

Abstract
Mitochondrial dysfunction has long been associated with Parkinson's disease (PD). Parkin and PINK1, two genes associated with familial PD, have been implicated in the degradation of depolarized mitochondria via autophagy (mitophagy). Here, we describe the involvement of parkin and PINK1 in a vesicular pathway regulating mitochondrial quality control. This pathway is distinct from canonical mitophagy and is triggered by the generation of oxidative stress from within mitochondria. Wild-type but not PD-linked mutant parkin supports the biogenesis of a population of mitochondria-derived vesicles (MDVs), which bud off mitochondria and contain a specific repertoire of cargo proteins. These MDVs require PINK1 expression and ultimately target to lysosomes for degradation. We hypothesize that loss of this parkin- and PINK1-dependent trafficking mechanism impairs the ability of mitochondria to selectively degrade oxidized and damaged proteins leading, over time, to the mitochondrial dysfunction noted in PD.
Keywords: MDV, mitochondria, mitophagy, Parkin, PINK1
See also: E Shlevkov & TL Schwarz (February 2014)
Introduction
Mitochondrial quality control, a term encompassing various cellular pathways overseeing the regulated turnover of mitochondrial proteins and lipids, ensures the functionality of the mitochondrial reticulum throughout the lifetime of the cell (Youle & van der Bliek, 2012). The deterioration of these mechanisms has been hypothesized to underlie the pathogeneses of several neurodegenerative diseases, most notably Parkinson's disease (PD), the second-most common neurodegenerative disorder worldwide (Tatsuta & Langer, 2008; Dawson et al, 2010; Schon & Przedborski, 2011). PINK1 and parkin, two PD-linked genes initially implicated in a common pathway regulating mitochondrial function in Drosophila, have been shown to mediate the degradation of dysfunctional mitochondria that have become depolarized by treatment of cells with the uncoupler CCCP (Greene et al, 2003; Clark et al, 2006; Park et al, 2006; Yang et al, 2006; Poole et al, 2008). In this paradigm, PINK1, a serine/threonine kinase, globally accumulates on depolarized mitochondria and recruits parkin from the cytosol to mitochondria in a manner dependent upon PINK1 phosphorylation activity (Narendra et al, 2008, 2010; Matsuda et al, 2010; Vives-Bauza et al, 2010; Greene et al, 2012). At depolarized mitochondria, parkin, an E3 ubiquitin ligase, catalyzes the polyubiquitination of several substrates, including the mitofusins and VDACs, and triggers whole-mitochondrial engulfment by autophagosomes and subsequent degradation through the autophagosome-lysosome pathway (a process known as mitophagy; Narendra et al, 2008; Geisler et al, 2010; Poole et al, 2010; Tanaka et al, 2010; Ziviani et al, 2010; Wang et al, 2011; Sun et al, 2012). Kinetic studies of mitophagic clearance of entire mitochondria by PINK1 and parkin have demonstrated that this process occurs on the scale of several hours to days (Chan et al, 2011; Yoshii et al, 2011).
Recently, we showed that a subpopulation of mitochondrial-derived vesicles (MDVs)—cargo-selective vesicles that bud off mitochondria independently of the mitochondrial fission machinery—play a role in mitochondrial quality control (Neuspiel et al, 2008; Soubannier et al, 2012a,b). We have shown that MDVs mediate transport between mitochondria and various other organelles in response to a variety of stimuli (Neuspiel et al, 2008; Braschi et al, 2010; Soubannier et al, 2012a). Correspondingly, upon generation of cytosolic oxidative stress, we observed the biogenesis of mitochondrial vesicles that ultimately targeted to lysosomes for degradation, on a scale of tens of minutes to an hour (Soubannier et al, 2012a). As in vitro reconstitution experiments demonstrated that these vesicles were enriched for oxidized proteins (Soubannier et al, 2012b), we hypothesized that certain populations of mitochondrial vesicles may regulate a form of mitochondrial quality control, kinetically faster than mitophagy, which would function prior to total depolarization in an effort to preserve the integrity of the organelle.
Here, we demonstrate the involvement of parkin and PINK1 in the generation of MDVs in response to mitochondrial oxidative stress. Parkin colocalizes with MDVs in a PINK1-dependent manner, and stimulates their formation in response to antimycin A, a potent generator of reactive oxygen species (ROS). Once formed, these vesicles target to lysosomes for degradation in a manner independent of canonical mitophagy. These findings implicate PINK1 and parkin in a mitochondrial quality control pathway that operates at an early stage in order to salvage mitochondria by selectively extracting damaged components via vesicular carriers. We propose that it is only once this first line of defense is overwhelmed—and that mitochondria become irreversibly damaged—that the entire organelle is targeted for degradation by mitophagy.
Results
Mitochondrial ROS generate mitochondrial vesicles in a parkin-dependent manner
To test whether parkin is involved in a mitochondrial vesicular trafficking pathway, we expressed GFP-parkin and a mitochondrial matrix-targeted DsRed2 (OCT-DsRed2) in HeLa cells, which express no endogenous parkin (Denison et al, 2003). Throughout this study, to ensure that we were examining MDVs rather than mitochondrial fragments generated by fission, Drp1-dependent fission was suppressed by either overexpressing a dominant-negative Drp1 mutant (CFP-Drp1K38E, Supplementary Fig S1; Harder et al, 2004) or by reducing endogenous Drp1 levels via siRNA. These cells, subjected to a variety of treatments, were fixed and immunostained against the outer mitochondrial membrane (OMM) protein TOM20 (Fig1A). In untreated cells, GFP-parkin remained predominantly cytosolic, and both mitochondrial markers overlapped almost perfectly (Fig1B, untreated). As expected, disruption of Δψm with CCCP led to recruitment of parkin en masse to the mitochondrial reticulum (Fig1B, CCCP), which remained elongated as Drp1-dependent fission was inhibited by CFP-Drp1K38E. In our previous study, the generation of ROS within mitochondria in COS7 cells (which express low levels of endogenous parkin), using the complex III inhibitor antimycin A, led to the formation of specific MDVs that incorporated selective cargo (Soubannier et al, 2012a). In vitro reconstitution followed by biochemical and ultrastructural analysis showed that these vesicles were enriched for oxidized cargo proteins, and that matrix cargo-containing MDVs, which excluded TOM20, were double-membrane structures containing both an inner and outer membrane (Soubannier et al, 2012b). Strikingly, treatment of HeLa cells with antimycin A for 2 h led to the recruitment of GFP-parkin from the cytosol to puncta containing the matrix-targeted DsRed2 but not TOM20 (Fig1C, boxes i to iii). Although we only observed these antimycin A-induced, matrix-positive/TOM20-negative MDVs in HeLa cells expressing GFP-parkin and not GFP alone (Fig2A), only 20–40% of these structures colocalized with parkin, often at the periphery of mitochondria on what appeared to be nascent, budding vesicles (Fig1C, arrowheads), as well as ones that had dissociated completely from mitochondria (Fig1C, arrows). Thus, whereas both parkin and mitochondrial ROS are required to induce this population of double-membraned mitochondrial vesicles, it appears that the association of parkin with these structures is transient and occurs initially at sites of MDV budding.
Figure 1.
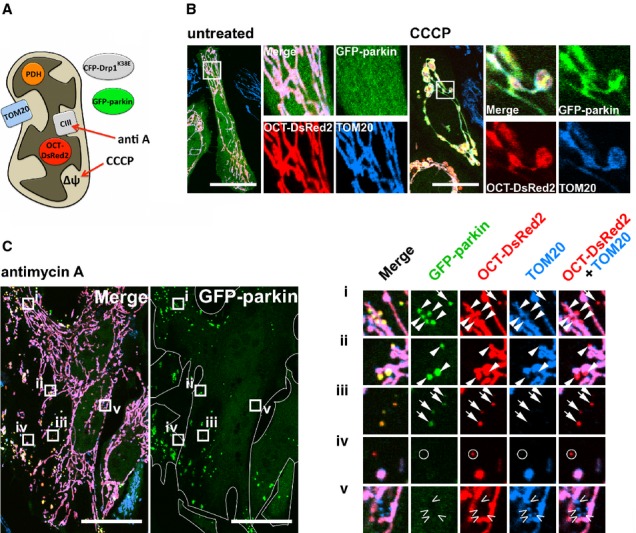
- Schematic depicting the location of relevant protein markers relative to the mitochondrion, as well as the site of action of pharmacological agents used.
