

Abstract
β-Hydrogen-containing alkyl Grignard reagents were used in a stereospecific nickel-catalyzed cross-coupling reaction to form sp3–sp3 carbon–carbon bonds. Aryl Grignard reagents were also utilized to synthesize 1,1-diarylalkanes. Several compounds synthesized by this method exhibited selective inhibition of proliferation of MCF-7 breast cancer cells.
Keywords: alkyl Grignard, breast cancer, β-hydride elimination, enantiospecific, Kumada cross-coupling, MCF-7, nickel
Alkyl–alkyl cross-coupling reactions have emerged as powerful transformations that provide a new disconnection for the synthesis of tertiary stereocenters that are difficult to construct using other methods.[1,2] However, due to the inherent reactivity of alkylmetal intermediates, sp3–sp3 coupling reactions are significantly less common than their sp2–sp2 counterparts. In particular, alkylmetal intermediates are prone to β-hydride elimination that results in non-productive pathways. In this manuscript we report nickel-catalyzed cross-coupling reactions of alkyl Grignard reagents containing β-hydrogens. We also utilize this method to prepare new compounds that display selective inhibition of breast cancer cell proliferation.
We have previously reported the stereospecific nickel-catalyzed Kumada cross-coupling of methylmagnesium bromide with benzylic ethers (Scheme 1a).[3,4] Furthermore, we were able to extend the utility of this reaction by employing arylmagnesium reagents for the stereospecific synthesis of triarylmethanes.[5] One class of nucleophiles that remained elusive was long-chain alkyl Grignard reagents (Scheme 1b). These reagents perform poorly under our reported cross-coupling reaction conditions as hydrogenolysis or elimination typically predominate. We hypothesized that these products result from β-hydride elimination of the organonickel intermediates, and that with fine-tuning of the catalyst we could identify a system that favors the desired cross-coupling pathway.
Scheme 1.
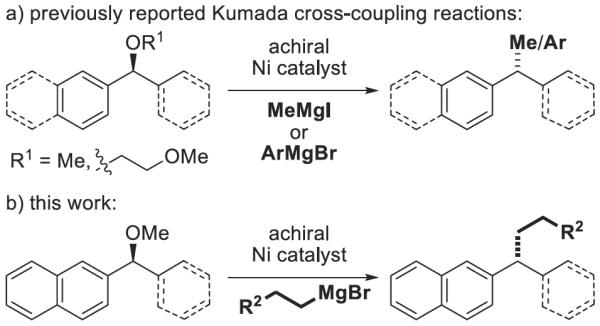
Ni-catalyzed Kumada cross-coupling reactions.
The bite angle and steric and electronic environment of a ligand can have profound effects on the relative rates of elementary steps in a catalytic cycle. We evaluated a series of catalysts (Table 1, entries 1–7) and determined that Ni(acac)2 in the presence of 1,2-bis(diphenylphosphino)ethane (dppe) afforded 2 as the major product in good conversion and modest enantiospecificity (entry 7).[6] To improve the yield of the cross-coupling reaction we systematically varied the ligand:metal stoichiometry. When the ratio of dppe:Ni(acac)2 was <2:1 the reaction showed little dependence on ligand loading (entries 7 and 8). When more than two equivalents of dppe were used with respect to Ni(acac)2 no desired product was detected and 1 was recovered quantitatively (entry 10). Interestingly, when 2:1 dppe:Ni(acac)2 was employed, the reaction gave highly variable results (entry 9); across seven experiments, four provided only recovered starting material and three provided >80% yield.[7] We propose that when the ligand loading is ≥2:1, an inactive complex, Ni(dppe)2, is formed quantitatively and the cross-coupling pathway is shut down. Consistent with this hypothesis, use of Ni(cod)2 in the presence of dppe provided no product, due to rapid formation of the Ni(dppe)2 complex.[8,9] To ensure strict control of ligand:Ni ratio, we evaluated the complex Ni(dppe)Cl2. The latter was a competent catalyst in the reaction affording 2 in good yield, albeit with slightly diminished enantiospecificity (entry 12). Additionally, this NiII salt is commercially available, inexpensive, and air- and moisture-stable.
Table 1.
