

Abstract
Hormones in the hypothalamus-pituitary-adrenal (HPA) axis mediate many of the bodily responses to stressors, yet there is not a clear relationship between the levels of these hormones and stress-associated mental illnesses such as post-traumatic stress disorder (PTSD). Therefore, other hormones are likely to be involved in this effect of stress. Here we used a rodent model of PTSD in which rats repeatedly exposed to a stressor display heightened fear learning following auditory Pavlovian fear conditioning. Our results show that stress-related increases in circulating ghrelin, a peptide hormone, are necessary and sufficient for stress-associated vulnerability to exacerbated fear learning and these actions of ghrelin occur in the amygdala. Importantly, these actions are also independent of the classic HPA stress axis. Repeated systemic administration of a ghrelin receptor agonist enhanced fear memory but did not increase either corticotropin releasing factor (CRF) or corticosterone. Repeated intra-amygdala infusion of a ghrelin receptor agonist produced a similar enhancement of fear memory. Ghrelin receptor antagonism during repeated stress abolished stress-related enhancement of fear memory without blunting stress-induced corticosterone release. We also examined links between ghrelin and growth hormone (GH), a major downstream effector of the ghrelin receptor. GH protein was upregulated in the amygdala following chronic stress, and its release from amygdala neurons was increased by ghrelin receptor stimulation. Virus-mediated overexpression of GH in the amygdala was also sufficient to increase fear. Finally, virus-mediated overexpression of a GH receptor antagonist was sufficient to block the fear enhancing effects of repeated ghrelin receptor stimulation. Thus, ghrelin requires GH in the amygdala to exert fear-enhancing effects. These results suggest that ghrelin mediates a novel branch of the stress response and highlight a previously unrecognized role for ghrelin and growth hormone in maladaptive changes following prolonged stress.
Keywords: Ghrelin, Growth Hormone, Stress, Fear, HPA axis, Adrenalectomy, Post-traumatic stress disorder (PTSD), Trauma-related Anxiety Disorders
INTRODUCTION
When a cue or event threatens the well-being of an organism, stress responses are engaged to promote coping and adaptation (1). Despite the utility of these responses, repeated or prolonged activation of the stress response causes detrimental effects, including increased susceptibility to mental illnesses such as depression, anxiety, or post-traumatic stress disorder (PTSD) (2–8). It is thought that the stress response is principally coordinated by hormones of the hypothalamic-pituitary-adrenal (HPA) axis, however strong correlations between stress-induced alterations in these hormones and mental illness are lacking, and not all effects of chronic stress can be simulated with exogenous administration of HPA hormones (9, 10). Furthermore, while excessive HPA activity has been linked to heightened fear and anxiety in rodents, there has been little success in the clinical application of these findings (11). Both high and low levels of HPA activity have been observed in humans with stress-sensitive mental disorders and, in some cases, patients respond positively to treatment with exogenous glucocorticoids, one of the adrenal stress hormones (12). Thus, there is a crucial need for novel biomarkers and therapeutic targets.
Recently, it has been found that ghrelin, a peptide hormone, is modulated by exposure to stress (13, 14). Ghrelin is produced by the stomach where it is activated by post-translational acylation before being transported into the blood stream. It can then cross the blood-brain barrier (13) where it binds to the Growth Hormone Secretagogue Receptor 1a (GHSR1a, or ghrelin receptor). Interestingly, the ghrelin receptor is found in the basolateral complex of the amygdala (BLA) (15), a brain region that regulates negative emotional states such as fear. Single infusions of exogenous ghrelin into the amygdala can alter behavior in tasks such as the elevated plus maze and inhibitory avoidance (16), but a relationship between emotional learning and endogenous ghrelin has not been explored. Furthermore, growth hormone (GH), which is released by cells in response to ghrelin receptor activation and altered in the brain by stress (17), is present in BLA neurons (18). The stress-sensitivity of ghrelin, together with the localization of its receptor and downstream signaling partner (GH) in BLA, make it an attractive candidate mechanism by which emotional memories may be altered following periods of stress.
PTSD and other trauma- and stress-related disorders can arise following a traumatic experience (19). Chronically stressed individuals are especially vulnerable to developing these disorders in response to trauma (3, 8, 20, 21). Humans with PTSD and other anxiety disorders exhibit hyperactivity in the amygdala (22, 23) and amygdala-dependent processes, such as fear learning to novel stimuli in laboratory settings, are enhanced in humans with PTSD (24). The “over-acquisition” of aversive memories is not merely a symptom of trauma-related disorders. It is also thought to contribute to the genesis of such disorders and may perpetuate distress after the trauma: humans with PTSD have extremely strong memories of the PTSD-inducing trauma and many symptoms of PTSD, such as social avoidance and sleep disturbance, are secondary to these memories (25–27). Thus, understanding why certain individuals, such as those with a significant lifetime stress burden, have dysregulated encoding of traumatic memories is critical for both preventing and treating PTSD and other trauma-related disorders. Also, by understanding how stress alters brain regions responsible for emotional memory such as the amygdala, we may better understand how stress increases susceptibility to the development of other psychiatric disorders.
In this paper, we used a rodent model of PTSD in which rats were repeatedly exposed to stress and then subjected to auditory Pavlovian fear conditioning. Relative to unstressed controls, these animals displayed enhanced fear learning, mimicking the stress-induced vulnerability to excessive learning about aversive experiences that is a key feature in the acquisition and symptomatology of PTSD. Our studies uncover an essential and novel role for ghrelin and growth hormone in stress-induced susceptibility to exacerbated fear.
MATERIALS AND METHODS
Subjects
All experiments used adult male Long Evans rats (250–350 g, Taconic, Germantown, NY), housed individually (68–72°F; 12-h light-dark cycle, 7AM lights on). Food and water (or 0.9% saline for adrenalectomy experiments) was provided ad libitum. Stressed and unstressed animals were housed in separate cubicles. All procedures were in accordance with the US National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and were approved by the MIT Institutional Animal Care and Use Committee and the Animal Care and Use Review Office of the USAMRMC.
Surgical Procedures
Some rats received adrenalectomy, cannulae implants, or virus infusions, as described in Supplementary Information Methods.
Virus Preparation
HSV-based amplicons for the overexpression of growth hormone were previously constructed and characterized (28). Virus was packaged with the 5dl1.2 helper virus and 2-2 cells using standard methods as described in Supplementary Information Methods (29). Virus was purified on a sucrose gradient, pelleted, and resuspended in 10% sucrose in D-PBS. Titers were ~1X108IU/ml.
AAV-based viruses were constructed as described in Supplementary Information Methods and sent to Virovek (Hayward, CA) for packaging with AAV1/2 chimeric pseudotyping. Purified viruses were suspended in 10% sucrose in D-PBS and titers were 2.4X1012vg/ml.
Drug preparation
For systemic drug delivery, rats were injected with 1ml/kg (i.p.) of the appropriate solution. All drugs were solubilized in 0.9% saline (vehicle) such that injection volumes remained constant for each experiment. MK-0677 (also known as ibutamoren mesylate; Merck; Whitehouse, NJ, or Axon Medchem, Groningen, Netherlands) is a highly specific GHSR1a agonist that readily crosses the blood-brain barrier and has a half-life of over six hours (30, 31). A concentration of 0.5 mg/ml, diluted in vehicle, was selected because it is well-tolerated and results in significant and prolonged increases in growth hormone release (31). D-Lys3-GHRP-6 (Tocris Biosciences; Minneapolis, MN) was diluted to 2.74μg/ml in vehicle. D-Lys3-GHRP-6 is a selective and potent inhibitor of GHSR 1a (32, 33) with an IC50 of 0.9 μM (34) (Tocris Bioscience literature). It also crosses the blood brain barrier (35). The only other known receptor class with affinity for D-Lys3-GHRP-6 is the melanocortin receptors but the Ki = 26–120 μM so the dilute dose used here would not be expected to affect these receptors. D-Lys3-GHRP-6 was injected within 30min of the start of immobilization stress or following handling.
For experiments using intra-BLA drug delivery, drugs were solubilized in physiological artificial cerebrospinal fluid (vehicle; pH=7.35). MK-0677 was solubilized to 0.5μg/μl. For bioactive ghrelin, a dose of 5 nmol/μl, diluted in vehicle was selected as it was previously shown to have behavioral effects following a single infusion into the amygdala(16).
Drug Infusion
For intra-cranial infusions, rats were placed in 18.9L buckets containing bedding. The dummy cannulae were removed and injectors (30G stainless steel cannulae; extending 1mm beyond the cannulae end) were inserted. The injectors were attached to Hamilton syringes (10μl; Hamilton Co., Reno, NV) via polyethylene tubing, and the syringes were mounted in a Harvard syringe pump (Harvard Apparatus; Holliston, MA). Infusions were given at a rate of 0.1μl/m for 5min for a total volume of 0.5μl/side, with 1min for diffusion, before the injectors were removed and new dummy cannulae were inserted.
