

Abstract
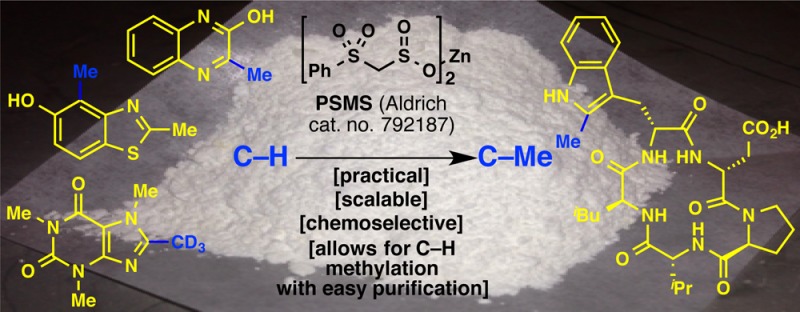
A practical C–H functionalization method for the methylation of heteroarenes is presented. Inspiration from Nature’s methylating agent, S-adenosylmethionine (SAM), allowed for the design and development of zinc bis(phenylsulfonylmethanesulfinate), or PSMS. The action of PSMS on a heteroarene generates a (phenylsulfonyl)methylated intermediate that can be easily separated from unreacted starting material. This intermediate can then be desulfonylated to the methylated product or elaborated to a deuteriomethylated product, and can divergently access medicinally important motifs. This mild, operationally simple protocol that can be conducted in open air at room temperature is compatible with sensitive functional groups for the late-stage functionalization of pharmacologically relevant substrates.
The concept of methylation plays a key role in epigenetics, as DNA and histone methylation is largely responsible for gene expression without affecting the gene sequence.1 Uracil methylation is important for DNA biosynthesis and RNA post-translational modification,2 and cysteine methylation can disrupt ubiquitin-chain binding, which in turn affects signaling pathways.3 Methylation is also fundamental in medicinal chemistry, as several biological and physical properties of a drug can be adversely or favorably affected with the mere addition of one methyl group to a lead compound.4 For example, a molecular unit as small as methyl can improve the IC50 value of a drug candidate 100-fold in a phenomenon called the “magic methyl effect”.5,6 In light of these merits, “a call for new C–H methylation reactions” was made recently to encourage academic and industrial chemistry research groups to study the topic in detail.6
To address this challenge, we designed a mild method for C–H methylation (Figure 1A) to enable laboratory methylation of tryptophan (1), cysteine (2), or even uracil (3). With inherently reactive functional groups such as the indole N–H, carboxyl O–H, and uracil N–H bonds present on 1–3, a radical reaction pathway, as opposed to an ionic/polar one, was sought. Our design plan was influenced by a key methylating agent found in Nature, S-adenosylmethionine (SAM, or 4).7 SAM (4) is known to methylate 1–3, with the case of tryptophan being particularly compelling since enzymological studies point to SAM as a source of methyl radical (Me•).7,8
Figure 1.