- HeLa cells expressing GFP-parkin (green), OCT-DsRed2 (red) and CFP-Drp1K38E were left untreated (untreated) or treated for 2 h with 10 μM CCCP (CCCP), then fixed and stained for TOM20 (blue). Scale bars, 30 μm.
- Cells prepared as in (B) were treated with 50 μM antimycin A for 2 h. Arrows indicate OCT-DsRed2-positive/TOM20-negative MDVs that colocalize with GFP-parkin, while circles indicate MDVs that are parkin-negative. Arrowheads indicate “nascent” vesicles, adjacent to mitochondria, exhibiting cargo selectivity and parkin recruitment. Open arrowheads indicate parkin-negative MDVs containing the reciprocal cargo (TOM20-positive/OCT-DsRed2-negative). Cell boundaries are delineated in the GFP-parkin single-channel image. Scale bars, 30 μm.
Figure 2.
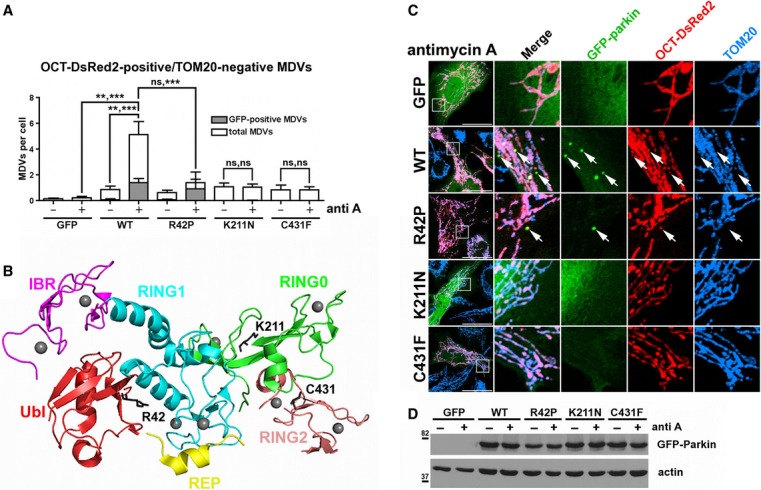
- Quantification of the number of OCT-DsRed2-positive/TOM20-negative vesicles in cells expressing GFP, GFP-parkinWT, GFP-parkinR42P, GFP-parkinK211N, or GFP-parkinC431F, treated with or without antimycin A (anti A) as in (C); both the total number (white bars) and the number colocalizing with GFP-parkin (gray bars) are indicated. Bars represent the mean ± s.e.m. P-values are given first for GFP-/GFP-parkin-positive vesicles, then for total vesicle number (n = 49–68 cells in 2–3 experiments); ns, not significant; **P < 0.01; ***P < 0.001).
- Structure of parkin, with PD-linked residues from (D) highlighted in dark gray (PDB ID: 4K95). Ubl, ubiquitin-like domain (red); RING0, RING0 domain (green); RING1, RING1 domain (cyan); IBR, in-between RING (magenta); REP, repressor element of parkin (yellow); RING2, RING2 domain (salmon).
- HeLa cells expressing various GFP-parkin mutant contructs (green), pOCT-DsRed2 (red) and CFP-Drp1K38E were treated with 50 μM antimycin A for 2 h, then fixed and immunostained against TOM20. Arrows indicate matrix-positive/TOM20-negative structures colocalizing with GFP-parkin. Scale bars, 30 μm.
- Immunoblot of whole-cell lysates of HeLa cells expressing various GFP-parkin constructs, treated with 50 μM antimycin A (anti A, +) or DMSO (−) for 2 h.
Importantly, we found that the antimycin A-induced vesicular recruitment of parkin was specific to this OCT-DsRed2-positive/TOM20-negative MDV population, as vesicles with the reciprocal cargo (TOM20-positive/OCT-DsRed2-negative) lacked parkin (Fig1C, open arrowheads). Moreover, treatment of GFP-parkin-exressing HeLa cells with antimycin A did not increase the overall number of this other, reciprocal vesicle population (Supplementary Fig S2A). When cells were treated with glucose oxidase, which induces ROS formation external to mitochondria, for 2 h—a treatment that triggers the robust formation of TOM20-positive/matrix-negative vesicles (Soubannier et al, 2012a)—we again did not observe the recruitment GFP-parkin to these MDVs (Supplementary Fig S2B, open arrowheads). Thus, parkin is recruited to a specific population of MDVs upon generation of mitochondrial ROS.
Next, we tested whether different PD-linked parkin mutants (depicted in Fig2B) could induce MDVs in response to antimycin A. The number of matrix-positive, TOM20-negative structures was quantified in antimycin A-treated HeLa cells expressing GFP, GFP-parkinWT, GFP-parkinR42P, GFP-parkinK211N or GFP-parkinC431F. Only upon expression of GFP-parkinWT did we observe the formation of matrix-positive MDVs (Fig2A and C). In contrast, both GFP-parkinK211N and GFP-parkinC431F failed to generate mitochondrial vesicles (Fig2A and C). The mutation of the catalytic C431 abolishes parkin ubiquitin ligase activity (Trempe et al, 2013), suggesting a role for ubiquitination in MDV biogenesis, and the GFP-parkinK211N has been previously reported to be completely deficient in depolarization-induced parkin translocation (Geisler et al, 2010). In the case of cells expressing GFP-parkinR42P, it was found that, although the antimycin A-dependent increase in total matrix-positive, TOM20-negative vesicles was not significant (Fig2A and C), parkin colocalized with most of the few mitochondrial vesicles present in these treated cells (Fig2A). The R42P mutation disrupts the folding of the Ubl domain but does not abolish CCCP-induced mitochondrial translocation (Safadi & Shaw, 2007; Geisler et al, 2010; Narendra et al, 2010). Moreover, as an intact Ubl is critical for parkin to interact with membrane trafficking partners (Fallon et al, 2006; Trempe et al, 2009), we hypothesize that a functional Ubl domain may facilitate interactions between parkin and trafficking proteins downstream of vesicle formation. Additionally, we do not disregard the possibility that GFP-parkinR42P-induced MDV formation may simply be kinetically slower in comparison to the wild-type (WT).
To further extend our findings and show they were not cell type-specific, instead of HeLa cells, we used U2OS cells stably expressing either GFP (U2OS:GFP) or GFP-parkin (U2OS:GFP-parkin) and, instead of CFP-Drp1K38E, we used Drp1 siRNA to inhibit mitochondrial fission (Fig3A). To determine whether the parkin- and antimycin A-induced structures could incorporate endogenous mitochondrial proteins, instead of using OCT-DsRed2, the cells were stained with antibodies raised against the E2 and E3bp subunits of the pyruvate dehydrogenase complex (PDH E2/E3bp, localized to the mitochondrial matrix; Fig1A) as well as TOM20. We again observed cargo-selective (PDH E2/E3bp-positive/TOM20-negative) structures that did (Fig3B, arrows) or did not (Fig3B, circles) colocalize with parkin after treatment. Expectedly, silencing of Drp1 had no effect on the total number of MDVs observed, confirming that MDVs are not a product of mitochondrial fission (Fig3C). Again, only a fraction of the parkin-induced structures colocalized with GFP-parkin (Fig3C) and those that did were predominantly adjacent to mitochondrial tubules (Fig3D and E). Next, we asked whether parkin is required for the formation of this class of MDVs. We had previously shown that, in COS7 cells, a population of PDH E2/E3 bp-positive, TOM20-negative MDVs was similarly induced by treatment with antimycin A (Soubannier et al, 2012a). Silencing of endogenous parkin with siRNA in these cells (Supplementary Fig S3A) significantly reduced the number of PDH E2/E3 bp-positive, TOM20-negative MDVs that formed in response to antimycin A (Supplementary Fig S3B). Thus, the ROS-induced biogenesis of this class of MDV requires endogenous parkin in COS7 cells and is stimulated by parkin expression under multiple conditions in a variety of cell lines.
