

Abstract
Somatic GNAQ mutations at codon 209 have been identified in approximately 50% of uveal melanomas (UM) and have been reported to be oncogenic through activating PLCβ-PKC-Erk1/2 pathways. We hypothesized that PKC may provide new opportunities for therapeutic targeting of UM carrying GNAQ mutations. To test this hypothesis, UM cells harboring wild type or mutant GNAQ were treated with the PKC inhibitor AEB071 (sotrastaurin) or infected with lentivirus expressing shRNAs targeting PKC isoforms. Notably, AEB071 at low micromolar concentrations significantly inhibited the growth of UM cells harboring GNAQ mutations through induction of G1 arrest and apoptosis. However, AEB071 had little effect on UM cells carrying wild type GNAQ. AEB071-mediated cell inhibition in the GNAQ mutated UM was accompanied by inhibition of Erk1/2 phosphorylation, NF-κB, decreased expression of cyclin D1, survivin, Bcl-xL and XIAP, and increased expression of cyclin-dependent kinase inhibitor p27Kip1. AEB071 suppressed the expression of PKC α, β, δ, ε and θ in GNAQ mutated UM cells. Our findings from shRNA-mediated knockdown studies revealed that these PKC isoforms are functionally important for UM cells harboring GNAQ mutations. Furthermore, inhibitors of Erk1/2 and NF-κB pathways reduced viability of UM cells. Together, our findings demonstrate that AEB071 exerts antitumor action on UM cells carrying GNAQ mutations via targeting PKC/Erk1/2 and PKC/NF-κB pathways. Targeted PKC inhibition with drugs such as AEB071 offers novel therapeutic potential for UM harboring GNAQ mutations.
Keywords: Melanoma, Uveal Melanoma, Protein Kinase C
INTRODUCTION
Uveal melanoma (UM) is the most common primary intraocular malignant tumor in adults, with an incidence of seven cases per million annually (1). Approximately half of UM patients develop metastases to the liver within 15 years of initial diagnosis. With no effective treatment modality available the median survival time of UM patients with metastasis is less than six months (2).
The etiology of UM has not been fully understood. Mutations in the GNAQ gene have been identified in approximately 50% of UM and 83% blue naevi (3–5). The GNAQ gene encodes for the α subunit of q class of heterotrimeric GTP binding proteins (G proteins) that are composed of three subunits (Gα, Gβ, and Gγ) and transduce signals from 7-transmembrane G-protein coupled receptors (GPCRs) to intracellular cascades (6). Activation of GPCRs results in exchange of GDP for GTP on the Gα subunit, resulting in the dissociation of the GTP bound form of Gα from Gβγ Both Gα and Gβγ can then activate downstream cellular signaling pathways. The signal is terminated when GTP is hydrolyzed to GDP by the intrinsic GTPase activity of the Gα subunit. The majority of GNAQ mutations occur at codon 209 within the GTPase catalytic domain, resulting in loss of the intrinsic GTPase activity and constitutively activation of GNAQ. Expression of mutated GNAQ results in melanocyte transformation and increased Erk1/2 phosphorylation, indicating that mutant GNAQ behaves as a dominant acting oncogene (4, 5). Currently, there are no available therapies targeting GNAQ.
The protein kinase C (PKC) family is a widely expressed group of serine/threonine kinases comprising multiple isoforms that can be divided into three structurally and functionally distinct subgroups (7, 8). These are the conventional PKCs (PKCα, PKCβ and PKCγ), which are activated by diacylglycerol (DAG) and phospholipid and are Ca2+ dependent; the novel PKCs (PKCδ, PKCε, PKCθ and PKCη), which are also activated by DAG and phospholipids, but are not Ca2+ dependent; and the atypical PKCs, which do not require DAG or Ca2+ for activation. PKCs regulate key biological processes including cell proliferation, apoptosis, differentiation, angiogenesis, tumor development, and chemoresistance (7, 9–15). PKCs are involved in GNAQ mediated activation of the MAPK/Erk1/2 pathways (6, 16). It has been known that GNAQ transduces signals from GPCRs to phospholipase Cβ (PLCβ)(6). PLCβ enzymes catalyze the hydrolysis of phosphatidylinositol biphosphate to release inositol trisphosphate and DAG that function as second messengers propagating and amplifying the Gα-mediated signal through activation of PKCs. Active PKCs further activate Erk1/2 through the RAF/MAPK/Erk1/2 pathway (16). Using shRNA mediated downregulation of PKC isoforms β, ε, and θ we have recently shown that these isoforms are functionally important for GNAQ mutated UM cells (17). The oncogenic properties of mutant GNAQ and the important PKC roles in GNAQ-mediated Erk1/2 activation and GNAQ mutated UM cells (4, 16) suggested that PKC may provide new opportunities for therapeutic intervention of UM carrying GNAQ mutations. To test this hypothesis, UM cells carrying wild type GNAQ or GNAQ mutated at codon 209 were treated with the PKC inhibitor AEB071 (sotrastaurin), a PKC inhibitor that has potent activity against classical and novel PKC isotypes (18). AEB071 selectively inhibited the growth of UM cells harboring GNAQ mutations by targeting PKC/Erk1/2 and PKC/NF-κB pathways.