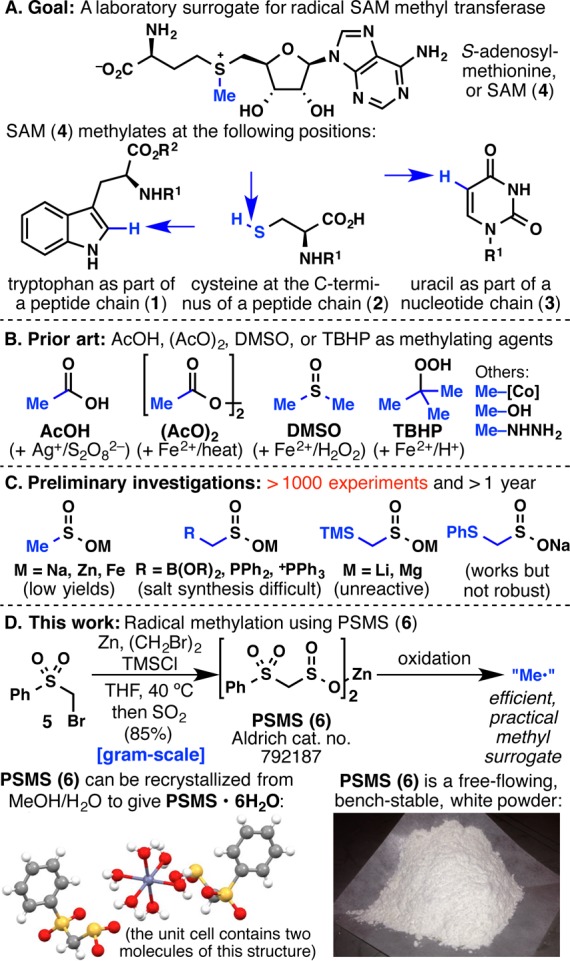
(A) S-Adenosylmethionine (SAM) as an inspiration for mild methylation. (B) Prior art and (C) preliminary investigations in methyl radical chemistry. (D) The design and development of a practical source of a methyl radical equivalent.
The idea of employing a methyl radical to methylate arenes and heteroarenes is not new, as many methyl radical sources are known (Figure 1B),9 including acetic acid,9a−9c diacetyl peroxide,9d−9f DMSO,9g−9j and TBHP.9k,9l These methyl radical sources are largely inefficient, or require highly acidic or high-temperature conditions for their generation. However, perhaps the greatest difficulty arises from the similar polarities of the starting material and the methylated product, which prevents proper isolation and purification of the product. With this in mind, our ultimate strategy involved the formation of a methyl radical surrogate—a reagent that installs a functional group that clearly differentiates the product from the starting material in terms of polarity while being easily unmasked to reveal a methyl group.
Based on our ongoing efforts in the synthesis and use of alkanesulfinate reagents for heteroarene C–H functionalization,10 investigations were centered around the use of methanesulfinate or substituted methanesulfinate reagents (Figure 1C). Sodium methanesulfinate, a commercially available reagent, only reacted with heterocycles such as caffeine (7a) in very low yields, and only after using Fe2+ salts as an additive. Zinc and iron methanesulfinates were then synthesized, but they only reacted with heterocycles in persistently low yields. Methanesulfinates substituted with boronate, phosphine, or phosphonium groups proved to be difficult to make, and although lithium and magnesium (trimethylsilyl)methanesulfinates were synthesized, they did not react with heteroarenes even in the presence of ZnCl2.10d
Mechanistic enzymology of SAM (4) points to the C–S bond as being an essential stabilizing force for the generation of a carbon-based radical that is eventually cleaved to reveal a methyl group. This logic was then incorporated into the design of the next methanesulfinate reagent. Thus, sodium (phenylthio)methanesulfinate was synthesized, which proved to react with an array of heterocycles, albeit with varying yields. With the desire to address this lack of robustness, zinc bis(phenylsulfonylmethanesulfinate) (PSMS, or 6; Sigma-Aldrich catalog no. 792187) was invented (Figure 1D). PSMS (6) can be synthesized in one step from bromomethyl phenyl sulfone (5)11 and is a free-flowing, bench-stable (stable at room temperature under air over 4 months), white powder whose structure was characterized by X-ray crystallography as the hexahydrate. PSMS (6) enables the installation of a (phenylsulfonyl)methyl group onto a heteroarene at both an early and late stage of the synthesis. This intermediate is easily separated from starting material, and it can be cleanly desulfonylated to give the methylated product or used divergently to generate a host of other useful products (vide infra). This chemistry has been field-tested at Pfizer and found to be an enabling method for SAR studies.
