

Abstract
Cochlear inner hair cells (IHCs) develop from pre-sensory pacemaker to sound transducer. Here, we report that this involves changes in structure and function of the ribbon synapses between IHCs and spiral ganglion neurons (SGNs) around hearing onset in mice. As synapses matured they changed from holding several small presynaptic active zones (AZs) and apposed postsynaptic densities (PSDs) to one large AZ/PSD complex per SGN bouton. After the onset of hearing (i) IHCs had fewer and larger ribbons; (ii) CaV1.3 channels formed stripe-like clusters rather than the smaller and round clusters at immature AZs; (iii) extrasynaptic CaV1.3-channels were selectively reduced, (iv) the intrinsic Ca2+ dependence of fast exocytosis probed by Ca2+ uncaging remained unchanged but (v) the apparent Ca2+ dependence of exocytosis linearized, when assessed by progressive dihydropyridine block of Ca2+ influx. Biophysical modeling of exocytosis at mature and immature AZ topographies suggests that Ca2+ influx through an individual channel dominates the [Ca2+] driving exocytosis at each mature release site. We conclude that IHC synapses undergo major developmental refinements, resulting in tighter spatial coupling between Ca2+ influx and exocytosis.
Keywords: active zone, Ca2+ channel, Ca2+ dependence, exocytosis, vesicle
Introduction
Inner hair cells (IHC) ribbon synapses are molecularly and morphologically specialized to transmit acoustic information at hundreds of Hz with sub-millisecond precision over long periods of time (recently reviewed in Matthews & Fuchs, 2010; Rutherford & Pangršič, 2012; Pangršič et al, 2012; Safieddine et al, 2012). The IHC receptor potential is coupled to glutamate exocytosis by Ca2+ influx through CaV1.3 Ca2+ channels (Platzer et al, 2000; Brandt et al, 2003; Dou et al, 2004). Tens of these channels are thought to cluster within the presynaptic density (Brandt et al, 2005; Frank et al, 2010; Zampini et al, 2013). One to two dozen synaptic vesicles are tethered at the plasma membrane within nanometer distance of the Ca2+ channel cluster and are thought to represent the putative readily releasable pool (RRP) of vesicles for exocytosis (Moser & Beutner, 2000; Zenisek et al, 2000; Lenzi et al, 2002; Rutherford & Roberts, 2006; Frank et al, 2010). Precisely how Ca2+ channels couple to exocytosis and which mechanism of exocytosis governs transmitter release is not well understood.
Exocytosis at individual IHC AZs has been suggested to be multivesicular (Glowatzki & Fuchs, 2002), whereby Ca2+ is thought to regulate the rate of release events without affecting the number of vesicles released per event. Ca2+-nanodomain coupling, in which exocytosis is controlled by very few nearby Ca2+ channels (Augustine et al, 1991; Stanley, 1993; Mintz et al, 1995; Wang et al, 2008; reviewed in Augustine et al, 2003; Moser et al, 2006; Neher & Sakaba, 2008; Eggermann et al, 2012), has been put forward to explain the physiological coupling of Ca2+ influx to RRP exocytosis in mature IHCs (Brandt et al, 2005; Goutman & Glowatzki, 2007; Zampini et al, 2013). A Ca2+ nanodomain may synchronize multivesicular release (Jarsky et al, 2010; Graydon et al, 2011). Alternatively, Ca2+ microdomain coupling (Heil & Neubauer, 2010), in which exocytosis is controlled by many channels cooperatively, and a linear intrinsic Ca2+ dependence of fusion (Johnson et al, 2010) have been suggested to explain the coupling of Ca2+ influx to RRP exocytosis in mature IHCs and these concepts, too, may coexist with multivesicular release.
Before the onset of hearing, exocytosis is evoked by Ca2+ action potentials (Beutner & Moser, 2001; Glowatzki & Fuchs, 2002; Johnson et al, 2005) with low “Ca2+ efficiency” (Beutner & Moser, 2001; Johnson et al, 2005). Non-mutually exclusive hypotheses for the increase in Ca2+ efficiency of exocytosis over development include (i) progressive confinement of Ca2+ influx to the AZ, (ii) establishment of nanodomain coupling between CaV1.3 channels and release sites, and (iii) a change in intrinsic Ca2+ dependence of the molecular exocytosis machinery. Upregulation of synaptotagmin IV during postnatal development has been proposed to “linearize” the intrinsic Ca2+ dependence of IHC exocytosis around the onset of hearing (Johnson et al, 2010), supporting hypothesis iii. However, this contrasts with Ca2+ uncaging experiments that found a supralinear intrinsic Ca2+ dependence of exocytosis in mature IHCs (Beutner et al, 2001). Moreover, in immature IHCs, a thorough biophysical analysis of the intrinsic Ca2+ dependence of exocytosis or of the spatial coupling between Ca2+ influx and exocytosis was lacking until now.
Here, we combined morphological approaches: transmission electron microscopy (TEM), immunohistochemistry, confocal and stimulated emission depletion (STED) microscopy; with physiological techniques: electrophysiology, confocal Ca2+ imaging, Ca2+ uncaging; and biophysical modeling of AZ function to study the IHC afferent synapse during postnatal maturation. We find the intrinsic Ca2+ dependence of fast exocytosis to be similar in immature and mature IHCs, arguing against hypothesis iii. On the other hand, we find a diminution of extrasynaptic Ca2+ channels (supporting hypothesis i) and indicate a developmental switch from Ca2+microdomain to Ca2+-nanodomain control of vesicle fusion around hearing onset (supporting hypothesis ii).
Results
Postnatal maturation of AZ ultrastructure in IHC afferent synapses
Afferent synaptic contacts between IHCs and type I SGNs are present in mice already at birth (Shnerson et al, 1981) and increase in number during the first postnatal week (Shnerson et al, 1981; Sobkowicz et al, 1982; recently reviewed in Bulankina & Moser, 2012). Around the onset of hearing (p12, Mikaelian et al, 1965), synaptic contacts mature (Sobkowicz et al, 1982) and the number of presynaptic ribbons and postsynaptic AMPA receptor clusters decreases (Huang et al, 2007, 2012; Sendin et al, 2007). By p21, IHC-afferent synapses are considered predominantly mature morphologically (Bulankina & Moser, 2012; Safieddine et al, 2012) and functionally (Khimich et al, 2005; Grant et al, 2010; Wong et al, 2013). We first investigated the molecular and structural changes within individual IHC-SGN contacts during the 2-week period around hearing onset.
We triple-labeled afferent IHC synapses in whole-mounts of the organ of Corti for (i) RIBEYE/CtBP2 (the major component of the synaptic ribbon (Schmitz et al, 2000; Khimich et al, 2005)), (ii) GluA2/3 (AMPA receptor subunits of the SGN PSD (Matsubara et al, 1996; Khimich et al, 2005)), and (iiia) bassoon (AZ scaffold contributing to ribbon anchorage (tom Dieck et al, 1998; Khimich et al, 2005)), or (iiib) the Na+/K+-ATPase α3 subunit marking the SGN membrane (McLean et al, 2009). Immature synapses displayed several small appositions of AZs and PSDs (p6, Fig 1A). These AZ/PSD complexes often appeared to form circular structures >1 μm in diameter (Fig 1B), larger than the ring-like GluA clusters of mature synapses (p20, Fig 1C; up to approximately 700 nm, Meyer et al, 2009). Na+/K+-ATPase labeling of SGN indicated that these circular structures localized to the periphery of the contact between each individual immature postsynaptic bouton and an IHC (Fig 1C,D, p6). Some of the AZ/PSD complexes lacked detectable RIBEYE immunofluorescence and likely represented ribbonless AZs (quantified in Supplementary Fig S2). After the onset of hearing each IHC-SGN synapse displayed a single AZ/PSD complex (p20, Fig 1).
Figure 1.
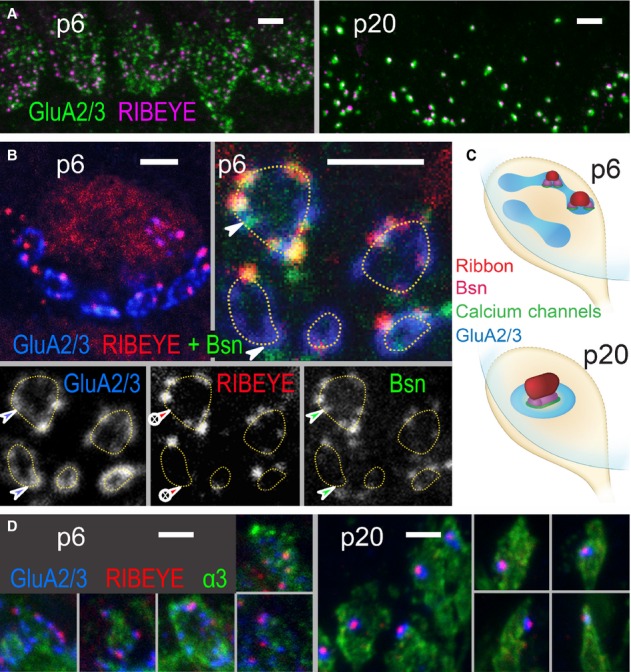
- Projection of confocal images showing GluA2/3 (green), RIBEYE (magenta) across the basal region of a row of IHCs at p6 (left) and p20 (right). Scale bar, 2 μm.
- Single confocal sections. Upper left: At p6, the basal pole of each IHC was contacted by several patches of postsynaptic glutamate receptors (GluA2/3, blue) forming circular structures (highlighted by dashed lines in upper right and lower left). Upper right: Each GluA2/3 spot was juxtaposed to an AZ spot, marked by protein bassoon (green) and in some cases by a ribbon (RIBEYE, red). Two ribbonless AZs marked by colocalizing bassoon and GluA2/3 puncta were highlighted by arrowheads. Individual fluorescence channels are presented separately in gray scale below. Scale bars, 2 μm.
- Schematized distribution of ribbon, bassoon, Ca2+ channels and GluA2/3-containing receptors on immature (p6, upper) and mature (p20, lower) IHC-SGN contacts. Immature IHC-SGN contacts hold multiple AZ/PSD complexes. Mature IHC-SGN contacts show a single and larger AZ/PSD complex.
- Projection of confocal images showing GluA2/3 (blue), RIBEYE (red), and Na+/K+-ATPase α3 subunit (green) at p6 (left) and p20 (right). The latter antibody labels the membranes of SGNs, each of which terminates onto the IHC with a bouton-type contact. In p6 preparations, the large circular structures formed by GluA spots partially encompass the perimeter of the bouton contacts. At p20, each postsynaptic bouton contains only a single condensed glutamate receptor cluster that is juxtaposed to a single presynaptic ribbon. Scale bars, 2 μm.
TEM confirmed the presence of extended immature synaptic contacts that included ribbon-occupied and ribbonless AZ/PSD appositions (Fig 2A, p6 & p9). An exemplary synapse reconstructed from serial ultrathin sections (Fig 2D, p6) illustrates three ribbon-occupied AZs and one ribbonless AZ (black arrowhead). Compared with more mature ribbons (Fig 2B, p15 & p20), immature ribbons assumed more spherical or ovular shapes (Fig 2C), were smaller (Fig 2E), associated with fewer vesicles (Fig 2E and tomogram in Supplementary Fig S1), and were juxtaposed to shorter PSDs (Fig 2E). After hearing onset, droplet-or wedge-shaped ribbons became prevalent (Fig 2C). Different ribbon shapes were observed in the same IHCs but we did not analyze their subcellular distribution. A typical p20 ribbon spanned four 70-nm-thin sections, compared to 1.5 sections on average for p6 ribbons. Floating ribbons tethering vesicles were observed near membrane-anchored ribbons at p6 (Fig 2A, green arrowhead). All preparations for TEM were fixed and processed at around noon time, minimizing possible effects of circadian changes (Hull et al, 2006).