Figure 3.

- Representative immunoblot of whole-cell lysates from U2OS:GFP and U2OS:GFP-parkin cells transfected with non-targeting siRNA or siRNA targeting Drp1 (siDrp1).
- U2OS:GFP-parkin cells transfected with siRNA targeting Drp1 were treated with DMSO (upper panels) or 25 μM antimycin A (anti A, lower panels) for 90 min, then fixed and immunostained against PDH (red) and TOM20 (blue). PDH E2/E3 bp-positive/TOM20-negative MDVs colocalizing with GFP-parkin (arrows) or not (circles) are indicated. Scale bars, 20 μm (first panels on left) and 2 μm.
- Quantification of PDH-positive/TOM20-negative vesicles in U2OS:GFP and U2OS:GFP-parkin cells transfected with the indicated siRNA, treated with DMSO or antimycin A (anti A) as in (B); both the total number (white bars) and the number colocalizing with GFP-parkin (gray bars) are indicated. Bars represent the mean ± s.e.m. P-values are given first for GFP-/GFP-parkin-positive vesicles, then for total vesicle number (n = 48–85 cells in 2–3 experiments); ns, not significant; ***P < 0.001).
- 5 μm-long profile of the parkin-positive vesicle and adjacent mitochondrial tubule depicted in the antimycin A-treated cell from (B) (left). Increasing position values on the x-axis of the fluorescence intensity plot (right) correspond to moving from the top to the bottom of the profile. *MDV indicated by an arrow in (B).
- Quantification of the distance between the centres of GFP-parkin-negative (black bar) or -positive (gray bar) vesicles and the edge of the nearest mitochondrial tubule for the vesicles quantified in antimycin A-treated U2OS:GFP-parkin cells transfected with siDrp1 in (C). Error bars represent the mean ± s.e.m.; *P < 0.05 (obtained by Student's t-test).
- U2OS:GFP-parkin cells transfected with siRNA targeting Drp1 (siDrp1) were treated with 25 μM antimycin A for 90 min prior to fixation. Samples were immunostained against TOM20 (blue) and the indicated mitochondrial marker (red). TOM20-negative MDVs containing the specified cargo and colocalizing with GFP-parkin (arrows) or not (circles) are indicated. Arrowheads show lack of colocalization between GFP-parkin and the indicated mitochondrial marker. Scale bars, 20 μm (first two columns of panels) and 2 μm (remaining columns of panels).
- Quantification of TOM20-negative structures that stained positively for the indicated cargo in U2OS:GFP (GFP) and U2OS:GFP-parkin (GFP-parkin) cells, transfected with siRNA targeting Drp1, treated with antimycin A (anti A); both the total number (white bars) and the number colocalizing with GFP-parkin (gray bars) are indicated. Error bars represent the mean ± s.e.m. P-values are given first for GFP-/GFP-parkin-positive vesicles, then for total vesicle number (n = 24–59 cells in two experiments; ns, not significant; ***P < 0.001).
Source data are available online for this figure.
To further characterize the endogenous cargo being carried by these vesicles, we also screened a variety of other mitochondrial proteins to determine if they, like PDH E2/E3 bp and OCT-DsRed2, were incorporated into the parkin-dependent MDVs in U2OS:GFP-parkin cells. Strikingly, we did not observe sorting of the E1α subunit of the PDH complex (PDH E1a) in parkin-dependent MDVs (Fig3F and G), suggesting that the megadalton PDH complex is at least partially disassembled before incorporation into vesicles. Moreover, while COXI (a transmembrane component of complex IV of the electron transport chain [ETC] located in the inner mitochondrial membrane [IMM]) was incorporated into the antimycin A/parkin-induced MDV population, several other mitochondrial proteins, including cytochrome c (intermembrane space [IMS]), TIM23 (IMM) and TRAP1 (IMS/matrix) were not (Fig3F and G). These data confirm that cargo selection extends beyond the exclusion of TOM20 and suggest that components from the same mitochondrial locale and complex sort differentially into vesicles, in line with what has been observed in in vitro reconstitution assays (Soubannier et al, 2012b).
Mitochondrially localized parkin has been shown to polyubiquitinate and mediate the degradation of OMM proteins in a manner dependent upon p97/VCP and the proteasome (Tanaka et al, 2010; Chan et al, 2011; Yoshii et al, 2011). Thus, it could be conceived that the exclusion of TOM20 from the observed parkin-dependent, antimycin A-induced MDV population may result from its proteasomal degradation. To test this, we treated HeLa cells expressing GFP-parkin with antimycin A in the presence of MG132 or epoxomicin, two proteasomal inhibitors (Supplementary Fig S4A). Under circumstances of proteasomal inhibition, we observed no decrease in the total number of antimycin A-induced PDH E2/E3 bp-positive/TOM20-negative structures, or those colocalizing with parkin (Supplementary Fig S4B). Additionally, we did not observe a change in the ratios of PDH E2/E3 bp-positive/TOM20-negative puncta (vesicles) with those that were doubly-positive (mitochondrial fragments) that colocalized with GFP-parkin (Supplementary Fig S4C), as would be expected if parkin were degrading TOM20 from the surface of these structures. We also did not observe any change in total levels of TOM20 in U2OS:GFP-parkin cells treated with antimycin A compared to those treated with DMSO (Supplementary Fig S4D and E). Finally, we silenced p97/VCP expression in U2OS:GFP-parkin cells (Supplementary Fig S4F), and observed no change in the total number of antimycin A-induced MDVs (Supplementary Fig S4G). Taken together with our data demonstrating differential sorting of endogenous cargo into vesicles (Fig3), these results implicate bona fide selective incorporation—rather than selective degradation—in the generation of parkin-dependent MDVs.
Parkin MDVs are targeted to lysosomes for degradation independently of autophagy
As our previous study had shown that ROS-induced mitochondrial vesicles are targeted to lysosomes for degradation (Soubannier et al, 2012a), we suspected the fate of parkin-dependent vesicles to be similar. To address this, we first silenced Drp1 expression in HeLa cells (Supplementary Fig S5A), and overexpressed either GFP or GFP-parkin. In cells expressing GFP-parkin, we saw an increase in PDH E2/E3 bp-positive/TOM20-negative structures after antimycin A treatment (Supplementary Fig S5B), similar to what we had observed in U2OS:GFP-parkin cells. We then incubated these HeLa cells with antimycin A in the presence of the V-ATPase inhibitor bafilomycin A1 or the lysosomal hydrolase inhibitors pepstatin A and E64d, inhibitors commonly used in the study of the lysosomal degradative pathway (Mizushima et al, 2010; Klionsky et al, 2012). Both the total number of PDH E2/E3 bp-positive/TOM20-negative MDVs, as well as the number of vesicles that colocalized with GFP-parkin, increased significantly in the presence of antimycin A and bafilomycin or pepstatin/E64d compared to treatment with antimycin A alone (Fig4A and B). The increase resulting from lysosomal inhibition was parkin-dependent, as it was not observed in cells expressing an empty GFP vector (Fig4B).
Figure 4.
