

1. INTRODUCTION
The RNA polymerase II (Pol II) C-terminal domain (CTD) is a repetitive disordered domain that extends from the catalytic core of the enzyme. This “tail” domain is heavily modified by phosphorylation, glycosylation and proline isomerization. In addition to the enzymes that modify the tail a number of RNA processing factors and chromatin modification factors interact with the CTD. Thus, this domain acts as a tether to bring into close proximity the machinery necessary to synthesize and process Pol II transcripts.
By definition the CTD consists of the amino acid sequences extending from the largest subunit, RPO21 or RPB1 in yeast and POLR2A in human. Specifically the CTD consists of sequences beyond the most C-terminal region that is conserved among the largest subunits of all multi-subunit RNA polymerases 1. This “H” homology region plays a role in positioning the catalytic core and provides surfaces for interactions with Rpb2 and Rpb6 2. The CTD thus consists of three regions (Figure 1). In order of proximity to the catalytic core these are the linker, the heptad repeats, and a C-terminal non-repeat or “tip” domain. The linker region is not conserved among different organisms but does contain an enrichment of amino acids found in the CTD.
Figure 1.
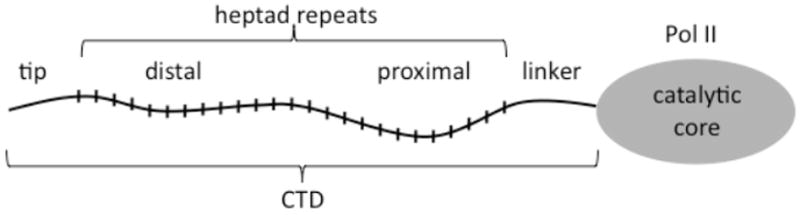
Diagram showing the three regions of the CTD.
The heptad repeat domain consists of multiple tandem repeats of the consensus sequence YSPTSPS. Since this sequences is tandemly repeated the consensus could be any one of seven permutations of this sequence. The selection of Tyr as position one is essentially arbitrary and other permutations have been proposed 1. Because the Tyr in position one is more universally adopted in the literature we will use this nomenclature in this review.
The discovery of the CTD in the mid-1980’s 3 explained the multiple forms of Pol II that were identified by ion exchange chromatography4,5. These forms, termed Pol IIO, IIA and IIB differed by the mobility in SDS gels of the largest subunit termed IIo, IIa and IIb, respectively (Figure 2). Comparing the amino acid content of these different forms to the coding sequence revealed that the most rapidly migrating form, IIb, is a proteolytic breakdown product lacking the CTD 3a. Forms IIo and IIa both contain the CTD but differ in that IIo is highly phosphorylated as could be shown by labeling with 32P or by conversion of IIo to IIa by phosphatase treatment 6. The ability to separate phosphorylated and unphosphorylated CTD species by SDS gel electrophoresis has allowed the characterization of CTD phosphorylation states established both in vitro and in vivo.
Figure 2.
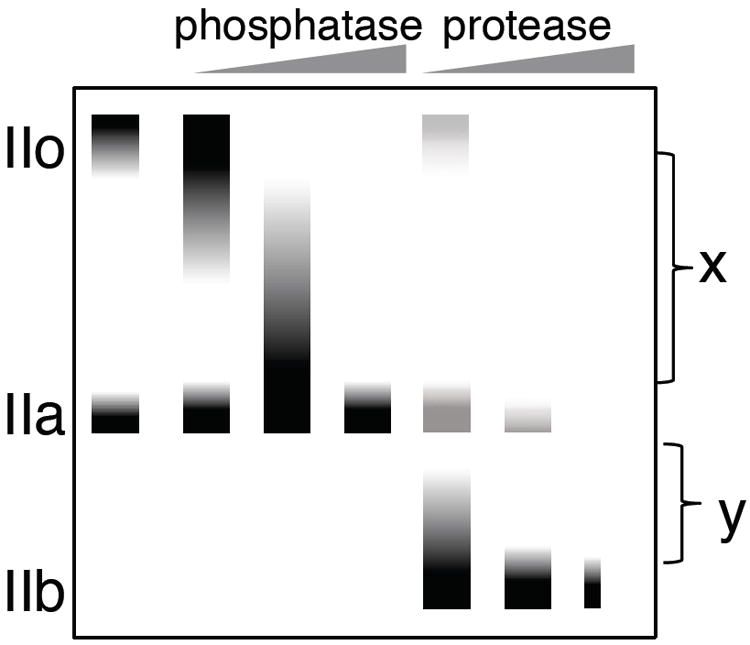
Idealized SDS PAGE separation of the Rpb1 subunit. Increasing phosphatase treatment increases the mobility of the IIo form while proteolysis increases the mobility of both forms. x refers to intermediate phosphorylation states while y refers to intermediates in CTD degradation.
The existence of both the IIO and IIA forms of Pol II in vivo led to the discovery that Pol II undergoes reversible phosphorylation with the unphosphorylated IIA form functioning in initiation and the phosphorylated IIO form carrying out the elongation step 7. Genetic and biochemical studies showed that multiple sites in the CTD heptad repeats could be phosphorylated and these sites were not functionally equivalent 8. Mapping different CTD phosphorylations to different chromatin sites in coding regions led to the “CTD code” hypothesis in which different phosphorylation states exist in the initiation, elongation and termination phases of the transcription cycle for the purpose of recruiting the appropriate factors to carry out the needed processing reactions 9.
In this review we will focus on the general properties of the CTD and its modification and interactions with CTD-binding proteins. Gene-specific aspects of CTD function will be discussed in accompanying reviews by Jeronimo et al. and Eick and Geyer. To set the stage for discussing the CTD we will consider the evolution of this domain. We will then discuss structural studies of the CTD. Genetic and gene expression effects of altering the CTD will then be considered. Finally, we will discuss the kinases phosphatases and proline isomerases that establish the code and the CTD-binding proteins that read the code.
2. EVOLUTION OF THE CTD
C-terminal extensions are specific to the largest subunit of RNA polymerase II and related subunits. No similar extension of the largest subunit is seen on Pol I or Pol III nor in any prokaryotic or archaeal largest subunit 10. This suggests that the CTD emerged as a Pol II specific adaptation after the duplication of subunit genes and the specialization of the three eukaryotic RNA polymerases. In plants, the existence of extensions from the largest subunits of RNA polymerases IV and V is consistent with this view as these plant-specific enzymes are most closely related to Pol II 11. In this section we will consider the evolutionary origin of the Pol II CTD and the conditions that have shaped the evolution of this domain.
2.1. Origin of the CTD
While virtually every Pol II largest subunit (Rpb1) has a sequence extending from the conserved “H” homology region 2 there is a wide difference in the sequence, repetitiveness, consensus, spacing and length of the CTD. Stiller and Hall derived an evolutionary tree based on the catalytic domains of Rpb1 and used this tree to distinguish between a CTD-clade that consists of organisms in which the CTD is fixed in the YSPTSPS consensus and primordial CTDs like those of Trypanosoma brucei, Giardia lamblia, and Trichomonas vaginalis 12 In T. brucei and G. lamblia there are extensions of 291 and 267 amino acids that are rich in Ser and Thr but with no discernable repeat (GenBank AAZ13503.1 and EDO76544.1). Trichomonas is even further from the CTD-clade and its extension of 320 amino acids is rich in Leu, Lys, Glu and Asp (GenBank EAY20967.1). Thus, in the most deeply branched eukaryotes there is little evidence for a repeated sequence. Chapman et al. 1 have proposed that in primordial CTDs the presence of sub-motifs SPXY and YSPX (where X is any amino acid) coalesced to form a heptad motif SPXYSPX. Amplification of this sequence could have provided selective advantages that enabled the development of more sophisticated gene regulatory mechanisms.
While it seems logical that CTD clade organisms containing the YSPTSPS repeat originated from a common ancestor, there are several organisms that deviate from this consensus suggesting the possibility that repetitive C-terminal repeat domains emerged more than once. Aspergillus oryzae has multiple repeats with Phe in position one (FSPTSPS) 13 while in Plasmodium falciparum the consensus is YSPTSPK 14. Mastigamoeba invertens is the most challenging to explain as this brown algae species contains repeats with the consensus YSPASPA 15. It is hard to imagine how two positions in multiple repeats could have been altered so completely through single amino acid substitution starting with the YSPTSPS consensus. One possibility is that changes in one repeat were duplicated and conferred a selective advantage over the YSPTSPS consensus that was lost over time. It remains difficult, however, to rule out the possibility that Mastigamoeba and the other non-consensus repeats evolved independently from a primordial Ser Pro rich C-terminal extension.
2.2. Expansion of the CTD
In organisms that evolved more than one repeat the stage was set for further expansion. There are two ways that repeated DNA sequences might lead to further duplication. For short repeats (3-4 nt) replication slippage is the predominant cause of insertion or deletion of repeats 16. For longer sequences like the 21 nt DNA repeat encoding the CTD heptad the mechanism of further duplication is unlikely to be replication slippage but rather out of register recombination between repeat sequences 17. The results of such unequal crossovers would lead to rapid expansion of the heptad repeat. An example of this process is provided in S. cerevisiae where eight consensus repeats are sufficient for viability but result in slow growth and sensitivity to extremes of temperature 18. Large colonies derived from this strain represent rapidly growing revertants that contain increased number of heptad repeats 18. Further culturing this strain for about 100 population doublings leads to the appearance of sub-populations with CTDs containing 13-19 repeats and a loss of the 8 repeat parental strain (Creamer and Corden, unpublished). This observation indicates that growth in laboratory culture provides sufficient selective pressure for expansion of the CTD.
Clearly, the re-construction of the S. cerevisiae CTD from identical 21 nt DNA oligos enhances the rate of recombination 18. Naturally occurring 21 nt sequences have diverged from one another likely stabilizing the CTD from rampant expansion and contraction by recombination. The presence of many Ser residues with 6 codons each has left an evolutionary clue, however, to the expansion process 1. In humans the distribution of Ser codons indicates that the consensus repeats (Figure 3) proximal to the catalytic core arose by tandem duplications. Thus, recombination among repeat domains can lead to the rapid evolution of longer CTDs.
Figure 3.
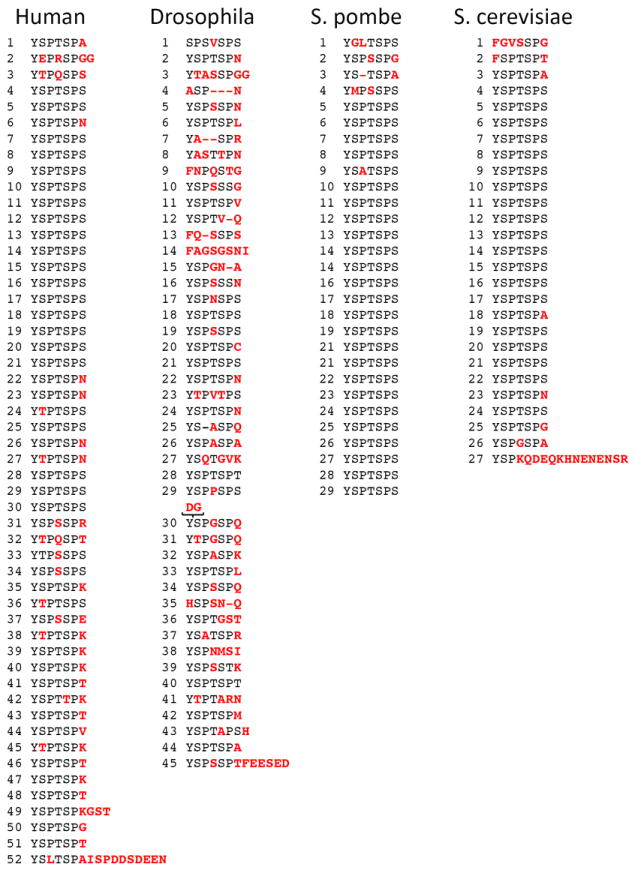
Sequences of CTDs from different organisms. The sequences are aligned to emphasize the heptad repeat. Residues in red deviate from the consensus.
2.3. The CTD Clade
The presence of consensus YSPTSPS repeats in the CTD of animals, plants and fungi led Stiller and colleagues to propose the CTD clade hypothesis. The crux of their argument is that the CTD provides an essential function that requires tandem repeats of YSPTSPS. Among these organisms there is wide variation in the number of repeats and the presence of non-consensus amino acids. There is little variation, however in the tandem arrangement of the heptad repeats. Within the CTD-clade the number of repeats varies noticeably from 52 repeats in vertebrates to ~ 20 repeats in fungi yielding a rough correlation between the complexity of the organism and the number of repeats 12,19.
In the Saccharomyces family including cerevisiae, bayanus, mikatae, castellii and kulyveri there are 27, 25, 26, 24 and 25 repeats respectively (Saccharomyces Genome Database). The positions of the few non-consensus amino acids also differ in these species. These differences in both number of repeats and the position of non-consensus repeats argues for considerable instability during the 20 million years since their last common ancestor (www.timetree.org 20). This instability is further supported by the observation that two commonly used in laboratory strains of S. cerevisiae (S288c and A364A) contain 27 and 26 repeats respectively and based on DNA sequence comparison this difference results from two changes; an insertion of two repeats and a deletion of one repeat 21. Adaptation by CTD repeat expansion is also seen with CTD truncation mutants that result in cold-sensitivity. Selection for spontaneous revertants of this phenotype yielded CTDs which contain more heptad repeats 22. These observations indicate that the naturally occurring Saccharomyces cerevisiae CTD is genetically unstable.
In more complicated multicellular organisms there are generally more repeats and considerable variation in the heptapeptide sequence. In Drosophila melanogaster the consensus heptad is YSPTSPS but only 2 of 45 repeats match the consensus (Figure 3). Despite this degree of degeneration the tandem register of repeats is largely maintained indicating that the repetitive nature of the CTD is more important than the actual sequence. Examining different Drosophila species (melanogaster, virilis, mojavensis, GenBank AAF48057.1, EDW66298.1 and EDW06178.1) indicates that the number of repeats is maintained although these CTDs differ at more than 20 amino acid positions over 40 million years of evolution 20. Comparing Drosophila melanogaster to another dipteran Aedes agypti that diverged about 250 million years ago 20 one observes considerable differences in sequence (GenBank: EJY57389.1). In total, the A. aegypti CTD amino acid sequence differs at over 100 positions in the CTD. Whereas the Drosophila CTD is very degenerate with only two consensus repeats, the A. aegypti CTD contains 11 consensus repeats. Despite these differences the melanogaster and aegypti CTDs contain the same number of repeats.
2.4. Vertebrate CTDs
The CTD of vertebrate animals contains 52 repeats with about half adhering to the consensus and with most of the non-consensus repeats distal to the catalytic core (Figure 3). Position seven is the most often substituted with Lys, Thr or Asn appearing in multiple repeats. Among mammals, the 52 repeats are identical (including non-consensus amino acids) in human, marmoset, rat, cow, elephant and opossum and differ by a single amino acid only in mouse (GenBank: NP_000928.1, XP_002724554.1, NP_001193242.1, XP_003416946.1, XP_001364837.1, NP_033115.1, respectively). This striking level of conservation spans about 175 million years of evolution 20. The number of repeats also appears to have been maintained from zebrafish (Daneo rerio, GenBank: XP_682682.1) to humans spanning more than 400 million years of vertebrate evolution. The zebrafish CTD differs from the mammalian CTD at only nine positions, mostly at the least conserved seventh position and most of these changes are conservative and/or create consensus repeats. This striking level of conservation suggests a powerful stabilizing selection among vertebrates.
2.5. Summary of CTD evolution
The extreme conservation of this domain argues for an essential function in gene regulation. Several possible reasons for conservation are that a specific structure is formed by this sequence. Alternatively the conservation could be due to co-evolution of proteins that interact with CTD. These possibilities are discussed in the following sections.
3. CTD STRUCTURE
3.1. Modeling
While predicting the three-dimensional structure of proteins based solely on sequence remains difficult, recent progress has been made in identifying sequences that are likely to be intrinsically disordered 23. Such disordered regions contain few bulky hydrophobic residues and a high percentage of polar or charged amino acids. The CTD fills this expectation with the possible exception of the high percentage of Tyr. Despite the presence of tyrosine, computer algorithms predict that the CTD is highly disordered 24.
Although the CTD consensus sequence is predicted to lack order, it has the potential to form secondary structures. Suzuki first proposed that the SPSY and SPTS motifs repeated in the CTD form ß-turns similar to the SPXX motifs found in histones 25. More elaborate CTD models consisting of helices comprised of ß-turns and proline helices have been proposed but no evidence supporting extensive formation of such helices in vivo has been obtained 26.
Another possible CTD structure derives from the recent observation that low complexity (LC) sequences in proteins that contain multiple copies of the motif [G/S]-Y-[G/S] are able to reversibly form amyloid-like fibers 27. These LC sequences are present in many RNA-binding proteins and McKnight and colleagues have shown that proteins like FUS, RBM3, hnRNP A2, CPEB2, TIA and hnRNP A1 are able to form both homotypic and heterotypic fibers 27. The LC sequence motif occurs once in each CTD repeat and preliminary results from McKnight and colleagues indicate that the CTD can form ameloid-like fibers with other LC sequences (Kwan and Kato et al., submitted). The conformation of proteins in amyloid-lke fibers is a cross-ß structure in which the peptide backbone folds to form a series of ß-sheets that stack upon each other 28. It is possible that multiple repeats within the CTD are able to form a ß-sheet that can stack with other complementary sheets to form a cross-ß steric zipper which acts as the building block of an amyloid-like fibril. In the case of the CTD this association is reversible, as phosphorylation of the CTD withing these structures releases the CTD (Kwan and Kato et al., submitted).
3.2. Solution structure
NMR studies of single repeat and multiple repeat peptides have indicated that the CTD is mainly disordered in solution but with a slight propensity to form ß-turns as indicated by a small fraction of folded structures 26b,29. Addition of the hydrogen bond promoting solvent trifluoroethanol (TFE) increases the population of CTD peptides adopting a ß-turn conformation 26b. Circular dichroism (CD) of CTD peptides also indicates a predominantly unordered conformation 26b,29b,30 but careful examination of the CD spectra of CTD peptides in water indicate small but measurable populations of polyproline II helix (PII) and ß-turns 30. When the CD spectra are measured in TFE the population of ß-turns is greatly increased. Taken together these data argue that the CTD in solution is a dynamic population with fluctuating elements of secondary structure. In the appropriate environment the CTD may contain more or less secondary structure.
In another NMR study focused on a single heptad repeat Dobbins et al 31 showed that altering the i+2 position (underlined) in the SPXX motif of the heptad to Ala or Gly stabilized turn formation. Thus, the natural occurrence of these non-consensus amino acids may have a structural role favoring the formation ß-turns. Woody and colleagues further showed using CD that altering Ser2 to Ala in each of eight repeats had little effect on solution structure in water or TFE 30. In contrast, Ser5 to Ala substitution leads to an increase in PII in water and a loss of ß-turn conformation in TFE 30. This latter effect of changing Ser5 suggests that some caution must be taken in interpreting the results of mutations that convert Ser to Ala in CTD repeats.
The structural effect of CTD phosphorylation has been addressed using CD. In this case phosphorylation of Ser2 appears to lead to increasing disorder or alteration of the ß-turn conformation 30. The increased disorder of a phosphorylated CTD is also consistent with an increase in the apparent Stokes radius of the CTD in solution upon phosphorylation 32. Whether these changes are due to altered backbone conformations or simply a shift in equilibrium between alternative backbone conformations due to charge repulsion is not known.
One limitation to structures that the CTD can adopt is that X-Pro peptide bonds can exist as either cis or trans isomers. In most proteins the trans isomer is favored because there is less steric clash between the Pro amide hydrogen and the preceding Cα atom. NMR spectroscopy has been used to show that for a thirteen residue CTD peptide containing four prolines the trans conformation is the most highly populated at about 70% 29b. If this is the same for the entire CTD then the mammalian CTD will contain about 30 cis Pro bonds at any one time. Since the conversion between cis and trans isomers is slow, this places a limit on the rate of folding of potential CTD structures 33. Folding can be accelerated by peptidyl prolyl isomerases that will be discussed in a later section.
The length of the CTD peptide also plays a role in its conformational stability. Tyr side chain ordering as determined by CD spectra is different in an eight repeat peptide compared to a two repeat peptides 30. This suggests that Tyr side chains interact over a distance of more than two repeats. Comparing the eight repeat peptide with the mouse CTD or with synthetic peptides containing ~90 repeats 34 revealed negligible difference suggesting that ~ eight repeats is sufficient to adopt any potential secondary structure.
3.3. Crystallographic analysis
A second line of evidence for CTD flexibility comes from X-ray and low-resolution electron crystallographic analysis of Pol II. Neither the CTD nor the 80 amino acid linker region are visible in the crystal structure of yeast Pol II although the base of the linker is stable and associates with Rpb7 near the RNA exit channel 2,35. While negative stain is able to fully penetrate the CTD in solution, the 2-dimensional crystallization of Pol II against a lipid layer enabled visualization of a low-density large volume region consistent with a high degree of CTD conformational mobility.
3.4. Potential dimensions in vivo
The inherent disorder of the CTD might suggest that this domain occupies a large volume in vivo. This volume may be subject to influences that could have opposing effects on the overall dimensions of the CTD. First, phosphorylation of the CTD is likely to produce a more extended structure as the negatively charged side chains will tend to repel one another. This is seen in vitro in an increased Stokes radius of the phosphorylated CTD 32 and an increase in susceptibility to proteolysis 36. If fully extended the mammalian CTD could reach over 500 Å, many times the diameter of the catalytic core of Pol II. When proteins are bound to the CTD this could relieve charge repulsion and allow the CTD to form a more compact structure. Indeed, EM images of the CTD bound to Mediator suggest that the CTD adopts a compact structure 37, although the exact dimensions of this structure were not determined.
4. GENETIC DISSECTION OF CTD FUNCTION
Genetic analysis of the CTD began shortly after its discovery and led to two fundamental insights. First, deletion of the CTD is lethal indicating that it plays an essential role in yeast 21,38, Drosophila 39 and mammalian cells in culture 40. This essential function is not RNA synthesis as CTD deletions leave the catalytic core intact and biochemical studies of Pol II in which the CTD was removed showed normal catalytic activity 3c,41. A second important result from deletion studies is that cells are able to grow with less than the natural number of heptad repeats 21,38,40a. This result indicates that the heptad repeats are functionally redundant.
Before discussing the results of CTD deletions and amino acid substitutions the different genetic systems used to carry out these experiments will be discussed. The bulk of the data have been obtained from the yeasts Saccharomyces cerevisiae and Schizzosaccharomyces pombe and in mammalian cells in tissue culture. In each of these three model systems there are slightly different approaches to testing viability of CTD mutations. In the case of S. cerevisiae the effect of CTD mutation can be assessed by plasmid shuffle 42. Starting with a diploid strain one copy of the RPB1 gene is deleted and the strain is then transformed with a plasmid containing the WT RPB1 gene and a selectable URA3 marker gene. This strain is then sporulated and a haploid strain containing the RPB1 on a plasmid and harboring a deletion of the endogenous RPB1 is selected. This strain is then transformed with a plasmid expressing a mutated version of rpb1 on a plasmid with a LEU2 marker. Growth in 5-fluoroorotic acid counter-selects against the URA2 plasmid with the WT RPB1 thus leaving the cell with only the mutant rpb1 gene for survival 18,21. In S. pombe there is no plasmid shuffle available and thus mutant CTDs along with a selectable marker gene are recombined into one copy of the RPB1 gene in a diploid strain. The effect of CTD mutation on viability is then assessed by sporulating the strain and scoring for the presence of the selectable marker linked to the mutant CTD 43. This same approach was also taken in S. cerevisiae 38. These two approaches, plasmid shuffle and sporulation, while broadly similar may exert slightly different selective pressure. For example, in plasmid shuffle there will usually be multiple copies of the plasmid and this may yield more of the mutant Rpb1 subunit. In sporulation, the colonies must emerge from spores and this may require transcriptional programs not assessed in the plasmid shuffle assay.
To test the role of the CTD in mammalian cells a co-dominant α-amanitin resistant form of the Rpb1 subunit is expressed from a plasmid. Mutation of the CTD repeats in this α-amanitin resistant gene are assessed by scoring α-amanitin-resistant colonies after transfection 40a or by co-selecting a second marker gene and then assessing the long-term ability to grow in the presence of α-amanitin 40c,44. Finally, the CTD can be altered by recombination in embryonic stem cells and creation of mice with deletion of CTD repeats 45. In this approach the effect of mutation is assessed after breeding to obtain mice homozygous for the deletion.
Functional analysis of CTD mutants must take into account sequences in the tip domain located C-terminal to the heptad repeats (Figure 1). This domain in mammals contains a number of acidic residues, is phosphorylated by casein kinase II 46 and binds to Abl1/2 tyrosine kinases 47. While the sequence of this tip domain is not critical, deletions that remove the tip render the Rpb1 subunit unstable 40c,48 making it difficult to interpret the CTD deletion phenotype. Instability due to deletion of the tip has not been demonstrated in yeast, although deletions that alter the reading frame leading to longer heterologous tip sequences are lethal 21. Interestingly, the S.pombe CTD does not contain a tip domain.
4.1 CTD Deletions in yeast
Yeast CTD deletion construction has taken several approaches. In the first studies exonuclease digestion was used to remove sequences progressively from the 3’-end of the Rpb1 gene 21. In this study, maintaining less than 10 of the most proximal repeats rendered cells inviable. CTDs containing the proximal 10 or 11 repeats were viable although grew poorly and CTDs with more than 11 proximal repeats grew normally. Similar results were obtained through a process that removed internal repeats and tested for viability in a sporulation assay 38. In a separate study the CTD was reconstructed from 21 nt oligonucleotides leaving the most distal and proximal sequences intact 18. In his case CTDs containing 10 or 11 repeats grew normally while shorter CTDs with 9 or 8 repeats grew slowly and were temperature and cold sensitive 18.
Similar truncation mutants have been made in S. pombe 43a. This study showed that 16 of 29 proximal heptad repeats are sufficient for normal growth. Deletions that leave 10, 11, 12 or 13 repeats grow increasingly poorly as the number of repeats is reduced. The shortest of these mutations fail to form colonies at high or low temperatures. Reducing the S. pombe CTD to the 8 proximal repeats is lethal. Thus, in both S. cerevisiae and S. pombe about two thirds of the heptad repeats are dispensable for growth.
4.2. Deletions in metazoa
Genetic analysis of the murine CTD was made possible by the isolation of an α-amanitin resistance mutation in the mouse RPB1 gene 49. This mutation serves as a selectable marker to test the effect of mutations in the CTD 40a. Growing cells in otherwise lethal concentrations of α-amanitin assessed the function of ectopically expressed amanitin-resistant CTD mutants. Using this approach deletion of the CTD is lethal while partial deletions that contain 29, 31 or 32 of 52 repeats were viable. Removing more than 23 repeats is lethal 40a,50.
The effect of different CTD deletions in mammalian systems is complicated by the presence of many non-consensus repeats in the distal part of the CTD (Figure 3). To determine whether the non-consensus repeats are functionally equivalent to the consensus repeats CTDs containing only consensus or non-consensus repeats were constructed. These experiments showed that CTDs with only non-consensus repeats were not viable even if more than the minimum number of consensus repeats were present 40a,40c,44.
Given the difference in function of consensus and non-consensus repeats it is difficult to assess the minimal number of repeats in mammalian systems as the non-consensus repeats, while not required for cell growth in culture, may have roles in development. Deletion of 13 repeats containing several non-consensus repeats in the mouse CTD was tested in this regard by constructing a targeted deletion in the mouse germline 45. Mice homozygous for this deletion were born at normal rates but exhibited a high rate of neonatal lethality and were significantly smaller then normal littermates. Thus, these non-consensus repeats likely play a role in normal growth and development.
Truncation of the Drosphila CTD through insertion of a transposon yielded an RPB1 allele expressing 20 of the proximal repeats and resulting in lethality although this polymerase was still able to support early embryogenesis and transcribe in vitro 3c,39. To what degree wild-type Rpb1 derived from maternally inherited mRNA contributed to embryonic development was not determined.
4.3. Mutation of the consensus heptad
The presence of mainly consensus repeats in budding and fission yeast, together with facile genetics provided an opportunity to examine the role of different amino acids within each heptad repeat. Rather than mutating each repeat individually, new CTDs have been constructed by the concatenation of DNA oligonucleotides such each repeat (or pair of repeats) contains the same substitution. The results of these analyses reveal amino acid substitutions that are allowed or disallowed at each position.
Most of the CTD mutations analyzed to date alter the phosphorylatable residues to non-phosphorylatable Ala or the phosphomimetic Glu. Replacement in S. cerevisiae of the phosphorylation sites, Ser2 or Ser5 with either Ala or Glu did not support viability 18. This result is consistent with essential roles for both the phosphorylated and unphosphorylated form of the CTD. Altering the order of Ser2 and Pro3 also is lethal indicating the need for correct spacing of the heptad Pro residues 18. Changing the Tyr residue in position one to Phe was also lethal arguing for a possible role for Tyr phosphorylation 18. Substitution of the S. cerevisiae CTD with that of M. invertans in which each Thr4 and Ser7 is replaced by Ala supports viability indicating that modification of these residues in not essential for viability 51. Finally, substitution of Ser7 with Glu is lethal suggesting that at some point this residue must be dephosphorylated 52. Phenotypes of these mutants are summarized in Figure 4.
Figure 4.

Phenotypes of CTD substitution mutants. Indicated phenotypes are due to substitution in each repeat. Red indicates lethal mutations while blue indicates viable mutations. Some mutations are listed as both viable and inviable depending one the organism as designated in subscripts (c = S. cerevisiae, p = S. pombe, and h = human).