The feasibility of our design was first tested by reacting PSMS (6) with TBHP and caffeine (7a), resulting in 2-[(phenylsulfonyl)methyl]caffeine (7b). A solvent screen revealed that trifluorotoluene (PhCF3) allows for the highest conversion of 7a, and further optimization of reaction conditions led to the use of 1.5 equiv of PSMS (6) and 5.0 equiv of TBHP in 5:2 PhCF3:H2O at room temperature for 24 h (see the Supporting Information for details). It is of note that air has no noticeable effect on this reaction, which in fact can be conducted in an open flask if desired.
With these standard conditions in hand, the substrate scope for the (phenylsulfonyl)methylation reaction was examined (Table 1). Caffeine (7a) and other xanthines gave the corresponding products 7b–9b in moderate to good yields. Electron-rich heterocycles such as pyrroles gave moderate to excellent yields of product (10b–13b), owing to the electrophilic nature of the radical. Imidazoles and electron-rich diazines gave good yields of product as well (14b–19b). Other heterocycles generally gave good yields of product in the case of indoles (20b–23b), azaindoles (24b), benzimidazoles (25b), benzothiazoles (26b, 27b), and indolizine-type systems (28b–32b). Notably, this reaction tolerates free O–H and N–H bonds, and the most electron-rich position of the substrate often reacts with PSMS (6) in a chemoselective fashion. Perhaps most importantly, this method allows for the easy separation of product from starting material when unreacted starting material remains (e.g., 9b, 16b), especially for very polar frameworks such as 18b, whose methylated product co-elutes with the starting material.
Table 1. Substrate Scope of the (Phenylsulfonyl)methylation Reaction.
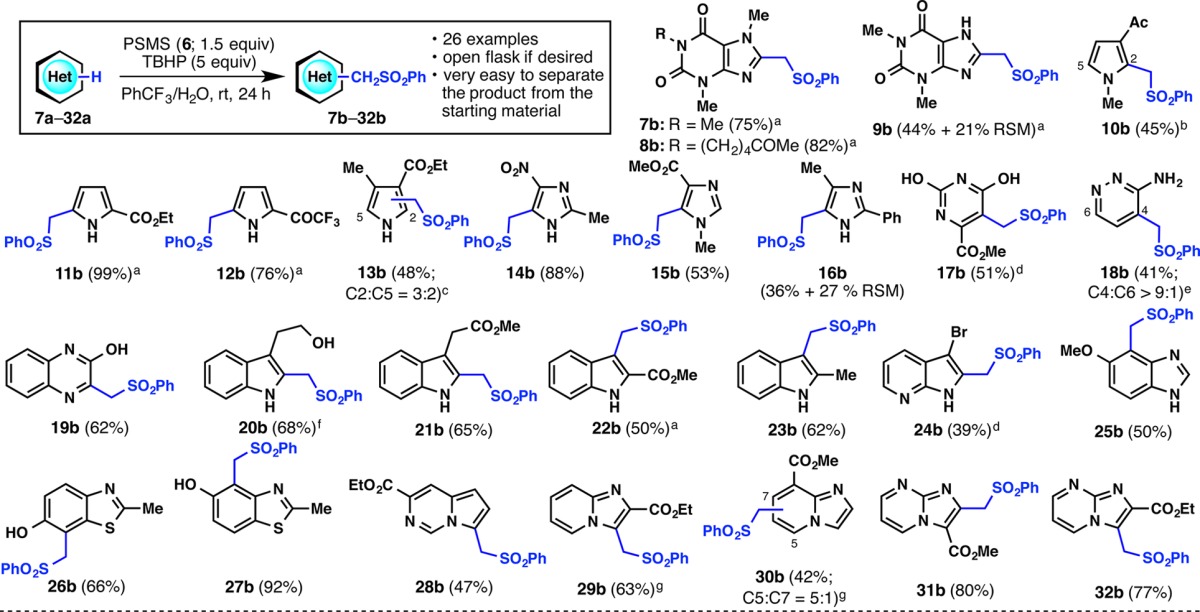
General conditions: heterocycle (0.2 mmol, 1.0 equiv), PSMS (6; 0.3 mmol, 1.5 equiv), TBHP (1.0 mmol, 5.0 equiv), PhCF3/H2O (1 mL/0.4 mL), rt; isolated yields are shown. RSM = recovered starting material.