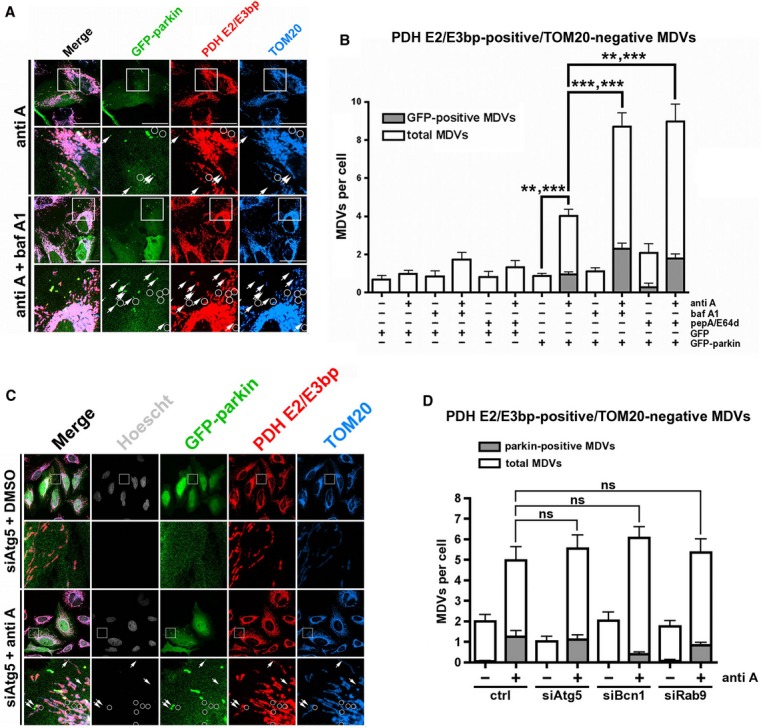
- HeLa cells, transfected with siRNA targeting Drp1 and GFP-parkin (green), were treated with 25 μM antimycin A for 90 min following a 30-min pretreatment with 50 nM bafilomycin A1, then fixed and immunostained against PDH E2/E3 bp (red) and TOM20 (blue). PDH E2/E3 bp-positive/TOM20-negative MDVs colocalizing with GFP-parkin (arrows) or not (circles) are indicated. Scale bar, 30 μm.
- Quantification of PDH E2/E3bp-positive/TOM20-negative vesicles in HeLa cells treated with antimycin A in the presence of the lysosomal inhibitors bafilomycin A1 or pepstatin A and E-64d; both total number (white bars) and the number colocalizing with GFP-parkin (gray bars) are indicated. Bars represent the mean ± s.e.m. P-values are given for total vesicle number (n = 19 to 120 cells in 2–3 experiments; **P < 0.01; ***P < 0.001).
- HeLa cells, transfected with GFP-parkin (green) and siRNA targeting Drp1 and Atg5, were treated with 25 μM antimycin A for 90 min, then fixed, immunostained for PDH E2/E3 bp (red) and TOM20 (blue), and counterstained for Hoescht (gray). PDH E2/E3 bp-positive/TOM20-negative MDVs colocalizing with GFP-parkin (arrows) or not (circles) are indicated. Scale bars, 20 μm.
- Quantification of PDH E2/E3 bp-positive/TOM20-negative vesicles from HeLa cells transfected with the indicated siRNA; both the total number (white bars) and the number colocalizing with GFP-parkin (gray bars) are indicated. Error bars represent the mean ± s.e.m. P-values are given for the total vesicle number (n = 42–64 cells in 2–3 experiments); ns, not significant.
Given that parkin-dependent mitochondrial turnover via autophagy requires mitochondrial fission (Twig et al, 2008; Tanaka et al, 2010) and that Drp1 was suppressed in our cells, it is unlikely that the parkin-induced MDV biogenesis that we observed involved canonical autophagy pathways. To definitively test whether the role of parkin in the formation of mitochondrial vesicles is distinct from its established role in mitophagy, MDV formation in autophagy-deficient cell systems was investigated. We observed antimycin A-induced mitochondrial vesicles in parkin-expressing, immortalized mouse embryonic fibroblasts (MEFs) generated from both WT mice and mice lacking Atg5 (an essential component of the canonical autophagic machinery, and required for parkin-dependent mitophagy; Supplementary Fig S6A). In HeLa cells, expression of Atg5, beclin-1 (a core component of the PI[3]K complex regulating autophagy) and Rab9 (implicated in an alternative autophagic pathway) were individually silenced using siRNA (Supplementary Fig S6B; Xie & Klionsky, 2007; Narendra et al, 2008; Nishida et al, 2009). In these cells, autophagy was severely inhibited, as evidenced by the lack of GFP-LC3-positive autophagosomes (Supplementary Fig S6C). When GFP-parkin was expressed in these autophagy-deficient HeLa cells, we again observed an antimycin A-dependent increase in the total number of PDH E2/E3 bp-positive/TOM20-negative vesicles (Fig4C and D). As these numbers remained unchanged between autophagy-deficient and control cells, it is unlikely that these autophagy-related proteins play a role in parkin-dependent MDV biogenesis or degradation, as, in these instances, the number of vesicles would be expected to decrease or increase, respectively.
PINK1 is required for parkin-dependent MDV biogenesis
Given that parkin-dependent mitophagy has been shown to require PINK1 protein stabilization on mitochondria (Geisler et al, 2010; Matsuda et al, 2010; Narendra et al, 2010; Vives-Bauza et al, 2010), the involvement of PINK1 in parkin-dependent MDV formation was investigated. PINK1 expression was silenced via siRNA-mediated knockdown in HeLa cells expressing GFP-parkin (Fig5A), and the number of PDH E2/E3 bp-positive/TOM20-negative structures was quantified after treatment with antimycin A. Interestingly, it was observed that, in cells lacking PINK1, antimycin A-induced, parkin-dependent MDV biogenesis was severely abrogated (Fig5B and C). This finding indicates that PINK1 and parkin regulate both mitophagy and mitochondrial vesicle formation, and may do so in response to distinct stimuli. ROS generation may induce the formation of MDVs in an effort to preserve organelle integrity, while mitophagy may occur only once this vesicular system is overwhelmed and Δψm has been lost.
Figure 5.
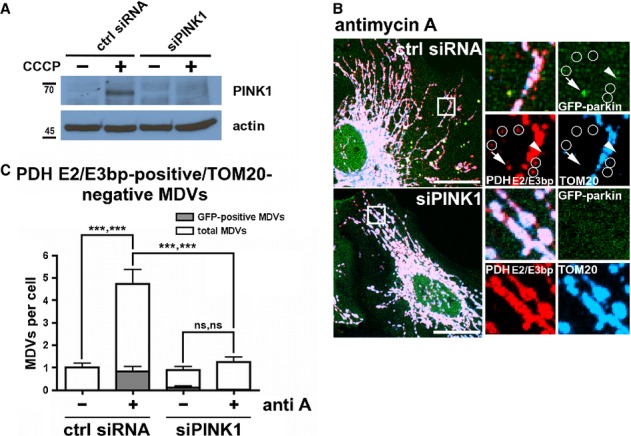
- Representative immunoblot of whole-cell lysates from HeLa cells transfected with non-targeting siRNA or siRNA targeting PINK1 (siPINK1), treated with 10 μM CCCP for 6 h in order to stabilize the PINK1 full-length band.
- HeLa cells transfected with GFP-parkin (green) and siRNA targeting PINK1 (siPINK1) or non-targeting control (ctrl siRNA), were treated with 25 μM antimycin A for 90 min, then fixed and immunostained for PDH E2/E3 bp (red) and TOM20 (blue). PDH E2/E3 bp-positive/TOM20-negative MDVs colocalizing with GFP-parkin (arrows) or not (circles) are indicated. Arrowheads indicate parkin-positive MDVs adjacent to mitochondria, possibly budding. Scale bar, 20 μm.
- Quantification of PDH E2/E3 bp-positive/TOM20-negative structures from (B); both the total number (white bars) and the number colocalizing with GFP-parkin (gray bars) are indicated. Error bars represent the mean ± s.e.m. P-values are given first for GFP-/GFP-parkin-positive vesicles, then for total vesicle number (n = 42–60 cells in three experiments); ns, not significant; ***P < 0.001).