Optimization for cross-coupling with n-pentyl Grignard.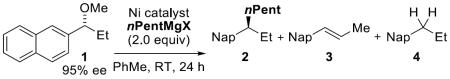
| Entry | Ni catalyst (mol %) | X | Yield 2 [%][a] | ee 2 [%][b] | es 2 [%][c] | Yield 3 [%][a] | Yield 4 [%][a] |
|---|---|---|---|---|---|---|---|
| 1 | no Ni, no ligand | I | <5[d] | – | – | <5[d] | <5[d] |
| 2 | Ni(acac)2 (10), no ligand | I | <5[d] | – | – | <5[d] | <5[d] |
| 3 | Ni(acac)2 (10), rac-BINAP (10) | I | <5[d] | – | – | <5[d] | <5[d] |
| 4 | Ni(acac)2(10), DPEphos (10) | I | 28 | 86 | 91 | 27 | 12 |
| 5 | Ni(acac)2 (10), PPh3 (10) | I | 8 | 82 | 86 | 55 | 14 |
| 6 | Ni(acac)2 (10), dppp (10) | I | 31 | 45 | 47 | 12 | 14 |
| 7 | Ni(acac)2 (10), dppe (10) | I | 69 | 66 | 69 | <5 | <5 |
| 8 | Ni(acac)2 (10), dppe (15) | I | 95 | 61 | 64 | <5 | <5 |
| 9 | Ni(acac)2 (10), dppe (20) | I | 0–90[e] | 83[f] | 87[f] | <5 | <5 |
| 10 | Ni(acac)2 (10), dppe (22) | I | <5[d] | – | – | <5[d] | <5[d] |
| 11 | Ni(cod)2 (10), dppe (10) | I | <5[d] | – | – | <5[d] | <5[d] |
| 12 | Ni(dppe)CI2 (10) | I | 89 | 55 | 58 | <5 | <5 |
| 13 | Ni(dppe)CI2 (10) | CI | 96 | 51 | 54 | 5 | <5 |
| 14 | Ni(dppe)CI2 (10) | Br | 95 | 91 | 96 | <5 | <5 |
| 15 | Ni(dppe)CI2 (5) | Br | 85 | 93 | 98 | <5 | <5 |
| 16 | Ni(dppe)CI2 (2) | Br | 97[g] | 96 | >99 | <5 | <5 |
Determined by 1H NMR analysis using internal standard (PhSiMe3).
Determined by chiral SFC.
[c] Enantiospecificity (es) = eeproduct/eestarting material × 100%.
Recovered unreacted 1.
Reaction was irreproducible: run 1: <5% yield; run 2: <5% yield; run 3: 90% yield, 85% ee, 89% es; run 4: <5% yield; run 5: <5% yield; run 6: 84% yield, 68% ee, 72% es; run 7: 90% yield, 94% ee, 99% es;
Average of runs 3, 6, and 7.
Isolated yield.
To improve the enantiospecificity of the reaction we investigated the importance of the identity of the organomagnesium reagent. Organomagnesium bromide proved to be superior to the respective chloride and iodide Grignard reagents, improving the modest es with nPentMgI (58%) to 96% es with nPentMgBr (entries 12–14). We next examined the impact of catalyst loading on es, since in related transformations our laboratory had observed an inverse correlation between catalyst loading and stereochemical fidelity.[10] We hypothesized that, in analogy to palladium-catalyzed allylic[11] and benzylic[12] substitution reactions, the key π-benzylnickel intermediate could be racemized by nucleophilic attack of a second nickel species (vide infra).[13] We were pleased to see that lower catalyst loadings do provide higher es; employing 2 mol % Ni(dppe)Cl2 afforded the desired product in >99% es without a drop in yield (entry 16).
We designed a series of chiral benzylic ethers to determine the scope of the transformation (Table 2). Enantioenriched ethers can be prepared by several routes. For example, CBS reduction[14] of the corresponding ketone or enantioselective alkylation[15] or arylation[16] of the requisite aldehyde typically provide robust strategies for their construction.[17]
Table 2.