3.0 equiv PSMS (6) was added in two portions.
2,5-Disubstituted product was obtained in 11% yield.
2,5-Disubstituted product was obtained in 26% yield.
Reaction ran in 2:1 DMSO:H2O.
Reaction ran in DMSO with 1.0 equiv TFA.
1.0 equiv PSMS (6) was used.
These substrates do not work at all using Fenton methylation conditions (DMSO/Fe2+/H2O2).
The chemoselectivity of this reaction was further apparent when it was applied to biologically relevant motifs (Table 2). In a biomimetic fashion,8N-Ac- and N-Cbz- l-tryptophan methyl esters were methylated at the indole C2 position without protecting the free N–H bond, giving rise to l-33b and l-34b in good yields. Without the need to protect the carboxyl O–H or the heterocyclic N–H bonds, N-Ac-cysteine provided S-substituted product 35b, and uracil provided 36b. The potential of this reaction for use in bioconjugation, pull-down experiments, and other native chemical tagging12 processes of relevance to chemical biology has not escaped our attention and is a current topic of study in our laboratory.
Table 2. Products from Biologically Relevant Substrates.
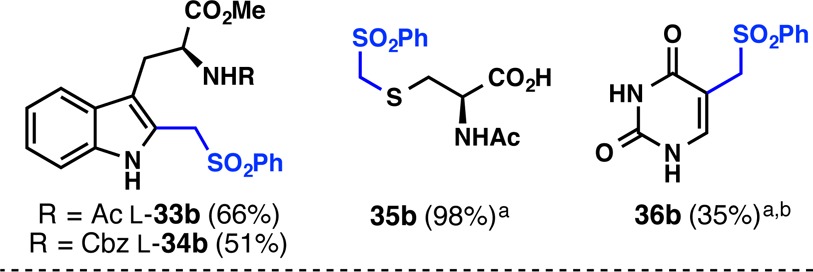
General conditions: substrate (0.2 mmol, 1.0 equiv), PSMS (6; 0.3 mmol, 1.5 equiv), TBHP (1.0 mmol, 5.0 equiv), PhCF3/H2O (1 mL/0.4 mL), rt; isolated yields are shown.
H2O was used instead of PhCF3/H2O.
3.0 equiv PSMS (6) was added in two portions.
To unveil the methyl group from the phenylsulfonyl mask, desulfonylation was attempted on 7b (Table 3). Three different (and orthogonal) conditions were identified (Mg in MeOH,13 SmI2 in H2O/THF,14 and Raney-Ni in EtOH15), and all gave >90% yield of 8-methylcaffeine (7c). These desulfonylating conditions were then applied to selected substrates. One of the three conditions always gave good (>73%) yields of product.
Table 3. Desulfonylation Reveals the Methyl Group.