Figure 2.

Maturation of ribbon synapse ultrastructure.
A, B Representative electron micrographs of IHC ribbon synapses in pre-hearing (A) and hearing (B) mice. Before the onset of hearing (A), at p6 and p9, multiple appositions of small discontinuous pre-and postsynaptic densities were found (magenta arrowheads) typically accompanied by one or more round-shaped ribbon(s)anchored close to the presynaptic membrane via two rootlets. In some cases ribbons were “floating” in the cytoplasm at a greater distance from the membrane (green arrowhead). After the onset of hearing (B), at p15 and p20, typically one ribbon occupied the presynaptic cytoplasm at the AZ. In comparison to before the onset of hearing, the mature pre-and postsynaptic densities were relatively extended and continuous. The sections were cut parallel to the ribbons' short axes, the perspective from which ribbon-shape was measured. Scale bar of 200 nm in (A) applies also to (B).
C Through development the ribbon-shape changed from predominantly round to oval, droplet-like, or wedge-like shapes, as shown in (A) and (B).
D 3D reconstructions from serial sections before (left, p6) and after (right, p20) the onset of hearing. Presynaptic densities connect the ribbon to the plasma membrane.
E Quantification of synaptic parameters from random EM sections. Number of ribbon-associated vesicles, size of synaptic ribbon as well as the extent of postsynaptic density (PSD) increase with age (student's t-test). Only sections parallel to the ribbons' short axes with sharply delineated PSD were analyzed. Ribbon size was measured as the length of the longest extent across the synaptic ribbon.
F Increase of ribbon size/content during development was corroborated by the increase in integrated puncta intensity from quantification of RIBEYE immunofluorescence. A developmental increase in full-width-of-half-maximum (FWHM) of Gaussian fits to puncta was also observed, Wilcoxon rank test.
G Decrease of number of synaptic ribbon during development estimated by immunohistochemistry (immuno) and live imaging (peptide), student's t-test.
Data information: *P < 0.05; **P < 0.01; ***P < 0.001. n.s., no significant difference.
Comparisons of confocal RIBEYE immunofluorescence between p6 and p20 revealed an approximate halving of ribbon number per IHC and an approximate doubling of the integrated pixel intensities per ribbon (Fig 2F,G and Supplementary Fig S2). An increase in full-width-at-half-maximum (FWHM, Fig 2F) of these RIBEYE spots is consistent with the increase in ribbon size observed in TEM. For GluA2/3, both the reduction in puncta number and the increase in integrated intensity per punctum were larger than for ribbons (Supplementary Fig S2C,E). We note that confocal measurements provide only a semi-quantitative comparison because immunolabeling may not be linear or equally efficient amongst puncta and because some ribbons or GluA2/3 puncta can be smaller than the resolution limit of confocal microscopy, especially in immature IHCs. Nevertheless, the confocal images indicate an increase in the ratio of ribbon and GluA2/3 puncta number from approximately 0.6 at p6 to nearly 1 for p20 IHCs (Supplementary Fig S2D), consistent with our observations of ribbonless AZs in immature IHCs and with the idea of a 1:1 correspondence between ribbons and receptor clusters per SGN bouton upon maturation (shown in Fig 1C,D, p20). Immature ribbons were anchored to the AZ membrane via up to two rootlets (presynaptic densities, Figs 2A and 3A,B). In contrast, a single continuous density attached the entire base of the mature ribbon to the AZ (Fig 3C,D). Using anti-bassoon immuno-EM (Fig 3F) we indicate that bassoon localizes to the presynaptic density underneath the ribbon, where the majority of immuno-gold was found (on average 1.4 ± 0.3 particles at the base and 0.3 ± 0.1 elsewhere at the ribbon, P < 0.001, n = 21 labeled synapses). This is consistent with a previous immuno-EM study on photoreceptor ribbon synapses (Dick et al, 2001) and also supported by present STED microscopy of hair cell synapses (see Fig 3G and below). The notion of ribbon-anchor consolidation was supported by the observation of fewer electron-dense connections between membrane and ribbon in random sections of ribbon-occupied AZs from p6 to p20 (Supplementary Fig S3).
Figure 3.

- TEM of an immature IHC ribbon synapse using high-pressure freezing and freeze substitution (HPF/FS).
- Corresponding serial 3D reconstruction of (A), showing the immature ribbon connected via two rootlets to the presynaptic membrane. For color code see (D)
- Representative TEM of a mature ribbon, connected with one continuous presynaptic density to the membrane, as demonstrated in the 3D reconstruction in (D).
- 3D reconstruction: ribbon: red; AZ membrane: blue; rootlet (immature)/presynaptic density (mature): magenta; membrane-proximal SVs: orange; ribbon-associated SVs: yellow.
- Schematics of immature (top) and mature (bottom) AZs inspired by exemplary 3D reconstructions and STED imaging of CaV1.3 immunofluorescence. Ca2+ channels formed small clusters and vesicles were less organized at the immature AZ. Stripe-like Ca2+-channel cluster with vesicles positioned regularly along each side at the mature AZ.
- Pre-embedding immunogold labeling for bassoon (arrowhead) was detected at the ribbon-anchoring presynaptic density of mature ribbons. Two primary antibodies specific to the N-terminal or C-terminal of bassoon, respectively, were used (see Materials and Methods). Scale bar, 200 nm.
- STED image of immunolabeled bassoon (magenta) and CaV1.3 channel clusters (green) in mature IHCs: stripe-like morphology and closely aligned immunofluorescence of bassoon and CaV1.3 reminiscent of the presynaptic density reconstructed from TEM (D). The image is a maximum intensity projection after smoothing with a Gaussian (σ = 1 pixel) in ImageJ. The color lookup was chosen to optimize contrast and visibility of dimmer structures. Pixel size, 20 nm. Scale bar, 2 μm. Insets, 700 × 700 nm.
Next, we studied the topography of bassoon and CaV1.3 at the mature AZ, employing dual-color STED microscopy of immunolabeled IHCs in whole-mounts. Bassoon and CaV1.3 immunofluorescence often formed elongated stripes that aligned intimately with each other (Fig 3G). Most p19 synapses had one Ca2+-channel stripe, some had more than one stripe in parallel, and others had round or more complex configurations (Fig 4B). Before the onset of hearing we observed only spot-like Ca2+-channel clusters (Fig 4A). These spots are reminiscent of the small presynaptic densities seen in immature AZs (Fig 2A), suggesting that Ca2+ channels cluster within the presynaptic density of immature IHCs, as they do in the stripe-like densities of mature IHCs (Frank et al, 2010). As depicted in Fig 3E, TEM analysis of membrane-proximal vesicles and presynaptic densities, and STED imaging of CaV1.3 channels suggest that synapse maturation leads to a more ordered topology of Ca2+ channels and membrane-proximal vesicles. In summary IHC-SGN synapses underwent major structural changes around the onset of hearing characterized by larger ribbons, continuously anchored to a single elongated presynaptic density, juxtaposed to a single postsynaptic receptor cluster.
Figure 4.
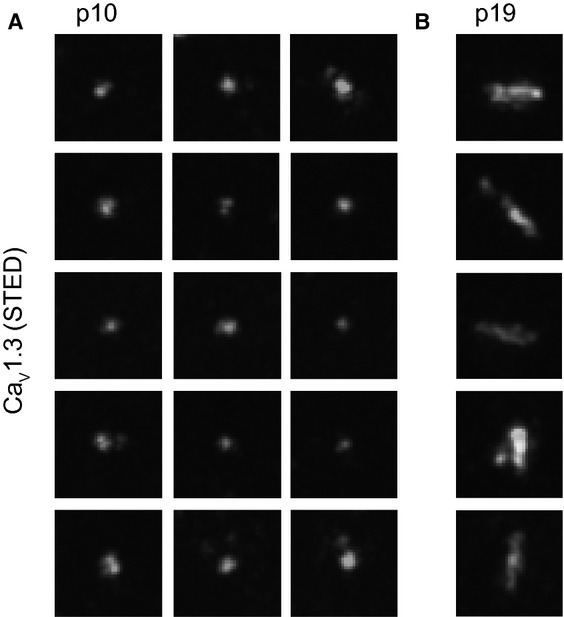
- Projections of STED sections of immunolabeled CaV1.3 clusters in the organ of Corti whole-mount at p10 (0.6 μm step size, total z covered: 4 μm). CaV1.3 clusters in pre-hearing IHCs appeared spot-like (A), in contrast to the stripe-like appearance in mature IHCs (B). Images were processed as in Fig 3G. Pixel size, 20 nm, 700 × 700 nm regions-of-interest centered at individual CaV1.3 clusters.
- Projections of STED sections of immunolabeled CaV1.3 clusters in the organ of Corti whole-mount at p19 (0.6 μm step size, total z covered: 4 μm). CaV1.3 clusters in pre-hearing IHCs appeared spot-like (A), in contrast to the stripe-like appearance in mature IHCs (B). Images were processed as in Fig 3G. Pixel size, 20 nm, 700 × 700 nm regions-of interest centered at individual CaV1.3 clusters.
Progressive confinement of CaV1.3 and Ca2+ influx to AZs over development
CaV1.3 channels conduct >90% of the voltage-gated Ca2+ influx in IHCs before and after the onset of hearing (Platzer et al, 2000; Brandt et al, 2003). Over development, we tracked changes in abundance and location of CaV1.3 with immunofluorescence and confocal Ca2+ imaging (Fig 5), in experiments run in parallel under identical conditions for the different age groups. Using CaV1.3-knockout mice as a negative control we observed non-specific immunoreactivity only near the cuticular plate at the apical pole of p9 IHCs (Supplementary Fig S4), as previously shown for IHCs of hearing mice (Brandt et al, 2005).
Figure 5.
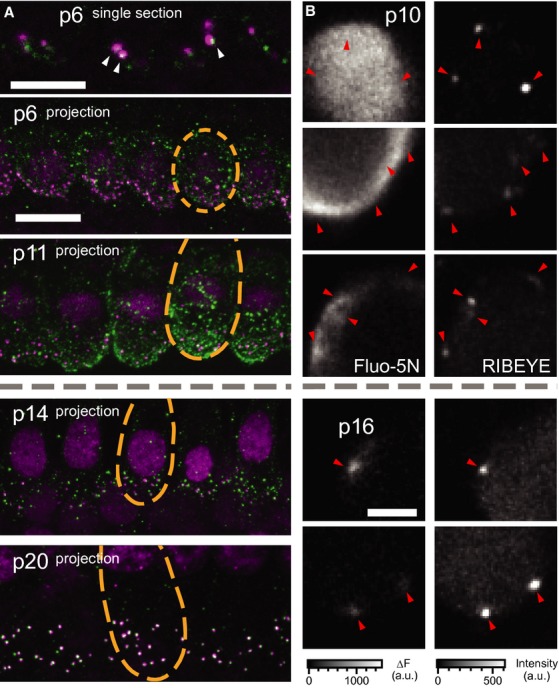
- Organs of Corti immunolabeled for CaV1.3 (green) and RIBEYE/CtBP2 (magenta). At p6 (top), a single section shows both ribbon-occupied (arrowheads) and ribbonless CaV1.3 spots as well as diffuse membrane staining. At p11 (middle, projection) puncta of CaV1.3 were observed throughout the basolateral membrane, especially the basal pole. By p14 the clusters of CaV1.3 were concentrated in the basal poles near synaptic ribbons, while the entire inner hair cell membrane remained diffusely labeled for CaV1.3. At p20 there was very little extrasynaptic labeling and the cell membrane was no longer diffusely immunoreactive for CaV1.3. Each synaptic ribbon was accompanied by a cluster of CaV1.3 and only few CaV1.3 clusters remained ribbonless. Scale bars, 10 μm.
