

Abstract
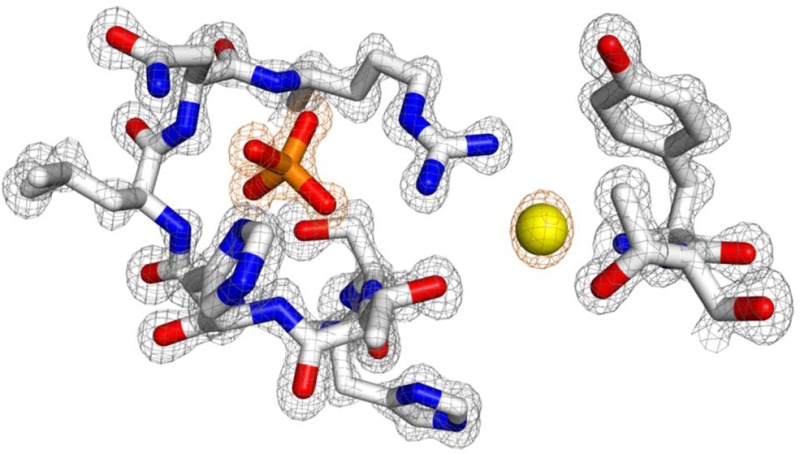
PIR1 is an atypical dual-specificity phosphatase (DSP) that dephosphorylates RNA with a higher specificity than phosphoproteins. Here we report the atomic structure of a catalytically inactive mutant (C152S) of the human PIR1 phosphatase core (PIR1-core, residues 29–205), refined at 1.20 Å resolution. PIR1-core shares structural similarities with DSPs related to Vaccinia virus VH1 and with RNA 5′-phosphatases such as the baculovirus RNA triphosphatase and the human mRNA capping enzyme. The PIR1 active site cleft is wider and deeper than that of VH1 and contains two bound ions: a phosphate trapped above the catalytic cysteine C152 exemplifies the binding mode expected for the γ-phosphate of RNA, and ∼6 Å away, a chloride ion coordinates the general base R158. Two residues in the PIR1 phosphate-binding loop (P-loop), a histidine (H154) downstream of C152 and an asparagine (N157) preceding R158, make close contacts with the active site phosphate, and their nonaliphatic side chains are essential for phosphatase activity in vitro. These residues are conserved in all RNA 5′-phosphatases that, analogous to PIR1, lack a “general acid” residue. Thus, a deep active site crevice, two active site ions, and conserved P-loop residues stabilizing the γ-phosphate of RNA are defining features of atypical DSPs that specialize in dephosphorylating 5′-RNA.
The protein tyrosine phosphatase (PTP) superfamily is broadly grouped into four subfamilies, of which the first and largest subfamily (Class I) includes 99 cysteine-based phosphatases.1,2 Class I PTPs are divided into classical PTPs, which dephosphorylate exclusively phospho-Tyr, and dual-specificity phosphatases (DSPs), which can hydrolyze phosphate groups of phospho-Tyr and phospho-Ser/Thr residues. Since the discovery of the first DSP, VH1, encoded by Vaccinia virus,3 61 VH1-like DSPs have been identified in all kingdoms of life. The human genome encodes 38 VH1-like DSPs (also termed “DUSPs”4) that function as critical signaling molecules, are central to cell physiology, and are involved in a myriad of pathological processes that lead to disease.5 VH1-like DSPs can be further divided into six subgroups on the basis of amino acid sequence conservation.4 One subgroup includes 19 “atypical” DSPs,6 which usually lack an N-terminal Cdc25 homology domain common to mitogen-activated protein kinase phosphatases (MKPs) and vary greatly in size from the 150 amino acids of VHZ (DUSP23)7 to the 1158 residues of DUSP27.8 Atypical DSPs possess broad substrate specificity and dephosphorylate peptidic and nonpeptidic substrates. For instance, laforin functions as a glycogen phosphatase,9 the mitochondrial phosphatase PTPMT1 dephosphorylates phosphatidylglycerol phosphate,10 and PIR1, described in this paper, is specific to RNA.11
PIR1 (phosphatase that interacts with RNA–ribonucleoprotein complex 1),11 also known as DUSP11,4 is a ubiquitous member of the VH1 superfamily whose phosphatase core shares a high level of sequence identity with the RNA 5′-phosphatase BVP from the Autographa californica nuclear polyhedrosis virus.11,12 Mainly localized to the cell nucleus, PIR1 was originally identified as being associated with RNA or ribonucleoprotein complexes.11In vitro, the PIR1 phosphatase activity is several orders of magnitude higher for RNA than phosphoprotein substrates, with a marked preference for phospho-Tyr over phospho-Ser/Thr residues.13 The protein has 5′-triphosphatase and diphosphatase activity for short RNAs but is less active toward mononucleotide triphosphates, suggesting its primary function in vivo is to dephosphorylate RNA 5′-ends.13 PIR1’s exact physiological substrate is unknown, but several lines of evidence link this phosphatase to RNA splicing. Using a yeast two-hybrid screen, it was found that PIR1 associates with splicing factors 9G8 and SRp30C.13 Both in vitro and in vivo, PIR1 binds to splicing factor SAM68, and this activity may contribute to p53-dependent inhibition of cell proliferation.14 PIR1 expression and phosphatase activity have also been linked to cancer. The PIR1 gene is overexpressed in colon carcinoma and glioblastoma tumor cell lines,15 and expression of PIR1 is induced in a p53-dependent manner after treatment with DNA-damaging agents. Ectopic expression of wild-type PIR1 in a cell culture leads to growth arrest, while silencing its expression increases the level of proliferation of normal and DNA-damaged cells in a tissue culture.14 In addition, the PIR1 transcript is differentially expressed in mucosal tissue from patients suffering from inflammatory bowel diseases.16 In Caenorhabditis elegans, PIR1 (also termed PIR-1)17 was identified as a cellular factor that specifically interacts with Dicer DCR-1. In both embryos and adult worms, PIR1 is essential for the development and RNA interference (RNAi): because of its unique ability to hydrolyze RNA 5′-γ- and β-phosphates, PIR1 was hypothesized to generate optimal 5′-monophosphate products for recognition by Dicer or Argonaute proteins involved in downstream steps of RNA silencing.17
In this paper, we report a high-resolution structure of the human PIR1 catalytic core and a biochemical characterization of active site residues involved in catalysis. Our work defines novel and conserved features of an atypical DSP that has RNA 5′-triphosphatase and diphosphatase activity.