Stress Exposure
Some rats were exposed to either immobilization stress or water stress, as described in Supplementary Information Methods.
Behavioral Testing
Some rats were subjected to either auditory Pavlovian fear conditioning or the elevated plus maze, as described in Supplementary Information Methods.
Histology
Following completion of the experiment, animals were anesthetized with an overdose of isoflurane and intracardially perfused with physiological saline followed by 4% formalin fixative in saline. Perimortem blood was collected from some rats, as described in Supplementary Information Methods. Brains were harvested and placed in 4% formalin for 24–72h. The brains were then transferred to a 30% sucrose/4% formalin solution for a minimum of 3 days. For brains infused with virus, solutions containing paraformaldehyde were used in lieu of formalin. Coronal sections (40μm) were made and mounted on gelatinized slides. Tissue that did not contain virus was stained with 0.1% cresyl violet. Slides were then assessed for cannulae position or GFP florescence. Animals with incorrect placements were excluded from all analyses.
Hormone Assays
Corticosterone, acylated ghrelin, growth hormone and CRF levels were determined using commercial ELISA kits, as described in Supplementary Information Methods.
Statistics
For each fear memory session, conditional freezing was assessed as a percentage of time spent freezing, a probability estimate that is amenable to analysis with parametric statistics. These probability estimates of freezing, along with other measures, were analyzed using ANOVA. Post hoc comparisons in the form of Fisher’s PLSD tests were performed after a significant omnibus F-ratio (p<0.05). Statistical trends are noted in the text when omnibus F-ratio did not reach p <0.05 but were p<0.10. All data where p>0.10 are identified as not significant (ns).
RESULTS
Stress-related changes in fear and ghrelin are independent of adrenal stress hormones
We used an animal model of PTSD in which rats were repeatedly exposed to immobilization stress (4h/d for 14d) and subsequently administered auditory fear conditioning. Many studies suggest that the development of affective illness following stress, including disorders involving fear (9, 10, 36), is due to repeated activation of the HPA axis, which results in elevated adrenal stress hormone release (6, 37). To determine whether stress-induced increases in fear learning require adrenal stress hormones, such as corticosterone or adrenaline, we examined the impact of adrenalectomy on stress-related enhancement of fear conditioning. Following adrenalectomy (ADX) or sham surgery (SHAM), animals were exposed to immobilization stress (STR) or daily handling (no stress, or NS). One subset of animals underwent auditory fear conditioning 24h after the final stress or handling session. Fear to the tone was assessed 48h post-conditioning. Though a slight enhancement of fear acquisition was seen in stressed rats, this did not reach statistical significance (Fig. 1a, stress: F(1, 22)=3.98, p<0.10). However, stress produced a robust enhancement of long-term fear memory when fear to the tone was later tested in a novel context (Fig. 1b, stress: F(1, 22)=12.17, p<0.01). Surprisingly, the fear-enhancing effect of stress was observed in the complete absence of adrenal stress hormones (Fig. 1b, Surgery X Stress interaction, F(1, 22)=1.3, p=ns; corticosterone verified as undetectable in all ADX animals, Fig. 1c.).
Figure 1. Stress-related changes in fear and ghrelin are independent of adrenal stress hormones.

Animals received adrenalectomy (ADX) or sham surgery (SHAM). After at least a week of recovery, animals received either 14 days (4h/d) immobilization stress (STR) or gentle handling (NS). (a) Some animals received auditory Pavlovian fear conditioning 24h after the last stress or handling session. (b) Fear to the tone was assessed 48h later in a novel context. In a separate group of animals, trunk blood was collected 24h after the lass stress session. Plasma level corticosterone (c) and acylated ghrelin (d) were determined with ELISA. All data are mean ±s.e.m. * p<0.05, **p<0.01,*** p<0.001,**** p<0.0001, ~ p<0.10 in planned comparisons.
The enhancement in long-term fear memory after stress cannot be explained by stress-related changes in extinction nor memory retrieval: no difference in extinction retention was observed in a second extinction test performed 48h after the initial extinction session (Supp. Fig. 1a; stress: F(1, 12)=2.15, p=ns), and stress administered after fear conditioning did not alter the expression of previously acquired fear memories (Supp. Fig. 1b; stress: F(1, 17)=.107, p=ns). The high levels of conditional freezing seen in rats in the STR or ADX groups also cannot be explained by non-specific decreases in locomotor activity (Supp. Fig. 2a; stress: F(1, 22)=1.52, p=ns and surgery: F(1, 22)=.56, p=ns) or increases in spontaneous freezing (Supp. Fig. 2b; stress: F(1, 22)=.37, p=ns and surgery: F(1, 22)=.23, p=ns).
Stress-related enhancement of fear required multiple sessions and was not due to the most recent stress session: a single session of immobilization stress was not sufficient to increase subsequent fear learning (Supp. Fig. 3a). Stress-related enhancement of fear also did not stem from delayed effects of the first stress exposure as has been shown in other aspects of stress (9, 38): a single exposure to immobilization stress did not affect fear conditioning administered 14d later (Supp. Fig. 3b, stress: F(1, 6)=2.90, p=ns). Rather, stress-related increases in fear memory appeared after cumulative stress exposure of a approximately five or more days for this particular stressor (Supp. Fig. 3a and c, group: F(5, 46)=5.01, p<0.01 and F(1, 13)=4.62, p<0.10), respectively). These results are consistent with findings that stress “load” and neuronal dysfunction are correlated (39).
Because hormones of the HPA axis coordinate so many aspects of the stress response, it is surprising that stress-enhanced fear learning is not mediated by glucocorticoids or adrenaline. Interestingly, this is consistent with the limited clinical benefit of pharmacological manipulations targeting adrenal hormone signaling in PTSD patients (12, 40). Importantly, these data also suggest that other stress hormones drive stress-related changes in aversive learning and memory.
Circulating acylated ghrelin is elevated following chronic stress (13), raising the possibility that it may function as a stress hormone but its relationship with HPA hormones is poorly defined. To clarify this, we examined the impact of adrenalectomy on stress-induced increases in acylated ghrelin. Animals were administered surgical and stress treatments as per the previous experiment but sacrificed 24h after the final stress or handling session for the collection of blood samples. This was performed during a narrow window surrounding the circadian trough of ghrelin release to minimize hunger-induced variability in ghrelin levels. Corticosterone was significantly elevated by immobilization stress in the SHAM group but undetectable in the ADX group, as expected (Fig. 1c, Stress X Surgery interaction: F(1, 17)=8.37, p<0.05). In contrast, acylated ghrelin was elevated by stress regardless of the presence or absence of the adrenal glands (Fig. 1d, stress: F(1,17)=13.19, p<0.01, and Stress X Surgery interaction: F(1,17)=2.99, p=ns). Interestingly, stress-related increases in acylated ghrelin were amplified by adrenalectomy (Fig. 1d, surgery: F(1,17)=9.97, p<0.01), showing that adrenal hormones actually inhibit, rather than facilitate, ghrelin release (41). Additionally, the elevation of ghrelin by stress in the absence of adrenal hormones raises the intriguing possibility that the ghrelin pathway mediates stress-related enhancement of fear.
Ghrelin is elevated not only by psychological stressors such as immobilization stress, but also by other stressors, including those induced by environmental factors (water stress, Supp. Fig. 4, stress: F(1,14)=33.46, p<0.0001) and social status [social defeat, (13)]. Also, just as a single immobilization session does not lead to enhanced fear learning 24h later, a single immobilization session also does not elevated circulating ghrelin (Supp. Fig. 5, stress: F(1,11)=.01, p=ns). Together, our data reveal that ghrelin is not simply a downstream effector of adrenal hormone recruitment during chronic stress, and may instead represent an independent hormonal pathway of the stress response, broadly recruited by different stressors.