- Examples of XY scans across the basolateral portion of IHCs before (upper, p10) and after (lower, p14) the onset of hearing. Left and right panels show the increase in Fluo-5N fluorescence during a step depolarization to −7 mV for 254 ms (2 mM [EGTA]i) and peptide-labeled synaptic ribbons (position marked with arrowheads). Note the relatively widespread submembraneous Ca2+ signal in immature IHCs as compared to the local Ca2+ rise in mature IHCs. Still, the highest [Ca2+] was usually observed at the ribbons-occupied AZs. Scale bar, 3 μm.
CaV1.3 immunofluorescence near ribbons was detectable already at p6 (arrowheads in Fig 5A) and increased until p14 (mean intensity around ribbons at p6: 6494 ± 366 a.u. versus p14: 7721 ± 265 a.u., P = 0.007). Thereafter, we did not find significant changes of synaptic CaV1.3 immunofluorescent spots except for a subtle increase in the long axis (p14: 0.336 ± 0.004 versus p20: 0.350 ± 0.003 μm, P = 0.005). Consistent with a previous report (Zampini et al, 2010) we also noted CaV1.3 immunofluorescence away from ribbons, appearing spot-like or diffuse along the entire basolateral plasma membrane of immature IHCs. Upon the onset of hearing, CaV1.3 immunoreactivity became largely confined to ribbon-occupied AZs (Fig 5A, lower panels). We quantified the density of CaV1.3 immunofluorescence immediately around ribbons (synaptic) versus distant from ribbons (extrasynaptic, see Materials and Methods). At p6, the density of extrasynaptic fluorescence was around 43% of that in synaptic regions, while by p20, it had decreased to only 9%.
Next, we studied changes in intracellular Ca2+ concentration ([Ca2+]i) elicited by step depolarizations (to –7 mV for 200 or 254 ms) in IHCs using fast confocal Ca2+ imaging before and after the onset of hearing (Fig 5B). To preferentially visualize sites of Ca2+ influx, we used the low-affinity Ca2+ indicator Fluo-5N (0.4 mM) in conjunction with the slow Ca2+ chelator EGTA (2 mM; Frank et al, 2009), and a fluorescently tagged RIBEYE-binding peptide (Francis et al, 2011) for identification of ribbon-occupied AZs. Different from the synapse-confined Ca2+ signals of mature IHCs (Fig 5B, p16), in immature IHCs we found a spatially extended rise of [Ca2+]i (Fig 5B, p10) consistent with substantial extrasynaptic Ca2+ influx and with immunohistochemistry (Fig 5A), and also compatible with potential Ca2+-induced Ca2+ release (Kennedy & Meech, 2002). Occasionally (two out of seven IHCs) we found wide-spread “global” Ca2+ signals (two uppermost rows in Fig 5B, and Supplementary Fig S5). In the other five immature IHCs the peak Ca2+ signal was “spot-like” around ribbons (third row in Fig 5B, and Supplementary Fig S5), further suggesting that CaV1.3 clusters at AZs already before the onset of hearing. We approximated the strength of the extrasynaptic Ca2+ signal as the ratio of ΔF away from the ribbon over ΔF at the ribbon, which was greatest in immature IHCs with “global” signals, smaller in immature IHCs with “spot-like” signals, and even smaller in IHCs after the onset of hearing (Supplementary Fig S5).
The intense, broadly distributed CaV1.3 immunofluorescence in immature IHCs suggests that the reduction of the whole-cell Ca2+ current around the onset of hearing reflects a reduction in the number of Ca2+ channels, primarily in the extrasynaptic membrane. We studied the IHC Ca2+ current before and after the onset of hearing using whole-cell patch-clamp recordings. We did not detect significant differences in the voltage dependence of activation (Supplementary Fig S6A,B) or in the extent or kinetics of inactivation (Supplementary Fig S6C) between p10 and p14 IHCs. Non-stationary fluctuation analysis of Ca2+-tail currents (Roberts et al, 1990; Frank et al, 2010) indicated that the smaller number of Ca2+ channels almost completely accounted for the reduction of the whole-cell current (P < 0.001, ANOVA, Supplementary Fig S6D,E). The single channel current (iCa) estimate did not change significantly (P = 0.1, ANOVA, Supplementary Fig S6F) and the estimated maximal open probability (Popen,max) decreased slightly (P < 0.001, ANOVA; Supplementary Fig S6F), whereas single channel recordings indicated a reduction in iCa and an increase in Popen over development (Zampini et al, 2010, 2013). While both approaches agree on the notion of much higher total Ca2+ channel numbers in immature cells, they differ in their estimates of iCa and Popen and also report opposite trends over development likely for technical reasons (see text accompanying Supplementary Fig S6 for discussion). In summary, immature IHCs contain thousands of synaptic and extrasynaptic CaV1.3 channels that together support presensory Ca2+ action potentials. Upon maturation extrasynaptic CaV1.3 Ca2+ channels are downregulated and Ca2+ influx becomes confined to AZs, supporting hypothesis 1 in introduction.
Maturation of the coupling between Ca2+ influx and exocytosis
The developmental confinement of Ca2+ influx to AZs together with the changes in ultrastructure and molecular nanoanatomy of the AZ prompted us to test for parallel changes in the coupling of Ca2+ influx to exocytosis. In particular, we tested hypothesis ii: tighter functional coupling between Ca2+ channels and the RRP, and hypothesis iii: a change in the intrinsic (biochemical) Ca2+ dependence of the molecular exocytosis machinery.
No change in the intrinsic Ca2+ dependence of fast exocytosis
First, we addressed the intrinsic Ca2+ dependence of IHC exocytosis before the onset of hearing, which had not been done previously. UV-laser photolysis of caged Ca2+ was used to elevate [Ca2+] homogenously throughout the cytosol in p6–8 IHCs and in p14–18 IHCs for comparison. [Ca2+] was monitored with ratiometric imaging and related to the resulting exocytic increase of membrane capacitance (ΔCm). Approximately half (22 out of 40) of p6–8 IHCs and nearly all (32 out of 33) of p14–18 IHCs responded with a Cm rise that comprised two kinetic components with time constants of a few milliseconds and tens of millisecond, respectively (Fig 6A,C). The total amplitude of the Cm rise was largely independent of [Ca2+] at both developmental stages (Fig 6D), as previously reported for p14–25 IHCs (Beutner et al, 2001). However, it was approximately 3.6 times smaller in p6–8 IHCs than in p14–18 IHCs (324 ± 24 fF; n = 40 versus 1161 ± 104 fF; n = 31; P = 1.5 × 10−13, Wilcoxon Rank Test), potentially relating to the lower number of synaptic vesicles found at the AZs of p6–7 IHCs (Fig 2E). Moreover, when both kinetic components were present in p6–8 IHCs we found a greater contribution of the slow one (69%), while the two components contributed equally in p14–18 IHCs (Supplementary Fig S7). In the other p6–8 IHCs the Cm rise was best approximated by a single exponential function with relatively slow time constants within the range of those of the slow component of the Cm rise in IHCs with bi-exponential responses (Fig 6C). Focusing on the fast component of exocytosis, we probed the intrinsic Ca2+ dependence of exocytosis by measuring the delay and rate constant of the Cm rise for a range of [Ca2+]i (Fig 6C,E). We found a similar supralinear intrinsic Ca2+ dependence in p6–8 and p14–18 IHCs, indicating that the Ca2+-binding properties of the molecule(s) mediating fast exocytosis do not change upon the onset of hearing. Because of the steep Ca2+ dependence we applied statistical comparison among p6–8 and p14–17 IHCs for the rate constants of the fast component and the exocytic delays within a narrow [Ca2+] range (15–25 μM, for which we found the best comparable representation of [Ca2+] changes). Neither the rate constants (P = 0.7, Student's t-test) nor the delays (P = 0.86, Student's t-test) were significantly different. The rate constants were larger and the delays shorter for the fast component in the present study (Fig 6C,E) than those observed previously at comparable [Ca2+], which we attribute to the use of shorter stimuli (100 μs laser stimulation versus 1 ms long arc lamp flashes) that achieved a faster rise of [Ca2+].
Figure 6.

- Representative examples of UV-laser evoked capacitance rise (ΔCm) of IHCs from mice of the specified ages. Upper: Intracellular [Ca2+] of a p14 (black), a p7 (magenta) and a p6 (blue) IHCs after UV-flash, measured ratiometrically with mag-fura-2. Lower: ΔCm of the corresponding IHCs (solid lines). Lighter dashed lines represent bi-exponential fits to the data and lighter solid and dotted lines represent the fast and slow components respectively.
- The average (±SEM) ΔCm of p14–18 (black) and p6–8 IHCs (magenta: all cells; grey: only cells with bi-exponential ΔCm).
- Rate constants from exponential fits to the ΔCm upon flash photolysis plotted against post-flash [Ca2+]i. Circles mark the rate constants for first (fast) component and rhombi mark those for the second (slow) component of the bi-exponential fits. Rate constants of responses in p6–8 IHCs, which were best fitted with a single exponential are also marked with rhombi. Note the overlap between pre-hearing (p6–8) and hearing (p14+) age groups. Black (p14–18) and magenta (p6–8) symbols are newly acquired data, while open circles represent previously published data on mature IHCs (Beutner et al, 2001; Pangršič et al, 2010).
- Total response amplitudes (sum of first and second component, in case of double exponential fits) were largely independent of post-flash [Ca2+]i.
- Delay of responses plotted against post-flash [Ca2+]i.
Data information: Solid lines in (C) and (E) are model fits from a previous publication (Beutner et al, 2001).
Linearization of the apparent Ca2+ dependence of exocytosis is limited to changes in the number of open Ca2+ channels
Next, we addressed the apparent Ca2+ dependence of exocytosis (Matveev et al, 2011) by studying the coupling between Ca2+ influx and RRP exocytosis in p6, p9–10, and p14–17 IHCs. We used strong (−17 mV) and brief (20 ms) depolarizations to evoke exocytosis of the RRP (Moser & Beutner, 2000; Beutner & Moser, 2001) in perforated-patch recordings. Following the framework of our previous analysis (Brandt et al, 2005) we studied the relationship between RRP exocytosis (ΔCm,20 ms) and the integrated Ca2+ influx (QCa) while either varying the single Ca2+ channel current (iCa) or changing the number of open CaV1.3 channels (NCa × Popen). The apparent Ca2+ cooperativity m was obtained by fitting the exocytosis-QCa relationship for each cell with a power function: ΔCm = A(QCa)m (Augustine et al, 1991), limited to the QCa range in which no obvious saturation of the exocytosis was observed. For Ca2+-microdomain control (see Introduction), m should be similar to the intrinsic Ca2+ cooperativity (4–5, Beutner et al, 2001), regardless of how the Ca2+ influx was manipulated. For Ca2+-nanodomain control (see Introduction), the m should differ between the two manipulations, because there, the number of released RRP vesicles is linearly determined by NCa × Popen (m ˜ 1, Augustine et al, 1991, see our biophysical model below).
To vary iCa, we slowly changed the extracellular [Ca2+] (Fig 7A,C) in both directions while supplementing divalent ions (Mg2+) at low Ca2+ concentration (< 1.3 mM [Ca2+]e). We observed a supralinear exocytosis-QCa relationship in the range of low QCa (Fig 7C, average of the maximal QCa used for fitting indicated by dashed vertical lines) before and after the onset of hearing (supralinear apparent Ca2+ dependence, p6–8: m = 3.10 ± 0.39, n = 6 IHCs; p14–17: m = 2.91 ± 0.38, n = 7 IHCs). Next, to gradually reduce NCa × Popen (Fig 7D) while maintaining iCa (Hess et al, 1984), we slowly bath-perfused preparations with the antagonistic dihydropyridine isradipine (10 μM). In immature IHCs, we found a supralinear apparent Ca2+ dependence (m = 2.35 ± 0.18, n = 7 IHCs) consistent with control of any given release site by several channels (“Ca2+ microdomain-like” control of exocytosis). The exponent m declined with maturation to a quasi-linear apparent Ca2+ dependence after the onset of hearing (p14-p17: 1.42 ± 0.13; n = 7 IHCs, see also Supplementary Fig S8 for corroborating data), suggesting a change towards “Ca2+ nanodomain-like” control of exocytosis.