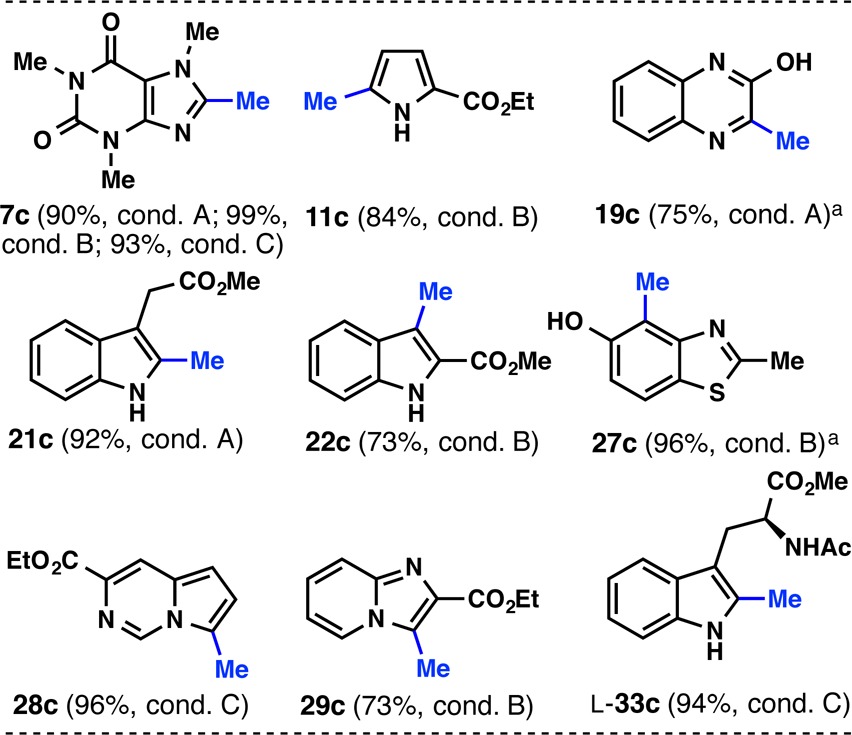
Reaction conditions: (A) Mg (40 equiv), MeOH, 50 °C, 2 h; (B) SmI2 (4 equiv), H2O (50 equiv), THF, rt, 30 min; (C) Raney-nickel, EtOH, reflux, 2 h.
The reaction was followed by bubbling with 1 atm O2 for 12 h.
The phenylsulfonyl moiety should not be considered a cumbersome protecting group that needs to be removed, but rather an enabling group that facilitates the isolation of the product, as well as a functional group that can lead to other useful motifs in medicinal chemistry (Scheme 1). Thus, caffeine (7a) was (phenylsulfonyl)methylated to give 7b (notably on gram-scale), which then reacted in four different ways other than mere desulfonylation. The adduct 7b was transformed into trideuteriomethylcaffeine 7d in a two-step reaction in 70% overall yield and with 91% deuterium incorporation. The phenylsulfonyl group in 7b can also be transformed into a difluoromethyl group (7e)16 and a xanthate (7f),17 as well as an alkene under Julia conditions (7g).18
Scheme 1. Synthetic Utility of the Phenylsulfonylated Product.
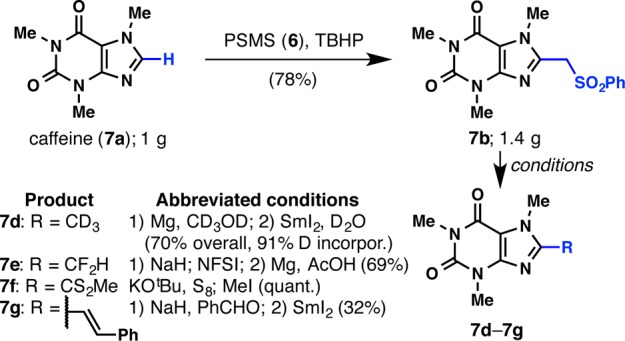
Reaction conditions: 7a to 7b, PSMS (6, 3 equiv), TBHP (10 equiv), PhCF3/H2O, rt, 24 h (78%); 7b to 7d, (1) Mg (40 equiv), CD3OD, reflux, 24 h; (2) SmI2 (6 equiv), THF:D2O (10:1), rt, 30 min, 70% (2 steps), 91% D incorporation; 7b to 7e, (1) NaH (10 equiv), NFSI (5 equiv), DMF, 0 °C to rt, 12 h, 94%; (2) Mg (30 equiv), 8 M HOAc:NaOAc (1:1) buffer solution, DMF, rt, 8 h, 73%. 7b to 7f, KOtBu, S8, THF; then MeI (quant.); 7b to 7g, (1) NaH, PhCHO, THF, 50 °C, 36 h; (2) SmI2, rt, 5 min (32%).
