tumor necrosis factor cytotoxicity. EMBO J. 11, 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaattela M.; Wissing D.; Kokholm K.; Kallunki T.; Egeblad M. (1998) Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 17, 6124–6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde M.; Daugaard M.; Jensen M. H.; Helin K.; Nylandsted J.; Jaattela M. (2005) Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 19, 570–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volloch V. Z.; Sherman M. Y. (1999) Oncogenic potential of Hsp72. Oncogene 18, 3648–3651. [DOI] [PubMed] [Google Scholar]
- McDermott K. M.; Crocker P. R.; Harris A.; Burdick M. D.; Hinoda Y.; Hayashi T.; Imai K.; Hollingsworth M. A. (2001) Overexpression of MUC1 reconfigures the binding properties of tumor cells. Int. J. Cancer 94, 783–791. [DOI] [PubMed] [Google Scholar]
- Singh P. K.; Wen Y.; Swanson B. J.; Shanmugam K.; Kazlauskas A.; Cerny R. L.; Gendler S. J.; Hollingsworth M. A. (2007) Platelet-derived growth factor receptor β-mediated phosphorylation of MUC1 enhances invasiveness in pancreatic adenocarcinoma cells. Cancer Res. 67, 5201–5210. [DOI] [PubMed] [Google Scholar]
- Singh P. K.; Behrens M. E.; Eggers J. P.; Cerny R. L.; Bailey J. M.; Shanmugam K.; Gendler S. J.; Bennett E. P.; Hollingsworth M. A. (2008) Phosphorylation of MUC1 by Met modulates interaction with p53 and MMP1 expression. J. Biol. Chem. 283, 26985–26995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y.; Caffrey T. C.; Wheelock M. J.; Johnson K. R.; Hollingsworth M. A. (2003) Nuclear association of the cytoplasmic tail of MUC1 and β-catenin. J. Biol. Chem. 278, 38029–38039. [DOI] [PubMed] [Google Scholar]
- Costa N. R.; Paulo P.; Caffrey T.; Hollingsworth M. A.; Santos-Silva F. (2011) Impact of MUC1 mucin downregulation in the phenotypic characteristics of MKN45 gastric carcinoma cell line. PLoS One 6, e26970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acton T. B.; Xiao R.; Anderson S.; Aramini J.; Buchwald W. A.; Ciccosanti C.; Conover K.; Everett J.; Hamilton K.; Huang Y. J.; Janjua H.; Kornhaber G.; Lau J.; Lee D. Y.; Liu G.; Maglaqui M.; Ma L.; Mao L.; Patel D.; Rossi P.; Sahdev S.; Shastry R.; Swapna G. V.; Tang Y.; Tong S.; Wang D.; Wang H.; Zhao L.; Montelione G. T. (2011) Preparation of protein samples for NMR structure, function, and small-molecule screening studies. Methods Enzymol. 493, 21–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R.; Anderson S.; Aramini J.; Belote R.; Buchwald W. A.; Ciccosanti C.; Conover K.; Everett J. K.; Hamilton K.; Huang Y. J.; Janjua H.; Jiang M.; Kornhaber G. J.; Lee D. Y.; Locke J. Y.; Ma L. C.; Maglaqui M.; Mao L.; Mitra S.; Patel D.; Rossi P.; Sahdev S.; Sharma S.; Shastry R.; Swapna G. V.; Tong S. N.; Wang D.; Wang H.; Zhao L.; Montelione G. T.; Acton T. B. (2010) The high-throughput protein sample production platform of the Northeast Structural Genomics Consortium. J. Struct. Biol. 172, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax A. (2011) Triple resonance three-dimensional protein NMR: Before it became a black box. J. Magn. Reson. 213, 442–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M.; Kay L. E.; Bax A. (1990) A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: Heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry 29, 4659–4667. [DOI] [PubMed] [Google Scholar]
- Kay L. E.; Ikura M.; Tschudin R.; Bax A. (2011) Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. 1990. J. Magn. Reson. 213, 423–441. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. (2005) The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 59, 687–696. [DOI] [PubMed] [Google Scholar]
- Bonvin A. M.; Rosato A.; Wassenaar T. A. (2010) The eNMR platform for structural biology. J. Struct. Funct. Genomics 11, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Lange O.; Delaglio F.; Rossi P.; Aramini J. M.; Liu G.; Eletsky A.; Wu Y.; Singarapu K. K.; Lemak A.; Ignatchenko A.; Arrowsmith C. H.; Szyperski T.; Montelione G. T.; Baker D.; Bax A. (2008) Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. U.S.A. 105, 4685–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Vernon R.; Baker D.; Bax A. (2009) De novo protein structure generation from incomplete chemical shift assignments. J. Biomol. NMR 43, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Tjandra N.; Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. (2009) TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nederveen A. J.; Doreleijers J. F.; Vranken W.; Miller Z.; Spronk C. A.; Nabuurs S. B.; Guntert P.; Livny M.; Markley J. L.; Nilges M.; Ulrich E. L.; Kaptein R.; Bonvin A. M. (2005) RECOORD: A recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 59, 662–672. [DOI] [PubMed] [Google Scholar]