Figure 7.
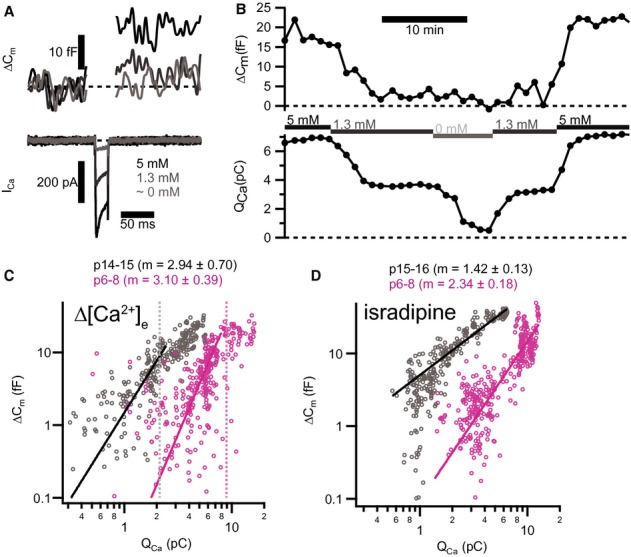
- Depolarizations (20 ms to −17 mV) elicited inward Ca2+ current (ICa, lower panel) and triggered ΔCm (upper panel) in a representative pre-hearing (p7) IHC. Increasing [Ca2+]e enlarged both ICa and ΔCm.
- Time course for ΔCm and integrated Ca2+ influx (QCa) of the same IHC in (A) for repetitive depolarizations (20 ms to −17 mV) at 60 s interval. Experiment started at 5 mM [Ca2+]e and periods where bath solution was slowly perfused with a different [Ca2+]e are marked by horizontal bars.
- Plotting ΔCm, 20 ms against QCa reveals a supralinear apparent Ca2+ dependence of exocytosis for low QCa. Solid lines represent best fit power functions (ΔCm = A(QCa)m) to the whole data set in this range. Each small symbol represents an individual response to depolarization (p15–16, grey, n = 7 IHCs; p6, magenta, n = 7 IHCs). Dashed vertical lines indicate the average maximal QCa limiting the QCa range used for fitting.
- Decreasing apparent Ca2+ cooperativity of exocytosis through blocking L-type Ca2+ channels with isradipine. Each small symbol represents an individual response to depolarization (p14–15, grey, n = 6 IHCs; p6–8, magenta, n = 7 IHCs).
Data information: In (C) and (D), solid lines show best fit power functions to the whole data set. Average values of the exponent m are indicated on top.
Linearization of the apparent Ca2+ dependence of exocytosis for NCa × Popen changes indicates tightening of Ca2+ influx-exocytosis coupling: insights from modeling
How precisely Ca2+ influx through nearby Ca2+ channels regulates the exocytosis of a given RRP vesicle depends on several parameters, including the number, distance and open probability of Ca2+ channels, the concentration and binding kinetics of Ca2+ buffers and the Ca2+-binding properties of the Ca2+ sensor of exocytosis (Moser et al, 2006; Matveev et al, 2011). Given this complexity we turned to biophysical modeling in order to reconcile the topography of membrane-proximal vesicles (here assumed to represent the RRP) and Ca2+ channels at the AZ with the apparent Ca2+ dependence of exocytosis observed in IHCs during changes of the number of open channels (NCa × Popen) or iCa before and after the onset of hearing. We based the models on morphological and biophysical information (e.g. data from EM, STED, Ca2+ channel analysis, Ca2+ uncaging of this study and published work: for parameters and computational methods see Ssupplementary Material S9). We then studied paradigmatic topographies of Ca2+ channels and Ca2+ sensors for mature AZ (scenarios “M1–3”, Fig 8A and Supplementary Material S9) and immature AZs (scenarios “IM”, Fig 8B and Supplementary Material S9).
Figure 8.
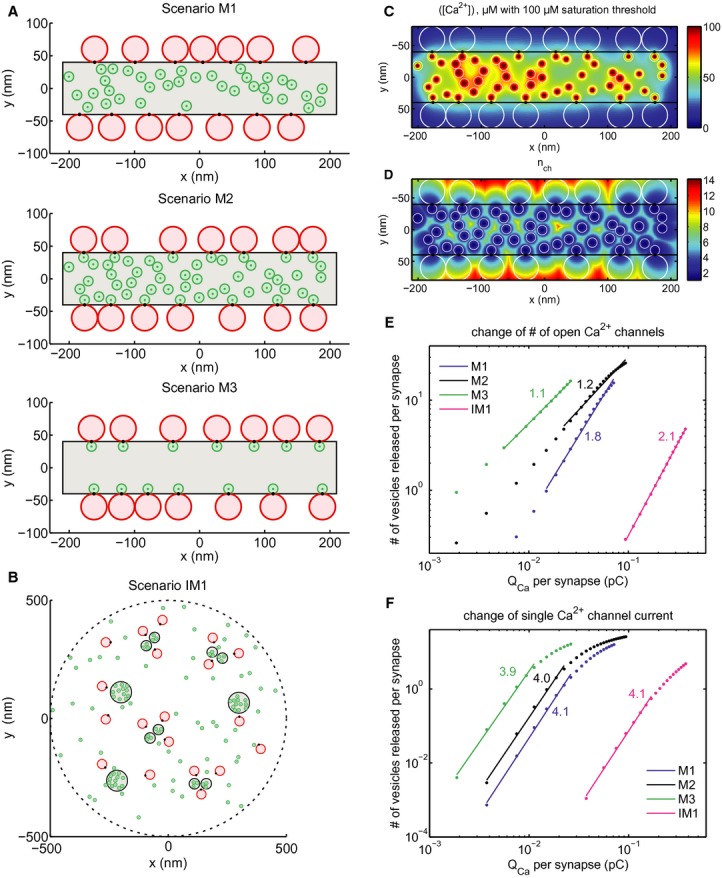
- Scenarios M1–3 of the mature AZ topography. Grey area: presynaptic density, red discs: RRP vesicles, green discs: Ca2+ channels, black spots: Ca2+ sensors of exocytosis. Scenario M1: random positioning of channels within the presynaptic density, Ca2+ sensor pointing towards the channel cluster. Scenario M2: introduction of “private channels” simulating molecular coupling of release site and channel. Scenario M3: only “private channels”, unrealistically low number of channels. Plots correspond to one out of a hundred realizations considered in the simulations.
- Scenario IM1 of the immature AZ topography. Legend as in (A). Note the more extended synaptic contact with several Ca2+ channel clusters, which were placed at the sites of presynaptic densities.
- Total mean steady state [Ca2+] profile at the AZ membrane for a particular realization of scenario M2.
- Effective number of channels contributing to total mean steady state [Ca2+] shown in (C).
- Average overall release (number of vesicles released) versus Ca2+ charge influx per synapse after the first 20 ms after the stimulus onset when NCa × Popen was changed by blocking channels (modeling the isradipine experiment). Dots correspond to the values averaged over different synapses and realizations as described in Supplementary Material S9. Straight lines are linear fits in log-log scale within the interval of fivefold decrease in QCa. Numbers correspond to the slope of fits, representing the apparent Ca2+ cooperativity of release, m. Blue—mature scenario M1, black—mature scenario M2, green—mature scenario M3, magenta—immature scenario IM1.
- The same as (E) but QCa was varied by manipulating iCa instead of NCa × Popen.
For the mature AZ, 14 vesicles (40-nm-diameter red disks in Fig 8A,B) were randomly distributed at the longer edges of an 80 × 420 nm presynaptic density (seven vesicles per one side), with the vesicular Ca2+ sensor (black spots in Fig 8A,B) in contact with the density in the plane of the plasma membrane. The size of the presynaptic density (grey area in Fig 8A,B) was chosen based on exemplary synapses reconstructed from serial EM sections (e.g. Fig 3D), and is consistent with CaV1.3 immunofluorescence in STED microscopy (Figs 4G and 5B). We placed 14–90 channels within the presynaptic density, representing different scenarios of channel-vesicle coupling. In scenario M1 (Fig 8A, upper panel), 36 channels were distributed randomly in the presynaptic density area. In scenario M2 (Fig 8A, middle panel), in addition to the 36 randomly distributed ones, 14 channels were placed in physical contact with the vesicle Ca2+ sensors, simulating molecular coupling (review in Eggermann et al, 2012) and nicknamed hereafter as “private channel” to highlight its dominance on the Ca2+ signal of a given vesicle. In scenario M3, only those 14 channels in physical contact with the vesicles were implemented (Fig 8A, lower panel). For each scenario, 100 different realizations of random channel and vesicle positions were considered. Analogous to experimental analysis, we compared the slope m of vesicle release against Ca2+ influx in double-logarithmic plots among scenarios of different degree of coupling. Scenarios M1–3 resulted in different exponents m for NCa × Popen change, all below 2: 1.8 for M1, 1.2 for M2 and 1.1 for M3 (Fig 8E), while m estimates for iCa change were all approximately 4 (Fig 8F). As another measure of Ca2+ influx to exocytosis coupling, we studied the effective number of contributing channels nch (Fig 8D for scenario M2), which is the ratio of the total mean steady state [Ca2+] (Fig 8C) to the mean steady state [Ca2+] elicited by the channel that contributed the most. With increasing dominance of the nearest channel, the effective number nch of channels contributing to [Ca2+] at the Ca2+ sensor went down from 4.4 for M1 to 2.1 for M2 and 1.2 for M3. Specifically, nch = 2.3 (1.2) in scenario M2 (M3) indicates that each “private channel” on average contributes 44% (83%) of [Ca2+] at the corresponding Ca2+ sensor, whereas nch = 4.4 in scenario M1 implies a contribution of only 23% by the nearest channel. Figure 8D illustrates for the case of scenario M2 how nch depends on the spatial position of channels and Ca2+ sensors in the plane of the plasma membrane. Simply moving the sensor 20 nm away from the “private channel” towards the vesicle center in M2 (scenario M2d, Supplementary Figs S9 and S14), increased nch to 5.8 and m to 2.5 for NCa × Popen changes, highlighting the strong effect of spatial proximity for the relevance of the nearest channel in controlling release of a given vesicle. In conclusion, modeling confirmed that the experimentally tractable Ca2+ cooperativity m during NCa × Popen changes is diagnostic for the relevance of the nearest channel in controlling fusion and support the notion of a Ca2+-nanodomain control of exocytosis in mature IHCs.
Despite the fact that implementing molecular coupling (M2–3) was sufficient to reproduce m near unity during NCa × Popen changes, this may not be the case if the channel density at the AZ is very high. Indeed, when we placed 40 additional randomly distributed Ca2+ channels in scenario M2 (a total of 90 channels, scenario M2b in Supplementary Fig S9), m increased from 1.2 to 2.0 and nch from 2.3 to 3.5. One possibility to preserve the dominance of the “private channels” is to implement an exclusion zone around them where no other channels can be located. Scenario M2c (Supplementary Figs S9 and S14, same density of channels outside the exclusion zone as in M2b) assumed circular exclusion zones with the width of a channel diameter and resulted in estimates of m = 1.2 and nch = 2.1 (very similar to those of scenario M2). On the other hand, if the density of the channels in the presynaptic density was low, m near unity could be reproduced even with two “private channels”. For example, addition of one more “private channel” per vesicle in M3 (scenario M3b in Supplementary Figs S9 and S14) led to m = 1.2 (NCa × Popen change) and nch = 2.7, close to the values obtained for scenario M2. Placing additional Ca2+ channels outside the presynaptic density (10/μm2, likely a strong overestimate of the average extrasynaptic density, see Brandt et al, 2005; Zampini et al, 2013) had negligible impact. In all above-mentioned scenarios m stayed high, approximately 4, when manipulating iCa.