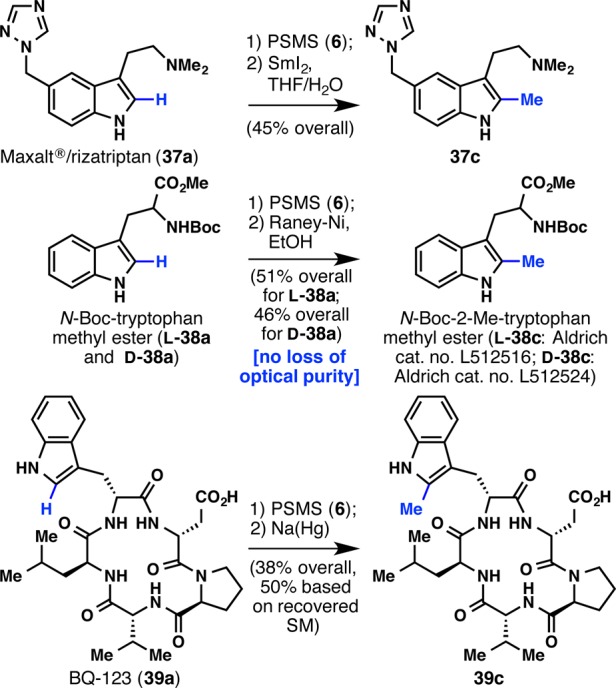
As a testament to the utility of PSMS (6), biologically important substrates can be methylated in a chemoselective manner, forming 37c, l-38c, d-38c, and 39c in good yields (Scheme 2). Maxalt (rizatriptan, 37a) is a successful commercial drug used to treat migraines, and this example showcases how this methylation strategy is amenable to the late-stage modification of important pharmaceutical agents. 2-Methyltryptophan is an intermediate in the biosynthesis of the antibiotic thiostrepton,8 and oligopeptides containing this privileged motif are endothelin receptor antagonists.19 However, its synthesis is difficult, and often resorts to the union of 2-methylindole with a protected aziridine19 or an α-aminoenoate.20 Sigma-Aldrich has now commercialized l-38c and d-38c using this chemistry. As for the methylation of a selective ETA endothelin receptor antagonist, BQ-123 (39a),19,21 selective methylation at the tryptophan C2 position would otherwise require a de novo synthesis starting with 2-methyltryptophan.
Scheme 2. Methylation of Biologically Important Substrates.
Finally, any chemical method is not without its limitations. In Tables 1 and 2, pyridine-containing substrates are not depicted. PSMS (6) behaves as an electrophilic radical and therefore reacts rather sluggishly with some electron-deficient heteroarenes.
In summary, a new reagent (PSMS, 6) was invented to provide practical access to methylated heteroarenes. This two-stage methylation, rather than direct methylation, is critical to allow for product purification. This method is efficient, can be conducted on gram scale and in an open flask, and can tolerate sensitive functional groups that ionic or polar reactions will damage. The formed (phenylsulfonyl)methylated intermediates can also divergently access many medicinally relevant motifs, including deuterated products. This strategy has already been field-tested at Pfizer (compounds 17b, 18b, and 24b are building blocks in a current program), and studies demonstrating its utility in more complex biological systems are currently underway.
Acknowledgments
Financial support for this work was provided by NIH/NIGMS (GM-106210), Sigma-Aldrich, SIOC/Zhejiang Medicine Co./Pharmaron (postdoctoral fellowships to J.G. and Q.Z.), and JSPS (postdoctoral fellowship for Y.Y.). The authors thank A. L. Rheingold and C. E. Moore for X-ray crystallographic analysis, J. Ewanicki and W. Wang for assistance with NMR spectroscopy, and K.-J. Xiao for assistance with chiral HPLC, as well as R. Gianatassio and J. A. Romero-Revilla for technical assistance in the early phases of the project.