Source data are available online for this figure.
Parkin-dependent MDV formation and turnover precedes mitophagy
In order to test our hypothesis that, relative to mitophagy, parkin-dependent vesicle biogenesis represents an early response to mitochondrial stress, we performed kinetic experiments in U2OS:GFP-parkin cells, a cell line which undergoes parkin-dependent mitophagy in response to loss of Δψm by CCCP (Lefebvre et al, 2013). As we decided to monitor both MDV formation and mitophagy, we did not manipulate Drp1 levels. In addition to using antimycin A and CCCP to generate mitochondrial ROS and ablate Δψm, respectively, we also chose to incubate cells with both antimycin A and oligomycin (a mitochondrial ATPase inhibitior). As oligomycin blocks both forward and reverse electron flow through the F1F0 ATP synthase, its use would prevent, during antimycin A-induced respiratory chain inhibition, the attempted repolarization of mitochondria through reverse ATP synthase activity (Hatefi, 1985).
We first monitored Δψm in these cells by TMRM fluorescence, and observed a sharp decline in TMRM fluorescence signal (down to 23.8% of DMSO-treated control cells) following CCCP treatment at 30 min (the first time point measured) that was maintained for the 6 h of the experiment (Fig6A, CCCP). As expected, antimycin A induced a much smaller decrease in TMRM signal after the first hour (67.0% of control at 60 min), which was then followed by a rebound in Δψm that, at 6 h, had recovered almost completely (90.5% of control; Fig6A, anti A). This finding is consistent with the phenomenon of reverse proton flow through the F1F0 ATPase induced by compromised mitochondrial respiration (Hatefi, 1985; Campanella et al, 2008). Indeed, combined inhibition with antimycin A and oligomycin prevented this rebound, and Δψm decreased steadily over time (to 50.9% of control at 6 h; Fig6A, anti A + oligo), although this decline was not as severe as cells treated with CCCP (17.2% of control at 6 h, Fig6A). Interestingly, we still observed global GFP-parkin recruitment in U2OS:GFP-parkin cells treated with both antimycin A and oligomycin for 2 h (Fig6B). This result supports previous data showing that perturbations in the Δψm-dependent import machinery can amplify effects on parkin recruitment to mitochondria (Greene et al, 2012), and is in line with a study demonstrating that F1F0 ATPase inhibition is sufficient to induce parkin recruitment in non-respiring cells (Suen et al, 2010).
Figure 6.
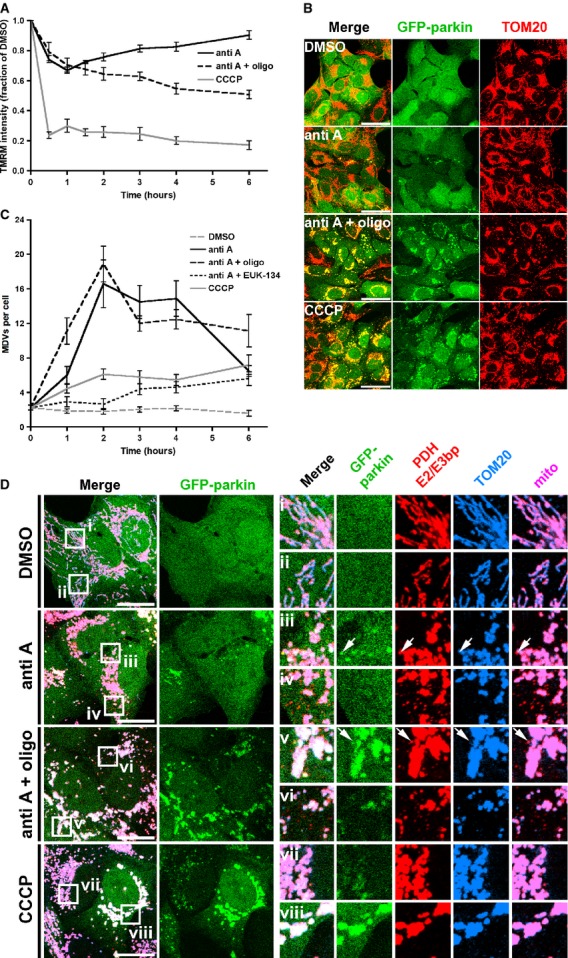
- Quantification of TMRM fluorescence by flow cytometry in U2OS:GFP-parkin cells treated with 25 μM antimycin A (anti A), 25 μM antimycin A and 10 μM oligomycin (anti A + oligo), and 20 μM CCCP (CCCP) for the indicated time, represented as a fraction of fluorescence intensity compared to DMSO-treated cells. Error bars represent the mean ± s.e.m. (n = 3 experiments).
- U2OS:GFP-parkin cells were treated with DMSO (DMSO), 25 μM antimycin A (anti A), 25 μM antimycin A and 10 μM oligomycin (anti A + oligo), or 20 μM CCCP (CCCP) for two hours, then fixed and immunostained for TOM20 (red). Scale bars, 50 μm.
- Quantification of the total number of PDH E2/E3 bp-positive/TOM20-negative structures in U2OS:GFP-parkin cells observed at various times for the indicated treatment. Error bars represent the mean ± s.e.m. (n = 46–166 cells in 2–3 experiments).
- U2OS:GFP-parkin cells were treated with DMSO (DMSO), 25 μM antimycin A (anti A), 25 μM antimycin A and 10 μM oligomycin (anti A + oligo), or 20 μM CCCP (CCCP) for two hours, then fixed and immunostained for PDH E2/E3 bp (red) and TOM20 (blue). A merged image of both mitochondrial markers (mito) is shown at the far right. Arrows indicate PDH E2/E3 bp-positive/TOM20-negative structures colocalizing with GFP-parkin (green). Scale bars, 20 μm.
Source data are available online for this figure.
We then addressed the kinetics of MDV formation by quantifying the total number of PDH E2/E3 bp-positive/TOM20-negative structures over time in U2OS:GFP-parkin cells treated with DMSO, CCCP, antimycin A, antimycin A and oligomycin, and antimycin A and the ROS scavenger EUK-134 (Fig6C and D). Both antimycin A and antimycin A combined with oligomycin triggered vesicle formation, which peaked at 2 h (Fig6C and D, anti A and anti A + oligo). Interestingly, the average number of vesicles per cell decreased after this time, likely due to their lysosomal turnover, as well as decreased biogenesis owing to inhibition of forward electron flow through the ETC mitigating the amount of ROS generated by antimycin A (Fig6A). Indeed, addition of EUK-134 decreased the observed number of antimycin A-induced MDVs, whereas incubation of cells with CCCP did not generate many vesicles (Fig6C, anti A + EUK-134 and CCCP), indicating that parkin MDV formation is a ROS-dependent process not induced by global depolarization.
Our observation that parkin-dependent MDV formation and turnover occur over a period of 1–4 h (Fig6C) contrasts this pathway with parkin-mediated mitochondrial turnover via autophagy, which occurs over a much larger scale of time (Narendra et al, 2008; Geisler et al, 2010). Indeed, depolarizing U2OS:GFP and GFP-parkin cells with CCCP or with antimycin A and oligomycin induced parkin-dependent mitophagy over a period of 4–24 h, as assayed by the decrease in levels of several mitochondrial markers (Fig7A and B and Supplementary Fig S7A). In contrast, no such effect was observed after treatment with antimycin A alone. Moreover, we observed cell-wide clearance of mitochondria in cells that had been depolarized for 12 h, but not in cells treated with antimycin A alone (Fig7C and D, Supplementary Fig S7B and C). These data support our view that parkin-dependent vesicle formation is a rapid response triggered by ROS, whereas turnover of the entire organelle is a relatively slow process initiated by depolarization.
Figure 7.