Repeated activation of the ghrelin receptor is sufficient for enhanced fear in the absence of stress and independent of the HPA axis
To determine whether increased activation of the ghrelin receptor is sufficient for enhancement of fear memory, we conducted experiments using pharmacological agonism of GHSR-1a in non-stressed animals. Stress-induced changes in acylated ghrelin were observed at the nadir of the diurnal ghrelin cycle, suggesting that stress-related increases in ghrelin persist throughout the day. Because the half-life of acylated ghrelin is short [~30m (42)], we used MK-0677 (also known as ibutamoren mesylate), a highly selective GHSR-1a agonist with a half-life of at least 5–6h (43), instead of exogenous acylated ghrelin in order to more closely model the prolonged stress-induced increases in GHSR activation by endogenous ghrelin. We systemically administered MK-0677 (MK: 5d) or saline (VEH: 5d) once a day for five consecutive days in non-stressed rats to determine whether repeated ghrelin receptor agonism in the absence of stress is sufficient to increase fear learning and whether HPA hormones may play a role in this effect. Five days of treatment were used because this reflects the minimum number of sessions our immobilization stress must be repeated to see stress-related enhancement of fear (Supp. Fig. 3a,c). A subset of animals was administered auditory fear conditioning 24h after the last injection. This drug regimen did not alter acquisition during conditioning (Fig. 2a, injection: F(1,31)=1.54, p=ns), but did significantly enhance long-term fear memory (Fig. 2b, injection: F(1,31)=4.21, p<0.05). This enhancement was similar to the effect of chronic immobilization stress and was not attributable to a drug-induced increase in spontaneous freezing (Supp. Fig. 6a, injection: F(1,31)=.25, p=ns) or a drug-induced decrease in locomotor activity (Supp. Fig. 6b and c, injection: F(1,31)=0.95, F(1,15)=2.44, p=ns all). Additionally, it was specific to associative aversive processing, as innate anxiety was not altered (Supp. Fig. 6d, treatment; F(1,15)=.15, p=ns).
Figure 2. Long-term pharmacological stimulation of ghrelin receptor activity enhances fear memory without altering other stress hormones.

Rats received daily systemic injections of MK-0677 (MK: 5d), a GHSR-1a agonist, or saline (VEH: 5d) for five days at the endogenous ghrelin signaling nadir. (a) One group underwent auditory fear conditioning 24h following the final injection. Fear acquisition was assessed by monitoring freezing levels. (b) Conditional freezing to the tone was assessed in a novel context 48h following fear conditioning. A separate group was sacrificed 24h following the final injection and microdissections of hypothalamus and amygdala performed. Brain CRF levels were measured using ELISA (c, hypothalamus, d, basolateral complex of the amygdala). * p<0.05 in planned comparisons.
Furthermore, just as we observed following chronic immobilization stress (Supp. Fig. 1b), fear expression was not altered following chronic ghrelin receptor agonism: previously acquired auditory fear memory was not affected by chronic ghrelin receptor agonism (Supp. Fig. 6e, injection: F(1,10)=.30, p=ns). Additionally, the enhancement of fear memory by repeated ghrelin receptor agonism cannot be attributed to effects of the most recent drug treatment (Supp. Fig. 7a, injection; F(1,19)=3.70, p<0.10) or delayed effects arising from the first drug treatment (Supp. Fig. 7b; injection: F(1,6)=.22, p=ns). Interestingly, there is a trend towards impairment, and not enhancement, of fear learning after a single dose of the ghrelin receptor agonist (Supp. Fig. 7a). This effect is similar to the effect of a single immobilization session (See Supp. Fig. 3a) and is also consistent with other reports that ghrelin may reduce aversive processing following acute stress (44). These data suggest that long-term activation of the ghrelin receptor is sufficient to enhance fear memory, while short-term activation of the ghrelin receptor may actually impair fear memory.
While stress-related increases in ghrelin are not triggered by the HPA axis, ghrelin could interact with the HPA axis in other ways to enhance fear. For example, the hypothalamic stress hormone corticotrophin-releasing factor (CRF) is secreted by neurons of the paraventricular nucleus, an area dense with GHSR-1a (45). Moreover, hypothalamic CRF neurons project to the amygdala and amygdalar CRF can modulate fear memory (46, 47). Thus, systemic ghrelin receptor agonism could mediate effects on fear learning by increasing CRF release in the amygdala. Additionally, ghrelin receptors have been identified in the adrenal cortex (48). Therefore systemic ghrelin receptor agonism could mediate effects on fear learning by increasing release of adrenal hormones. To determine whether the effects of ghrelin on fear learning are mediated through the HPA axis, we examined CRF peptide levels in both the hypothalamus and the amygdala of animals treated as above. There was no change in hypothalamic CRF (Fig. 2c, injection: F(1,21)=.20, p=ns) or amygdalar CRF (Fig. 2d, injection: F(1,21)=1.3, p=ns). In a third group of animals, we examined adrenal weights following a more prolonged period of ghrelin receptor agonism. Increased adrenal weight is seen following prolonged recruitment of adrenocorticotrophin (ACTH) from the pituitary because it triggers enhanced glucocorticoid and adrenaline production and release from the adrenal glands. Animals received systemic administration of MK-0677 (MK: 14d) or saline (VEH: 14d) once a day for 14 days. Repeated systemic ghrelin receptor agonism did not alter adrenal weight (Supp. Fig. 8, injection: F(1,14)=1.24, p=ns). This suggests that repeated ghrelin receptor agonism at the doses used here does not stimulate the HPA axis.
Fear memory requires plasticity in numerous brain regions but the basolateral complex of the amygdala (BLA) is particularly important for both formation and storage of learned fear. Recently, it has also been identified as the region of the amygdala with the highest density of ghrelin receptors (15). To determine whether repeated ghrelin receptor activation in the BLA is sufficient to enhance fear memory, we infused either MK-0677 (MK-Inf: 5d) or artificial cerebrospinal fluid (vehicle, VEH-Inf: 5d) directly into the BLA daily for five days prior to auditory fear conditioning. Freezing during fear conditioning was not altered by the treatment (Fig. 3a, infusion: F(1,8)=.36, p=ns) but long-term fear memory was significantly enhanced (Fig. 3b, infusion: F(1,8)=13.75, p<0.01). A similar potentiation of fear memory was observed when acylated ghrelin (GHR) was infused into the BLA daily for five days (Supp. Fig. 9; infusion: F(1,8)=6.07, p<0.05). Collectively, these data show that repeated activation of the ghrelin receptor directly in BLA is sufficient for heightened fear memory. The ability of the repeated intra-amygdala infusions to fully recapitulate the effects of repeated systemic ghrelin agonist delivery strongly suggests that stress-induced increases in circulating ghrelin enhance fear through direct actions in the BLA. Additionally, because direct intra-BLA manipulations are unlikely to increase either CRF or ACTH (49), this provides further support for the claim that ghrelin alters fear by direct actions in the amygdala, rather than through interactions with the HPA axis or ghrelin receptors in the periphery.
Figure 3. Long-term pharmacological stimulation of ghrelin receptor activity in the amygdala enhances fear memory.

Rats were implanted with bilateral cannulae aimed at the basolateral amygdala (BLA). The arrow indicates the tip of the injector within a representative coronal brain section. Following recovery, intra-BLA infusions of either MK-0677 (MK-Inf: 5d) or aCSF (VEH-Inf: 5d) were administered daily for five consecutive days and, 24h following the final infusion, (a) auditory fear conditioning was administered. (b) Fear memory was assessed in a novel context 48h following fear conditioning. Brain illustration adapted from (88). All data are mean ±s.e.m. ** p<0.01 in planned comparisons.
The ghrelin pathway is necessary for stress-induced vulnerability to fear during chronic stress
To determine whether ghrelin signaling is necessary for stress-related enhancement of fear memory, we blocked ghrelin receptor signaling during repeated stress sessions. Rats were administered immobilization stress (STR) or daily handling (NS) and given either a systemic injection of D-[Lys3]-GHRP-6 (DLys3), a highly specific inverse agonist of GHSR-1a that crosses the blood-brain barrier, or saline (VEH) at the start of each session (43). Twenty-four hours following the final stress or handling session, we administered auditory fear conditioning and assessed fear to the tone in a subsequent session. Stress enhanced fear acquisition in the saline treated group (Fig. 4a; for Trial 3, stress: F(1,27)=6.75, p<0.05, and planned comparisons, STR-VEH vs. NS-VEH, p<0.05) but this effect was not seen when the ghrelin receptor was antagonized during immobilization stress (Fig. 4a, Trial 3, Stress X Injection interaction: F(1,27)=3.94, p<0.10 and planned comparisons, STR-Dlys3 vs. STR-VEH). Long-term fear memory was enhanced by stress in saline-treated control animals as seen previously (Fig. 1b, Supp. Fig. 3a), demonstrating that any stress from injections did not alter this effect (50), but DLys3 completely reversed stress-enhanced fear (Fig. 4b; Injection X Stress interaction: F(1,27)=6.36, p<0.05 and planned comparisons, STR- Dlys3 vs. STR-VEH). In contrast, ghrelin receptor antagonism had no effect on fear memory in non-stressed controls (Fig. 4b; planned comparison, NS-SAL vs. NS-Dlys3). Moreover, DLys3 treatment did not blunt stress-induced HPA activation, demonstrated by similar levels of stress-induced corticosterone between DLys3- and vehicle-injected rats (Fig. 4c; injection: F(1,5)=.10, p=ns). These data show that ghrelin-mediated signaling is necessary for stress-related enhancement of fear and suggest, surprisingly, that other peripheral or central stress hormones are not sufficient to mediate this effect in the absence of heightened ghrelin signaling.
