

Significance
Wilson disease (WD) is a disorder of copper overload whose variable presentation poses diagnostic and treatment challenges. WD is caused by mutations in ATP7B, a transporter that loads Cu(I) onto newly synthesized cupro-enzymes in the trans-Golgi network (TGN) and exports excess copper by trafficking from the TGN to the plasma membrane. This multidisciplinary study established that a patient mutation, ATP7B-S653Y, has Cu(I) transport activity in the TGN, but completely disrupts Cu(I)-responsive trafficking. ATP7B-S653Y perturbs long-range interdomain interactions mediated by transmembrane segments TM1/TM2, suggesting a new functional role for this region. ATP7B-S653Y is the best-characterized example of a new functional class of WD mutants. We suggest that functional classification of WD mutations may facilitate targeted therapy for patients.
Keywords: ceruloplasmin, interdomain interactions, molecular dynamics
Abstract
Wilson disease (WD) is a monogenic autosomal-recessive disorder of copper accumulation that leads to liver failure and/or neurological deficits. WD is caused by mutations in ATP7B, a transporter that loads Cu(I) onto newly synthesized cupro-enzymes in the trans-Golgi network (TGN) and exports excess copper out of cells by trafficking from the TGN to the plasma membrane. To date, most WD mutations have been shown to disrupt ATP7B activity and/or stability. Using a multidisciplinary approach, including clinical analysis of patients, cell-based assays, and computational studies, we characterized a patient mutation, ATP7BS653Y, which is stable, does not disrupt Cu(I) transport, yet renders the protein unable to exit the TGN. Bulky or charged substitutions at position 653 mimic the phenotype of the patient mutation. Molecular modeling and dynamic simulation suggest that the S653Y mutation induces local distortions within the transmembrane (TM) domain 1 and alter TM1 interaction with TM2. S653Y abolishes the trafficking-stimulating effects of a secondary mutation in the N-terminal apical targeting domain. This result indicates a role for TM1/TM2 in regulating conformations of cytosolic domains involved in ATP7B trafficking. Taken together, our experiments revealed an unexpected role for TM1/TM2 in copper-regulated trafficking of ATP7B and defined a unique class of WD mutants that are transport-competent but trafficking-defective. Understanding the precise consequences of WD-causing mutations will facilitate the development of advanced mutation-specific therapies.
Copper is essential for the normal development and function of human cells because it serves as a cofactor for many important metabolic enzymes. However, intracellular levels of copper must be tightly regulated (1, 2) because excess copper is toxic. Inborn mutations in the Cu(I)-ATPases, ATP7A [Online Mendelian Inheritance in Man (OMIM) accession no.*606882] or ATP7B (OMIM *300011) result in either systemic copper deficiency or copper accumulation in several tissues, causing Menkes disease or Wilson disease (WD), respectively. WD (OM#277900) is an autosomal-recessive disorder with a heterogeneous clinical presentation; in the absence of family history, diagnosis of WD requires multiple clinical and laboratory studies (3). The large number of rare mutations in the ATP7B gene [>500, (www.hgmd.cf.ac.uk/ac/gene.php?gene=ATP7B)] contribute to the difficulty in making genotype-phenotype associations. Presently, about two dozen mutations found in WD patients have been characterized in detail (4–7). These studies revealed that the most common effect of a WD mutation is ATP7B misfolding, which results in retention of newly synthesized ATP7B in the endoplasmic reticulum (ER), a marked decrease in protein stability, and loss of Cu(I)-transport activity (8, 9). Destabilization and inactivation of ATP7B explain the common phenotypic manifestations in WD, such as impaired copper export from the liver and the lack of copper incorporation into secreted cuproenzymes, such as ceruloplasmin (CPN).
ATP7B and the highly homologous ATP7A (Menkes disease protein) play central roles in maintaining copper levels in cells. These proteins belong to the evolutionarily conserved family of P1B-ATPases, which use the energy of ATP hydrolysis to transport copper from the cytosol across cellular membranes (Fig. 1A). ATP7A and ATP7B load copper onto newly synthesized cupro-proteins in the late Golgi and remove excess copper from the cytosol after relocating to vesicles, which in turn traffic to the plasma membrane to release copper into the extracellular milieu. In low and basal copper, ATP7A and ATP7B are located predominantly in a subcompartment of the trans-Golgi network (TGN) marked by syntaxin 6 (10). When copper levels increase, ATP7A and ATP7B exit the TGN in distinct vesicles; ATP7B vesicles move to the apical region in polarized epithelia, whereas ATP7A vesicles move to the basolateral region. Because copper-dependent ATP7B trafficking is a complex process, the precise sequence of events and the function of trafficking determinants in ATP7B’s structure are yet to be fully understood.
Fig. 1.

Hypothetical ATP7B model and multiple species alignment of the conserved regions, amino acids 621–668, in the two Cu-ATPases. (A) A hypothetical ATP7B ribbon model, generated by UCSF Chimera, showing the conserved core organization (20). The two large cytoplasmic loops in the core structure are: the A domain (actuator, green), between TM4 and TM5, which contains the phosphatase activity; and the N and P domains (nucleotide binding and phosphorylation, red) between TM6 and TM7, which bind ATP (N), catalyzing formation of a phosphorylated intermediate (P) as part of the catalytic cycle. The eight TMs (yellow are): TM1 (amino acids 645–670), TM2 (including the platform helix, amino acids 694–722), TM3 (amino acids 729–749), TM4 (amino acids 765–786), TM5 (amino acids 916–942), TM6 (amino acids 967–1004), TM7 (amino acids 1307–1345), and TM8 (amino acids 1352–1373). The six N-terminal MBDs (blue, N-MBDs, also referred to as the N-terminal domain of ATP7B, N-ATP7B) (61, 62) were manually positioned onto the published model. The box approximates the region of the multiple species alignment shown in B. (B) A multiple species alignment of human ATP7B sequence 621–668 (Upper) and ATP7A sequence 621–668 (Lower). WD patient mutations are underlined and in bold. The ATP7B S653 position is marked with an asterisk (bold). Portions of MBD6 and TMD 1 are bracketed. In ATP7A, the bracketed sequence shows the deleted region that is replaced with two amino acids (IR) in a patient with Occipital Horn Syndrome. The deleted sequence of ATP7A is underlined and in bold (35). Alignments were obtained using ClustalW (63). Amino acids that are identical (*), conserved (:), and semiconserved (.) are shown.
We previously developed a comprehensive set of cell-based assays that use both polarized hepatic cells and fibroblasts lacking ATP7B and ATP7A (derived from a Menkes disease patient) to evaluate the activity, stability, and trafficking of ATP7B and its mutants (11, 12). In this study, we combined these assays with additional mutational analysis and computational studies to dissect the molecular phenotype of WD mutations found in a highly conserved region of ATP7B, G621-S668. We demonstrate that the S653Y mutation has a distinct “transport-competent/trafficking-defective” phenotype. We also show that the transmembrane segment (TM) that harbors S653 has an important and previously unanticipated role in regulating exit of ATP7B from the TGN in response to copper elevation.
Results
WD Patients with the H1069Q/S653Y Genotype Have Variable Levels of Serum CPN.
Because of the well-documented phenotypic heterogenity of WD, multiple clinical and laboratory studies are used to provide an accurate diagnosis (13–15). A confounding observation has been the presence of normal serum CPN levels in 5–15% of patients (16, 17). We identified a group of WD patients from two unrelated families, in which one family had reduced serum CPN and a patient from the other family had normal serum CPN (Table 1). All patients had p.H1069Q in one ATP7B allele and an uncharacterized missense mutation, c.1958C > A [p.S653Y] in the second allele. Previous studies have demonstrated that the H1069Q mutation results in significant mislocalization, instability (8, 18), and low residual activity of ATP7B protein (9), which may account for some active copper-loaded CPN (holo-CP). It is also known that heterozygous individuals with one wild-type and one p.H1069Q allele do not have WD (Table 1) (19), indicating that in patients with the p.H1069Q/S653Y genotype, ATP7BS653Y is not functionally competent to prevent WD. Because ATP7BS653Y has not been previously validated in either biochemical or cellular assays, we focused our studies on this variant.