Experimental Procedures
Molecular Biology and Biochemical Techniques
A synthetic gene encoding human PIR1 was cloned in expression vector pGEX-6P-1 (Novagen) between restriction sites BamHI and XhoI. Deletion constructs PIR1-coreFED (residues 29–222), PIR1-core (residues 29–205), and PIR1-DSP (residues 66–205) were generated by polymerase chain reaction. Point mutations C152S, H154A, N157A, R192K, and H119G and double mutant H154A/N157A were introduced by site-directed mutagenesis. The fidelity of the DNA sequence for all constructs generated in this study was confirmed by DNA sequencing. Because of its toxicity, all constructs of catalytically active PIR1 (containing C152) were expressed in the NEB Express Iq Escherichia coli strain that produces large quantities of LacI repressor, whereas constructs of catalytically inactive PIR1 (containing C152S) were expressed in the E. coli BL21(DE3) strain. In both cases, cells were grown for 6 h at 25 °C after induction with 0.4 mM IPTG at an OD600 of ∼0.6. Cells were lysed by sonication in lysis buffer [50 mM HEPES (pH 7.5), 200 mM sodium chloride, 5 mM β-mercaptoethanol, 0.1 mM PMSF, and 0.25% DDM], and GST–PIR1 fusion proteins were purified by sequential passages over GST beads (GenScript). GST was cleaved off with PreScission Protease (GE Healthcare) overnight at 4 °C followed by size exclusion chromatography on a Superdex-75 column (GE Healthcare) equilibrated in 200 mM sodium chloride, 20 mM HEPES (pH 7.5), 5 mM β-mercaptoethanol, and 0.1 mM PMSF. PIR1 constructs used for crystallization were concentrated to ∼20.0 mg/mL using a 10K molecular weight Vivaspin 15 concentrator (Sartorius).
Thermal Stability
Thermal stability measurements were recorded using a Jasco J-810 spectropolarimeter equipped with a Neslab RTE7 refrigerated recirculator, as previously described.18,19 PIR1-C152S-coreFED dissolved at a final concentration of 17.0 μM in 20 mM sodium phosphate (pH 7.4) and 50 mM NaCl was measured using a 1 mm long × 12.5 mm wide quartz cuvette (Starna Cells, Inc.), which holds 0.4 mL. Variations in ellipticity at 222 nm as a function of temperature were measured in 0.2 °C increments between 25 and 90 °C. Slow cooling to 25 °C followed by a CD scan at 222 nm to assess the presence of secondary structures demonstrated that PIR1-C152S-coreFED unfolds irreversibly.
Crystallization, Data Collection, and Structure Determination
Crystals of PIR1-C152S-coreFED were obtained using the hanging-drop vapor diffusion method by mixing together 2 μL of gel filtration-purified protein at a concentration of 20.0 mg/mL with 1 μL of 0.1 M Bis-Tris (pH 5.5), 0.2 M sodium chloride, and 22% (w/v) polyethylene glycol 3350, at 18 °C. Addition of 5 mM AMP to the crystallization droplet increased the crystal size and reproducibility. Crystals of the smaller construct PIR1-C152S-core were obtained by mixing together 2 μL of gel filtration-purified protein at a concentration of 20.0 mg/mL with 2 μL of 0.1 M potassium thiocyanate and 34% (w/v) polyethylene glycol monomethyl ether 2000, also at 18 °C. Crystals were harvested in nylon cryo-loops, cryo-protected using 27% (w/v) ethylene glycol, and flash-frozen in liquid nitrogen. Diffraction data were collected on beamlines X6A and X29, at the National Synchrotron Light Source (NSLS) on ADSC Quantum 270 and Quantum 315r CCD detectors, respectively. Data indexing, integration, and scaling were conducted with the HKL2000 software package.20 PIR1-C152S-coreFED crystals belong to space group P213 with one copy in the asymmetric unit and are complete to 1.85 Å resolution (Table 1). In contrast, PIR1-C152S-core crystallized in a primitive orthorhombic space group (P212121) with a trimer in the asymmetric unit. Diffraction data were collected to 1.20 Å resolution, although the completeness of the data is modest in the outer shell: X-ray data are 55.5% complete between 1.24 and 1.20 Å, 74.9% complete between 1.29 and 1.24 Å, and >95% complete between 1.29 and 15.0 Å (Table 1). The structure of PIR1-C152S-coreFED was determined by molecular replacement using the structure of BVP [Protein Data Bank (PDB) entry 1YN9]21 as the search model, as implemented in PHASER.22 This initial phasing model was subjected to manual rebuilding using COOT23 followed by refinement with phenix.refine,24 using cycles of positional and isotropic B factor refinement with six distinct translation/libration/screw (TLS) groups. The PIR1-C152S-coreFED final model contains residues 29–207 of human PIR1, five N-terminal residues from the expression vector (sequence of GPLGS), 181 water molecules, a phosphate, and a chloride ion and was refined to Rwork and Rfree values of 16.85 and 18.75%, respectively, using all diffraction data between 15 and 1.85 Å resolution (Rfree was calculated using 1129 randomly chosen reflections, corresponding to ∼5% of all diffraction data) (Table 1). This model was then used to determine the orthorhombic crystal form of PIR1-C152S-core that contains a trimer in the asymmetric unit. An initial solution obtained using PHASER22 was first refined isotropically with Refmac_5.8.004925 until Rfree dropped below 20%. At this point, the solvent was built using phenix.refine,24 riding hydrogens were added to protein atoms, and the model was further subjected to several cycles of positional and anisotropic B factor refinement in Refmac_5.8.0049, using all diffraction data between 6.0 and 1.20 Å (Table 1). The PIR1-C152S-core final atomic model contains residues 29–205 for chains A–C (and five N-terminal residues, GPLGS, from the expression vector are visible for chains A and B), 973 water molecules, three phosphates, and three chloride ions and was refined to Rwork and Rfree values of 13.73 and 16.99%, respectively (Rfree was calculated using ∼5% of the diffraction data). Stereochemistry was checked using PROCHECK:26 atomic models of PIR1-C152S-coreFED and PIR1-C152S-core have outstanding geometry, with root-mean-square deviations (rmsds) from ideal bonds and angles of 0.012 Å and 1.378° and 0.009 Å and 1.472°, respectively, and >91% of residues in the most favored regions of the Ramachandran plot and no disallowed residues. Atomic coordinates and experimental structure factors of PIR1-C152S-coreFED and PIR1-C152S-core have been deposited in the Protein Data Bank as entries 4MBB and 4NYH, respectively.
Table 1. Crystallographic Data Collection and Refinement Statistics.
| PIR1-C152S-coreFED | PIR1-C152S-core | |
|---|---|---|
| Data Collectiona | ||
| wavelength (Å) | 0.9789 | 1.070 |
| space group | P213 | P212121 |
| unit cell dimensions | a = 92.7 Å, b = 92.7 Å, c = 92.7 Å | a = 51.0 Å, b = 62.7 Å, c = 178.8 Å |
| α = β = γ = 90° | α = β = γ = 90° | |
| resolution range (Å) | 15.0–1.85 | 15.0–1.20 |
| Wilson B factor (Å2) | 35.0 | 23.8 |
| total no. of observations | 163375 | 2783883 |
| no. of unique observations | 22788 | 163044 |
| completeness (%) | 99.1 (100) | 91.0 (55.2) |
| Rsymb (%) | 6.6 (51.5) | 6.3 (48.2) |
| ⟨I⟩/⟨σ(I)⟩ | 57.3 (5.0) | 20.1 (3.0) |
| Refinement | ||
| resolution (Å) | 15.0–1.85 | 6.0–1.20 |
| no. of reflections | 22769 | 153934 |
| Rwork, Rfreec (%) | 16.85, 18.75 | 13.73, 16.99 |
| no. of copies in the asymmetric unit | 1 | 3 |
| no. of water molecules | 181 | 973 |
| B value of the model (Å2) | ||
| protein | 43.0 | 22.5 |
| water | 50.0 | 38.1 |
| Pi | 29.5 | 15.4 |
| Cl | 27.1 | 14.1 |
| rmsd from ideal | ||
| bond lengths (Å) | 0.012 | 0.009 |
| bond angles (deg) | 1.378 | 1.472 |
| Ramachandran plot (%) | ||
| core | 90.4 | 91.1 |
| allowed | 9.6 | 8.9 |
| generously allowed | 0.0 | 0.0 |
| disallowed | 0.0 | 0.0 |
In parentheses are statistics for the outer resolution shell (1.92–1.85 and 1.24–1.20 Å).