More recently, similar mutations have been made in the S. pombe CTD 43b,c. In this organism Ala substitutions at Pro3, Ser5 and Pro6 were found to be lethal but, in contrast to the S. cerevisiae CTD, Ser2 to Ala or Tyr1 to Phe substitutions were not lethal (Figure 4). In the case of Ser2 to Ala substitutions in 12 repeats there is a mating defect 43b and a failure of septation following mitosis 53. Substituting either Val or Ala at Thr4 was not lethal indicating that phosphorylation of this residues in S. pombe is not essential. Substituting Thr for Ser2 or Ser5 yielded different results. In the case of Ser2, Thr substitution is not lethal but a Ser5 to Thr is lethal indicating that at this position the presence of an extra methyl group interferes with CTD conformation or interaction with modifying enzymes. Finally, Gly substitution at Pro3 is lethal while substitution at Pro6 is not, although these cells grow slowly 43b.
One difference between the S. cerevisiae and S. pombe experiments is that the S. pombe constructs all contained four degenerate repeats proximal to the catalytic core. It is possible that these repeats supply partial function compensating for the loss of Ser2 or Tyr1. An alternate explanation is that Ser2 is generally a less important residue. Individual deletion or mutation of several Mediator subunit genes allows the growth of S. cerevisiae containing Ala in position 2 demonstrating that phosphorylation of this residue is not essential in all genetic backgrounds 8b.
Mutation of the consensus heptad has not been extensively studied in mammals. Chapman et al. have examined the effect of substituting Ser7 with various amino acids in a reconstructed 48 repeat CTD. Only the substitution with Ala was viable while substitution of each repeat with Glu or Thr/Lys in alternating repeats were not viable. This indicates that phosphorylation of Ser7 may not be essential and is consistent with CTD mutants that replaced the consensus repeats with the distal non-consensus repeats 40a,40c,44. In separate studies, the CTD of human Rpb1 was replaced with a CTD in which Thr4 was substituted with either Val or Ala. In each case the mutant Rpb1 subunit was not able to support growth 54. This result is consistent with an essential function for Thr4 perhaps requiring phosphorylation (see later section).
4.4. Spacing of heptad repeats
The tandem nature of the CTD suggests that the heptad repeat is the functional unit. However, mutations in which the spacing of repeats is altered has led to the realization that the functional unit comprises more than one repeat. Stiller and colleagues first showed that addition of a single alanine residue in each heptad repeat is lethal in each of four permutations. In contrast, inserting a single alanine between Ser7-Tyr1 or Tyr1-Ser2 in every other repeat supported viability 55. Shuman and colleagues extended this result in S. pombe showing that insertion of a single alanine in all seven permutations of a di-heptad repeat is not lethal 43c. Taken together, these results provide strong evidence that the functional unit of the CTD lies with two tandem heptad repeats.
Tandem repeats of di-heptads in which residues of the conserved consensus repeats are deleted or substituted with Ala have further defined the essential unit of CTD function. In pombe, substituting positions 5-7 of the distal di-heptad (YSPTSPSYSPTAAA)7 had no discernable effect on growth 43c. Changing one or two more residues to Ala in the distal repeat resulted in impaired growth. Taken together, these results indicate that the Ser5-Pro6 sequence is not required in every repeat. Substitutions of residues in the proximal repeat of a tandem di-heptad further refine the functional unit. Replacing the first three residues in pombe is lethal as is replacing Tyr1 in every other repeat. However, the Pro3 to Ala change in the proximal repeat was not lethal indicating that Ser2-Pro3 need not be present in each repeat. Taking these results together leads to the minimal functional unit in pombe of YSPTSPSYSP43c. This result confirmed an earlier result in S. cerevisiae in which a tandem repeat of a partial di-heptad lacking the last three residues of the distal heptad grows normally. Thus, Stiller and colleagues proposed minimal “252” CTD functional unit consisting of three SP units Ser2-Pro3 – Ser5-Pro6 – Ser2-Pro3 56.
The spacing of consecutive CTD functional units is not critical as up to five Ala residues between di-heptads is viable although cells grow slowly 57. Insertion of seven Ala residues between di-heptads is lethal but between tri-heptads is not 57. The tendency of poly-alanine to form α-helices indicates that the helical nature of the insert may alter the conformation of adjacent heptads. This was directly demonstrated by substituting two Pro residues in a seven Ala insert. The helix breaking residues restored viability to this CTD 57. Helical inserts may alter the functional unit when each heptad is adjacent to a helical domain but in the case of the tri-heptad the internal repeat would be shielded.
4.5. Proximal vs distal consensus repeats
The S. cerevisiae CTD consists almost entirely of consensus repeats that are redundant in their function. It is therefore unexpected that sequence requirements for the distal repeats should differ from those of the proximal repeats. This is the case, however, as Ser2 to Glu substitutions in the proximal repeats are viable while the same substitutions in the distal repeats are lethal 18. The converse is observed with Ser5 to Glu substitutions; proximal substitutions are lethal while distal substitutions are viable 18. Together these results indicate that distal and proximal repeats have at least partially non-redundant functions.
4.9 CTD mutation summary
Mutations in the CTD of a variety of organisms have shown that the CTD performs an essential function. The genetic malleability of this domain indicates a degree of functional redundancy that is somewhat surprising given the evolutionary conservation, especially in mammals. Insertion of residues between heptad repeats has shown that the functional unit of the CTD extends beyond a single heptad and can be separated by non-consensus residues. Finally, the effect of substitutions of individual residues in each heptad varies from organism to organism. The wide variety of available CTD mutants has greatly aided the functional dissection of the CTD.
5. Functional analysis of CTD mutants
Cells harboring mutant CTDs have been exploited to identify the genetic pathways that are perturbed thereby providing information about CTD functions. Genetic suppression of the conditional growth phenotype of the most severe CTD mutants has been used to identify factors that interact with the CTD. In addition, the effect of CTD mutations on transcription of endogenous or reporter genes has been used to identify the step(s) in transcription that are effected by mutation of the CTD.
5.1. Suppression of CTD mutations
In S. cerevisiae the most severe CTD truncation mutants grow slowly at 30°C and not at all at low or high temperature 21. The Young laboratory used the inability of strains with CTD of 10 or 11 repeats to grow at low temperature to isolate spontaneous suppressors 22,58 of this phenotype. This screen yielded nine different SRB (suppressors of RNA polymerase B) genes. Characterization of Srb proteins led eventually to the identification of the mediator complex that contains the Srbs and a number of other transcription regulators. The association between Mediator and the CTD will be discussed in a later section but the identification of Srbs led to a number of experiments aimed at understanding the role of the CTD in transcription.
Suppressor screens were also used to show that Ser2 and Ser5 have different functions in the CTD 8b. Substitution of yeast Ser2 with Ala can be suppressed by a number of srb mutations but Ser5 to Ala substitutions cannot 8b. This implies that the function impaired in CTD truncation is similar to the defect caused by lack of Ser2 and suggests that these Srbs may have a repressive function that counters a positive function provided by Ser2.
5.2. Changes in transcription and processing due to CTD mutation
The slow growth of CTD truncation mutants suggests an underlying defect in some aspect of Pol II function and examination of transcription in these cells has pointed to the involvement of the CTD at many steps in the transcription cycle. In this section we will discuss data implicating the CTD in various stages in the biogenesis of Pol II transcripts. The historical progress of this endeavor follows the transcription cycle from early work on initiation to the more recent work on Pol II termination.
5.2.1. Activation and initiation
Yeast cells harboring truncated CTDs show defects in activation of some genes like GAL10 and INO1 but not others like HIS4 59. The similar binding of activators to the UASs of the effected genes suggested that the CTD might act through directly contacting the activation domains of transcription factors like Gal4. Deletion of the transcription suppressor SIN1 suppresses a CTD truncation mutant suggesting that the CTD may also be involved in removing repressors to allow transcription activation 60. Defects in the ability to respond to activators are also observed in CTD truncation mutants in mammalian systems 61.
Studies in mammalian transcription extracts gave mixed results concerning the role of the CTD in transcription initiation. Transcription in vitro from the adenovirus-2 major late promoter and the Drosophila HSP70 and actin promoters apparently does not require the CTD 3c,41,62. In contrast, transcription from the mouse DHFR gene does require the CTD 63. This latter result is consistent with other studies indicating that monoclonal antibodies against the CTD could inhibit transcription of the adenovirus MPL and DHFR promoters 64.
One possible role of the CTD in initiation would be to recruit Pol II to the pre-initiation complex through interaction with the general transcription factors. In this regard several reports showed interaction between the CTD and the TATA Box binding protein TBP 65 or its larger TFIID complex 66. While direct interactions with TBP have not been further pursued recent data indicates that the CTD can interact with the Taf15 component of TFIID (Kwan and Kato et al., in preparation).
Interest in interactions between the CTD and GTFs began to subside with the discovery that the Srb proteins form a complex that associates with Pol II 58a. This complex was later named the Mediator after its biochemical function in mediating the activation of Pol II transcription in an activator-dependent reaction Purified Mediator also stimulates the phosphorylation of the CTD by TFIIH kinase 67. The CTD was shown to interact with the Mediator 68 but whether this interaction is defective in CTD truncations was not explored. Only recently has the basis for this interaction been determined 37 and this interaction will be discussed more fully in a later section.
5.2.2. Elongation
The transition from initiation to elongation was shown to correlate with phosphorylation of the CTD 46,69. Two CTD phosphorylation dependent steps have been identified. First, phosphorylation has been proposed in yeast to release the CTD from essential contacts with the initiation complex, particularly with the Mediator. As will be discussed in a later section this release is provided by a CTD kinase associated with the general transcription factor TFIIH.
A second step requiring CTD phosphorylation is the transition between the early, non-processive elongation complex and the processive elongation complex. In mammalian cells the CTD is required for the transition from promoter proximal pausing to productive elongation 50,70. In yeast, the positive role of CTD phosphorylation on transcription elongation was first documented by Lee and Greenleaf who showed that the yeast CTD kinase I (CTDK-I) enhances elongation but not initiation in vitro. Inhibition of CTD phosphorylation has the opposite effect on elongation. Price and colleagues who discovered P-TEFb showed that this kinase phosphorylates the CTD in the early elongation complex. Inhibition of P-TEFb or removal of the CTD prohibit the transition from the early elongation complex to processive elongation 71. The details of P-TEFb phosphorylation are discussed in a later section.
5.2.3. RNA processing: capping
The 5’-ends of eukaryotic pre-mRNAs are modified by addition of a non-templated methylated guanyl cap through a process that takes place in three steps. First, the 5’ γ phosphate is removed by an RNA triphosphatase (RTase). Second, GMP is added to the 5’ β phosphate by a GTP-dependent guanylyltransferase (GTase). Finally, a methyl group is added at position 7 of the 5’ cap guanine by a methyltransferase. Capping is the initial step in processing nascent Pol II transcripts occurring when the nascent transcript has just emerged from the elongating Pol II 72.
CTD deletion mutants are synthetically lethal with mutations in the capping enzymes suggesting a role for the CTD in this process 73. The GTase enzyme has been shown to interact directly with the CTD in both yeast and mammalian cells 73-74 and this interaction requires that the CTD is phosphorylated 9a,75. The structure of GTase bound to the CTD will be discussed in a later section.
Recruitment of the capping enzymes to the CTD ensures that Pol II transcripts are preferentially modified. Mutations in the CTD that disrupt this interaction are expected to have deleterious effects on capping and this may have further consequences. For example, if CTD mutants like Ser5 to Ala fail to cap normally this would render transcripts unstable and lead to premature termination through the Rat1 pathway 76. Whether CTD truncation mutations are lethal because of a failure to cap is not clear, however. As few as two heptads are required for recognition of the CTD by the mammalian capping enzyme 77 suggesting that deletions removing only half of the repeats will still be capped normally.
5.2.4. RNA processing: splicing
CTD truncation markedly reduces the efficiency of splicing in mammalian cells 78. Bentley and colleagues used an α-amanitin-resistant Pol II with a CTD truncated to 5 heptad repeats to drive expression of reporter genes containing either SV40 or ß-globin introns 78a. In this system transcription is reduced in the presence of α-amanitin and the transcripts that are synthesized display a 3-5-fold reduction in the percentage of spliced transcript 78a. This observation fits nicely with CTD binding studies indicating that the CTD interacts with SR-like proteins and other splicing factors 78b,79.
Various mechanisms have been proposed to explain the necessity of the CTD for efficient splicing in vivo. One possible role of the CTD is to sequester the upstream exon close to the elongating Pol II so that when the downstream exon is synthesized it will be in close proximity to its partner 80. The fact that in vitro splicing takes place in the absence of Pol II indicates that the CTD is not required for the actual splicing reactions. However, in vitro splicing is much slower than in vivo splicing and cannot efficiently join exons that are more than a few hundred base pairs apart. In contrast, long introns are efficiently removed in vivo 81 supporting the idea that the CTD tethers the upstream exon to the Pol II elongation complex. Furthermore, the addition of Pol II with a phosphorylated CTD (or the phosphorylated CTD alone) to an in vitro reaction can stimulate splicing 82 although short CTD peptides are inhibitory 79b.
Several proteins have been proposed as the functional link between the spliceosome and the CTD. In yeast, the U1 snRNP protein Prp40 has been demonstrated to bind the CTD through its WW domain 83. This observation suggested that the recognition of 5’ splice sites could be facilitated through recruitment of the U1 snRNP. Deletion of the Prp40 WW domains is not lethal, however, and does not results in any observable splicing defect 84.
More recent work has identified a CTD-dependent splicing activity consisting of a complex containing U2AF65 and Prp19C 85. U2AF65 binds directly to the CTD phosphorylated on both Ser2 and Ser5 85a. Transcripts produced from a CTD mutant in which Ser2 is substituted in all repeats with Ala is defective for splicing suggesting that this CTD phosphoisoform, present in the middle and 3’-end of genes is required for interaction with the splicosome 86.
The CTD has also been shown to play a role in alternative splicing 87. The inclusion of an alternative exon in the fibronectin gene is inhibited by recruitment of the SRp20 SR protein by the CTD 88. Transcription by a Pol II lacking most of the CTD results in inclusion of this exon 88. Although SRp20 co-immunoprecipitates with Pol II 89 the mechanism of SR protein recruitment has not been established. In addition to splicing factor recruitment alternative splicing is also regulated by chromatin modification and Pol II elongation rates, both of which may be regulated by the CTD 87. A more comprehensive discussion of the role of the CTD in splicing is found in an accompanying review by Eick and Geyer (this issue).
5.2.5. RNA processing: 3’-end formation and termination
CTD truncation mutants are also defective in 3’-end processing and termination 90. The first evidence connecting the CTD to 3’-end formation came from the same experiments showing the CTD-dependence of splicing 78a. In these experiments transcripts synthesized by Pol II with a truncated CTD readthrough the 3’ processing signals and fail to cleave and polyadenylate the nascent transcript. In support of a role for the CTD in 3’-end formation a number of cleavage/polyadenylation factors have been shown to interact with the CTD 78a,91. One of the most important factors in the 3’-end machinery is the protein Pcf11. This protein is part of pre-mRNA cleavage complex II and binds directly to the Ser2 phosphorylated form of the CTD that is prominent at the 3’-end of genes 92.
The CTD is also required for proper termination of yeast non-polyadenylated Pol II transcripts through the Nrd1-Nab2-Sen1 pathway 91e,93. Nrd1 and Nab3 are RNA-binding proteins that act as sensors binding to terminator elements in nascent transcripts 93-94. Nrd1 binds that Ser5 phosphorylated CTD 95 through it CTD-interacting domain (CID) and this leads to termination of Pol II in a process that directs the 3’ end of the transcript to the nuclear exosome 96. CTD truncation mutants are defective in this pathway and many snoRNA and other non-coding RNAs fail to terminate properly 93,97. In addition, a number of protein coding genes are regulated by attenuation through the Nrd1-Nab3-Sen1 pathway 98. These genes are over expressed in CTD truncation mutants. No similar non-poly(A) termination pathway has been discovered in metazoa although the mammalian SCAF8/RBM16 gene encodes a protein with similarity to Nrd1 79b, co-localizes with sites of transcription 99 and binds Ser2P + Ser5P CTD fusion proteins and peptides 99-100.
5.2.6. RNA transport
Several studies have indicated that transport of mRNA from the nucleus to the cytoplasm involves proteins that interact with the CTD. The S. cerevisiae proteins Npl3 and Yra1 have been implicated in different stages of mRNA biogenesis; transcription elongation, splicing, mRNA transport and translation.
Npl3 is similar to mammalian SR proteins containing tandem RRMs and a Ser/Arg-rich domain. Npl3 co-purifies with Pol II, interacts with the Pol II CTD, is recruited to chromatin early in the transcription cycle 101 and mutations of Npl3 that reduce RNA binding lead to a reduced elongation rate 102. After transcription termination Npl3 remains bound to the mRNA and facilitates its export to the cytoplasm 103. Once in the cytoplasm Npl3 plays a role in translation through interaction with ribosomal proteins and the mRNA poly(A)-binding protein Pab1104. Furthermore, npl3 mutants display impaired translation suggesting a repressive role for Npl3 104a. In more recent studies Npl3 has been shown to play a role in translation termination fidelity 105. Npl3 has also been shown to repress translation through an interaction between its RGG domain and the translation initiation factor eIF4G 106. The roles of Npl3 thus span the mRNA biogenesis pathway and this implies that conditions that alter CTD interactions with Npl3 could have consequences for translation in the cytoplasm.
Yra1 is a second yeast transport protein that binds the CTD 107. This RNA-binding protein is homologous to the mammalian Aly/REF export factor 108. Yra1 is cotranscriptionally bound to a subset of nascent mRNAs 101a,109 and interacts with the mRNA export factor Mex67 that escorts the mRNP to the nuclear pore 110. Mutations in YRA1 lead to nuclear retention of completed transcripts 111. Greenleaf and colleagues have shown that Yra1 binds to doubly phosphorylated Ser2P + Ser5P CTD present in transcription elongation complexes. What role this binding plays is not clear, but mutations that impair CTD binding but not RNA binding have a slight growth phenotype and a minor transport defect under stress conditions 107b. Given that Yra1 binds as many as a thousand mRNAs 101a,109 the sum of these minor processing defects could have wide-ranging deleterious effects. Clearly, more work will be needed to establish the role of the CTD in establishing the unique mRNP signatures of pre- and mature mRNAs.
5.2.7. Chromatin modification
Nucleosomes present a barrier to Pol II elongation and histone modification and nucleosome remodeling play critical roles in transcriptional regulation 112. While Pol II lacking most of the CTD can transcribe transfected reporter gene shortly after transfection 61a,78a transcription of endogenous genes is blocked early in the transcription cycle 40b,50,70. One difference in these classes of genes is that endogenous genes are assembled into chromatin while the transiently transfected DNA is likely not fully assembled into native chromatin. The first indication of a connection between the CTD and chromatin was the observation that histone methyltransferses Set1 and Set2 are recruited to actively transcribed genes 113.
The Set1 histone methyltransferase is part of the COMPASS complex that methylates histone 3 on lysine 4 (H3K4) 114. This modification requires Ser5P and the PAF complex 113c and is localized to the 5’ ends of genes 115. Recruitment of Set1/COMPASS also requires Bur1 which is surprising since this kinase phosphorylates Ser2 (discussed in a later section). One possible explanation is that Bur1 phosphorylates a component of COMPASS to stimulate the H3K4Me3 modification 116. What role modification by Set1 plays in transcription remains unclear as mutation of SET1 or H3K4 have only minor effects on gene expression 117. Because Set1 is required for repression of Ty1 and PHO84 in yeast and both transcripts are regulated by cryptic unstable transcripts it is possible that H3K4Me plays a role in repression of Pol II transcription by trans-acting RNAs 97,118.
Set2 methyltransferase methylates histone H3 on K36 119 and this modification localizes to the middle and 3’-end of genes 115 through interaction of Set2 with the elongating form of Pol II phosphorylated on both Ser2 and Ser5 of the CTD 113a,b,120. Deletion of the SET2 gene is not lethal but acetylated histones accumulate over coding regions leading to the expression from cryptic promoters 121. This set2 defect is due to the failure of the histone acetylase complex Rpd3s to deacetylate histone in the wake of elongating Pol II 121-122. Surprisingly, Rpd3s is not recruited by the H3K36me mark but rather through interaction directly with the Ser2+Ser5 phosphorylated CTD 123. The H3K36me mark is, however, required for deacetylase activity arguing for an allosteric mechanism of the H3K36me mark on HDAC1.
Spt6 is a factor required for positioning nucleosomes in transcribed regions 124. Working together with Spt4 and Spt5, Spt6 is also required for transcription elongation 125. The C-terminal region of Spt6 contains several SH2 domains that facilitate recruitment of Spt6 to the transcription elongation complex 126. Spt6 in this complex is also involved in H3K36 methylation through its interaction with IWS1/Spn1 that recruits the mammalian Set2 to coding regions 126a.
Other chromatin proteins have been shown to interact with the CTD including heterochromatin protein 1 (HP1) that acts to recruit FACT to the CTD 127. The Chromodomain-helicase-DNA-binding protein 8 (CHD8) protein has also been shown to associate with the CTD 128. Taken together, these results indicate that the CTD plays an important role in modulating the structure of the chromatin template.
5.2.8. DNA replication and repair
The first indication of a link between the CTD and DNA replication came from the identification of the replication factor mini chromosome maintenance (MCM) helicase complex in high molecular weight Pol II holoenzyme complexes from Xenopus oocytes and HeLa cells 129. This connection was supported by genetic interactions between a mcm5 ts mutation and a CTD truncation 130. In addition, CTD truncation mutations show increased instability of minichromosomes 130. More recent experiments have suggested that stalled Pol II elongation complexes recruit components of the origin replication complex (ORC) through interactions with the CTD 131. This model is consistent with genome-wide mapping studies showing a correlation between Pol II promoters and ORC complexes 132.
The CTD is also involved in DNA repair. Truncation of the yeast CTD results in a reduced DNA damage response 133 and deletion of the CTD kinase catalytic subunit gene CTK1 renders cells sensitive to chemical and irradiative damage 134. Furthemore, a number of proteins that bind to the phosphorylated CTD are involved in the DNA damage response pathway 134.
Errors in processing of nascent transcripts can lead to R-loop formation and this has been linked to an increased rate of recombination and genome instability 135. While the Nrd1-Nab3-Sen1 complex that associates with the CTD has been implicated in this process 135e the detailed mechanisms have not been established. Another factor that could couple transcription and repair is the DNA helicase RecQ5ß. This protein is required for the maintenance of genome integrity 136 and has been shown to interact with the CTD 137. The presence of DNA repair components in Pol II elongation complexes could enable the enzyme to respond rapidly to encounters with damaged DNA or in the event of collisions with the DNA replication machinery.
6. WRITING THE CTD CODE: CTD KINASES
The strong selective pressures that maintain the CTD through evolution have been proposed to originate through co-selection of the proteins that interact with the CTD 138. Among the most co-conserved proteins are the kinases and phosphatases that modify the CTD and the RNA processing factors that bind the CTD and couple transcription and processing have fixed the CTD structure in its present state. In this section we will describe the CTD kinases that modify the CTD. There are a number of different CTD kinases and a number of different possible phosphorylation sites in the CTD heptad. The preferential phosphorylation of different residues in the heptad repeat at different stages of the transcription cycle has been termed the CTD code 9b,43c,92b,139. While there does not seem to be a strict correlation between the phosphorylation state and the function of the CTD there are definite correlations between different patterns of CTD phosphorylation and the position of Pol II in the transcription cycle. In this section we first discuss the analysis of CTD phosphorylation. We will then discuss the kinases individually and finally address the interplay among these kinases.
6.1. CTD phosphorylation
6.1.1. Characterization of CTD phosphorylation sites
Consensus CTD heptad peptide substrates phosphorylated by kinases in vitro and subsequently sequenced by Edman degradation established that the human Cdc2 kinase phosphorylates both Ser2 and Ser5 8a. Mass spec analysis has also been used to identify phosphorylation sites on in vitro phosphorylated model CTD substrates 140 and in vivo phosphorylated CTD as part of a phosphoproteome analysis 141. These approaches are useful for identifying which of the consensus residues is phosphorylated but identifying which of the many repeats is phosphorylated is limited to the non-consensus heptads.
The most commonly used method for mapping CTD phosphorylation sites employs antibodies against phosphorylated CTD epitopes. The earliest experiments consisted of polyclonal antibodies raised against a CTD fusion protein phosphorylated in vitro by CTDK-I 142. This antibody was affinity purified and used to show that Pol II on transcription puffs contained a phosphorylated CTD while Pol II near the promoter contained an unphosphorylated CTD 143. The problem with this approach is that the epitopes generated by CTDK-I, although unknown at the time, consisted of both Ser2P and Ser5P. Thus, while this approach led to the first in vivo evidence supporting a CTD code 143, the polyclonal nature of the antibody prep limited its utility in identifying specific phosphorylation sites.
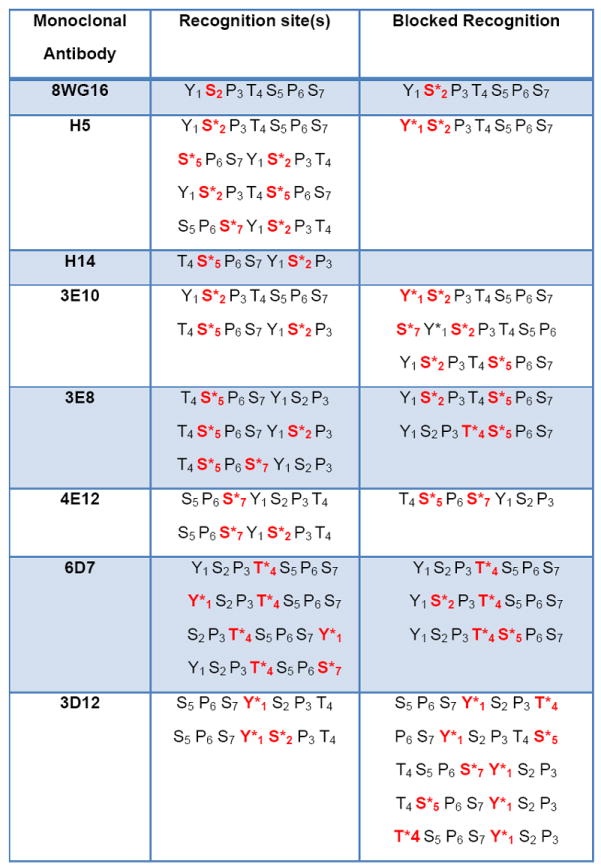
Monoclonal antibodies (mAbs) are able to discriminate between different phosphoepitopes. The first phosphoepitope-specific mAbs were generated against mixtures of proteins and the target of individual mAbs was determined by characterizing the bound protein. In this way two mAbs recognizing different CTD phosphoepitopes were identified 144. The identity of the phosphoepitope (H5 = Ser2P and H14 = Ser5P) was determined by the ability of each antibody to identify phosphorylated CTD fusion proteins containing either Ser2 or Ser5 to Ala substitutions 144b. More recently, Eick and colleagues have used CTD antigens consisting of peptides phosphorylated on specific residues to generate mAbs and their specificity has been confirmed using reactivity to a similar bank of phosphorylated CTD peptides and/or CTD fusion proteins containing mutations in the consensus heptad repeat 44,54b,145. Monoclonal antibodies against various CTD isoforms have standardized the mapping of in vivo phosphorylation patterns although the binding of these antibodies can be blocked by modification of adjacent residues introducing a degree of uncertainty in the resulting maps. These antibodies are described in Table 1.
Table 1.
Anti-CTD monoclonal antibody specificities
|
The asterisks indicate phosphorylated residues. The data in this table was derived from work published by the Eick laboratory in Munich.
6.1.2. Chemical genetic approach to kinase function
The characterization of CTD phosphorylation patterns in vivo has benefitted greatly from a chemical genetic manipulation of CTD kinase subunits 146. In this approach the ATP binding site of the kinase of interest is genetically altered to allow binding of a bulky ATP analog inhibitor that does not bind to any other kinase. The assumption is that altering the ATP binding pocket does not alter the specificity of the kinase. This has been demonstrated in at least one case 147. Using cell-permeable inhibitors like 1NM-PP1 or 1NA-PP1 allows selective in vivo inhibition of a single kinase. This selectivity can be assured by demonstrating a lack of effect on the wild-type cell. For in vitro analyses the use of radioactive bulky ATP analogs can be used to identify kinase targets in complex mixtures of potential substrates 148. These chemical genetic approaches have proven extremely useful in clarifying the roles of different CTD kinases.
6.1.3. Limitations to characterization of phosphorylation sites
Given the positive aspects of the above approaches to mapping phosphorylation sites in the CTD there are certain limitations that must be taken into account in the interpretation of many experiments. For example, there are some problems with anti-phospho-CTD antibodies that are not always adequately addressed and thus some caution must be used in the interpretation of CTD phosphorylation patterns. First, the recognition of the epitope may be altered by phosphorylation of adjacent sites. This looks to be the case for H5 that recognizes Ser2P but binds more efficiently if the adjacent Ser5 is also phosphorylated 149. mAb 3E10 that also recognizes Ser2P, in contrast does not bind if there is a Ser5P in the next heptad 44. Another problem is that the reactivity on a Western blot may not be linearly dependent on the level of phosphorylation. This is particularly problematic for the pentavalent IgM antibodies H14 and H5. IgM antibodies generally have lower affinity that is compensated for by their increased valency. Thus H14 and H4 may not detect low levels of CTD phosphorylation preferring to bind to the most highly phosphorylated CTDs containing multiple closely spaced epitopes. The valency problem also arises in immunoprecipitation, for example in ChIP experiments. Titrating the amount of antibody can yield different results due to ratio of antibody binding sites and the density of the epitopes 150.
Another problem with analysis of CTD phosphorylation state using antibodies is in determining whether all modifications occur on the same CTD or whether transcribing Pol IIs are heterogeneous with some CTDs having mainly one phosphoepitope and another Pol II predominantly a different epitope. Immunoprecipitation of Pol II phosphorylated in vivo with the anti-Thr4P mAb 6D7 left substantial Pol IIO in the supernatant 54b. This Pol IIO contained Ser5P and Ser7P but little Ser2P. Conversely, precipitation of Pol II with a Ser5P-specific mAb left Ser2P and Thr4P containing Pol IIO in the supernatant. These results suggest the presence of three types of Pol IIO containing primarily Ser5P, Ser5P+Ser7P, and Ser2P+Thr4P 54b. While these classes of Pol IIO may be separable by IP it is far from certain how discrete their phosphorylation patterns are. Some sets of repeats may be differentially modified and this may lead to optimal IP conditions while other repeats in the same CTD may have a different pattern of phosphorylation. In the future it will be important to map the different Pol IIO subforms on the genome to determine their functional role(s).
