

Abstract
Aspergillus fumigatus is often associated in asthmatic patients with the exacerbation of asthma symptoms. The pathomechanism of this phenomenon has not been fully understood. Here, we evaluated the immunological mechanisms and the role of the prostaglandin D2/ Chemoattractant Receptor-Homologous Molecule Expressed on Th2 Cells (CRTH2) pathway in the development of Aspergillus-associated asthma exacerbation. We studied the effects of A. fumigatus on airway inflammation and bronchial hyper-responsiveness in a rat model of chronic asthma. Inhalation delivery of A. fumigatus conidia increased the airway eosinophilia and bronchial hyper-responsiveness in ovalbumin-sensitized, challenged rats. These changes were associated with prostaglandin D2 synthesis and CRTH2 expression in the lungs. Direct inflammation occurred in ovalbumin-sensitized, challenged animals, whereas pre-treatment with an antagonist against CRTH2 nearly completely eliminated the A. fumigatus-induced worsening of airway eosinophilia and bronchial hyper-responsiveness. Our data demonstrate that production of prostaglandin D2 followed by eosinophil recruitment into the airways via a CRTH2 receptor are the major pathogenic factors responsible for the A. fumigatus-induced enhancement of airway inflammation and responsiveness.
Keywords: airway responsiveness, Aspergillus fumigatus, asthma, CRTH2, eosinophil, prostaglandin D2
Introduction
Asthma is an inflammatory disease of the airways that may be worsened by numerous extrinsic factors. The most common trigger is continuous exposure to allergens of which fungal agents are important factors. There is overwhelming evidence for the presence of fungal sensitization in patients with asthma. There is also a strong association between fungal exposure and severity of asthma. Epidemiological studies have associated mould exposure with the development, persistence and severity of asthma. However, the mechanism of these changes has not been established. Several lines of evidence suggest that eosinophils play an important role in patients with asthma during fungal infection-induced exacerbation.1–9 However, little is known regarding the mechanisms of recruitment and activation of eosinophils in asthmatic airways and enhancement of airway responsiveness during fungal infection.
Infection with fungus immediately activates the innate immune system via several pathways.10 Aspergillus fumigatus, the most common species colonizing the lungs of patients with chronic lung disease, is recognized by membrane or cytosolic receptors, such as cognate C-type lectin and Toll-like receptors.11–13 In asthmatic patients, the changed milieu in the airways facilitates the adhesion of A. fumigatus spores, which allows the colonization and eventual release of spores.14 This process activates signalling pathways linked to the production of inflammatory cytokines and chemokines, as well as the production of reactive oxygen. The activation of these pattern-recognition receptors by A. fumigatus triggers the synthesis of interleukins15,16 and other cytokines/chemokines in airway epithelial cells, dendritic cells and other immune cells.17 However, little is known about its impact on the axis of lipid mediators and their receptors.
Recent studies have shown that prostaglandin D2 (PGD2), a major cyclooxygenase 3 metabolite synthesized in activated mast cells and macrophages, acting as a potent chemoattractant of eosinophils, is an important mediator involved in eosinophilic airway inflammation.18–23 The bioactivity of PGD2 is mediated by two G protein-coupled receptors, DP (DP1) and Chemoattractant Receptor-Homologous Molecule Expressed on Th2 Cells (CRTH2) (DP2), and both in vitro and in vivo eosinophil trafficking is mediated mostly by CRTH2, which is preferentially expressed on eosinophils, basophils and T helper type 2 lymphocytes.20,21,24–26 Reports recently showed that CRTH2 agonists administered into the trachea led to translocation of eosinophils from the bloodstream into the airway,27–29 and that CRTH2-deficient mice exhibited decreased infiltration of eosinophils and other inflammatory cells during chronic allergic skin inflammation.30
As reported herein, we developed rat models mimicking the enhanced asthmatic responses induced by A. fumigatus. In these models, we used the inhalation delivery of A. fumigatus conidia to simulate fungal exposure and showed that it worsened allergen-induced eosinophilic airway inflammation and airway hyper-responsiveness (AHR). In these experiments, we also establish that PGD2, acting via the CRTH2 receptor, is the essential modulator of eosinophilic airway inflammation and bronchial hyper-responsiveness induced by A. fumigatus spore inhalation.
Materials and methods
Animals
Specific pathogen-free 5-to 6-week-old, male Wistar rats weighing between 140 and 180 g were supplied by the Shanghai Laboratory Animal Centre (Shanghai, China). The animals were maintained in a 12/12 hr light/dark cycle at a room temperature of 23° and relative humidity 40%; they were provided with standard laboratory rat chow and water ad libitum. The experimental protocol was reviewed and approved by the Shanghai Jiaotong University School of Medicine Ethical Committee for Laboratory Animals.
Protocols for allergen exposure and A. fumigatus inhalation
A total of 40 male Wistar rats were randomly divided into five groups (n = 8/group): control group (NS); A. fumigatus spore-exposed group (NS + AF); ovalbumin (OVA)-sensitized and OVA-challenged group (OVA); OVA-sensitized, OVA-challenged and A. fumigatus spore-exposed group (OVA + AF); and CRTH2 antagonist OC00459-treated group (OVA + AF + Treat). Saline (0·1 ml) was intraperitoneally administered to a control group (NS) and an AF group (NS + AF) on days 0 and 7. The other groups were sensitized and challenged with OVA. Briefly, rats were sensitized by an intraperitoneal injection of 1 mg OVA (Albumin Chicken egg, Grade V; Sigma Chemical Co., St. Louis, MO) with 100 mg aluminium hydroxide (Sigma Chemical) in 1 ml saline on days 0 and 7. Rats were challenged via the airways with aerosolized 2% OVA for 30 min, 3 days per week, from days 14 to 37 by an ultrasonic nebulizer (PARI-BOY N037; PARI, Starnberg, Germany). Five days after the last challenge (day 42), rats were given via the airways aerosolized A. fumigatus spore suspension 5 days per week from days 42 to 67 (Fig. 1). Non-sensitized and non-challenged rats receiving aerosolized saline were treated in the same fashion. From days 42 to 67, a single oral dose of 5 mg/kg OC000459 by gavage in 10% DMSO/saline solution was administered to an antagonist-treated group (OVA + AF + Treat). Airway hyper-responsiveness was assessed 72 h after the last inhalation, and tissues and cells were obtained for further assays.
Figure 1.
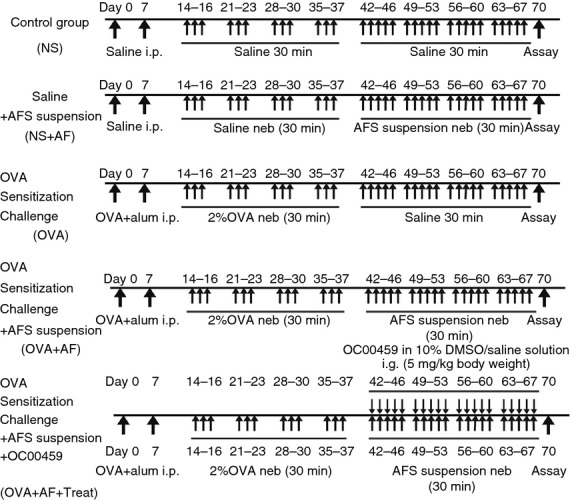
Experimental protocols. Rats were divided into five groups: non-sensitized control group (NS); Aspergillus fumigatus spore-exposed group (NS + AF); ovalbumin (OVA)-sensitized and OVA-challenged group (OVA); OVA-sensitized, OVA-challenged and A. fumigatus spore-exposed group (OVA + AF); and CRTH2 antagonist OC00459-treated group (OVA + AF + Treat). In the OVA-sensitized and OVA-challenged receiving saline (OVA) group, rats were sensitized by two intraperitoneal injections of OVA/aluminium hydroxide and then received three consecutive days of an aerosolized OVA challenge from days 14 to 37 for 4 weeks. To evaluate the effect of A. fumigatus, OVA-sensitized and OVA-challenged mice received 5 consecutive days of an aerosol inhalation of 1 × 106 colony-forming unit/ml spore suspension from days 42 to 67 for 4 weeks. The OVA + AF + Treat group received OC000459 at a dose of 5 mg/kg daily. Non-sensitized and non-challenged mice receiving saline or A. fumigatus represented the NS and NS + AF groups, respectively. Values are means±SEM. *P < 0·05.