Modelling the apparent Ca2+ dependence of immature IHCs based on AZ topography is more challenging because (i) of the large fraction of extrasynaptic Ca2+ channels (possibly driving extrasynaptic exocytosis) and (ii) the fact that half of the IHCs lacked the fast component of exocytosis elicited by Ca2+ uncaging (which was used for modelling). However, higher m predictions for NCa × Popen changes than found for the mature scenarios M2–3 were readily obtained with models based on realistic AZ topography (IM1), such as that depicted in Fig 8B. There, a presynaptic density area similar to that of M1–3 was distributed within 1 μm diameter disk in the form of separate patches corresponding to either pairs of smaller or single bigger densities (consistent with EM reconstructions), wherein 60 Ca2+ channels were distributed with similar average density as in M2. Scenario IM1 did not force physical contact of Ca2+ channel and vesicular Ca2+ sensor, which was placed in shortest distance to the nearest presynaptic density. In addition, 60 channels were distributed randomly outside the presynaptic density patches. The predicted m (NCa × Popen change) was 2.1, which is similar to the experimental observation (2.3) and considerably higher than the m (NCa × Popen change) estimates of M2 or M3 (Fig 8E,F). On the other hand, m (iCa change) was 4.1, which is very similar to those of M1–3: m = 3.9–4.1 (Fig 8F). However, m considerably decreased for NCa × Popen (m = 1.1) but not for iCa changes (m = 4.0) when implementing “private channels” as in scenarios M2 and M3 (scenario IM1b, Supplementary Material S9, Supplementary Figs S9 and S14).
In our model, we realized that RRP depletion (fusion exceeding vesicle replenishment, see Supplementary Material S9) could cause exocytosis to saturate with increasing Ca2+ influx depending on the model parameters (Figs 8D,E and Supplementary Fig S10). Saturation would lead to underestimation of m if data of this QCa range was included into the fit. Release leveled-off more in the case of iCa variation, where, different from NCa × Popen changes (Fig 7D, see also Supplementary Fig S8), saturation was also observed experimentally and fitting was restricted to the non-saturating range (Fig 7C). Our treatment of this “release saturation” is described in Supplementary Material S9. Using simulations, we also explored the QCa range in which the number of open channels per cluster is very low, which is difficult to achieve experimentally because of the approximately 20% residual current with isradipine block. As expected, m approached unity regardless of the topography, because release becomes dependent on Ca2+ influx through single openings of the few available channels. In contrast, for iCa changes m values remained near constant even at considerably lowered QCa outside the saturation region. In conclusion, biophysical modeling supports the experimentally derived notion of a maturational tightening of Ca2+ channel-exocytosis coupling.
Discussion
Here we studied the maturation of afferent IHC synapses around the onset of hearing. We found a transformation from an immature synaptic contact containing multiple small and peripheral spot-like AZ/PSD complexes into a consolidated contact with a single large AZ/PSD complex per SGN bouton. Ca2+ channels and Ca2+ influx became progressively confined to synaptic AZs over development. CaV1.3 channels first formed spot-like clusters whereas, following AZ remodeling, they formed an extended stripe alongside bassoon protein within the presynaptic density. Synaptic ribbons were larger in size and more flat in shape after the onset of hearing. Ca2+ uncaging triggered a fast component of exocytosis in only half of the immature IHCs, where it was smaller than in mature IHCs but had a similar intrinsic Ca2+ dependence. A “linearization” of the apparent Ca2+ dependence of exocytosis observed for NCa × Popen changes, together with computational modeling of immature and mature AZ topographies, indicated a developmental switch from more Ca2+-microdomain-like to more Ca2+nanodomain-like coupling between CaV1.3 channels and vesicular release sites.
Maturation of molecular nanoanatomy of IHC afferent synapses
High-resolution immunofluorescence and electron microscopy jointly demonstrated major structural changes of IHC synapses around the onset of hearing, schematized in Figs 1C and 3E. We propose that this change to a more ordered stripe-like AZ topography, together with the dramatic reduction of extrasynaptic CaV1.3 channels, enhances the Ca2+ efficiency of exocytosis and tightens the spatial coupling between CaV1.3 channels and vesicular release sites (Figs 7 and 8). We consider two not mutually exclusive candidate mechanisms for the structural refinement of the synapse over development: (i) merging of the individual synaptic specializations within a contact and (ii) pruning of all but one AZ/PSD complex per contact.
Merging may be mediated by interactions of pre-and/or post-synaptic scaffold molecules and could involve trans-synaptic regulation. Such cluster-cluster interactions may join multiple small ribbons, Ca2+-channel clusters, or GluA clusters into single large structures. Several lines of evidence indicate an important role of bassoon in this process. First, the involvement of cytomatrix of the AZ proteins in synaptogenesis was demonstrated for other synapses (Zhai et al, 2000, 2001; Shapira et al, 2003) whereby bassoon also recruits other proteins to the AZ (Shapira et al, 2003; Takao-Rikitsu et al, 2004; Maas et al, 2012). Second, bassoon resides in the stripe-like presynaptic density of the mature IHC synapse of wild-type mice (Fig 3) and also in the immature presynaptic density (Fig 1). Third, while synapses are still formed in IHCs lacking functional bassoon (Khimich et al, 2005), the presynaptic density and Ca2+-channel clusters remain spot-like (Frank et al, 2010). Notably, around hearing onset bassoon mRNA is upregulated in the mouse organ of Corti under control of thyroid hormone (Sendin et al, 2007). Perhaps bassoon facilitates AZ merging via interactions with CAST (Takao-Rikitsu et al, 2004), RIBEYE (tom Dieck et al, 2005), piccolo (Wang et al, 2009), and other proteins. The fusion of neighboring AZ-anchored ribbons is plausible given the strong RIBEYE-RIBEYE interactions (Magupalli et al, 2008), reports of ribbon-ribbon fusion during retinal synaptogenesis (Regus-Leidig et al, 2009), their stimulus-dependent structural plasticity (Spiwoks-Becker et al, 2004), and our observation of floating ribbons near anchored ones at immature IHC AZs (Fig 2).
Alternatively, or in parallel with merging, all but one of the AZs may be degraded. Pruning is a well-established mechanism for synaptic refinement in general (Goda & Davis, 2003). In the organ of Corti, pruning is thought to refine SGN afferent connectivity at the level of fibers and contacts (Sobkowicz et al, 1982; Huang et al, 2007, 2012; Sendin et al, 2007) but its role in development within a synaptic contact is not clear. Molecularly, the pruning of AZs might involve protein degradation, which appears to be counteracted by bassoon and piccolo via negative regulation of ubiquitin-ligase activity (Waites et al, 2013), such that their greater abundance might protect the largest of the initially formed AZs. In addition, a mutual regulation of ribbon size and Ca2+-channel clusters by Ca2+ influx and RIBEYE expression levels, respectively, was indicated in zebrafish lateral line hair cells during early development (Sheets et al, 2011, 2012). Super-resolution live-imaging of fluorescently-tagged ribbons and other AZ/PSD components in organotypic organ of Corti cultures will be required to directly evaluate the hypotheses of merging and pruning in future studies.
Maturation of Ca2+ influx-exocytosis coupling in IHCs
So far, neither the intrinsic Ca2+ dependence of exocytosis nor the Ca2+ influx-exocytosis coupling in immature IHCs was well understood. We characterized the intrinsic Ca2+ dependence of exocytosis and the spatial coupling between Ca2+ influx and exocytosis in IHCs before and after the onset of hearing. We note that the amplitude of the fast Cm rise evoked by Ca2+ uncaging, as used for studying the intrinsic Ca2+ dependence of exocytosis, was about 50 times larger than that reflecting the exocytosis of the RRP, analyzed for apparent Ca2+ dependence. Moreover, the mediating vesicle population(s) remain to be elucidated and might involve parallel extrasynaptic fusion of vesicles or vesicle priming and subsequent fusion (Fuchs et al, 2003). We assume that fast Cm rise reports synaptic vesicle fusion and the Ca2+ dependence of transmitter release, based on the observations of (i) partial cross-depletion of exocytosis elicited by Ca2+ influx and uncaging (Beutner et al, 2001) and (ii) the abolition of fast exocytosis in the absence of the putative priming factor and Ca2+ sensor of hair cell exocytosis otoferlin (Roux et al, 2006; Pangršič et al, 2010). The Ca2+ dependent kinetics of the fast component were, when present, comparable between IHCs before and after the onset of hearing, suggesting a common Ca2+ sensor of exocytosis, most likely otoferlin. It will be interesting to study the molecular changes underlying the emergence of the fast component in immature IHCs in future experiments.
What then accounts for the major developmental increase in efficiency of whole-cell Ca2+ influx to evoke exocytosis? In agreement with a recent single-channel study (Zampini et al, 2010) we find that immature IHCs express several thousands of CaV1.3 channels. Upon maturation and the cessation of Ca2+ action potential firing (Kros et al, 1998; Brandt et al, 2007), Ca2+ influx was largely restricted to AZs (Fig 5B). This reduction of extrasynaptic Ca2+ channels likely accounts for much of the dramatic developmental increase in the efficiency of Ca2+ influx to drive exocytosis (Beutner & Moser, 2001; Johnson et al, 2005) and lowers the metabolic expenditure for clearing Ca2+. Within synapses, Ca2+ channels appear to be retained and consolidated and, likely due to stabilizing interactions with scaffolding proteins.
Biophysical experiments and modeling indicated tighter spatial coupling between Ca2+ influx and RRP exocytosis after the onset of hearing. In agreement with our own and other published work, m was near unity for manipulation of NCa × Popen in mature IHCs (Fig 7D). This means that release scaled linearly with the number of open channels, suggesting either a Ca2+-nanodomain-like coupling or a linear intrinsic Ca2+-dependence of exocytosis. However, we found a larger m when manipulating iCa, consistent with a nonlinear Ca2+-dependence also seen with Ca2+ uncaging. On the other hand, in immature IHCs we found a supralinear apparent Ca2+ dependence of exocytosis when manipulating iCa or NCa × Popen, consistent with previous reports (Johnson et al, 2005) and indicative of a more Ca2+-microdomain-like coupling with a cooperative Ca2+ sensor. Together, these findings argue against the hypothesis that the Ca2+ dependence of IHC exocytosis is “linearized” upon maturation as a result of a change in the Ca2+ sensor of fusion (Johnson et al, 2010). Moreover, the “linearization” of Ca2+ dependence of exocytosis observed in whole-cell Cm recordings is unlikely to result from summation of heterogeneously supralinear AZs (Heil & Neubauer, 2010) because in such a scenario the apparent Ca2+ dependence should be linear no matter how it was manipulated.
Does the experimentally observed “linearization” of apparent Ca2+ dependence during manipulation of NCa × Popen indeed reflect the establishment of a Ca2+-nanodomain control of exocytosis in mature IHCs? After all, these protocols are thought to apply only to initial release at stimulus onset when pool depletion has not commenced (Augustine et al, 1991; Stanley, 1993; Mintz et al, 1995). Here, however, we had to accept considerable release of the RRP when probing exocytosis with 20 ms depolarizations in elevated [Ca2+]e to obtain a good signal to noise ratio. Therefore, we performed biophysical modeling to critically assess the consistency of our conclusions and also to gain further insights into the mechanism of IHC stimulus-secretion coupling. Combining data from a variety of different experimental approaches, we carefully investigated the effects of release saturation, resulting primarily from RRP depletion, on the predicted apparent Ca2+ dependence of exocytosis. Saturation was most pronounced when increasing iCa and less prevalent during manipulation of NCa × Popen. Importantly, RRP depletion is counteracted by fast vesicle replenishment at the IHC AZ (Pangršič et al, 2010; Goutman 2012), and for experimental m estimation during iCa manipulation we restricted the QCa range outside the saturating range. The m estimates derived from full and reduced QCa range did not differ much for NCa × Popen (Supplementary Fig S8). Because m was much lower for manipulation of NCa × Popen than for iCa, opposite of the expected result if saturation impacted our analysis, we trust that it indicates Ca2+-nanodomain control of exocytosis at mature AZs. The m estimate of approximately 1.4 placed strong constrains on the topography of the AZ models. Within our modeling framework and explored parameter space this was reproduced best when favoring control of exocytosis by “private channels”, which may indeed reflect molecular coupling (e.g. Liu et al, 2011).