There are also limitations in the use of analog sensitive kinases. One problem is that the inhibitors may not work at 100% efficiency and thus remaining phosphorylation may not be due to phosphorylation by other kinases but rather by the low level of kinase activity remaining in the presence of inhibitor. Another possibility is that the specificity of the analog sensitive kinase may be different. This has been checked for some kinases on some substrates but has not been thoroughly explored.
Finally, the use of in vitro kinase reactions on peptide or fusion protein substrates may yield specificities that are not evident in vivo. For example, Ctk1 can phosphorylate Ser5 inefficiently on an unphosphorylated substrate 149 but in vivo prefers substrates already phosphorylated on Ser5 by CDK7 or CDK8.
6.2. Cyclin-dependent kinases targeting the CTD
The CTD code is primarily written by members of the cyclin-dependent kinase (CDK) class of enzymes that were first identified as controllers of cell cycle transitions leading to the initiation of DNA synthesis and entry into mitosis 151. Later work identified transcription-related CDKs as integral components of the Pol II transcription machinery 152. These two functions in metazoans are carried out by two sets of CTKs. CDK1, CDK2, CDK4, and CDK6 play central roles in cell cycle control while CDK8, CDK9, CDK12 and CDK13 play different roles in transcription through phosphorylation of the CTD.
The CDK catalytic subunits are not active as single polypeptide chains but need to be activated. This takes place through the interaction with regulatory cyclin subunits and through phosphorylation of a Thr residue in the activation segment (T-loop) 153. The cyclin subunit can also provide a degree of substrate specificity. The transcription-related CDKs from human and fission and budding yeast are listed in Table 2 and discussed individually in a following section.
Table 2.
CTD-Associated Cyclin-Dependant Kinases
| human | S.cerevisiae | S.pombe | AKA | CTD P-sit | |
|---|---|---|---|---|---|
| CDK7 | Cdk7/cyclin H/Mat1 | Kin28/Ccl1/Tfb3 | Mcs6/Msc2/Pmh1 | TFFIIH kinase | Ser5, S |
| CAK = CDK7 | CAK = Cak1 | CAK=Csk1 | |||
| CDK9 | Cdk9/cyclin T | Bur1/Bur2 | Cdk9/Pch1 | P-TEFb | Ser2, Ser5 |
| CAK=CDK7 | CAK = Cak1 | CAK = Csk1 | |||
| CDK12 | Cdk12/cyclin K Cdk13/cyclin K |
Ctk1/Ctk2/Ctk3 | Lsk1/Lsc1 | CTDK-I | Ser2, Ser5 |
| CAK = CDK7 | CAK = Cak1 | CAK = Csk1 | |||
| CDK8 | Cdk8/cyclin C/MED12/MED13 | Ssn3/Ssn8/Srb8/Ssn2 | Cdk8/CycC/Med12/Med13 | Mediator CDK module (CKM) | Ser2, S |
| CAK = none? | CAK = none? | CAK = none? |
The specificity of cell cycle CDKs was initially established through the use of libraries of potential peptide substrates 154. Using this approach the preferred motif of CDK1 is S/T-P-X-R/K. With lower specificity CDK1 can recognize and phosphorylate Ser or Thr in the S/T-P di-amino acid. Only the minimal CDK recognition motif is present in the CTD at Ser2 and Ser5. For some cell cycle substrates a nearby RXL motif that binds to a hydrophobic patch on the cyclin can enhance phosphorylation 155 but this motif is not present in the CTD.
Given the caveats concerning the specificity of kinases and the characterization of CTD phosphorylation sites we will now discuss the kinases that have been shown to phosphorylate the CTD. This discussion will focus mainly on the role of these kinases in the transcription cycle in human cells and in the yeasts S. pombe and S. cerevisiae. This restriction is due to the elegant recent work employing analog sensitive kinases that have helped to clarify earlier work. The review by Jeronimo et al. (this issue) provides a more in-depth discussion of the genomic distribution of kinases and the resulting genome-wide CTD phosphorylation pattern in the accompanying review. After covering the CTD kinases individually the interrelationships of these kinases and their CTD targets will be addressed.
6.2.1. CDK7: structure and biochemistry
This CTD kinase is comprised of Kin28/Ccl1 in S. cerevisiae, Mcs6/Mcs2 in S. pombe and CDK7/cyclin H in metazoa. In metazoa CDK7 was initially identified as the CDK-activating kinase CAK that phosphorylates the T-loop in the cell cycle CDKs to render them catalytically active 151,156. In yeast, this activity is provided by an independent kinase Cak1 that is distantly related to CDKs 157. Subsequently, Cdk7 was shown to form a heterotrimeric complex with the ring finger protein Mat1 and this complex is part of the general transcription factor TFIIH that phosphorylates the CTD 158.
CDK7 can apparently execute these dual functions by recognizing different classes of substrate. CDKs are known to prefer Ser-Pro recognition sites and this site is clearly present in the CTD at Ser2 and Ser5. The activation loop sequence in mammalian CDKs is TXXVVTL in which the underlined T is the phospho-acceptor. The first X residue in this sequence in Cdk1, Cdk2 and Cdk6 is not proline. Thus, CDK7 can recognize and phosphorylate a number of non-standard amino acid sequences.
Two mechanisms have been proposed to explain the diversity of CDK7 phosphorylation sites. First, CDK7 may be promiscuous in recognizing phosphorylation target sequences. This could either be intrinsic to the active site or could depend on conformational arrangement of non-consensus targets. A second explanation is that CDK7 has more than one way of interacting with substrates but both modes are very specific. Using an analog sensitive version of human CDK7 in in vitro reactions Fisher and colleagues showed that the specificity for CTD required the presence of Mat1 as the dimeric complex of Cdk7/cyclin H did not efficiently phosphorylate the CTD 159. This is consistent with the observation that Mat1 is required in vivo for stimulation of CTD phosphorylation by CDK7 160. The difference between CTD-like and T-loop substrates is also seen in the ability of Cdk7 to phosphorylate peptides in solution. CTD peptides are effective substrates while T-loop substrates are not. Presumably additional contacts with the CDK polypeptide are required to achieve T-loop phosphorylation.
In vitro phosphorylation reactions on CTD substrates were initially used to show that human CDK7 prefers to phosphorylate Ser5 158d,161. Roy et al first showed that mammalian Cdk7 phosphorylates a peptide with Ala2 substitutions but cannot phosphorylate a similar peptide with Ser5 converted to Ala 158d. Similar results were obtained with fusion proteins containing multiple repeats in which each Ser2 or Ser5 is converted to Ala 161c. Peptides with Ser7 converted to Ala are also effective Cdk7 substrates 161b,161d. CDK7 can phosphorylate a peptide already phosphorylated on Ser2 but cannot phosphorylate a similar peptide phosphorylated on Ser5 161d. The S. cerevisiae Kin28/Ccl1/Mat complex also phosphorylates Ser5 in vitro 162. Taken together, these in vitro results indicate that CDK7 has a strong preference for Ser5.
With the discovery of Ser7 phosphorylation 44 the hunt began for the kinase that deposits this mark. Surprisingly, CDK7 was shown to be responsible for at least the promoter proximal deposition of this mark 150,163. The Ser7 consensus diverges markedly from the S/T-P motif common to CDKs raising the question of how this phosphoacceptor site is recognized by the kinase catalytic center. Two possible explanations are that the kinase is promiscuous or that two distinct recognition modes are present in the catalytic center. Evidence for the latter explanation has come from in vivo work of Fisher and colleagues 159. Whether Ser7 phosphorylation falls into this second category is not known but in vitro the efficiency of this phosphorylation is less than that for Ser5 150.
Phosphorylation of peptides or recombinant CTDs of different lengths indicate a preference for CTDs longer than eight repeats suggesting that Cdk7 may recognize some type of secondary structure not present in shorter peptides 161b,c. CTD length is particularly important for phosphorylation of Ser7 as CTDs with fewer than 24 repeats are poorly phosphorylated 44. Perhaps secondary structure favored in the longer CTD positions the non-standard Ser7 phosphoacceptor side chain in the Cdk7 active site.
Mammalian CTD fusions containing either the conserved proximal repeats or the distal non-conserved repeats are phosphorylated with similar kinetics. When specific non-consensus repeat peptides are used as substrates, however, an approximately four-fold preference is observed for peptides with Lys7 in place of Ser7 161c. This substitution occurs in nine of the distal mammalian repeats. No similar preference was observed for Asn7 or Thr7 peptides. The basic nature of Lys located +2 residues from the phosphoacceptor is similar to the consensus of the cell cycle CDKs suggesting that a kinase like CDK1 may have an unappreciated role in CTD phosphorylation.
6.2.2. CDK7: Function and genetics
CDK7 is the first kinase to phosphorylate the CTD within the PIC 158. This TFIIH kinase activity is stimulated by the Mediator complex that binds to the unphosphorylated form of the CTD and helps recruit Pol II to the pre-initiation complex (PIC) 67. Phosphorylation of Ser5 and Ser7 residues in the CTD 150,163 disrupts this interaction 162a,164 permitting promoter escape and entry into the elongation phase of the transcription cycle. The early stages of elongation coincide with assembly of the elongation complex that consists of factors promoting processive elongation, chromatin modification and RNA processing 165. This stage of the transcription cycle differs in yeasts and metazoa. In yeast, the Pol II elongation complex is rapidly assembled and the transition to elongation occurs without an apparent delay. In metazoa there is a delay in this transition leading to a paused Pol II early elongation complex about 30 nt downstream of the transcription start site 166. CTD kinases play an important role in these early elongation complexes and thus, although these kinases are similar in yeast and metazoa their functions are subtly different.
Despite its proposed central role in releasing Pol II from the PIC and the fact that essential genes encode CDK7, transcription of some genes occurs in the absence of CDK7 activity. This was first observed in vitro where neither basal nor activated transcription was reduced in the absence of TFIIH kinase activity 62c,167. Later in vivo experiments supported this observation. Inhibition of analog-sensitive Kin28 has only a minimal effect on global Pol II transcription 168. In the first case, Kanin et al showed no decline in global transcription of Pol II genes 168b. In the second case Hong et al normalized their data differently to show that steady state levels of 58% of Pol II transcripts were sensitive to Kin28 inhibition 168a. Despite this decrease in steady state RNA the occupancy of Pol II on chromatin did not appreciably decline. Phosphorylation of Ser5 did decrease while that of Ser2 remained nearly constant. Taking these results together, a picture emerges that Kin28 phosphorylation provides Ser5P for the purpose of recruiting the capping enzymes. In the absence of this phosphorylation capping is reduced and, although transcription continues the uncapped transcripts are unstable. CDK8/Srb10 and/or Cdc2/Cdc28 likely provide some residual Ser5 phosphorylation.
Global transcription seems to be independent of CDK7 in S. pombe and humans cells. Microarray analysis of transcription in a mcs6 temperature sensitive mutant grown at the non-permissive temperature for a short period demonstrated only a minor effect on global transcription 169. Similarly, there is little effect on global transcription in a human cell line expressing an analog sensitive Cdk7 gene 163c. In this latter case the accumulation of Pol II near the 5’ end of genes is reduced suggesting that promoter proximal pausing is dependent on CDK7 163c. This effect on pausing is most likely not due to changes in CTD phosphorylation but rather to changes in phosphorylation of TFIIE and/or DSIF 170.
Although Cdk7 is essential for organismal viability there has been some difficulty in discriminating between its physiological roles in transcription and cell cycle control. In C. elegans loss of Cdk7 effects both transcription and mitosis 171. In Drosophila, adult animals homozygous for a cdk7ts mutation are viable for over 40 days at the non-permissive temperature arguing against an essential role in transcription. A role in cell cycle control is suggested by a reduction in the number of rapidly dividing follicle cells in females. Mice homozygous for a Cdk7 deletion die early in embryogenesis 172 but demonstrate little effect on Pol II transcription. The major effect in these cells is in cessation of cell division brought on by the lack of Cak activity. Tissue specific knockouts of Cdk7 in adults cause the loss of proliferating cells but little change in non-proliferating cells. This result argues that TFIIH kinase is not required for transcription in post-mitotic cells. Furthermore, inhibition of Rb family proteins with ectopic expression of the SV40 T antigen restored growth to cdk7 null embryonic fibroblasts 172. Taken together these results indicate that in some cells, Cdk7 is not essential for transcription. The likely explanation is that other CDKs can provide the missing CTD kinase activity.
6.2.3. Cdk8
CDK8 is the catalytic subunit of the CDK8 kinase module (CKM) that associates dynamically with the Mediator complex 173. The CKM consists of Cdk8, cyclin C (CycC), Med12 and Med13 in human and Srb8/9/10/11 in S. cerevisiae 58c,174. Med12 is required for CTD kinase activity but MED13 is not 175. About one third of the CDK module is not associated with the Mediator complex suggesting that some CDK8 functions may be attributable to activities that occur apart from the transcription initiation or elongation complexes 173a,174f,175.
In vitro phosphorylation of CTD substrates indicates that CDK8 phosphorylates both Ser2 and Ser5 with a slight preference for Ser5 58c,161c,d,162a. Unlike CDK7, CKM displays no preference for heptad repeats that contain Lys in position seven 161c. Consistent with this finding CDK8 displays a preference for phosphorylating the more conserved mammalian proximal repeats 161d.
Early work on the kinase catalytic subunit Srb10 in S. cerevisiae suggested a repressive function for this kinase in phosphorylating the CTD before PIC formation thereby preventing a stable PIC to form 162a. This was supported by transcription profiling of an srb10 deletion strain that displays up regulation of over 150 genes 176. Later work employing analog sensitive kinases showed that Srb10 has a positive role in transcription both in vivo and in vitro 177. Inhibition of srb10-as reduced transcription but not to the extent as inhibition of Cdk7. Inhibiting both kinases together yielded even more inhibition suggesting that both of these kinases have similar, partially overlapping positive roles in transcription 177. These roles are clearly not fully redundant as deletion of Cdk7 is lethal while deletion of Srb10 is not.
6.2.4. Cdk9: Structure and Biochemistry
Cdk9 and its regulatory cyclin subunit form the positive transcription elongation factor b (P-TEFb) 71,178. The closest relative to Cdk9 in S.cerevisiae is the Bur1/Bur2 kinase complex 179. In S. pombe the equivalent kinase is comprised of a Cdk9/Pch1 heterodimer that co-purifies with components of the capping complex 53,180. In both yeasts this Cdk9 heterodimer is essential. In yeasts there is a second gene encoding a non-essential Cdk9-related kinase (Ctk1 in S. cerevisiae and Lsk1 in S. pombe, Table 2) and two additional kinases Cdk12 and Cdk13 in metazoa. These kinases will be discussed in a later section.
Mammalian CDK9 was initially cloned as a cdc2-related kinase and named PITALRE after a unique sequence motif that occurs in this kinase 181. There are two isoforms of this subunit, Cdk9p42 and Cdk9p55 that differ in size due to alternative transcription start sites 182. These catalytic subunits form a complex with one of four cyclin subunits T, T2a or T2b creating a diverse set of heterodimers that add a degree of functional complexity beyond the scope of this review
Whereas Ser2 is thought to be the main in vivo target of P-TEFb in vitro biochemical analysis indicates that this kinase phosphorylates primarily Ser5. CTD peptide substrates pre-phosphorylated at Ser5 are not phosphorylated by CDK9 while substrates phosphorylated at Ser2 are 161d. CTD peptides with Ala substitutions at Ser2 are phosphorylated while those substituted as Ser5 are not. Furthermore, Western blot analysis with mAbs against different phosphorylated forms of the CTD generated by P-TEFb revealed primarily Ser5P with a small amount of Ser7P and virtually no Ser2P 140a. Interestingly, Ser7P substrates are phosphorylated more rapidly than unphosphorylated substrates indicating that Ser7P may prime phosphorylation by P-TEFb 140a,183.
The S. cerevisiae Bur1/Bur2 kinase phosphorylates a GST-CTD fusion protein on mainly on Ser5, although phosphorylation of Ser7 and Ser2 has also been observed 162b,179b,184. Ser2 is only slightly phosphorylated when probed with mAb 3E10 that recognizes Ser2P but when the same substrate is probed with mAb H5 that recognizes Ser2P along with an adjacent Ser5P there is an increase in reactivity. These observations suggest that Cdk9/Bur1 kinases phosphorylate Ser2 on CTD substrates that are already phosphorylated on Ser5 and Ser7 CDK7 185. Once Ser5 is phosphorylated Ser2 may become a preferred substrate. This could also be a priming reaction at the substrate level or could be a change in substrate specificity brought about by phosphorylation of the CDK9 T-loop by CDK7 170. In this connection it should be noted that the in vitro assays often employ immunoprecipitated or tagged kinases. These kinases may not be phosphorylated on the T-loop as this phosphorylation takes place on chromatin 170.
6.2.5. Cdk9: Function and genetics
In metazoans Pol II pauses about 30 nt downstream of many promoters 166. This was first observed at Drosophila heat shock genes by Lis and colleagues 186. More recent genome-wide experiments have shown that many genes in eukaryotes display a peak of Pol II occupancy just downstream of the start site 187. Work done by the Price and Handa labs led to the identification of proteins that establish this paused elongation complex 178g,188. The DRB-sensitivity-inducing-factor (DSIF) consisting of Spt4 and Spt5 along with the negative-elongation-factor (NELF) and the newly discovered protein GDOWN1 189 are involved in blocking the early elongation complex. P-TEFb is recruited to this complex in part through interaction with transcription factors or the bromodomain protein Brd4 187d,190.
The transition from the paused complex to productive elongation requires P-TEFb kinase activity 178a. Among the P-TEFb targets are Spt5 which is converted from an elongation inhibitor to a processivity factor and the CTD that is further phosphorylated to create binding sites for elongation, processing and chromatin modification factors 178g. Inhibition of P-TEFb with flavopiridol blocks this transition but does not inhibit Pol II that has already entered the productive elongation phase 187d,189,191. This observation argues that P-TEFb is required only near the promoter.
The activity of P-TEFb within the paused Pol II elongation complex is stimulated in several ways. First, previous phosphorylation of the CTD on Ser7 primes the kinase for phosphorylation of Ser2 140a. In addition, the Cdk9 activation loop is phosphorylated either by Cdk7 170 or Brd4 kinases 192. The activation of P-TEFb is likely to be an important regulatory step 166.
Recruitment of CDK9 in S. pombe and Bur1/Bur2 in S. cerevisiae takes place through a different mechanism. NELF and GDOWN1 are not present in yeast 193 an observation that is consistent with a lack of promoter proximal pausing 194. Despite the lack of promoter proximal pausing there is evidence for a sequential action of CDKs at yeast promoters. Recruitment of Cdk9 or Bur1 requires the activity of Cdk7 180b,185. In the case of Bur1 there is a domain that interacts with Ser5P sites produced by Cdk7 185. In S. pombe previous phosphorylation of Ser7 primes Cdk9 activity 180b. Cdk9 and Bur1 thus provide the initial Ser2 phosphorylation on the early elongation complex. This process may also involve the atypical Brd4 kinase in mammalian cells (see later section). The phosphorylation of Ser2 on downstream elongation complexes involves another family of CDKs discussed in the next section.
6.2.6. Cdk12
This family of non-essential CTD kinases was initially discovered in S. cerevisiae as CTDK-I, the first CTD kinase identified 142,188d,195. For many years it was thought that the functions of P-TEFb were split between Bur1 and Ctk1 in S.cerevisiae 188d,196. With the discovery that metazoan Cdk12 is a CTD kinase this has clarified the situation and the Cdk12-related kinases are now recognized as distinct from Cdk9 providing a CTD kinase activity that functions later in the transcription cycle 188d. The S. pombe CTDK-I kinase is comprised of Lsk1, the catalytic subunit and Lsc1 the cyclin subunit and Lsg1 corresponds to Ctk3 197. The human CDK12 and CDK13 genes are closely related by sequence and are the closest relative of the S. cerevisae Ctk1 and S. pombe Lsk1 catalytic subunits. Recent work from the Greenleaf lab has shown that CDK12/13 are CTD kinases both in vitro and in vivo 198.
In yeast, the CDK12 homolog CTDK-I is comprised of the catalytic subunit Ctk1, the cyclin subunit Ctk2 and Ctk3, a subunit of unknown function 188d. CTDK-I is the major Ser2 kinase in S. cerevisiae. Bur1 and Ctk1 make approximately equal contributions to Ser2P near promoters but deletion of Ctk1 reduces SerP in the downstream region by 90% 185. Similar inhibition of Bur1 reduces Ser2P by 50%, much greater than the expected 10% remaining after Ctk1 inhibition 185. This result suggests that Bur1 phosphorylation enhances Ctk1 phosphorylation and loss of Bur1 activity also leads to a reduction in Ctk1 activity.
The role of these kinases in depositing the Ser2P mark is apparently to encourage the recruitment of factors that bind Ser2P or the dually phosphorylated Ser5P-Ser2P heptad repeats. This includes splicing and termination factors as well as factors that alter chromatin. These factors are discussed in a later section.
6.2.7. Cdk1
The cyclin-dependent kinase I (Cdk1) is also known as Cdc2 in metazoa and S. pombe or Cdc28 in S. cerevisiae. The catalytic subunit assembles with cyclin B to form the active CDK. The primary function of this kinase complex is to regulate cell cycle 151 but several recent reports suggest a role in transcription 199.
The first evidence that Cdk1 is a CTD kinase came several decades ago when it was identified as the first metazoan CTD kinase 200. With the identification of other, transcription-specific CDKs it was assumed that Cdk1 phosphorylation of the CTD was an artifact of promiscuous in vitro activity. Recent results, however, have suggested that the yeast Cdc28 kinase plays an important role in CTD phosphorylation and transcription. Enserink and colleagues have used ChIP assays to show that Cdc28 is present on some but not all genes 199a. Using an analog sensitive Cdc28 they show that inhibition of this kinase results in lower Pol II density on these genes and show that the overall level of Ser5P is reduced. Their work further shows that Cdc28 acts together with Kin28 to provide Ser5P. Inhibition of either kinase produces a modest decrease in Ser5P while inhibition of both kinases causes a major reduction. These experiments suggest that Cdc28 may play a similar role to Kin28, providing a boost in this function as cells enter the cell cycle.
6.3. Other CTD Kinases
6.3.1. MAP kinases
Mitogen-activated protein kinases (MAPKs) are Ser/Thr kinases most closely related to the CDKs 201. The MAPKs are activated by mitogens, osmotic stress and heat shock 202. Bensaude and colleagues showed that serum stimulation of resting fibroblasts causes a marked increase in the highly phosphorylated IIO form of Rpb1 203. The enhanced CTD kinase activity in these cells co-purified with MAP kinases and purified MAP kinases were shown to phosphorylate the CTD in vitro 203. The MAPKs ERK1 and ERK2 were later shown to specifically phosphorylate Ser5 in CTD peptides 161b.
6.3.2. DNA-PK
DNA-dependent protein kinase (DNA-PK) is a DNA-activated protein kinase that has been implicated in DNA repair processes like homologous recombination and non-homologous end joining 204. Although initial studies indicated that DNA-PK preferentially targets S/T-Q or Q-S/T phosphoacceptor sites several studies have shown that DNA-PK phosphorylates CTD fusion proteins or peptides 161b,205. Surprisingly, DNA-PK can phosphorylate all three Ser residues in the CTD 206. DNA-PK was identified as part of the Pol II holoenzyme 207 and has recently been implicated in transcription of the HIV genome 206 and in transcription-coupled repair 208. Phosphorylation of the CTD by DNA-PK depends not only on DNA but is also stimulated by transcription activators 209.
6.3.3. BRD4
BRD4 is a bromodomain protein that has been implicated in a variety of human diseases 210. Recently Singer and colleagues have shown that BRD4 is an atypical protein kinase that phosphorylates the CTD on Ser2 211. Whereas P-TEFb phosphorylation of the CTD generates epitopes recognized by both mAbs H5 and 3E10, BRD4 generates only the Ser2P-specific 3E10 epitope. This difference is possibly due to the ability of P-TEFb to phosphorylate both Ser2 and Ser5 whereas BRD4 is apparently restricted to Ser2. The BRD4 kinase activity differs from P-TEFb in that it is insensitive to the inhibitor flavopiridol but is inhibited by apigenin 211. Inhibition of P-TEFb with flavopiridol in an in vitro transcription reaction did not substantially reduce Ser2 phosphorylation while apigenin reduced SerP levels by 90% 211. Over expression of BRD4 in vivo leads to a marked increase in Ser2P while drugs that block the acetylated histone binding of the BRD4 bromodomain reduce the in vivo level of Ser2P. These results indicate that BRD4 is likely to play an important role in the transcription cycle potentially acting at the transition between initiation and elongation, before the recruitment of pTEFb 210.
6.3.4. Plk3
Polo-like kinase 3 is a member of the polo Ser/Thr kinase family 212. Plk3 is unique to mammalian cells and is widely expressed and localized to the nucleus 212. Like other members of this family Plk3 contains a polo box domain (PBD) that binds phosphoserine/threonine containing motif 213. Plks phosphorylate Ser or Thr embedded in acidic sequences, preferring acidic residues in all positions between +4 and -4 from the phosphoacceptor 214. There are no amino acid sequences in the CTD that correspond to this motif but phosphorylation of multiple serines could enhance the phosphorylation of adjacent sites by Plk3.
Eick and colleagues identified an antibody (6D7) that recognizes Thr4P in the CTD and have shown that in human cells Plk3 deposits this mark 54b,215. In vitro, phosphorylation occurs on Thr4 and can convert Pol IIA to Pol IIO. Thr4P is found in vivo on a subset of Pol IIO containing Ser2P but lacking Ser5P and Ser7P 54b. Plk3 is activated by oxidative stress 216 and treating HeLa cells with H2O2 results in a marked increase in Thr4P in vivo 54b. Ser2P peaks before Thr4P along a gene indicating that Ser2P is a prerequisite for Thr4P 54b. This is consistent with the known preference for acidic phosphoacceptor sites. Phosphorylation of Thr4 has also been observed in budding 54b and fission 217 yeast but the kinase(s) responsible for this modification have not been identified.
6.3.5. Tyrosine kinases
While phosphoserine and phosphothreonine are the most abundant phosphoamino acids obtained from Rpb1 immunoprecipitated from in vivo extracts small amounts of phosphotyrosine are also obtained 47b. In vitro phosphorylation of a GST-CTD fusion protein by c-Abl or the Arg kinase suggested that the origin of the phosphotyrosine was Tyr1 of the CTD 47.
Recently Eick and colleagues have described a monoclonal antibody that is specific for Tyr1P in the CTD although reactivity is blocked by phosphorylation of adjacent Ser5 residues 215. This antibody detects tyrosine phosphorylation both in metazoa and yeast. ChIP using this antibody has been used to show that Tyr1P CTD is widely distributed throughout the genome being low at promoters, increasing toward the middle of genes and reduced in abundance at the polyadenylation site 145. The abundance of Ser5P near the 5’-end may lead to an underestimation of promoter-proximal Y1P. Tyr1P-containing peptides do not bind to the CTD interacting domains (CIDs) of several termination factors. Taken together these results suggest a role for Tyr1P in regulating access of termination factors to the CTD during elongation 145. Kinase(s) that deposit this mark in yeast have not yet been identified.
6.3.6. Casein kinase II
Casein kinase II (CK2) is a Ser/Thr kinase that phosphorylates acidic target sites with the consensus S/T-X-X-D/E 218. This sequence occurs several times in the mammalian CTD and has been reported to be phosphorylated by CK2 152. Although this kinase cannot phosphorylate the consensus repeats 152 it is possible that the presence of phosphates deposited by other kinases can prime phosphorylation by CK2.
6.4. Summary of CTD kinases
The broad outline of the sequential phosphorylation of CTD targets is now becoming clear and we can see the similarities between the sequential chain of protein kinases that regulate the cell cycle and those that regulate the transcription cycle219. Emerging evidence indicates that many of the CTD kinases interact in ways that enhance or inhibit CTD phosphorylation. These interactions fall into three main categories. First, the kinases can phosphorylate each other to either inhibit of activate CTD phosphorylation. Second, phosphorylation of some CTD sites may prime phosphorylation by other kinases. Third, phosphorylation may facilitate recruitment of the kinase to the transcription complex. All of these possible interactions have been shown to occur and the remaining challenge is to determine how the interplay between kinases functions on individual genes.
While the CTD code offer a solid foundation for understanding the regulation of the transcription cycle and its coupling with processing of the nascent transcript we still lack details about many steps in this process One problem is that the tools used to characterize the CTD phosphorylation pattern are not fully understood and are not able to distinguish between heptads within a CTD. In addition, the genetic tools used to alter the function of different CTD kinases have limitations. For example, there are likely to be many other substrates of the CTD kinases and some of these may play critical roles in regulating transcription. Future studies will need to address the spectrum of substrates of the CTD kinases.
6.5. CTD glycosylation
The CTD in mammalian cells is post-synthetically modified by the addition of O-linked N-acetylglucosamine by the enzyme O-GlcNAc-transferase (OGT) 220. Removal of O-GlcNAc is catalyzed by an enzyme named N-acetyl-β-D-glucosaminidase (OGA) 221. For almost two decades the significance of CTD glycosylation remained elusive but the recent development of inhibitors of OGT 222 and OGA 223 has allowed the characterization of CTD glycosylation demonstrating that this modification plays a significant role in regulation of transcription in higher eukaryotes.