MATERIALS AND METHODS
Cell lines
The sources and GNAQ mutational status of UM cell lines C918, Ocm1, Ocm3, Mel285, Mel202, 92.1 and Omm1.3 have been described previously (19). UM cells were cultured in RPMI 1640 containing 10% FBS, 50 μg/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. These cell lines were recently authenticated by short tandem repeat PCR analysis at Biosynthesis Inc. Human epidermal melanocytes were purchased from Lifeline Cell Technology (Walkersville, MD) and grown in the medium provided by the company.
Viability assay
Cells were seeded in 96-well plates at 2×103 cells/well and incubated over night followed by treatment with AEB071 (provided by Novartis) for three days. Cell viability was measured as previously described (17).
Cell cycle analysis
Cells were collected by trypsinization and fixed in cold ethanol. After incubation with RNase A and PI, cell cycle distribution was determined by flow cytometry analysis (FACS).
Analysis of apoptosis
Apoptotic cells were detected by Annexin V-FITC staining and FACS as described previously (17). Compensation for AEB071 autofluorescence was performed.
Knockdown of PKC isotypes by shRNA
The constructs (pLKO.1-puro) containing shRNA target sequences for PKCα, PKCδ, or GFP were provided by Dana-Farber Cancer Institute shRNA Core Facility. Lentivirus expressing PKC shRNA was produced as describe previously (17). Cells were infected with virus for three days and cell viability was determined using MTS assay.
Immunoblotting
Preparation of whole cell lysates and immunoblotting have been described previously (19). Antibodies against PKC isoforms were: PKCα (Cell Signaling #2056), PKCδ (Cell Signaling #2058), PKCθ (BD Biosciences #610090); PKCθ Thr538 (Cell Signaling #9377); PKCθ/δ Ser643/676 (Cell Signaling #9376); PKCε (BD Biosciences #610085). Antibodies against Akt, phospho-Akt, Erk1/2, phospho-Erk1/2, cyclin D1, Bcl-xL, XIAP, survivin, HADC1 and GAPDH were purchased from Cell Signaling Technology. Antibodies against PKCβII, p27Kip1, and RelA (p65) were purchased from Santa Cruz Biotechnology. Actin antibody was purchased from Sigma-Aldrich (St. Louis, MI). Protein signal intensity was measured using NIH ImageJ software and normalized that of actin.
Nuclear extract preparation and electrophoresis mobility shift assay (EMSA)
Nuclear extraction and NF-κB EMSA kits purchased from Signosis (Sunnyvale, CA) were used to isolate nuclear extracts from UM cells and perform EMSA. The instructions provided by the manufacturer were followed.
Measurement of IL-6 in cell culture medium
Cells were treated with DMSO or 5 μM AEB071 for 72 h in 6-well plates. Medium was collected and centrifuged to remove cells. IL-6 in the supernatant was determined using the Human IL-6 Quantikine ELISA Kit purchased from R&D Systems (Minneapolis, MN).
Statistical analysis
Data are presented as mean ± SD. Differences between treated and control groups was analyzed using Student’s t-Test and considered significant with P ≤ 0.05.