Scope of cross-coupling reaction of alkylmagnesium bromides.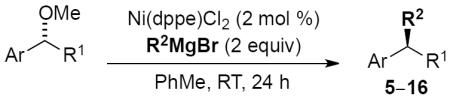
| Entry | Product | Yield [%][a] | S.M. ee [%][b] | Prod ee [%][b] | es [%] | ||
|---|---|---|---|---|---|---|---|
| 1 |
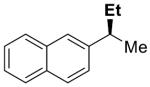
|
5 | 80 | 92 | 91 | 99 | |
| 2 | 6 | R2 = nPr | 93 | 97 | 97 | >99 | |
| 3 | 7 | R2 = nPent | 91 | 97 | 97 | >99 | |
| 4 |
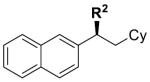
|
8 | R2 = (CH2)3Ph | 88 | 97 | 97 | >99 |
| 5 [c] | 9 | R2 = 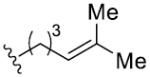
|
81 | 97 | 97 | >99 | |
| 6[d] | 10 | R2 = (CH2)3CF3 | 68 | >99 | 97 | 97 | |
| 7 | 11 | R2 = iBu | 40 | 97 | 90 | 93 | |
| 8 |
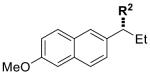
|
12 | R2 = nPr | 80 | 93 | 92 | 99 |
| 9 | 13 | R2 = (CH2)3Ph | 88 | 93 | 93 | >99 | |
| 10[c] |
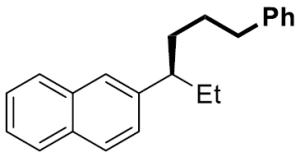
|
14 | 93 | 97 | 97 | >99 | |
| 11[e] |
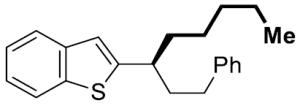
|
15 | 54 | 99 | 96 | 97 | |
| 12[f] |
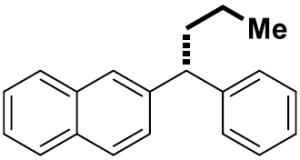
|
16 | 67 | >99 | 91 | 91 | |
Calculated yield after silica gel chromatography.
Determined by chiral SFC chromatography, S.M. = starting material, Prod. = product.
5 mol % Ni(dppe)CI2.
10 mol % Ni(dppe)CI2.
Ni(dppe)CI2 was added in two aliquots of 5 mol %; see SI for details.
5 °C; 48 h; 15% 2-benzylnapthalene byproduct.
Reaction of each substrate and Grignard reagent was first evaluated under our standard reaction conditions employing 2 mol % catalyst. A range of primary alkyl Grignard reagents afforded cross-coupled product in good yields and excellent enantiospecificities (entries 1–4). The cross-coupling reactions proceed with high stereochemical fidelity and inversion at the stereogenic center.[18] A trisubstituted olefin was well tolerated in the reaction, affording a product containing a convenient synthetic handle for further functionalization (entry 5). β-Substitution on the alkylmagnesium reagent resulted in a low yield of product 11 due to the formation of large amounts of elimination byproduct, yet the reaction proceeded with satisfactory es (entry 7). An electron donating methoxy group on the naphthyl ring was well tolerated without loss of stereospecificity (entries 8 and 9).
For challenging coupling reactions, we could typically improve enantiospecificity or yield by modifying catalyst loading and reaction temperature. For example, an electron-poor fluorinated alkylmagnesium reagent reacted sluggishly; increasing the catalyst loading to 10 mol % provided good yield and maintained high es (entry 6). Substrates containing heterocyclic moieties also required higher catalyst loading, presumably due to coordination and deactivation of the catalyst (entry 11). For this substrate, addition of Ni(dppe)Cl2 in two portions over the course of the reaction permitted use of higher catalyst loadings without compromising es. Diarylmethanol derivatives proved to be a more challenging class of substrates: reactions with primary alkyl Grignard reagents resulted in increased hydrogenolysis (21%) and low enantiospecficity (77% es).[19] For diarylmethanol derivatives that were prone to racemization, lowering the temperature generally increased the enantiospecificity (entry 12, 91% es).
To investigate the generality of this methodology and evaluate its applicability to other classes of Grignard reagents we chose to examine the use of arylmagnesium reagents as a strategy for synthesis of chiral 1,1-diarylalkanes, pharmacophores found in a range of bioactive compounds.[20–23] Our laboratory has previously developed stereospecific cross-coupling of aryl Grignard reagents with benzhydryl alcohol derivatives to provide triarylmethanes.[5] However, this method failed to afford satisfactory yields with simple benzylic alcohol derivatives such as 1, and therefore was not amenable to 1,1-diarylalkane synthesis. To address this bond construction, we examined a variety of substituted aryl Grignard reagents (Table 3). Under standard reaction conditions the cross-coupling proceeded in modest yield with significant byproduct from elimination.[24] Improvement was observed when overall reaction concentration was decreased 2.5-fold, affording 17 in 67% yield with only 16% elimination byproduct (entry 1). Furthermore, we noted that the use of more than 2 equivalents of phenylmagnesium bromide was detrimental to the cross-coupling reaction.[25] The electron rich aniline and anisole-derived organomagnesium reagents afforded the corresponding products in good yields and in the case of 19 excellent stereospecificity (entries 2 and 3). Electron deficient 4-fluorophenyl- and 2-thienylmagnesium bromides were also competent in this reaction (entries 4 and 5). Electrophiles containing the benzofuran and benzothiophene moieties were tolerated in the cross-coupling, affording 24 and 25 respectively in good yields and excellent stereospecificity.