Early experiments showed that the unphosphorylated IIa form of Rpb1 is O-GlcNAcylated at Thr4 and Ser5 220a. The stoichiometry of the modification was very low, however, leaving open the question of function. More recent work has used an OGA inhibitor to stabilize the O-GlcNAcylated CTD 224. These studies showed the presence of a highly modified form of the unphosphorylated CTD that was termed Pol IIγ 224. The retarded mobility suggests that this form is multiply GlcNAcalated but the stoichiometry was not determined. In vitro modification of GST-CTD fusion proteins containing Ser5 or Ser7 to Ala substitution are poor substrates for OGT. Taken together with the earlier identification of Ser5 and Thr4 as the targets site for modification this indicates the Ser5 is the most likely site of CTD GlcNAcalation.
OGT and OGA inhibitors block transcription indicating that O-GlcNAc modification is an essential step in transcription. Since both the addition and removal of the modification are essential this has led to model in which O-GlcNAc cycles on and off the CTD in the PIC 224. What role this “γ-cycle” may play is unclear and the possibility that the prevalence of hexosaminidases removes the GlcNAc from the CTD rapidly in vitro could mean that the unmodified IIa form of Rpb1 is a rare intermediate. In this view Pol II would cycle in vivo between the IIO and IIγ forms. The rationale for modifying phosphorylation sites with O-GlcNAc may be to protect these sites from inadvertent phosphorylation.
6.6. Other CTD modifications
Several other CTD modifications have been observed in mammalian cells. These modifications generally target non-consensus amino acids like the methylation of the arginine residue in position seven of the 31st repeat. This modification is carried out by the coactivator-associated arginine methyl transferase I (CARM1). The methylated CTD acts to inhibit expression of snRNAs and snoRNAs 225. Methylation of this CTD arginine is suppressed when the CTD is phosphorylated on nearby Ser2 and Ser5 residues 225.
7. CTD CODE ERASERS: CTD PHOSPHATASES
CTD phosphorylation is a dynamic process in vivo with different sites phosphorylated and dephosphorylated during the transcription cycle to provide binding sites for proteins involved in processing the nascent transcript. In addition, dephosphorylation is required to recycle Pol II after transcription termination providing an unphosphorylated Pol IIA for assembly of new PICs. In this section we will discuss the phosphatases that act to counter the CTD kinases and thus shape the changing CTD phosphorylation pattern.
7.1. The Fcp1 CTD phosphatase family
The Fcp1 CTD phosphatase was first identified biochemically as an enzyme activity that converts the phosphorylated IIO form of Pol II to the unphosphorylated IIA form 226. This phosphatase is essential in yeast 227 and highly conserved in evolution. Fcp1 is a multi-domain protein with an N-terminal catalytic domain and a breast cancer protein related C-terminal (BRCT) domain 228. The catalytic domain contains a DXDX(T/V) motif commonly found in phosphotransferases and hydrolases 229. In addition to Fcp1 animal cells contain a related family of small CTD phosphatases (SCPs) encoded by separate genes and consisting of an Fcp1-related catalytic domain but lacking the BRCT domain 230. The human SCPs act to silence neuronal genes in non-neuronal tissues 231.
The first structure of an FCP1-like phosphatase to be solved was human SCP1 232. These studies showed that the active site is located in a depression on the surface that normally contains a Mg++ ion. The first Asp in the conserved motif DXDX(T/V) acts as a phosphoacceptor for the phosphatase reaction and mutation of this residue to Ala in the yeast Fcp1 protein results in an inactive enzyme 228a,232a,233. There is a fundamental difference in the specificities of Scp1 and Fcp1. The small phosphatase Scp1 recognizes Ser5P about 60-fold better than Ser2P 232b. In contrast, while Fcp1 will dephosphorylate both Ser2P and Ser5P it displays a preference for Ser2P of about 6-fold 234. In both cases the preferred substrate contains an SP sequence in the trans conformation 235. The difference in specificity between Scp1 and Fcp1 lies in the particular arrangement of amino acids that lie in the active site groove and by the presence of additional domains in the Fcp1 enzyme 234a. Scp1 makes specific contacts with Tyr1, Pro3 and Thr4 thus positioning Ser5P in the catalytic center. Fcp1 cannot make the same contacts as many of the contact residues are buried in the Fcp1 structure. Structure-activity experiments show that mutations in the Fcp1 CTD-binding pocket alter specificity for either Ser2P or Ser5P. Most importantly, mutations that reduce Ser5P specificity have no effect on the growth of S. cerevisiae strongly supporting the idea that Fcp1 is primarily a Ser2P-specific phosphatase 236.
Fcp1 interacts with the catalytic core of Pol II 237 and this docking site includes the Rpb4/7 subunits 227b,232a located adjacent to the CTD linker domain 35,238. In addition, Fcp1 interacts with TFIIF and this interaction stimulates the phosphatase activity 227b,239. These interactions are not essential for phosphatase activity however, as Fcp1 can dephosphorylate CTD peptides 233 and Pol II that is not engaged in transcription 240.
The CTD is dynamically phosphorylated in vivo and Fcp1 plays an important role in this process. A major question is whether CTD Ser2P dephosphorylation occurs continuously along a gene as it is being transcribed or alternatively is restricted to the 3’ end where it could serve to recycle recently terminated Pol II. Consistent with an ongoing role in elongation fcp1 mutants show an increase in the level of phosphorylation of Ser2 detected by mAb H5 184,236. Surprisingly, the level of reactivity of the Ser2P specific antibody 3E10 is not changed arguing that the role of Fcp1 may not be able to remove all Ser2P but rather those adjacent to Ser5P 184.
Transcription both in vivo and in vitro is stimulated by Fcp1 activity 228a,228c,236,241. One mechanism leading to higher levels of transcription is the removal of CTD phosphates resulting in a higher level of transcription initiation. Recent work by Lis and colleagues have shown that Fcp1 depletion in Drosophila does not change the phosphorylation state of chromatin bound Pol II but leads instead to an increase in the amount of non-chromatin bound phosphorylated Pol II 241. This result suggests a role for Fcp1 in providing the pool of unphosphorylated initiation competent Pol II 241.
7.2. CTD phosphatase Ssu72
The SSU72 gene was initially identified as a modifier of a mutation in the gene encoding the general transcription factor TFIIB that causes a change in Pol II start-site selection 242. The presence of a CX5R sequence motif in Ssu72 suggested that this protein is related to protein tyrosine phosphatases and this prediction was confirmed through biochemical and genetic analyses 243.
Ssu72 is present in Pol II complexes throughout the transcription cycle. It plays a role in start site selection through interaction with TFIIB 244. Mutations in ssu72 alter the sensitivity of yeast to 6-azauracil arguing for a role in elongation 244c. Ssu72 also plays a role in 3’end formation as a part of the cleavage and polyadenylation complex 91e,245. Finally, Ssu72 brings the transcription cycle full circle by promoting interactions between the 5’ and 3’ ends in a process called gene looping 246.
Biochemical studies of Ssu72 show that this phosphatase preferentially removes Ser5P from the CTD 234b,247. Rather than acting at the 5’ end of genes where Ser5P predominates, Ssu72 has recently been shown to remove Ser5P that persists into the 3’ end. Mutation of Ssu72 causes the persistence of Ser5P and the termination of Pol II that retains Ser5P 184. Ser7P mirrors Ser5P at the 3’ end and mutation of ssu72 leads to a build up of this phosphorylation mark suggesting that Ssu72 is a dual specificity phosphatase or that it indirectly affects removal of phosphate from Ser7P.
The activity of Ssu72 is enhanced by the peptidyl prolyl isomerases Ess1 248 and structural studies of Ssu72-CTD substrates show that the phosphosphatase preferentially interacts with the cis configuration of the Ser5P-Pro6 bond 249. Mutation of ess1 leads to a retention of both Ser5P and Ser7P on Pol II mapping to the 3’ ends of genes. Taken together, these results indicate that Ssu72 plays an important role in recycling Pol II.
7.3. Rtr1/RPAP2 plays a role in CTD dephosphorylation
Yeast Rtr1 is related to the human Pol II-associated protein RPAP2 250. The yeast Rtr1 protein was identified as a protein that interacts with Pol II and in yeast localizes to the coding region of genes in a ChIP assay 251. Deletion of Rtr1 leads to an increase in Ser5 CTD phosphorylation suggesting that Rtr1 is a CTD phosphatase and two different groups have reported CTD phosphatase activity for purified Rtr1 or Rpap2 251-252.
Although poorly conserved overall, the present of a conserved Zn-finger motif suggested this might be a phosphatase active site. However, the structure of Rtr1 does not contain an active site cleft with conserved residues at the bottom and bacterially expressed Rtr1 does not display any CTD phosphatase activity suggesting that Rtr1 may play a regulatory role in CTD dephosphorylation 253. Future studies will need to address the precise role of Rtr1 in CTD dephosphorylation.
8. REARRANGING THE CTD CODE: PROLINE ISOMERASES
Prolyl peptide bonds can adopt either of two conformations designated cis and trans. Isomerization between the cis and trans conformations in proteins has an activation energy barrier of about 20-25 Kcal mol-1 and thus the isomerization reaction is slow and is often the rate-limiting step in protein folding 254. In globular proteins the percentage of cis X-P bonds is low and depends on the preceding amino acid and on the structural context 33. Given these constraints, the majority of cis bonds in proteins occur in turn motifs 33. In more disordered protein domains like the CTD the Ser-Pro bond is less constrained. If 30% of the S-P bonds in the mammalian CTD were to adopt the cis conformation 29b this would mean that on average every CTD would contain about 30 cis S-P bonds. The rate of cis-trans isomerization is greatly enhanced by peptidyl-prolyl-cis-trans-isomerases (PPIases) 255. Several PPIases have been shown to interact with the CTD including Pin1, Ess1 and Rrd1.
8.1. Pin1
Pin1 was the first PPIase shown to alter the conformation of the CTD 256. This metazoan enzyme was initially identified as a protein that interacts with NIMA to regulate cell cycle 257 and together with its yeast homolog Ess1 are members of the parvulin class of PPIases 258. These PPIases do not bind immunosuppressive drugs and thus are distinct from the immunophilin PPIases 259. Pin1 is dependent on phosphorylation of its target site, Ser or Thr followed by Pro 260. While it is impossible to measure the isomerization state of the CTD in vivo, genetic manipulations of PPIase gene function cause changes in the CTD modification state and thus one can infer a role for proline isomerization in this process. Pin1 inhibits dephosphorylation of the CTD by Fcp1 261. Over expression of Pin1 causes Pol II to dissociate from the chromatin template and a hyperphosphorylated form designated Pol IIoo accumulates in speckle structures in the nucleus 262. Depletion of Pin1 enhances transcription and Xu and Manley have shown that Pin1-induced transcription inhibition occurs early in the transcription cycle at the transition between initiation and elongation 262.
8.2. Ess1
Ess1 is the yeast homolog of Pin1 and is the only essential PPIase gene in Saccharomyces cerevisiae 263. Like Pin1, Ess1 has been shown to interact with the CTD 264. While Pin1 has been shown to isomerize many target proteins 265, Ess1 has only been shown to act on the CTD. In vitro Ess1 isomerizes the Ser5-Pro6 bond about 5-times more effectively than the Ser2-Pro3 bond 266. Ess1 plays several roles in RNA synthesis and processing. Hani et al identified ESS1 (PTF1) in a screen for mutants that failed to terminate transcription at the ADH1 3’-end 258,267. Subsequent work from Hanes and colleagues has expanded this to show that Ess1 plays a role in both mRNA and ncRNA termination and processing 184,268. One possible mechanism for Ess1 function is to enable the Ser5P-Pro6 bond to adopt the cis conformation and thus enable Ssu72 to bind and dephosphorylate this residue 249,269. Pcf11 prefers to bind Ser2P and the continued presence of Ser5P could reduce Ser2 phosphorylation 91c. In addition, failing to remove Ser5P would lead to the persistence of Nrd1 binding and thus enhance premature termination of many transcripts without polyadenylation 91e.
8.3. Rrd1
Rrd1 is an S. cerevisiae PPIase that is distinct from the Pin1/Ess1 family in that it is sensitive to the immunosuppressive drug rapamycin. This non-essential proline isomerase was initially identified by its similarity to the human phosphotyrosyl phosphatase activator PTPA 270. Deletion of Rrd1 confers resistance to rapamycin 270 and subsequent studies demonstrated that Rrd1 is a PPIase 271. Rdr1 has been shown to act at the transcriptional level 272. Genes that are normally activated at the diauxic shift are not properly up regulated in an rrd1 mutant and expression of genes like the ribosomal protein genes that are normally down-regulated are not reduced in an rrd1 mutant 272-273.
While the mechanism of Rrd1 has not been elucidated several possible targets have been identified. Rrd1 binds to the yeast PP2A-like phosphatase Sit4 274. One possibility is that Rrd1 isomerizes and thereby activates this phosphatase and this leads to changes in the phosphorylation state of transcription activators and repressors. A second potential target is the CTD. Recent studies have indicated that Rrd1 binds to chromatin in vivo and to the CTD and isomerizes it both in vitro and in vivo 273,275. Like the case of Pin1 this leads to removal of Pol II from the chromatin template 275. In contrast to the case with Ess1 there is no evidence that Rdr1-induced changes in CTD conformation alter the posphorylation state of the CTD 275.
9. READING THE CTD CODE
The highly conserved tandem arrangement of CTD heptad repeats initially suggested that the CTD interacts with a regularly repeating structure like DNA or RNA and some experiments indicated such interactions can occur 25b,60,276. However, the functions of these interactions have not been forthcoming and the emphasis is now on proteins that interact with specific sequences or modifications within the CTD to execute CTD functions.
CTD-interacting proteins have been identified both by in vivo genetic approaches 79b and through in vitro biochemical techniques 107a,277. This has led to identification of a large number of potential interacting proteins but in vivo verification of these interactions is more limited. In this section we will focus on a subset of these interactions that have been verified as interacting directly with the CTD. Much of our knowledge of the stereospecificity of these interactions has come from three-dimensional structures of CTD-binding proteins bound to CTD peptides. A number of proteins that bind to different phosphorylated forms of the CTD are involved in various stages of the transcription cycle and several of these will be discussed in general terms as to their roles. The reader is directed to the reviews by Jeronimo et al. and Eick and Geter in this series for a further discussion of gene-specific functions of CTD-binding proteins.
9.1. Mediator
The CTD undergoes reversible phosphorylation during the transcription cycle 278. The unphosphorylated Pol IIA form assembles in the pre-initiation complex 36,46 and is converted to the IIO form by phosphorylation on Ser5 by the TFIIH kinase 158a,158c,161b. This phosphorylation event coincides with promoter clearance leading to the idea that phosphorylation of the CTD releases Pol II from the PIC allowing the transition to elongation. While elements of the model are correct, the situation is likely more complex.
Early genetic and biochemical approaches suggested that the CTD interacts with the Mediator complex that enables regulation of Pol II transcription 173b,c,279. The genes encoding Mediator subunits were initially identified as suppressors of CTD truncation mutants 22 and later were shown to be components of the megaDalton Mediator complex 67. Mediator contains over 30 subunits in four different sub-modules. The tail module interacts with activators and repressors, while the head and middle modules interact with Pol II. The CKM containing the Ckd8 CTD kinase activity reversibly associates with the middle module. Mediator can bind to the uphosphorylated CTD in vitro 68 and can be displaced from the Pol II holoenzyme complex by mAb 8WG16 67,280 that binds to the unphosphorylated CTD 144b. Truncation of the CTD weakens the response to activators that function through the Mediator and lengthening the CTD acts to suppress mutations in Mediator genes 281. Together, these results pointed to a functional interaction between Mediator and the CTD.
Purification of the mediator using an antibody to the CTD strongly suggests that the CTD interacts directly with the CTD but early efforts to identify interacting subunits were unsuccessful. The reason for this failure has recently become apparent as new studies have shown that the CTD makes multiple contacts with the Mediator including both the head 282 and the middle module 37. The strongest interactions occur between the CTD and the middle module and are thought to represent the initial stages of holoenzyme formation 37. Although the precise interaction site has not been identified, the CTD binding site on the middle module overlaps with the CKM binding site and the presence of CKM on the Mediator interferes with Pol II binding and holoenzyme formation 37. This nicely explains the identification of the CKM genes as suppressors of CTD truncation mutations. Presumably truncation of the CTD weakens interaction with Mediator and this weak interaction can be suppressed by mutations that weaken the competitive interaction of CKM with Mediator 37.
Once the initial complex between the CTD and the middle module forms it has been proposed that the conformation of the pre-holoenzyme changes, positioning the CTD on the head module. In the head module the CTD contact surface spreads over 73Å and involves almost four heptad repeats in an extended conformation282. Specific contacts are made with highly conserved regions of Med6, Med8 and Med17. The distributed nature of these interactions may explain why single mutations are unlikely to eliminate CTD interactions with the Mediator.
What releases the CTD from Mediator contacts? In vitro experiments first showed that TFIIH is able to disrupt holoenzyme complexes containing Mediator and Pol II assembled on promoters in vitro 283. The recent finding that in vivo inhibition of analog sensitive TFIIH kinase does not inhibit transcription leaves open the possibility that CTD phosphorylation is not necessary for releasing Pol II from the PIC. The Mediator makes non-CTD contacts with Pol II 284 and the act of initiation may alter the conformation of the catalytic core in a way that weakens the affinity for Mediator 285 without the need for CTD phosphorylation by TFIIH. An alternative explanation is that other CTD kinases are able to substitute for TFIIH and by phosphorylating the CTD release Pol II from the PIC. In vitro, Mediator can be released by CTDK-I arguing that phosphorylation itself may be sufficient for release and the Ser2P or Ser5P are sufficient to loosen the CTD-Mediator interactions 283b. Precisely which contacts are disrupted are not clear but it is interesting to note that CTD phosphorylation may induce a conformational change in the Mediator 286.
9.2. Capping complex
The m7GpppN 5’ cap is added to Pol II transcripts early in the transcription process 72b,287 and serves to protect the nascent transcript from premature decay and direct further processing and translation of the mRNA (reviewed in Ghosh and Lima, 2010288). Targeting of the capping reaction to Pol II transcripts is generated through the interaction of the capping complex with Pol II phosphorylated on Ser5 in the early elongation complex 73-74,77,288. How the capping machinery interacts with the CTD has been addressed through crystallographic analysis of complexes formed between the guanyltransferase enzymes and CTD peptides.
Lima, Shuman and colleagues crystallized the nucleotidyl transferase domain of the Candida albicans Cgt1 GTase subunit bound to a Ser5 phosphorylated CTD peptide containing four repeats 289. The GTase domain adopts a mixed α/β fold with an interaction surface consisting of a 40Å long channel in which 17 non-contiguous amino acids of the CTD peptide lie in an extended ß-like conformation. Within this channel there are two CTD docking sites (CDS1 and CDS2, Figure 5) that make specific contacts with CTD side chains. In each of the docking sites there is a positively charged depression that binds the phosphate on Ser5 of the first and third heptad repeats. Critical interactions are also made with tyrosine residues both through hydrophobic interaction (CDS1) and hydrogen bonding (CDS2). All of these interactions are fully supported by genetic structure-activity experiments 289.
Figure 5.
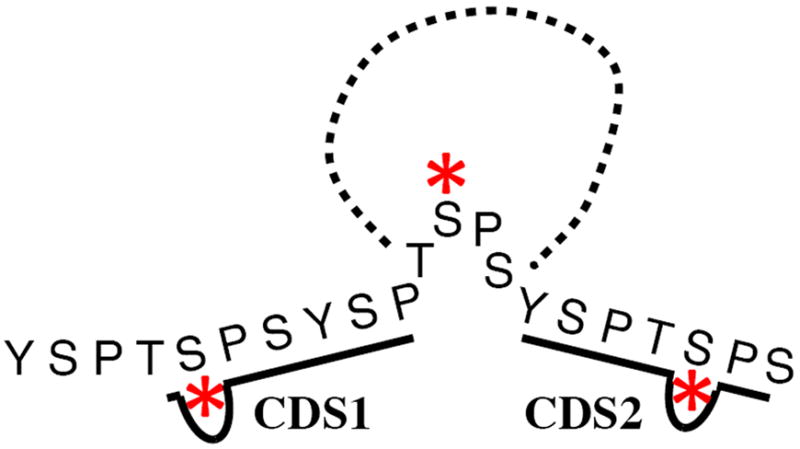
Diagram showing the recognition of adjacent heptad repeats by the C. albicans capping enzyme CDS1 and CDS2 binding pockets. The dotted line indicates the potential position of additional repeats if the CDS1 and CDS2 binding pockets recognize heptads that are not adjacent but separated by one or more heptad repeat.
The four residues between CDS1 and CDS2 do not make contact and form a bend that protrudes from the surface of the protein. This feature of the CTD-Cgt1 complex has important implications for the interaction not only of Cgt1 but for other CTD-binding proteins. First, the Ser5P in the second repeat makes no contacts with the protein indicating that Ser5 on contiguous heptads need not be phosphorylated to interact with Cgt1. Thus, low levels of Ser5 might be sufficient to recruit the capping complex. Second, the looping out of one or more repeats may results in CTD heptads adopting conformations that are stabilized relative to CTD repeats free in solution. This may enhance further interactions or modifications.
The structure of the human Cgt1 homolog Mce1 has recently been solved in a complex with the same phosphorylated peptide 290. The CTD in this structure is also in an extended ß-like conformation but there are important differences in the details of the interactions compared to the yeast complex. Mce1 interacts directly with only six amino acids, S*PSYS*P in which Ser2 and Ser5 are phosphorylated (asterisks). In this structure both Pro3 and Pro6 adopt a trans conformation and Tyr1 is packed into a hydrophobic pocket with its hydroxyl is hydrogen-bonded to a Glu residue at the bottom of the pocket. The Ser5P in this structure makes direct contact with two Arg side chains but the Ser2P does not contact the enzyme. Interactions with Tyr and Ser5 are supported by earlier mutational studies showing that mutations that alter these two CTD positions fail to bind the enzyme 291. The observation that Ser2P makes no direct contacts and points away from the Mce1-CTD interface is inconsistent with previous binding studies showing that Mce1 binds equally to CTD peptides phosphorylated at either Ser2 or Ser5. These binding studies indicate a second Ser2P binding site on Mce1 but the nature of this interaction remains unclear. Given the difference in the manner of interaction with the CTD in Mce1 and Cgt1 it is not surprising that the Mce side chains that interact with the CTD are not conserved in yeast. The one conserved aspect of the interaction with the CTD is the dependence of both structures on interactions with Ser5P and Tyr1. These interactions serve to direct one of the most important CTD functions, coupling transcription to 5’-end capping.
9.3 The WW domain interacts with phosphorylated CTD repeats
WW domains are approximately 40 amino acid domains characterized by a pair of conserved tryptophan residues 292. These domains interact specifically with proline-rich peptides 293 and in the case of type IV WW domains with phosphorylated SerP/ThrP-Pro diamino acids. The function of WW domains is typically in providing an interaction module for proteins involved in signal transduction.
9.3.1 Pin1/Ess1
The proline isomerases Pin1 and its yeast homolog Ess1 have been shown to interact with the phosphorylated CTD 256,264a,267,294. These proteins contain two domains; an N-terminal WW domain and a C-terminal peptidyl prolylisomerase (PPIase) domain of the parvulin family 295. The PPIase of these enzymes show a weak preference for binding to phosphorylated Ser/Thr-Pro sequences but the WW domain has a higher affinity and thus is the major determinant of substrate specificity 296.
The structure of the Pin1 WW domain has been determined by X-ray crystallography both in the absence of a peptide in the binding site 295a and in a complex with a canonical CTD heptapeptide phosphorylated at both Ser2 and Ser5 296b. The CTD peptide lies in a cavity between the PPIase and the WW domain making contacts on the surface of the WW domain but not with the PPIase domain. The CTD peptide adopts an extended coil conformation in which the last five residues (PTS*PS) make contacts. Ser5P makes contacts with three residues that form a phosphate binding module. Ser2P, in contrast, makes no contacts. The proline residues are in the trans conformation making both hydrophobic and van der Waals interactions and the backbone of the TS*P sequence is clamped in place by a pair of aromatic residues. Interestingly, the Tyr1 residue makes no contacts and is apparently not involved in binding. Solution binding studies296b, however, do indicate that Tyr1 is important. An alternative interpretation is that longer peptides may bind through interactions with multiple phosphates as well as the Tyr residue.
9.4 CTD-interacting domains (CIDs) and transcription termination
The CTD-interacting domain (CID) was initially identified by its ability to interact with the CTD in a yeast 2-hybrid assay and by direct binding to CTD fusion proteins 79b,99. The CID is an approximately 140 amino acid domain with similarity to the VHS domain 26c,297 and like this domain is commonly found at the N-terminus of proteins. The first identified CID-containing proteins were mammalian SCAF8/RBM16 and SCAF4/SFRS1, RNA-binding proteins of unknown function 79b. More recently mammalian proteins RPRD1A and RPRD1B have been shown to contain a CID and to play an unspecified role in regulating the CTD phosphorylation state 298. Yeast proteins containing this domain; Nrd1 93,299, PCF11 91a,300, and Rtt103 76,301 have been implicated in transcription termination through their interaction with the CTD 91e. Each of these CIDs has a slightly different specificity for CTD phosphorylation states and the structures of CIDs from several of these proteins have been described and shown to interact with the CTD in a variety of different ways (Figure 6a).
Figure 6.
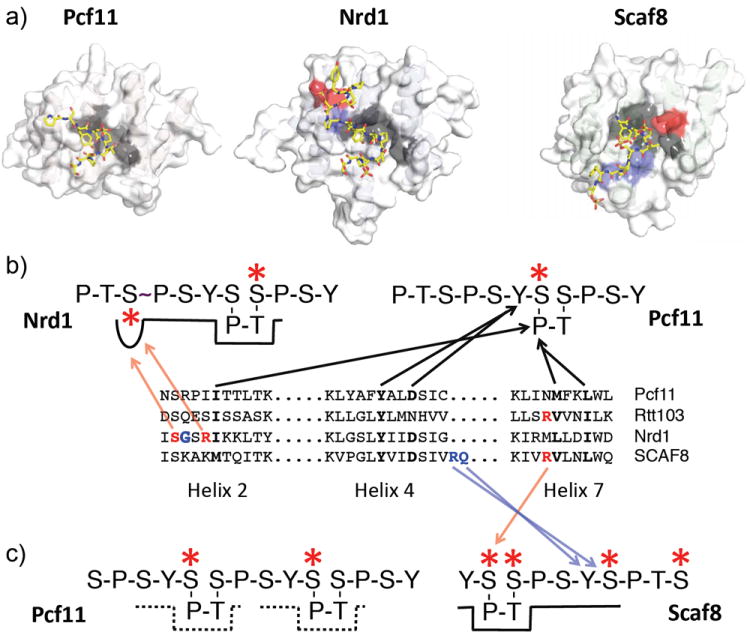
The CTD-interacting domain (CID). (a) Structures of the CID of Pcf11, Nrd1 and Scaf8 bound to CTD peptides. (b) Diagram showing the interaction of CID side chains with CTD residues. Black arrows indicate interactions with the ß-turn and preceding Tyr residue. Blue arrows indicate Scaf8 interactions with the downstream Tyr residue. Red arrows indicate interactions with phosphorylated Ser residues by Nrd1 and Scaf8. The position of these interactions is indicated by the same colors in part A. The wavey bond in the CTD peptide bound to Nrd1 indicates the cis conformation of this paptide bond. (c) Dotted lines indicate alternative binding modes for Pcf11 bound to a doubly phosphorylated CTD peptide. The solid line represents the interaction of SCAF8 with a doubly phosphorylated CTD repeat.
9.4.1. PCF11
PCF11 is a subunit of the cleavage and polyadenylation machinery that processes the 3’-ends of mRNAs 91e,302. The amino-terminal CID of PCF11 binds directly to the CTD 91a,300. Although this interaction favors Ser2 phosphorylated CTD repeats 91c binding to other phosphoisoforms is nearly as strong 301. Pcf11 also binds RNA and has been implicated in removal of Pol II from the template during termination 303. The structure of the Pcf11 CID has been determined both by crystallography and NMR spectroscopy 26c,29b.
The Pcf11 CID module consists of eight α-helices that form a compact right-handed superhelix. The helices are linked by short well-ordered turns and the module is stabilized by a large hydrophobic core. The CTD peptide in this structure lays in a groove formed by conserved residues in helices 2, 4 and 7 and the CTD adopts a ß-turn structure consisting of Ser2P-Pro3-Thr4-Ser5 while flanking residues are in an extended conformation 26c. Within this channel seven hydrogen bonds are formed between the CTD backbone and CID side chains (Figure 6b). Pro3 of the CTD ß-turn is buried in a hydrophobic pocket as is the upstream Tyr1 side chain. This Tyr1 residue also makes a hydrogen bond with a conserved Asp residue possibly explaining why Tyr1 to Phe mutations in the CTD are lethal in S. cerevisiae 18. Surprisingly, the Ser2P does not make any contact with the CID.
The interaction between the Pcf11 CID and the CTD has also been studied in solution using chemical shift mapping. This approach finds interactions similar to those in the crystal structure but extending over a broader area (Figure 6C). Using a longer peptide containing three repeats two of the turn structures are able to interact with the CID. Because no evidence was obtained for simultaneous binding of two CIDs this indicates that the CTD peptide can alternate between two distinct binding modes (dotted lines) with adjacent binding sites alternatively occupying the CID binding pocket. This type of fuzzy complex 304 has interesting implications for movement of CTD binding proteins along the CTD.
A major question concerning the binding of the CTD is whether interacting proteins recognize pre-formed structures or rather the interaction induces a conformation that interacts specifically. Using both calorimetry and NMR Taylor and colleagues measured the binding properties of the Pcf11 CID with this two repeat peptide containing a single Ser2P 29b. The Kd (equilibrium dissociation constant) for this interaction was about 10-4 M-1 indicating a short lifetime for the interaction. The entropic term of the interaction is unfavorable (TdS = -4.3 kCal mol-1, 293.14K) suggesting an induced-fit binding mode rather than docking of a pre-formed turn 23a,29b.