Rsym = ∑i,h|I(i,h) – ⟨I(h)⟩|/∑i,h|I(i,h)|, where I(i,h) and ⟨I(h)⟩ are the ith and mean measurements of intensity of reflection h, respectively.
The Rfree value was calculated using 5% of randomly chosen reflections.
Structure Analysis and Molecular Docking
All ribbon diagrams and surface representations presented here were prepared using Pymol (Delano Scientific). Nonlinear Poisson–Boltzmann electrostatic calculations were performed using APBS Tools.27 The topological diagram was generated using PDBsum,28 and structural superimpositions were conducted in Coot.23 Analysis of cavities was done using VOIDOO,29 as described previously.30 Solvent accessibility surface (SAS) areas were calculated using Chimera.31 In this program, the SAS is defined as the locus of the center of a probe sphere (representing the solvent molecule) as it rolls over the van der Waals surface of the protein. Analysis of the interactions between ClAS and the PIR1 active site was conducted using LigPlot.32 Docking of ATP and ADP inside PIR1-core was performed using AutoDock Vina33 and HADDOCK.34
In Vitro Phosphatase Assay
Hydrolysis of 200 μM ATP or ADP substrate (Sigma-Aldrich) was assessed in dephosphorylation buffer [50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 2 mM EDTA, and 5 mM DTT] in 60 μL reaction mixtures containing 5 μg of purified PIR1-coreFED, at 22 °C. Phosphatase reactions were terminated after 50 min via addition of 60 μL of 0.1 M N-ethylmaleimide and 120 μL of Malachite green reagent, as previously described.35 Fifteen minutes after the reaction had been quenched, the absorbance was measured at 620 nm, a blank was subtracted, and the exact quantity of released phosphate was determined using a phosphate standard curve. The enzymatic activity of PIR1-coreFED carrying alanine substitutions at H154, N157, and both H154 and N157 was monitored using the phospho-substrate OMFP (Sigma-Aldrich), at a final concentration ranging from 10 to 300 μM, as previously described.36 The dephosphorylation reaction was initiated by addition of purified PIR1-coreFED constructs to the reaction mix at a final concentration of 1 μM. Dephosphorylation of OMFP was monitored at 477 nm using a Tecan Infinite M1000 PRO plate reader (Tecan), at 22 °C. For each concentration of OMFP, the initial linear rate of product formation (or initial velocity) was calculated from the variation in absorbance over the first 120 s of the reaction. For each PIR1 construct, initial velocities were plotted against the concentration of OMFP, and Km, Vmax, and kcat values were determined by nonlinear regression using GraphPad (GraphPad Software, Inc.). A list of all kinetic parameters is given in Table 2.
Table 2. Enzymatic Activity of PIR1-coreFED Constructs toward OMFP.
| PIR1-coreFED construct | Km (μM) | Vmax (μM min–1) | kcat/Km (min–1 M–1) |
|---|---|---|---|
| C152 | 73.9 ± 17.3 | 1.8 ± 0.15 | 2.4 × 104 |
| H154A | 88.8 ± 33.3 | 0.6 ± 0.12 | 6.7 × 103 |
| N157A | 98.8 ± 20.5 | 0.65 ± 0.1 | 6.5 × 103 |
| DM | 92.0 ± 21.2 | 0.30 ± 0.1 | 3.2 × 103 |
| C152S | not available | not available | not available |
Results
Domain Organization of Human PIR1
Human PIR1 is a 330-residue nuclear protein with a predicted molecular mass of ∼38939 Da. Bioinformatics analysis identified a conserved DSP domain between residues 66 and 205, which is 41% identical to the RNA 5′-phosphatase domain of BVP.11,12 The PIR1 catalytic core is flanked by two regions with low levels of complexity. At the N-terminus, residues 12–26 contain a putative bipartite nuclear localization signal (NLS)37 (12-GRRRDFSGRSSAKKK-26) (Figure 1A), consistent with PIR1 nuclear localization.11 The C-terminus of the DSP domain is a long noncatalytic extension (residues 206–330) that contains two short predicted helices (h10 and h11) between residues 235–240 and 263–270 (Figure 1A). This C-terminal extension is not required for RNA 5′-phosphatase activity, and its deletion enhances PIR1 phosphatase activity in vitro.13
Figure 1.
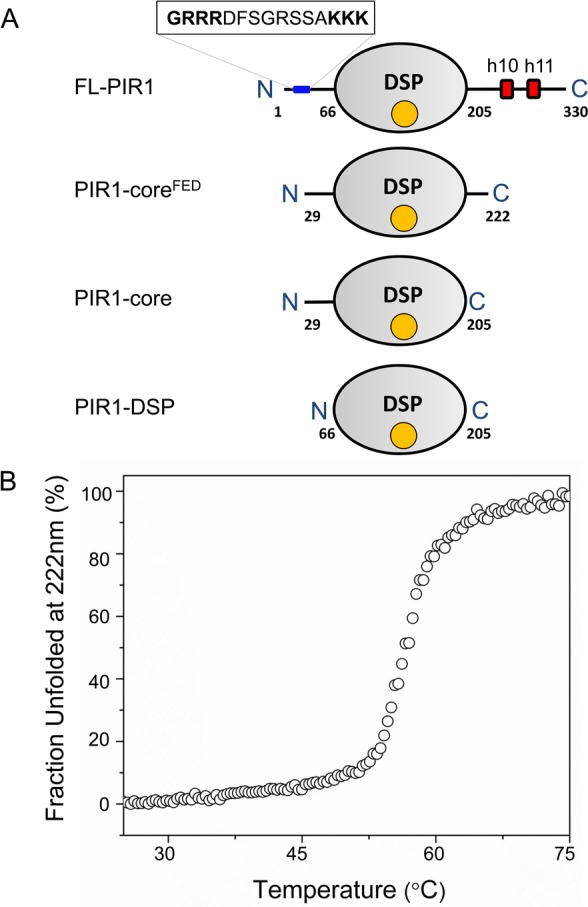
Domain organization and stability of human PIR1. (A) Schematic diagram of the PIR1 domain organization and deletion constructs generated in this study. The DSP domain (residues 66–205) is colored gray and flanked by an N-terminal putative NLS and two predicted C-terminal α-helices (h10 and h11) (colored red). (B) Thermal denaturation of PIR1-C152S-coreFED monitored by measuring temperature-induced CD changes in ellipticity at 222 nm. PIR1-C152S-coreFED unfolded irreversibly with an appTm of ∼56.7 °C.