The notion of a developmental tightening of the spatial Ca2+channel release site coupling is further supported by a decrease of the EGTA-sensitivity of RRP exocytosis (6% ΔCm reduction with 5 mM [EGTA]i compared with perforated-patch in p14–25 IHCs versus 37% in p6 IHCs for 10 ms depolarization, Beutner & Moser, 2001). A similar conclusion was reached at the calyx of Held synapse (Wang et al, 2008), another auditory synapse with high rates of synaptic transmission and high fidelity. There, developmental downregulation of the AZ component septin 5 seems critically involved in the transition from Ca2+-microdomain control before to Ca2+-nanodomain control after the onset of hearing (Yang et al, 2010). The precise molecular mechanism governing the maturation of coupling between Ca2+ channels and release sites at the IHC AZ remains to be elucidated in future studies.
Materials and Methods
Animals
C57Bl/6 mice (aged 6–30 days) mice were used for experiments. All experiments complied with national animal care guidelines and were approved by the University of Göttingen Board for animal welfare and the animal welfare office of the state of Lower Saxony.
Patch-clamp and confocal Ca2+ imaging
IHCs from apical coils of freshly dissected organs of Corti were patch-clamped as described (Moser & Beutner, 2000). The standard pipette solution contained (in mM): 115 Cs-glutamate, 13 TEA-Cl, 20 CsOH-HEPES, 1 MgCl2, 2 MgATP, 0.3 NaGTP, 10 EGTA, 0.4 Fluo-5N (Penta-K+ salt, Invitrogen), and carboxytetramethyl-rhodamine(TAMRA)-conjugated RIBEYE-binding dimer peptide (2 μM, Francis et al, 2011) for Ca2+ imaging. The pipette solution for perforated patch experiments contained (in mM): 135 Cs-gluconate, 10 TEA-Cl, 10 4-aminopyridine, 10 CsOH-HEPES, 1 MgCl2, and 250 μg/ml amphotericin. The extracellular solution contained (in mM): 104 NaCl, 35 TEA-Cl, 2.8 KCl, 5 CaCl2, 1 MgCl2, 10 NaOH-HEPES, 10 D–glucose, pH 7.3. Extracellular [Ca2+] was varied between nominally Ca2+-free to 10 mM for experiments manipulating iCa. In all cases, NaCl concentration was adjusted for osmolarity of solutions with different CaCl2 concentrations. Currents were low-pass filtered at 2.9 kHz (8.5 kHz for fluctuation analysis) and sampled at 50 kHz (100 kHz for fluctuation analysis). Cells with holding current greater than −50 pA were discarded. An EPC-9 amplifier or an EPC-10 amplifier and “Patchmaster” software (HEKA Elektronik, Lambrecht, Germany) was used for measurements. Whole-cell capacitance measurement was performed in perforated-patch configuration as previously described (Moser & Beutner, 2000), except that ΔCm was estimated as the difference of the mean Cm over 100 ms after the end of the depolarization (the initial 40 ms were skipped). All voltages were corrected for liquid-junction potentials and voltage-drops across series resistance. Ca2+ currents were further isolated from background current using a P/n protocol. Confocal Ca2+ imaging was performed as described (Frank et al, 2009). In brief, presynaptic Ca2+signal of IHCs were observed as changes of Ca2+ indicator fluorescence in XY scans using long (200–254 ms) step depolarizations to −7 mV.
UV-laser photolysis of caged calcium
UV-laser photolysis of caged calcium was performed as previously described (Nouvian et al, 2011). Briefly, to obtain step-wise increases in intracellular calcium, 100 μs of pulsed laser light from a DPSL-355/1000 UV laser (Rapp OptoElectronic, Hamburg, Germany) were applied after achieving the whole-cell configuration. [Ca2+]i was measured by ratiometric imaging using the calcium indicator dye mag-fura-2 (Invitrogen, Darmstadt, Germany). The dye was excited by a monochromatic light source alternating between 340 and 380 nm, and imaged using a CCD camera (TILL Photonics, Gräfelfing, Germany). [Ca2+]i was determined as previously described (Beutner et al, 2001). The pipette solution for flash-photolysis contained (in mM): 83 Cs-gluconate, 16 TEA-Cl, 18 Cs-HEPES (pH 7.2), 0.3 mag-fura-2, 10 DM-nitrophen (gift of A. Leonov and C. Griesinger, Göttingen; or Calbiochem, Darmstadt, Germany), 5 1,3-diaminopropan-2-ol-tetraacetic acid and 10 CaCl2. The extracellular solution for flash-photolysis contained (in mM): 97 NaCl, 35 TEA-Cl, 2.8 KCl, 10 CaCl2, 1 MgCl2, 10 Na-HEPES, 1 CsCl, 11.1 D–glucose (pH 7.2). A double exponential function with variable delay was used to fit the change in Cm, from which rate constants and response delay were extracted. In cases where the rate constants of the two components differed by less than a factor of four, the traces were re-fitted with a single exponential function. IHCs in which no Cm responses could be elicited were excluded from analysis.
Immunohistochemistry and immunofluorescence microscopy
Apical cochlear turns were fixed for 25 min in 99% methanol at −20°C. Primary antibodies: mouse anti-CtBP2 (1:200; BD Biosciences), rabbit anti-GluA2/3 (1:200; Chemicon), rabbit anti-CaV1.3 (1:50; Alomone Labs), goat anti-CtBP2 (1:150; Santa Cruz Biotech), mouse anti-GluA2 (1:75 Chemicon), mouse anti-Sap7f407 to bassoon (1:1,000; Abcam), mouse anti-Na+/K+ATPase α3 subunit (1:200; Thermo Scientific). Secondary antibodies: AlexaFluor488, 594, and 647 (1:200; Molecular Probes). Images were acquired with an SP5 confocal microscope (Leica) with a 100× oil-immersion objective (NA = 1.4). Experiments were repeated until all antigens were stained with relative uniformity in at least one preparation from each age group processed in batch. Each preparation yielded several images. Those with readily-apparent bleaching were discarded. Each image had 5–7 IHCs. Stacks from two preparations per age, stained and acquired in parallel across age groups, were analysed with Igor Pro 6 (Wavemetrics). Intensity per synapse was calculated in the optical section with the peak intensity. A region-of-interest (ROI) was determined by fitting a 2D Gaussian function on a 1 μm2 region surrounding each CtBP2 or CaV1.3 immunofluorescent spot (Frank et al, 2010). Comparison of plasma membrane versus ribbon-associated CaV1.3 immunofluorescence was performed by first connecting all ribbons within 3 μm of each other in 3D, and then using the connecting lines to define the axes of cylinders of 800 nm diameter to demarcate a volume of basal plasma membrane. The average intensity of this basal plasma membrane was compared with that of the ribbon-proximal region, defined as 3D spheres of 800 nm diameter centered at all connected ribbons. Three IHCs per age group were analyzed like this. Average voxel intensity of the stack was subtracted as background.
Two-color STED images were acquired on a custom setup with a stage-scanner (Mad City Labs) using Atto590-and Atto647N-conjugated secondary antibodies (Atto-Tec) excited with pulsed diode lasers (PicoQuant) at 595 and 640 nm, temporally interleaved at 50 ns intervals. After excitation, the fluorescence of dyes in the periphery of the excited spot was suppressed through stimulated emission by the STED laser, emitting 1.2 ns pulses of 775 nm light at 20 MHz (IPG Photonics). The STED beam was guided through a polymeric phase plate (vortex pattern; RPC Photonics) and circularly-polarized before coupling into the objective. Because emitted fluorescence is confined to the zero intensity center of the STED beam, using one STED-beam for both dyes ensured that a small misalignment of the excitation beams would not show in the resulting images. The fluorescence was detected at 600–640 nm (Atto590) and 650–690 nm (Atto 647N) with SPCM-AQRH13 fiber-coupled photon-counting modules (Perkin Elmer). The fiber core acted as a confocal pinhole of 1.1 Airy disks.
Transmission electron microscopy
Cochleae were explanted around noon and perfusion-fixed on ice with 4% PFA and 0.5% glutaraldehyde in 1× PBS, pH 7.2. After 1 h of incubation, the apical cochlear turns were explanted in 1× PBS and fixed overnight on ice with secondary fixative comprising 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.2. The samples were washed in sodium cacodylate buffer and postfixed on ice for 1 h with 1% osmium tetroxide ((v/v) in 0.1 M sodium cacodylate buffer), followed by a 1 h washing step in sodium cacodylate buffer and three brief washing steps in distilled water. The samples were stained en bloc with 1% (v/v) uranyl acetate in distilled water for 1 h on ice. After a brief wash with distilled water, samples were dehydrated at room temperature in increasing ethanol concentrations, infiltrated in Epon resin (100% EtOH/Epon 1:1 (v/v), 30 and 90 min; 100% Epon, overnight), and embedded for 48 h at 70°C. Following conventional embedding 65–75 nm sections were obtained approaching from the anterior edge using an Ultracut E (Reichert-Jung). Slices were postfixed and-stained with uranyl acetate/lead citrate following standard protocols. Micrographs were taken with a JEOL electron microscope (JEM 1011) equipped with a Gatan Orius 1200A camera using the Digital Micrograph software package at an 8000-fold magnification.
Quantitative image analysis was performed as follows: For the size of ribbons, the longest axis of each ribbon in a section, excluding the membrane-bound rootlet region, was measured. For ribbon-associated synaptic vesicles the first row of vesicles around the ribbon were counted per section. For analysis, Student's t-test was used, if not otherwise indicated. Three-dimensional reconstruction was performed on 4–7 serial 70 nm sections with the free software Reconstruct (Fiala, 2005).
For electron-tomography, 250 nm conventionally embedded sections were applied to Formvar-coated 100 copper mesh grids and stained with 4% uranyl acetate and Reynold's lead citrate, subsequently 10 nm gold particles were applied to the grid. A single tilt series was acquired at a JEOL JEM 2100 electron microscope at 200 kV from −48 to +60° with 1° increment using Serial-EM software. The tomogram was generated using the IMOD package etomo and model was rendered using 3dmod (bio3d.colorado.edu/imod/).
High-pressure freezing
For high-pressure freezing organs of Corti were explanted as described above and placed in aluminium specimen carrier of 200 μm (type A) depth filled with inhibiting solution (Pangršič et al, 2010). The aluminium lid (type B) has been dipped in hexadecen (Sigma-Aldrich) before placing onto the sample. Samples were frozen immediately using the HPM010 (Bal-tec) and rapidly transfered into liquid nitrogen for storage. Freeze substitution was performed in an EM AFS2 (Leica) according to (Rostaing et al, 2006; Siksou et al, 2007). Briefly, samples were incubated for 4 days in 0.1% tannic acid in acetone at −90°C. Before raising the temperature to −20°C (5°C/h) and three washing steps with aceton 2% osmium tetroxide in acetone has been added and incubated for 16 h. Finally temperature has been raised to 4°C (10°C/h) and samples were washed in acetone and warmed to RT. Finally samples were infiltrated and embedded in Epon resin.