9.4.2. Nrd1 CID
Nrd1 is an S. cerevisiae RNA-binding protein that forms a complex with another RNA-binding protein Nab3 and the RNA helicase Sen1 299b,305. This complex is required for termination of non-coding Pol II transcripts like snoRNAs, snRNAs and cryptic unstable transcripts (CUTs) 93,96-98,306. The N-terminal CID of Nrd1 binds to the CTD phosphorylated on Ser5 rather than Ser2 95 consistent with its termination function near the promoter where Ser5P predominates 9,165. Ser7 phosphorylation slightly weakens this interaction 307.
The Nrd1 CID binds the same ß-turn (Ser2-Pro3-Thr4-Ser5P) that is the central feature of other CID-CTD interactions 307. This turn binds in a hydrophobic pocket created by helices 2, 4 and 7. Where Nrd1 differs from Pcf11 is in binding to a second upstream repeat (Figure 6b). This interaction occurs between residues at the tip of helix 2 and the upstream Ser5P. Specific contacts are made with Nrd1 residues Ser25 and Arg28. Interestingly, the CTD Ser5P-Pro6 bond in this upstream repeat is found in the cis conformation and this feature allows the Ser5P to fit into an electropositive pocket formed by a Gly26 307. This position in the CID is occupied by more bulky side chains in other CIDs making Nrd1 unique in its specificity for Ser5P.
9.4.3 SCAF8/RBM16 CID
SCAF8/RBM16 is a mammalian RNA-binding protein that was identified in a two-hybrid screen for proteins that interact with the CTD 79b. The sequence organization of SCAF8 and a related mammalian protein SCAF4/SFRS15 are most closely related to Nrd1 in that they contain a single RRM domain. The function of SCAF8/RBM16 has not yet been elucidated but this protein has been shown to co-localize with active transcription sites in nuclei 99. The CID of this protein binds selectively to CTD fusion proteins phosphorylated on both Ser2 and Ser5 99. More recent binding assays have supported this preference for Ser2P-Ser5P and have shown that Ser2P is the dominant feature recognized by SCAF8 100.
Meinhart and colleagues have solved the structure of the SCAF8 CID complexed with various phosphorylated CTD peptides 100. The structure of the SCAF8 CID-CTD complexes are generally similar to that of other CID-CTD complexes but show several key differences that can explain the altered specificity for doubly phosphorylated heptad repeats. In the complex containing two repeats of the consensus sequence phosphorylated on both Ser2 and Ser5 of both repeats the first repeat forms the Ser2P-Pro3-Thr4-Ser5P ß-turn common to other CID-CTD structures. This structure brings Ser2P and Ser5P into closer juxtaposition than might be expected when considering the negative charge on the phosphates (Figure 6c). The SCAF8 CID deals with this in two ways. First, distinct from other complexes the SCAF8 CID interacts specifically with Ser2P through Arg-118 (Figure 6b). Mutation of this residue reduces the affinity of SCAF8 for the peptide by an order of magnitude to a level exhibited by Pcf11 which does not specifically recognize this residue. Second, the charge on Ser5P is further compensated for by a water molecule (hydronium ion) in the crystal structure 100. A third distinct feature of this complex is contacts between the C-terminal tip of helix 4 and the downstream heptad repeat. These additional contacts are only seen when Ser2 and Ser5 doubly phosphorylated peptides are bound. Taken together, the SCAF8 CID is adapted to recognize a heavily phosphorylated form of the CTD seen in transcription elongation complexes located in the coding regions of genes.
9.4.4. Rtt103 CID
Rtt103 was initially identified in a screen for regulators of Ty transposition in yeast 308 and later shown to interact with Rat1 and Rai1 and to play a role in transcription termination of Pol II 76. Like PCF11 and Nrd1, Rtt103 has an N-terminal CID but unlike these other CID-containing proteins there is no evidence for an RNA-binding domain. The Rtt103 CID binds exclusively to heptad repeats phosphorylated on Ser2 76. The structure of the Rtt103 CID-bound to a CTD peptide containing two repeats both phosphporylated on Ser2 has been determined by NMR spectroscopy and the overall structure of the CTD peptide is similar to that seen in the Pcf11-CTD complex with a ß-turn consisting of Ser2-Pro3-Thr4-Ser5.
9.4.5. CID-CTD interaction summary
The CID is a key binding module on several proteins that direct termination and processing of nascent Pol II transcripts. Although these CIDs share a common structural framework there is sufficient diversity that many different phosphoisomers can be selectively bound. Solution binding studies argue that the interaction of the CTD with CID domains takes place through an induced fit mechanism and such interactions are characteristically of low affinity 23a. Several features of this type of interaction allow for an increase in specificity. First, as seen with Pcf11 the CID can rapidly shift registers along a CTD containing multiple binding sites. This not only serves to increase binding affinity but may also play a role in movement of this factor along the CTD. A second feature seen primarily in Rtt103 is the ability of individual CIDs to cooperatively bind to the CTD 301. This would serve to increase the local concentration of key RNA processing factors.
9.5. The SH2 domain of SPT6 interacts with the CTD
The SH2 (Src homology 2) domain is a conserved structural module of about 100 amino acids that binds phosphorylated Tyr and plays significant roles in signal transduction pathways 309. Spt6 has two SH2 domains that are unusual in recognizing phosphoserine rather than phosphotyrosine. While a single Ser2P is sufficient for binding, a second SerP in the peptide enhances binding. The basis of this specificity is thought to lie in the phospho-binding pockets of the two non-canonical SH2 domains. In each case the deep pocket that normally accommodates a phosphotyrosine is shallower and more appropriate for a phosphoserine. The consequences of mutation of key residues in these pockets are consistent with this view but the details of the CTD-SH2 interactions await a crystal structure of the complex with a CTD peptide. Nonetheless, the interaction of Spt6 with Ser2 phosphorylated CTD is consistent with genome-wide ChIP experiments that place Spt6 in the body and especially near the 3’-ends of genes, regions that display the highest levels of Ser2 phosphorylation 92c.
9.6. The histone methylase Set2-Rpb1 interacting (SRI) domain
The histone methyltransferases Set1 and Set2 are part of the Pol II elongation complex 310. Set2 interacts directly with Pol II and ChIP experiments place this protein in the coding region of transcribed genes 113b,119a,120b. Recruitment of Set2 requires the CTD kinase CTDK-I suggesting that this recruitment requires Ser2 phosphorylation 113b,120b,311. Set2 contains a C-terminal domain of about 100 amino acids that is required for the interaction with Ser2 phosphorylated CTD. This SRI domain consists of the three-helix bundle that resembles the s2 domain of bacterial sigma factors 312. 313The Set2 SRI domain binds a two repeat CTD phosphopeptide with affinities in the low micromolar range 113a,312. Maximum binding requires two repeats phosphorylated on both Ser2 and Ser5. While the structure of the CTD in this complex was not determined, the interaction covers a broad region and involved interactions with two Tyr residues in the CTD.
9.7. FF domain proteins
The FF domain is a 50-60 amino acid phosphopeptide interaction module characterized by conserved phenylalanine residues near the N- and C-termini 314. The domain is comprised of three α-helices arranged in an orthogonal bundle with a 310 helix connecting the second and third helices 315. FF domains are usually found in repeated arrays of 4-6 domains separated by linkers of variable length 314.
Three proteins with FF domains have been shown to bind to the CTD; the human transcription factor TCERG1 (CA150), and the yeast splicing factor Prp40 and mammalian HYPA/FBP1183,316. Each of these factors falls into a family of RNA-processing proteins that contain a pair of WW domains followed by four FF domains. TCERG1/CA150 is a factor that regulates transcription elongation and pre-mRNA splicing 317. This factor interacts with the phosphorylated CTD through multiple FF domains 316. Interestingly, the interaction of FF domains 4-6 with a variety of phosphorylated CTD peptides shows that this cluster of FF domains requires phosphorylation of Ser2, Ser5 and Ser7, a phosphorylation state that is observed in the middle of genes where co-transcriptional splicing occurs 318.
9.8. Fus and related proteins
A recent report has indicated that the RNA-binding protein FUS (fused in sarcoma) can interact with the CTD both in vivo and in vitro 319. This protein is of great interest as it is mutated in several neurological diseases 320. FUS is a member of the FET family of RNA-binding proteins that include EWS (Ewing sarcoma) and the TFIID subunit TAF15 320a,321. These proteins all contain a central conserved RNA-recognition motif (RRM) and a C-terminal Zn-finger motif 320a. In addition, each protein contains a N-terminal low-complexity domain rich in Gln, Gly, Ser and Tyr residues. Recent work from McKnight and colleagues has shown that low complexity sequences in the FET proteins contain multiple copies of the motif [G/S]-Y-[G/S] and this sequence allows these domains to form both homotypic and hetertypic ameloid-like cross-ß structures 27. The CTD also contains this motif and a recent study shows that the CTD can associate with FET proteins through the formation of heterotypic cross-ß structures (Kwan and Kato et al., submitted).
9.9. Other CTD-interacting proteins
The proteins we have discussed in this section have really only scratched the surface of the family of CTD interacting proteins. A large number of proteins identified in systematic binding assays have not yet been subjected to further study 8c,107a,277. In addition a variety of proteins like Bur1 185, RecQ5 137, Cdc73 185, and the MCM 129-130 proteins have been shown to bind the CTD but have not been extensively studied. Future work will illuminate the roles these proteins play in elaborating the CTD code.
9.10. Summary of CTD interactions
The large network of interactions formed by the CTD is based on two basic principles. First, the CTD is structurally flexible and thus can adopt a wide variety of conformations. Second, CTD-interacting proteins are likely to bind using an induced-fit mode. Such coupled folding and binding yields highly specific interactions but with relatively low affinity 23a. One advantage of conformational flexibility is that the CTD can bind both to modifying enzymes and to the readers of these modifications using overlapping sites that can adopt different conformations. The adaptability of CTD structure is seen in the variety of conformations the CTD adopts in complexes with CTD-binding proteins. These interactions are weak and thus have a short lifetime but are boosted by the number of different binding motifs present in the CTD.
10. GLOBAL CHANGES IN CTD PHOSPHORYLATION
While the CTD code primarily addresses the regulatory processes occurring on the CTD during the transcription cycle there are other instances where transcription within the cell is altered on a global scale. Several examples of this global alteration involve changes in CTD phosphorylation.
10.1. Stress-induced changes in CTD phosphorylation
Sudden changes in environmental conditions like temperature, pH, osmolarity, oxidative state or the presence of DNA damaging agents can induce rapid and widespread changes in gene expression 322. In S.cerevisiae, changing the temperature from 25°C to 37 °C alters expression of about one third of all genes 322b. This change in gene expression allows the cell to respond to the changing condition by expressing a small set of genes including heat shock proteins (chaparones) that enable cells to deal with the effect of temperature on protein folding and assembly 322c. In addition, transcription of highly expressed genes like those encoding ribosomal proteins and glycolytic enzymes are rapidly shut off. Since these genes recruit most of the active Pol II in the cell, heat shock is expected to alter the transcriptional state of almost all Pol II.
Bensaude and colleagues first observed that in HeLa cells heat shock results in the accumulation of the phosphorylated IIo form of the largest subunit 323. Further work showed that this change is brought about, not necessarily directly, by stress-activated kinases 324, inhibition of cyclin-dependent kinases and inactivation of CTD phosphatase FCP1 325. The increased reactivity toward the CC3 mAb suggests that the increase in IIo abundance is due to phosphorylation of Ser2 144b. When S. cerevisiae cultures are shifted from 25°C to 37°C there is a rapid but transient increase in the level of Ser2P 144b,326. The timing of this change correlates with the rapid changes seen in steady state RNAs by microarray 322b. As with the case in HeLa cells this increase is mainly due to Ser2P as there is no change in the reactivity to mAbs that recognize Ser5P.
Similar changes in CTD phosphorylation are also seen in other stress conditions. Yeast cells in logarithmic growth transcribe genes necessary for glycolysis and protein synthesis at high frequency. As glucose is depleted the Ras and Tor signaling pathways combine to alter expression of a large set of genes in what has been termed the diauxic shift 327. The end result of this shift is a reduction in expression of glycolytic and protein synthesis genes and activation of genes required for oxidative phosphorylation. During the diauxic shift there is an increase in the phosphorylation of Ser2 as detected by the H5 mAb 144b.
In S. pombe a similar increase in Ser2 is observed as cells are starved for nitrogen 197b. This change in CTD phosphorylation pattern has been linked to a shift in gene expression leading to sporulation. As in S. cerevisiae the kinase that phosphorylates the Ser2 sites is the Cdk1 homolog Lsk1 which is activated through a MAP kinase cascade. Global changes in CTD phosphorylation are also brought about by DNA damaging agents 328. In this case as well an increase in Ser2P is observed and this increase is mediated by CTDK-I.
A common theme of the stress response is an increase in the apparent level of Ser2 phosphorylation. This change in the global CTD phosphorylation pattern may be accomplished in several ways. First, there could be an increase in Ser2 phosphorylation through activation of Cdk9, Cdk12 or the S. cerevisiae homologs Bur1 or Ctk1. In S. pombe the Ctk1 homolog Lsk1 is activated through the MAP kinase signaling pathway 197b. An alternative method for increasing Ser2P is through inhibition of Fcp1 241. Finally, the inhibition of Ser5 phosphorylation may increase the reactivity of Ser2P to antibodies that are blocked by adjacent Ser5.
What is the rationale for increasing global Ser2P levels? For most genes initiation requires unphosphorylated Pol II that becomes phosphorylated on Ser5 while in the PIC. An increase in the level of Ser2 phosphorylated Pol II would have the effect of depleting the pool of Pol II available for initiation. In the case of stress response this is a desired result as most gene expression must be reduced. Ser2 phosphorylation correlates with promoter escape and the transition to productive elongation but for stress-related genes this paradigm may not apply. Transcription of heat shock genes is less dependent on TFIIH than are other genes 329. In Drosophila, Pol II on heat shock genes appears to be less phosphorylated than on developmentally induced genes 143a. In a similar manner the S. pombe small heat shock protein hsp9 is up regulated by Ser2 phosphorylation197b. Taken together this indicates that a subset of genes involved in responding to stress are transcribed through a CTD code that differs from the bulk of growth promoting genes.
10.2. Growth stimulation and CTD phosphorylation
In addition to changes in stress, the phosphorylation state of the CTD also changes in response to the sudden availability of nutrients and/or growth factors. Quiescent fibroblasts contain equal amounts of the IIA and IIO forms of Pol II. When these cells are stimulated to grow by the addition of serum there is a marked increase in the level of Pol IIO that persists for several hours and then subsides 203. This change in phosphorylation state is not transcription dependent as a CTD fusion protein is similarly phosphorylated in response to serum stimulation. MAP kinases co purify with the serum induced CTD kinase activity and are thought to carry out this global modification 203. Stimulation of quiescent lymphocytes yields a five-fold increase in TFIIH kinase resulting an a similar increase in the level of CTD Ser5 phosphorylation 330. This change in CTD phosphorylation is coincident with an increase in global promoter melting as PICs with unphosphorylated CTDs are converted to open complexes and execute promoter escape.
A recent report has also indicated that in yeast, Cdc28 (Cdc2) is able to phosphorylate the CTD of highly transcribed genes 199. Since Cdc2 activity is increased as cells enter the cell cycle after serum stimulation it is entirely possible that in mammalian cells Cdc2 plays a role in generating the high level of Pol IIo seen after growth stimulation.
10.3. CTD phosphorylation in early development
Oocytes from different organisms contain large amounts of maternally derived Pol II that is held in store for use in the early cell division cycles 331. The CTD of this stored maternal Pol II is phosphorylated despite the fact that it is not engaged in transcription. In Xenopus oocyte development the CTD is largely unphosphorylated despite the fact that these cells are transcriptionally active 332. At the final stage of maturation the CTD becomes hyperphosphorylated through the action of MAP kinase. Within one hour after fertilization the level of phosphorylation is reduced 332a. A similar hyperphosphorylated form of the CTD is present in mammalian oocytes 333. As with Xenopus, the hypophosphorylated IIa form of Rbp1 can be detected several hours after fertilization 333. What role CTD phosphorylation plays in the storage and mobilization of Pol II in oocytes and early zygotes is not known.
In a variety of organisms the specification of germ cell fate is based on transcription repression in a process that is thought to involve inhibition of CTD phosphorylation 334. This was first observed in C. elegans where Seydoux and colleagues showed that the level of both Ser5P and Ser2P are reduced in germ line nuclei compared to somatic nuclei 335. They went on to show that PIE-1, a maternal protein that segregates with germ cells is capable of inhibiting Ser2 and Ser5 phosphorylation 336.
The inhibition of Ser2 phosphorylation takes place through a sequence in PIE-1, YAPMAPT that acts like a CTD heptad repeat binding to the cyclin T subunit of P-TEFb and blocking CTD phosphorylation 337. PIE-1 deleted for this sequence fails to repress P-TEFb in germ cells leading to elevated levels of Ser2P 338. In a more recent study it was shown that Cdk12 is responsible for the bulk of Ser2 phosphorylation in germ cells 339. It is possible that PIE-1 also inhibits Cdk12 or alternatively, the small amount of Ser2P remaining after Cdk12 knockdown may represent a small number of genes where Cdk9 provides Ser2 phosphorylation. Further work will be required to delineate that roles of Cdk9 and Cdk12 in the C. elegans soma and germline.
Inhibition of P-TEFb in germ cells also occurs in early Drosophila development. In this case the polar granule component Pgc is expressed in germ cell precursors that contain low levels of Ser2P 340. Pgc forms an inactive complex with P-TEFb in these cells and over expression of P-TEFb creates a similar phenotype as a pgc mutant. These results suggest that similar to PIE-I the Pgc protein is able to inhibit Ser2 phosphorylation. Pgc1 is not related to PIE-I indicating convergent evolution of this process.
Germline specification in mammals is different from C. elegans and Drosophila in that the earliest embryonic cells (epiblasts) are equally likely to contribute to the germline 341. Only after about one week of mouse development are PGCs identifiable and as these cells migrate to the gonad they lose both Ser2 and Ser5 phosphorylation and RNA synthesis in these cells is markedly reduced 342. The common theme of these observations is that inhibition of CTD Ser2 and Ser5 phosphorylation in germ cells is important in specifying germ cell fate. Since Ser2P and Ser5P are usually associated with active transcription by Pol II the implication is that many genes are inactive in germ cells. Clearly there are genes that escape this regulation and maintain the viability of germ cells. How these genes escape is not know but may involve an override of the CTD code.
11. CTD MODIFICATION AND POL II SUBNUCLEAR LOCALIZATION
Pol II forms large complexes that contain all of the components necessary to synthesize and process nascent transcripts. Pulse labeling RNA with Br-UTP first showed that RNA synthesis in HeLa cells is localized to 300-500 sites that have been termed transcription factories 343. Each of these factories contain an average of eight Pol II molecules 344 with an apparent mass of >8 M Da 343d. In metazoa these factories are bound to a nuclear substructure 345. Whether such transcription factories exist in yeast has not been shown.
HeLa cell transcription factories only contain about one quarter of the total Pol II in the cell, the remainder is not localized and apparently not engaged in transcription 344,345b. Within factories two different CTD phosphorylation states have been observed; those with Ser5P only and those with both Ser2P and Ser5P 346. The former are proposed contain primarily poised Pol II complexes while the later contain actively elongating complexes 346. Recent experiments have shown that the Ser5P factories contain Cdk9 and are distinct from the Ser2P factories 347.
In addition to transcription factories several studies have shown that Pol II is localized to sub-nuclear substructures that are not associated with transcription including speckles interchromatin granule clusters) 79a,144a,348, paraspeckles 348b, and Cajal bodies (CBs) 332b,349. Speckles are irregular shaped punctae found in the nucleoplasm of mammalian cells and consisting of splicing factors and other RNA processing proteins 350. Although transcription does not occur in speckles, the Pol II found in these structures is phosphorylated preferentially on Ser2 79a,144a,348b. This observation suggests that CTD phosphorylation may have an alternative function in storage or assembly of Pol II complexes. Pol II has also been identified in Cajal bodies (CBs). These are small spherical structures 0.1-1μm in diameter located in the nucleoplasm 351. There are typically only a few (~5) CBs in most cells although more are seen in Xenopus germinal vesicles. CBs contain many transcription and processing proteins and have been proposed as sites of assembly of the transcription and pre-mRNA processing machinery 352. Pol II in CBs is phosphorylated primarily on Ser5 which is surprising since there is no evidence for ongoing transcription in these structures. The CTD may have a role in localization of Pol II to CBs. A GST-CTD fusion protein expressed in Xenopus germinal vesicles is rapidly phosphorylated and localized to CBs332b. Phosphorylation of this domain is not required, however, as a fusion protein with Ser2 and Ser5 converted to Ala is also localized to CBs. The localization of Pol II within the nucleus plays an as yet poorly defined role in transcription regulation and the localization of Pol II with different phosphorylated CTD isoforms to different subnuclear structures will be an important area of future research.
12. CONCLUDING REMARKS
The last three decades since the discovery of the Pol II CTD have seen a dramatic expansion in our understanding of this enigmatic domain. Research has focused both on the enzymes that establish the wide array of modification patterns and the myriad of proteins that recognize these marks. The overarching theme of these studies is the central role that the CTD plays in integrating Pol II transcription with processing of the nascent transcript and the interaction of Pol II with the chromatin template.
The nature of the CTD structure is well adapted to its role in tethering multiple functional activities to the Pol II catalytic core. The extended structure enables the CTD to interact with other proteins over a distance of up to a thousand angstroms. The flexible nature of the CTD allows different repeats to adopt different conformations in response to interactions with CTD modifiers or RNA processing proteins. While the number of tandem repeats seems to be far in excess of that necessary for viability in a number of different organisms CTD deletions have only been tested in a limited number of conditions and it seems certain that the natural length of the CTD confers a selective advantage under some conditions. Similarly, while the consensus heptad repeats are found in tandem in most organisms, mutations that insert amino acids between every other repeat are viable indicating that the minimal functional unit is contained within two repeats 43c,56. Why then is the tandem register maintained in evolution? There must be some interaction that the CTD makes that covers more than a pair of repeats and this type of interaction must confer a selective advantage. One possibility is that the CTD makes contacts with other low complexity regions in proteins. Such interactions are often based on cross-beta structures in which polypeptide chains are in a fully extended conformation 27. If the CTD were to make such interactions either with other proteins or within the CTD itself then this could explain the need to maintain the tandem heptad register.
Dynamic modification of the CTD enables changes in the tethered activities as Pol II proceeds through the transcription cycle. This is the basis of the CTD code that has emerged as a unifying concept in transcription regulation. According to this model particular CTD modifications specify particular outcomes for the transcription machinery, the chromatin template and the nascent transcript. These dynamic changes are inferred from ChIP experiments showing that Pol II at different points along a template carries different CTD modifications. In the simplest form of the code the CTD is unphosphorylated when Pol II enters the PIC allowing the CTD to interact with the Mediator. Once initiation occurs the CTD is phosphorylated on Ser5 and Ser7 breaking the contacts with the Mediator and allowing promoter escape. Ser5P also serves to recruit the capping machinery ensuring that Pol II transcripts are modified at the 5’-end. Once capping is completed Pol II enters a stable elongation complex and the CTD phosphorylation pattern changes with an increase in Ser2P, Thr4P, Tyr1P and a decrease in Ser5P. These changes allow the recruitment of splicing factors and enzymes that modify the chromatin template. Finally, as Pol II nears the 3’-end there is a further decrease in Ser5P, Ser7P and Thr4P allowing Ser2P to predominate. This modification recruits the cleavage and polyadenylation machinery and, following 3’-end formation and termination the CTD is further modified by complete dephosphorylation thus regenerating initiation-competent Pol II.
The CTD code model has been very effective in focusing attention on the role of CTD modifications. The prevailing model is that dynamic changes in CTD modification underlie many of the steps in the transcription cycle. Much of this model is based on static data, however, in which the Pol II machinery is frozen in place following formaldehyde treatment. This type of data yields snapshots of Pol II along the gene and some inference is necessary to paint a dynamic picture. This picture is also clouded by the fact that ChIP yields a population average of the CTD modifications at a particular point along a given gene. For example, near the 5’-end most genes have more Ser5P than Ser2P. This could be due to the presence of high levels of Ser5P and a small amount of Ser2P on each CTD or rather could mean that most of the Pol II near the 5’-end has only Ser5P and a small number of Pol II molecules have mainly Ser2P. This issue becomes more critical in the middle of genes where both modifications are present in similar amounts.
Further complicating the CTD code are the uncertain specificities and binding properties of the antibodies used to derive the ChIP data. The specificity of some of these antibodies are masked by adjacent phosphorylations. Other antibodies preferentially bind to multiply phosphorylated repeats. Finally, and perhaps most importantly, these antibodies are multi-valent and thus bind to CTDs modified at multiple sites. Part of the affinity is thus not dependent solely on modification of a single repeat but is due to the probability that a nearby repeat is also modified. Until these antibody properties are fully understood the CTD code will only be a low-resolution model. To increase the resolution it will be necessary to carry out sequential immunoprecipitations to characterize the distribution of different CTD phosphoisomers. This has been done to a limited extent using western analysis and this has revealed subpopulations of Pol II with CTDs either phosphorylated on Ser5 and Ser7 or on Ser2 and Thr4 54b. Net-seq or PAR-CLIP using this type of sequential precipitation will allow a more detailed view of the distribution of CTD phosphoisomers on the genome. This will also address the issue of whether the CTD code is the same for all genes.
Another emerging theme in the CTD code is the degree to which different modifications can interact. One example is the priming of one CTD kinase by phosphorylation by another CTD kinase. This can take place either through modification of the CTD substrate or by the direct phosphorylation of one kinase by another140a,170,183,185,192 thus establishing an ordered series of phosphorylation reactions during the transcription cycle much like the series of phosphorylations that govern the cell cycle219. Another emerging principle is the allosteric effect of CTD phosphorylation marks. Ser5P has been shown to enhance the capping activity 77,291,353. Isomerization of the Ser-Pro bonds in the CTD also cooperates with different CTD binding and modification steps. Ess1 and/or Pin1 are necessary for full activity of the Ssu72 phosphatase that recognizes Ser5P in the context of a cis peptide bond 249,269. Finally, the binding of Nrd1 to the CTD requires one of three S-P bonds to be in the cis conformation 307. All of these interactions work together to provide a set of sequential binding complexes that direct the processing of nascent Pol II transcripts. The picture that this paints is of a series of complexes that are recruited to a moving Pol II elongation complex and fall off once they have executed their required function.
The problem with this picture is that the various CTD –interacting proteins bind weakly to the CTD and are unlikely to remain bound long enough to complete a transcription cycle. The most well studied interactions with the CTD involve the CTD-interacting domain (CID) found in Pcf11, Rtt103, SCAF8 and Nrd1. This domain binds to the same basic structure in the CTD but details of the interaction vary among the proteins leading to specificity for Ser2P, Ser5P or both. The key feature recognized by these proteins is a ß-turn comprising Ser2-Pro3-Thr4-Ser5. This turn is not abundant in the CTD in solution and binding thus requires a disorder to order transition in the CTD. Such coupled folding and binding reactions typically are of low affinity and high specificity and that seems to hold for many CTD interactions. Some CTD interactors may overcome low binding affinity by cooperative interactions 301. Alternatively, stable interaction may not be required. Because there are many potential binding sites within the CTD it may not be necessary for an “interacting” protein to be stably bound. In this case the CTD could act to increase the local concentration of CTD-binding sites and thus increase the local concentration of any interacting proteins 354.
In this later model the CTD is not so much acting as a tether, but as a fly-casting apparatus luring the RNA-processing proteins closer to the site of their function. Appendages like the CTD are not seen on Pol I or Pol III perhaps indicating that their processing need not be tethered or otherwise concentrated. One possible reason for not tethering the processing factors for Pol I is that transcription by this polymerase is sequestered in the nucleolus. Pol II transcription, while not restricted to a subnuclear organelle is, however, localized in transcription factories. Whether the CTD is involved in localizing Pol II in such structures is an open question. Several lines of investigation suggest that the CTD may target Pol II to structures in the nucleus. For example, the interaction with SCAFs (CTD associated SR-like factors) that co-localize to sites of active transcription may direct Pol II to sub-nuclear structures 79b,99. Another possibility is that the CTDs from different Pol II molecules could interact with each other or with a common low complexity factor to form a cluster of polymerases that form a transcription factory.
Many challenges remain before the role of the CTD is understood in detail. As is apparent from the studies cited here the CTD is involved in many steps in the synthesis and processing of Pol II transcripts. Despite key insights into CTD function there are still many details that are not clear. Part of the problem in teasing out detailed mechanisms is the degree to which transcription and RNA processing are coupled. Thus, defects in one step caused by a CTD mutation may yield defects in subsequent or preceding RNA processing steps. Future advances will depend on our ability to biochemically reconstitute these coupled steps in transcription and processing and assess the role of the CTD at each step along the way. The large number of CTD mutations already constructed will be a valuable asset in this endevour.
Acknowledgments
The author thanks Dr. Anton Meinhart for Figure 6a. This work was funded by NIH grant GM066108 to J.L.C.
Biography
Jeffry Corden received his PhD in Biochemistry and Biophysics from Oregon State University for his work on viral chromatin with Drs. George Pearson and Ken van Holde. He then joined the laboratory of Pierre Chambon at the Faculte de Medcine of the University of Strasbourg, France where he worked to define RNA polymerase II promoter elements. Since 1982 he has been on the faculty at Johns Hopkins Medical School where his laboratory investigates the function of the RNA polymerase II CTD.