Table 1.
WD patient clinical data summary
| Patient –WD mutation first allele | WD mutation second allele | Sex (#) | Age of WD onset | WD type (#) | CPN mg/dL* (normal: 25–45) | Cu serum μg/dL* (normal: 70–140) | Cu urine μg/24 h* (normal: <50) | KF ring (%) |
| Normal | None | F(50) | — | — | 35 ± 10.3† | 96.7 ± 24.7† | ND | ND |
| M(50) | ||||||||
| Heterozygous‡-H1069Q | None | 31.8 ± 6.7† | 100.4 ± 24† | 18.5 | — | |||
| Homozygous§-H1069Q (median IQR) | H1069Q | F(95) | 27.9 ± 9.9† | N(89) | 13.9 [9.2] | 61.2 [34] | 163 [255] | + (28%) |
| M(80) | H(57) | |||||||
| pre(25)¶ | ||||||||
| KM‖-S653Y | H1069Q | F | ND | pre | 9.2 | 35 | 135 | — |
| KG‖-S653Y | H1069Q | M | 22 | H | 9.8 | 35 | 110 | — |
| KJ‖-S653Y | H1069Q | M | 21 | N | 3.5 | 27 | 1050 | + |
| BP‖-S653Y | H1069Q | M | 29 | H | 46.4 | 131 | 162 | + |
| BP**-S653Y | H1069Q | M | 32 | H | 14 | 250 | 3335 | + |
KF, Keyser Fleisher ring; H, hepatic; pre, presymptomatic; SD standard deviation; N, neurological; ND, not determined; IQR, interquartile range. Mean ± SD or median (IQR) are given when individual variables had either normal or abnormal distributions, respectively, in the analyzed population.
Normal laboratory values are given.
Mean ± SD.
Clinical data for these individuals was reported in ref. 19.
Data were compiled from the Wilson Disease Database of the Institute of Psychiatry and Neurology, Second Department of Neurology, Warsaw, Poland.
Only 171 of the 175 patients with a WD diagnosis had sufficient data to designate the WD type.
Mutation analysis was reported in ref. 64.
Clinical data before liver transplant was reported in ref. 65.
ATP7BS653Y Is Located Within a Conserved Region (G621-S668) Following Metal Binding Domain 6.
Although structural information for individual metal binding domains (MBDs) is available, the precise nature of their linkage to the core ATP7B sequence is unknown (20). Therefore, it was particularly interesting that ATP7BS653Y is located within a region (G621-S668) following MBD6 and encompassing TM1 that is highly conserved in vertebrates (Fig. 1B). A multiple-species sequence alignment illustrates that the regions linking individual MBDs to one another are more divergent than the region linking MBD6 to TM1 (Fig. S1), suggesting it is functionally important. Analysis of the WD database (www.wilsondisease.med.ualberta.ca/database.asp) and the clinical literature revealed that G621-S668 is a hot spot for WD missense mutations (Fig. 1B and Table S1). In addition to p.S653Y, mutations p.G626A and p.M645R were reported with p.H1069Q as the second allele, and one patient was homozygous for p.D642H (Table S1). The biochemical properties of p.G626A and p.M645R mutants were previously studied in vitro, where both variants showed diminished but significant copper transport activity (21). We reasoned that mutations in this region might disrupt Cu-specific functions of ATP7B characteristic of vertebrates, such as Cu-dependent trafficking. Because the intracellular characteristics of these mutants have not been explored, we studied their properties in detail.
All Six Patient Mutations Exhibit Copper Transport Activity.
The following patient mutations (G626A, H639Y, L641S, D642H, M645R, S653Y) (Table S2) were introduced into mGFP-ATP7B, and their ability to transport copper into the secretory pathway was evaluated using a cell-based assay that monitors activation of the copper-dependent enzyme, tyrosinase (10). The YST fibroblasts used for these measurements lack endogenous copper-transport activity because of an inactivating mutation in ATP7A. Transfection of YST cells with a plasmid encoding apo-tyrosinase yields inactive tyrosinase, as judged by the lack of any intracellular black reaction product (Fig. 2A). Cells cotransfected with plasmids encoding tyrosinase and the wild-type GFP-ATP7B showed copper-dependent activation of tyrosinase (Fig. 2B and Table 2), as expected. The GFP-ATP7BS653Y variant was also active in this assay (Fig. 2C and Table 2). As a second control, we used the GFP-ATP7B858TGE860>AAA mutant that lacked copper transport activity in the Saccharomyces cerevisiae ccc2Δ strain (22) and was present in vesicles independent of cellular copper levels in mammalian cells (22). This mutant did not activate apo-tyrosinase (Fig. 2D), confirming the specificity of our assay. Transfection of plasmids encoding other patient mutations resulted in tyrosinase activation in all cases (Table 2). Thus, patient mutations within the G621-S668 segment do not cause gross misfolding or mistargeting of ATP7B and allow for detectable copper transport activity in our assay system.
Fig. 2.
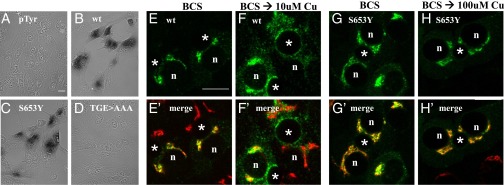
wtATP7B and ATP7BS653Y have similar Cu(I) transport activity yet show different trafficking behaviors in polarized hepatic cells. (C) ATP7BS653Y exhibits Cu(I) transport activity. YST cells were cotransfected with pTyrosinase (pTyr) and the indicated plasmid as described in Methods. The black reaction product reflects copper-dependent tyrosinase activity. Negative controls (A and D) and wtATP7B (B) were always included. (Magnification: A–D, 40×.) WIF-B cells were infected with wtATP7B and ATP7BS653Y adenovirus, cultured overnight in 10 μM BCS, then kept in the chelator (E, E′, G, and G′), or incubated in 10 μM CuCl2 (F and F′) or 100 μM CuCl2 (H and H′) for 1 h, fixed, stained with antibodies to GFP (green in E, F, G, and H) and TGN38 (red in E′, F′, G′, and H′), and imaged by confocal microscopy. Single optical sections are shown. Exogenous wtATP7B redistributes from the TGN (E and E′) to vesicles and the apical membrane when copper levels are elevated (F and F′). In contrast, ATP7BS653Y remains in the TGN in the absence (G and G′) or presence of copper (H and H′). n, nucleus; *, apical space. (Scale bars, 10 μm.)
Table 2.
Summary of protein phenotypes
| Assay construct | Tyrosinase activation* | Protein expression† | Anterograde trafficking‡ | Retrograde trafficking‡ | ||
| +Chelator | +Cu | +Cu | +Chelator | |||
| Wild-type 7B | + | + | TGN§ | TGN/ves/ap§ | TGN/ves/ap§ | TGN§ |
| 7B Patient missense substitutions | ||||||
| G626A | + | + | Wild-type | Wild-type | Wild-type | Wild-type |
| H639Y | + | + | Wild-type | Wild-type | Wild-type | Wild-type |
| L641S | + | + | Wild-type | Wild-type | Wild-type | Wild-type |
| D642H | + | + | Wild-type | Wild-type | Wild-type | Wild-type |
| M645R | + | + | Wild-type | Wild-type | Wild-type | Wild-type |
| S653Y | + | + | Wild-type | TGN | TGN | ND |
| Engineered mutations | ||||||
| S653A | + | + | Wild-type | Wild-type | Wild-type | Wild-type |
| S653F | + | + | Wild-type | TGN | TGN | ND |
| S653T | + | + | Wild-type | Wild-type | Wild-type | Wild-type |
| S653C | + | + | Wild-type | Wild-type | Wild-type | Wild-type |
| S653D | + | + | Wild-type | TGN | TGN | ND |
| S653E | + | + | Wild-type | TGN | TGN | ND |
Performed in YST cells (SI Methods).
Performed in YS cells (SI Methods).
Performed in WIF-B cells (SI Methods).