Immunogold pre-embedding
Immunogold pre-embedding labeling on p14 wild-type mice was performed according to (Nieratschker et al, 2009), using the guinea pig anti-bassoon antibody (anti-bassoon (Cterm) in Fig 3F; Synaptic Systems; 1:500), mouse anti-Sap7f407 antibody (anti-bassoon (Nterm) in Fig 3F, 1:400; Abcam) and goat anti-guinea pig or anti-mouse nanogold IgG from Nanoprobes. For silver enhancement the Nanoprobes HQ Silver™ enhancement kit (Nanoprobes) was used.
Data analysis and statistical tests
Data are presented as mean ± SEM, unless otherwise specified. Normality was assessed with the Jarque–Bera test. F-test was used to assess equality of variance in normally distributed data sets. Unpaired, two-tailed Wilcoxon rank test (also known as Mann–Whitney U-test) was used to compare data of non-normal distribution or when variances of experimental groups were unequal. In case of normally-distributed equal-variance data, Student's unpaired two-tailed t-test was used to compare two samples (* indicates P < 0.05). Comparison of dispersion was performed with a modified Levene's test (Brown–Forsythe test, Brown & Forsythe, 1974), using median instead of mean for improved robustness under non-normality. One-way ANOVA followed by Tukey's test was used to detect differences in multiple comparisons for FA.
Computational model
Supplementary Material S9 describes the model implementation in detail. In brief, the spatial positioning of RRP vesicles and Ca2+ channels (AZ topography) at mature and immature synapses was based on functional (fluctuation analysis, Ca2+ imaging, published single channel recording data) and morphological (EM and STED) estimates. Ca2+ channel gating was modelled as a three state Markov chain with gating rates based on the experimental data by (Neef et al, 2009). The spatial [Ca2+] profile was assumed to equilibrate instantaneously upon channel opening and closing. [Ca2+] levels were estimated by treating open channels as hemispherical Ca2+ sources and by using the linearized buffer approximation (Naraghi & Neher, 1997). Ca2+ diffusion was constrained by the reflective plasma membrane but considered unaffected by synaptic vesicles or the synaptic ribbon. Ca2+ profile was assumed to be shaped by the following mobile Ca2+ buffers: calretinin, calbindin, parvalbumin, and ATP. The concentrations of endogeneous Ca2+ buffers were taken from (Hackney et al, 2005) and [ATP] was set to 2 mM. Ca2+ triggered fusion of the readily releasable pool vesicles followed the seven state Markov chain model proposed by (Beutner et al, 2001) for both mature and immature synapses. The refilling of the vesicle docking sites was treated as a single step process with a fixed rate. The value of the refilling rate was set based on the experimental data by (Pangršič et al, 2010). Channel gating and vesicle fusion-replenishment dynamics were propagated by using Gillespie's algorithm (Gillespie, 1977) and its extended version for time dependent reaction rates. All calculations were performed in MATLAB. To decrease the computation time to convenient level, 100 CPU cores were used for simulating a chosen synapse scenario.
Acknowledgments
We thank G. Hoch for developing image analysis routines and S. Gerke, C. Senger-Freitag for expert technical assistance. We thank Nikolai M. Chapochnikov for inspiring discussions and contributions to the modelling approach used in this study and Drs A. Bulankina, T. Dresbach, and E. Neher for critical reading of a previous version of the MS. A.B.W. was supported by a Lichtenberg fellowship of the state of Lower Saxony (through the “Neurosenses” PhD program). This work was supported by grants of the German Research Foundation through the collaborative research center 889 (projects A2 to T.M., A7 to C.W. and C6 to F.W.) and the Center for Nanoscopy and Molecular Physiology of the Brain (to T.M. and S.H.) and the German Federal Ministry of Education and Research (through the Bernstein Center grant 01GQ0433 to T.M. and 01GQ0811 to F.W.).
Author contributions
ABW, MAR, CW and TM designed the study. ABW performed electrophysiology, Ca2+ imaging and analysis of immunohistochemical data. ABW and TP performed calcium uncaging experiments. MAR performed immunohistochemistry, confocal and STED microscopy. MG and FW constructed the biophysical model with input from ABW, MAR, CW and TM. All numerical simulations were performed and analyzed by MG. FG built the two-channel STED microscope. TF performed non-stationary fluctuation analysis. CW performed electron microscopy, SM did part of the immuno-EM. ABW, MG, MAR, CW and TM prepared the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Augustine GJ, Adler EM, Charlton MP. The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann N Y Acad Sci. 1991;635:365–381. doi: 10.1111/j.1749-6632.1991.tb36505.x. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Beutner D, Moser T. The presynaptic function of mouse cochlear inner hair cells during development of hearing. J Neurosci. 2001;21:4593. doi: 10.1523/JNEUROSCI.21-13-04593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner D, Voets T, Neher E, Moser T. Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 2001;29:681–690. doi: 10.1016/s0896-6273(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Brandt A, Khimich D, Moser T. Few CaV1. 3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J Neurosci. 2005;25:11577. doi: 10.1523/JNEUROSCI.3411-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T. CaV1. 3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt N, Kuhn S, Münkner S, Braig C, Winter H, Blin N, Vonthein R, Knipper M, Engel J. Thyroid hormone deficiency affects postnatal spiking activity and expression of Ca2+ and K+ channels in rodent inner hair cells. J Neurosci. 2007;27:3174–3186. doi: 10.1523/JNEUROSCI.3965-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MB, Forsythe AB. Robust tests for the equality of variances. J Am Stat Assoc. 1974;69:364–367. [Google Scholar]
- Bulankina AV, Moser T. Neural circuit development in the mammalian cochlea. Physiology (Bethesda) 2012;27:100–112. doi: 10.1152/physiol.00036.2011. [DOI] [PubMed] [Google Scholar]
- Dick O, Hack I, Altrock WD, Garner CC, Gundelfinger ED, Brandstätter JH. Localization of the presynaptic cytomatrix protein Piccolo at ribbon and conventional synapses in the rat retina: comparison with Bassoon. J Comp Neurol. 2001;439:224–234. doi: 10.1002/cne.1344. [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Altrock WD, Kessels MM, Qualmann B, Regus H, Brauner D, Fejtová A, Bracko O, Gundelfinger ED, Brandstätter JH. Molecular dissection of the photoreceptor ribbon synapse: physical interaction of Bassoon and RIBEYE is essential for the assembly of the ribbon complex. J Cell Biol. 2005;168:825–836. doi: 10.1083/jcb.200408157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tom Dieck S, Sanmartí-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kämpf U, Fränzer JT, Stumm M, Garner CC, Gundelfinger ED. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol. 1998;142:499–509. doi: 10.1083/jcb.142.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Vazquez AE, Namkung Y, Chu H, Cardell EL, Nie L, Parson S, Shin H-S, Yamoah EN. Null mutation of alpha1D Ca2+channel gene results in deafness but no vestibular defect in mice. J Assoc Res Otolaryngol. 2004;5:215–226. doi: 10.1007/s10162-003-4020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci. 2012;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Francis AA, Mehta B, Zenisek D. Development of new peptide-based tools for studying synaptic ribbon function. J Neurophysiol. 2011;106:1028–1037. doi: 10.1152/jn.00255.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank T, Khimich D, Neef A, Moser T. Mechanisms contributing to synaptic Ca2+ signals and their heterogeneity in hair cells. Proc Natl Acad Sci USA. 2009;106:4483. doi: 10.1073/pnas.0813213106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank T, Rutherford MA, Strenzke N, Neef A, Pangršič T, Khimich D, Fejtova A, Gundelfinger ED, Liberman MC, Harke B, Bryan KE, Lee A, Egner A. Bassoon and the synaptic ribbon organize Ca2+ channels and vesicles to add release sites and promote refilling. Neuron. 2010;68:724–738. doi: 10.1016/j.neuron.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs PA, Glowatzki E, Moser T. The afferent synapse of cochlear hair cells. Curr Opin Neurobiol. 2003;13:452–458. doi: 10.1016/s0959-4388(03)00098-9. [DOI] [PubMed] [Google Scholar]
- Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J Phys Chem. 1977;81:2340–2361. [Google Scholar]
- Glowatzki E, Fuchs PA. Transmitter release at the hair cell ribbon synapse. Nat Neurosci. 2002;5:147–154. doi: 10.1038/nn796. [DOI] [PubMed] [Google Scholar]
- Goda Y, Davis GW. Mechanisms of synapse assembly and disassembly. Neuron. 2003;40:243–264. doi: 10.1016/s0896-6273(03)00608-1. [DOI] [PubMed] [Google Scholar]
- Goutman JD. Transmitter release from cochlear hair cells is phase locked to cyclic stimuli of different intensities and frequencies. J Neurosci. 2012;32:17025–17036. doi: 10.1523/JNEUROSCI.0457-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutman JD, Glowatzki E. Time course and calcium dependence of transmitter release at a single ribbon synapse. Proc Natl Acad Sci USA. 2007;104:16341–16346. doi: 10.1073/pnas.0705756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant L, Yi E, Glowatzki E. Two modes of release shape the postsynaptic response at the inner hair cell ribbon synapse. J Neurosci. 2010;30:4210–4220. doi: 10.1523/JNEUROSCI.4439-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graydon CW, Cho S, Li G-L, Kachar B, von Gersdorff H. Sharp Ca2+ nanodomains beneath the ribbon promote highly synchronous multivesicular release at hair cell synapses. J Neurosci. 2011;31:16637–16650. doi: 10.1523/JNEUROSCI.1866-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney CM, Mahendrasingam S, Penn A, Fettiplace R. The concentrations of calcium buffering proteins in mammalian cochlear hair cells. J Neurosci. 2005;25:7867. doi: 10.1523/JNEUROSCI.1196-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil P, Neubauer H. Summing across different active zones can explain the quasi-linear Ca2+-dependencies of exocytosis by receptor cells. Front Synaptic Neurosci. 2010;2:148. doi: 10.3389/fnsyn.2010.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- Huang L-C, Barclay M, Lee K, Peter S, Housley GD, Thorne PR, Montgomery JM. Synaptic profiles during neurite extension, refinement and retraction in the developing cochlea. Neural Dev. 2012;7:1–17. doi: 10.1186/1749-8104-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L-C, Thorne PR, Housley GD, Montgomery JM. Spatiotemporal definition of neurite outgrowth, refinement and retraction in the developing mouse cochlea. Development. 2007;134:2925–2933. doi: 10.1242/dev.001925. [DOI] [PubMed] [Google Scholar]
- Hull C, Studholme K, Yazulla S, von Gersdorff H. Diurnal changes in exocytosis and the number of synaptic ribbons at active zones of an ON-type bipolar cell terminal. J Neurophysiol. 2006;96:2025–2033. doi: 10.1152/jn.00364.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarsky T, Tian M, Singer JH. Nanodomain control of exocytosis is responsible for the signaling capability of a retinal ribbon synapse. J Neurosci. 2010;30:11885–11895. doi: 10.1523/JNEUROSCI.1415-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Franz C, Kuhn S, Furness DN, Ruttiger L, Munkner S, Rivolta MN, Seward EP, Herschman HR, Engel J, Knipper M, Marcotti W. Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nat Neurosci. 2010;13:45–52. doi: 10.1038/nn.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Marcotti W, Kros CJ. Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol. 2005;563:177–191. doi: 10.1113/jphysiol.2004.074740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy HJ, Meech RW. Fast Ca2+ signals at mouse inner hair cell synapse: a role for Ca2+-induced Ca2+ release. J Physiol. 2002;539:15–23. doi: 10.1113/jphysiol.2001.013171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khimich D, Nouvian R, Pujol R, tom Dieck S, Egner A, Gundelfinger ED, Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP, Rüsch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature. 1998;394:281–284. doi: 10.1038/28401. [DOI] [PubMed] [Google Scholar]