Measurement of AHR
Airway hyper-responsiveness was assessed by a change in airway function after the challenge with aerosolized methacholine (Sigma-Aldrich, St Louis, MO) using barometric plethysmography (Buxco Electronics Inc., Troy, NY) as previously described.31,32 Briefly, pressure differences were measured between the main chamber of the plethysmograph, which contained conscious, spontaneously breathing animals, and the reference chamber (box pressure signal). Rats were challenged with aerosolized saline for baseline measurements or with methacholine (3·125–50 mg/ml) for 3 min, and readings were taken and averaged for 3 min after each nebulization. Data were expressed using the dimensionless parameter, enhanced pause (Penh).
Sample collection
Rats were anaesthetized with an intraperitoneal injection of 1% sodium pentobarbital. Approximately 10 ml of blood was collected by heart bleed. The serum was isolated and stored at −80° until assay.
The trachea was cannulated and the left lungs were lavaged via the tracheal cannula with 3·5 ml PBS (pH 7·2) three times. Lavage fluid was recovered by gentle aspiration with a syringe. The collected bronchoalveolar lavage fluid was then centrifuged at 401 g for 7 min at 4°. The supernatant from the first wash was collected and stored at −80° for cytokine determination.
The right lower lobe was harvested, inflated with 10% neutral buffered formalin, fixed in formalin for 16 hr, and paraffin-embedded for histological and immunohistochemical analyses. The other lobe was stored in liquid nitrogen for further examination.
Quantification of eosinophils in the bronchoalveolar lavage
After supernatant removal, cells were resuspended in 0·5 ml PBS (pH 7·4). Total cell numbers were counted on a haemocytometer and 1 × 103 to 5 × 103 cells were spun onto glass microscope slides (cytospin 3; Shandon Scientific, Runcorn, UK). The cell slides were air dried for 24–36 hr, fixed and stained with a Wright–Giemsa stain set (Jiancheng Bioengineering Institute, Nanjing, China). Differential cell counts of at least 300 cells in duplicate slides were counted in a blinded fashion according to morphological criteria. The number of eosinophils recovered was calculated and expressed as absolute cell numbers.
Detection of PGD2
Concentration of PGD2 in serum and bronchoalveolar lavage fluid supernatants was measured by the Prostaglandin D2-MOX EIA Kit (Detection Limit 80% B/B0 −3·1 pg/ml; Cayman Chemical, Ann Arbor, MI). This assay is based on the competition between sample PGD2 and a PGD2-acetylcholinesterase conjugate (PGD2 tracer) for a limited number of PGD2 monoclonal antibody binding sites. Sample extraction procedures for protein removal and eicosanoid stabilization were performed according to the provider's instructions, and the PGD2 concentration was determined following the manufacturer's instructions. The assay has a detection range between 250 and 2 pg/ml, and intra-and inter-assay coefficient of variation was < 10%.
Immunohistochemical analysis of lung CRTH2 expression
Sections of lung specimens underwent immunoperoxidase staining using antibodies directed against CRTH2 (1 : 400) (PAB13243; Abnova, Taipei City, Taiwan). The sections were deparaffinized in xylene and rehydrated in methanol. Endogenous peroxidases were blocked by 5% H2O2 treatment. For better antigen retrieval of CRTH2, the samples were boiled in a microwave oven for 2–3 min in citrate buffer. Samples were then washed with PBS. Samples were then incubated with the primary antibody at room temperature for 1 hr. After washing, the revelation was performed with the use of appropriate secondary antibody, the Ready-to-go SABC-AP anti-rabbit IgG kit (Boster, Wuhan, Hubei, China) according to the supplier's recommendations. Immunoreactivity was visualized by a treatment with diaminobenzidine (Sigma-Aldrich), and the slides were counterstained with Mayer's haematoxylin.
Measurement of total CRTH2 protein expression
Preparation of protein extracts
For preparation of protein extracts, tissue was crushed in the frozen state in a cryotube by shaking with a sterile steel ball. The crushing was resuspended in cold RIPA lysis buffer (Tris–HCl pH 7·6 25 mm; NaCl 150 mm; Nonidet-P40 1%; sodium deoxycholate 1%; sodium dodecyl sulphate 0·1%; Pierce, Rockford, IL) supplemented with protease inhibitors (Complete, Roche, Basel, Switzerland) and subsequently swirled for 10 min on ice. The extracts were then centrifuged at 14 000 g for 15 min at 4°. The supernatants were analysed for protein content by the Bio-Rad protein assay based on the Bradford method (Bio-Rad, Hercules, CA).
Western blotting
Samples were separated on a 10% SDS–PAGE and transferred to a polyvinylidene difluoride membrane (Roche). The anti-CRTH2 rabbit polyclonal antibody (PAB13243; Abnova) was used at a 1 : 1000 dilution. The secondary anti-rabbit antibody coupled with horseradish peroxidase (HSA0003) at a 1 : 3000 dilution was detected by chemiluminescence with the ECL system (Beyotime, Haimen City, Jiangsu, China), using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as loading control.
Reverse transcription-PCR analysis of CRTH2 mRNA
Total RNA was isolated using the TRIzol® reagent (TaKaRa, Yasu, Shiga, Japan) according to the manufacturer's instructions. To remove contaminating DNA, isolated RNA was treated with RNase-free DNase I (Takara Bio Inc). The RNA was quantified using the RiboGreen RNA quantification kit (Molecular Probes, Eugene, OR), dissolved in diethylpyrocarbonate-treated H2O and stored at −80° until use. One microgram of total RNA was reverse-transcribed using PrimeScript™ Master Mix (Takara Bio Inc.) as detailed in the manufacturer's guidelines. One-fiftieth of the reverse transcriptase reaction was analysed by real-time quantitative PCR. The following primers were used: GAPDH, sense 5′-CAC CCg CgA gTA CAA CCT TC-3′ and antisense 5′-CCC ATA CCC ACC ATC ACA CC-3′; CRTH2, sense 5′-gCT TCC AAA CCA CAg CAA CT-3′ and antisense 5′-CCA CCA CAA ACA ggA TgA gTC-3′.
Quantitative PCR analysis was performed in a total volume of 20 μl containing template DNA, 0·05 nm of sense and antisense primers, 10 μl SYBR®Premix Ex Taq (Tli RNaseH Plus; Takara Bio Inc.). After incubation at 95° for 30 seconds (initial denaturation), the mixtures were subjected to 40 amplification cycles (5 seconds at 95° for denaturation and 30 seconds for annealing and extension at 60°). Incorporation of SYBR® Green dye into PCR products was monitored in real time using a CFX96™ Real-Time PCR Detection System (Applied Biosystems, Foster City, CA) allowing determination of the threshold cycle (CT) at which exponential amplification of PCR products begins. After PCR, dissociation curves were generated with one peak, indicating the specificity of the amplification. A threshold cycle (CT value) was obtained from each amplification curve using the software provided by the manufacturer (Applied Biosystems).