Figure 4. Ghrelin receptor antagonism during chronic stress abolishes stress-related enhancement of fear memory without affecting corticosterone release.
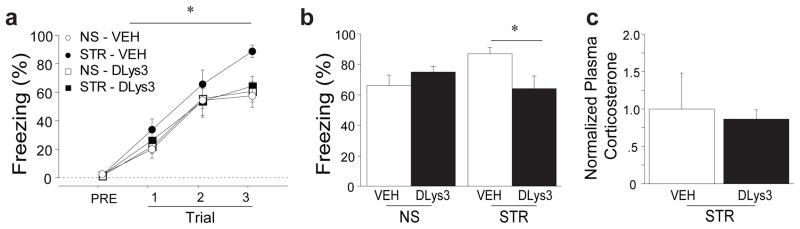
Rats received either daily handling (NS) or immobilization stress (STR). Each day, a systemic injection of either D-lys3-GHRP-6 (Dlys3), an antagonist of GHSR-1a, or saline (VEH) was administered within 30m of handling or stress initiation. (a) Animals received auditory fear conditioning 24h after the last stress or handling session. (b) Fear memory was assessed 48h after the conditioning session by placing the animals in a novel context and measuring conditional freezing during tone presentation. (c) In a subset of animals in the STR group, tail bleeds were performed during the final 30m of the final stress session and plasma corticosterone levels were measured using ELISA. All data are mean ±s.e.m. * p<0.05 in planned comparisons.
Growth hormone, a major effector of the ghrelin receptor, interacts with ghrelin in the amygdala to enhance fear memory
One of the best-characterized consequences of ghrelin receptor activation is release of GH (51). While the pituitary expresses the highest levels of GH, it is also expressed in other brain regions, including the BLA (18). In one limbic region, the hippocampus, GH levels have been shown to increase following acute stress (17). However, it is not known how prolonged stress alters GH in the BLA. To test this, we examined the impact of repeated immobilization stress (STR) or daily handling (NS) on GH levels in the BLA. We found that GH was readily detected in BLA homogenate and significantly upregulated 24h after chronic stress (Fig. 5a, group: F(1,16)=6.44, p<0.05), the time point at which we observe increases in circulating ghrelin and fear conditioning. This suggests that GH-mediated signaling in the BLA may be amplified following stress.
Figure 5. Amygdalar growth hormone is increased by chronic stress, is sufficient to enhance fear memory, and is necessary for the fear-potentiating effects of ghrelin receptor stimulation.

(a) Rats received either daily handling (NS) or immobilization stress (STR) for 14d. Animals were sacrificed 24h after the last stress or handling session and the BLA was dissected. Growth hormone (GH) levels in the BLA were measured using ELISA. (b) The HSV-based viral vectors (28) expressing either GFP (CON) or recombinant rat GH (rGH) was infused in the BLA and expression was assessed after behavioral testing was complete. Representative GFP expression from two rats is shown. (c) Auditory Pavlovian fear conditioning was performed 3 days after HSV virus infusions. (d) Long-term fear memory was assessed by placing the animals in a novel context and measuring conditional freezing following tone presentation 48h after the conditioning session. (e) Short-term BLA cell cultures were used to measure GH release following treatment with a ghrelin receptor agonist (MK) or vehicle (VEH). (f) AAV constructs were generated to examine the contribution of GH-mediated signaling in the BLA to ghrelin-induced potentiation of fear. (g) Following infusion of the AAVs into the BLA and recovery, rats received ten days of systemic injection of either a ghrelin receptor agonist (MK) or vehicle (VEH). Twenty-four hours later, auditory Pavlovian fear conditioning was administered to all rats. (h) Long-term fear memory was assessed by placing the animals in a novel context and measuring conditional freezing following tone presentation 48h after the conditioning session. Scale bar is 500 microns. All data are mean ±s.e.m. * p<0.05, ** p<0.01in planned comparisons.
GH can induce synaptic plasticity (52) and is increased in response to learning (53), but it is unclear how it affects amygdala function. Herpes simplex virus (HSV)-based viral vectors were used to express recombinant rat GH (rGH) and a green fluorescent protein (GFP) reporter or GFP only (28). Naive rats received intra-BLA infusions of either the rGH virus (rGH) or the GFP-only control virus (CON) (Fig. 5b). After three days, when HSV-mediated transgene expression is at its maximum (54), auditory fear conditioning was administered. Fear to the tone was assessed 48h later. Overexpression of rGH did not alter fear acquisition (Fig. 5c; Infusion X Trial interaction: F(4,52)=.57, p=ns) but did enhance long-term fear memory (Fig. 5d, infusion: F(1, 13)=9.97, p<0.01). These data demonstrate that high levels of GH in the BLA are sufficient to enhance fear learning, an effect which is similar to the effect of repeated intra-BLA ghrelin receptor stimulation (Fig. 3 and Supp. Fig. 9).
To determine whether ghrelin receptor stimulation in amygdala triggers the release of GH as it does from the pituitary (51), we generated short-term cultures of BLA cells and measured GH protein in the media following treatment with either MK-0677 or vehicle. Stimulation of the ghrelin receptor in BLA cells led to significantly elevated release of GH (Fig. 5e; treatment: F(1,8)=8.24, p<0.05). These results show that ghrelin receptor stimulation can trigger GH release from the amygdala.
Because ghrelin and GH can interact in amygdala, we next sought to determine whether GH is a necessary downstream signaling partner for the fear-enhancing effects of repeated ghrelin receptor stimulation. We generated an AAV viral construct to overexpress a mutant form of the rat GH protein (GHA) that acts as a functional antagonist to endogenous GH (55, 56). Following infusion of AAV to overexpress either GHA or a control protein (GFP) (Fig. 5f), rats were permitted to recover for five weeks. After recovery, rats that were infused with GHA received daily injections of either the ghrelin receptor agonist MK-0677 (MK) or saline (SAL) for ten days. Rats that were infused with GFP received daily injections of SAL for ten days. Twenty-four hours after the final injection, all rats were subjected to auditory fear conditioning, followed by assessment of long-term auditory fear memory in a subsequent session 48h later. While no differences were observed between any group during fear conditioning (Fig. 5g; group: F(2,13)=.07; p=ns), differences in long-term fear memory were observed (Fig. 5h; group: F(2,13)=4.32, p<0.05). Specifically, antagonizing the activity of GH prevented the fear-enhancing effects of repeated ghrelin receptor agonism (Fig. 5h; planned comparisons between GFP-SAL vs. GFP-MK and GFP-MK vs. GHA-MK). These data reveal that repeated ghrelin receptor stimulation requires GH-dependent signaling in the amygdala to exert its fear-enhancing effects.
DISCUSSION
In conclusion, we first show that ghrelin acts in parallel to the HPA axis: adrenalectomy does not affect the ability of stress to enhance fear learning or increase circulating acylated ghrelin. This finding indicates that these effects of stress are not simply downstream from glucocorticoids or adrenal catecholamines. We also show that increased ghrelin receptor activity is sufficient and necessary for stress-enhanced fear and is dissociable from HPA activity. Repeated activation of ghrelin receptors in non-stressed animals significantly enhances fear learning without elevating HPA stress hormones, while systemic blockade of the ghrelin receptor during chronic stress prevents stress-related enhancement of fear, even in the presence of elevated adrenal stress hormones. We demonstrate that the amygdala, a brain region that displays enhanced function in chronically stressed animals and in patients with trauma-related disorders, is likely the locus of the fear-enhancing effects of repeated ghrelin receptor stimulation. Finally, we show that GH, a downstream effector of ghrelin receptor activation, is increased in the BLA by chronic stress, is sufficient to enhance fear learning, and plays a necessary role in the fear-potentiating effects of ghrelin. Thus, ghrelin and growth hormone act together in the amygdala to enhance fear.
Our study is the first to explicitly examine the effects of protracted exposure to elevated ghrelin, as observed following chronic stress. We show that there are profound differences in the behavioral consequences of ghrelin exposure following different exposure durations, similar to the cumulative nature of stress. We also provide the first evidence to link prolonged exposure to elevated ghrelin with a specific, detrimental consequence of stress, enhanced fear memory, which typifies trauma-induced anxiety disorders such as PTSD. Because PTSD is a multi-faceted disorder producing many symptoms, including those related to avoidance and hyperarousal, it will be interesting to determine whether chronically elevated ghrelin contributes to these sequelae of PTSD in addition to promoting changes in fear learning and memory.
Our study is also the first to show that GH is a critical downstream mediator of the effects of ghrelin in amygdala. Such a relationship between ghrelin and GH has not been described outside of the pituitary (51). We also provide the first evidence to link elevated amygdala GH with chronic stress and enhanced fear memory. Taken together, our data reveal that the amygdala may be especially sensitive to ghrelin-mediated effects of stress because chronic stress amplifies both ghrelin and GH.