This subcellular localization is designated as wild-type trafficking for each treatment.
The ATP7BS653Y Mutation Inhibits Exit of ATP7B from the TGN in Polarized Hepatic Cells.
Because ATP7B is predominantly expressed in liver (23) and exports copper in a polarized fashion (24, 25) (i.e., via the apical membrane), we used the polarized hepatic WIF-B cells to evaluate copper-dependent trafficking of ATP7B variants. We have previously shown that these cells express endogenous ATP7B, which responds to changes in cellular copper levels (10). Because WIF-B cells are recalcitrant to transfection, recombinant adenoviruses were generated for all mutant proteins, which were tagged with GFP at the N terminus, and conditions were optimized to evaluate their expression and trafficking in these cells. Each of the viruses produced a GFP-protein of expected size (Fig. S2A) and, in the copper chelator bathocuproinedisulfonic acid (BCS), the wild-type ATP7B and all mutants were targeted to the TGN, as indicated by their colocalization with a TGN marker (Fig. 2 E and E′ and Table 2). (Note: GFP-tagged ATP7B constructs will be called ATP7B hereafter.) Thus, consistent with the functional data in YST cells, the mutations do not negatively affect protein structure, allowing them to pass the ER’s quality control and move to the TGN.
We next evaluated the ability of mutants to undergo copper-responsive trafficking in polarized WIF-B cells. In the presence of 10 μM CuCl2, ATP7B was found in vesicles and at the apical surface (Fig. 2 F and F′ and Table 2). Quantification revealed that ATP7B trafficked to the apical surface in nearly 90% of expressing cells (anterograde trafficking) (Fig. 3C). Five of the six mutants (G626A, H639Y, L641S, D642H, M645R) exhibited trafficking behavior that was quantitatively and qualitatively similar to the copper-responsive trafficking of wild-type ATP7B (Fig. 3 and Table 2). It is important to note that these five mutations are predicted to be located in the region preceding TM1 of ATP7B (Fig. 1A). However, ATP7BS653Y, with a substitution of serine predicted to be in TM1, showed defective trafficking. The S653Y mutant failed to exit the TGN even when cells were incubated in 50- to 100-μM copper (i.e., concentrations that are ∼5- to 10-fold higher than physiologic) (Fig. 2 G and G′ and Table 2). The presence of ATP7BS653Y in the TGN did not alter the half-life of the protein (Fig. S2B).
Fig. 3.

Qualitative and quantitative evidence that five WD patient mutations show normal trafficking but ATP7BS653Y does not. (A and B) WIF-B cells infected with adenoviruses encoding wtATP7B and each of the patient mutations were processed for anterograde and retrograde trafficking as described in Methods. (A and A′) In the presence of copper, GFP-ATP7BG626A fluorescence is seen at the apical membrane and in vesicles; it does not overlap with the TGN marker (red). (B and B′) After copper chelation, the protein is no longer seen at the apical membrane and overlaps strongly with the TGN marker. (C) The numbers of polar cells with ATP7B protein fluorescence at the apical surface in the presence and after copper chelation were determined and expressed as a percentage of the total polarized cells expressing the ATP7B protein. Five of the mutant ATP7B showed wild-type trafficking in copper and after copper chelation. However, the ATP7BS653Y mutant was not found at the apical surface under any copper condition; it was not evaluated using the retrograde assay. The numbers of polar cells evaluated for anterograde/retrograde trafficking, respectively, were: G626A: 310/152; H639Y: 193/172; L641: 202/118; D642H: 242/168; M645R: 228/123; and S653Y: 228. (Scale bar, 10 μm.)
To evaluate the return (retrograde) trafficking of the five mutants whose exit from TGN was normal, a duplicate set of CuCl2-treated hepatic cells were rinsed and transferred to medium containing the copper chelator ammonium tetrathiomolybdate (TTM) for 2 h. Cells were fixed and the number of expressing cells with ATP7B remaining at the apical surface was determined. All five of the patient mutants behaved similarly to wild-type ATP7B and returned to the TGN after copper depletion (Fig. 3C and Fig. S2C). These five mutations were not deleterious to either retrograde or anterograde trafficking of ATP7B. Although we concluded that three mutants (p.G626A, p.D642H, and p.M645R) were disease-causing (Table S1), their biochemical defect remains unexplained. Consequently, we focused on the S653Y substitution that had activity but was unable to exit from the TGN, an ATP7B phenotype that is uncharacterized.
Bulky or Charged Amino Acid Substitutions of S653 Inhibit TGN Exit.
The phenotype of ATP7BS653Y was especially interesting, not only because it was unusual for WD, but also because it offered an opportunity to better understand factors influencing ATP7B exit from TGN in response to copper elevation. Because ATP7BS653Y had transport activity (Fig. 2C) but was unable to traffic in response to copper, we hypothesized that TM1 harboring S653 played a specific and important role in regulated trafficking of ATP7B. To better understand what this role could be, we examined structural characteristics of this region using the recently generated homology model of ATP7B (20). Analysis of the ATP7B model (Fig. 4A) and calculations of a solvent accessible surface area (SASA) (Table 3, fourth column) showed that S653 is located within TM1 and is buried, as indicated by a SASA exposure of 9%. Thus, the direct involvement of Ser653 residue in protein–protein or protein–lipid interaction is unlikely. We then hypothesized that tight packing of the TM1-TM2 region may be necessary for ATP7B to adopt a conformational state necessary for trafficking.
Fig. 4.

The ATP7B structural model shows that Ser653 in TM1 interacts with different TM2 residues than does the patient mutation, Tyr653. (A) A rotated view of the ATP7B ribbon model shows the full atom sphere of S653 (magenta) and the platform helix (yellow). (B and C) Expanded views show interatomic distances among selected residues nearby Ser653 (B) and Tyr653 (C). (B) In Ser653, there is a 3 Å distance between the hydroxyl group γ-oxygen (red) of Ser653 (magenta) and the carbonyl oxygen (red) of Gly710 (magenta) just before the platform helix (yellow). Tyr713 (magenta) is part of the platform helix (yellow). (C) However, Tyr653 (blue) is rotated away from Gly710 (magenta) and shows a large interatomic distance of 6.8 Å with it. Instead, Tyr653 interacts with Tyr713 (magenta) as revealed by an interatomic distance of 3.7 Å between their functional groups (red). All models were generated using PyMOL (The PyMOL Molecular Graphics System, v1.5.0.4; Schrödinger).
Table 3.
Computed structural properties
| Residue | Interatomic distance* Å from 653 residue to Gly-710 | SASA of fully exposed residue† Å2 | Static structure SASA† Å2 (% exposed) | Molecular synamic SASA averages‡ Å2 (% exposed) | ||
| 653 residue | Tyr713 | 653 residue | Tyr713 | |||
| Ser (wild-type) γ oxygen | 3.0 | 80.0 | 7.2 (9%) | 78.40 (42%) | 40.38 (50%) | 88.53 (47%) |
| Ala | N/A | 67 | 5.40 (8%) | 77.50 (41%) | 40.21 (60%) | 96.58 (51%) |
| Cys γ sulfur | 2.7 | 104 | 6.10 (6%) | 77.70 (41%) | 61.79 (60%) | 99.26 (53%) |
| Thr γ1 oxygen | 4.2 | 102 | 8.20 (8%) | 76.30 (41%) | 25.92 (25%) | 87.66 (47%) |
| Tyr hydroxyl | 6.8 | 187 | 47.3 (25%) | 67.3 (36%) | 125.25 (70%) | 136.81 (73%) |
| Phe | N/A | 175 | 34.10 (19%) | 70.40 (38%) | 102.85 (59%) | 65.13 (35%) |
| Glu ε1 oxygen/ε2 Oxygen | 2.9/4.1 | 138 | 14.10 (10%) | 71.60 (38%) | 11.21 (8%) | 71.12 (38%) |
| Asp δ1 oxygen/δ2 Oxygen | 5.2/3.9 | 106 | 13.70 (13%) | 75.40 (40%) | 38.96 (37%) | 72.66 (38%) |
NA, not applicable because of lack of a functional group.