Relative mRNA expression in chondrocytes was determined using the ΔΔCT method, as detailed in the manufacturer's guidelines (Applied Biosystems). A ΔCT value was first calculated by subtracting the CT value for the housekeeping gene GAPDH from the CT value for each sample. A ΔΔCT value was then calculated by subtracting the ΔCT value of the control from the ΔCT value of each treatment. Fold changes compared with the control were then determined by raising 2 to the – ΔΔCT power. Each PCR generated only the expected specific amplicon as shown by the melting-temperature profiles of the final product and by gel electrophoresis of test PCRs. Each PCR was performed in triplicate on two separate occasions for each independent experiment.
Statistical analysis
Statistical analysis was performed by one-way analysis of variance and Student–Newman–Keuls test. P value <0·05 was considered significant.
Results
A. fumigatus increases airway responsiveness in OVA-sensitized rats only
Analysis of the OVA + AF and OVA + NS groups showed significant differences in Penh (Fig. 2a). The inhalation of A. fumigatus significantly increased Penh in OVA + AF group rats relative to OVA + NS in an methacholine dose-dependent manner (Fig. 2a). Analysis of NS + AF and OVA + AF groups showed significant differences in Penh between these groups (Fig. 2a). To determine if AF without exposure to OVA has an effect on airway responsiveness, we compared Penh in the NS + AF group with Penh in the NS + NS group. No significant difference in Penh was observed in the NS + AF group when compared with the NS + NS group (Fig. 2a). Significant reduction in Penh was observed in the OVA + AF + Treat group when compared with the OVA + AF group (Fig. 2a).
Figure 2.
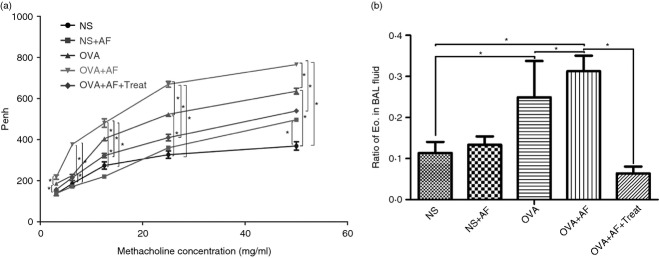
Inhalation of Aspergillus fumigatus increased bronchial hyper-responsiveness (a) and recruited eosinophils into the airways (b) of ovalbumin (OVA)-sensitized and OVA-challenged rats. Treatment with OC000459 prevents the development of airway hyper-responsiveness (AHR) (a) and eosinophil accumulation in bronchoalveolar lavage (BAL) fluid (b). Enhanced pause (Penh) values for increasing concentrations of inhaled methacholine and the ratio of eosinophils in BAL fluid were measured in the five groups: non-sensitized control group (NS); Aspergillus fumigatus spore-exposed group (NS + AF); ovalbumin (OVA)-sensitized and OVA-challenged group (OVA); OVA-sensitized, OVA-challenged and A. fumigatus spore-exposed group (OVA + AF); and CRTH2 antagonist OC00459-treated group (OVA + AF + Treat)., 72 hr after the last inhalation. Eo, eosinophil. Values are means ± SEM. *P < 0·05.
A. fumigatus increases lung eosinophilia in OVA-sensitized rats
Significant enhancement of the eosinophil ratio in bronchoalveolar lavage fluid was generally seen in OVA-sensitized, OVA-challenged rats (NS 11·34 ± 2·71% versus OVA 24·93 ± 8·81%, P < 0·05). Inhalation of A. fumigatus in NS-sensitized, challenged rats did not increase the ratio of eosinophils in bronchoalveolar lavage fluid (NS + AF 13·34 ± 2·05%, P = 0·425 versus NS 11·34 ± 2·71%) but produced a significant increase in the ratio of eosinophils in bronchoalveolar lavage fluid of OVA-sensitized and OVA-challenged rats (OVA + AF 31·24 ± 3·81% versus OVA 24·93 ± 8·81%, P < 0·05; Fig. 2b). In bronchoalveolar lavage fluid from the treated group, the ratio of eosinophils was significantly lower compared with the OVA + AF group (OVA + AF + Treat 6·38 ± 1·68% versus OVA + AF 31·24 ± 3·81% P < 0·05; Fig. 2b).
Role of PGD2/CRTH2 receptor in A. fumigatus-induced exacerbation of eosinophilic airway inflammation in OVA-sensitized rats
In the present study we observed that A. fumigatus increased the concentration of PGD2 in the bronchoalveolar lavage fluid of OVA-exposed animals significantly, though not in bronchoalveolar lavage fluid of NS-exposed animals (Fig. 3). To determine the mechanisms involved in PGD2-mediated eosinophil chemotaxis we first examined the expression of PGD2 receptor CRTH2. Expression of CRTH2 was identified in lung tissue by immunohistochemical analysis (Fig. 4). Microscopic examination of the lung immunostained slides showed higher expression for CRTH2 in samples from OVA-sensitized rats compared with NS group (Fig. 4). CRTH2 expression was lower in the OVA group than in the OVA + AF group (Fig. 4) and lower in the treated rats than in the OVA + AF group (Fig. 4). These results indicate the involvement of CRTH2 in the pathophysiology of eosinophilic inflammation in asthma.
Figure 3.
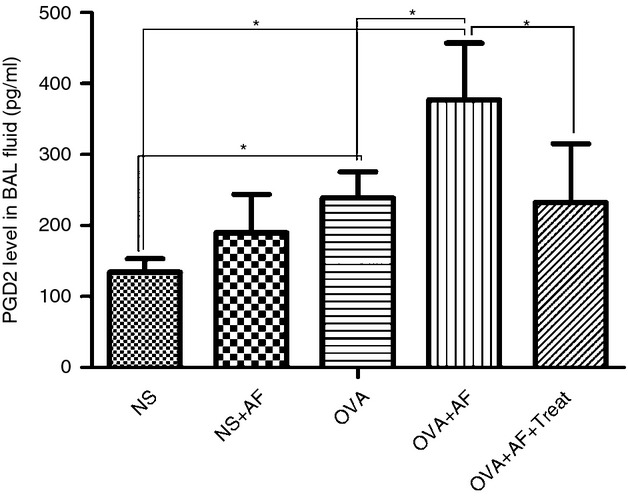
Prostaglandin D2 (PGD2) levels measured by using ELISA in bronchoalveolar lavage (BAL) fluid samples. Administration of Aspergillus fumigatus alters PGD2 levels in BAL fluid. Levels of PGD2 in BAL fluid were measured in the five groups: non-sensitized control group (NS); Aspergillus fumigatus spore-exposed group (NS + AF); ovalbumin (OVA)-sensitized and OVA-challenged group (OVA); OVA-sensitized, OVA-challenged and A. fumigatus spore-exposed group (OVA + AF); and CRTH2 antagonist OC00459-treated group (OVA + AF + Treat), 72 hr after the last challenge. Values are means ± SEM. *P < 0·05.
Figure 4.