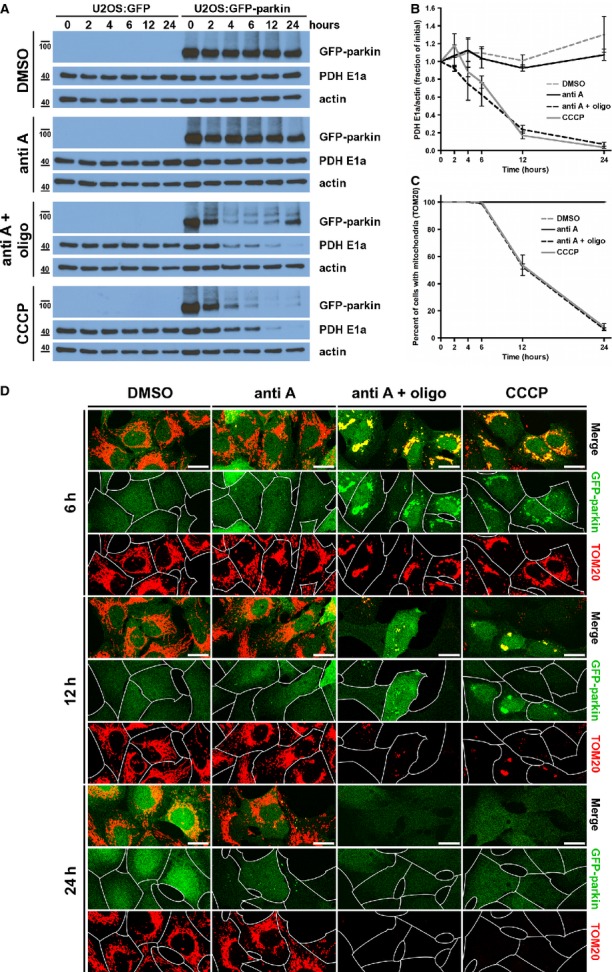
- Representative immunoblot of whole-cell lysates from U2OS:GFP and GFP-parkin cells treated with DMSO, 25 μM antimycin A (anti A), 25 μM antimycin A with 10 μM oligomycin (anti A + oligo), or 20 μM CCCP for the indicated time period.
- Quantification of PDH E1a signal intensity relative to that of actin in immunoblots from (A). Error bars represent the mean ± s.e.m. (n = 3 experiments).
- Quantification of mitochondrial clearance in U2OS:GFP-parkin cells treated as in (A), fixed and immunostained for TOM20. Data are shown as percentage of cells containing mitochondria, by TOM20 staining, visualized by fluorescence microscopy. Error bars represent the mean ± s.e.m. (n = 3 experiments, with at least 85 cells quantified per condition, per experiment).
- U2OS:GFP-parkin cells were treated as in (A) for the indicated time period, then fixed and immunostained for TOM20 (red). Cell boundaries are delineated in single-channel images. Scale bars, 20 μm.
Source data are available online for this figure.
Discussion
Parkin function has been implicated in both vesicular trafficking and mitochondrial quality control (Fallon et al, 2002, 2006; Joch et al, 2007; Narendra et al, 2008; Trempe et al, 2009; Youle & Narendra, 2011). We recently identified MDV transport as a delivery mechanism of selective, oxidized cargo to the late endosome (Soubannier et al, 2012a,b). With this study, we greatly expand our understanding of this pathway with the identification of both parkin and PINK1 as core machinery required for the generation of double-membrane, matrix-containing MDVs. Thus, parkin and PINK1 sit at the crossroads of two distinct mitochondrial quality control mechanisms: the shuttling of selective cargo to lysosomes by autophagy-independent vesicular carriers, and the delivery of entire organelles to the autophagosome via autophagy. In contrast to mitophagy, parkin- and PINK1-dependent vesicles form in the absence of Drp1-dependent fission and are generated by ROS, not global mitochondrial depolarization. Additionally, these vesicles, whose formation can be induced by endogenous levels of parkin, preferentially incorporate certain endogenous mitochondrial cargo while excluding others, and target to lysosomes in a manner that is autophagy-independent. Kinetic studies demonstrate that much of the vesicle biogenesis and turnover occurs within the first few hours of treatment, whereas mitophagic clearance of mitochondria occurs over a period of days.
Both parkin-dependent mitophagy and vesicle formation may contribute to mitochondrial quality control, albeit it at differing levels. MDV formation in vitro shows that about 4% of the selected mitochondrial cargo can be sorted into vesicles per hour (Soubannier et al, 2012b), which is similar to the 6–12% of protein turnover generated by mitochondrial proteases in yeast (Augustin et al, 2005). Our data suggest that parkin may mediate the extraction of damaged respiratory proteins and chaperones by vesicular carriers that transport them to lysosomes for degradation, thus preserving the integrity of the whole mitochondrion at the steady state. If, however, these damaged proteins accumulate beyond a certain threshold, the dissipation of Δψm would trigger the autophagic engulfment and degradation by parkin-dependent mitophagy.
PINK1 may play a crucial role in the detection of this threshold. While CCCP triggers a massive PINK1 increase due to global import failure (Matsuda et al, 2010; Narendra et al, 2010; Greene et al, 2012; Lazarou et al, 2012), antimycin A-induced ROS may cause PINK1 buildup at local or individual import channels. This interplay between local and global PINK1 accumulation may regulate the magnitudes of subsequent parkin recruitment and ubiquitin ligase activity. This latter point is intriguing, as while mitochondrial polyubiquitination has been implicated in autophagy (Geisler et al, 2010; Tanaka et al, 2010; Chan et al, 2011), we have previously shown that interactions between parkin and binding partners involved in endocytic trafficking and membrane curvature are regulated by monoubiquitination (Fallon et al, 2006; Joch et al, 2007; Trempe et al, 2009). Additionally, monoubiquitination and ubiquitin cycling are common themes in the endocytic system (d'Azzo et al, 2005). In our study, a role for ubiquitination in MDV formation is supported by the observation that vesicle formation is abolished upon expression of the ligase-dead parkin mutant.
Mounting evidence implicates the dysregulation of membrane trafficking pathways, as well as mitochondrial dysfunction, in the pathogenesis of PD. Recently, two exome sequencing analyses linked the retromer subunit Vps35 to PD (Vilarino-Guell et al, 2011; Zimprich et al, 2011). Vps35, along with Vps26 and Vps29, forms the so-called cargo recognition complex of the retromer, which cooperates with sorting nexins to mediate cargo selection and membrane curvature, respectively (Bonifacino & Hurley, 2008; Cullen & Korswagen, 2012). Interestingly, Vps35 localizes to mitochondria and is required for the formation of a population of mitochondrial vesicles that targets to peroxisomes (Braschi et al, 2010). Clearly, identification of the machinery regulating cargo selection and membrane dynamics, downstream of PINK1-dependent parkin recruitment to bud sites, requires further elucidation.
It is becoming evident that, in neurons, continued deregulation of mitochondrial quality control can lead to neurodegeneration (Rugarli & Langer, 2012). The existence of two distinct, yet complimentary, mitochondrial quality control pathways involving parkin and PINK1 is supported by a recent study in Drosophila, in which parkin/PINK1 mitochondrial quality control was shown to have both autophagy-dependent and -independent components at steady-state in vivo (Vincow et al, 2013). Additionally, Vincow et al found that the turnover rates of individual subunits from the same multiprotein complex vary significantly, supporting our finding that different PDH subunits are sorted differentially into the MDV population that we observed.
In PD patients harbouring defects in parkin or PINK1, a loss of mitochondrial quality control mechanisms may lead to a buildup of mitochondrial damage, and ultimately mitochondrial dysfunction. This is supported by the observation that leukocytes collected from PD patients carrying parkin mutations have decreased mitochondrial respiratory capacity (Muftuoglu et al, 2004), similar to what has been observed in the substantia nigra of sporadic PD patients (Mann et al, 1992). Elucidating the precise role of PD-linked genes in the regulation of mitochondrial quality control, and how the loss of these processes may lead to the death of dopaminergic neurons, may point to specific degenerative mechanisms shared between genetic and sporadic forms of PD. Thus, further investigation of the exact role played by parkin and PINK1 in MDV biogenesis will enrich existing knowledge regarding mitochondrial quality control mechanisms and their relation to PD, and prospectively shed light on therapeutic possibilities regarding this debilitating, aging disease.