Footnotes
The author declares no competing financial interest
References
- 1.Chapman RD, Heidemann M, Hintermair C, Eick D. Trends Genet. 2008;24:289. doi: 10.1016/j.tig.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Cramer P, Bushnell DA, Kornberg RD. Science. 2001;292:1863. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 3.(a) Corden JL, Cadena DL, Ahearn JM, Jr, Dahmus ME. Proc Natl Acad Sci U S A. 1985;82:7934. doi: 10.1073/pnas.82.23.7934. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Allison LA, Moyle M, Shales M, Ingles CJ. Cell. 1985;42:599. doi: 10.1016/0092-8674(85)90117-5. [DOI] [PubMed] [Google Scholar]; (c) Zehring WA, Lee JM, Weeks JR, Jokerst RS, Greenleaf AL. Proc Natl Acad Sci U S A. 1988;85:3698. doi: 10.1073/pnas.85.11.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Roeder RG. In: RNA Polymerase. Losick R, Chamberlin M, editors. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1976. [Google Scholar]; (b) Dahmus ME. J Biol Chem. 1983;258:3956. [PubMed] [Google Scholar]
- 5.Greenleaf AL, Haars R, Bautz EK. FEBS Lett. 1976;71:205. doi: 10.1016/0014-5793(76)80932-5. [DOI] [PubMed] [Google Scholar]
- 6.Cadena DL, Dahmus ME. J Biol Chem. 1987;262:12468. [PubMed] [Google Scholar]
- 7.Dahmus ME. Biochim Biophys Acta. 1995;1261:171. doi: 10.1016/0167-4781(94)00233-s. [DOI] [PubMed] [Google Scholar]
- 8.(a) Zhang J, Corden JL. J Biol Chem. 1991;266:2290. [PubMed] [Google Scholar]; (b) Yuryev A, Corden JL. Genetics. 1996;143:661. doi: 10.1093/genetics/143.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Phatnani HP, Greenleaf AL. Genes Dev. 2006;20:2922. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 9.(a) Komarnitsky P, Cho EJ, Buratowski S. Genes Dev. 2000;14:2452. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Buratowski S. Nat Struct Biol. 2003;10:679. doi: 10.1038/nsb0903-679. [DOI] [PubMed] [Google Scholar]
- 10.Werner F, Grohmann D. Nat Rev Microbiol. 2011;9:85. doi: 10.1038/nrmicro2507. [DOI] [PubMed] [Google Scholar]
- 11.Haag JR, Pikaard CS. Nat Rev Mol Cell Biol. 2011;12:483. doi: 10.1038/nrm3152. [DOI] [PubMed] [Google Scholar]
- 12.Stiller JW, Hall BD. Proc Natl Acad Sci U S A. 2002;99:6091. doi: 10.1073/pnas.082646199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakajima K, Chang YC, Suzuki T, Jigami Y, Machida M. Biosci Biotechnol Biochem. 2000;64:641. doi: 10.1271/bbb.64.641. [DOI] [PubMed] [Google Scholar]
- 14.Li WB, Bzik DJ, Gu HM, Tanaka M, Fox BA, Inselburg J. Nucleic Acids Res. 1989;17:9621. doi: 10.1093/nar/17.23.9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stiller JW, Duffield EC, Hall BD. Proc Natl Acad Sci U S A. 1998;95:11769. doi: 10.1073/pnas.95.20.11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirkin SM. Nature. 2007;447:932. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 17.(a) Jeffreys AJ, Royle NJ, Wilson V, Wong Z. Nature. 1988;332:278. doi: 10.1038/332278a0. [DOI] [PubMed] [Google Scholar]; (b) Jeffreys AJ, Wilson V, Thein SL. Nature. 1985;314:67. doi: 10.1038/314067a0. [DOI] [PubMed] [Google Scholar]
- 18.West ML, Corden JL. Genetics. 1995;140:1223. doi: 10.1093/genetics/140.4.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Corden JL. Trends Biochem Sci. 1990;15:383. doi: 10.1016/0968-0004(90)90236-5. [DOI] [PubMed] [Google Scholar]; (b) Corden JL, Ingles CJ. In: Transcriptional Regulation. McKnight SL, Y KR, editors. Vol. 1 Cold Spring Harbor Laboratory Press; Plainview, N.Y.: 1992. [Google Scholar]
- 20.Hedges SB, Dudley J, Kumar S. Bioinformatics. 2006;22:2971. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- 21.Nonet M, Sweetser D, Young RA. Cell. 1987;50:909. doi: 10.1016/0092-8674(87)90517-4. [DOI] [PubMed] [Google Scholar]
- 22.Nonet ML, Young RA. Genetics. 1989;123:715. doi: 10.1093/genetics/123.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Dyson HJ, Wright PE. Nat Rev Mol Cell Biol. 2005;6:197. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]; (b) Tompa P. Trends Biochem Sci. 2002;27:527. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Genome Inform Ser Workshop Genome Inform. 1999;10:30. [PubMed] [Google Scholar]
- 25.(a) Suzuki M. J Mol Biol. 1989;207:61. doi: 10.1016/0022-2836(89)90441-5. [DOI] [PubMed] [Google Scholar]; (b) Suzuki M. Nature. 1990;344:562. doi: 10.1038/344562a0. [DOI] [PubMed] [Google Scholar]; (c) Suzuki M, Yagi N. Proc Biol Sci. 1991;246:231. doi: 10.1098/rspb.1991.0149. [DOI] [PubMed] [Google Scholar]
- 26.(a) Matsushima N, Creutz CE, Kretsinger RH. Proteins. 1990;7:125. doi: 10.1002/prot.340070204. [DOI] [PubMed] [Google Scholar]; (b) Cagas PM, Corden JL. Proteins. 1995;21:149. doi: 10.1002/prot.340210209. [DOI] [PubMed] [Google Scholar]; (c) Meinhart A, Cramer P. Nature. 2004;430:223. doi: 10.1038/nature02679. [DOI] [PubMed] [Google Scholar]
- 27.(a) Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL. Cell. 2012;149:768. doi: 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]; (b) Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell. 2012;149:753. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Nature. 2007;447:453. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]; (b) Fitzpatrick AW, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang L, Ladizhansky V, Muller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TP, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, Dobson CM. Proc Natl Acad Sci U S A. 2013;110:5468. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Harding MM. J Med Chem. 1992;35:4658. doi: 10.1021/jm00103a002. [DOI] [PubMed] [Google Scholar]; (b) Noble CG, Hollingworth D, Martin SR, Ennis-Adeniran V, Smerdon SJ, Kelly G, Taylor IA, Ramos A. Nat Struct Mol Biol. 2005;12:144. doi: 10.1038/nsmb887. [DOI] [PubMed] [Google Scholar]
- 30.Bienkiewicz EA, Moon Woody A, Woody RW. J Mol Biol. 2000;297:119. doi: 10.1006/jmbi.2000.3545. [DOI] [PubMed] [Google Scholar]
- 31.Dobbins JR, Murali N, Long EC. Int J Pept Protein Res. 1996;47:260. doi: 10.1111/j.1399-3011.1996.tb01354.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Corden JL. J Biol Chem. 1991;266:2297. [PubMed] [Google Scholar]
- 33.Lu KP, Finn G, Lee TH, Nicholson LK. Nat Chem Biol. 2007;3:619. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 34.Nishi N, Ohiso I, Sakairi N, Tokura S, Tsunemi M, Oka M. Biochem Biophys Res Commun. 1995;206:981. doi: 10.1006/bbrc.1995.1139. [DOI] [PubMed] [Google Scholar]
- 35.Armache KJ, Mitterweger S, Meinhart A, Cramer P. J Biol Chem. 2005;280:7131. doi: 10.1074/jbc.M413038200. [DOI] [PubMed] [Google Scholar]
- 36.Laybourn PJ, Dahmus ME. J Biol Chem. 1989;264:6693. [PubMed] [Google Scholar]
- 37.Tsai KL, Sato S, Tomomori-Sato C, Conaway RC, Conaway JW, Asturias FJ. Nat Struct Mol Biol. 2013;20:611. doi: 10.1038/nsmb.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allison LA, Wong JK, Fitzpatrick VD, Moyle M, Ingles CJ. Mol Cell Biol. 1988;8:321. doi: 10.1128/mcb.8.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brickey WJ, Greenleaf AL. Genetics. 1995;140:599. doi: 10.1093/genetics/140.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Bartolomei MS, Halden NF, Cullen CR, Corden JL. Mol Cell Biol. 1988;8:330. doi: 10.1128/mcb.8.1.330. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meininghaus M, Eick D. FEBS Lett. 1999;446:173. doi: 10.1016/s0014-5793(99)00184-2. [DOI] [PubMed] [Google Scholar]; (c) Chapman RD, Conrad M, Eick D. Mol Cell Biol. 2005;25:7665. doi: 10.1128/MCB.25.17.7665-7674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim WY, Dahmus ME. J Biol Chem. 1989;264:3169. [PubMed] [Google Scholar]
- 42.Boeke JD, Trueheart J, Natsoulis G, Fink GR. Methods Enzymol. 1987;154:164. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- 43.(a) Schneider S, Pei Y, Shuman S, Schwer B. Mol Cell Biol. 2010;30:2353. doi: 10.1128/MCB.00116-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schwer B, Shuman S. Mol Cell. 2011;43:311. doi: 10.1016/j.molcel.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schwer B, Sanchez AM, Shuman S. Proc Natl Acad Sci U S A. 2012;109:18024. doi: 10.1073/pnas.1208995109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, Eick D. Science. 2007;318:1780. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 45.Litingtung Y, Lawler AM, Sebald SM, Lee E, Gearhart JD, Westphal H, Corden JL. Mol Gen Genet. 1999;261:100. doi: 10.1007/s004380050946. [DOI] [PubMed] [Google Scholar]
- 46.Payne JM, Laybourn PJ, Dahmus ME. J Biol Chem. 1989;264:19621. [PubMed] [Google Scholar]
- 47.(a) Baskaran R, Chiang GG, Mysliwiec T, Kruh GD, Wang JY. J Biol Chem. 1997;272:18905. doi: 10.1074/jbc.272.30.18905. [DOI] [PubMed] [Google Scholar]; (b) Baskaran R, Dahmus ME, Wang JY. Proc Natl Acad Sci U S A. 1993;90:11167. doi: 10.1073/pnas.90.23.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chapman RD, Palancade B, Lang A, Bensaude O, Eick D. Nucleic Acids Res. 2004;32:35. doi: 10.1093/nar/gkh172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartolomei MS, Corden JL. Mol Cell Biol. 1987;7:586. doi: 10.1128/mcb.7.2.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meininghaus M, Chapman RD, Horndasch M, Eick D. J Biol Chem. 2000;275:24375. doi: 10.1074/jbc.M001883200. [DOI] [PubMed] [Google Scholar]
- 51.Stiller JW, McConaughy BL, Hall BD. Yeast. 2000;16:57. doi: 10.1002/(SICI)1097-0061(20000115)16:1<57::AID-YEA509>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 52.Zhang DW, Mosley AL, Ramisetty SR, Rodriguez-Molina JB, Washburn MP, Ansari AZ. J Biol Chem. 2012;287:8541. doi: 10.1074/jbc.M111.335687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karagiannis J, Balasubramanian MK. PLoS One. 2007;2:e433. doi: 10.1371/journal.pone.0000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.(a) Hsin JP, Sheth A, Manley JL. Science. 2011;334:683. doi: 10.1126/science.1206034. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hintermair C, Heidemann M, Koch F, Descostes N, Gut M, Gut I, Fenouil R, Ferrier P, Flatley A, Kremmer E, Chapman RD, Andrau JC, Eick D. EMBO J. 2012;31:2784. doi: 10.1038/emboj.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stiller JW, Cook MS. Eukaryot Cell. 2004;3:735. doi: 10.1128/EC.3.3.735-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu P, Greenleaf AL, Stiller JW. Mol Biol Evol. 2008;25:719. doi: 10.1093/molbev/msn017. [DOI] [PubMed] [Google Scholar]
- 57.Liu P, Kenney JM, Stiller JW, Greenleaf AL. Mol Biol Evol. 2010;27:2628. doi: 10.1093/molbev/msq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.(a) Thompson CM, Koleske AJ, Chao DM, Young RA. Cell. 1993;73:1361. doi: 10.1016/0092-8674(93)90362-t. [DOI] [PubMed] [Google Scholar]; (b) Hengartner CJ, Thompson CM, Zhang J, Chao DM, Liao SM, Koleske AJ, Okamura S, Young RA. Genes Dev. 1995;9:897. doi: 10.1101/gad.9.8.897. [DOI] [PubMed] [Google Scholar]; (c) Liao SM, Zhang J, Jeffery DA, Koleske AJ, Thompson CM, Chao DM, Viljoen M, van Vuuren HJ, Young RA. Nature. 1995;374:193. doi: 10.1038/374193a0. [DOI] [PubMed] [Google Scholar]
- 59.(a) Allison LA, Ingles CJ. Proc Natl Acad Sci U S A. 1989;86:2794. doi: 10.1073/pnas.86.8.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Scafe C, Chao D, Lopes J, Hirsch JP, Henry S, Young RA. Nature. 1990;347:491. doi: 10.1038/347491a0. [DOI] [PubMed] [Google Scholar]
- 60.Peterson CL, Kruger W, Herskowitz I. Cell. 1991;64:1135. doi: 10.1016/0092-8674(91)90268-4. [DOI] [PubMed] [Google Scholar]
- 61.(a) Gerber HP, Hagmann M, Seipel K, Georgiev O, West MA, Litingtung Y, Schaffner W, Corden JL. Nature. 1995;374:660. doi: 10.1038/374660a0. [DOI] [PubMed] [Google Scholar]; (b) Chun RF, Jeang KT. J Biol Chem. 1996;271:27888. doi: 10.1074/jbc.271.44.27888. [DOI] [PubMed] [Google Scholar]
- 62.(a) Buratowski S, Sharp PA. Mol Cell Biol. 1990;10:5562. doi: 10.1128/mcb.10.10.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zehring WA, Greenleaf AL. J Biol Chem. 1990;265:8351. [PubMed] [Google Scholar]; (c) Serizawa H, Conaway JW, Conaway RC. Nature. 1993;363:371. doi: 10.1038/363371a0. [DOI] [PubMed] [Google Scholar]
- 63.Kang ME, Dahmus ME. J Biol Chem. 1993;268:25033. [PubMed] [Google Scholar]
- 64.(a) Dahmus ME, Kedinger C. J Biol Chem. 1983;258:2303. [PubMed] [Google Scholar]; (b) Moyle M, Lee JS, Anderson WF, Ingles CJ. Mol Cell Biol. 1989;9:5750. doi: 10.1128/mcb.9.12.5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Usheva A, Maldonado E, Goldring A, Lu H, Houbavi C, Reinberg D, Aloni Y. Cell. 1992;69:871. doi: 10.1016/0092-8674(92)90297-p. [DOI] [PubMed] [Google Scholar]
- 66.(a) Koleske AJ, Buratowski S, Nonet M, Young RA. Cell. 1992;69:883. doi: 10.1016/0092-8674(92)90298-q. [DOI] [PubMed] [Google Scholar]; (b) Conaway RC, Bradsher JN, Conaway JW. J Biol Chem. 1992;267:8464. [PubMed] [Google Scholar]
- 67.Kim YJ, Bjorklund S, Li Y, Sayre MH, Kornberg RD. Cell. 1994;77:599. doi: 10.1016/0092-8674(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 68.Myers LC, Gustafsson CM, Bushnell DA, Lui M, Erdjument-Bromage H, Tempst P, Kornberg RD. Genes Dev. 1998;12:45. doi: 10.1101/gad.12.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.(a) Laybourn PJ, Dahmus ME. J Biol Chem. 1990;265:13165. [PubMed] [Google Scholar]; (b) Arias JA, Peterson SR, Dynan WS. J Biol Chem. 1991;266:8055. [PubMed] [Google Scholar]
- 70.Lux C, Albiez H, Chapman RD, Heidinger M, Meininghaus M, Brack-Werner R, Lang A, Ziegler M, Cremer T, Eick D. Nucleic Acids Res. 2005;33:5139. doi: 10.1093/nar/gki802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marshall NF, Peng J, Xie Z, Price DH. J Biol Chem. 1996;271:27176. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- 72.(a) Coppola JA, Field AS, Luse DS. Proc Natl Acad Sci U S A. 1983;80:1251. doi: 10.1073/pnas.80.5.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rasmussen EB, Lis JT. Proc Natl Acad Sci U S A. 1993;90:7923. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cho EJ, Takagi T, Moore CR, Buratowski S. Genes Dev. 1997;11:3319. doi: 10.1101/gad.11.24.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.(a) McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Shuman S, Bentley DL. Genes Dev. 1997;11:3306. doi: 10.1101/gad.11.24.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yue Z, Maldonado E, Pillutla R, Cho H, Reinberg D, Shatkin AJ. Proc Natl Acad Sci U S A. 1997;94:12898. doi: 10.1073/pnas.94.24.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ho CK, Sriskanda V, McCracken S, Bentley D, Schwer B, Shuman S. J Biol Chem. 1998;273:9577. doi: 10.1074/jbc.273.16.9577. [DOI] [PubMed] [Google Scholar]
- 75.Schroeder SC, Schwer B, Shuman S, Bentley D. Genes Dev. 2000;14:2435. doi: 10.1101/gad.836300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S. Nature. 2004;432:517. doi: 10.1038/nature03041. [DOI] [PubMed] [Google Scholar]
- 77.Ho CK, Shuman S. Mol Cell. 1999;3:405. doi: 10.1016/s1097-2765(00)80468-2. [DOI] [PubMed] [Google Scholar]
- 78.(a) McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson SD, Wickens M, Bentley DL. Nature. 1997;385:357. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]; (b) Misteli T, Spector DL. Mol Cell. 1999;3:697. doi: 10.1016/s1097-2765(01)80002-2. [DOI] [PubMed] [Google Scholar]
- 79.(a) Mortillaro MJ, Blencowe BJ, Wei X, Nakayasu H, Du L, Warren SL, Sharp PA, Berezney R. Proc Natl Acad Sci U S A. 1996;93:8253. doi: 10.1073/pnas.93.16.8253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yuryev A, Patturajan M, Litingtung Y, Joshi RV, Gentile C, Gebara M, Corden JL. Proc Natl Acad Sci U S A. 1996;93:6975. doi: 10.1073/pnas.93.14.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vincent M, Lauriault P, Dubois MF, Lavoie S, Bensaude O, Chabot B. Nucleic Acids Res. 1996;24:4649. doi: 10.1093/nar/24.23.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim E, Du L, Bregman DB, Warren SL. J Cell Biol. 1997;136:19. doi: 10.1083/jcb.136.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.(a) Greenleaf AL. Trends Biochem Sci. 1993;18:117. doi: 10.1016/0968-0004(93)90016-g. [DOI] [PubMed] [Google Scholar]; (b) Corden JL, Patturajan M. Trends Biochem Sci. 1997;22:413. doi: 10.1016/s0968-0004(97)01125-0. [DOI] [PubMed] [Google Scholar]; (c) Goldstrohm AC, Greenleaf AL, Garcia-Blanco MA. Gene. 2001;277:31. doi: 10.1016/s0378-1119(01)00695-3. [DOI] [PubMed] [Google Scholar]; (d) Dye MJ, Gromak N, Proudfoot NJ. Mol Cell. 2006;21:849. doi: 10.1016/j.molcel.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 81.Singh J, Padgett RA. Nat Struct Mol Biol. 2009;16:1128. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.(a) Hirose Y, Tacke R, Manley JL. Genes Dev. 1999;13:1234. doi: 10.1101/gad.13.10.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zeng C, Berget SM. Mol Cell Biol. 2000;20:8290. doi: 10.1128/mcb.20.21.8290-8301.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Millhouse S, Manley JL. Mol Cell Biol. 2005;25:533. doi: 10.1128/MCB.25.2.533-544.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morris DP, Greenleaf AL. J Biol Chem. 2000;275:39935. doi: 10.1074/jbc.M004118200. [DOI] [PubMed] [Google Scholar]
- 84.Gornemann J, Barrandon C, Hujer K, Rutz B, Rigaut G, Kotovic KM, Faux C, Neugebauer KM, Seraphin B. RNA. 2011;17:2119. doi: 10.1261/rna.02646811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.(a) David CJ, Boyne AR, Millhouse SR, Manley JL. Genes Dev. 2011;25:972. doi: 10.1101/gad.2038011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) David CJ, Manley JL. Transcription. 2011;2:221. doi: 10.4161/trns.2.5.17272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gu B, Eick D, Bensaude O. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Munoz MJ, de la Mata M, Kornblihtt AR. Trends Biochem Sci. 2010;35:497. doi: 10.1016/j.tibs.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 88.de la Mata M, Kornblihtt AR. Nat Struct Mol Biol. 2006;13:973. doi: 10.1038/nsmb1155. [DOI] [PubMed] [Google Scholar]
- 89.Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R. Mol Cell. 2007;26:867. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 90.(a) Proudfoot N. Curr Opin Cell Biol. 2004;16:272. doi: 10.1016/j.ceb.2004.03.007. [DOI] [PubMed] [Google Scholar]; (b) Buratowski S. Curr Opin Cell Biol. 2005;17:257. doi: 10.1016/j.ceb.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 91.(a) Barilla D, Lee BA, Proudfoot NJ. Proc Natl Acad Sci U S A. 2001;98:445. doi: 10.1073/pnas.98.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kyburz A, Sadowski M, Dichtl B, Keller W. Nucleic Acids Res. 2003;31:3936. doi: 10.1093/nar/gkg478. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Licatalosi DD, Geiger G, Minet M, Schroeder S, Cilli K, McNeil JB, Bentley DL. Mol Cell. 2002;9:1101. doi: 10.1016/s1097-2765(02)00518-x. [DOI] [PubMed] [Google Scholar]; (d) Hirose Y, Manley JL. Nature. 1998;395:93. doi: 10.1038/25786. [DOI] [PubMed] [Google Scholar]; (e) Kuehner JN, Pearson EL, Moore C. Nat Rev Mol Cell Biol. 2011;12:283. doi: 10.1038/nrm3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.(a) Kim H, Erickson B, Luo W, Seward D, Graber JH, Pollock DD, Megee PC, Bentley DL. Nat Struct Mol Biol. 2010;17:1279. doi: 10.1038/nsmb.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tietjen JR, Zhang DW, Rodriguez-Molina JB, White BE, Akhtar MS, Heidemann M, Li X, Chapman RD, Shokat K, Keles S, Eick D, Ansari AZ. Nat Struct Mol Biol. 2010;17:1154. doi: 10.1038/nsmb.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mayer A, Lidschreiber M, Siebert M, Leike K, Soding J, Cramer P. Nat Struct Mol Biol. 2010;17:1272. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- 93.Steinmetz EJ, Conrad NK, Brow DA, Corden JL. Nature. 2001;413:327. doi: 10.1038/35095090. [DOI] [PubMed] [Google Scholar]
- 94.(a) Carroll KL, Ghirlando R, Ames JM, Corden JL. RNA. 2007;13:361. doi: 10.1261/rna.338407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Carroll KL, Pradhan DA, Granek JA, Clarke ND, Corden JL. Mol Cell Biol. 2004;24:6241. doi: 10.1128/MCB.24.14.6241-6252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Porrua O, Hobor F, Boulay J, Kubicek K, D’Aubenton-Carafa Y, Gudipati RK, Stefl R, Libri D. EMBO J. 2012;31:3935. doi: 10.1038/emboj.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.(a) Gudipati RK, Villa T, Boulay J, Libri D. Nat Struct Mol Biol. 2008;15:786. doi: 10.1038/nsmb.1460. [DOI] [PubMed] [Google Scholar]; (b) Vasiljeva L, Kim M, Mutschler H, Buratowski S, Meinhart A. Nat Struct Mol Biol. 2008;15:795. doi: 10.1038/nsmb.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.(a) Arigo JT, Eyler DE, Carroll KL, Corden JL. Mol Cell. 2006;23:841. doi: 10.1016/j.molcel.2006.07.024. [DOI] [PubMed] [Google Scholar]; (b) Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, Libri D. Mol Cell. 2006;23:853. doi: 10.1016/j.molcel.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 97.Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Regnault B, Devaux F, Namane A, Seraphin B, Libri D, Jacquier A. Cell. 2005;121:725. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 98.(a) Arigo JT, Carroll KL, Ames JM, Corden JL. Mol Cell. 2006;21:641. doi: 10.1016/j.molcel.2006.02.005. [DOI] [PubMed] [Google Scholar]; (b) Kuehner JN, Brow DA. Mol Cell. 2008;31:201. doi: 10.1016/j.molcel.2008.05.018. [DOI] [PubMed] [Google Scholar]; (c) Jenks MH, O’Rourke TW, Reines D. Mol Cell Biol. 2008;28:3883. doi: 10.1128/MCB.00380-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Thiebaut M, Colin J, Neil H, Jacquier A, Seraphin B, Lacroute F, Libri D. Mol Cell. 2008;31:671. doi: 10.1016/j.molcel.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 99.Patturajan M, Wei X, Berezney R, Corden JL. Mol Cell Biol. 1998;18:2406. doi: 10.1128/mcb.18.4.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Becker R, Loll B, Meinhart A. J Biol Chem. 2008;283:22659. doi: 10.1074/jbc.M803540200. [DOI] [PubMed] [Google Scholar]
- 101.(a) Lei EP, Krebber H, Silver PA. Genes Dev. 2001;15:1771. doi: 10.1101/gad.892401. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lei EP, Silver PA. Genes Dev. 2002;16:2761. doi: 10.1101/gad.1032902. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dermody JL, Dreyfuss JM, Villen J, Ogundipe B, Gygi SP, Park PJ, Ponticelli AS, Moore CL, Buratowski S, Bucheli ME. PLoS One. 2008;3:e3273. doi: 10.1371/journal.pone.0003273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.(a) Bucheli ME, Buratowski S. EMBO J. 2005;24:2150. doi: 10.1038/sj.emboj.7600687. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bucheli ME, He X, Kaplan CD, Moore CL, Buratowski S. RNA. 2007;13:1756. doi: 10.1261/rna.607207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Henry M, Borland CZ, Bossie M, Silver PA. Genetics. 1996;142:103. doi: 10.1093/genetics/142.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.(a) Windgassen M, Sturm D, Cajigas IJ, Gonzalez CI, Seedorf M, Bastians H, Krebber H. Mol Cell Biol. 2004;24:10479. doi: 10.1128/MCB.24.23.10479-10491.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sachs AB, Varani G. Nat Struct Biol. 2000;7:356. doi: 10.1038/75120. [DOI] [PubMed] [Google Scholar]
- 105.Estrella LA, Wilkinson MF, Gonzalez CI. J Mol Biol. 2009;394:410. doi: 10.1016/j.jmb.2009.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.(a) Rajyaguru P, Parker R. Cell Cycle. 2012;11 doi: 10.4161/cc.20716. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rajyaguru P, She M, Parker R. Mol Cell. 2012;45:244. doi: 10.1016/j.molcel.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.(a) Phatnani HP, Jones JC, Greenleaf AL. Biochemistry. 2004;43:15702. doi: 10.1021/bi048364h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mackellar AL, Greenleaf AL. J Biol Chem. 2011;286:36385. doi: 10.1074/jbc.M111.268144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim VN, Dreyfuss G. Mol Cells. 2001;12:1. [PubMed] [Google Scholar]
- 109.Hieronymus H, Silver PA. Nat Genet. 2003;33:155. doi: 10.1038/ng1080. [DOI] [PubMed] [Google Scholar]
- 110.Kohler A, Hurt E. Nat Rev Mol Cell Biol. 2007;8:761. doi: 10.1038/nrm2255. [DOI] [PubMed] [Google Scholar]
- 111.(a) Strasser K, Hurt E. EMBO J. 2000;19:410. doi: 10.1093/emboj/19.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zenklusen D, Vinciguerra P, Strahm Y, Stutz F. Mol Cell Biol. 2001;21:4219. doi: 10.1128/MCB.21.13.4219-4232.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.(a) Li B, Carey M, Workman JL. Cell. 2007;128:707. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]; (b) Rando OJ. Curr Opin Genet Dev. 2012;22:148. doi: 10.1016/j.gde.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rando OJ, Winston F. Genetics. 2012;190:351. doi: 10.1534/genetics.111.132266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.(a) Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. Mol Cell Biol. 2005;25:3305. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, Shilatifard A, Buratowski S, Greenblatt J. Mol Cell Biol. 2003;23:4207. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, Greenblatt JF, Shilatifard A. Mol Cell. 2003;11:721. doi: 10.1016/s1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]; (d) Ng HH, Robert F, Young RA, Struhl K. Mol Cell. 2003;11:709. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 114.(a) Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. Proc Natl Acad Sci U S A. 2001;98:12902. doi: 10.1073/pnas.231473398. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML. Proc Natl Acad Sci U S A. 2002;99:90. doi: 10.1073/pnas.221596698. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF. EMBO J. 2001;20:7137. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. Cell. 2005;122:517. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 116.Laribee RN, Krogan NJ, Xiao T, Shibata Y, Hughes TR, Greenblatt JF, Strahl BD. Curr Biol. 2005;15:1487. doi: 10.1016/j.cub.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 117.(a) Venkatasubrahmanyam S, Hwang WW, Meneghini MD, Tong AH, Madhani HD. Proc Natl Acad Sci U S A. 2007;104:16609. doi: 10.1073/pnas.0700914104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiang H, Shukla A, Wang X, Chen WY, Bernstein BE, Roeder RG. Cell. 2011;144:513. doi: 10.1016/j.cell.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lenstra TL, Benschop JJ, Kim T, Schulze JM, Brabers NA, Margaritis T, van de Pasch LA, van Heesch SA, Brok MO, Groot Koerkamp MJ, Ko CW, van Leenen D, Sameith K, van Hooff SR, Lijnzaad P, Kemmeren P, Hentrich T, Kobor MS, Buratowski S, Holstege FC. Mol Cell. 2011;42:536. doi: 10.1016/j.molcel.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.(a) Camblong J, Beyrouthy N, Guffanti E, Schlaepfer G, Steinmetz LM, Stutz F. Genes Dev. 2009;23:1534. doi: 10.1101/gad.522509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Berretta J, Pinskaya M, Morillon A. Genes Dev. 2008;22:615. doi: 10.1101/gad.458008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.(a) Strahl BD, Grant PA, Briggs SD, Sun ZW, Bone JR, Caldwell JA, Mollah S, Cook RG, Shabanowitz J, Hunt DF, Allis CD. Mol Cell Biol. 2002;22:1298. doi: 10.1128/mcb.22.5.1298-1306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schaft D, Roguev A, Kotovic KM, Shevchenko A, Sarov M, Neugebauer KM, Stewart AF. Nucleic Acids Res. 2003;31:2475. doi: 10.1093/nar/gkg372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.(a) Rao B, Shibata Y, Strahl BD, Lieb JD. Mol Cell Biol. 2005;25:9447. doi: 10.1128/MCB.25.21.9447-9459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, Strahl BD. Genes Dev. 2003;17:654. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.(a) Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. Cell. 2005;123:581. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]; (b) Joshi AA, Struhl K. Mol Cell. 2005;20:971. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 122.Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, Boone C, Emili A, Weissman JS, Hughes TR, Strahl BD, Grunstein M, Greenblatt JF, Buratowski S, Krogan NJ. Cell. 2005;123:593. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 123.(a) Drouin S, Laramee L, Jacques PE, Forest A, Bergeron M, Robert F. PLoS Genet. 2010;6:e1001173. doi: 10.1371/journal.pgen.1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Govind CK, Qiu H, Ginsburg DS, Ruan C, Hofmeyer K, Hu C, Swaminathan V, Workman JL, Li B, Hinnebusch AG. Mol Cell. 2010;39:234. doi: 10.1016/j.molcel.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.(a) Kaplan CD, Laprade L, Winston F. Science. 2003;301:1096. doi: 10.1126/science.1087374. [DOI] [PubMed] [Google Scholar]; (b) Adkins MW, Tyler JK. Mol Cell. 2006;21:405. doi: 10.1016/j.molcel.2005.12.010. [DOI] [PubMed] [Google Scholar]; (c) Bortvin A, Winston F. Science. 1996;272:1473. doi: 10.1126/science.272.5267.1473. [DOI] [PubMed] [Google Scholar]