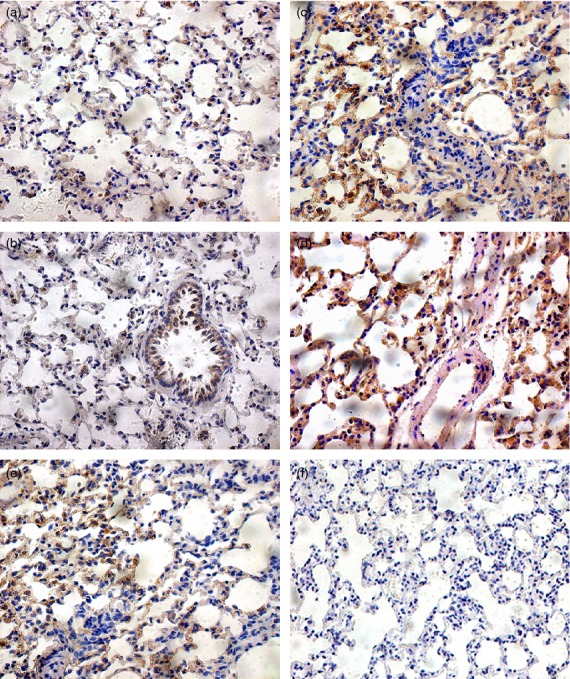
Immunohistochemical staining of CRTH2 in lung tissue from the five groups; non-sensitized, non-challenged saline inhaled rats (NS) (a), non-sensitized, non-challenged and Aspergillus fumigatus spores inhaled rats (NS + AF) (b), OVA-sensitized, OVA-challenged and saline-inhaled rats (OVA) (c), OVA-sensitized, OVA-challenged and A. fumigatus spores inhaled rats (OVA + AF) (d), and OVA-sensitized, OVA-challenged and A. fumigatus spores inhaled rats receiving OC000459 (OVA + AF + Treat) (e). (f) PBS negative control.
We also investigated the effect of A. fumigatus on the expression of CRTH2 by quantitative PCR (Fig. 5). In comparison with the NS group, CRTH2 mRNA levels were significantly elevated in all OVA-sensitized and challenged groups (Fig. 5). CRTH2 mRNA levels were significantly decreased in OVA + AF + Treat group versus the OVA + AF group, and the lowest level was found in the NS + AF group (Fig. 5).
Figure 5.
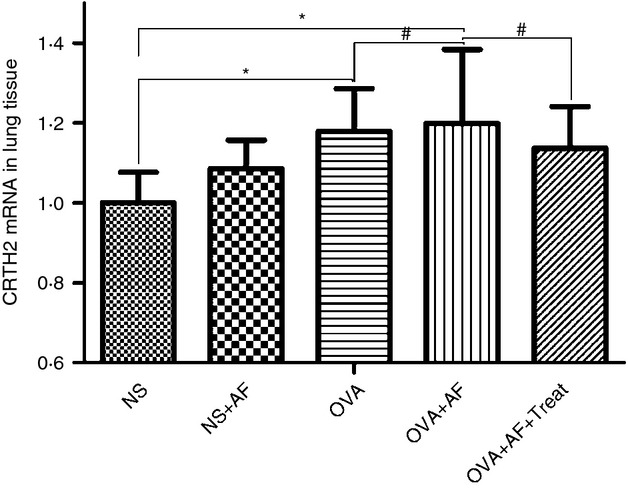
qRT-PCR of CRTH2 mRNA transcripts in lung tissue. Results were expressed as the number of CRTH2 gene copies normalized to the number of glyceraldehyde-3-phosphate dehydrogenase gene copies (housekeeping gene). Values are means ± SEM. *P < 0·05. #The difference between groups tended to be different (P = 0·05) but did not reach significance.
These results were confirmed by Western blot analysis using specific anti-CRTH2 antibodies (Fig. 6). Higher expression was observed in OVA-sensitized with respect to NS rats. Inhalation of A. fumigatus increased the total CRTH2 protein contents. As seen in Fig. 6, after treatment the expression of CRTH2 protein significantly decreased. Equal loading of protein in tissue homogenates was also determined by Western blot using an anti-GAPDH antibody. All blots were repeated three times and one representative image is shown.
Figure 6.
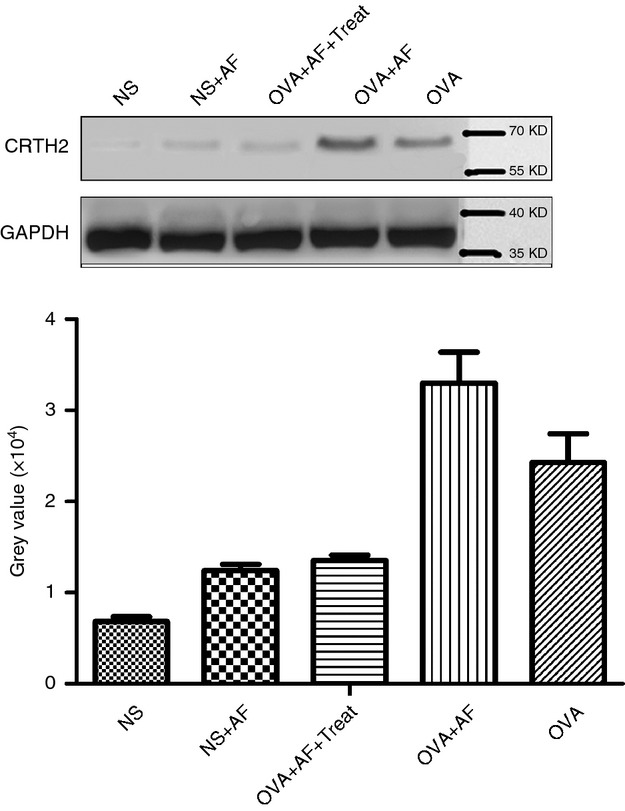
Expression of CRTH2 in lung tissue. Western blot was performed on whole tissue extracts from the five groups: non-sensitized control group (NS); Aspergillus fumigatus spore-exposed group (NS + AF); ovalbumin (OVA)-sensitized and OVA-challenged group (OVA); OVA-sensitized, OVA-challenged and A. fumigatus spore-exposed group (OVA + AF); and CRTH2 antagonist OC00459-treated group (OVA + AF + Treat).; GAPDH was used as control.
These results indicate that infiltrated cells express functional CRTH2 and suggest that PGD2 might modulate eosinophil chemotaxis through activation of CRTH2, which may be mediated by an increase of transcription at the CRTH2 locus.
Discussion
Eosinophilia is a hallmark in allergic asthma, and is blamed for the chronicity of the disease. Exacerbations of asthma are known to be associated with eosinophilic inflammation in children and adults.33–35 For example, asthmatics with refractory eosinophilic inflammation have significantly higher exacerbation rates,33 whereas eosinophil-targeted therapies such as anti-interleukin-5 significantly reduce asthma exacerbation rates.33–35 In murine models of allergen-induced AHR, depletion of eosinophils protects mice from developing mucus accumulation, AHR and/or airway remodelling, suggesting that eosinophilia plays a role in the pathogenesis of asthma.36,37
The immunomodulatory effect of A. fumigatus and its components have been well documented.38–41 Exposure to A. fumigatus extracts leads to eosinopoiesis and the subsequent increase in peripheral blood eosinophils, reiterating that fungal antigens are responsible for the recruitment of eosinophils into the affected tissues.42 In our previous studies we showed that prolonged exposure of OVA-sensitized and OVA-challenged rats to A. fumigatus spores resulted in enhanced levels of serum IgE, pulmonary eosinophils and bronchial hyper-responsiveness.14,43 Although some authors reported accumulation of eosinophils following A. fumigatus inhalation,44–47 in this study only a few eosinophils were observed after A. fumigatus exposure alone. However, when A. fumigatus inhalation was combined with OVA allergen challenge of sensitized mice, the number of eosinophils was also significantly enhanced. Similarly, studies in humans have also shown significant blood eosinophilia, as a result of infection with A. fumigatus.48–50 Therefore, the current interest in airway eosinophilic inflammation is to discover the molecular events resulting in eosinophil recruitment and hence to develop an effective mode of therapy.