Table 3.
Scope of cross-coupling reaction of arylmagnesium bromides.
| Entry | Product | Ni(dppe)CI2 [mol %] | Yield [%][a] | S.M. ee [%][b] | Prod ee [%][b] | es [%] | ||
|---|---|---|---|---|---|---|---|---|
| 1 |
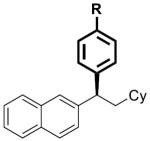
|
17 | R = H | 2 | 67 | >99 | 91 | 92 |
| 2 | 18 | R = NMe2 | 5 | 80 | >99 | nd[c] | nd[c] | |
| 3 | 19 | R = OMe | 2 | 86[d] | >99 | 97 | 97 | |
| 4 | 20 | R = F | 2 | 82[d] | >99 | 87 | 88 | |
| 5 |
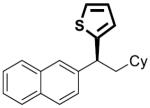
|
21 | 10 | 76 | >99 | 93 | 94 | |
| 6 |
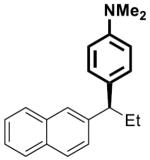
|
22 | 5 | 80 | >99 | 85 | 86 | |
| 7 |
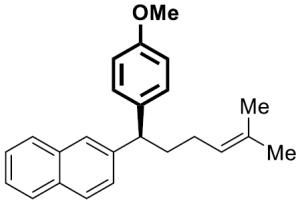
|
23 | 4 | 92[d] | 96 | 88 | 92 | |
| 8 |
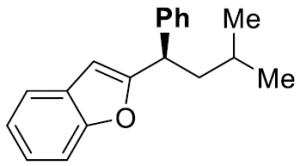
|
24 | 10 | 71 | 97 | 95 | 98 | |
| 9[e] |
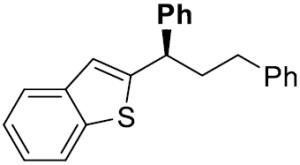
|
25 | 20 | 76 | 99 | 93 | 94 | |
During our initial optimization of the reaction and development of the scope, we noted that increased enantiospecificity could be obtained at lower catalyst loading (vide supra). We hypothesized that the loss of fidelity in the transfer of stereochemical information resulted from racemization of the enantioenriched π-benzylnickel intermediate (S)-27 by reaction with a low-valent nickel species (Figure 1a). This mechanism contrasts alternatives where stereochemical information is eroded during a competitive radical oxidative addition reaction or homolysis of the carbon-nickel bond in 27.[13,26] Consistent with our hypothesis, experiments performed in the presence of 1 equivalent of TEMPO afforded no improvement or erosion of the enantiospecificity of the reaction. We sought to obtain experimental evidence to further support or refute the bimolecular racemization mechanism. Based on our mechanistic hypothesis, the formation of the major and minor enantiomers should be first- and second-order with respect to catalyst concentration, respectively. Derivation of rate laws indicates that if that is the case, the ratio of the two enantiomers would be directly proportional to 1/[catalyst].[27] Indeed, a plot of [(S)-17]/[(R)-17] versus 1/[Ni(dppe)Cl2] yielded a good fit for a linear equation (Figure 1b). The data are consistent with a mechanism where the formation of the minor enantiomer is second order with respect to catalyst concentration, as shown in Figure 1a.
Figure 1.

Ni-catalyzed racemization of π-benzylnickel intermediate.