The interatomic distance between the functional group of the 653 residue and Gly-710 carbonyl oxygen were calculated using the PyMOL program.
SASA within the structural model of wtATP7B and SWISS-MODEL generated models containing the substitutions were calculated using the AREAIMOL program. The SASA or cavity surface area is defined as the surface traced by the center of a sphere with the radius of a water molecule (0.15 mm) as it is rolled over the surface of a molecular model of the solution (66). The SASA for each amino acid side chain is given when the Gly-X-Gly is an extended conformation (67). These values are taken to represent the maximum SASA and are provided for comparison and as a reference point to the SASA reported for the static structure.
Average SASAs were calculated using the molecular dynamics calculations, as described in Methods.
We engineered six ATP7B mutants, where we varied the size, hydophobicity, and charge of the amino acid at position 653 (Table S2). The Ser-to-Tyr substitution introduces an amino acid that has a volume ∼twice that of serine [193.6 Å3 vs. 89 Å3, respectively (26)]. To test whether the size of the side-chain at position 653 in ATP7B is critical for the transporter’s ability to exit the TGN, we generated recombinant ATP7B with Phe or Ala substitutions [residue volumes of 189.9 Å3 and 88.6 Å3, respectively (26)]. Both mutants migrated with the correct mobility on SDS/PAGE (Fig. S3A) and were catalytically active (Table 2 and Fig. S3B). In the trafficking assay, ATP7B with the large substitution (Phe) remained in the TGN when copper was elevated to 10–100 μM (Fig. 5A, Left, and Table 2), whereas ATP7B with a small substitution (Ala) behaved like wtATP7B (Fig. 5 B and C and Table 2). To determine the volume limitations, we produced Thr653 and Cys653 substitutions with volumes of 116.1 Å3 and 108.5 Å3, respectively. Both ATP7BS653T and ATP7BS653C behaved like wtATP7B in all respects (Fig. 5C and Table 2).
Fig. 5.
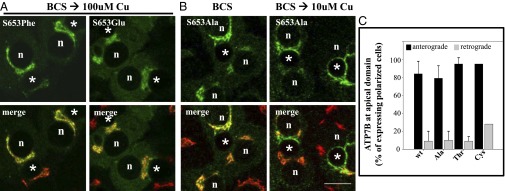
ATP7BS653F and ATP7BS653E mutant proteins are retained in the TGN in 100-μM copper, whereas ATP7BS653A mutant protein exhibits wild-type trafficking. WIF-B cells were infected with the indicated ATP7B adenovirus, cultured, and processed as described in Methods. Single confocal sections are shown. (A) Substitution of S653 with either phenylalanine (F, Phe) or glutamate (E, Glu) blocked TGN-exit of ATP7B in 100 μM copper. A cytoplasmic green haze found in ATP7BS653E-expressing cells may reflect its degradation (Fig. S3A). (B) In contrast, ATP7BS653A redistributed from the TGN (B, Left) to vesicles and the apical membrane when copper levels were elevated (B, Right). n, nucleus; *, apical space. (Scale bar,10 μm.) (C) Quantitative evidence that ATP7BS653A, ATP7BS653T and ATP7BS653C substitutions show wild-type trafficking in polar hepatic cells. WIF-B cells infected with adenoviruses encoding wtATP7B and three 653 substitutions were cultured, processed, and quantitatively analyzed for trafficking as described in Fig. 3. Like wtATP7B, ATP7BS653A, ATP7BS653T, and ATP7BS653C variants exited the TGN (anterograde) in 10-μM copper and returned (retrograde) when copper levels were lowered. The number of polar cells evaluated for anterograde/retrograde trafficking, respectively, were Ala: 108/115; Thr: 98/142; and Cys: 158/77.
We next explored the effect of charged residues in position 653 of ATP7B. Glu and Asp have volumes similar to those of Cys and Ser, respectively (26), but they are negatively charged. Introduction of these residues does not significantly change the predicted surface exposure of residue 653 (Table 3, fourth column), nor did it disrupt copper transport, as both ATP7BS653E and ATP7BS653D activated tyrosinase (Table 2 and Fig. S3B). However, neither the ATP7BS653E (Fig. 5A, Right, and Table 2) nor the ATP7BS653D mutant was able to exit the TGN, even when cells were treated with 100 μM CuCl2 (Table 2). Thus, both volume and side-chain functionality at the 653 position are critical for copper-dependent TGN-exit of ATP7B.
TM1 Orientation and Charge Are Critical for ATP7B Exit from TGN.
Inspection of the ATP7B structure using PyMOL revealed that the hydroxyl group γ-oxygen of Ser653 in TM1 and the carbonyl oxygen of Gly710 in TM2 show an interatomic hydrogen bond distance of 3.0 Å, which is “moderate to mostly electrostatic” with an energy of ∼5–6 kcal/mol and is consistent with proton sharing (Fig. 4B and Table 3, second column) (27, 28). To examine this region further, we predicted the local structural effects for each of the Ser653 substitutions using the SWISS-MODEL structural bioinformatics server. Analyses of the Ala, Cys, and Thr substitutions revealed only minor structural perturbations of the static structure, which is consistent with the normal behavior of the corresponding ATP7B mutants (Table 3, fourth column). In contrast, the ATP7BS653Y model revealed that the side chain of Tyr653 was rotated away from the protein core, resulting in an increased SASA (Table 3, fourth column, exposure 25%). Additionally, the interatomic distance between Tyr653 hydroxyl and Gly710 was 6.8 Å (Fig. 4C and Table 3, second column), ruling out proton sharing. We also observed that the exposure of Tyr713 in TM2 was decreased in ATP7BS653Y relative to wt (36% vs. 42%, respectively) (Table 3, fifth column). This change could be because of the increased volume of the 653 tyrosyl moiety in the region or because of stacking interactions of the π electrons from the two conjugated rings. A Phe653 substitution, which has an aromatic ring and comparable bulk, had a similar effect on Tyr713, albeit to a lesser extent than the ATP7BS653Y substitution (Fig. 4 and Table 3, fifth column).
We next examined the electrostatic potential of the wtATP7B static structure using adaptive Poisson-Boltzmann surface analysis (APBS) and found a pocket adjacent to TM1 Ser653, TM2 Gly710 and the platform helix of TM2 (Fig. S4 A and B). A combination of both neutral and positive charges was observed in this region (shown as blue and white surfaces, Fig. S4B). Interestingly, in ATP7BS653Y, the pocket was less accessible and the surface charges more neutral (Fig. S4C). The charged 653 substitutions, Glu653 and Asp653, also showed intriguing surface charge changes (Fig. S4 D and E). The Asp653 substitution had a strong influence upon the region around the pocket, reflected by an increase in negatively charged surfaces (red in Fig. S4D). The Glu653 substitution lacked a strong negatively charged surface, indicating that the residue is deeply buried (10% exposure) (Fig. S4E).
To evaluate the dynamic behavior of the mutants, we subjected the generated models to molecular dynamic simulations using the generalized-born simple-switching approach (Fig. 6). In this approach, the transmembrane portion of the model was inserted into a dielectric slab mimicking the membrane bilayer, a reasonable substitution that significantly reduces computation time. The simulation studies confirmed our initial findings and provided additional insight into the surface area changes in a dynamic context. Ser653, Ala653, and Cys653 behaved in a similar manner and were more exposed than Thr653 (Fig. 6A and Table 3, sixth column) which is consistent with their normal trafficking behavior. However, relative to Ser653, the Glu653 charged substitution was deeply buried and Asp653 was less so (8% and 37% exposed, respectively) (Fig. 6A and Table 3, sixth column). These observations are consistent with the APBS of the static model (Fig. S4) and suggest that charge has a distinct effect on ATP7B structure that impedes TGN exit (Table 2). In contrast, the Tyr653 substitution that also failed to exit the TGN was more exposed as revealed by the increased SASA (Fig. 6A and Table 3, sixth column), and this had a substantial effect on Tyr713, the exposure of which was also increased relative to that determined for the other mutations (Fig. 6B and Table 3, seventh column). The Phe653 substitution showed a phenotype intermediate between Ser653 and Tyr653 (Table 3, sixth column). These results imply that a dominant steric hindrance, which is most likely because of repulsion between the two bulky groups, Tyr653 and Tyr713, introduces a persistent change in the TM1/TM2 segment of the mutant protein. Taken together, the calculations revealed that increased bulk or a negative charge at the 653 position had distinct effects upon ATP7B structure, both of which prevented ATP7B exit from the TGN.