Supporting Information Available
Experimental procedures and analytical data (1H and 13C NMR, MS) for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ J.G. and Q.Z. contributed equally to this paper.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Jones P. A.; Takai D. Science 2001, 293, 1068. [DOI] [PubMed] [Google Scholar]; b Jaenisch R.; Bird A. Nat. Genet. 2003, 33, 245. [DOI] [PubMed] [Google Scholar]; c Sasaki H.; Matsui Y. Nat. Rev. Genet. 2008, 9, 129. [DOI] [PubMed] [Google Scholar]
- Mishanina T. V.; Koehn E. M.; Kohen A. Bioorg. Chem. 2012, 43, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Ding X.; Cui J.; Xu H.; Chen J.; Gong Y.-N.; Hu L.; Zhou Y.; Ge J.; Lu Q.; Liu L.; Chen S.; Shao F. Nature 2012, 481, 204. [DOI] [PubMed] [Google Scholar]
- Leung C. S.; Leung S. S. F.; Tirado-Rives J.; Jorgensen W. L. J. Med. Chem. 2012, 55, 4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamovsky I.; de Graaf C.; Alderin L.; Bengtsson M.; Bladh H. k.; Börjesson L.; Connolly S.; Dyke H. J.; van den Heuvel M.; Johansson H.; Josefsson B.-G. r.; Kristoffersson A.; Linnanen T.; Lisius A.; Männikkö R.; Nordén B.; Price S.; Ripa L.; Rognan D.; Rosendahl A.; Skrinjar M.; Urbahns K. J. Med. Chem. 2009, 52, 7706. [DOI] [PubMed] [Google Scholar]
- Schönherr H.; Cernak T. Angew. Chem., Int. Ed. 2013, 52, 12256. [DOI] [PubMed] [Google Scholar]
- a Zhang Q.; Liu W. J. Biol. Chem. 2011, 286, 30245. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang Q.; van der Donk W. A.; Liu W. Acc. Chem. Res. 2012, 45, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fujimori D. G. Curr. Opin. Chem. Biol. 2013, 17, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chan K. K. J.; Thompson S.; O’Hagan D. ChemBioChem 2013, 14, 675. [DOI] [PubMed] [Google Scholar]
- a Zhou P.; O’Hagan D.; Mocek U.; Zeng Z.; Yuen L. D.; Frenzel T.; Unkefer C. J.; Beale J. M.; Floss H. G. J. Am. Chem. Soc. 1989, 111, 7274. [Google Scholar]; b Kelly W. L.; Pan L.; Li C. J. Am. Chem. Soc. 2009, 131, 4327. [DOI] [PubMed] [Google Scholar]; c Pierre S.; Guillot A.; Benjdia A.; Sandström C.; Langella P.; Berteau O. Nat. Chem. Biol. 2012, 8, 957. [DOI] [PubMed] [Google Scholar]
- a Minisci F.; Bernardi R.; Bertini F.; Galli R.; Perchinummo M. Tetrahedron 1971, 27, 3575. [Google Scholar]; b Minisci F.; Mondelli R.; Gardini G. P.; Porta O. Tetrahedron 1972, 28, 2403. [Google Scholar]; c Sugimori A.; Yamada T. Bull. Chem. Soc. Jpn. 1986, 59, 3911. [Google Scholar]; d Levy M.; Szwarc M. J. Am. Chem. Soc. 1955, 77, 1949. [Google Scholar]; e Janda M.; Šrogl J.; Stibor I.; Němec M.; Vopatrná P. Tetrahedron Lett. 1973, 14, 637. [Google Scholar]; f Araki M.; Maeda M.; Kawazoe Y. Tetrahedron 1976, 32, 337. [Google Scholar]; g Bertilsson B.-M.; Gustafsson B.; Kühn I.; Torssell K. Acta Chem. Scand. 