Having synthesized a variety of enantioenriched alkanes and diarylalkanes, we set out to evaluate these compounds for biological activity. Compounds containing the 1,1-diarylalkane scaffold have demonstrated bioactivity against a wide range of indications, including breast cancer.[21] The cross-coupling products in Tables 2 and 3 were tested for selective anti-breast cancer activity against the MCF-7 breast cancer cell line relative to the normal MCF-10A stromal cell line using a proliferation-based procedure. Selected results of the broad compound screen are shown in Figure 2. Several compounds demonstrated selectivity for the inhibition of breast cancer cell proliferation; results were compared to those obtained with estrogen receptor antagonist faslodex (ICI 182,780).[28] Thiophene-containing diarylalkane (+)-21 inhibited MCF-7 cell proliferation with an EC50 of 5.3 μM. We observed that (−)-21 (EC50 = 6.5 μM) and the racemic mixture (EC50 = 7.3 μM) were both nearly as efficacious as the (+)-enantiomer. Interestingly, the structurally analogous diarylalkane 25 exhibited a similar level of inhibition. Control experiments confirmed that thiophene (28) and benzothiophene (29) did not inhibit cell growth. Furthermore, while replacing the thiophene moiety with different aryl groups, such as phenyl (17), para-methoxy (19), or para-fluoro (20) resulted in similar selective inhibition of cancer cell proliferation, compounds containing hydrocarbon chains (9 and 7) were much less potent. These results provide new lead compounds with selective inhibition of breast cancer cell growth.
Figure 2.
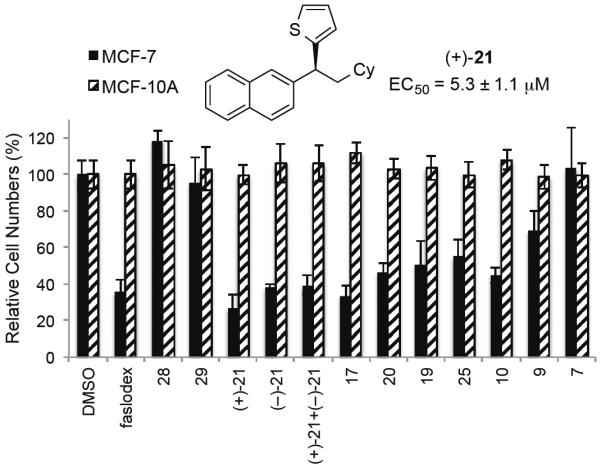
Anti-breast cancer activity of compounds at 10 μM screened against breast cancer (MCF-7) and normal breast cell lines (MCF-10A). Cell proliferation is represented as relative cell numbers after treatment, where a low percentage indicates potent anti-cancer activity for that compound. All data are normalized to the DMSO vehicle control.
In conclusion, we have developed a stereospecific nickel-catalyzed Kumada cross-coupling reaction that tolerates Grignard reagents containing extended alkyl chains. This catalytic system is also amenable to reactions of aryl- and heteroarylmagnesium reagents for the synthesis of 1,1-diarylalkanes. Reactions typically provide higher es at lower catalyst loading, and mechanistic experiments are consistent with racemization of the π-benzylnickel intermediate. Biological testing of compounds synthesized using this methodology identified several promising leads that exhibit selective inhibition of breast cancer cell proliferation in the low micromolar range.
Supplementary Material
Footnotes
This work was supported by NIH NIGMS (R01GM100212), the UC Cancer Research Coordinating Committee, NCI Award P30CA062203, NCI Award F31CA177212 (C.A.O.), and the LaVerne Noyes Fellowship (A.G.J.). We thank R. J. Ochoa for advice and assistance with biochemical assays, Prof. J. A. Prescher, Prof. X. Zi, and C. A. Blair for helpful discussions, and Prof. M. J. Buchmeier for use of the fluorimeter. Prof. S. D. Rychnovsky and A. J. Wagner are acknowledged for confirming absolute configuration of enantioenriched alcohol intermediates by their CEC method. Dr. J. Greaves is acknowledged for mass spectrometry data.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- [1].For reviews of asymmetric variants, see: Swift EC, Jarvo ER. Tetrahedron. 2013;69:5799–5817. doi: 10.1016/j.tet.2013.05.001. Taylor BLH, Jarvo ER. Synlett. 2011:2761–2765. Jana R, Pathak TP, Sigman MS. Chem. Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. Glorius F. Angew. Chem. 2008;120:8474–8476. Angew. Chem. Int. Ed. 2008;47:8347–8349. doi: 10.1002/anie.200803509.
- [2].For a lead reference of stereoconvergent alkyl–alkyl cross-coupling, see: Cordier CJ, Lundgren RJ, Fu GC. J. Am. Chem. Soc. 2013;135:10946–10949. doi: 10.1021/ja4054114.