- Huang Y. J.; Powers R.; Montelione G. T. (2005) Protein NMR Recall, Precision, and F-measure Scores (RPF Scores): Structure Quality Assessment Measures Based on Information Retrieval Statistics. J. Am. Chem. Soc. 127, 1665–1674. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A.; Tejero R.; Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795. [DOI] [PubMed] [Google Scholar]
- Eisenberg D.; Luthy R.; Bowie J. U. (1997) VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 277, 396–404. [DOI] [PubMed] [Google Scholar]
- Sippl M. J. (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355–362. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. III; Snoeyink J.; Richardson J. S.; Richardson D. C. (2007) MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., and Higgins D. G. (2002) Multiple sequence alignment using ClustalW and ClustalX. Current Protocols in Bioinformatics, Chapter 2, Unit 2, 3, Wiley, New York. [DOI] [PubMed] [Google Scholar]
- Pellecchia M.; Szyperski T.; Wall D.; Georgopoulos C.; Wuthrich K. (1996) NMR structure of the J-domain and the Gly/Phe-rich region of the Escherichia coli DnaJ chaperone. J. Mol. Biol. 260, 236–250. [DOI] [PubMed] [Google Scholar]
- Qian Y. Q.; Patel D.; Hartl F. U.; McColl D. J. (1996) Nuclear magnetic resonance solution structure of the human Hsp40 (HDJ-1) J-domain. J. Mol. Biol. 260, 224–235. [DOI] [PubMed] [Google Scholar]
- Gao X. C.; Zhou C. J.; Zhou Z. R.; Wu M.; Cao C. Y.; Hu H. Y. (2012) The C-terminal helices of heat shock protein 70 are essential for J-domain binding and ATPase activation. J. Biol. Chem. 287, 6044–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig B.; Nicholls A. (1995) Classical electrostatics in biology and chemistry. Science 268, 1144–1149. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H.; Erez E.; Martz E.; Pupko T.; Ben-Tal N. (2010) ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier K. A.; Germer K.; Powers R. (2006) Design and characterization of a functional library for NMR screening against novel protein targets. Comb. Chem. High Throughput Screening 9, 515–534. [DOI] [PubMed] [Google Scholar]
- Mercier K. A.; Powers R. (2005) Determining the optimal size of small molecule mixtures for high throughput NMR screening. J. Biomol. NMR 31, 243–258. [DOI] [PubMed] [Google Scholar]
- Mercier K. A.; Baran M.; Ramanathan V.; Revesz P.; Xiao R.; Montelione G. T.; Powers R. (2006) FAST-NMR: Functional annotation screening technology using NMR spectroscopy. J. Am. Chem. Soc. 128, 15292–15299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers R.; Mercier K. A.; Copeland J. C. (2008) The application of FAST-NMR for the identification of novel drug discovery targets. Drug Discovery Today 13, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett D.; Seok Y.-J.; Peterkofsky A.; Clore G. M.; Gronenborn A. M. (1997) Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry 36, 4393–4398. [DOI] [PubMed] [Google Scholar]
- Huey R.; Morris G. M.; Olson A. J.; Goodsell D. S. (2007) A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 28, 1145–1152. [DOI] [PubMed] [Google Scholar]
- Morris G.; Goodsell D.; Halliday R.; Huey R. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662. [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner M. F. (1999) Python: A programming language for software integration and development. J. Mol. Graphics Modell. 17, 57–61. [PubMed] [Google Scholar]
- Stark J.; Powers R. (2008) Rapid protein-ligand costructures using chemical shift perturbations. J. Am. Chem. Soc. 130, 535–545. [DOI] [PubMed] [Google Scholar]
- Chen Y. R.; Tan T. H. (2000) The c-Jun N-terminal kinase pathway and apoptotic signaling (review). Int. J. Oncol. 16, 651–662. [DOI] [PubMed] [Google Scholar]