Fig. 6.

Molecular dynamic simulation of Ser653 mutant proteins reveals fluctuations in the SASA. (A) Box plots show fluctuations of SASAs at the 653 position in wild-type and mutant (Ala, Cys, Thr, Tyr, Phe, Glu, and Asp) substituted models. Tyr and Phe mutants are more exposed and Glu is buried within the structure. (B) Box plots showing fluctuations of SASAs at the 713 tyrosine in wild-type and mutant (Ala, Cys, Thr, Tyr, Phe, Glu, and Asp) substituted models. Solid line represents median; dashed line with dot represents average; upper dot represents value at 95%; lower dot represents value at 5%; error bars are shown. The 95th and 5th percentile datapoints represent the sampling extremes.
Second-Site Mutational Analysis Reveals That the S653Y Mutation Alters ATP7B Structure.
How could small changes in the properties of TM1 result in such a significant effect on trafficking of ATP7B? Previous biochemical studies with recombinant peptides showed that ATP7B’s N-terminal cytosolic domain independently interacted with two other cytosolic domains, the nucleotide binding (N) domain in low Cu conditions (29) and the actuator (A) domain in elevated Cu (30) (Fig. 1A). Because TM1 links the cytosolic, regulatory N-terminal domain of ATP7B to the protein core, we hypothesized that the intramembrane substitution at the 653 position of TM1 might have a long-range effect on the cytosolic interdomain associations, and that in addition to the regulatory and catalytic roles, the cytosolic domains may be required for copper-dependent protein trafficking. To test this hypothesis, we introduced the S653Y mutation into two ATP7B mutants that have been previously shown to constitutively leave the TGN irrespective of copper levels (11, 31).
ATP7B858TGE860>AAA is locked in an acyl-phosphorylated state as a consequence of inactivation of the intrinsic phosphatase in the cytosolic A domain of ATP7B (31). Because this mutant protein is found out of the TGN and in vesicles irrespective of copper levels, it is thought to represent a structurally favorable state for TGN-exit (22, 31). To test whether the S653Y mutation in TM1 would be dominant or permissive to the structural changes induced by the TGE>AAA substitution, which is ∼50 Å away, we made a double mutant ATP7B858TGE860>AAA/S653Y. Recombinant adenoviruses of ATP7B858TGE860>AAA and ATP7B858TGE860>AAA/S653Y expressed full-length proteins, but neither mutant protein exhibited activity in our tyrosinase activation assay (which requires multiple rounds of copper transport per ATP7B molecule to yield detectable amounts of active tyrosinase) (Fig. S3 C and D). Studies in WIF-B cells revealed that the single mutant (ATP7B858TGE860>AAA) was present in vesicles and at the apical surface when copper was either chelated or elevated (Fig. 7 A, A′, B, and B′). In contrast, the double-mutant (ATP7B858TGE860>AAA/S653Y) was found in the ER when copper was chelated, as well as at the apical surface (Fig. 7 C and C′). Vesicles were also present in some cells. After addition of copper, this mutant was found predominantly in vesicles and the apical surface, a phenotype that resembled the single mutant, ATP7B858TGE860>AAA (Fig. 7 D and D′). Interestingly, the presence of copper appears to promote ATP7BS653Y/858TGE860>AAA exit from the ER, as has been reported for the ATP7BG875R variant (30). We conclude that the S653Y was not dominant in the context of the TGE>AAA mutation.
Fig. 7.

ATP7BS653Y exhibits a dominant effect upon the trafficking of ATP7BY44A but not ATP7BTGE>AAA. WIF-B cells were infected with ATP7BTGE>AAA (A, A′, B, and B′), ATP7BS653Y/TGE>AAA (C, C′, D, and D′), ATP7BY44A (E, E′, F, and F′), and ATP7BS653Y/Y44A (G, G′, H, and H′) adenoviruses, cultured overnight in 10 μM BCS, then either maintained in BCS (A, A′, C, C′, E, E′, G, and G′) or incubated in 10 μM CuCl2 (B, B′, D, D′, F, F′, H, and H′) for 1 h before being fixed and processed as described in Fig. 2. (A and B) ATP7BTGE>AAA trafficked to vesicles and the apical membrane in the absence (A and A′) or presence of copper (B, B′). Quantification showed that in 85% (n = 118) and 91% (n = 107) of polar cells expressing ATP7BTGE>AAA irrespective of copper’s presence or absence, the protein was in vesicles and the apical surface. (C and D) The double-mutant, ATP7BS653Y/TGE>AAA, was also found in vesicles and the apical surface in the absence (C and C′) or presence of copper (D and D′). In the absence but not the presence of copper (C and C′), ATP7BS653Y/TGE>AAA was also found in the ER in 55% of expressing, polar cells (n = 62). (E and F) ATP7BY44A trafficked to the basolateral PM (76% of cells n = 59) in the absence (E and E′) or presence of copper (F and F′). (G and H) Introduction of S653Y in ATP7BY44A changed the resulting double-mutant protein’s behavior. ATP7BS653Y/Y44A was found predominantly in the TGN in the absence of copper (G and G′) and in its presence (H and H′). Only 22% (n = 60) of polar cells expressing ATP7BS653Y/Y44A showed basolateral PM fluorescence. n, nucleus; *, apical space. (Scale bar, 10 μm.)
Finally, we hypothesized that the Tyr653 substitution might have a long-range effect on the large N-terminal domain of ATP7B. We previously demonstrated that Y44A, which is located in an essential apical targeting signal, F(37)AFDNVGYE(45) within the N-terminal domain of ATP7B, disrupts TGN retention and constitutively targets ATP7BY44A to the wrong (basolateral) membrane domain independent of copper levels (11). Therefore, we introduced a secondary mutation, Y44A, generating the double-mutant, ATP7BY44A/S653Y. Expression of the double-mutant ATP7BY44A/S653Y in fibroblasts revealed it to be catalytically active and of full-length (Fig. S3 C and D). However, unlike the ATP7BY44A single mutation (Fig. 7 E, E′, F, and F′), the ATP7BY44A/S653Y double-mutant failed to exit the TGN when copper was chelated or elevated (Fig. 7 G, G′, H, and H′). We confirmed this difference by quantitating the number of cells exhibiting basolateral labeling in the presence of copper (Fig. 7). Such a dominant effect of S653Y upon constitutive exit from the TGN indicates that small structural changes within TM1 can have a marked long-range effect on the cytosolic N-terminal domain of ATP7B, making the apical targeting signal unavailable.
Discussion
Our comprehensive analysis of a WD mutation, ATP7BS653Y, located within a conserved sequence, G621-S668, of ATP7B demonstrates very clearly that Cu(I) transport activity into the TGN lumen can occur in WD patients in the absence of the protein’s trafficking, which is required for Cu(I) export. CPN in serum is commonly used as a diagnostic test of WD because it reflects ATP7B’s metallating function, which occurs within the Golgi apparatus of hepatocytes. However, 5–15% of WD patients have normal serum CPN (16, 32, 33). We speculate that the transport-active/trafficking-defective phenotype of the ATP7BS653Y mutants accounts for some WD cases where serum CPN is normal. Further study of the relationship between ATP7B and serum CPN is required. Clinical assays are confounded by several factors, including hyperestrogenemia, the inflammatory state of the liver, or stage of liver disease; most assays do not differentiate the holo- and apo- forms of the protein.