- [3].a) Taylor BLH, Swift EC, Waetzig JD, Jarvo ER. J. Am. Chem. Soc. 2011;133:389–391. doi: 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]; b) Greene MA, Yonova IM, Williams FJ, Jarvo ER. Org. Lett. 2012;14:4293–4296. doi: 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]
- [4].Stereospecific Negishi couplings: Wisniewska HM, Swift EC, Jarvo ER. J. Am. Chem. Soc. 2013;135:9083–9090. doi: 10.1021/ja4034999.
- [5].Taylor BLH, Harris MR, Jarvo ER. Angew. Chem. 2012;124:7910–7913. Angew. Chem. Int. Ed. 2012;51:7790–7793. doi: 10.1002/anie.201202527. stereospecific Suzuki couplings of arylboronic esters: Harris MR, Hanna LE, Greene MA, Moore CE, Jarvo ER. J. Am. Chem. Soc. 2013;135:3303–3306. doi: 10.1021/ja311783k. Zhou Q, Srinivas HD, Dasgupta S, Watson MP. J. Am. Chem. Soc. 2013;135:3307–3310. doi: 10.1021/ja312087x.
- [6].Nap = 2-naphthyl, dppe = 1,2-bis(diphenylphosphino)ethane.
- [7].We hypothesized that weighing error was responsible for the high variability in these small scale reactions. See: Wernerova M, Hudlicky T. Synlett. 2010:2701–2707.
- [8].31P NMR studies indicated formation of Ni(dppe)2, an inactive complex in this cross-couplings; see SI; characterization of Ni(dppe)2: Fisher KJ, Alyea EC. Polyhedron. 1989;8:13–15. for a similar observation: Yamashita K.-i., Takeda H, Kashiwabara T, Hua R, Shimada S, Tanaka M. Tetrahedron Lett. 2007;48:6655–6659.
- [9].An alternative Ni0 source, Ni(PPh3)4, afforded 6 in 73% yield. See SI.
- [10].Taylor BLH. Order No. 3540398 University of California, Irvine. ProQuest.; Ann Arbor: 2012. “Development of Nickel-Catalyzed Stereospecific Cross-Coupling Reactions.”. Web. 17 July 2013. [Google Scholar]
- [11].a) Murahashi SI, Taniguchi Y, Imada Y, Tanigawa Y. J. Org. Chem. 1989;54:3292–3303. [Google Scholar]; (b) Granberg KL, Bäckvall JE. J. Am. Chem. Soc. 1992;114:6858–6863. [Google Scholar]
- [12].Legros J-Y, Toffano M, Fiaud J-C. Tetrahedron. 1995;51:3235–3246. [Google Scholar]
- [13].This mechanism was proposed, and discounted in favor of a radical oxidative addition reaction, to rationalize stereoablative reactions of benzylic bromides with nickel complexes. See: Stille JK, Cowell AB. J. Organomet. Chem. 1977;124:253–261.
- [14].a) Corey EJ, Helal CJ. Angew. Chem. 1998;110:2092–2118. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; b) Li JJ, Limberakis C, Pflum DA. Modern Organic Synthesis in the Laboratory: A Collection of Standard Experimental Procedures. Oxford University Press; New York: 2007. “Reductions”; pp. 96–97. [Google Scholar]
- [15].a) Noyori R, Kitamura M. Angew. Chem. 1991;103:34–55. [Google Scholar]; Angew. Chem. Int. Ed. 1991;30:49–69. [Google Scholar]; b) Zhang F-Y, Yip C-W, Rong C, Chan ASC. Tetrahedron Asymmetry. 1997;8:585–589. [Google Scholar]
- [16].a) Bolm C, Rudolph J. J. Am. Chem. Soc. 2002;124:14850–14851. doi: 10.1021/ja028518l. [DOI] [PubMed] [Google Scholar]; b) Braga AL, Paixao MW, Westermann B, Schneider PH, Wessjohann LA. J. Org. Chem. 2008;73:2879–2882. doi: 10.1021/jo702413n. [DOI] [PubMed] [Google Scholar]
- [17].The competing enantiomer conversion method can be used to assign absolute configuration of secondary alcohols: Wagner AJ, Rychnovsky SD. J. Org. Chem. 2013;78:4594–4598. doi: 10.1021/jo400432q.
- [18].See Supporting Information for details.
- [19].At room temperature, 16 was obtained in 71% yield and 77% ee and 2-benzylnaphthalene was formed in 21% yield.