Aiming at structural studies, we generated several PIR1 constructs lacking the N-terminal putative NLS and containing various deletions of the C-terminal noncatalytic extension. We found that a construct encompassing residues 29–205 (termed PIR1-core) and a slightly longer construct containing a C-terminal “FED” motif (PIR1-coreFED, residues 29–222) had the highest solubility in vitro (Figure 1A). However, because of its in vivo toxicity, it was difficult to purify large quantities of catalytically active PIR1-core for structural studies. Replacing the active site C152 with a serine reduced in vivo toxicity and yielded a protein less aggregation-prone in vitro. Circular dichroism (CD) spectra of PIR1-C152S-coreFED recorded at pH 7.4 were consistent with a folded protein containing a mixture of α-helices and β-stands and <30% of its residues in a random coil conformation (Figure S1 of the Supporting Information). To determine the structural stability of PIR1-C152S-coreFED, we measured heat-induced denaturation by monitoring variations in ellipticity at 222 nm as a function of temperature (Figure 1B). PIR1-C152S-coreFED unfolded irreversibly in a highly cooperative manner, with an apparent melting temperature (appTm) of ∼56.7 °C, comparable to that of VH1 (appTm ∼ 60 °C)38 but significantly lower than that of the DUSP26 catalytic core (appTm ∼ 68 °C).39 An identical appTm was measured for PIR1-C152S-core (Figure S2 of the Supporting Information), suggesting that the C-terminal extension spanning residues 206–222 does not affect protein stability. Thus, PIR1-core adopts a folded and stable conformation suitable for structural analysis.
Atomic Structure of the PIR1 Catalytic Core
We crystallized PIR1-C152S-coreFED and PIR1-C152-core over a wide range of pH values between 5.5 and 8.5. At a lower pH, PIR1-C152S-coreFED yielded cubic crystals containing a monomer in the asymmetric unit that diffracted to ∼1.85 Å resolution. In contrast, PIR1-C152S-core crystallized as a trimer in a primitive orthorhombic space group, and the best crystals diffracted past 1.20 Å resolution. We determined both crystal forms by molecular replacement using BVP as a search model and refined PIR1-C152S-coreFED to Rwork and Rfree values of 16.85 and 18.75%, respectively, at 1.85 Å resolution and PIR1-C152S-core to Rwork and Rfree values of 13.73 and 16.99%, respectively, at 1.20 Å resolution (Table 1). The structures of the two PIR1 constructs are essentially identical (rmsd of ∼0.32 Å) (Figure S3 of the Supporting Information), and because there is no evidence of PIR1 oligomerization in solution, the trimeric arrangement seen in the orthorhombic crystal form may reflect a crystallization artifact. In this paper, we will refer to the structure of PIR1-C152S-core, which was refined to 1.20 Å resolution (Figure 2A), for structural analysis and comparison to other DSPs.
Figure 2.

Atomic structure of PIR1-C152S-core determined at 1.20 Å resolution. (A) Ribbon diagram of PIR1-C152S-core with S152 shown as a red sphere. The active site phosphate and chloride ions are colored orange and yellow, respectively. (B) Topological diagram of PIR1-C152S-core. The central β-sheet formed by strands β1−β5 is highlighted with a light blue background. Insertion elements not found in VH1 are colored red. (C) Superimposition of PIR1-S152C-core (cyan) and VH1 (gray) with helices shown as cylinders (for the sake of clarity, the VH1 N-terminal helix spanning residues 1–20 was omitted). Colored red are the structural elements that render the PIR1 catalytic cleft significantly deeper than in VH1.
PIR1-C152S-core is built by a quasi-globular cysteine-phosphatase domain of 45 Å × 45 Å × 60 Å, with N- and C-termini projecting in opposite directions (Figure 2A). The phosphatase core consists of a central five-stranded β-sheet (β1−β5) (highlighted with a light blue background in Figure 2B) sandwiched between two clusters of five (h1–h5) and four (h6–h9) helices that make contacts with the solvent (Figure 2B). A search using the Dali server40 identified baculovirus BVP (PDB entry 1YN9)21 and the RNA 5′-phosphatase domain of the mouse mRNA capping enzyme (PDB entry 1I9S)41 as the phosphatases that were most structurally similar to PIR1-core (rmsds of 1.4 and 1.5 Å, respectively). PIR1-C152S-core can also be superimposed on VH1 (PDB entry 3CM3) (Figure 2C) and VHR (PDB entry 1VHR) with rmsds of 2.5 and 2.2 Å, respectively, but presents four noticeable differences from the classical VH1-like DSP-core (colored red in Figure 2B,C). First, it contains an N-terminal extension upstream of the DSP-core (residues 29–40), partially folded into a 3/10 helix (h1) (Figure 2B,C). Second, a helical hairpin containing helices h2 and h3 inserts between strand β2 and helix h4 of the PIR1 phosphatase core, forcing helix h4 to adopt a position orthogonal to its counterpart in VH1 (Figure 2B,C). Third, the PIR1 “WPD loop” (or “general acid loop”, residues 116–122) is two residues shorter than in most phosphoprotein phosphatases.38 Fourth, the loop connecting stands β3 and β4 (known as the “E-loop”, residues 96–101) is significantly longer than in VH1 and folds into a 3/10 helix (h5) between residues 102 and 104 (Figure 2B,C). Together, these subtle variations on the classical DSP fold affect the PIR1 active site conformation and, potentially, substrate specificity. Most noticeably, the PIR1 N-terminal extension packs against the insertion hairpin and makes contacts with the E-loop, surrounding the P-loop (Figure 2A,C). This generates a remarkably wider and deeper (Figure 3A,D) active site cleft than in VH1 (Figure 3C,F).38 The PIR1 active site crevice is also deeper than in BVP (Figure 3B,E),21 mainly because of the conformation adopted by the E-loop, which faces inward in PIR1 and projects outward in BVP. PIR1 catalytic residue C152 (S152 in our structure) sits at the bottom of this deep crevice, buried ∼10 Å below the enzyme surface: the calculated SAS area for C152 is ∼1.45 Å2, significantly smaller than that of C119 in BVP (∼2.18 Å2) or C110 in VH1 (∼1.61 Å2).
Figure 3.

Depth of the PIR1 catalytic cleft. Surface representations of (A) human PIR1-C152S-core, (B) baculovirus BVP (PDB entry 1YN9), and (C) Vaccinia virus monomeric VH1 (PDB entry 3CM3) reveal striking differences in the width of the active site pocket. Magnified cut-through views of (D) PIR1, (E) BVP, and (F) VH1 catalytic pockets reveal differences in the depth of the active site crevice.