1970, 24, 3590. [Google Scholar]; h Giordano C.; Minisci F.; Tortelli V.; Vismara E. J. Chem. Soc., Perkin Trans. 2 1984, 293. [Google Scholar]; i Crean C.; Geacintov N. E.; Shafirovich V. J. Phys. Chem. B 2009, 113, 12773. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Kawai K.; Li Y.-S.; Song M.-F.; Kasai H. Bioorg. Med. Chem. Lett. 2010, 20, 260. [DOI] [PubMed] [Google Scholar]; k Maeda M.; Nushi K.; Kawazoe Y. Tetrahedron 1974, 30, 2677. [Google Scholar]; l Hix S.; Da Silva Morais M.; Augusto O. Free Radical Biol. Med. 1995, 19, 293. [DOI] [PubMed] [Google Scholar]
- a Ji Y.; Brueckl T.; Baxter R. D.; Fujiwara Y.; Seiple I. B.; Su S.; Blackmond D. G.; Baran P. S. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14411. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fujiwara Y.; Dixon J. A.; Rodriguez R. A.; Baxter R. D.; Dixon D. D.; Collins M. R.; Blackmond D. G.; Baran P. S. J. Am. Chem. Soc. 2012, 134, 1494. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fujiwara Y.; Dixon J. A.; O’Hara F.; Funder E. D.; Dixon D. D.; Rodriguez R. A.; Baxter R. D.; Herlé B.; Sach N.; Collins M. R.; Ishihara Y.; Baran P. S. Nature 2012, 492, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhou Q.; Ruffoni A.; Gianatassio R.; Fujiwara Y.; Sella E.; Shabat D.; Baran P. S. Angew. Chem., Int. Ed. 2013, 52, 3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Deeming A. S.; Russell C. J.; Hennessy A. J.; Willis M. C. Org. Lett. 2014, 16, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rocke B. N.; Bahnck K. B.; Herr M.; Lavergne S.; Mascitti V.; Perreault C.; Polivkova J.; Shavnya A. Org. Lett. 2014, 16, 154. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Gui J.; Pan C.-M.; Albone E.; Cheng X.; Suh E. M.; Grasso L.; Ishihara Y.; Baran P. S. J. Am. Chem. Soc. 2013, 135, 12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C.; Carpino L. A. J. Org. Chem. 1985, 50, 1749. [Google Scholar]
- Kumar V.; Ramesh N. G. Chem. Commun. 2006, 4952. [DOI] [PubMed] [Google Scholar]
- Sadanandan E. V.; Srinivasan P. C. Synthesis 1992, 648. [Google Scholar]
- Ni C.; Hu J. Tetrahedron Lett. 2005, 46, 8273. [Google Scholar]
- Abrunhosa I.; Gulea M.; Masson S. Synthesis 2004, 928. [Google Scholar]
- Keck G. E.; Savin K. A.; Weglarz M. A. J. Org. Chem. 1995, 60, 3194. [Google Scholar]
- Fukami T.; Yamakawa T.; Niiyama K.; Kojima H.; Amano Y.; Kanda F.; Ozaki S.; Fukuroda T.; Ihara M.; Yano M.; Ishikawa K. J. Med. Chem. 1996, 39, 2313. [DOI] [PubMed] [Google Scholar]
- a Angelini E.; Balsamini C.; Bartoccini F.; Lucarini S.; Piersanti G. J. Org. Chem. 2008, 73, 5654. [DOI] [PubMed] [Google Scholar]; b Kieffer M. E.; Repka L. M.; Reisman S. E. J. Am. Chem. Soc. 2012, 134, 5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatola A. F.; Crozet Y.; deWit D.; Yanagisawa M. J. Med. Chem. 1996, 39, 3842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.