Materials and Methods
Reagents
MG132 and epoxomicin were obtained from Boston Biochem. EUK-134 was purchased from Cayman Chemical. Tetramethylrhodamine methyl ester (TMRM) was purchased from Molecular Probes. All other reagents were purchased from Sigma-Aldrich.
Plasmids
pEGFP-C3 (Clontech), pEGFP-parkin, pECFP-Drp1K38E (Harder et al, 2004), pEGFP-LC3B and mito-DsRed2/pOCT-DsRed2 (Harder et al, 2004) were used in this study. pEGFP-parkinR42P, pEGFP-parkinK211N and pEGFP-parkinC431F were generated from pEGFP-parkinWT using the QuikChange II site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions.
siRNA oligonucleotides
Stealth RNAi siRNA oligonucleotides were purchased from Invitrogen. These included siRNA duplexes targeting Atg5 [5′-AACCUUUGGCCUAAGAAGAAAUGGA-3′ (Chen et al, 2007)], beclin-1 [1:1 mixture of 5′-CAGUUUGGCACAAUCAAUAACUUCA-3′ and 5′-CAGGAACUCACAGCUCCAUUACUUA-3′ (Hoyer-Hansen et al, 2005)], Drp1 [5′-ACUAUUGAAGGAACUGCAAAAUAUA-3′ (Taguchi et al, 2007)] and Rab9 [5′-AAGUUUGAUACCCAGCUCUUCCAUA-3′ (Ganley et al, 2004)]. Non-targeting siRNA was also obtained from Invitrogen. siRNA targeting parkin (ON-TARGETplus SMARTpool), PINK1 (ON-TARGETplus J-004030-07) and p97/VCP (ON-TARGETplus SMARTpool) were purchased from Dharmacon.
Cell culture and transfection
HeLa cells (ATCC), COS7 cells (ATCC), Atg5+/+ and Atg5−/− MEFs (RIKEN BioResource Center, Ibaraki, Japan), and U2OS:GFP and GFP-parkin cells (Rob Screaton, University of Ottawa) were cultured in DMEM (Multicell) supplemented with 10% fetal bovine serum, L-glutamine, penicillin and streptomycin at 37°C with 5% CO2. HeLa cells and MEFs were plated on coverslips in 24-well plates (at 5–6 × 104 cells per well). Cells were simultaneously infected with the Drp1K38E-CFP adenovirus (MOI of 300) and transfected with 0.3–0.5 μg/ml of the indicated plasmid using jetPRIME transfection reagent (Polypus Transfection, Illkirch, France) according to the manufacturer's instructions. In the case of subsequent siRNA and DNA transfections, cells were first plated in 6-well or 10-cm plates, and transfected with 25–50 nM of each siRNA oligo using jetPRIME. At 24 h post-transfection, cells were plated in 6-well plates or on coverslips in 24-well plates (at 2.5 × 105 and 5–6 × 104 cells per well, respectively). The following day (48 h post-siRNA transfection), cells were transfected with DNA as indicated above. U2OS:GFP and GFP-parkin cells were plated for siRNA transfection, and then replated in 6-well plates or on coverslips in 24-well plates (at 2.5 × 105 and 4 × 104 cells per well, respectively) at 24 h post-transfection. For the time-course experiment, U2OS:GFP-parkin cells were plated on coverslips in 24-well plates (at 8 × 104 cells per well) the day prior to treatment.
Cell treatments
Unless otherwise specified, treatments were performed 24 h after DNA transfection and/or 72 h following siRNA transfection. Typically, cells were treated with 100 mU/ml glucose oxidase, 10 μM CCCP or 25–50 μM antimycin A for 90–120 min before fixation or lysis. In assays requiring lysosomal or proteasomal inhibition, cells were preincubated with 2 μM epoxomicin, 10 μM MG132, 50 nM bafilomycin A1 or 10 μg/ml pepstatin A and 10 μg/ml E-64d for 30 to 60 minutes, followed by an additional 90 min of the indicated inhibitor with antimycin A or DMSO. In the time-course experiment, cells were treated with DMSO, 25 μM antimycin A, 25 μM antimycin A and 10 μM oligomycin, 25 μM antimycin A and 50 μM EUK-134, and 20 μM CCCP for the indicated time prior to fixation.
Cell lysis and immunoblotting
Cells were rinsed in ice-cold PBS and lysed in lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40 substitute, 1% sodium deoxycholate, and a protease inhibitor cocktail [aprotinin, leupeptin, benzamidine and PMSF]) on ice. Protein content of the lysates was determined by BCA assay (Pierce/Thermo Scientific). Fifteen to 45 μg of protein were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Primary antibodies were diluted in 5% milk or 3% BSA in PBS-Tween, and incubations with primary antibody were performed overnight at 4°C. Primary antibodies used in this study included anti-actin (Millipore, MAB1501, 1:100,000), anti-Apg5 (Santa Cruz Biotechnology, sc-8667. 1:1,000), anti-beclin-1 (BD Biosciences, 612113, 1:1,000), anti-Drp1 (BD, 611113, 1:5,000), anti-GAPDH (Novus Biologicals, NB300-320, 1:50,000), anti-parkin (Santa Cruz Biotechnology, sc-32282, 1:2,000 [endogenous parkin] or 1:100,000 [ectopic parkin]), anti-PINK1 (Novus, BC100-494, 1:2,500), anti-Rab9 (Abcam, ab2810, 1:1,000), anti-TIM23 (BD, 611222, 1:40,000), anti-TOM20 (Santa Cruz Biotechnology, sc-11415, 1:50,000), anti-p97/VCP (Abcam, ab11433, 1:10,000), anti-PDH E1a (Abcam, ab110330, 1:5,000), anti-TRAP1 (Abcam, ab2721, 1:5,000), anti-α-tubulin (Santa Cruz Biotechnology, sc-23948, 1:50,000) and anti-VDAC1 (Abcam, ab14734, 1:40,000). HRP-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, and were diluted in 5% milk in PBS-Tween. Incubations with secondary antibodies were performed at room temperature for 1 h. Western Lightning ECL and Plus-ECL kits (PerkinElmer) were used to detect protein bands according to the manufacturer's instructions.
Immunofluorescence
Following treatments, cells were fixed in 6% paraformaldehyde and quenched with 50 mM NH4Cl in PBS. Cells were then permeabilized in PBS containing 0.25% Triton X-100 and blocked in 10% FBS in PBS. Primary antibodies were diluted in PBS containing 5% FBS. Primary antibodies used in this study included anti-COXI (Abcam, ab14705, 1:50), anti-cytochrome c (BD, 556432, 1:500), anti-PDH E1a (Abcam, ab110330, 1:50), anti-PDH E2/E3 bp (Abcam, ab110333, 1:1,000), anti-TIM23 (BD, 611222, 1:50), anti-TOM20 (Santa Cruz Biotechnology, sc-11415, 1:1,000) and anti-TRAP1 (Abcam, ab2721, 1:100). Alexa Fluor 555- and 647-conjugated donkey anti-mouse and anti-rabbit secondary antibodies were purchased from Molecular Probes, and a DyLight 405-conjugated donkey anti-rabbit secondary antibody was purchased from BioLegend. Both primary and secondary antibody incubations were performed at room temperature for 1 h. In certain cases, coverslips were counterstained with Hoescht 33342 (Molecular Probes) prior to mounting on glass slides using Aqua Poly/Mount (Polysciences Inc.).