Our mechanistic studies revealed that bulky or charged substitutions at position 653 mimicked the phenotype of the patient mutation, whereas small neutral replacements did not. Molecular modeling and molecular dynamic simulation suggest that bulky substitutions induce local distortions within TM1 and alter TM1 interaction with TM2. Introduction of second-site mutations confirmed that TM1 regulates long-range interdomain interactions required for TGN exit. Taken together, our experiments reveal two new aspects of WD and ATP7B: (i) a molecular mechanism underlying a class of WD patients who might benefit from previously unidentified approaches to treatment; and (ii) an unexpected role for TM1/TM2 in copper-regulated trafficking of ATP7B. Many questions remain. For example, are there other patient mutations that behave similarly to ATP7BS653Y? Is there a hierarchy of conformations or signals in ATP7B that dictates its localizations separate from the conformations taking place during its reaction cycle? Finally, will better genotype/phenotype correlations lead to improved approaches to WD patient management?
Does ATP7BS653Y Typify a Transport Competent/Trafficking Defective Class of WD Mutation?
Review of WD patient mutations revealed two more common patient mutations, ATP7BG710S in TM2 and ATP7BG943S in TM5, which may behave similarly to ATP7BS653Y. Both mutants were reported to localize to the Golgi in basal medium and exhibit some Cu(I) transport activity (5, 18, 21). Trafficking studies in fibroblasts showed that ATP7BG943S failed to exit the TGN in elevated copper. We suggest that ATP7BS653Y is the best-characterized example of what appears to be a previously unidentified functional class of trafficking mutants that may also include G710S and G943S.
Several Menkes disease-causing mutations in ATP7A have phenotypes similar to that of ATP7BS653Y. Although no functional studies have been presented on ATP7AS653Y (34), we predict that its phenotype will be similar to that of ATP7BS653Y. Another ATP7A patient mutation, in which 26 aa were deleted in the conserved region before TM1 (E624-Q649 of ATP7B) (Fig. 1B), was reported to be functionally active yet did not exit the TGN in elevated copper (35). The finding that deleting 26 residues in ATP7A affected its TGN exit, yet five single amino acid substitutions in the same region of ATP7B did not, suggests that the region may function as a flexible linker between more distant domains of these proteins (Table 2).
Interactions Within TM1 + TM2 Are Important for TGN-exit of ATP7B and Its Cu(I)-Dependent Trafficking.
The molecular dynamics calculations revealed important changes in TM packing interactions indicated by the diversity of the SASAs for each substitution as was first suggested in the homology models. Furthermore, the calculations show that neither the wild-type nor the mutants exhibit a single dominant conformation but are a dynamic mix of structures revealed by changes in the free energy surface (Fig. 6). The computed structural properties of the patient mutation, ATP7BS653Y, showed the greatest effect on both the SASA of Tyr653 in TM1 and Tyr713 in TM2 and indicate that TM1/TM2 packing interactions are altered. We speculate that the nature of the change is both structural and dynamic because of a shift in the stable state coupled with dynamic changes in the accessible nearby energy states and may underlie the observed trafficking defect.
Our analysis also showed that introducing charge at the 653 position had a modest effect on TM1/TM2 packing interactions. Because neither ATP7BS653D nor ATP7BS653E were able to exit the TGN, we speculate that long-range TM packing interactions were altered, which may influence the structural state required for TGN-exit. By the use of experimental mutational analysis and biophysical computation, we show that the SASA and charge of the amino acids at position 653 are important structural determinants. Thus, we propose that interaction between Ser653and Gly710 of the TM2 kink is critical for copper-dependent structural changes that allow for TGN-exit.
Is There a Hierarchy of Conformations and Signals That Regulates TGN-Exit and Copper-Dependent Trafficking of ATP7B?
Copper-ATPase trafficking is a complex process that is not yet understood and the factors regulating TGN-exit of ATP7B into a specialized anterograde compartment are just being identified. In a recent study, the involvement of the clathrin adaptor, AP1 was implicated (36), as has the small cytoplasmic protein, Murr1/COMMD1, which was identified in Bedlington terriers with WD symptoms. The latter protein’s function is still unclear despite considerable effort (37, 38). Finally, we know little about the components of ATP7B’s specialized vesicles, although the chloride channel, ClC-4, has been implicated in ATP7B’s copper-transport function in the Golgi (39).
In addition to the cellular components required for copper-dependent ATP7B trafficking, studies have shown that molecular signals within the protein, as well as intramolecular interactions, play important roles (30, 40). Two well-characterized trafficking signals encoded within the primary sequences of both copper-ATPases have been identified: (i) F37-E45 in ATP7B, which is required for retention of ATP7B in the TGN (−Cu) and targeting to the apical region in polarized hepatic cells (+Cu, anterograde trafficking) (11); and (ii) di- and trileucines in ATP7A and ATP7B’s C-termini, which are needed for retrograde trafficking to the TGN (−Cu) (12, 41, 42). Trafficking also requires MBD5 or MBD6 (10), the C terminus (12), nucleotide binding-domain stability (43), and ATPase-dependent copper-transport activity (31).
Previous studies demonstrated that mutation of the phosphatase domain (ATP7B858TGE860>AAA) prevents completion of the catalytic cycle, yet the mutant constitutively exits the TGN irrespective of cellular copper levels (22, 31). Our study confirmed this dominant effect upon wtATP7B and showed that TGE>AAA is also dominant over ATP7BS653Y. Although our study demonstrates that the catalytic cycle and trafficking are not coupled, we speculate that the acyl-ATP7B858TGE860>AAA conformation is the signal that initiates TGN-exit. We propose that in high copper there is an equilibrium shift such that a greater number of ATP7B molecules are in the acyl-phosphorylated state which is permissive for TGN-exit.
Will Better Genotype/Phenotype Correlations Benefit WD Patients?
WD can be lethal if left untreated and requires lifelong management. Current treatments focus on controlling the amount of copper in the body with chelating agents, which have a long list of side effects (44, 45); however, new approaches that focus on correcting defective proteins could provide a novel treatment strategy (46). Recent homology-modeling studies have provided a convenient tool for in silico structure/function analysis of the ATP7A (47) and ATP7B cores (20, 47). In an effort to improve functional predictions of WD patient mutations located within the core region, a grading strategy was developed based on the ATP7B model/structure (20) and suggested as a guide for selecting relevant assays useful for elucidating functional defects of WD missense mutations. Although epigenetics, modifier loci, and environment likely contribute to the range of WD clinical phenotypes (48), we propose that a combination of clinical and genetic studies together with in depth analyses would allow for functional classification of WD mutations similar to the classification system used for Cystic Fibrosis mutations (49), and might allow for the development of other treatment strategies, which may possibly correct a functionally defective protein and improve patient management.
Methods
Additional details are in SI Methods.
Subjects and Clinical Laboratory Tests.
WD patient data in Table 1 were compiled from the Wilson Disease Database of the Institute of Psychiatry and Neurology, Second Department of Neurology, Warsaw, Poland.
ATP7B Mutants.
QuikChange II XL Site-Directed Mutagenesis kit (Stratagene) was used with pYG7 as a template to create GFP tagged mutants (Table S2) (10).
Trafficking Assays.
Anterograde trafficking of ATP7B from the TGN was induced by BCS→Cu shift. Specifically, 16–24 h after infection, WIF-B cells cultured in 10 μM BCS were rinsed, one set fixed (BCS), and a duplicate set incubated for 1.5 h in 10–100 μM CuCl2 (BCS→Cu). Cells were fixed, processed for indirect immunofluorescence, and imaged.
Retrograde trafficking (return to TGN) was induced by incubating copper-pretreated cells with a copper chelator TTM (Cu→TTM). Specifically, 16–24 h after infection, WIF-B cells cultured in 10 μM BCS were rinsed, incubated for 1.5 h in 10 μM CuCl2 (BCS→Cu), then rinsed twice, one set fixed (BCS→Cu), and a duplicate set incubated in 25 μM TTM for 2 h (Cu→TTM). Cells were fixed, processed for indirect immunofluorescence, and imaged as described below.
Indirect Immunofluorescence, Imaging, and Quantification (50).