In contrast to our findings which link ghrelin to a pathological condition, prior studies have argued that ghrelin promotes adaptive changes during stress, including antidepressant effects (13) and reduction in anxiety (44). However, these studies are problematic because they either focused exclusively on acute ghrelin manipulations, which we show can have profoundly different effects from repeated ghrelin manipulations, or used short- and long-term ghrelin manipulations interchangeably. Additionally, the alterations in ghrelin levels were achieved through artificial states: heightened ghrelin levels were attained by extreme food deprivation or a single bolus injection of the short-lived peptide. The antidepressant effect of ghrelin requires extremely high levels of ghrelin, as found in food-restricted rodents after 10–15% weight loss (13). We find that this level of food deprivation leads to increased exploratory motor activity (Supp. Fig. 10; F(1, 13)=7.51, p<0.05). A recent study has also reported similar motor effects following acute ghrelin manipulations (57). These motor effects can be a significant confound for measures that require locomotor activity, such as social interaction or exploration. Thus, the ghrelin may alleviate the psychomotor effects of depression in a manner similar to amphetamine (58). It is also important to note that the antidepressant effect of ghrelin reported following a single injection of exogenous ghrelin was only a mild improvement of a stress-related impairment in social interaction (13); enhanced ghrelin signaling did not promote “normal” function following stress. Indeed, our results reported here are consistent with limited human data showing that patients with treatment-resistant major depressive disorder have higher ghrelin levels than control patients (59).
Here we demonstrate changes in endogenous ghrelin following stress and also use a low dose, long-acting agonist to replicate the naturally occurring ghrelin state. We also provide clear evidence that acute and chronic ghrelin receptor manipulations have profoundly different effects. It is important to note that the changes in fear reported here occurred following small, but persistent, changes in ghrelin signaling, and all were in the absence of any locomotor effects. We suggest that the utility of ghrelin in the stress response may be similar to glucocorticoids: under “normal” conditions, there is an optimal level of the hormone (60) and too little (61, 62) or too much hormonal signaling (16) can lead to dysfunction in neuronal circuits. Repeated activation of ghrelin and glucocorticoid pathways together contributes to stress-induced “load” on the body. In this regard, heightened ghrelin signaling may have both advantageous and undesirable consequences, but these must be carefully considered with respect to the length and level of elevated ghrelin exposure.
It is important to acknowledge the limitations of our rodent model of PTSD. In our experiments, rats were repeatedly exposed to immobilization stress to generate a vulnerability to high levels of fear learning and memory following Pavlovian fear conditioning. The unstressed rats that received fear conditioning displayed normal, adaptive levels of fear, and did not express PTSD-like changes. It is not clear whether repeated exposure to stress leads to PTSD per se in the absence of a specific aversive experience such as fear conditioning. For example, when rats are administered a single stressor repeatedly, many aspects of the stress response show habituation over the course of stress, suggesting that later stress exposures are less “stressful” and perhaps less likely to give rise to PTSD (63). Additionally, the Diagnostic and Statistical Manual of Mental Disorders IV requires that a diagnosis of PTSD be tied to a single traumatic event (19), and not a history of aversive experiences, such as a gradual accumulation of significant life stressors such as low socioeconomic status or abuse. However, populations with greater life stress accumulation demonstrate higher risk of both the occurrence of a traumatic event and the development of PTSD after trauma (7, 64). In animal models, when a single stressor is repeated, small changes during delivery of the stress, such as shifts in the environmental context in which it occurs (65), are sufficient to relieve habituation. Thus, it is possible that even when a single type of stress is repeated over days or weeks, prior exposure to that stress can lead to changes in neural circuits that facilitate the mnemonic encoding of subsequent experiences of the stress. Consistent with this, rats exposed to chronic stress show physiological changes that are consistent with PTSD, including enhanced corticosterone responses to novel stressors (66), sleep fragmentation (67), decreased hippocampal volume (68), anhedonia (69), and enhanced amygdala excitability (70). As biomarkers for PTSD become better defined, it will be interesting to determine whether repeated stress exposure alone is sufficient to produce changes associated with PTSD before additional trauma exposure occurs.
The source of ghrelin that is important for modulating fear is unclear. The majority of ghrelin is synthesized and released by specialized endocrine cells lining the stomach, with a lesser quantity released by the small intestine (71). However, significantly smaller amounts have been detected in a variety of other tissues, including the pancreas, lungs, and kidneys (72). Thus peripheral tissues likely contribute to the circulating levels of acylated ghrelin that we report here. Ghrelin may also be synthesized by small populations of neurons in the hypothalamus (73), the cerebral cortex, and the brainstem (74) where it may act as a paracrine hormone rather than being secreted into the blood stream. However, immunoreactive ghrelin-containing fibers have never been reported in amygdala. Thus, it seems that the most likely source of bioactive ghrelin affecting fear lies in the periphery, though a role for centrally-derived ghrelin cannot be fully eliminated.
Our results strongly suggest that the fear-modulating actions of repeated ghrelin receptor activation occur via direct actions in the lateral or basolateral divisions of the amygdala (BLA, together). Some studies have found that intra-hypothalamic application of ghrelin can enhance activity in the amygdala, suggesting an indirect mechanism by which ghrelin can affect amygdala output, but this increased activity is observed only in the central nucleus of the amygdala (75), which is downstream of the BLA. We show here that repeated direct intra-BLA application of a ghrelin receptor agonist was sufficient to mimic the fear-potentiating effects of chronic stress. While the GHSR1a is expressed throughout multiple nuclei of the amygdala, it is densest in the lateral nucleus and is expressed by the majority of cells in this region (15). It is not clear whether this expression is restricted only to excitatory pyramidal neurons, or whether it may also be found in inhibitory interneurons; this is an exciting area for future research which will facilitate our understanding of the mechanistic underpinnings of ghrelin-related fear enhancement.
It is notable that our data show that ghrelin receptor antagonism did not modulate fear memory in unstressed animals. From a clinical standpoint, this is important because it suggests that anti-ghrelin therapies aimed at reducing the stress response may have minimal impact on “normal”, adaptive fear learning. Our finding contrasts with one study, in which genetic ablation of the ghrelin receptor, GHSR-1a, produced a mild impairment of contextual fear memory when tested one month following fear conditioning (76). The reason for this discrepancy is unclear. It may be that genetic ablation of the ghrelin receptor represents an extreme level of functional “antagonism” of ghrelin signaling, and we might have observed slight fear memory deficits in unstressed animals if we used a high dose of the ghrelin receptor antagonist. Alternatively, the knockout of the ghrelin receptor from birth may have led to compensatory changes through development that were not present in our study.
It is not clear why acute and repeated ghrelin receptor stimulation have opposite effects on fear learning. While GHSR-1a activation engages excitatory Gq-dependent molecular cascades, the GHSR-1 also exhibits an extremely high level of constitutive activity in the absence of bound ligand (77). Accordingly, transient stimulation of GHSR-1a leads to rapid desensitization and internalization of the receptor which is slow to recover (78). Such a change is consistent with the decreased fear learning we observed 24h after a single injection of ghrelin receptor agonist. It is also consistent with the observation that transient bath application of ghrelin to lateral amygdala slices leads to decreased excitatory neurotransmission (15). The electrophysiological changes elicited by chronic ghrelin receptor stimulation in amygdala are completely unexplored, but our work suggests that the change must be opposite to that seen after acute ghrelin receptor stimulation. We suggest that the internalization of the ghrelin receptor may habituate (63) following either chronic administration of ghrelin receptor agonist or chronic stress exposure. The differences in receptor kinetics following acute versus chronic ghrelin receptor stimulation represent an especially promising area for future research.
It is also not clear how ghrelin receptor activation changes the amygdala to produce changes in fear learning. Repeated ghrelin receptor stimulation enhances neuronal spine density and facilitates long-term potentiation (61), effects that are also observed following chronic stress (79, 80). GH has also been shown to enhance long-term potentiation (52, 81), yet links between GH and spine density have never been reported. It will be interesting to determine whether ghrelin requires GH for its effects on synaptic plasticity, as it does for its effects on fear learning.