- 125.(a) Swanson MS, Winston F. Genetics. 1992;132:325. doi: 10.1093/genetics/132.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hartzog GA, Wada T, Handa H, Winston F. Genes Dev. 1998;12:357. doi: 10.1101/gad.12.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.(a) Yoh SM, Cho H, Pickle L, Evans RM, Jones KA. Genes Dev. 2007;21:160. doi: 10.1101/gad.1503107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun M, Lariviere L, Dengl S, Mayer A, Cramer P. J Biol Chem. 2010;285:41597. doi: 10.1074/jbc.M110.144568. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dengl S, Mayer A, Sun M, Cramer P. J Mol Biol. 2009;389:211. doi: 10.1016/j.jmb.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 127.Kwon SH, Florens L, Swanson SK, Washburn MP, Abmayr SM, Workman JL. Genes Dev. 2010;24:2133. doi: 10.1101/gad.1959110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rodriguez-Paredes M, Ceballos-Chavez M, Esteller M, Garcia-Dominguez M, Reyes JC. Nucleic Acids Res. 2009;37:2449. doi: 10.1093/nar/gkp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yankulov K, Todorov I, Romanowski P, Licatalosi D, Cilli K, McCracken S, Laskey R, Bentley DL. Mol Cell Biol. 1999;19:6154. doi: 10.1128/mcb.19.9.6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gauthier L, Dziak R, Kramer DJ, Leishman D, Song X, Ho J, Radovic M, Bentley D, Yankulov K. Genetics. 2002;162:1117. doi: 10.1093/genetics/162.3.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mayan MD. PLoS One. 2013;8:e53405. doi: 10.1371/journal.pone.0053405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cadoret JC, Meisch F, Hassan-Zadeh V, Luyten I, Guillet C, Duret L, Quesneville H, Prioleau MN. Proc Natl Acad Sci U S A. 2008;105:15837. doi: 10.1073/pnas.0805208105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wong JM, Ingles CJ. Mol Gen Genet. 2001;264:842. doi: 10.1007/s004380000374. [DOI] [PubMed] [Google Scholar]
- 134.Winsor TS, Bartkowiak B, Bennett CB, Greenleaf AL. PLoS One. 2013;8:e60909. doi: 10.1371/journal.pone.0060909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.(a) Aguilera A. EMBO J. 2002;21:195. doi: 10.1093/emboj/21.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gonzalez-Aguilera C, Tous C, Gomez- Gonzalez B, Huertas P, Luna R, Aguilera A. Mol Biol Cell. 2008;19:4310. doi: 10.1091/mbc.E08-04-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li X, Manley JL. Cell. 2005;122:365. doi: 10.1016/j.cell.2005.06.008. [DOI] [PubMed] [Google Scholar]; (d) Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P. Genes Dev. 2012;26:163. doi: 10.1101/gad.179721.111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mischo HE, Gomez-Gonzalez B, Grzechnik P, Rondon AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ. Mol Cell. 2011;41:21. doi: 10.1016/j.molcel.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.(a) Branzei D, Foiani M. Genes Dev. 2007;21:3019. doi: 10.1101/gad.1624707. [DOI] [PubMed] [Google Scholar]; (b) Hu Y, Lu X, Barnes E, Yan M, Lou H, Luo G. Mol Cell Biol. 2005;25:3431. doi: 10.1128/MCB.25.9.3431-3442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, Jasin M, Vogel H, Sung P, Luo G. Genes Dev. 2007;21:3073. doi: 10.1101/gad.1609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kanagaraj R, Huehn D, MacKellar A, Menigatti M, Zheng L, Urban V, Shevelev I, Greenleaf AL, Janscak P. Nucleic Acids Res. 2010;38:8131. doi: 10.1093/nar/gkq697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Guo Z, Stiller JW. BMC Genomics. 2004;5:69. doi: 10.1186/1471-2164-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.(a) Egloff S, Dienstbier M, Murphy S. Trends Genet. 2012;28:333. doi: 10.1016/j.tig.2012.03.007. [DOI] [PubMed] [Google Scholar]; (b) Egloff S, Murphy S. Trends Genet. 2008;24:280. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]; (c) Heidemann M, Hintermair C, Voss K, Eick D. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbagrm.2012.08.013. [DOI] [PubMed] [Google Scholar]; (d) Jasnovidova O, Stefl R. Wiley Interdiscip Rev RNA. 2012 doi: 10.1002/wrna.1138. [DOI] [PubMed] [Google Scholar]
- 140.(a) Czudnochowski N, Bosken CA, Geyer M. Nat Commun. 2012;3:842. doi: 10.1038/ncomms1846. [DOI] [PubMed] [Google Scholar]; (b) Morris DP, Stevens RD, Greenleaf AL. Biochem Biophys Res Commun. 1998;245:53. doi: 10.1006/bbrc.1998.8373. [DOI] [PubMed] [Google Scholar]
- 141.(a) Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F. Nucleic Acids Res. 2011;39:D261. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gnad F, Gunawardena J, Mann M. Nucleic Acids Res. 2011;39:D253. doi: 10.1093/nar/gkq1159. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. Nucleic Acids Res. 2012;40:D261. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee JM, Greenleaf AL. Proc Natl Acad Sci U S A. 1989;86:3624. doi: 10.1073/pnas.86.10.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.(a) Weeks JR, Hardin SE, Shen J, Lee JM, Greenleaf AL. Genes Dev. 1993;7:2329. doi: 10.1101/gad.7.12a.2329. [DOI] [PubMed] [Google Scholar]; (b) O’Brien T, Hardin S, Greenleaf A, Lis JT. Nature. 1994;370:75. doi: 10.1038/370075a0. [DOI] [PubMed] [Google Scholar]
- 144.(a) Bregman DB, Du L, van der Zee S, Warren SL. J Cell Biol. 1995;129:287. doi: 10.1083/jcb.129.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patturajan M, Schulte RJ, Sefton BM, Berezney R, Vincent M, Bensaude O, Warren SL, Corden JL. J Biol Chem. 1998;273:4689. doi: 10.1074/jbc.273.8.4689. [DOI] [PubMed] [Google Scholar]
- 145.Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, Kremmer E, Eick D, Cramer P. Science. 2012;336:1723. doi: 10.1126/science.1219651. [DOI] [PubMed] [Google Scholar]
- 146.(a) Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. Nature. 2000;407:395. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]; (b) Knight ZA, Shokat KM. Cell. 2007;128:425. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 147.Witucki LA, Huang X, Shah K, Liu Y, Kyin S, Eck MJ, Shokat KM. Chem Biol. 2002;9:25. doi: 10.1016/s1074-5521(02)00091-1. [DOI] [PubMed] [Google Scholar]
- 148.(a) Shah K, Liu Y, Deirmengian C, Shokat KM. Proc Natl Acad Sci U S A. 1997;94:3565. doi: 10.1073/pnas.94.8.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shah K, Shokat KM. Chem Biol. 2002;9:35. doi: 10.1016/s1074-5521(02)00086-8. [DOI] [PubMed] [Google Scholar]
- 149.Jones JC, Phatnani HP, Haystead TA, MacDonald JA, Alam SM, Greenleaf AL. J Biol Chem. 2004;279:24957. doi: 10.1074/jbc.M402218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kim M, Suh H, Cho EJ, Buratowski S. J Biol Chem. 2009;284:26421. doi: 10.1074/jbc.M109.028993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Morgan DO. Annu Rev Cell Dev Biol. 1997;13:261. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 152.Bregman DB, Pestell RG, Kidd VJ. Front Biosci. 2000;5:D244. doi: 10.2741/bregman. [DOI] [PubMed] [Google Scholar]
- 153.Pavletich NP. J Mol Biol. 1999;287:821. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- 154.Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC. Curr Biol. 1994;4:973. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 155.Chen J, Saha P, Kornbluth S, Dynlacht BD, Dutta A. Mol Cell Biol. 1996;16:4673. doi: 10.1128/mcb.16.9.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.(a) Fesquet D, Labbe JC, Derancourt J, Capony JP, Galas S, Girard F, Lorca T, Shuttleworth J, Doree M, Cavadore JC. EMBO J. 1993;12:3111. doi: 10.1002/j.1460-2075.1993.tb05980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Matsuoka M, Kato JY, Fisher RP, Morgan DO, Sherr CJ. Mol Cell Biol. 1994;14:7265. doi: 10.1128/mcb.14.11.7265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Solomon MJ, Lee T, Kirschner MW. Mol Biol Cell. 1992;3:13. doi: 10.1091/mbc.3.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.(a) Thuret JY, Valay JG, Faye G, Mann C. Cell. 1996;86:565. doi: 10.1016/s0092-8674(00)80130-0. [DOI] [PubMed] [Google Scholar]; (b) Kaldis P, Sutton A, Solomon MJ. Cell. 1996;86:553. doi: 10.1016/s0092-8674(00)80129-4. [DOI] [PubMed] [Google Scholar]
- 158.(a) Feaver WJ, Gileadi O, Li Y, Kornberg RD. Cell. 1991;67:1223. doi: 10.1016/0092-8674(91)90298-d. [DOI] [PubMed] [Google Scholar]; (b) Feaver WJ, Svejstrup JQ, Henry NL, Kornberg RD. Cell. 1994;79:1103. doi: 10.1016/0092-8674(94)90040-x. [DOI] [PubMed] [Google Scholar]; (c) Lu H, Zawel L, Fisher L, Egly JM, Reinberg D. Nature. 1992;358:641. doi: 10.1038/358641a0. [DOI] [PubMed] [Google Scholar]; (d) Roy R, Adamczewski JP, Seroz T, Vermeulen W, Tassan JP, Schaeffer L, Nigg EA, Hoeijmakers JH, Egly JM. Cell. 1994;79:1093. doi: 10.1016/0092-8674(94)90039-6. [DOI] [PubMed] [Google Scholar]; (e) Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, Morgan DO, Reinberg D. Nature. 1995;374:283. doi: 10.1038/374283a0. [DOI] [PubMed] [Google Scholar]
- 159.Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. Nat Struct Mol Biol. 2006;13:55. doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- 160.Yankulov KY, Bentley DL. EMBO J. 1997;16:1638. doi: 10.1093/emboj/16.7.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.(a) Gebara MM, Sayre MH, Corden JL. J Cell Biochem. 1997;64:390. [PubMed] [Google Scholar]; (b) Trigon S, Serizawa H, Conaway JW, Conaway RC, Jackson SP, Morange M. J Biol Chem. 1998;273:6769. doi: 10.1074/jbc.273.12.6769. [DOI] [PubMed] [Google Scholar]; (c) Rickert P, Corden JL, Lees E. Oncogene. 1999;18:1093. doi: 10.1038/sj.onc.1202399. [DOI] [PubMed] [Google Scholar]; (d) Ramanathan Y, Rajpara SM, Reza SM, Lees E, Shuman S, Mathews MB, Pe’ery T. J Biol Chem. 2001;276:10913. doi: 10.1074/jbc.M010975200. [DOI] [PubMed] [Google Scholar]
- 162.(a) Hengartner CJ, Myer VE, Liao SM, Wilson CJ, Koh SS, Young RA. Mol Cell. 1998;2:43. doi: 10.1016/s1097-2765(00)80112-4. [DOI] [PubMed] [Google Scholar]; (b) Keogh MC, Podolny V, Buratowski S. Mol Cell Biol. 2003;23:7005. doi: 10.1128/MCB.23.19.7005-7018.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.(a) Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, Ansari AZ. Mol Cell. 2009;34:387. doi: 10.1016/j.molcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boeing S, Rigault C, Heidemann M, Eick D, Meisterernst M. J Biol Chem. 2010;285:188. doi: 10.1074/jbc.M109.046565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL. Mol Cell Biol. 2009;29:5455. doi: 10.1128/MCB.00637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.(a) Akoulitchev S, Makela TP, Weinberg RA, Reinberg D. Nature. 1995;377:557. doi: 10.1038/377557a0. [DOI] [PubMed] [Google Scholar]; (b) Jiang Y, Yan M, Gralla JD. Mol Cell Biol. 1996;16:1614. doi: 10.1128/mcb.16.4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Buratowski S. Mol Cell. 2009;36:541. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Adelman K, Lis JT. Nat Rev Genet. 2012;13:720. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.(a) Makela TP, Parvin JD, Kim J, Huber LJ, Sharp PA, Weinberg RA. Proc Natl Acad Sci U S A. 1995;92:5174. doi: 10.1073/pnas.92.11.5174. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tirode F, Busso D, Coin F, Egly JM. Mol Cell. 1999;3:87. doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- 168.(a) Hong SW, Hong SM, Yoo JW, Lee YC, Kim S, Lis JT, Lee DK. Proc Natl Acad Sci U S A. 2009;106:14276. doi: 10.1073/pnas.0903642106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kanin EI, Kipp RT, Kung C, Slattery M, Viale A, Hahn S, Shokat KM, Ansari AZ. Proc Natl Acad Sci U S A. 2007;104:5812. doi: 10.1073/pnas.0611505104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lee KM, Miklos I, Du H, Watt S, Szilagyi Z, Saiz JE, Madabhushi R, Penkett CJ, Sipiczki M, Bahler J, Fisher RP. Mol Biol Cell. 2005;16:2734. doi: 10.1091/mbc.E04-11-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. Nat Struct Mol Biol. 2012;19:1108. doi: 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wallenfang MR, Seydoux G. Proc Natl Acad Sci U S A. 2002;99:5527. doi: 10.1073/pnas.082618399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ganuza M, Saiz-Ladera C, Canamero M, Gomez G, Schneider R, Blasco MA, Pisano D, Paramio JM, Santamaria D, Barbacid M. EMBO J. 2012;31:2498. doi: 10.1038/emboj.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.(a) Galbraith MD, Donner AJ, Espinosa JM. Transcription. 2010;1:4. doi: 10.4161/trns.1.1.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Conaway RC, Conaway JW. Curr Opin Genet Dev. 2011;21:225. doi: 10.1016/j.gde.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Malik S, Roeder RG. Nat Rev Genet. 2010;11:761. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.(a) Borggrefe T, Davis R, Erdjument-Bromage H, Tempst P, Kornberg RD. J Biol Chem. 2002;277:44202. doi: 10.1074/jbc.M207195200. [DOI] [PubMed] [Google Scholar]; (b) Wang G, Cantin GT, Stevens JL, Berk AJ. Mol Cell Biol. 2001;21:4604. doi: 10.1128/MCB.21.14.4604-4613.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Samuelsen CO, Baraznenok V, Khorosjutina O, Spahr H, Kieselbach T, Holmberg S, Gustafsson CM. Proc Natl Acad Sci U S A. 2003;100:6422. doi: 10.1073/pnas.1030497100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Leclerc V, Tassan JP, O’Farrell PH, Nigg EA, Leopold P. Mol Biol Cell. 1996;7:505. doi: 10.1091/mbc.7.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Larschan E, Winston F. Mol Cell Biol. 2005;25:114. doi: 10.1128/MCB.25.1.114-123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ. Genes Dev. 2009;23:439. doi: 10.1101/gad.1767009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Knuesel MT, Meyer KD, Donner AJ, Espinosa JM, Taatjes DJ. Mol Cell Biol. 2009;29:650. doi: 10.1128/MCB.00993-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Cell. 1998;95:717. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 177.Liu Y, Kung C, Fishburn J, Ansari AZ, Shokat KM, Hahn S. Mol Cell Biol. 2004;24:1721. doi: 10.1128/MCB.24.4.1721-1735.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.(a) Marshall NF, Price DH. J Biol Chem. 1995;270:12335. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]; (b) Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH. Genes Dev. 1997;11:2622. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peng J, Marshall NF, Price DH. J Biol Chem. 1998;273:13855. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]; (d) Peng J, Zhu Y, Milton JT, Price DH. Genes Dev. 1998;12:755. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. Cell. 1998;92:451. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]; (f) Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, Flores O. Genes Dev. 1997;11:2633. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Peterlin BM, Price DH. Mol Cell. 2006;23:297. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 179.(a) Prelich G, Winston F. Genetics. 1993;135:665. doi: 10.1093/genetics/135.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Murray S, Udupa R, Yao S, Hartzog G, Prelich G. Mol Cell Biol. 2001;21:4089. doi: 10.1128/MCB.21.13.4089-4096.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.(a) Pei Y, Du H, Singer J, Stamour C, Granitto S, Shuman S, Fisher RP. Mol Cell Biol. 2006;26:777. doi: 10.1128/MCB.26.3.777-788.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Viladevall L, St Amour CV, Rosebrock A, Schneider S, Zhang C, Allen JJ, Shokat KM, Schwer B, Leatherwood JK, Fisher RP. Mol Cell. 2009;33:738. doi: 10.1016/j.molcel.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Grana X, De Luca A, Sang N, Fu Y, Claudio PP, Rosenblatt J, Morgan DO, Giordano A. Proc Natl Acad Sci U S A. 1994;91:3834. doi: 10.1073/pnas.91.9.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.(a) Shore SM, Byers SA, Dent P, Price DH. Gene. 2005;350:51. doi: 10.1016/j.gene.2005.01.015. [DOI] [PubMed] [Google Scholar]; (b) Shore SM, Byers SA, Maury W, Price DH. Gene. 2003;307:175. doi: 10.1016/s0378-1119(03)00466-9. [DOI] [PubMed] [Google Scholar]
- 183.St Amour CV, Sanso M, Bosken CA, Lee KM, Larochelle S, Zhang C, Shokat KM, Geyer M, Fisher RP. Mol Cell Biol. 2012;32:2372. doi: 10.1128/MCB.06657-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, Robert F. Mol Cell. 2012;45:158. doi: 10.1016/j.molcel.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 185.Qiu H, Hu C, Hinnebusch AG. Mol Cell. 2009;33:752. doi: 10.1016/j.molcel.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lis J. Cold Spring Harb Symp Quant Biol. 1998;63:347. doi: 10.1101/sqb.1998.63.347. [DOI] [PubMed] [Google Scholar]
- 187.(a) Core LJ, Waterfall JJ, Lis JT. Science. 2008;322:1845. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, Lis JT. Genes Dev. 2011;25:742. doi: 10.1101/gad.2005511. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, Adelman K. Science. 2010;327:335. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. Cell. 2010;141:432. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA. Nat Genet. 2007;39:1512. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. Cell. 2007;130:77. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. Nature. 2005;436:876. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.(a) Kephart DD, Marshall NF, Price DH. Mol Cell Biol. 1992;12:2067. doi: 10.1128/mcb.12.5.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, Sugimoto S, Yano K, Hartzog GA, Winston F, Buratowski S, Handa H. Genes Dev. 1998;12:343. doi: 10.1101/gad.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J, Handa H. Cell. 1999;97:41. doi: 10.1016/s0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]; (d) Bartkowiak B, Greenleaf AL. Transcription. 2011;2:115. doi: 10.4161/trns.2.3.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cheng B, Li T, Rahl PB, Adamson TE, Loudas NB, Guo J, Varzavand K, Cooper JJ, Hu X, Gnatt A, Young RA, Price DH. Mol Cell. 2012;45:38. doi: 10.1016/j.molcel.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.(a) Eberhardy SR, Farnham PJ. J Biol Chem. 2002;277:40156. doi: 10.1074/jbc.M207441200. [DOI] [PubMed] [Google Scholar]; (b) Wu SY, Chiang CM. J Biol Chem. 2007;282:13141. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 191.Ni Z, Saunders A, Fuda NJ, Yao J, Suarez JR, Webb WW, Lis JT. Mol Cell Biol. 2008;28:1161. doi: 10.1128/MCB.01859-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Devaiah BN, Singer DS. J Biol Chem. 2012;287:38755. doi: 10.1074/jbc.M112.412015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.(a) Li T, Price D. Transcription. 2012;3:177. doi: 10.4161/trns.20600. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Narita T, Yamaguchi Y, Yano K, Sugimoto S, Chanarat S, Wada T, Kim DK, Hasegawa J, Omori M, Inukai N, Endoh M, Yamada T, Handa H. Mol Cell Biol. 2003;23:1863. doi: 10.1128/MCB.23.6.1863-1873.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Steinmetz EJ, Warren CL, Kuehner JN, Panbehi B, Ansari AZ, Brow DA. Mol Cell. 2006;24:735. doi: 10.1016/j.molcel.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 195.(a) Lee JM, Greenleaf AL. Gene Expr. 1991;1:149. [PMC free article] [PubMed] [Google Scholar]; (b) Sterner DE, Lee JM, Hardin SE, Greenleaf AL. Mol Cell Biol. 1995;15:5716. doi: 10.1128/mcb.15.10.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wood A, Shilatifard A. Cell Cycle. 2006;5:1066. doi: 10.4161/cc.5.10.2769. [DOI] [PubMed] [Google Scholar]
- 197.(a) Liu J, Kipreos ET. Mol Biol Evol. 2000;17:1061. doi: 10.1093/oxfordjournals.molbev.a026387. [DOI] [PubMed] [Google Scholar]; (b) Sukegawa Y, Yamashita A, Yamamoto M. PLoS Genet. 2011;7:e1002387. doi: 10.1371/journal.pgen.1002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, Greenleaf AL. Genes Dev. 2010;24:2303. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.(a) Chymkowitch P, Eldholm V, Lorenz S, Zimmermann C, Lindvall JM, Bjoras M, Meza-Zepeda LA, Enserink JM. Proc Natl Acad Sci U S A. 2012;109:10450. doi: 10.1073/pnas.1200067109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chymkowitch P, Enserink JM. Transcription. 2012;4 doi: 10.4161/trns.22456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.(a) Cisek LJ, Corden JL. Nature. 1989;339:679. doi: 10.1038/339679a0. [DOI] [PubMed] [Google Scholar]; (b) Cisek LJ, Corden JL. Methods Enzymol. 1991;200:301. doi: 10.1016/0076-6879(91)00148-p. [DOI] [PubMed] [Google Scholar]
- 201.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 202.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Endocr Rev. 2001;22:153. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 203.Dubois MF, Nguyen VT, Dahmus ME, Pages G, Pouyssegur J, Bensaude O. EMBO J. 1994;13:4787. doi: 10.1002/j.1460-2075.1994.tb06804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.(a) Kong X, Shen Y, Jiang N, Fei X, Mi J. Cell Signal. 2011;23:1273. doi: 10.1016/j.cellsig.2011.04.005. [DOI] [PubMed] [Google Scholar]; (b) Anderson CW, Carter TH. Curr Top Microbiol Immunol. 1996;217:91. doi: 10.1007/978-3-642-50140-1_7. [DOI] [PubMed] [Google Scholar]
- 205.Dvir A, Stein LY, Calore BL, Dynan WS. J Biol Chem. 1993;268:10440. [PubMed] [Google Scholar]
- 206.Tyagi S, Ochem A, Tyagi M. J Gen Virol. 2011;92:1710. doi: 10.1099/vir.0.029587-0. [DOI] [PubMed] [Google Scholar]
- 207.Maldonado E, Shiekhattar R, Sheldon M, Cho H, Drapkin R, Rickert P, Lees E, Anderson CW, Linn S, Reinberg D. Nature. 1996;381:86. doi: 10.1038/381086a0. [DOI] [PubMed] [Google Scholar]
- 208.An J, Yang T, Huang Y, Liu F, Sun J, Wang Y, Xu Q, Wu D, Zhou P. BMC Biochem. 2011;12:2. doi: 10.1186/1471-2091-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Peterson SR, Jesch SA, Chamberlin TN, Dvir A, Rabindran SK, Wu C, Dynan WS. J Biol Chem. 1995;270:1449. doi: 10.1074/jbc.270.3.1449. [DOI] [PubMed] [Google Scholar]
- 210.Devaiah BN, Singer DS. Transcription. 2012;4 doi: 10.4161/trns.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ, 3rd, Singer DS. Proc Natl Acad Sci U S A. 2012;109:6927. doi: 10.1073/pnas.1120422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.de Carcer G, Manning G, Malumbres M. Cell Cycle. 2011;10:2255. doi: 10.4161/cc.10.14.16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. Cell. 2003;115:83. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 214.Johnson EF, Stewart KD, Woods KW, Giranda VL, Luo Y. Biochemistry. 2007;46:9551. doi: 10.1021/bi7008745. [DOI] [PubMed] [Google Scholar]
- 215.Heidemann M, Eick D. RNA Biol. 2012;9 doi: 10.4161/rna.21726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wang L, Gao J, Dai W, Lu L. J Biol Chem. 2008;283:25928. doi: 10.1074/jbc.M801326200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sakurai H, Ishihama A. Genes Cells. 2002;7:273. doi: 10.1046/j.1365-2443.2002.00522.x. [DOI] [PubMed] [Google Scholar]
- 218.Kuenzel EA, Mulligan JA, Sommercorn J, Krebs EG. J Biol Chem. 1987;262:9136. [PubMed] [Google Scholar]
- 219.Sanso M, Fisher RP. Transcription. 2013;4 doi: 10.4161/trns.25146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.(a) Kelly WG, Dahmus ME, Hart GW. J Biol Chem. 1993;268:10416. [PubMed] [Google Scholar]; (b) Haltiwanger RS, Blomberg MA, Hart GW. J Biol Chem. 1992;267:9005. [PubMed] [Google Scholar]
- 221.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. J Biol Chem. 2001;276:9838. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- 222.Gross BJ, Kraybill BC, Walker S. J Am Chem Soc. 2005;127:14588. doi: 10.1021/ja0555217. [DOI] [PubMed] [Google Scholar]
- 223.Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. J Biol Chem. 2005;280:25313. doi: 10.1074/jbc.M413819200. [DOI] [PubMed] [Google Scholar]
- 224.Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, Lewis BA. J Biol Chem. 2012;287:23549. doi: 10.1074/jbc.M111.330910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sims RJ, 3rd, Rojas LA, Beck D, Bonasio R, Schuller R, Drury WJ, 3rd, Eick D, Reinberg D. Science. 2011;332:99. doi: 10.1126/science.1202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chambers RS, Dahmus ME. J Biol Chem. 1994;269:26243. [PubMed] [Google Scholar]