The published structural model of ATP7B (20) was used as a template to model the S653 substitutions using SWISS-MODEL (51). The solvent accessible surface area for the functional groups in positions 653, 710, and 713 was calculated using the AREAIMOL program from the CCP4i suite (52). The interatomic distances were measured using PyMOL Molecular Graphics System, v1.4.1, (Schrödinger). Visual inspection of the ATP7B model/structure was also performed using University of California at San Francisco (UCSF) CHIMERA, www.cgl.ucsf.edu/chimera (53).
Molecular Dynamics Simulations.
The structural model of ATP7B (20) and the Ser653 substitutions generated by SWISS-MODEL were inserted into the implicit bilayer of 34 Å in depth by translating the molecule along the z axis with the center of the transmembrane region kept at z = 0 (54–57). CHARMM [vs. 37 b1 (58, 59)] was used to perform the molecular dynamics simulation on the KEENELAND resource (XSEDE-GaTECH) equipped with Intel Sandy Bridge (NVIDIA 3 M2090) GPU accelerators. The 8-ns trajectory files were analyzed using visual molecular dynamics (60) and the SASAs were calculated using the SURF feature (RPRObe -radius 1.4 Å – Correll.doc). The datasets were plotted using SigmaPlot 11 (Systat Software).
Supplementary Material
Acknowledgments
We thank Dr. Michael J. Schell and Dr. Arnab Gupta for many helpful discussions and critical reading of the manuscript; Dr. Yuta Hatori for rendering ATP7B in Fig. 1A; Dr. Janine Buettner for patient DNA sequence analysis; Professor Anna Członkowska, chief of Second Department of Neurology, Institute of Psychiatry and Neurology in Warsaw, Poland; The Synthesis and Sequencing Facility at The Johns Hopkins University School of Medicine for sequencing of plasmids and viral DNA; and the Imaging Core of Hopkins Digestive Diseases Basic Research Development Center for services. This work was supported by National Institutes of Health Grants P01 DK-072084, P01-GM067166, R24 DK-064388, and the Polish National Ministry of Education and Science Award N N402 375239.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314161111/-/DCSupplemental.
References
- 1.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87(3):1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 2.Lutsenko S, Gupta A, Burkhead JL, Zuzel V. Cellular multitasking: The dual role of human Cu-ATPases in cofactor delivery and intracellular copper balance. Arch Biochem Biophys. 2008;476(1):22–32. doi: 10.1016/j.abb.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferenci P, Roberts EA. Defining Wilson disease phenotypes: From the patient to the bench and back again. Gastroenterology. 2012;142(4):692–696. doi: 10.1053/j.gastro.2012.02.035. [DOI] [PubMed] [Google Scholar]
- 4.Forbes JR, Cox DW. Functional characterization of missense mutations in ATP7B: Wilson disease mutation or normal variant? Am J Hum Genet. 1998;63(6):1663–1674. doi: 10.1086/302163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forbes JR, Cox DW. Copper-dependent trafficking of Wilson disease mutant ATP7B proteins. Hum Mol Genet. 2000;9(13):1927–1935. doi: 10.1093/hmg/9.13.1927. [DOI] [PubMed] [Google Scholar]
- 6.Hsi G, et al. Sequence variation in the ATP-binding domain of the Wilson disease transporter, ATP7B, affects copper transport in a yeast model system. Hum Mutat. 2008;29(4):491–501. doi: 10.1002/humu.20674. [DOI] [PubMed] [Google Scholar]
- 7.Morgan CT, Tsivkovskii R, Kosinsky YA, Efremov RG, Lutsenko S. The distinct functional properties of the nucleotide-binding domain of ATP7B, the human copper-transporting ATPase: Analysis of the Wilson disease mutations E1064A, H1069Q, R1151H, and C1104F. J Biol Chem. 2004;279(35):36363–36371. doi: 10.1074/jbc.M404553200. [DOI] [PubMed] [Google Scholar]
- 8.Payne AS, Kelly EJ, Gitlin JD. Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc Natl Acad Sci USA. 1998;95(18):10854–10859. doi: 10.1073/pnas.95.18.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsivkovskii RER, Lutsenko S. The role of the invariant His-1069 in folding and function of the Wilson's disease protein, the human copper-transporting ATPase ATP7B. J Biol Chem. 2003;278(15):13302–13308. doi: 10.1074/jbc.M300034200. [DOI] [PubMed] [Google Scholar]
- 10.Guo Y, Nyasae L, Braiterman LT, Hubbard AL. NH2-terminal signals in ATP7B Cu-ATPase mediate its Cu-dependent anterograde traffic in polarized hepatic cells. Am J Physiol Gastrointest Liver Physiol. 2005;289(5):G904–G916. doi: 10.1152/ajpgi.00262.2005. [DOI] [PubMed] [Google Scholar]
- 11.Braiterman L, et al. Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am J Physiol Gastrointest Liver Physiol. 2009;296(2):G433–G444. doi: 10.1152/ajpgi.90489.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braiterman L, Nyasae L, Leves F, Hubbard AL. Critical roles for the COOH terminus of the Cu-ATPase ATP7B in protein stability, trans-Golgi network retention, copper sensing, and retrograde trafficking. Am J Physiol Gastrointest Liver Physiol. 2011;301(1):G69–G81. doi: 10.1152/ajpgi.00038.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferenci P, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23(3):139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 14.Roberts EA, Schilsky ML. American Association for Study of Liver Diseases (AASLD) Diagnosis and treatment of Wilson disease: An update. Hepatology. 2008;47(6):2089–2111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]
- 15.Weiss KH, Stremmel W. Evolving perspectives in Wilson disease: Diagnosis, treatment and monitoring. Curr Gastroenterol Rep. 2012;14(1):1–7. doi: 10.1007/s11894-011-0227-3. [DOI] [PubMed] [Google Scholar]
- 16.Steindl P, et al. Wilson’s disease in patients presenting with liver disease: A diagnostic challenge. Gastroenterology. 1997;113(1):212–218. doi: 10.1016/s0016-5085(97)70097-0. [DOI] [PubMed] [Google Scholar]
- 17.Pfeiffer RF. Wilson’s disease. Handb Clin Neurol. 2011;100:681–709. doi: 10.1016/B978-0-444-52014-2.00049-5. [DOI] [PubMed] [Google Scholar]
- 18.Huster D, et al. Defective cellular localization of mutant ATP7B in Wilson’s disease patients and hepatoma cell lines. Gastroenterology. 2003;124(2):335–345. doi: 10.1053/gast.2003.50066. [DOI] [PubMed] [Google Scholar]
- 19.Gromadzka G, et al. Middle-aged heterozygous carriers of Wilson’s disease do not present with significant phenotypic deviations related to copper metabolism. J Genet. 2010;89(4):463–467. doi: 10.1007/s12041-010-0065-3. [DOI] [PubMed] [Google Scholar]
- 20.Schushan M, Bhattacharjee A, Ben-Tal N, Lutsenko S. A structural model of the copper ATPase ATP7B to facilitate analysis of Wilson disease-causing mutations and studies of the transport mechanism. Metallomics. 2012;4(7):669–678. doi: 10.1039/c2mt20025b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huster D, et al. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology. 2012;142(4):947–956. doi: 10.1053/j.gastro.2011.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cater MA, La Fontaine S, Mercer JF. Copper binding to the N-terminal metal-binding sites or the CPC motif is not essential for copper-induced trafficking of the human Wilson protein (ATP7B) Biochem J. 2007;401(1):143–153. doi: 10.1042/BJ20061055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5(4):327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 24.Frommer DJ. Defective biliary excretion of copper in Wilson’s disease. Gut. 1974;15(2):125–129. doi: 10.1136/gut.15.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts EA, Sarkar B. Liver as a key organ in the supply, storage, and excretion of copper. Am J Clin Nutr. 2008;88(3):851S–854S. doi: 10.1093/ajcn/88.3.851S. [DOI] [PubMed] [Google Scholar]
- 26.Zamyatnin AA. Protein volume in solution. Prog Biophys Mol Biol. 1972;24(0):107–123. doi: 10.1016/0079-6107(72)90005-3. [DOI] [PubMed] [Google Scholar]