PIR1 Active Site Residues
The 1.20 Å structure of PIR1-C152S-core, the highest resolution ever achieved for a human dual-specificity phosphatase, provides a detailed view of the enzyme active site (Figure 4A). The PIR1 catalytic cysteine, C152 (replaced with a serine in our structure), and the general base R158 are part of a P-loop that can be structurally superimposed with the P-loop of VH1-related DSPs.38 The invariant arginine, R158, is 4.6 Å from the C152 sulfur atom, slightly closer than in VH1, where this distance is 5.1 Å. Unlike classical PTPs and most DSPs, PIR1 does not contain a general acid, which is usually located on a separate loop (the general acid loop), near the top of the active site.42 Furthermore, the PIR1 active site is occupied by two ions. The first ion, a phosphate as in VH138 or DUSP27,8 resembles the γ-phosphate of RNA and makes close contacts with main chain atoms of P-loop residues 153–158 and the side chains of S152, H154, N157, and R158 (Figure 4A). The second ion, a chloride (termed “active site Cl” or ClAS) is visible as an ∼8σ peak of positive electron density in an Fo – Fc difference map (Figure 4A). ClAS is coordinated by the side chain atoms of T96, R158, and T153, as well as the backbone atoms of T96 and Y95 and a water molecule (Figure 4B). The approximate distance between PIR1 atoms and ClAS is 3.2 Å, in good agreement with the distance expected for a chloride atom interacting with a protein, given a van der Waals radius of 1.81 Å.30 Although ClAS is only 6 Å from the phosphate ion, the two ions interact only indirectly through the guanidinium group of R158. The refined B factor of ClAS is only 14.1 Å2, comparable to that of the active site phosphate (∼15.4 Å2) but significantly lower than the B factors of both protein (∼22.5 Å2) and solvent (∼38.1 Å2) atoms (Table 1). To rule out the possibility that ClAS represents an artifact of crystallization resulting from the presence of 200 mM NaCl in the protein buffer, we also determined a 2.2 Å crystal structure of PIR1-C152S-core crystallized in only 35 mM NaCl, the lowest salt concentration at which PIR1 yields crystals (Figure S4 of the Supporting Information). Despite an ∼6-fold lower NaCl concentration (35 mM vs 200 mM), an identical peak of density corresponding to ClAS was observed in the electron density, suggesting this metal ion binds specifically to the PIR1 active site in a dedicated binding pocket (Figure 4B).
Figure 4.
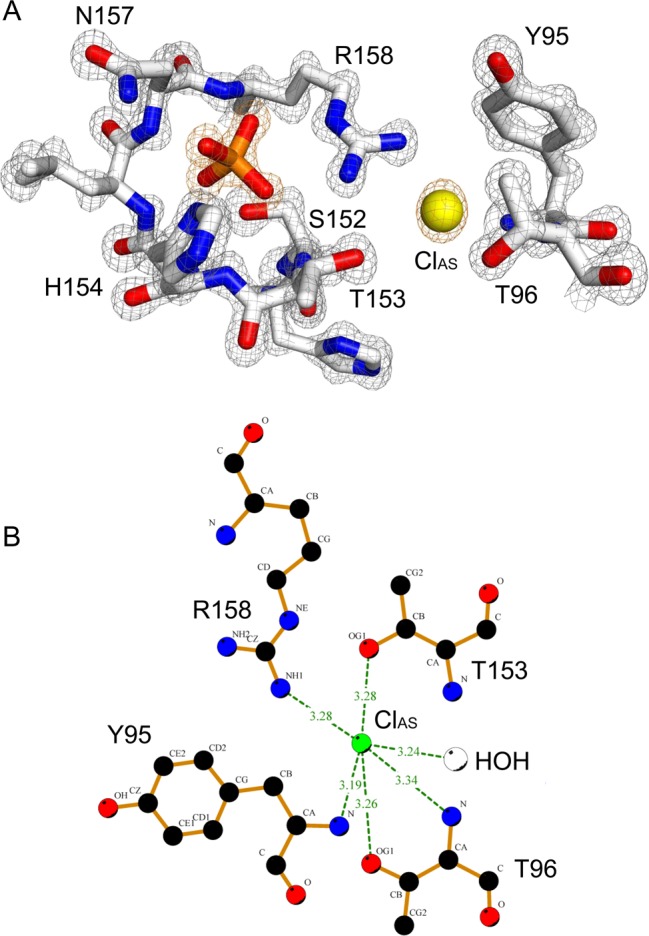
Architecture of the PIR1 P-loop. (A) Magnified view of the PIR1 final 2Fo – Fc electron density map calculated at 1.20 Å resolution and overlaid with the refined model of the PIR1 active site (shown as sticks). The density is colored gray and contoured 2.2σ above background. An Fo – Fc electron density map calculated after omitting all active site ions is overlaid with the PIR1 phosphate and ClAS; the map is colored orange and contoured 8σ above background. (B) LigPlot diagram of the interactions between ClAS and the PIR1 active site. Hydrogen bonds are indicated by dashed lines between the atoms involved.
Docking a Ribonucleotide inside the PIR1 Catalytic Pocket
Attempts to cocrystallize PIR1-core with polyribonucleotides were unsuccessful. Addition of 5 mM AMP (also a ribonucleotide) to PIR1-S152-coreFED during crystallization enhanced the crystal size and reproducibility, but no density for this ribonucleotide was observed in the final electron density. We then attempted in silico docking of ATP and ADP into the PIR1-S152C-core crystal structure using AutoDock Vina.33 This program readily placed the triphosphate tail of ATP inside the PIR1 positively charged crevice (Figure 5A), forcing the triphosphate backbone negative charge to be neutralized by PIR1 (Figure 5B). A comparable docking model was also obtained using HADDOCK,34 which uses structural constraints from the phosphate ion observed crystallographically to model the position of the γ-phosphate of ATP. The predicted free energy for binding of ATP to PIR1-core (ΔGbind) was −7.0 kcal/mol, slightly higher than that of ADP (−6.6 kcal/mol). In this model, the ATP triphosphate tail, ∼10 Å in length, fits perfectly inside the PIR1 catalytic pocket, while the nucleobase (an adenine) faces the catalytic cleft, making contacts with residues in the WPD loop (Figure 5A,B). Interestingly, this model predicts that the position of RNA in the catalytic cleft is compatible with the presence of ClAS (Figure 5B), which is not likely displaced during catalysis. The high degree of electrostatic complementarity between the PIR1 basic surface (blue in Figure 5A,B) and the negative charge of RNA is likely to play a pivotal role in molecular recognition.
Figure 5.