The accumulation of eosinophils in the lung is regulated by chemokines such as eotaxin, as well as a number of different cytokines such as interleukin-5, granulocyte–macrophage colony-stimulating factor and granulocyte colony-stimulating factor.51–53 Mast-cell-derived PGD2 is also an important mediator involved in eosinophilic airway inflammation19,23 and plays a substantial role in persistent eosinophil accumulation in airways repeatedly exposed to allergens. Current evidence showed that A. fumigatus exposure significantly increased both chemoattractants (data not shown), but the levels of PGD2 were also increased.
In the present study, we developed an experimental model that mimics the pathological and physiological changes observed in the airways of asthmatic patients during exacerbations induced by fungus exposure. The experiment focused on the effect of A. fumigatus on a Wistar rat model of chronic asthma in rats sensitized and challenged by OVA antigen. Herein, we show that the PGD2/CRTH2 pathway, which has been reported to be up-regulated in lung tissues of subjects with allergic asthma, has the ability to be up-regulated with A. fumigatus. We have shown, in this study, that (i) the administration of A. fumigatus into the airways increased the synthesis of PGD2 in OVA-exposed rats only, (ii) inhalation delivery of A. fumigatus conidia recruited eosinophils into the airways of OVA-exposed rats and increased bronchial hyper-responsiveness, (iii) the pharmacological blockade of CRTH2, a PGD2 receptor, prevented the increase in eosinophil accumulation and/or bronchial hyper-responsiveness induced by the administration of A. fumigatus in rats, and (iv) the administration of A. fumigatus into the airways increased the expression of CRTH2 in OVA-exposed lungs and its effect on the level of CRTH2 mRNA suggesting that the enhancing effect of A. fumigatus is mediated by an increase of transcription at the CRTH2 locus. All of these observations strongly indicate that PGD2 and its receptor CRTH2 is the essential system for the exacerbation of eosinophilic airway inflammation induced by A. fumigatus.
Studies in pre-clinical models of allergic disease support the view that CRTH2 plays a central role in leucocyte recruitment,30,54–57 AHR57 and the production of cytokines,58,59 mucus54 and IgE.30,58,60 Independently, it has been shown that PGD2 promotes chemotaxis of eosinophils through a receptor unrelated to DP1 and this was designated CRTH2.20 The ability of PGD2 to promote eosinophil accumulation in the airways of pre-clinical species is mimicked by selective CRTH2 agonists but not DP1 agonists28,29 and is inhibited by the CRTH2 antagonist.28 The effect of the mast cell supernatants on migration of T helper type 2 lymphocytes was blocked by a CRTH2 antagonist,61 as was the effect of mast cell supernatants on activation of eosinophils (S.L. Gyles, L. Xue, E.R. Townsend, F. Wettey, R. Pettipher, unpublished observations). Taken together, these data suggest that mast cell-dependent activation of eosinophils and T helper type 2 cells is mediated by PGD2 acting on CRTH2.
The observations linking CRTH2 to the development of allergic inflammation has spurred interest in identifying more potent, selective and orally bioavailable antagonists of this receptor for treating asthma and related disorders. A number of chemical series have been described that antagonize CRTH2, including tetrahydroquinolone derivatives, carbazole derivatives, indole acetic acids, azaindole-3-acetic acids, phenoxyacetic acids, phenylacetic acids, thiazoleacetic acids, 3-indolyl sultams, and other series as reviewed in detail by Ulven and Kostenis.62 OC000459, an indole-1-acetic acid derivative that is an orally active CRTH2 antagonist, which, in proof-of-concept phase IIb clinical trials has been shown to be highly potent and selective in inhibiting PGD2-mediated activation of T helper type 2 cells and eosinophils and is an ideal tool to explore the clinical utility of CRTH2 antagonism.63 The clinical efficacy findings with OC000459 are consistent with the results of pre-clinical studies in CRTH2-deficient mice and with small-molecule antagonists where reduction in eosinophil accumulation54–57 has been observed. It is also effective in inhibiting mast cell-dependent activation of T helper type 2 cells and eosinophils and airway eosinophilia in rats and guinea-pigs. It is concluded that OC000459 shows promise in the treatment of asthma and warrants further investigation in this indication.
These data demonstrate an important role for CRTH2 in the pathogenesis of allergic inflammation. Combined with our observation that exacerbation of allergic airway inflammation and AHR induced by A. fumigatus is reduced in rats treated with OC000459, these findings suggest that the CRTH2 antagonist may find utility in the prevention or treatment of fungal exacerbations in asthmatic subjects. It is tempting to speculate that CRTH2 receptor antagonism might be useful as a preventive or therapeutic agent in the exacerbations of asthma. Although CRTH2 receptor antagonists have been found to be effective to suppress inflammation, CRTH2-deficient mice developed more severe inflammation of the airways compared with wild-type mice,64 which makes the role of the CRTH2 receptor on the pathogenesis of allergic inflammation controversial. The mechanisms are still not well understood and await further investigation.
One limitation of the model is that we did not directly measure bronchoconstriction, but we assessed changes in ventilation by unrestrained whole body plethysmography. Penh (Enhanced pause), which derived from unrestrained whole body plethysmography, is being widely used as a method to study bronchial responsiveness in various animal models of lung disease.65–72 This technique, and particularly the use of Penh as an index of airway resistance, has been heavily criticized recently.73–75 Whether an increased Penh is a consequence of changes in the mechanical properties of the respiratory system has not been investigated thoroughly; nevertheless, there is little doubt that increases in Penh do reflect alterations in ventilatory pattern that are compatible with airway obstruction. Other observations in our laboratory support correlation between the Penh and pulmonary resistance (RL – lung resistance, a well recognized pulmonary function parameter; Figs S1 and S2). So it is very difficult to interpret the correlation between airway obstructive character and Penh. Independently of the physiological meaning of Penh, we infer that some degree of correlation between Penh and bronchoconstriction is expected. This index was indeed measuring changes in the pulmonary function, at least in our experimental conditions. However, whether Penh can be used as a surrogate for RL is unclear and requires further research.
Our study showed that prolonged exposure of OVA-sensitized and OVA-challenged rats to A. fumigatus spores resulted in enhanced levels of pulmonary eosinophils and bronchial hyper-responsiveness, pharmacological blockade of CRTH2 successfully suppressed pulmonary eosinophilia and AHR. Although eosinophils tend to parallel AHR, they did not appear to be the definitive causative process in this response. It is generally accepted that airway inflammation contributes to the presence and severity of AHR, but the association between allergen-induced pulmonary eosinophilia and the development of AHR has been, at best, a collection of confusing and often contradictory observations. A dissociation between eosinophil numbers and AHR has been shown in guinea-pigs,76 Brown Norway rats77 and mice.78 Clinical studies in human allergic asthma have also shown dissociation between airway inflammation, AHR and the late asthmatic reaction.79 These studies are not definitive; however, they do cast a shadow on the (cellular) inflammation AHR link. Some researchers point out that the lack of symptom improvement in asthma patients after administration of antibodies to interleukin-5 exemplifies the ambiguous character of clinical studies that attempt to ablate eosinophils. Mouse models purporting to ablate eosinophils are also ambiguous, as they either do not completely eliminate pulmonary eosinophils or they elicit the loss of eosinophils by mechanisms that do not differentiate between effects on eosinophils and other potentially important cellular targets. In each of these studies,76–78 eosinophil numbers in the experimental mice are low but elevated relative to control animals, the small numbers of eosinophils present may actually be sufficient to elicit AHR. And measurements of lung function after OVA sensitization/aerosol challenge of eosinophil-deficient mice (a transgenic line of mice that are specifically devoid of eosinophils but otherwise have a full complement of hematopoietically derived cells, PHIL mice) showed that methacholine-induced AHR was dependent on the presence of eosinophils.36 Therefore, to identify the contributions of inflammation to AHR, it is important to consider the many aspects of this process, including the location of the cells (i.e. lumen versus bronchial wall) and cell type as well as the many mediators associated with the injury to the airway. The present data are not sufficient to make a definitive conclusion, cellular mechanisms involved in the development of AHR need further study.