- 227.(a) Archambault J, Chambers RS, Kobor MS, Ho Y, Cartier M, Bolotin D, Andrews B, Kane CM, Greenblatt J. Proc Natl Acad Sci U S A. 1997;94:14300. doi: 10.1073/pnas.94.26.14300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kimura M, Suzuki H, Ishihama A. Mol Cell Biol. 2002;22:1577. doi: 10.1128/mcb.22.5.1577-1588.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.(a) Kobor MS, Archambault J, Lester W, Holstege FC, Gileadi O, Jansma DB, Jennings EG, Kouyoumdjian F, Davidson AR, Young RA, Greenblatt J. Mol Cell. 1999;4:55. doi: 10.1016/s1097-2765(00)80187-2. [DOI] [PubMed] [Google Scholar]; (b) Archambault J, Pan G, Dahmus GK, Cartier M, Marshall N, Zhang S, Dahmus ME, Greenblatt J. J Biol Chem. 1998;273:27593. doi: 10.1074/jbc.273.42.27593. [DOI] [PubMed] [Google Scholar]; (c) Cho H, Kim TK, Mancebo H, Lane WS, Flores O, Reinberg D. Genes Dev. 1999;13:1540. doi: 10.1101/gad.13.12.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Collet JF, Stroobant V, Pirard M, Delpierre G, Van Schaftingen E. J Biol Chem. 1998;273:14107. doi: 10.1074/jbc.273.23.14107. [DOI] [PubMed] [Google Scholar]
- 230.Yeo M, Lin PS, Dahmus ME, Gill GN. J Biol Chem. 2003;278:26078. doi: 10.1074/jbc.M301791200. [DOI] [PubMed] [Google Scholar]
- 231.Yeo M, Lee SK, Lee B, Ruiz EC, Pfaff SL, Gill GN. Science. 2005;307:596. doi: 10.1126/science.1100801. [DOI] [PubMed] [Google Scholar]
- 232.(a) Kamenski T, Heilmeier S, Meinhart A, Cramer P. Mol Cell. 2004;15:399. doi: 10.1016/j.molcel.2004.06.035. [DOI] [PubMed] [Google Scholar]; (b) Zhang Y, Kim Y, Genoud N, Gao J, Kelly JW, Pfaff SL, Gill GN, Dixon JE, Noel JP. Mol Cell. 2006;24:759. doi: 10.1016/j.molcel.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hausmann S, Shuman S. J Biol Chem. 2002;277:21213. doi: 10.1074/jbc.M202056200. [DOI] [PubMed] [Google Scholar]
- 234.(a) Ghosh A, Shuman S, Lima CD. Mol Cell. 2008;32:478. doi: 10.1016/j.molcel.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hausmann S, Koiwa H, Krishnamurthy S, Hampsey M, Shuman S. J Biol Chem. 2005;280:37681. doi: 10.1074/jbc.M505292200. [DOI] [PubMed] [Google Scholar]
- 235.Zhang M, Wang XJ, Chen X, Bowman ME, Luo Y, Noel JP, Ellington AD, Etzkorn FA, Zhang Y. ACS Chem Biol. 2012;7:1462. doi: 10.1021/cb3000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Cho EJ, Kobor MS, Kim M, Greenblatt J, Buratowski S. Genes Dev. 2001;15:3319. doi: 10.1101/gad.935901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.(a) Chambers RS, Wang BQ, Burton ZF, Dahmus ME. J Biol Chem. 1995;270:14962. doi: 10.1074/jbc.270.25.14962. [DOI] [PubMed] [Google Scholar]; (b) Suh MH, Ye P, Zhang M, Hausmann S, Shuman S, Gnatt AL, Fu J. Proc Natl Acad Sci U S A. 2005;102:17314. doi: 10.1073/pnas.0507987102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Bushnell DA, Westover KD, Davis RE, Kornberg RD. Science. 2004;303:983. doi: 10.1126/science.1090838. [DOI] [PubMed] [Google Scholar]
- 239.(a) Wostenberg C, Kumar S, Noid WG, Showalter SA. J Phys Chem B. 2011;115:13731. doi: 10.1021/jp208008m. [DOI] [PubMed] [Google Scholar]; (b) Kobor MS, Simon LD, Omichinski J, Zhong G, Archambault J, Greenblatt J. Mol Cell Biol. 2000;20:7438. doi: 10.1128/mcb.20.20.7438-7449.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Palancade B, Dubois MF, Dahmus ME, Bensaude O. Mol Cell Biol. 2001;21:6359. doi: 10.1128/MCB.21.19.6359-6368.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Fuda NJ, Buckley MS, Wei W, Core LJ, Waters CT, Reinberg D, Lis JT. Mol Cell Biol. 2012;32:3428. doi: 10.1128/MCB.00247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Sun ZW, Hampsey M. Mol Cell Biol. 1996;16:1557. doi: 10.1128/mcb.16.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.(a) Ganem C, Devaux F, Torchet C, Jacq C, Quevillon-Cheruel S, Labesse G, Facca C, Faye G. EMBO J. 2003;22:1588. doi: 10.1093/emboj/cdg141. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meinhart A, Silberzahn T, Cramer P. J Biol Chem. 2003;278:15917. doi: 10.1074/jbc.M301643200. [DOI] [PubMed] [Google Scholar]
- 244.(a) Wu WH, Pinto I, Chen BS, Hampsey M. Genetics. 1999;153:643. doi: 10.1093/genetics/153.2.643. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pappas DL, Jr, Hampsey M. Mol Cell Biol. 2000;20:8343. doi: 10.1128/mcb.20.22.8343-8351.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dichtl B, Blank D, Ohnacker M, Friedlein A, Roeder D, Langen H, Keller W. Mol Cell. 2002;10:1139. doi: 10.1016/s1097-2765(02)00707-4. [DOI] [PubMed] [Google Scholar]
- 245.(a) He X, Khan AU, Cheng H, Pappas DL, Jr, Hampsey M, Moore CL. Genes Dev. 2003;17:1030. doi: 10.1101/gad.1075203. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nedea E, He X, Kim M, Pootoolal J, Zhong G, Canadien V, Hughes T, Buratowski S, Moore CL, Greenblatt J. J Biol Chem. 2003;278:33000. doi: 10.1074/jbc.M304454200. [DOI] [PubMed] [Google Scholar]; (c) Steinmetz EJ, Brow DA. Mol Cell Biol. 2003;23:6339. doi: 10.1128/MCB.23.18.6339-6349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.(a) Ansari A, Hampsey M. Genes Dev. 2005;19:2969. doi: 10.1101/gad.1362305. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) O’Sullivan JM, Tan-Wong SM, Morillon A, Lee B, Coles J, Mellor J, Proudfoot NJ. Nat Genet. 2004;36:1014. doi: 10.1038/ng1411. [DOI] [PubMed] [Google Scholar]; (c) Singh BN, Hampsey M. Mol Cell. 2007;27:806. doi: 10.1016/j.molcel.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 247.Krishnamurthy S, He X, Reyes-Reyes M, Moore C, Hampsey M. Mol Cell. 2004;14:387. doi: 10.1016/s1097-2765(04)00235-7. [DOI] [PubMed] [Google Scholar]
- 248.Krishnamurthy S, Ghazy MA, Moore C, Hampsey M. Mol Cell Biol. 2009;29:2925. doi: 10.1128/MCB.01655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Werner-Allen JW, Lee CJ, Liu P, Nicely NI, Wang S, Greenleaf AL, Zhou P. J Biol Chem. 2011;286:5717. doi: 10.1074/jbc.M110.197129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.(a) Gibney PA, Fries T, Bailer SM, Morano KA. Eukaryot Cell. 2008;7:938. doi: 10.1128/EC.00042-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Therien C, Bergeron D, Bourassa S, Greenblatt J, Chabot B, Poirier GG, Hughes TR, Blanchette M, Price DH, Coulombe B. Mol Cell. 2007;27:262. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Mosley AL, Pattenden SG, Carey M, Venkatesh S, Gilmore JM, Florens L, Workman JL, Washburn MP. Mol Cell. 2009;34:168. doi: 10.1016/j.molcel.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Egloff S, Zaborowska J, Laitem C, Kiss T, Murphy S. Mol Cell. 2012;45:111. doi: 10.1016/j.molcel.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Xiang K, Manley JL, Tong L. Nat Commun. 2012;3:946. doi: 10.1038/ncomms1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Schiene C, Fischer G. Curr Opin Struct Biol. 2000;10:40. doi: 10.1016/s0959-440x(99)00046-9. [DOI] [PubMed] [Google Scholar]
- 255.Fanghanel J, Fischer G. Front Biosci. 2004;9:3453. doi: 10.2741/1494. [DOI] [PubMed] [Google Scholar]
- 256.Albert A, Lavoie S, Vincent M. J Cell Sci. 1999;112(Pt 15):2493. doi: 10.1242/jcs.112.15.2493. [DOI] [PubMed] [Google Scholar]
- 257.Lu KP, Hanes SD, Hunter T. Nature. 1996;380:544. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 258.Hani J, Stumpf G, Domdey H. FEBS Lett. 1995;365:198. doi: 10.1016/0014-5793(95)00471-k. [DOI] [PubMed] [Google Scholar]
- 259.Arevalo-Rodriguez M, Wu X, Hanes SD, Heitman J. Front Biosci. 2004;9:2420. doi: 10.2741/1405. [DOI] [PubMed] [Google Scholar]
- 260.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Science. 1997;278:1957. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 261.(a) Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. Genes Dev. 2003;17:2765. doi: 10.1101/gad.1135503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Palancade B, Marshall NF, Tremeau-Bravard A, Bensaude O, Dahmus ME, Dubois MF. J Mol Biol. 2004;335:415. doi: 10.1016/j.jmb.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 262.Xu YX, Manley JL. Genes Dev. 2007;21:2950. doi: 10.1101/gad.1592807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Hanes SD, Shank PR, Bostian KA. Yeast. 1989;5:55. doi: 10.1002/yea.320050108. [DOI] [PubMed] [Google Scholar]
- 264.(a) Morris DP, Phatnani HP, Greenleaf AL. J Biol Chem. 1999;274:31583. doi: 10.1074/jbc.274.44.31583. [DOI] [PubMed] [Google Scholar]; (b) Wu X, Chang A, Sudol M, Hanes SD. Curr Genet. 2001;40:234. doi: 10.1007/s00294-001-0257-8. [DOI] [PubMed] [Google Scholar]; (c) Wu X, Wilcox CB, Devasahayam G, Hackett RL, Arevalo-Rodriguez M, Cardenas ME, Heitman J, Hanes SD. EMBO J. 2000;19:3727. doi: 10.1093/emboj/19.14.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.(a) Lu KP, Zhou XZ. Nat Rev Mol Cell Biol. 2007;8:904. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]; (b) Yeh ES, Means AR. Nat Rev Cancer. 2007;7:381. doi: 10.1038/nrc2107. [DOI] [PubMed] [Google Scholar]
- 266.Gemmill TR, Wu X, Hanes SD. J Biol Chem. 2005;280:15510. doi: 10.1074/jbc.M412172200. [DOI] [PubMed] [Google Scholar]
- 267.Hani J, Schelbert B, Bernhardt A, Domdey H, Fischer G, Wiebauer K, Rahfeld JU. J Biol Chem. 1999;274:108. doi: 10.1074/jbc.274.1.108. [DOI] [PubMed] [Google Scholar]
- 268.Ma Z, Atencio D, Barnes C, DeFiglio H, Hanes SD. Mol Cell Biol. 2012;32:3594. doi: 10.1128/MCB.00672-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.(a) Singh N, Ma Z, Gemmill T, Wu X, Defiglio H, Rossettini A, Rabeler C, Beane O, Morse RH, Palumbo MJ, Hanes SD. Mol Cell. 2009;36:255. doi: 10.1016/j.molcel.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xiang K, Nagaike T, Xiang S, Kilic T, Beh MM, Manley JL, Tong L. Nature. 2010;467:729. doi: 10.1038/nature09391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Rempola B, Kaniak A, Migdalski A, Rytka J, Slonimski PP, di Rago JP. Mol Gen Genet. 2000;262:1081. doi: 10.1007/pl00008651. [DOI] [PubMed] [Google Scholar]
- 271.Jordens J, Janssens V, Longin S, Stevens I, Martens E, Bultynck G, Engelborghs Y, Lescrinier E, Waelkens E, Goris J, Van Hoof C. J Biol Chem. 2006;281:6349. doi: 10.1074/jbc.M507760200. [DOI] [PubMed] [Google Scholar]
- 272.(a) Douville J, David J, Lemieux KM, Gaudreau L, Ramotar D. Genetics. 2006;172:1369. doi: 10.1534/genetics.105.046110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Heitman J, Movva NR, Hall MN. Science. 1991;253:905. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]; (c) Powers T, Walter P. Mol Biol Cell. 1999;10:987. doi: 10.1091/mbc.10.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rohde J, Heitman J, Cardenas ME. J Biol Chem. 2001;276:9583. doi: 10.1074/jbc.R000034200. [DOI] [PubMed] [Google Scholar]
- 273.Jouvet N, Poschmann J, Douville J, Bulet L, Ramotar D. BMC Mol Biol. 2010;11:92. doi: 10.1186/1471-2199-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.(a) Douville J, David J, Fortier PK, Ramotar D. Curr Genet. 2004;46:72. doi: 10.1007/s00294-004-0513-9. [DOI] [PubMed] [Google Scholar]; (b) Mitchell DA, Sprague GF., Jr Mol Cell Biol. 2001;21:488. doi: 10.1128/MCB.21.2.488-500.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Van Hoof C, Martens E, Longin S, Jordens J, Stevens I, Janssens V, Goris J. Biochem J. 2005;386:93. doi: 10.1042/BJ20040887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Poschmann J, Drouin S, Jacques PE, El Fadili K, Newmarch M, Robert F, Ramotar D. PLoS One. 2011;6:e23159. doi: 10.1371/journal.pone.0023159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kaneko S, Manley JL. Mol Cell. 2005;20:91. doi: 10.1016/j.molcel.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 277.Phatnani HP, Greenleaf AL. Methods Mol Biol. 2004;257:17. doi: 10.1385/1-59259-750-5:017. [DOI] [PubMed] [Google Scholar]
- 278.(a) Dahmus ME. J Biol Chem. 1996;271:19009. doi: 10.1074/jbc.271.32.19009. [DOI] [PubMed] [Google Scholar]; (b) Svejstrup JQ. Biochim Biophys Acta. 2004;1677:64. doi: 10.1016/j.bbaexp.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 279.(a) Kornberg RD. Trends Biochem Sci. 2005;30:235. doi: 10.1016/j.tibs.2005.03.011. [DOI] [PubMed] [Google Scholar]; (b) Myers LC, Kornberg RD. Annu Rev Biochem. 2000;69:729. doi: 10.1146/annurev.biochem.69.1.729. [DOI] [PubMed] [Google Scholar]
- 280.Svejstrup JQ, Li Y, Fellows J, Gnatt A, Bjorklund S, Kornberg RD. Proc Natl Acad Sci U S A. 1997;94:6075. doi: 10.1073/pnas.94.12.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Scafe C, Nonet M, Young RA. Mol Cell Biol. 1990;10:1010. doi: 10.1128/mcb.10.3.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Robinson PJ, Bushnell DA, Trnka MJ, Burlingame AL, Kornberg RD. Proc Natl Acad Sci U S A. 2012;109:17931. doi: 10.1073/pnas.1215241109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.(a) Yudkovsky N, Ranish JA, Hahn S. Nature. 2000;408:225. doi: 10.1038/35041603. [DOI] [PubMed] [Google Scholar]; (b) Sogaard TM, Svejstrup JQ. J Biol Chem. 2007;282:14113. doi: 10.1074/jbc.M701345200. [DOI] [PubMed] [Google Scholar]
- 284.(a) Asturias FJ, Jiang YW, Myers LC, Gustafsson CM, Kornberg RD. Science. 1999;283:985. doi: 10.1126/science.283.5404.985. [DOI] [PubMed] [Google Scholar]; (b) Davis JA, Takagi Y, Kornberg RD, Asturias FA. Mol Cell. 2002;10:409. doi: 10.1016/s1097-2765(02)00598-1. [DOI] [PubMed] [Google Scholar]
- 285.Takagi Y, Kornberg RD. J Biol Chem. 2006;281:80. doi: 10.1074/jbc.M508253200. [DOI] [PubMed] [Google Scholar]
- 286.Naar AM, Taatjes DJ, Zhai W, Nogales E, Tjian R. Genes Dev. 2002;16:1339. doi: 10.1101/gad.987602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Hagler J, Shuman S. Science. 1992;255:983. doi: 10.1126/science.1546295. [DOI] [PubMed] [Google Scholar]
- 288.Ghosh A, Lima CD. Wiley Interdiscip Rev RNA. 2010;1:152. doi: 10.1002/wrna.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Fabrega C, Shen V, Shuman S, Lima CD. Mol Cell. 2003;11:1549. doi: 10.1016/s1097-2765(03)00187-4. [DOI] [PubMed] [Google Scholar]
- 290.Ghosh A, Shuman S, Lima CD. Mol Cell. 2011;43:299. doi: 10.1016/j.molcel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Pei Y, Hausmann S, Ho CK, Schwer B, Shuman S. J Biol Chem. 2001;276:28075. doi: 10.1074/jbc.M102170200. [DOI] [PubMed] [Google Scholar]
- 292.(a) Andre B, Springael JY. Biochem Biophys Res Commun. 1994;205:1201. doi: 10.1006/bbrc.1994.2793. [DOI] [PubMed] [Google Scholar]; (b) Sudol M, Chen HI, Bougeret C, Einbond A, Bork P. FEBS Lett. 1995;369:67. doi: 10.1016/0014-5793(95)00550-s. [DOI] [PubMed] [Google Scholar]
- 293.(a) Chen HI, Sudol M. Proc Natl Acad Sci U S A. 1995;92:7819. doi: 10.1073/pnas.92.17.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sudol M. Prog Biophys Mol Biol. 1996;65:113. doi: 10.1016/s0079-6107(96)00008-9. [DOI] [PubMed] [Google Scholar]
- 294.Myers JK, Morris DP, Greenleaf AL, Oas TG. Biochemistry. 2001;40:8479. doi: 10.1021/bi0027884. [DOI] [PubMed] [Google Scholar]
- 295.(a) Ranganathan R, Lu KP, Hunter T, Noel JP. Cell. 1997;89:875. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]; (b) Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. Cell. 1997;91:961. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 296.(a) Lu PJ, Zhou XZ, Shen M, Lu KP. Science. 1999;283:1325. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]; (b) Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Nat Struct Biol. 2000;7:639. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 297.(a) Lohi O, Poussu A, Mao Y, Quiocho F, Lehto VP. FEBS Lett. 2002;513:19. doi: 10.1016/s0014-5793(01)03287-2. [DOI] [PubMed] [Google Scholar]; (b) Misra S, Puertollano R, Kato Y, Bonifacino JS, Hurley JH. Nature. 2002;415:933. doi: 10.1038/415933a. [DOI] [PubMed] [Google Scholar]
- 298.Ni Z, Olsen JB, Guo X, Zhong G, Ruan ED, Marcon E, Young P, Guo H, Li J, Moffat J, Emili A, Greenblatt JF. Transcription. 2011;2:237. doi: 10.4161/trns.2.5.17803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.(a) Steinmetz EJ, Brow DA. Mol Cell Biol. 1996;16:6993. doi: 10.1128/mcb.16.12.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Conrad NK, Wilson SM, Steinmetz EJ, Patturajan M, Brow DA, Swanson MS, Corden JL. Genetics. 2000;154:557. doi: 10.1093/genetics/154.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Sadowski M, Dichtl B, Hubner W, Keller W. EMBO J. 2003;22:2167. doi: 10.1093/emboj/cdg200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Lunde BM, Reichow SL, Kim M, Suh H, Leeper TC, Yang F, Mutschler H, Buratowski S, Meinhart A, Varani G. Nat Struct Mol Biol. 2010;17:1195. doi: 10.1038/nsmb.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Gross S, Moore C. Proc Natl Acad Sci U S A. 2001;98:6080. doi: 10.1073/pnas.101046598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.(a) Hollingworth D, Noble CG, Taylor IA, Ramos A. RNA. 2006;12:555. doi: 10.1261/rna.2304506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Z, Fu J, Gilmour DS. Genes Dev. 2005;19:1572. doi: 10.1101/gad.1296305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Fuxreiter M, Tompa P. Adv Exp Med Biol. 2012;725:1. doi: 10.1007/978-1-4614-0659-4_1. [DOI] [PubMed] [Google Scholar]
- 305.Vasiljeva L, Buratowski S. Mol Cell. 2006;21:239. doi: 10.1016/j.molcel.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 306.Steinmetz EJ, Brow DA. Proc Natl Acad Sci U S A. 1998;95:6699. doi: 10.1073/pnas.95.12.6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kubicek K, Cerna H, Holub P, Pasulka J, Hrossova D, Loehr F, Hofr C, Vanacova S, Stefl R. Genes Dev. 2012;26:1891. doi: 10.1101/gad.192781.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Scholes DT, Banerjee M, Bowen B, Curcio MJ. Genetics. 2001;159:1449. doi: 10.1093/genetics/159.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Liu BA, Nash PD. Philos Trans R Soc Lond B Biol Sci. 2012;367:2556. doi: 10.1098/rstb.2012.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.(a) Gerber M, Shilatifard A. J Biol Chem. 2003;278:26303. doi: 10.1074/jbc.R300014200. [DOI] [PubMed] [Google Scholar]; (b) Hampsey M, Reinberg D. Cell. 2003;113:429. doi: 10.1016/s0092-8674(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 311.Li B, Howe L, Anderson S, Yates JR, 3rd, Workman JL. J Biol Chem. 2003;278:8897. doi: 10.1074/jbc.M212134200. [DOI] [PubMed] [Google Scholar]
- 312.Vojnic E, Simon B, Strahl BD, Sattler M, Cramer P. J Biol Chem. 2006;281:13. doi: 10.1074/jbc.C500423200. [DOI] [PubMed] [Google Scholar]
- 313.Gross CA, Chan C, Dombroski A, Gruber T, Sharp M, Tupy J, Young B. Cold Spring Harb Symp Quant Biol. 1998;63:141. doi: 10.1101/sqb.1998.63.141. [DOI] [PubMed] [Google Scholar]
- 314.Bedford MT, Leder P. Trends Biochem Sci. 1999;24:264. doi: 10.1016/s0968-0004(99)01417-6. [DOI] [PubMed] [Google Scholar]
- 315.Allen M, Friedler A, Schon O, Bycroft M. J Mol Biol. 2002;323:411. doi: 10.1016/s0022-2836(02)00968-3. [DOI] [PubMed] [Google Scholar]
- 316.Carty SM, Goldstrohm AC, Sune C, Garcia-Blanco MA, Greenleaf AL. Proc Natl Acad Sci U S A. 2000;97:9015. doi: 10.1073/pnas.160266597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.(a) Sune C, Garcia-Blanco MA. Mol Cell Biol. 1999;19:4719. doi: 10.1128/mcb.19.7.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sune C, Hayashi T, Liu Y, Lane WS, Young RA, Garcia-Blanco MA. Mol Cell Biol. 1997;17:6029. doi: 10.1128/mcb.17.10.6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Liu J, Fan S, Lee CJ, Greenleaf AL, Zhou P. J Biol Chem. 2013 doi: 10.1074/jbc.M113.460238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ, Cech TR. Genes Dev. 2012;26:2690. doi: 10.1101/gad.204602.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.(a) Kovar H. Sarcoma. 2011;2011:837474. doi: 10.1155/2011/837474. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mackenzie IR, Neumann M. Brain Res. 2012;1462:40. doi: 10.1016/j.brainres.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 321.Bertolotti A, Lutz Y, Heard DJ, Chambon P, Tora L. EMBO J. 1996;15:5022. [PMC free article] [PubMed] [Google Scholar]
- 322.(a) Chen D, Toone WM, Mata J, Lyne R, Burns G, Kivinen K, Brazma A, Jones N, Bahler J. Mol Biol Cell. 2003;14:214. doi: 10.1091/mbc.E02-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Mol Biol Cell. 2000;11:4241. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Morimoto RI. Genes Dev. 2008;22:1427. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.(a) Dubois MF, Bensaude O, Morange M. C R Acad Sci III. 1991;313:165. [PubMed] [Google Scholar]; (b) Dubois MF, Bellier S, Seo SJ, Bensaude O. J Cell Physiol. 1994;158:417. doi: 10.1002/jcp.1041580305. [DOI] [PubMed] [Google Scholar]; (c) Venetianer A, Dubois MF, Nguyen VT, Bellier S, Seo SJ, Bensaude O. Eur J Biochem. 1995;233:83. doi: 10.1111/j.1432-1033.1995.083_1.x. [DOI] [PubMed] [Google Scholar]
- 324.Legagneux V, Morange M, Bensaude O. Eur J Biochem. 1990;193:121. doi: 10.1111/j.1432-1033.1990.tb19312.x. [DOI] [PubMed] [Google Scholar]
- 325.Dubois MF, Marshall NF, Nguyen VT, Dahmus GK, Bonnet F, Dahmus ME, Bensaude O. Nucleic Acids Res. 1999;27:1338. doi: 10.1093/nar/27.5.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Patturajan M, Conrad NK, Bregman DB, Corden JL. J Biol Chem. 1999;274:27823. doi: 10.1074/jbc.274.39.27823. [DOI] [PubMed] [Google Scholar]
- 327.(a) Herman PK. Curr Opin Microbiol. 2002;5:602. doi: 10.1016/s1369-5274(02)00377-6. [DOI] [PubMed] [Google Scholar]; (b) DeRisi JL, Iyer VR, Brown PO. Science. 1997;278:680. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 328.(a) Jeong SJ, Kim HJ, Yang YJ, Seol JH, Jung BY, Han JW, Lee HW, Cho EJ. J Microbiol. 2005;43:516. [PubMed] [Google Scholar]; (b) Ostapenko D, Solomon MJ. Eukaryot Cell. 2003;2:274. doi: 10.1128/EC.2.2.274-283.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.(a) Lee D, Lis JT. Nature. 1998;393:389. doi: 10.1038/30770. [DOI] [PubMed] [Google Scholar]; (b) McNeil JB, Agah H, Bentley D. Genes Dev. 1998;12:2510. doi: 10.1101/gad.12.16.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kouzine F, Wojtowicz D, Yamane A, Resch W, Kieffer-Kwon KR, Bandle R, Nelson S, Nakahashi H, Awasthi P, Feigenbaum L, Menoni H, Hoeijmakers J, Vermeulen W, Ge H, Przytycka TM, Levens D, Casellas R. Cell. 2013;153:988. doi: 10.1016/j.cell.2013.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.(a) Latham KE, Solter D, Schultz RM. Dev Biol. 1992;149:457. doi: 10.1016/0012-1606(92)90300-6. [DOI] [PubMed] [Google Scholar]; (b) Roeder RG. J Biol Chem. 1974;249:249. [PubMed] [Google Scholar]; (c) Toyoda T, Wolffe AP. Dev Biol. 1992;153:150. doi: 10.1016/0012-1606(92)90099-3. [DOI] [PubMed] [Google Scholar]
- 332.(a) Bellier S, Dubois MF, Nishida E, Almouzni G, Bensaude O. Mol Cell Biol. 1997;17:1434. doi: 10.1128/mcb.17.3.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Doyle O, Corden JL, Murphy C, Gall JG. J Struct Biol. 2002;140:154. doi: 10.1016/s1047-8477(02)00547-6. [DOI] [PubMed] [Google Scholar]
- 333.Bellier S, Chastant S, Adenot P, Vincent M, Renard JP, Bensaude O. EMBO J. 1997;16:6250. doi: 10.1093/emboj/16.20.6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Nakamura A, Seydoux G. Development. 2008;135:3817. doi: 10.1242/dev.022434. [DOI] [PubMed] [Google Scholar]
- 335.Seydoux G, Dunn MA. Development. 1997;124:2191. doi: 10.1242/dev.124.11.2191. [DOI] [PubMed] [Google Scholar]
- 336.Batchelder C, Dunn MA, Choy B, Suh Y, Cassie C, Shim EY, Shin TH, Mello C, Seydoux G, Blackwell TK. Genes Dev. 1999;13:202. doi: 10.1101/gad.13.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Zhang F, Barboric M, Blackwell TK, Peterlin BM. Genes Dev. 2003;17:748. doi: 10.1101/gad.1068203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Ghosh D, Seydoux G. Genetics. 2008;178:235. doi: 10.1534/genetics.107.083212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Bowman EA, Bowman CR, Ahn JH, Kelly WG. Development. 2013 doi: 10.1242/dev.095778. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.(a) Hanyu-Nakamura K, Sonobe-Nojima H, Tanigawa A, Lasko P, Nakamura A. Nature. 2008;451:730. doi: 10.1038/nature06498. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deshpande G, Calhoun G, Schedl P. Development. 2004;131:1247. doi: 10.1242/dev.01004. [DOI] [PubMed] [Google Scholar]; (c) Martinho RG, Kunwar PS, Casanova J, Lehmann R. Curr Biol. 2004;14:159. doi: 10.1016/j.cub.2003.12.036. [DOI] [PubMed] [Google Scholar]
- 341.Tam PP, Zhou SX. Dev Biol. 1996;178:124. doi: 10.1006/dbio.1996.0203. [DOI] [PubMed] [Google Scholar]
- 342.Seki Y, Yamaji M, Yabuta Y, Sano M, Shigeta M, Matsui Y, Saga Y, Tachibana M, Shinkai Y, Saitou M. Development. 2007;134:2627. doi: 10.1242/dev.005611. [DOI] [PubMed] [Google Scholar]
- 343.(a) Jackson DA, Hassan AB, Errington RJ, Cook PR. EMBO J. 1993;12:1059. doi: 10.1002/j.1460-2075.1993.tb05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chakalova L, Fraser P. Cold Spring Harb Perspect Biol. 2010;2:a000729. doi: 10.1101/cshperspect.a000729. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cook PR. J Mol Biol. 2010;395:1. doi: 10.1016/j.jmb.2009.10.031. [DOI] [PubMed] [Google Scholar]; (d) Melnik S, Deng B, Papantonis A, Baboo S, Carr IM, Cook PR. Nat Methods. 2011;8:963. doi: 10.1038/nmeth.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rieder D, Trajanoski Z, McNally JG. Front Genet. 2012;3:221. doi: 10.3389/fgene.2012.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Jackson DA, Iborra FJ, Manders EM, Cook PR. Mol Biol Cell. 1998;9:1523. doi: 10.1091/mbc.9.6.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.(a) Jackson DA, Cook PR. EMBO J. 1985;4:919. doi: 10.1002/j.1460-2075.1985.tb03719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kimura H, Tao Y, Roeder RG, Cook PR. Mol Cell Biol. 1999;19:5383. doi: 10.1128/mcb.19.8.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Ferrai C, Xie SQ, Luraghi P, Munari D, Ramirez F, Branco MR, Pombo A, Crippa MP. PLoS Biol. 2010;8:e1000270. doi: 10.1371/journal.pbio.1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Ghamari A, van de Corput MP, Thongjuea S, van Cappellen WA, van Ijcken W, van Haren J, Soler E, Eick D, Lenhard B, Grosveld FG. Genes Dev. 2013;27:767. doi: 10.1101/gad.216200.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.(a) Mintz PJ, Patterson SD, Neuwald AF, Spahr CS, Spector DL. EMBO J. 1999;18:4308. doi: 10.1093/emboj/18.15.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xie SQ, Martin S, Guillot PV, Bentley DL, Pombo A. Mol Biol Cell. 2006;17:1723. doi: 10.1091/mbc.E05-08-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Xie SQ, Pombo A. Histochem Cell Biol. 2006;125:21. doi: 10.1007/s00418-005-0064-2. [DOI] [PubMed] [Google Scholar]
- 350.Spector DL, Lamond AI. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Gall JG. Nat Rev Mol Cell Biol. 2003;4:975. doi: 10.1038/nrm1262. [DOI] [PubMed] [Google Scholar]
- 352.(a) Gall JG. FEBS Lett. 2001;498:164. doi: 10.1016/s0014-5793(01)02461-9. [DOI] [PubMed] [Google Scholar]; (b) Kiss T. J Cell Sci. 2004;117:5949. doi: 10.1242/jcs.01487. [DOI] [PubMed] [Google Scholar]
- 353.Cho EJ, Rodriguez CR, Takagi T, Buratowski S. Genes Dev. 1998;12:3482. doi: 10.1101/gad.12.22.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Pontius BW. Trends Biochem Sci. 1993;18:181. doi: 10.1016/0968-0004(93)90111-y. [DOI] [PubMed] [Google Scholar]