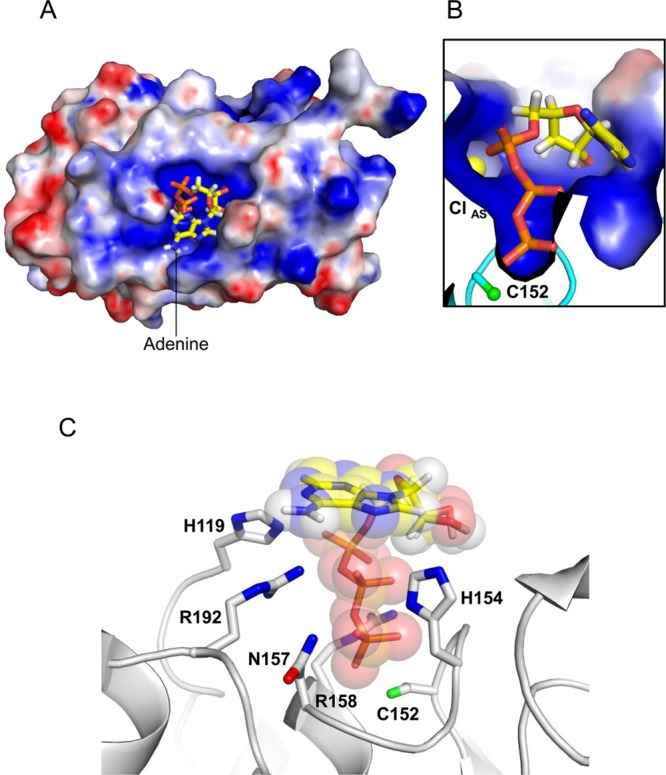
Modeling ATP inside the PIR1-core active site. (A) Model of ATP docked onto the electrostatic potential molecular surface of PIR1-C152S-core computed at neutral pH. (B) Magnified view of the ATP triphosphate tail docked inside the PIR1 active site crevice. (C) Ribbon diagram of PIR1 (gray) bound to ATP (shown as sticks overlaid with transparent spheres corresponding to van der Waals radii). PIR1 residues involved in ATP binding and subjected to site-directed mutagenesis are indicated (for the sake of clarity, hydrogen atoms are shown for only ATP). In all panels, ATP is colored by element, with carbon, hydrogen, nitrogen, oxygen, and phosphate atoms colored yellow, white, blue, red, and orange, respectively.
Structure-Based Site-Directed Mutagenesis
The high-resolution structure of PIR1-C152S-core bound to phosphate (Figure 4A) and the docking model in Figure 5 revealed two distinctive features of PIR1 substrate recognition. First, the nonaliphatic side chains of P-loop residues H154 and N157 make close contacts with the active site phosphate: H154 nitrogen atom ND1 and N157 carboxamide are only ∼2.7 Å from the phosphate O3 atom (Figure 4A). These close contacts are not commonly seen in DSPs that dephosphorylate phosphoprotein monoesters, where the P-loop is usually occupied by small side chain-bearing amino acids.39 Second, residues R192 and H119 of PIR1 (Figure 5C) are positioned <3.0 Å from the β-phosphate of RNA and possibly involved in β-phosphatase activity. Interestingly, these residues (H119 and R192) are conserved in BVP, also a β-phosphatase, where H119 is replaced with a glutamine (Q88 and R159, respectively), but are missing in the RNA phosphatase domain of the mouse (PDB entry 1I9S) and human (PDB entry 2C46) mRNA capping enzyme, which lack β-phosphatase activity and have a glycine and a lysine at equivalent positions (G95 and K166, respectively). To test the role of these residues in PIR1 catalytic activity, we generated five mutants of PIR1-coreFED carrying H154A, N157A, H154A/N157A (DM), R192K, and H119G substitutions. All mutants were expressed and purified under identical conditions, and their phosphatase activity was measured in vitro using PIR1-C152-coreFED and PIR1-C152S-coreFED as positive and negative controls, respectively. To determine if mutations preferentially affected PIR1 triphosphatase versus diphosphatase activity, all mutants were incubated with ATP and ADP and hydrolyzed phosphate was quantified with Malachite green reagent35 (Figure 6A). This assay revealed that removing the side chains of H154 and N157, as well as combining the two mutations (DM), dramatically disrupted phosphatase activity: all P-loop mutants were catalytically inactive in vitro, like PIR1-C152S-core (Figure 6A). Alanine substitution of H154 and N157 disrupted phosphatase activity irrespective of the phospho-substrate used in the assay (ATP or ADP), suggesting an essential role in catalysis. In contrast, replacing R192 with a lysine (to mimic the mRNA capping enzyme) reduced the level of hydrolysis of ADP β-phosphate by 80% while the level of ATP dephosphorylation was only halved (Figure 6A). Finally, mutating H119 to glycine had no impact on phosphatase activity under our experimental conditions, being the quantity of inorganic phosphate released by PIR1-H119G-coreFED comparable to that of wild-type PIR1-coreFED (Figure 6A).
Figure 6.

In vitro phosphatase assay. (A) Dephosphorylation of 200 μM ATP and ADP by various constructs of PIR1-coreFED. The amount of phosphate released upon hydrolysis was measured at 620 nm using Malachite green reagent and quantified using a phosphate standard curve. Error bars are based on three independent repeats. (B) Michaelis–Menten saturation kinetics of OMFP in the presence of 1 μM PIR1-coreFED, PIR1-H154A-coreFED, PIR1-N157A-coreFED, PIR1-DM-coreFED, and PIR1-C152S-coreFED. Table 2 lists all the kinetic parameters measured in this experiment.
To determine if P-loop mutations at H154 and N157 affect Km (substrate binding) or kcat/Km (catalytic efficiency), we also set up an in vitro dephosphorylation assay using the phosphatase substrate OMFP (Figure 6B). We found that alanine mutations at H154 and N157 minimally affected PIR1’s affinity for OMFP (<1.5-fold increase in Km) but reduced the kcat/Km by 3.5–7.5-fold relative to that of PIR1-C152-core (Table 2). Together, these data suggest that the nonaliphatic side chains of H154 and N157 play an important role in phosphatase activity and, when mutated to alanine, PIR1 is still able to bind substrate molecules with high affinity, but the enzyme–substrate complex converts a smaller proportion of bound substrate into product.
Discussion
Dual-specificity phosphatases are important signaling enzymes whose misregulation is intimately linked to cancer, diabetes, inflammation, and Alzheimer’s disease.43 Inhibiting DSPs is emerging as a potential therapeutic strategy in pharmacology.44 In particular, significant interest is devoted to understanding how the well-conserved and apparently simple PTP fold accounts for the remarkable substrate specificity present in a cell and governing signaling pathways. Knowledge of molecular determinants involved in substrate recognition is essential to the development of potent and specific phosphatase inhibitors. For classical PTPs and DSPs that hydrolyze phospho-amino acids, the depth of the active site pocket has been identified as a key determinant in substrate specificity.45−47 High-resolution structures of human VHR,46 VHZ,7Vaccinia virus VH1,36,38 and many other VH1-like phosphatases have revealed a shallow active site pocket (∼6 Å in depth). Less is known about atypical DSPs6 that dephosphorylate highly diverse, nonpeptidic substrates such as carbohydrates,48 lipids,10 and RNA.10 The 1.20 Å crystal structure of PIR1-core presented in this paper sheds light on novel and conserved structural features of a human RNA 5′-phosphatase.