In conclusion, OVA-induced inflammation led to an increased susceptibility of the airways to A. fumigatus and increased their likelihood to develop eosinophilic inflammation and bronchial hyper-responsiveness via the activation of the PGD2/CRTH2 pathway. The responsiveness to A. fumigatus that we observed appears enhanced in the inflamed airways, because (i) they have a higher capacity to produce PGD2 in response to A. fumigatus and (ii) eosinophils are more likely to be recruited into the inflamed airways in response to PGD2 through a CRTH2 receptor. The immunomodulatory effects of A. fumigatus and the relationship between allergen dosage and this effect pose interesting problems for future study.
Acknowledgments
Funding for the study was obtained from Basic Research Project of Shanghai Science and Technology Commission. The sponsors played no part in the design or interpretation of the study. We would like to acknowledge the staff in the Animal Resources Centre for their invaluable assistance in the performance of this study.
Disclosures
The authors have no conflicts of interest to disclose.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Airway responsiveness measured by barometric plethysmography in spontaneously breathing animals (Penh) (A) and plethysmography in mechanically ventilated animals (RL) (B).
Comparison of RL and Penh in Wistar rats challenged with methacholine (MCh) (0.025, 0.05, 0.1, 0.2, 0.4 mg/kg, i.v.). Data are presented as % increase from base level. RL and Penh are correlative measurements of airway reactivity (r2 = 0.842). Solid line: regression line.
References
- 1.Yoon J, Ponikau JU, Lawrence CB, Kita H. Innate antifungal immunity of human eosinophils mediated by a β2 integrin, CD11b. J Immunol. 2008;181:2907–15. doi: 10.4049/jimmunol.181.4.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams DL. Overview of (1→3)-β-d-glucan immunobiology. Mediators Inflamm. 1997;6:247–50. doi: 10.1080/09629359791550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ponikau JU, Sherris DA, Kita H. The role of ubiquitous airborne fungi in chronic rhinosinusitis. Clin Allergy Immunol. 2007;20:177–84. [PubMed] [Google Scholar]
- 4.Matsuwaki Y, Wada K, White TA, et al. Recognition of fungal protease activities induces cellular activation and eosinophil-derived neurotoxin release in human eosinophils. J Immunol. 2009;183:6708–16. doi: 10.4049/jimmunol.0901220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuwaki Y, Wada K, White T, Moriyama H, Kita H. Alternaria fungus induces the production of GM-CSF, interleukin-6 and interleukin-8 and calcium signaling in human airway epithelium through protease-activated receptor 2. Int Arch Allergy Immunol. 2012;158(Suppl. 1):19–29. doi: 10.1159/000337756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurup VP, Seymour BW, Choi H, Coffman RL. Particulate Aspergillus fumigatus antigens elicit a TH2 response in BALB/c mice. J Allergy Clin Immunol. 1994;93:1013–20. doi: 10.1016/s0091-6749(94)70050-8. [DOI] [PubMed] [Google Scholar]
- 7.Kumar R. Mild, moderate, and severe forms of allergic bronchopulmonary aspergillosis: a clinical and serologic evaluation. Chest. 2003;124:890–2. doi: 10.1378/chest.124.3.890. [DOI] [PubMed] [Google Scholar]
- 8.Inoue Y, Matsuwaki Y, Shin SH, Ponikau JU, Kita H. Nonpathogenic, environmental fungi induce activation and degranulation of human eosinophils. J Immunol. 2005;175:5439–47. doi: 10.4049/jimmunol.175.8.5439. [DOI] [PubMed] [Google Scholar]
- 9.Denning DW, O'Driscoll BR, Hogaboam CM, Bowyer P, Niven RM. The link between fungi and severe asthma: a summary of the evidence. Eur Respir J. 2006;27:615–26. doi: 10.1183/09031936.06.00074705. [DOI] [PubMed] [Google Scholar]
- 10.Mirkov I, Stosic-Grujicic S, Kataranovski M. Host immune defense against Aspergillus fumigatus: insight from experimental systemic (disseminated) infection. Immunol Res. 2012;52:120–6. doi: 10.1007/s12026-012-8274-x. [DOI] [PubMed] [Google Scholar]
- 11.Segal BH. Aspergillosis. N Engl J Med. 2009;360:1870–84. doi: 10.1056/NEJMra0808853. [DOI] [PubMed] [Google Scholar]
- 12.Brown GD. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol. 2011;29:1–21. doi: 10.1146/annurev-immunol-030409-101229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrett NA, Maekawa A, Rahman OM, Austen KF, Kanaoka Y. Dectin-2 recognition of house dust mite triggers cysteinyl leukotriene generation by dendritic cells. J Immunol. 2009;182:1119–28. doi: 10.4049/jimmunol.182.2.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao FS, Gao YY, Liu MJ, Liu YQ. Chronic Aspergillus fumigatus exposure upregulates the expression of mucin 5AC in the airways of asthmatic rats. Exp Lung Res. 2012;38:256–65. doi: 10.3109/01902148.2012.676705. [DOI] [PubMed] [Google Scholar]
- 15.Werner JL, Metz AE, Horn D, et al. Requisite role for the dectin-1 β-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol. 2009;182:4938–46. doi: 10.4049/jimmunol.0804250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Said-Sadier N, Padilla E, Langsley G, Ojcius DM. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS ONE. 2010;5:e10008. doi: 10.1371/journal.pone.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bozza S, Clavaud C, Giovannini G, et al. Immune sensing of Aspergillus fumigatus proteins, glycolipids, and polysaccharides and the impact on Th immunity and vaccination. J Immunol. 2009;183:2407–14. doi: 10.4049/jimmunol.0900961. [DOI] [PubMed] [Google Scholar]
- 18.Woodward DF, Hawley SB, Williams LS, Ralston TR, Protzman CE, Spada CS, Nieves AL. Studies on the ocular pharmacology of prostaglandin D2. Invest Ophthalmol Vis Sci. 1990;31:138–46. [PubMed] [Google Scholar]
- 19.O'Sullivan S, Roquet A, Dahlen B, Dahlen S, Kumlin M. Urinary excretion of inflammatory mediators during allergen-induced early and late phase asthmatic reactions. Clin Exp Allergy. 1998;28:1332–9. doi: 10.1046/j.1365-2222.1998.00368.x. [DOI] [PubMed] [Google Scholar]
- 20.Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. 2001;98:1942–8. doi: 10.1182/blood.v98.6.1942. [DOI] [PubMed] [Google Scholar]
- 21.Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255–61. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emery DL, Djokic TD, Graf PD, Nadel JA. Prostaglandin D2 causes accumulation of eosinophils in the lumen of the dog trachea. J Appl Physiol. 1989;67:959–62. doi: 10.1152/jappl.1989.67.3.959. [DOI] [PubMed] [Google Scholar]
- 23.Bochenek G, Nizankowska E, Gielicz A, Swierczynska M, Szczeklik A. Plasma 9α,11β-PGF2, a PGD2 metabolite, as a sensitive marker of mast cell activation by allergen in bronchial asthma. Thorax. 2004;59:459–64. doi: 10.1136/thx.2003.013573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagata K, Tanaka K, Ogawa K, et al. Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol. 1999;162:1278–86. [PubMed] [Google Scholar]