Our data show that stress-related changes in amygdala-based aversive processing are not dependent on HPA activity and that ghrelin plays an important role in stress-related affective dysfunction by actions independent of the HPA axis. It is important to acknowledge that we did not determine whether the fear-potentiating effect of stress is mediated via HPA-independent actions of ghrelin in adrenalectomized animals. For our purposes, the most important issue was to address the mechanisms by which chronic stress enhances fear under “normal” physiological circumstances, rather than how stress enhances fear in animals lacking adrenal hormones. We do not discount the role of the HPA axis in coordinating other aspects of the stress response, nor the importance of adrenal hormones in regulating amygdala function in animals without a history of prolonged stress exposure. It is clear that the HPA hormonal cascade mediates many of the consequences of stress [for review see (82)] and that signaling through glucocorticoid and adrenergic receptors is important for regulating amygdala function [see (83, 84) for review]. However, this work may need to be re-examined through the lens of putative parallel stress pathways such as ghrelin. Future work will be needed to explore the possible synergistic effects of co-activation of the HPA axis and the ghrelin system during chronic stress.
No current treatments exist for preventing stress-related affective disorders, suggesting that our most intriguing and important finding is that blockade of ghrelin signaling during stress is sufficient to prevent stress-related vulnerability to excessive fear. This raises the possibility that such a strategy might reduce or prevent the development of stress-sensitive affective disorders like PTSD during prolonged or extreme stress load. To date, very few studies have examined ghrelin or its receptor in the development of affective mental illness, however polymorphisms in the ghrelin gene have been associated with panic disorder (85) and serum ghrelin levels are higher in patients with treatment-resistant forms of major depressive disorder or panic disorder (59). While there are some non-HPA molecules which might be targeted in the treatment of stress-sensitive mental illnesses [such as brain-derived neurotrophic factor, tissue plasminogen activator, or FKBP5; for review, see (86)], the dysregulation of these molecules in rodent models of these diseases is brain region-specific. To effectively treat mental illness, pharmaceuticals for these molecules would need to cross the blood-brain barrier and act in a brain region-specific manner to minimize off-target side effects. Furthermore, there are no pharmaceuticals that can readily affect these molecules in humans. In contrast, because ghrelin is a peripheral hormone, it can be targeted using therapeutics that act in the periphery. Finally, many anti-ghrelin treatments already have undergone rigorous safety testing in humans because of the putative role of ghrelin in the development of obesity (87). Thus, the discovery that ghrelin plays a role in stress-related affective dysregulation reveals an especially attractive target for treating stress-sensitive mental disorders.
Supplementary Material
Summary.
Stress-related increases in bioactive ghrelin are necessary and sufficient for stress-induced enhancement of fear memory
Acknowledgments
We thank Merck & Co., Inc. for providing the MK-0677 used in some of these studies. We thank Stephanie P. Chen, Stanley P. Gill, Anita Y. Lin, Sarah Gomez, and Junmei Yao for their assistance in data collection and analysis. We thank Rebecca G. Canter and Drs. Ann Graybiel, Charles Jennings, Matt Wilson, Richard Wurtman, Tyler Brown, and Jill Crittenden for comments. Funding for undergraduate participation in this research was provided by Bob and Robyn Metcalfe to K.A.G., and by the MIT UROP office. Funding for graduate participation in this research was provided by the Hugh Hampton Young Memorial Fund, Schoemaker Fellowship, Singleton Presidential Fund, and James R. Killian Fellowship to R.M.M. This research was funded by NIMH (R01 MH084966), and the U.S. Army Research Office and the Defense Advanced Research Projects Agency (grant W911NF-10-1-0059) to K.A.G.
Footnotes
Author Contributions
RMM conceived experiments, designed protocols, performed experiments, and completed histology and data analyses for Figs 1, 2, 3, 4, S1, S2, S3c, S4, S5, S6, S7 and S8. ABR performed experiments, histology, and data analyses for Fig 5b–d, f–h. EL designed and performed experiments and completed data analysis for Fig S3a–b. SSC conceived, designed, performed, and analyzed the experiment in Fig S10. KAG conceived and designed experiments, prepared GH virus, performed the experiment in Fig 5a,e–h and S9, and performed data analysis. RMM and KAG designed figures and wrote the manuscript.
Conflict of Interest
The authors declare no competing financial interests.
Supplementary information is available at the website for Molecular Psychiatry.
Supplementary Information accompanies the paper on the Molecular Psychiatry website www.nature.com/mp
References
- 1.McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998 Jan 15;338(3):171–179. doi: 10.1056/NEJM199801153380307. [DOI] [PubMed] [Google Scholar]
- 2.Lederbogen F, Kirsch P, Haddad L, Streit F, Tost H, Schuch P, et al. City living and urban upbringing affect neural social stress processing in humans. Nature. 2011 Jun 23;474(7352):498–501. doi: 10.1038/nature10190. [DOI] [PubMed] [Google Scholar]
- 3.Mazure C. Does Stress Cause Psychiatric Illness? American Psychiatric Press, Inc; Washington, D.C: 1995. p. 281. [Google Scholar]
- 4.Belanoff JK, Flores BH, Kalezhan M, Sund B, Schatzberg AF. Rapid reversal of psychotic depression using mifepristone. J Clin Psychopharmacol. 2001 Oct;21(5):516–521. doi: 10.1097/00004714-200110000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Juster RP, Bizik G, Picard M, Arsenault-Lapierre G, Sindi S, Trepanier L, et al. A transdisciplinary perspective of chronic stress in relation to psychopathology throughout life span development. Dev Psychopathol. 2011 Aug;23(3):725–776. doi: 10.1017/S0954579411000289. [DOI] [PubMed] [Google Scholar]
- 6.Miller MM, McEwen BS. Establishing an agenda for translational research on PTSD. Ann N Y Acad Sci. 2006 Jul;1071:294–312. doi: 10.1196/annals.1364.023. [DOI] [PubMed] [Google Scholar]
- 7.McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003 Aug 1;54(3):200–207. doi: 10.1016/s0006-3223(03)00177-x. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz AC, Bradley RL, Sexton M, Sherry A, Ressler KJ. Posttraumatic stress disorder among African Americans in an inner city mental health clinic. Psychiatr Serv. 2005 Feb;56(2):212–215. doi: 10.1176/appi.ps.56.2.212. [DOI] [PubMed] [Google Scholar]
- 9.Mitra R, Sapolsky RM. Acute corticosterone treatment is sufficient to induce anxiety and amygdaloid dendritic hypertrophy. Proc Natl Acad Sci U S A. 2008 Apr 8;105(14):5573–5578. doi: 10.1073/pnas.0705615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conrad CD, MacMillan DD, 2nd, Tsekhanov S, Wright RL, Baran SE, Fuchs RA. Influence of chronic corticosterone and glucocorticoid receptor antagonism in the amygdala on fear conditioning. Neurobiol Learn Mem. 2004 May;81(3):185–199. doi: 10.1016/j.nlm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Frodl T, O’Keane V. How does the brain deal with cumulative stress? A review with focus on developmental stress, HPA axis function and hippocampal structure in humans. Neurobiol Dis. 2012 Mar 9; doi: 10.1016/j.nbd.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Searcy CP, Bobadilla L, Gordon WA, Jacques S, Elliott L. Pharmacological prevention of combat-related PTSD: a literature review. Mil Med. 2012 Jun;177(6):649–654. doi: 10.7205/milmed-d-11-00390. [DOI] [PubMed] [Google Scholar]
- 13.Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, et al. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 2008 Jul;11(7):752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng J, Dobner A, Babygirija R, Ludwig K, Takahashi T. Effects of repeated restraint stress on gastric motility in rats. Am J Physiol Regul Integr Comp Physiol. 2009 May;296(5):R1358–1365. doi: 10.1152/ajpregu.90928.2008. [DOI] [PubMed] [Google Scholar]
- 15.Alvarez-Crespo M, Skibicka KP, Farkas I, Molnar CS, Egecioglu E, Hrabovszky E, et al. The amygdala as a neurobiological target for ghrelin in rats: neuroanatomical, electrophysiological and behavioral evidence. PLoS One. 2012;7(10):e46321. doi: 10.1371/journal.pone.0046321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlini VP, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR. Differential role of the hippocampus, amygdala, and dorsal raphe nucleus in regulating feeding, memory, and anxiety-like behavioral responses to ghrelin. Biochem Biophys Res Commun. 2004 Jan 16;313(3):635–641. doi: 10.1016/j.bbrc.2003.11.150. [DOI] [PubMed] [Google Scholar]