Design Principles of a VH1-like RNA 5′-Phosphatase
The atomic structure of PIR1-core suggests that specificity for RNA is conferred by at least three distinct structural determinants. First, subtle variations of the VH1 fold (Figure 2B,C) render the PIR1-core active site remarkably wide and deep. An N-terminal extension (residues 29–40) common to RNA 5′-phosphatases that bear an amino-proximal DSP domain packs against the insertion hairpin (h2–h3) and makes contact with the top of the E-loop (β3−β4 loop), surrounding the P-loop (Figure 2C). A shorter acidic loop lacking a general acid (Glu or Asp) exerts reduced steric hindrance on the active site, making the PIR1 active site crevice both wider and deeper than in DSPs specific to peptidic substrates38 (Figure 3A,D). Both the helical hairpin and the N-terminal extension are also present in BVP21 and the human mRNA capping enzyme41 and thus represent general features of RNA 5′-phosphatases. Second, two residues in PIR1 P-loop, H154 and N157, make close side chain contacts with the active site phosphate (postulated to exemplify RNA γ-phosphate), stabilizing this ion in the deep active site pocket (Figure 4A). The imidazole group of H154 located two positions C-terminal of the catalytic cysteine makes close contacts with the phosphate O3 atom, which is also contacted by N157. The nonaliphatic side chains of P-loop residues H154 and N157 are strictly conserved in other RNA 5′-phosphatases like BVP and the mRNA capping enzyme, and substitution with alanine disrupts phosphatase activity (Figure 6A,B).21,41,49 Thus, as suggested by Changela et al.,21 the P-loop signature sequence of RNA 5′-phosphatases is HCXHXXNR(S/T). Third, the PIR1 active site contains an active site Cl ion that stabilizes the position of the general base R158 and makes contact with P-loop T153, immediately next to the catalytic cysteine (Figure 4B). Interestingly, a phosphate ion occupies an equivalent position in BVP21 (Figure 3B,E), suggesting that the presence of a second active site anion may represent a conserved feature of RNA 5′-phosphatases.
Structural Basis for RNA γ- and β-Phosphatase Activity
Phospho-monoesterase reactions catalyzed by classical PTPs and most DSPs proceed via the formation of a covalent enzyme–phospho-substrate intermediate, where the initial Cys attack benefits from protonation of the bridging oxygen leaving group by a general acid (such as Asp). Instead, cleavage of phosphoanhydrides by RNA-specific cysteine phosphatases like BVP, the mRNA capping enzyme, and PIR1 does not require a general acid residue, possibly because of the low pKa of the leaving group.21,49 The docking study presented in this paper suggests a perfect complementarity between the PIR1 deep catalytic cleft and the triphosphate tail of RNA. The high degree of electrostatic complementarity between the PIR1 basic surface (Figure 5A,B) and the negatively charged RNA backbone likely promotes insertion of the RNA 5′-end inside the PIR1 active site. The docked conformation of ATP brings the γ-phosphate within 3 Å of the catalytic cysteine, in a conformation suitable for phosphate hydrolysis (Figure 5C). On the basis of this model, we speculate that the PIR1 catalytic pocket is not likely to undergo substrate-induced conformational changes to hydrolyze the γ-phosphate of RNA. Our docking model, however, does not explain how PIR1 can dephosphorylate the β-phosphate of RNA, which is ∼6 Å from C152, likely too far for the catalytic cysteine, postulated to exist as a thiolate in the ground state, to make a nucleophilic attack.42 Furthermore, analogous to BVP,13 β-phosphate hydrolysis by PIR1 proceeds efficiently in the absence of γ-phosphate, arguing against a model in which the Cys−γ-Pi intermediate functions as the nucleophile that attacks the β-phosphorus of an RNA substrate. It is possible that the PIR1 catalytic pocket undergoes a conformational change to hydrolyze the β-phosphate of RNA. Analogous to classical DSPs that are thought to shrink their active site pocket to accommodate phospho-substrates with different steric hindrance,46 a conformational change in PIR1 would facilitate β-phosphate hydrolysis. In this respect, we identified R192 as a putative residue in PIR1 that is important for diphosphatase activity: a conservative mutation of R192 to lysine (as found in the mRNA capping enzyme that lacks β-phosphatase activity41) reduced the level of ADP hydrolysis by ∼80%, while ATP γ-phosphate hydrolysis was affected by only 50% (Figure 6A).
In conclusion, we have described the structural organization of the human PIR1 catalytic core. This work is a step forward in the molecular characterization of atypical DSPs that dephosphorylate nonpeptidic substrates. Future studies will have to identify additional residues outside the PIR1 P-loop that take part in β-phosphate stabilization and facilitate its hydrolysis as well as address how the PIR1 noncatalytic C-terminal extension (residues 206–330) affects enzymatic activity,13 likely by regulating association with RNA and other important physiological binding partners.
Acknowledgments
We are thankful to Vivian Stojanoff and the scientific staff at NSLS beamlines X6A and X29 for assistance and help in data collection.
Glossary
Abbreviations
- PIR1
phosphatase that interacts with RNA–ribonucleoprotein complex 1
- DSP
dual-specificity phosphatase
- PTP
protein tyrosine phosphatase
- P-loop
phosphate-binding loop
- MW
molecular weight
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- DDM
n-dodecyl β-d-maltoside
- PMSF
phenylmethanesulfonyl fluoride
- GST
glutathione S-transferase
- MKP
mitogen-activated protein kinase phosphatases
- RNAi
RNA interference
- CD
circular dichroism
- OMFP
3-O-methylfluorescein phosphate
- NLS
nuclear localization signal
- appTm
apparent melting temperature
- rmsd
root-mean-square deviation.
Supporting Information Available
Four additional figures presenting supplementary analysis of PIR1-core stability, structure, and binding to chloride. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported in part by a Weizmann-Thomas Jefferson University Collaborative Pilot Program and National Institutes of Health Grant 1R01GM100888-01A1 (to G.C.). Research in this publication includes work conducted at the Kimmel Cancer Center X-ray Crystallography and Molecular Interaction Facility, which is supported in part by National Cancer Institute Cancer Center Support Grant P30 CA56036.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Alonso A.; Sasin J.; Bottini N.; Friedberg I.; Osterman A.; Godzik A.; Hunter T.; Dixon J.; Mustelin T. (2004) Protein tyrosine phosphatases in the human genome. Cell 117, 699–711. [DOI] [PubMed] [Google Scholar]
- Tonks N. K. (2006) Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846. [DOI] [PubMed] [Google Scholar]
- Guan K. L.; Broyles S. S.; Dixon J. E. (1991) A Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature 350, 359–362. [DOI] [PubMed] [Google Scholar]
- Patterson K. I.; Brummer T.; O’Brien P. M.; Daly R. J. (2009) Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 418, 475–489. [DOI] [PubMed] [Google Scholar]
- Tautz L.; Critton D. A.; Grotegut S. (2013) Protein tyrosine phosphatases: Structure, function, and implication in human disease. Methods Mol. Biol. 1053, 179–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayon Y.; Alonso A. (2010) Atypical DUSPs: 19 phosphatases in search of a role. Emerging Signaling Pathways in Tumor Biology 185–208. [Google Scholar]