- 27.Schuster P. LCAO-MO-studies on intramolecular hydrogen bonding. Monatshefte für Chemie. 1969;100(6):2084–2095. [Google Scholar]
- 28.Jeffery GA. Hydrogen bonds and molecular recognition. Food Chem. 1996;56(3):241–246. [Google Scholar]
- 29.Tsivkovskii R, MacArthur BC, Lutsenko S. The Lys1010-Lys1325 fragment of the Wilson’s disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J Biol Chem. 2001;276(3):2234–2242. doi: 10.1074/jbc.M003238200. [DOI] [PubMed] [Google Scholar]
- 30.Gupta A, et al. Cellular copper levels determine the phenotype of the Arg875 variant of ATP7B/Wilson disease protein. Proc Natl Acad Sci USA. 2011;108(13):5390–5395. doi: 10.1073/pnas.1014959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petris MJ, et al. Copper-regulated trafficking of the Menkes disease copper ATPase is associated with formation of a phosphorylated catalytic intermediate. J Biol Chem. 2002;277(48):46736–46742. doi: 10.1074/jbc.M208864200. [DOI] [PubMed] [Google Scholar]
- 32.Kojimahara N, Nakabayashi H, Shikata T, Esumi M. Defective copper binding to apo-ceruloplasmin in a rat model and patients with Wilson’s disease. Liver. 1995;15(3):135–142. doi: 10.1111/j.1600-0676.1995.tb00660.x. [DOI] [PubMed] [Google Scholar]
- 33.Merle U, et al. Truncating mutations in the Wilson disease gene ATP7B are associated with very low serum ceruloplasmin oxidase activity and an early onset of Wilson disease. BMC Gastroenterol. 2010;10:8. doi: 10.1186/1471-230X-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gourdon P, et al. Crystal structure of a copper-transporting PIB-type ATPase. Nature. 2011;475(7354):59–64. doi: 10.1038/nature10191. [DOI] [PubMed] [Google Scholar]
- 35.Kim BE, Smith K, Petris MJ. A copper treatable Menkes disease mutation associated with defective trafficking of a functional Menkes copper ATPase. J Med Genet. 2003;40(4):290–295. doi: 10.1136/jmg.40.4.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirst J, et al. Distinct and overlapping roles for AP-1 and GGAs revealed by the “knocksideways” system. Curr Biol. 2012;22(18):1711–1716. doi: 10.1016/j.cub.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Bie P, et al. Distinct Wilson’s disease mutations in ATP7B are associated with enhanced binding to COMMD1 and reduced stability of ATP7B. Gastroenterology. 2007;133(4):1316–1326. doi: 10.1053/j.gastro.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fieten H, Leegwater PA, Watson AL, Rothuizen J. Canine models of copper toxicosis for understanding mammalian copper metabolism. Mamm Genome. 2012;23(1–2):62–75. doi: 10.1007/s00335-011-9378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang T, Weinman SA. Involvement of chloride channels in hepatic copper metabolism: ClC-4 promotes copper incorporation into ceruloplasmin. Gastroenterology. 2004;126(4):1157–1166. doi: 10.1053/j.gastro.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 40.Hasan NM, et al. Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. J Biol Chem. 2012;287(43):36041–36050. doi: 10.1074/jbc.M112.370403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Francis MJ, et al. Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane. J Cell Sci. 1999;112(Pt 11):1721–1732. doi: 10.1242/jcs.112.11.1721. [DOI] [PubMed] [Google Scholar]
- 42.Petris MJ, Mercer JF. The Menkes protein (ATP7A; MNK) cycles via the plasma membrane both in basal and elevated extracellular copper using a C-terminal di-leucine endocytic signal. Hum Mol Genet. 1999;8(11):2107–2115. doi: 10.1093/hmg/8.11.2107. [DOI] [PubMed] [Google Scholar]
- 43.Dmitriev OY, Bhattacharjee A, Nokhrin S, Uhlemann EM, Lutsenko S. Difference in stability of the N-domain underlies distinct intracellular properties of the E1064A and H1069Q mutants of copper-transporting ATPase ATP7B. J Biol Chem. 2011;286(18):16355–16362. doi: 10.1074/jbc.M110.198101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor RM, Chen Y, Dhawan A. EuroWilson Consortium Triethylene tetramine dihydrochloride (trientine) in children with Wilson disease: Experience at King’s College Hospital and review of the literature. Eur J Pediatr. 2009;168(9):1061–1068. doi: 10.1007/s00431-008-0886-8. [DOI] [PubMed] [Google Scholar]
- 45.Ferenci P. Review article: Diagnosis and current therapy of Wilson’s disease. Aliment Pharmacol Ther. 2004;19(2):157–165. doi: 10.1046/j.1365-2036.2003.01813.x. [DOI] [PubMed] [Google Scholar]
- 46.van den Berghe PV, et al. Reduced expression of ATP7B affected by Wilson disease-causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology. 2009;50(6):1783–1795. doi: 10.1002/hep.23209. [DOI] [PubMed] [Google Scholar]
- 47.Gourdon P, Sitsel O, Karlsen Jesper L, Møller Lisbeth B, Nissen P. Structural models of the human copper P-type ATPases ATP7A and ATP7B. Biol Chem. 2012;393(4):205–216. doi: 10.1515/hsz-2011-0249. [DOI] [PubMed] [Google Scholar]
- 48.Członkowska A, Gromadzka G, Chabik G. Monozygotic female twins discordant for phenotype of Wilson’s disease. Mov Disord. 2009;24(7):1066–1069. doi: 10.1002/mds.22474. [DOI] [PubMed] [Google Scholar]
- 49.MacDonald KD, McKenzie KR, Zeitlin PL. Cystic fibrosis transmembrane regulator protein mutations: ‘Class’ opportunity for novel drug innovation. Paediatr Drugs. 2007;9(1):1–10. doi: 10.2165/00148581-200709010-00001. [DOI] [PubMed] [Google Scholar]
- 50.Ihrke G, et al. Apical plasma membrane proteins and endolyn-78 travel through a subapical compartment in polarized WIF-B hepatocytes. J Cell Biol. 1998;141(1):115–133. doi: 10.1083/jcb.141.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 52.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 53.Pettersen EF, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 54.Im W, Feig M, Brooks CL., 3rd An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys J. 2003;85(5):2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Im W, Lee MS, Brooks CL., 3rd Generalized born model with a simple smoothing function. J Comput Chem. 2003;24(14):1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 56.Feig M, Im W, Brooks CL., 3rd Implicit solvation based on generalized Born theory in different dielectric environments. J Chem Phys. 2004;120(2):903–911. doi: 10.1063/1.1631258. [DOI] [PubMed] [Google Scholar]
- 57.Feig M, et al. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem. 2004;25(2):265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 58.Brooks BR, et al. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 59.Brooks BR, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33–38, 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 61.Achila D, et al. Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake. Proc Natl Acad Sci USA. 2006;103(15):5729–5734. doi: 10.1073/pnas.0504472103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Banci L, Bertini I, Cantini F, Rosenzweig AC, Yatsunyk LA. Metal binding domains 3 and 4 of the Wilson disease protein: Solution structure and interaction with the copper(I) chaperone HAH1. Biochemistry. 2008;47(28):7423–7429. doi: 10.1021/bi8004736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chenna R, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31(13):3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gromadzka G, et al. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin Genet. 2005;68(6):524–532. doi: 10.1111/j.1399-0004.2005.00528.x. [DOI] [PubMed] [Google Scholar]
- 65.Litwin T, Gromadzka G, Członkowska A. Neurological presentation of Wilson’s disease in a patient after liver transplantation. Mov Disord. 2008;23(5):743–746. doi: 10.1002/mds.21913. [DOI] [PubMed] [Google Scholar]
- 66.Lee B, Richards FM. The interpretation of protein structures: Estimation of static accessibility. J Mol Biol. 1971;55(3):379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 67.Miller S, Janin J, Lesk AM, Chothia C. Interior and surface of monomeric proteins. J Mol Biol. 1987;196(3):641–656. doi: 10.1016/0022-2836(87)90038-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.