- [20].Representative examples: as ligands for nuclear receptors, see: Kainuma M, Kasuga J.-i., Hosoda S, Wakabayashi K.-i., Tanatani A, Nagasawa K, Miyachi H, Makishima M, Hashimoto Y. Bioorg. Med. Chem. Lett. 2006;16:3213–3218. doi: 10.1016/j.bmcl.2006.03.075. as combretastatin analogs for colon cancer, see: Messaoudi S, Hamze A, Provot O, Tréguier B, Rodrigo De Losada J, Bignon J, Liu J-M, Wdzieczak-Bakala J, Thoret S, Dubois J, Brion J-D, Alami M. ChemMedChem. 2011;6:488–497. doi: 10.1002/cmdc.201000456. prostate cancer: Hu QZ, Yin LN, Jagusch C, Hille UE, Hartmann RW. J. Med. Chem. 2010;53:5049–5053. doi: 10.1021/jm100400a. diabetes: Kim RM, Parmee ER, Tan Q, Yang C, Lins AR. U. S. Patent 12/227,030. 2007 May 11;
- [21].For breast cancer: Pathak TP, Gligorich KM, Welm BE, Sigman MS. J. Am. Chem. Soc. 2010;132:7870–7871. doi: 10.1021/ja103472a. Pathak TP, Osiak JG, Vaden RM, Welm BE, Sigman MS. Tetrahedron. 2012;68:5203–5208. doi: 10.1016/j.tet.2012.03.075.
- [22].For similar disconnection, see: Lopez-Perez A, Adrio J, Carretero JC. Org. Lett. 2009;11:5514–5517. doi: 10.1021/ol902335c. Imao D, Glasspoole BW, Laberge VS, Crudden CM. J. Am. Chem. Soc. 2009;131:5024–5025. doi: 10.1021/ja8094075. Li J, Burke MD. J. Am. Chem. Soc. 2011;133:13774–13777. doi: 10.1021/ja205912y. Maity P, Shacklady-McAtee DM, Yap GPA, Sirianni ER, Watson MP. J. Am. Chem. Soc. 2013;135:280–285. doi: 10.1021/ja3089422.
- [23].For representative alternative strategies for enantioselective synthesis of 1,1-diarylalkanes, see: ref 21a. Saini V, Liao L, Wang Q, Jana R, Sigman MS. Org. Lett. 2013;15:5008–5011. doi: 10.1021/ol4023358. Wang X, Guram A, Caille S, Hu J, Preston JP, Ronk M, Walker S. Org. Lett. 2011;13:1881–1883. doi: 10.1021/ol200422p. Fessard TC, Andrews SP, Motoyoshi H, Carreira EM. Angew. Chem. 2007;119:9492–9495. doi: 10.1002/anie.200702995. Angew. Chem. Int. Ed. 2007;46:9331–9334. doi: 10.1002/anie.200702995.
- [24].17 was formed in 54% yield, 92% ee, 95% es, with 34% of elimination byproduct.
- [25].When 4 equiv of PhMgBr were used, 17 was obtained in 19% yield with 37% unreacted starting material and 34% elimination byproduct.
- [26].For evidence of a radical oxidative addition in cross-coupling reactions of alkyl halides, see: Kochi JK. Pure App. Chem. 1980;52:571–605. Jones GD, Martin JL, McFarland C, Allen OR, Hall RE, Haley AD, Brandon RJ, Konovalova T, Desrochers PJ, Pulay P, Vicic DA. J. Am. Chem. Soc. 2006;128:13175–13183. doi: 10.1021/ja063334i. Choi J, Fu GC. J. Am. Chem. Soc. 2012;134:9102–9105. doi: 10.1021/ja303442q. Lin X, Phillips DL. J. Org. Chem. 2008;73:3680–3688. doi: 10.1021/jo702497p. for evidence of Ni–C bond homolysis after a polar oxidative addition, see Nielsen DK, Huang C-Y, Doyle AG. J. Am. Chem. Soc. 2013;135:13605–13609. doi: 10.1021/ja4076716.
- [27].For full details of the derivation, see SI.
- [28].For a discussion of faslodex and estrogen receptor antagonists, see: Howell A. Endocr. Relat. Cancer. 2006;13:689–706. doi: 10.1677/erc.1.00846. Wakeling AE, Dukes M, Bowler J. Cancer Res. 1991;51:3867–3873.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.