- Agarwal R.; Burley S. K.; Swaminathan S. (2008) Structure of human dual specificity protein phosphatase 23, VHZ, enzyme-substrate/product complex. J. Biol. Chem. 283, 8946–8953. [DOI] [PubMed] [Google Scholar]
- Lountos G. T.; Tropea J. E.; Waugh D. S. (2011) Structure of human dual-specificity phosphatase 27 at 2.38 Å resolution. Acta Crystallogr. D67, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci V. S.; Turnbull J.; Wang W.; Girard J. M.; Zhao X.; Skurat A. V.; Delgado-Escueta A. V.; Minassian B. A.; Depaoli-Roach A. A.; Roach P. J. (2007) Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 19262–19266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J.; Engel J. L.; Zhang J.; Chen M. J.; Manning G.; Dixon J. E. (2011) Structural and functional analysis of PTPMT1, a phosphatase required for cardiolipin synthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 11860–11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Li D. M.; Sun H. (1998) PIR1, a novel phosphatase that exhibits high affinity to RNA·ribonucleoprotein complexes. J. Biol. Chem. 273, 20347–20353. [DOI] [PubMed] [Google Scholar]
- Takagi T.; Taylor G. S.; Kusakabe T.; Charbonneau H.; Buratowski S. (1998) A protein tyrosine phosphatase-like protein from baculovirus has RNA 5′-triphosphatase and diphosphatase activities. Proc. Natl. Acad. Sci. U.S.A. 95, 9808–9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande T.; Takagi T.; Hao L.; Buratowski S.; Charbonneau H. (1999) Human PIR1 of the protein-tyrosine phosphatase superfamily has RNA 5′-triphosphatase and diphosphatase activities. J. Biol. Chem. 274, 16590–16594. [DOI] [PubMed] [Google Scholar]
- Caprara G.; Zamponi R.; Melixetian M.; Helin K. (2009) Isolation and characterization of DUSP11, a novel p53 target gene. J. Cell. Mol. Med. 13, 2158–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardousis K.; Voolstra C.; Roengvoraphoj M.; Sekandarzad A.; Mesghenna S.; Winkler J.; Ko Y.; Hescheler J.; Sachinidis A. (2007) Identification of differentially expressed genes involved in the formation of multicellular tumor spheroids by HT-29 colon carcinoma cells. Mol. Ther. 15, 94–102. [DOI] [PubMed] [Google Scholar]
- Hasler R.; Kerick M.; Mah N.; Hultschig C.; Richter G.; Bretz F.; Sina C.; Lehrach H.; Nietfeld W.; Schreiber S.; Rosenstiel P. (2011) Alterations of pre-mRNA splicing in human inflammatory bowel disease. Eur. J. Cell Biol. 90, 603–611. [DOI] [PubMed] [Google Scholar]
- Duchaine T. F.; Wohlschlegel J. A.; Kennedy S.; Bei Y.; Conte D. Jr.; Pang K.; Brownell D. R.; Harding S.; Mitani S.; Ruvkun G.; Yates J. R. III; Mello C. C. (2006) Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell 124, 343–354. [DOI] [PubMed] [Google Scholar]
- Olia A. S.; Bhardwaj A.; Joss L.; Casjens S.; Cingolani G. (2007) Role of gene 10 protein in the hierarchical assembly of the bacteriophage P22 portal vertex structure. Biochemistry 46, 8776–8784. [DOI] [PubMed] [Google Scholar]
- Bhardwaj A.; Olia A. S.; Walker-Kopp N.; Cingolani G. (2007) Domain organization and polarity of tail needle GP26 in the portal vertex structure of bacteriophage P22. J. Mol. Biol. 371, 374–387. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Changela A.; Martins A.; Shuman S.; Mondragon A. (2005) Crystal structure of baculovirus RNA triphosphatase complexed with phosphate. J. Biol. Chem. 280, 17848–17856. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2004) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. A.; Steiner R. A.; Lebedev A. A.; Potterton L.; McNicholas S.; Long F.; Murshudov G. N. (2004) REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D60, 2184–2195. [DOI] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D50, 760–763. [DOI] [PubMed] [Google Scholar]
- Dolinsky T. J.; Nielsen J. E.; McCammon J. A.; Baker N. A. (2004) PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A. (2009) PDBsum new things. Nucleic Acids Res. 37, D355–D359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt G. J.; Jones T. A. (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D50, 178–185. [DOI] [PubMed] [Google Scholar]
- Olia A. S.; Casjens S.; Cingolani G. (2009) Structural plasticity of the phage P22 tail needle gp26 probed with xenon gas. Protein Sci. 18, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Wallace A. C.; Laskowski R. A.; Thornton J. M. (1995) LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. (2009) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries S. J.; van Dijk M.; Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897. [DOI] [PubMed] [Google Scholar]
- Worby C. A.; Gentry M. S.; Dixon J. E. (2006) Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 281, 30412–30418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koksal A. C.; Cingolani G. (2011) Dimerization of Vaccinia virus VH1 is essential for dephosphorylation of STAT1 at tyrosine 701. J. Biol. Chem. 286, 14373–14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott K.; Cingolani G. (2011) The importin β binding domain as a master regulator of nucleocytoplasmic transport. Biochim. Biophys. Acta 1813, 1578–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koksal A. C.; Nardozzi J. D.; Cingolani G. (2009) Dimeric quaternary structure of the prototypical dual specificity phosphatase VH1. J. Biol. Chem. 284, 10129–10137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokareddy R. K.; Bhardwaj A.; Cingolani G. (2013) Atomic structure of dual-specificity phosphatase 26, a novel p53 phosphatase. Biochemistry 52, 938–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L.; Sander C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Changela A.; Ho C. K.; Martins A.; Shuman S.; Mondragon A. (2001) Structure and mechanism of the RNA triphosphatase component of mammalian mRNA capping enzyme. EMBO J. 20, 2575–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M. D.; Denu J. M. (2001) Molecular reactions of protein phosphatases: Insights from structure and chemistry. Chem. Rev. 101, 2313–2340. [DOI] [PubMed] [Google Scholar]
- Ducruet A. P.; Vogt A.; Wipf P.; Lazo J. S. (2005) Dual specificity protein phosphatases: Therapeutic targets for cancer and Alzheimer’s disease. Annu. Rev. Pharmacol. Toxicol. 45, 725–750. [DOI] [PubMed] [Google Scholar]
- Lazo J. S.; Wipf P. (2009) Phosphatases as targets for cancer treatment. Curr. Opin. Invest. Drugs 10, 1297–1304. [PubMed] [Google Scholar]
- Di Cristofano A.; Kotsi P.; Peng Y. F.; Cordon-Cardo C.; Elkon K. B.; Pandolfi P. P. (1999) Impaired Fas response and autoimmunity in Pten+/– mice. Science 285, 2122–2125. [DOI] [PubMed] [Google Scholar]
- Yuvaniyama J.; Denu J. M.; Dixon J. E.; Saper M. A. (1996) Crystal structure of the dual specificity protein phosphatase VHR. Science 272, 1328–1331. [DOI] [PubMed] [Google Scholar]
- Jia Z.; Barford D.; Flint A. J.; Tonks N. K. (1995) Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 268, 1754–1758. [DOI] [PubMed] [Google Scholar]
- Nitschke F.; Wang P.; Schmieder P.; Girard J. M.; Awrey D. E.; Wang T.; Israelian J.; Zhao X.; Turnbull J.; Heydenreich M.; Kleinpeter E.; Steup M.; Minassian B. A. (2013) Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell Metab. 17, 756–767. [DOI] [PubMed] [Google Scholar]
- Martins A.; Shuman S. (2000) Mechanism of phosphoanhydride cleavage by baculovirus phosphatase. J. Biol. Chem. 275, 35070–35076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.