- Lenzi D, Crum J, Ellisman MH, Roberts WM. Depolarization redistributes synaptic membrane and creates a gradient of vesicles on the synaptic body at a ribbon synapse. Neuron. 2002;36:649–659. doi: 10.1016/s0896-6273(02)01025-5. [DOI] [PubMed] [Google Scholar]
- Liu KS, Siebert M, Mertel S, Knoche E, Wegener S, Wichmann C, Matkovic T, Muhammad K, Depner H, Mettke C, Bückers J, Hell SW, Müller M, Davis GW, Schmitz D, Sigrist SJ. RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science. 2011;334:1565–1569. doi: 10.1126/science.1212991. [DOI] [PubMed] [Google Scholar]
- Maas C, Torres VI, Altrock WD, Leal-Ortiz S, Wagh D, Terry-Lorenzo RT, Fejtova A, Gundelfinger ED, Ziv NE, Garner CC. Formation of Golgi-derived active zone precursor vesicles. J Neurosci. 2012;32:11095–11108. doi: 10.1523/JNEUROSCI.0195-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magupalli VG, Schwarz K, Alpadi K, Natarajan S, Seigel GM, Schmitz F. Multiple RIBEYE-RIBEYE interactions create a dynamic scaffold for the formation of synaptic ribbons. J Neurosci. 2008;28:7954–7967. doi: 10.1523/JNEUROSCI.1964-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP. Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci. 1996;16:4457. doi: 10.1523/JNEUROSCI.16-14-04457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G, Fuchs P. The diverse roles of ribbon synapses in sensory neurotransmission. Nat Rev Neurosci. 2010;11:812–822. doi: 10.1038/nrn2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveev V, Bertram R, Sherman A. Calcium cooperativity of exocytosis as a measure of Ca2+ channel domain overlap. Brain Res. 2011;1398:126–138. doi: 10.1016/j.brainres.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean WJ, Smith KA, Glowatzki E, Pyott SJ. Distribution of the Na, K-ATPase α subunit in the rat spiral ganglion and organ of corti. J Assoc Res Otolaryngol. 2009;10:37–49. doi: 10.1007/s10162-008-0152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AC, Frank T, Khimich D, Hoch G, Riedel D, Chapochnikov NM, Yarin YM, Harke B, Hell SW, Egner A, Moser T. Tuning of synapse number, structure and function in the cochlea. Nat Neurosci. 2009;12:444–453. doi: 10.1038/nn.2293. [DOI] [PubMed] [Google Scholar]
- Mikaelian D, Alford BR, Ruben RJ. Cochlear potentials and 8 nerve action potentials in normal and genetically deaf mice. Ann Otol Rhinol Laryngol. 1965;74:146–157. doi: 10.1177/000348946507400113. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Moser T, Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci USA. 2000;97:883–888. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Neef A, Khimich D. Mechanisms underlying the temporal precision of sound coding at the inner hair cell ribbon synapse. J Physiol. 2006;576:55–62. doi: 10.1113/jphysiol.2006.114835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef J, Gehrt A, Bulankina AV, Meyer AC, Riedel D, Gregg RG, Strenzke N, Moser T. The Ca2+ channel subunit beta2 regulates Ca2+ channel abundance and function in inner hair cells and is required for hearing. J Neurosci. 2009;29:10730–10740. doi: 10.1523/JNEUROSCI.1577-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Nieratschker V, Schubert A, Jauch M, Bock N, Bucher D, Dippacher S, Krohne G, Asan E, Buchner S, Buchner E. Bruchpilot in ribbon-like axonal agglomerates, behavioral defects, and early death in SRPK79D kinase mutants of Drosophila. PLoS Genet. 2009;5:e1000700. doi: 10.1371/journal.pgen.1000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouvian R, Neef J, Bulankina AV, Reisinger E, Pangršič T, Frank T, Sikorra S, Brose N, Binz T, Moser T. Exocytosis at the hair cell ribbon synapse apparently operates without neuronal SNARE proteins. Nat Neurosci. 2011;14:411–413. doi: 10.1038/nn.2774. [DOI] [PubMed] [Google Scholar]
- Pangršič T, Lasarow L, Reuter K, Takago H, Schwander M, Riedel D, Frank T, Tarantino LM, Bailey JS, Strenzke N, Brose N, Müller U, Reisinger E, Moser T. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010;13:869–876. doi: 10.1038/nn.2578. [DOI] [PubMed] [Google Scholar]
- Pangršič T, Reisinger E, Moser T. Otoferlin: a multi-C2 domain protein essential for hearing. Trends Neurosci. 2012;35:671–680. doi: 10.1016/j.tins.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Regus-Leidig H, tom Dieck S, Specht D, Meyer L, Brandstätter JH. Early steps in the assembly of photoreceptor ribbon synapses in the mouse retina: the involvement of precursor spheres. J Comp Neurol. 2009;512:814–824. doi: 10.1002/cne.21915. [DOI] [PubMed] [Google Scholar]
- Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostaing P, Real E, Siksou L, Lechaire J-P, Boudier T, Boeckers TM, Gertler F, Gundelfinger ED, Triller A, Marty S. Analysis of synaptic ultrastructure without fixative using high-pressure freezing and tomography. Eur J Neurosci. 2006;24:3463–3474. doi: 10.1111/j.1460-9568.2006.05234.x. [DOI] [PubMed] [Google Scholar]
- Roux I, Safieddine S, Nouvian R, Grati M, Simmler M-C, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Rutherford MA, Pangršič T. Molecular anatomy and physiology of exocytosis in sensory hair cells. Cell Calcium. 2012;52:327–337. doi: 10.1016/j.ceca.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Rutherford MA, Roberts WM. Frequency selectivity of synaptic exocytosis in frog saccular hair cells. Proc Natl Acad Sci USA. 2006;103:2898. doi: 10.1073/pnas.0511005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieddine S, El-Amraoui A, Petit C. The auditory hair cell ribbon synapse: from assembly to function. Annu Rev Neurosci. 2012;35:509–528. doi: 10.1146/annurev-neuro-061010-113705. [DOI] [PubMed] [Google Scholar]
- Schmitz F, Königstorfer A, Südhof TC. RIBEYE, a component of synaptic ribbons: a protein's journey through evolution provides insight into synaptic ribbon function. Neuron. 2000;28:857–872. doi: 10.1016/s0896-6273(00)00159-8. [DOI] [PubMed] [Google Scholar]
- Sendin G, Bulankina AV, Riedel D, Moser T. Maturation of ribbon synapses in hair cells is driven by thyroid hormone. J Neurosci. 2007;27:3163–3173. doi: 10.1523/JNEUROSCI.3974-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger ED, Ziv NE, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron. 2003;38:237–252. doi: 10.1016/s0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Sheets L, Kindt KS, Nicolson T. Presynaptic CaV1.3 channels regulate synaptic ribbon size and are required for synaptic maintenance in sensory hair cells. J Neurosci. 2012;32:17273–17286. doi: 10.1523/JNEUROSCI.3005-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets L, Trapani JG, Mo W, Obholzer N, Nicolson T. Ribeye is required for presynaptic CaV1.3a channel localization and afferent innervation of sensory hair cells. Development. 2011;138:1309–1319. doi: 10.1242/dev.059451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shnerson A, Devigne C, Pujol R. Age-related changes in the C57BL/6J mouse cochlea. II. Ultrastructural findings. Brain Res. 1981;254:77–88. doi: 10.1016/0165-3806(81)90060-2. [DOI] [PubMed] [Google Scholar]
- Siksou L, Rostaing P, Lechaire J-P, Boudier T, Ohtsuka T, Fejtová A, Kao H-T, Greengard P, Gundelfinger ED, Triller A, Marty S. Three-dimensional architecture of presynaptic terminal cytomatrix. J Neurosci. 2007;27:6868–6877. doi: 10.1523/JNEUROSCI.1773-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobkowicz HM, Rose JE, Scott GE, Slapnick SM. Ribbon synapses in the developing intact and cultured organ of Corti in the mouse. J Neurosci. 1982;2:942. doi: 10.1523/JNEUROSCI.02-07-00942.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiwoks-Becker I, Glas M, Lasarzik I, Vollrath L. Mouse photoreceptor synaptic ribbons lose and regain material in response to illumination changes. Eur J Neurosci. 2004;19:1559–1571. doi: 10.1111/j.1460-9568.2004.03198.x. [DOI] [PubMed] [Google Scholar]
- Stanley EF. Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron. 1993;11:1007–1011. doi: 10.1016/0896-6273(93)90214-c. [DOI] [PubMed] [Google Scholar]
- Takao-Rikitsu E, Mochida S, Inoue E, Deguchi-Tawarada M, Inoue M, Ohtsuka T, Takai Y. Physical and functional interaction of the active zone proteins, CAST, RIM1, and Bassoon, in neurotransmitter release. J Cell Biol. 2004;164:301–311. doi: 10.1083/jcb.200307101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites CL, Leal-Ortiz SA, Okerlund N, Dalke H, Fejtova A, Altrock WD, Gundelfinger ED, Garner CC. Bassoon and Piccolo maintain synapse integrity by regulating protein ubiquitination and degradation. EMBO J. 2013;32:954–969. doi: 10.1038/emboj.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-Y, Neher E, Taschenberger H. Synaptic vesicles in mature calyx of Held synapses sense higher nanodomain calcium concentrations during action potential-evoked glutamate release. J Neurosci. 2008;28:14450–14458. doi: 10.1523/JNEUROSCI.4245-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hu B, Zieba A, Neumann NG, Kasper-Sonnenberg M, Honsbein A, Hultqvist G, Conze T, Witt W, Limbach C, Geitmann M, Danielson H, Kolarow R, Niemann G, Lessmann V, Kilimann MW. A protein interaction node at the neurotransmitter release site: domains of Aczonin/Piccolo, Bassoon, CAST, and rim converge on the N-terminal domain of Munc13-1. J Neurosci. 2009;29:12584–12596. doi: 10.1523/JNEUROSCI.1255-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AB, Jing Z, Rutherford MA, Frank T, Strenzke N, Moser T. Concurrent maturation of inner hair cell synaptic Ca2+ influx and auditory nerve spontaneous activity around hearing onset in mice. J Neurosci. 2013;33:10661–10666. doi: 10.1523/JNEUROSCI.1215-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y-M, Fedchyshyn MJ, Grande G, Aitoubah J, Tsang CW, Xie H, Ackerley CA, Trimble WS, Wang L-Y. Septins regulate developmental switching from microdomain to nanodomain coupling of Ca2+ influx to neurotransmitter release at a central synapse. Neuron. 2010;67:100–115. doi: 10.1016/j.neuron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampini V, Johnson SL, Franz C, Knipper M, Holley MC, Magistretti J, Masetto S, Marcotti W. Burst activity and ultrafast activation kinetics of CaV1.3 Ca2+ channels support presynaptic activity in adult gerbil hair cell ribbon synapses. J Physiol. 2013;591:3811–3820. doi: 10.1113/jphysiol.2013.251272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampini V, Johnson SL, Franz C, Lawrence ND, Münkner S, Engel J, Knipper M, Magistretti J, Masetto S, Marcotti W. Elementary properties of CaV1.3 Ca(2+) channels expressed in mouse cochlear inner hair cells. J Physiol. 2010;588:187–199. doi: 10.1113/jphysiol.2009.181917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- Zhai R, Olias G, Chung WJ, Lester RAJ, tom Dieck S, Langnaese K, Kreutz MR, Kindler S, Gundelfinger ED, Garner CC. Temporal appearance of the presynaptic cytomatrix protein bassoon during synaptogenesis. Mol Cell Neurosci. 2000;15:417–428. doi: 10.1006/mcne.2000.0839. [DOI] [PubMed] [Google Scholar]
- Zhai R, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.