- 25.Nagata K, Hirai H, Tanaka K, Ogawa K, Aso T, Sugamura K, Nakamura M, Takano S. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s) FEBS Lett. 1999;459:195–9. doi: 10.1016/s0014-5793(99)01251-x. [DOI] [PubMed] [Google Scholar]
- 26.Gervais FG, Cruz RP, Chateauneuf A, Gale S, Sawyer N, Nantel F, Metters KM, O'neill GP. Selective modulation of chemokinesis, degranulation, and apoptosis in eosinophils through the PGD2 receptors CRTH2 and DP. J Allergy Clin Immunol. 2001;108:982–8. doi: 10.1067/mai.2001.119919. [DOI] [PubMed] [Google Scholar]
- 27.Spik I, Brenuchon C, Angeli V, Staumont D, Fleury S, Capron M, Trottein F, Dombrowicz D. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. 2005;174:3703–8. doi: 10.4049/jimmunol.174.6.3703. [DOI] [PubMed] [Google Scholar]
- 28.Shiraishi Y, Asano K, Nakajima T, et al. Prostaglandin D2-induced eosinophilic airway inflammation is mediated by CRTH2 receptor. J Pharmacol Exp Ther. 2005;312:954–60. doi: 10.1124/jpet.104.078212. [DOI] [PubMed] [Google Scholar]
- 29.Almishri W, Cossette C, Rokach J, Martin JG, Hamid Q, Powell WS. Effects of prostaglandin D2, 15-deoxy-δ12,14-prostaglandin J2, and selective DP1 and DP2 receptor agonists on pulmonary infiltration of eosinophils in Brown Norway rats. J Pharmacol Exp Ther. 2005;313:64–9. doi: 10.1124/jpet.104.079079. [DOI] [PubMed] [Google Scholar]
- 30.Satoh T, Moroi R, Aritake K, et al. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol. 2006;177:2621–9. doi: 10.4049/jimmunol.177.4.2621. [DOI] [PubMed] [Google Scholar]
- 31.Hirano A, Kanehiro A, Ono K, et al. Pirfenidone modulates airway responsiveness, inflammation, and remodeling after repeated challenge. Am J Respir Cell Mol Biol. 2006;35:366–77. doi: 10.1165/rcmb.2005-0452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ito W, Kanehiro A, Matsumoto K, et al. Hepatocyte growth factor attenuates airway hyperresponsiveness, inflammation, and remodeling. Am J Respir Cell Mol Biol. 2005;32:268–80. doi: 10.1165/rcmb.2004-0058OC. [DOI] [PubMed] [Google Scholar]
- 33.Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–84. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, Wardlaw AJ, Pavord ID. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–21. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 35.Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, Ortega H, Chanez P. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–9. doi: 10.1016/S0140-6736(12)60988-X. [DOI] [PubMed] [Google Scholar]
- 36.Lee JJ, Dimina D, Macias MP, et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;305:1773–6. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- 37.Humbles AA, Lloyd CM, McMillan SJ, et al. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–9. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 38.Free SJ. Fungal cell wall organization and biosynthesis. Adv Genet. 2013;81:33–82. doi: 10.1016/B978-0-12-407677-8.00002-6. [DOI] [PubMed] [Google Scholar]
- 39.Scharf DH, Heinekamp T, Remme N, Hortschansky P, Brakhage AA, Hertweck C. Biosynthesis and function of gliotoxin in Aspergillus fumigatus. Appl Microbiol Biotechnol. 2012;93:467–72. doi: 10.1007/s00253-011-3689-1. [DOI] [PubMed] [Google Scholar]
- 40.Dagenais TR, Keller NP. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev. 2009;22:447–65. doi: 10.1128/CMR.00055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balloy V, Chignard M. The innate immune response to Aspergillus fumigatus. Microbes Infect. 2009;11:919–27. doi: 10.1016/j.micinf.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 42.Murali PS, Dai G, Kumar A, Fink JN, Kurup VP. Aspergillus antigen-induced eosinophil differentiation in a murine model. Infect Immun. 1992;60:1952–6. doi: 10.1128/iai.60.5.1952-1956.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao FS, Qiao JO, Zhang Y, Jin XQ. Chronic intranasal administration of Aspergillus fumigatus spores leads to aggravation of airway inflammation and remodelling in asthmatic rats. Respirology. 2009;14:360–70. doi: 10.1111/j.1440-1843.2009.01482.x. [DOI] [PubMed] [Google Scholar]
- 44.Samarasinghe AE, Hoselton SA, Schuh JM. A comparison between intratracheal and inhalation delivery of Aspergillus fumigatus conidia in the development of fungal allergic asthma in C57BL/6 mice. Fungal Biol. 2011;115:21–9. doi: 10.1016/j.funbio.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murdock BJ, Falkowski NR, Shreiner AB, Sadighi Akha AA, McDonald RA, White ES, Toews GB, Huffnagle GB. Interleukin-17 drives pulmonary eosinophilia following repeated exposure to Aspergillus fumigatus conidia. Infect Immun. 2012;80:1424–36. doi: 10.1128/IAI.05529-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsuse H, Fukushima C, Fukahori S, Tsuchida T, Kawano T, Nishino T, Kohno S. Differential effects of dexamethasone and itraconazole on Aspergillus fumigatus-exacerbated allergic airway inflammation in a murine model of mite-sensitized asthma. Respiration. 2013;85:429–35. doi: 10.1159/000345861. [DOI] [PubMed] [Google Scholar]
- 47.Epstein VA, Bryce PJ, Conley DB, Kern RC, Robinson AM. Intranasal Aspergillus fumigatus exposure induces eosinophilic inflammation and olfactory sensory neuron cell death in mice. Otolaryngol Head Neck Surg. 2008;138:334–9. doi: 10.1016/j.otohns.2007.11.029. [DOI] [PubMed] [Google Scholar]
- 48.Greenberger PA. Chapter 18: Allergic bronchopulmonary aspergillosis. Allergy Asthma Proc. 2012;33(Suppl. 1):S61–3. doi: 10.2500/aap.2012.33.3551. [DOI] [PubMed] [Google Scholar]
- 49.Agarwal R, Khan A, Aggarwal AN, Varma N, Garg M, Saikia B, Gupta D, Chakrabarti A. Clinical relevance of peripheral blood eosinophil count in allergic bronchopulmonary aspergillosis. J Infect Public Health. 2011;4:235–43. doi: 10.1016/j.jiph.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 50.Morita H, Oguri T, Uemura T, Iwashima Y, Nakamura A, Sato S. [Case of chronic necrotizing pulmonary aspergillosis complicated by elevated eosinophils and serum IgE] Nihon Kokyuki Gakkai Zasshi. 2010;48:842–6. [PubMed] [Google Scholar]
- 51.Simon H, Alam R. Regulation of eosinophil apoptosis: transduction of survival and death signals. Int Arch Allergy Immunol. 1999;118:7–14. doi: 10.1159/000024025. [DOI] [PubMed] [Google Scholar]
- 52.Simon HU. Regulation of eosinophil and neutrophil apoptosis – similarities and differences. Immunol Rev. 2001;179:156–62. doi: 10.1034/j.1600-065x.2001.790115.x. [DOI] [PubMed] [Google Scholar]