Confocal microscopy and vesicle quantification
Confocal images (∽0.8 μm-thick slices) were acquired either on an LSM 510 Meta confocal microscope (Zeiss) through a 100×, 1.4 NA objective or on an LSM 710 (Zeiss) through either a 40×, 1.3 NA or a 63×, 1.4 NA objective using excitation wavelengths of 405, 458, 488, 543 and 633 nm. Image files were analyzed using ImageJ (NIH, Bethesda, MD, USA). Vesicles were identified by eye, based on cargo selectivity (i.e. the presence of one cargo and the absence of another) and quantified in two to three independent, blinded experiments. Typically, vesicles within 20–50 cells were counted per experiment.
Determination of Δψm by fluorescence-activated cell sorting
To quantitatively measure Δψm, we modified a protocol established by Chazotte (Chazotte, 2011). U2OS:GFP-parkin cells were plated in 12-well plates (at 1.3 × 105 cells per well) 12 h prior to incubation with TMRM. Cells were then pulsed with 600 nM TMRM diluted in culture medium for 30 min, rinsed with PBS and incubated in culture medium containing 150 nM TMRM. All treatments were performed in the presence of 150 nM TMRM. Following treatment with various inhibitors, cells were trypsinized for 2–3 min at 37°C, collected in culture medium without TMRM and then kept on ice. Fluorescence-activated cell sorting measurements were performed with a FACSCalibur flow cytometer (BD Biosciences). Data was analyzed using FlowJo software (Tree Star Inc., Ashland, OR, USA).
Statistical analysis
Histograms were generated from the mean number of vesicles per cell, obtained from two to three independent experiments in which the MDVs in at least 20 cells per experiment were counted. Error bars represent the mean ± s.e.m., which was calculated using the total number of cells quantified as the n-value. Statistical significance was determined by one- or two-way ANOVA followed by a Bonferroni post-hoc analysis (Prism 5; GraphPad Software, La Jolla, CA, USA). Differences were considered significant if P < 0.05.
Acknowledgments
We thank Miguel A. Aguileta and Marty Loignon (Montreal Neurological Institute) for the generation of the pEGFP-parkin mutants and technical assistance with the collection and analysis of FACS data, respectively. The Rab9 antibody was a gift from Peter S. McPherson (McGill University). The pEGFP-parkin and pEGFP-LC3 plasmids were provided by Michael G. Schlossmacher (University of Ottawa) and Ron R. Kopito (Stanford), respectively. The Atg5+/+ and Atg5−/− MEFs were provided by Noboru Mizushima (Tokyo Medical and Dental University). The U2OS:GFP and GFP-parkin cell lines were a gift from Rob Screaton (University of Ottawa). This work was supported by the Brain Repair Program from Neuroscience Canada as well as Operating Grants from the Canadian Institutes of Health Research (CIHR) to E.A.F. and H.M.M. G.L.M. has received support from the Fonds de la recherche du Québec—Santé (FRQS), Parkinson Society Canada, and the CIHR. E.A.F. is a Chercheur National of the FRQS.
Author contributions
GLM, VS, HMM and EAF conceived and planned the experiments, and interpreted the data. GLM, VS and CXC acquired and analyzed the data. GLM carried out the quantification and statistical analyses. GLM and EAF wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Legends
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S5
Source Data for Supplementary Figure S6
Source Data for Supplementary Figure S7
Source Data for Figures 2A, 3C, 3G, 4B, 4D, 5B, 6C, 7B, 7C and Supplementary Figures S2A, S4B, S4E, S4G, S7B
Review Process File
Source Data for Figure 2D
Source Data for Figure 3A
Source Data for Figure 5A
Source Data for Figure 7A
References
- Augustin S, Nolden M, Muller S, Hardt O, Arnold I, Langer T. Characterization of peptides released from mitochondria: evidence for constant proteolysis and peptide efflux. J Biol Chem. 2005;280:2691–2699. doi: 10.1074/jbc.M410609200. [DOI] [PubMed] [Google Scholar]
- d'Azzo A, Bongiovanni A, Nastasi T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic. 2005;6:429–441. doi: 10.1111/j.1600-0854.2005.00294.x. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol. 2010;20:1310–1315. doi: 10.1016/j.cub.2010.05.066. [DOI] [PubMed] [Google Scholar]
- Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazotte B. Labeling mitochondria with TMRM or TMRE. Cold Spring Harb Protoc. 2011;2011:895–897. doi: 10.1101/pdb.prot5641. [DOI] [PubMed] [Google Scholar]
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci. 2007;120:4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Korswagen HC. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat Cell Biol. 2012;14:29–37. doi: 10.1038/ncb2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison SR, Wang F, Becker NA, Schule B, Kock N, Phillips LA, Klein C, Smith DI. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene. 2003;22:8370–8378. doi: 10.1038/sj.onc.1207072. [DOI] [PubMed] [Google Scholar]
- Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, Brice A, van Bergen En Henegouwen PM, Fon EA. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- Fallon L, Moreau F, Croft BG, Labib N, Gu WJ, Fon EA. Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. J Biol Chem. 2002;277:486–491. doi: 10.1074/jbc.M109806200. [DOI] [PubMed] [Google Scholar]
- Ganley IG, Carroll K, Bittova L, Pfeffer S. Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol Biol Cell. 2004;15:5420–5430. doi: 10.1091/mbc.E04-08-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem. 1985;54:1015–1069. doi: 10.1146/annurev.bi.54.070185.005055. [DOI] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jaattela M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005;12:1297–1309. doi: 10.1038/sj.cdd.4401651. [DOI] [PubMed] [Google Scholar]
- Joch M, Ase AR, Chen CX, MacDonald PA, Kontogiannea M, Corera AT, Brice A, Seguela P, Fon EA. Parkin-mediated monoubiquitination of the PDZ protein PICK1 regulates the activity of acid-sensing ion channels. Mol Biol Cell. 2007;18:3105–3118. doi: 10.1091/mbc.E05-11-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V, Du Q, Baird S, Ng AC, Nascimento M, Campanella M, McBride HM, Screaton RA. Genome-wide RNAi screen identifies ATPase inhibitory factor 1 (ATPIF1) as essential for PARK2 recruitment and mitophagy. Autophagy. 2013;9:1770–1779. doi: 10.4161/auto.25413. [DOI] [PubMed] [Google Scholar]
- Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease. Brain. 1992;115(Pt 2):333–342. doi: 10.1093/brain/115.2.333. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M, Elibol B, Dalmizrak O, Ercan A, Kulaksiz G, Ogus H, Dalkara T, Ozer N. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord. 2004;19:544–548. doi: 10.1002/mds.10695. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. 2008;18:102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safadi SS, Shaw GS. A disease state mutation unfolds the parkin ubiquitin-like domain. Biochemistry. 2007;46:14162–14169. doi: 10.1021/bi7016969. [DOI] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012a;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE. 2012b;7:e52830. doi: 10.1371/journal.pone.0052830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA. 2010;107:11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem. 2012;287:40652–40660. doi: 10.1074/jbc.M112.419721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Chen CX, Grenier K, Camacho EM, Kozlov G, McPherson PS, Gehring K, Fon EA. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol Cell. 2009;36:1034–1047. doi: 10.1016/j.molcel.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K. Structure of Parkin Reveals Mechanisms for Ubiquitin Ligase Activation. Science. 2013;340:1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, Maccoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci USA. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- Yang JS, Zhang L, Lee SY, Gad H, Luini A, Hsu VW. Key components of the fission machinery are interchangeable. Nat Cell Biol. 2006;8:1376–1382. doi: 10.1038/ncb1503. [DOI] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci USA. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Legends
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S5
Source Data for Supplementary Figure S6
Source Data for Supplementary Figure S7
Source Data for Figures 2A, 3C, 3G, 4B, 4D, 5B, 6C, 7B, 7C and Supplementary Figures S2A, S4B, S4E, S4G, S7B
Review Process File
Source Data for Figure 2D
Source Data for Figure 3A
Source Data for Figure 5A
Source Data for Figure 7A