- Shaulian E. (2010) AP-1—The Jun proteins: Oncogenes or tumor suppressors in disguise?. Cell. Signalling 22, 894–899. [DOI] [PubMed] [Google Scholar]
- Weston C. R.; Davis R. J. (2007) The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149. [DOI] [PubMed] [Google Scholar]
- Wisdom R.; Johnson R. S.; Moore C. (1999) c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 18, 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S. U.; Milner J. (2009) Basal cancer cell survival involves JNK2 suppression of a novel JNK1/c-Jun/Bcl-3 apoptotic network. PLoS One 4, e7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R.; Ricci R.; Kenner L.; Zenz R.; David J. P.; Rath M.; Wagner E. F. (2003) Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell 112, 181–192. [DOI] [PubMed] [Google Scholar]
- Mathas S.; Hinz M.; Anagnostopoulos I.; Krappmann D.; Lietz A.; Jundt F.; Bommert K.; Mechta-Grigoriou F.; Stein H.; Dorken B.; Scheidereit C. (2002) Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-κB. EMBO J. 21, 4104–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S.; Corbley M. J.; Roberts T. M. (1995) Reconstitution of signal transduction from the membrane to the nucleus in a baculovirus expression system: Activation of Raf-1 leads to hypermodification of c-jun and c-fos via multiple pathways. Oncogene 11, 427–438. [PubMed] [Google Scholar]
- Binetruy B.; Smeal T.; Karin M. (1991) Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature 351, 122–127. [DOI] [PubMed] [Google Scholar]
- Wodrich W.; Volm M. (1993) Overexpression of oncoproteins in non-small cell lung carcinomas of smokers. Carcinogenesis 14, 1121–1124. [DOI] [PubMed] [Google Scholar]
- Shin S.; Asano T.; Yao Y.; Zhang R.; Claret F. X.; Korc M.; Sabapathy K.; Menter D. G.; Abbruzzese J. L.; Reddy S. A. (2009) Activator protein-1 has an essential role in pancreatic cancer cells and is regulated by a novel Akt-mediated mechanism. Mol. Cancer Res. 7, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessari G.; Ferrara C.; Poletti A.; Dubrovich A.; Corsini A.; Del Favero G.; Naccarato R. (1999) The expression of proto-oncogene c-jun in human pancreatic cancer. Anticancer Res. 19, 863–867. [PubMed] [Google Scholar]
- Jolly C.; Morimoto R. I. (2000) Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 92, 1564–1572. [DOI] [PubMed] [Google Scholar]
- Mosser D. D.; Caron A. W.; Bourget L.; Meriin A. B.; Sherman M. Y.; Morimoto R. I.; Massie B. (2000) The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol. Cell. Biol. 20, 7146–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama S.; Reed J. C.; Homma S. (2003) Heat-shock proteins as regulators of apoptosis. Oncogene 22, 9041–9047. [DOI] [PubMed] [Google Scholar]
- Sousa R.; Jiang J.; Lafer E. M.; Hinck A. P.; Wang L.; Taylor A. B.; Maes E. G. (2012) Evaluation of competing J domain:Hsp70 complex models in light of existing mutational and NMR data. Proc. Natl. Acad. Sci. U.S.A. 109, E734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A.; Bhattacharya A.; McDonald R. A.; Cordes M.; Ellington B.; Bertelsen E. B.; Zuiderweg E. R. (2011) Heat shock protein 70 kDa chaperone/DnaJ cochaperone complex employs an unusual dynamic interface. Proc. Natl. Acad. Sci. U.S.A. 108, 18966–18971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuiderweg E. R. P.; Ahmad A. (2012) Reply to Sousa et al.: Evaluation of competing J domain:Hsp70 complex models in light of methods used. Proc. Natl. Acad. Sci. U.S.A. 109, E735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene M. K.; Maskos K.; Landry S. J. (1998) Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc. Natl. Acad. Sci. U.S.A. 95, 6108–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokranjac D.; Sichting M.; Neupert W.; Hell K. (2003) Tim14, a novel key component of the import motor of the TIM23 protein translocase of mitochondria. EMBO J. 22, 4945–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokranjac D.; Bourenkov G.; Hell K.; Neupert W.; Groll M. (2006) Structure and function of Tim14 and Tim16, the J and J-like components of the mitochondrial protein import motor. EMBO J. 25, 4675–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]