- 53.Tai PC, Sun L, Spry CJ. Effects of IL-5, granulocyte/macrophage colony-stimulating factor (GM-CSF) and IL-3 on the survival of human blood eosinophils in vitro. Clin Exp Immunol. 1991;85:312–6. doi: 10.1111/j.1365-2249.1991.tb05725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uller L, Mathiesen JM, Alenmyr L, et al. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir Res. 2007;8:16. doi: 10.1186/1465-9921-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pettipher R. The roles of the prostaglandin D2 receptors DP1 and CRTH2 in promoting allergic responses. Br J Pharmacol. 2008;153(Suppl. 1):S191–9. doi: 10.1038/sj.bjp.0707488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oiwa M, Satoh T, Watanabe M, Niwa H, Hirai H, Nakamura M, Yokozeki H. CRTH2-dependent, STAT6-independent induction of cedar pollen dermatitis. Clin Exp Allergy. 2008;38:1357–66. doi: 10.1111/j.1365-2222.2008.03007.x. [DOI] [PubMed] [Google Scholar]
- 57.Lukacs NW, Berlin AA, Franz-Bacon K, et al. CRTH2 antagonism significantly ameliorates airway hyperreactivity and downregulates inflammation-induced genes in a mouse model of airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2008;295:L767–79. doi: 10.1152/ajplung.90351.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nomiya R, Okano M, Fujiwara T, et al. CRTH2 plays an essential role in the pathophysiology of Cry j 1-induced pollinosis in mice. J Immunol. 2008;180:5680–8. doi: 10.4049/jimmunol.180.8.5680. [DOI] [PubMed] [Google Scholar]
- 59.Boehme SA, Franz-Bacon K, Chen EP, Sasik R, Sprague LJ, Ly TW, Hardiman G, Bacon KB. A small molecule CRTH2 antagonist inhibits FITC-induced allergic cutaneous inflammation. Int Immunol. 2009;21:81–93. doi: 10.1093/intimm/dxn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boehme SA, Chen EP, Franz-Bacon K, Sasik R, Sprague LJ, Ly TW, Hardiman G, Bacon KB. Antagonism of CRTH2 ameliorates chronic epicutaneous sensitization-induced inflammation by multiple mechanisms. Int Immunol. 2009;21:1–17. doi: 10.1093/intimm/dxn118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gyles SL, Xue L, Townsend ER, Wettey F, Pettipher R. A dominant role for chemoattractant receptor-homologous molecule expressed on T helper type 2 (Th2) cells (CRTH2) in mediating chemotaxis of CRTH2+ CD4+ Th2 lymphocytes in response to mast cell supernatants. Immunology. 2006;119:362–8. doi: 10.1111/j.1365-2567.2006.02440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ulven T, Kostenis E. Novel CRTH2 antagonists: a review of patents from 2006 to 2009. Expert Opin Ther Pat. 2010;20:1505–30. doi: 10.1517/13543776.2010.525506. [DOI] [PubMed] [Google Scholar]
- 63.Barnes N, Pavord I, Chuchalin A, et al. A randomized, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy. 2012;42:38–48. doi: 10.1111/j.1365-2222.2011.03813.x. [DOI] [PubMed] [Google Scholar]
- 64.Chevalier E, Stock J, Fisher T, et al. Cutting edge: chemoattractant receptor-homologous molecule expressed on Th2 cells plays a restricting role on IL-5 production and eosinophil recruitment. J Immunol. 2005;175:2056–60. doi: 10.4049/jimmunol.175.4.2056. [DOI] [PubMed] [Google Scholar]
- 65.Bergren DR. Chronic tobacco smoke exposure increases airway sensitivity to capsaicin in awake guinea pigs. J Appl Physiol (1985) 2001;90:695–704. doi: 10.1152/jappl.2001.90.2.695. [DOI] [PubMed] [Google Scholar]
- 66.Claridge JA, Enelow RI, Young JS. Hemorrhage and resuscitation induce delayed inflammation and pulmonary dysfunction in mice. J Surg Res. 2000;92:206–13. doi: 10.1006/jsre.2000.5899. [DOI] [PubMed] [Google Scholar]
- 67.Djuric VJ, Cox G, Overstreet DH, Smith L, Dragomir A, Steiner M. Genetically transmitted cholinergic hyperresponsiveness predisposes to experimental asthma. Brain Behav Immun. 1998;12:272–84. doi: 10.1006/brbi.1998.0538. [DOI] [PubMed] [Google Scholar]
- 68.Finotto S, Sanctis De GT, Lehr HA, et al. Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J Exp Med. 2001;193:1247–60. doi: 10.1084/jem.193.11.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finotto S, Neurath MF, Glickman JN, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295:336–8. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 70.Hamada K, Goldsmith CA, Goldman A, Kobzik L. Resistance of very young mice to inhaled allergen sensitization is overcome by coexposure to an air-pollutant aerosol. Am J Respir Crit Care Med. 2000;161:1285–93. doi: 10.1164/ajrccm.161.4.9906137. [DOI] [PubMed] [Google Scholar]
- 71.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156:766–75. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 72.Park JK, Kim YK, Lee SR, Cho SH, Min KU, Kim YY. Repeated exposure to low levels of sulfur dioxide (SO2) enhances the development of ovalbumin-induced asthmatic reactions in guinea pigs. Ann Allergy Asthma Immunol. 2001;86:62–7. doi: 10.1016/S1081-1206(10)62358-7. [DOI] [PubMed] [Google Scholar]
- 73.Bates JH, Irvin CG. Measuring lung function in mice: the phenotyping uncertainty principle. J Appl Physiol (1985) 2003;94:1297–306. doi: 10.1152/japplphysiol.00706.2002. [DOI] [PubMed] [Google Scholar]
- 74.Lundblad LK, Irvin CG, Adler A, Bates JH. A reevaluation of the validity of unrestrained plethysmography in mice. J Appl Physiol (1985) 2002;93:1198–207. doi: 10.1152/japplphysiol.00080.2002. [DOI] [PubMed] [Google Scholar]
- 75.Petak F, Habre W, Donati YR, Hantos Z, Barazzone-Argiroffo C. Hyperoxia-induced changes in mouse lung mechanics: forced oscillations vs. barometric plethysmography. J Appl Physiol (1985) 2001;90:2221–30. doi: 10.1152/jappl.2001.90.6.2221. [DOI] [PubMed] [Google Scholar]
- 76.Mauser PJ, Pitman A, Witt A, et al. Inhibitory effect of the TRFK-5 anti-IL-5 antibody in a guinea pig model of asthma. Am Rev Respir Dis. 1993;148:1623–7. doi: 10.1164/ajrccm/148.6_Pt_1.1623. [DOI] [PubMed] [Google Scholar]
- 77.Elwood W, Lotvall JO, Barnes PJ, Chung KF. Effect of dexamethasone and cyclosporin A on allergen-induced airway hyperresponsiveness and inflammatory cell responses in sensitized Brown-Norway rats. Am Rev Respir Dis. 1992;145:1289–94. doi: 10.1164/ajrccm/145.6.1289. [DOI] [PubMed] [Google Scholar]
- 78.Hogan SP, Matthaei KI, Young JM, Koskinen A, Young IG, Foster PS. A novel T cell-regulated mechanism modulating allergen-induced airways hyperreactivity in BALB/c mice independently of IL-4 and IL-5. J Immunol. 1998;161:1501–9. [PubMed] [Google Scholar]
- 79.Crimi E, Spanevello A, Neri M, Ind PW, Rossi GA, Brusasco V. Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am J Respir Crit Care Med. 1998;157:4–9. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Airway responsiveness measured by barometric plethysmography in spontaneously breathing animals (Penh) (A) and plethysmography in mechanically ventilated animals (RL) (B).
Comparison of RL and Penh in Wistar rats challenged with methacholine (MCh) (0.025, 0.05, 0.1, 0.2, 0.4 mg/kg, i.v.). Data are presented as % increase from base level. RL and Penh are correlative measurements of airway reactivity (r2 = 0.842). Solid line: regression line.