- 17.Donahue CP, Kosik KS, Shors TJ. Growth hormone is produced within the hippocampus where it responds to age, sex, and stress. Proc Natl Acad Sci U S A. 2006 Apr 11;103(15):6031–6036. doi: 10.1073/pnas.0507776103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pacold ST, Kirsteins L, Hojvat S, Lawrence AM. Biologically active pituitary hormones in the rat brain amygdaloid nucleus. Science. 1978 Feb 17;199(4330):804–806. doi: 10.1126/science.203034. [DOI] [PubMed] [Google Scholar]
- 19.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4. Washington, DC: 2000. text rev. [Google Scholar]
- 20.Khoury L, Tang YL, Bradley B, Cubells JF, Ressler KJ. Substance use, childhood traumatic experience, and Posttraumatic Stress Disorder in an urban civilian population. Depress Anxiety. 2010 Dec;27(12):1077–1086. doi: 10.1002/da.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gillespie CF, Bradley B, Mercer K, Smith AK, Conneely K, Gapen M, et al. Trauma exposure and stress-related disorders in inner city primary care patients. Gen Hosp Psychiatry. 2009 Nov-Dec;31(6):505–514. doi: 10.1016/j.genhosppsych.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bremner JD, Vermetten E, Schmahl C, Vaccarino V, Vythilingam M, Afzal N, et al. Positron emission tomographic imaging of neural correlates of a fear acquisition and extinction paradigm in women with childhood sexual-abuse-related post-traumatic stress disorder. Psychol Med. 2005 Jun;35(6):791–806. doi: 10.1017/s0033291704003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, et al. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012 Nov;13(11):769–787. doi: 10.1038/nrn3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glover EM, Phifer JE, Crain DF, Norrholm SD, Davis M, Bradley B, et al. Tools for translational neuroscience: PTSD is associated with heightened fear responses using acoustic startle but not skin conductance measures. Depress Anxiety. 2011 Dec 21;28(12):1058–1066. doi: 10.1002/da.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ehlers A, Hackmann A, Michael T. Intrusive re-experiencing in post-traumatic stress disorder: phenomenology, theory, and therapy. Memory. 2004 Jul;12(4):403–415. doi: 10.1080/09658210444000025. [DOI] [PubMed] [Google Scholar]
- 26.Hackmann A, Ehlers A, Speckens A, Clark DM. Characteristics and content of intrusive memories in PTSD and their changes with treatment. J Trauma Stress. 2004 Jun;17(3):231–240. doi: 10.1023/B:JOTS.0000029266.88369.fd. [DOI] [PubMed] [Google Scholar]
- 27.Diamond DM, Campbell AM, Park CR, Halonen J, Zoladz PR. The temporal dynamics model of emotional memory processing: a synthesis on the neurobiological basis of stress-induced amnesia, flashbulb and traumatic memories, and the Yerkes-Dodson law. Neural Plast. 2007;2007:60803. doi: 10.1155/2007/60803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vander Weele CM, Saenz C, Yao J, Correia SS, Goosens KA. Restoration of hippocampal growth hormone reverses stress-induced hippocampal impairment. Frontiers in Neuroscience. 2013 doi: 10.3389/fnbeh.2013.00066. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim F, Neve R. Current Protocols in Neuroscience. John Wiley and Sons; 2001. Generation of High-Titer Defective HSV-1 Vectors; pp. 4.13.11–14.13.17. [DOI] [PubMed] [Google Scholar]
- 30.Chang CH, Rickes EL, McGuire L, Frazier E, Chen H, Barakat K, et al. Growth hormone (GH) and insulin-like growth factor I responses after treatments with an orally active GH secretagogue L-163,255 in swine. Endocrinology. 1996 Nov;137(11):4851–4856. doi: 10.1210/endo.137.11.8895356. [DOI] [PubMed] [Google Scholar]
- 31.Jacks T, Smith R, Judith F, Schleim K, Frazier E, Chen H, et al. MK-0677, a potent, novel, orally active growth hormone (GH) secretagogue: GH, insulin-like growth factor I, and other hormonal responses in beagles. Endocrinology. 1996 Dec;137(12):5284–5289. doi: 10.1210/endo.137.12.8940347. [DOI] [PubMed] [Google Scholar]
- 32.Pinilla L, Barreiro ML, Tena-Sempere M, Aguilar E. Role of ghrelin in the control of growth hormone secretion in prepubertal rats: interactions with excitatory amino acids. Neuroendocrinology. 2003 Feb;77(2):83–90. doi: 10.1159/000068652. [DOI] [PubMed] [Google Scholar]
- 33.Sethumadhavan K, Veeraragavan K, Bowers CY. Demonstration and characterization of the specific binding of growth hormone-releasing peptide to rat anterior pituitary and hypothalamic membranes. Biochem Biophys Res Commun. 1991 Jul 15;178(1):31–37. doi: 10.1016/0006-291x(91)91775-8. [DOI] [PubMed] [Google Scholar]
- 34.Traebert M, Riediger T, Whitebread S, Scharrer E, Schmid HA. Ghrelin acts on leptin-responsive neurones in the rat arcuate nucleus. J Neuroendocrinol. 2002 Jul;14(7):580–586. doi: 10.1046/j.1365-2826.2002.00810.x. [DOI] [PubMed] [Google Scholar]
- 35.Asakawa A, Inui A, Kaga T, Katsuura G, Fujimiya M, Fujino MA, et al. Antagonism of ghrelin receptor reduces food intake and body weight gain in mice. Gut. 2003 Jul;52(7):947–952. doi: 10.1136/gut.52.7.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jovanovic T, Norrholm SD, Blanding NQ, Phifer JE, Weiss T, Davis M, et al. Fear potentiation is associated with hypothalamic-pituitary-adrenal axis function in PTSD. Psychoneuroendocrinology. 2010 Jul;35(6):846–857. doi: 10.1016/j.psyneuen.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012 Jan;13(1):22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S. Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc Natl Acad Sci U S A. 2005 Jun 28;102(26):9371–9376. doi: 10.1073/pnas.0504011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marin MF, Lord C, Andrews J, Juster RP, Sindi S, Arsenault-Lapierre G, et al. Chronic stress, cognitive functioning and mental health. Neurobiol Learn Mem. 2011 Nov;96(4):583–595. doi: 10.1016/j.nlm.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 40.Hoge EA, Worthington JJ, Nagurney JT, Chang Y, Kay EB, Feterowski CM, et al. Effect of acute posttrauma propranolol on PTSD outcome and physiological responses during script-driven imagery. CNS Neurosci Ther. 2012 Jan;18(1):21–27. doi: 10.1111/j.1755-5949.2010.00227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otto B, Tschop M, Heldwein W, Pfeiffer AF, Diederich S. Endogenous and exogenous glucocorticoids decrease plasma ghrelin in humans. Eur J Endocrinol. 2004 Jul;151(1):113–117. doi: 10.1530/eje.0.1510113. [DOI] [PubMed] [Google Scholar]
- 42.Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000 Oct 19;407(6806):908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 43.Smith RG, Cheng K, Schoen WR, Pong SS, Hickey G, Jacks T, et al. A nonpeptidyl growth hormone secretagogue. Science. 1993 Jun 11;260(5114):1640–1643. doi: 10.1126/science.8503009. [DOI] [PubMed] [Google Scholar]
- 44.Spencer SJ, Xu L, Clarke MA, Lemus M, Reichenbach A, Geenen B, et al. Ghrelin regulates the hypothalamic-pituitary-adrenal axis and restricts anxiety after acute stress. Biol Psychiatry. 2012 Sep 15;72(6):457–465. doi: 10.1016/j.biopsych.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 45.Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res. 1997 Aug;48(1):23–29. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- 46.Isogawa K, Bush DE, Ledoux JE. Contrasting Effects of Pretraining, Posttraining, and Pretesting Infusions of Corticotropin-Releasing Factor into the Lateral Amygdala: Attenuation of Fear Memory Formation but Facilitation of its Expression. Biol Psychiatry. 2012 Oct 1; doi: 10.1016/j.biopsych.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roozendaal B, Schelling G, McGaugh JL. Corticotropin-releasing factor in the basolateral amygdala enhances memory consolidation via an interaction with the beta-adrenoceptor-cAMP pathway: dependence on glucocorticoid receptor activation. J Neurosci. 2008 Jun 25;28(26):6642–6651. doi: 10.1523/JNEUROSCI.1336-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Papotti M, Ghe C, Cassoni P, Catapano F, Deghenghi R, Ghigo E, et al. Growth hormone secretagogue binding sites in peripheral human tissues. J Clin Endocrinol Metab. 2000 Oct;85(10):3803–3807. doi: 10.1210/jcem.85.10.6846. [DOI] [PubMed] [Google Scholar]
- 49.Cabral A, Suescun O, Zigman JM, Perello M. Ghrelin Indirectly Activates Hypophysiotropic CRF Neurons in Rodents. PLoS One. 2012;7(2):e31462. doi: 10.1371/journal.pone.0031462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rao RP, Anilkumar S, McEwen BS, Chattarji S. Glucocorticoids protect against the delayed behavioral and cellular effects of acute stress on the amygdala. Biol Psychiatry. 2012 Sep 15;72(6):466–475. doi: 10.1016/j.biopsych.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999 Dec 9;402(6762):656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 52.Mahmoud GS, Grover LM. Growth hormone enhances excitatory synaptic transmission in area CA1 of rat hippocampus. J Neurophysiol. 2006 May;95(5):2962–2974. doi: 10.1152/jn.00947.2005. [DOI] [PubMed] [Google Scholar]
- 53.Donahue CP, Jensen RV, Ochiishi T, Eisenstein I, Zhao M, Shors T, et al. Transcriptional profiling reveals regulated genes in the hippocampus during memory formation. Hippocampus. 2002;12(6):821–833. doi: 10.1002/hipo.10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, et al. Regulation of cocaine reward by CREB. Science. 1998 Dec 18;282(5397):2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- 55.Chen WY, Wight DC, Mehta BV, Wagner TE, Kopchick JJ. Glycine 119 of bovine growth hormone is critical for growth-promoting activity. Mol Endocrinol. 1991 Dec;5(12):1845–1852. doi: 10.1210/mend-5-12-1845. [DOI] [PubMed] [Google Scholar]
- 56.Chen WY, Wight DC, Wagner TE, Kopchick JJ. Expression of a mutated bovine growth hormone gene suppresses growth of transgenic mice. Proc Natl Acad Sci U S A. 1990 Jul;87(13):5061–5065. doi: 10.1073/pnas.87.13.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hansson C, Shirazi RH, Naslund J, Vogel H, Neuber C, Holm G, et al. Ghrelin influences novelty seeking behavior in rodents and men. PLoS One. 2012;7(12):e50409. doi: 10.1371/journal.pone.0050409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stotz G, Woggon B, Angst J. Psychostimulants in the therapy of treatment-resistant depression Review of the literature and findings from a retrospective study in 65 depressed patients. Dialogues Clin Neurosci. 1999 Dec;1(3):165–174. doi: 10.31887/DCNS.1999.1.3/gstotz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishitobi Y, Kohno K, Kanehisa M, Inoue A, Imanaga J, Maruyama Y, et al. Serum ghrelin levels and the effects of antidepressants in major depressive disorder and panic disorder. Neuropsychobiology. 2012;66(3):185–192. doi: 10.1159/000339948. [DOI] [PubMed] [Google Scholar]
- 60.Carlini VP, Perez MF, Salde E, Schioth HB, Ramirez OA, de Barioglio SR. Ghrelin induced memory facilitation implicates nitric oxide synthase activation and decrease in the threshold to promote LTP in hippocampal dentate gyrus. Physiol Behav. 2010 Aug 4;101(1):117–123. doi: 10.1016/j.physbeh.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 61.Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006 Mar;9(3):381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 62.Davis JF, Choi DL, Clegg DJ, Benoit SC. Signaling through the ghrelin receptor modulates hippocampal function and meal anticipation in mice. Physiol Behav. 2011 Apr 18;103(1):39–43. doi: 10.1016/j.physbeh.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grissom N, Bhatnagar S. Habituation to repeated stress: get used to it. Neurobiol Learn Mem. 2009 Sep;92(2):215–224. doi: 10.1016/j.nlm.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jovanovic T, Blanding NQ, Norrholm SD, Duncan E, Bradley B, Ressler KJ. Childhood abuse is associated with increased startle reactivity in adulthood. Depress Anxiety. 2009;26(11):1018–1026. doi: 10.1002/da.20599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grissom N, Iyer V, Vining C, Bhatnagar S. The physical context of previous stress exposure modifies hypothalamic-pituitary-adrenal responses to a subsequent homotypic stress. Horm Behav. 2007 Jan;51(1):95–103. doi: 10.1016/j.yhbeh.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 66.McGuire J, Herman JP, Horn PS, Sallee FR, Sah R. Enhanced fear recall and emotional arousal in rats recovering from chronic variable stress. Physiol Behav. 2010 Nov 2;101(4):474–482. doi: 10.1016/j.physbeh.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kinn AM, Gronli J, Fiske E, Kuipers S, Ursin R, Murison R, et al. A double exposure to social defeat induces sub-chronic effects on sleep and open field behaviour in rats. Physiol Behav. 2008 Nov 28;95(4):553–561. doi: 10.1016/j.physbeh.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992 Aug 21;588(2):341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- 69.Muscat R, Willner P. Suppression of sucrose drinking by chronic mild unpredictable stress: a methodological analysis. Neurosci Biobehav Rev. 1992 Winter;16(4):507–517. doi: 10.1016/s0149-7634(05)80192-7. [DOI] [PubMed] [Google Scholar]
- 70.Rosenkranz JA, Venheim ER, Padival M. Chronic stress causes amygdala hyperexcitability in rodents. Biol Psychiatry. 2010 Jun 15;67(12):1128–1136. doi: 10.1016/j.biopsych.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology. 2000 Nov;141(11):4255–4261. doi: 10.1210/endo.141.11.7757. [DOI] [PubMed] [Google Scholar]
- 72.Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, et al. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab. 2002 Jun;87(6):2988. doi: 10.1210/jcem.87.6.8739. [DOI] [PubMed] [Google Scholar]
- 73.Sato T, Fukue Y, Teranishi H, Yoshida Y, Kojima M. Molecular forms of hypothalamic ghrelin and its regulation by fasting and 2-deoxy-d-glucose administration. Endocrinology. 2005 Jun;146(6):2510–2516. doi: 10.1210/en.2005-0174. [DOI] [PubMed] [Google Scholar]
- 74.Hou Z, Miao Y, Gao L, Pan H, Zhu S. Ghrelin-containing neuron in cerebral cortex and hypothalamus linked with the DVC of brainstem in rat. Regul Pept. 2006 May 15;134(2–3):126–131. doi: 10.1016/j.regpep.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 75.Olszewski PK, Grace MK, Billington CJ, Levine AS. Hypothalamic paraventricular injections of ghrelin: effect on feeding and c-Fos immunoreactivity. Peptides. 2003 Jun;24(6):919–923. doi: 10.1016/s0196-9781(03)00159-1. [DOI] [PubMed] [Google Scholar]
- 76.Albarran-Zeckler RG, Brantley AF, Smith RG. Growth hormone secretagogue receptor (GHS-R1a) knockout mice exhibit improved spatial memory and deficits in contextual memory. Behav Brain Res. 2012 Jun 15;232(1):13–19. doi: 10.1016/j.bbr.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. High constitutive signaling of the ghrelin receptor--identification of a potent inverse agonist. Mol Endocrinol. 2003 Nov;17(11):2201–2210. doi: 10.1210/me.2003-0069. [DOI] [PubMed] [Google Scholar]
- 78.Camina JP, Carreira MC, El Messari S, Llorens-Cortes C, Smith RG, Casanueva FF. Desensitization and endocytosis mechanisms of ghrelin-activated growth hormone secretagogue receptor 1a. Endocrinology. 2004 Feb;145(2):930–940. doi: 10.1210/en.2003-0974. [DOI] [PubMed] [Google Scholar]
- 79.Vyas A, Jadhav S, Chattarji S. Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience. 2006 Dec 1;143(2):387–393. doi: 10.1016/j.neuroscience.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 80.Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002 Aug 1;22(15):6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zearfoss NR, Alarcon JM, Trifilieff P, Kandel E, Richter JD. A molecular circuit composed of CPEB-1 and c-Jun controls growth hormone-mediated synaptic plasticity in the mouse hippocampus. J Neurosci. 2008 Aug 20;28(34):8502–8509. doi: 10.1523/JNEUROSCI.1756-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rodrigues SM, LeDoux JE, Sapolsky RM. The influence of stress hormones on fear circuitry. Annu Rev Neurosci. 2009;32:289–313. doi: 10.1146/annurev.neuro.051508.135620. [DOI] [PubMed] [Google Scholar]
- 83.Roozendaal B, McGaugh JL. Memory modulation. Behav Neurosci. 2011 Dec;125(6):797–824. doi: 10.1037/a0026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krugers HJ, Karst H, Joels M. Interactions between noradrenaline and corticosteroids in the brain: from electrical activity to cognitive performance. Front Cell Neurosci. 2012;6:15. doi: 10.3389/fncel.2012.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hansson C, Annerbrink K, Nilsson S, Bah J, Olsson M, Allgulander C, et al. A possible association between panic disorder and a polymorphism in the preproghrelingene. Psychiatry Res. 2013 Mar 30;206(1):22–25. doi: 10.1016/j.psychres.2012.09.051. [DOI] [PubMed] [Google Scholar]
- 86.Mahan AL, Ressler KJ. Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci. 2012 Jan;35(1):24–35. doi: 10.1016/j.tins.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seim I, El-Salhy M, Hausken T, Gundersen D, Chopin L. Ghrelin and the brain-gut axis as a pharmacological target for appetite control. Curr Pharm Des. 2012;18(6):768–775. doi: 10.2174/138161212799277806. [DOI] [PubMed] [Google Scholar]
- 88.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates - The New Coronal Set. 5. Elsevier Academic Press; San Diego: 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.