Results
The PKC inhibitor AEB071 selectively inhibits growth of UM cells harboring GNAQ mutations
We first evaluated the effect of AEB071 (Figure 1A) on viability of a panel of 7 UM cell lines (Figure 1B). Sequencing analysis confirmed that GNAQ is wild type in cell lines C918, Ocm1, Ocm3 and Mel285, while codon 209 of GNAQ is mutated from CAA (glutamine) to CTA (leucine) in cell lines Mel202 and 92.1 and to CCA (proline) in cell line Omm1.3. Cell lines Ocm1 and Ocm3 have BRAF V600E mutation (19, 20). AEB071 at low micromolar concentrations significantly decreased viability of all three GNAQ mutated cell lines (Figure 1C). The IC50 of AEB071 was approximately 0.8, 3, and 4 μM for 92.1, Omm1.3 and Mel202 cells respectively. AEB071 had little effect on viability of three GNAQ wild type cells up to 10 μM (Figure 1C and Supplemental Figure S1). In addition, the viability of human epidermal melanocytes was not reduced by AEB071 (Figure 1C), and may have been modestly increased by the inhibitor. Along with decreased viability, microscopic examination found morphological alterations in AEB071 treated cells harboring GNAQ mutations but not wild type GNAQ or normal melanocytes (Figure 1D). Treated Mel202, 92.1 and Omm1.3 cells lost spindle shape and became flattened with increased size or round floating dead cells with condensed cytoplasm.
Figure 1.

Effect of AEB071 on GNAQ wild type and mutated UM cells. (A) Molecular structure of AEB071 (Sotrastaurin) (adapted from (43)). (B) UM cell lines used and their GNAQ mutational status. (C) AEB071 selectively reduced viability of UM cells harboring GNAQ mutations. Cells were treated with varying amount of AEB071 for 72 hours. Data are presented as mean±SD of 4 or 5 independent experiments. NM = normal melanocytes; WT = GNAQ wild type; MT = GNAQ mutation. (D) AEB071 caused morphological alterations in GNAQ mutated cells. After AEB071 treatment for 3 days, Mel202, 92.1 and Omm1.3 cells lost spindle shape and became flattened with increased size or round floating dead cells with condensed cytoplasm. The morphology of GNAQ wild type UM cells was not altered by AEB071.
AEB071 induces G1 arrest in GNAQ mutated UM cells
To better understand growth inhibitory effect of AEB071 on GNAQ mutated cells, we investigated whether cell cycle progression was altered by drug exposure. AEB071 markedly increased the G1 phase population while decreasing the S phase population in UM cells harboring GNAQ mutations (Figures 2A and 2B). There was no significant change in the cell cycle pattern for cell lines carrying wild type GNAQ (C918, Mel285 and Ocm3). A decrease in the S phase population with a concomitant increase in the G2/M phase population was observed in Ocm1 cells. In agreement with this G1 arrest, AEB071 also significantly increased the accumulation of p27Kip1, while decreasing the expression of cylin D1 in all three GNAQ mutated cell lines tested (Figures 2C). In comparison, the expression of p27Kip1 and cyclin D1 was not significantly altered by AEB071 in GNAQ wild type cells. These findings suggest that AEB071 selectively induced G1 arrest in GNAQ mutated cells through altering the expression of regulators critical for the G1 to S transition.
Figure 2.

AEB071 induces G1 arrest in UM cells harboring GNAQ mutations. (A) UM cells were treated with DMSO or 5 μM AEB071 for 24 hours and subjected to cell cycle analysis. (B) Bar graphs of percentages of G1, S and G2/M populations. (C) AEB071 selectively increased p27 and decreased cyclin D1 expression in GNAQ mutant UM cells. Cells were treated with 0, 2, or 5 μM AEB071 for 72 hours and analyzed by immunoblot. WT = GNAQ wild type; MT = GNAQ mutation.
AEB071 induces apoptosis in GNAQ mutated UM cells
We next examined whether AEB071 promoted apoptosis in UM cells. Treatment with 2 and 5 μM AEB071 for 72 hours significantly increased Annexin V positive (apoptotic) populations in GNAQ mutated 92.1 and Omm1.3 cells (Figure 3A). Further demonstration of apoptosis was observed with caspase-3 cleavage in treated cells (Figure 3B). For Mel202 cells, only a minimal increase in the Annexin V positive cell population was observed after AEB071 treatment (Figure 3A), but significant caspase-3 cleavage was induced (Figure 3B). In contrast, AEB071 did not increase Annexin V positive cell populations or caspase-3 cleavage in UM cells harboring wild type GNAQ (Figures 3A and B). AEB071 further inhibited the expression of the anti-apoptotic proteins survivin, Bcl-xL and XIAP in a dose-dependent manner in Mel202, 92.1 and Omm1.3 cells (Figure 3C). Under the same conditions, survivin, Bcl-xL and XIAP were not affected or were modestly increased in GNAQ wild type UM cells (Figure 3C). These results indicate that AEB071 selectively induced apoptosis in UM cells harboring GNAQ mutations.
Figure 3.
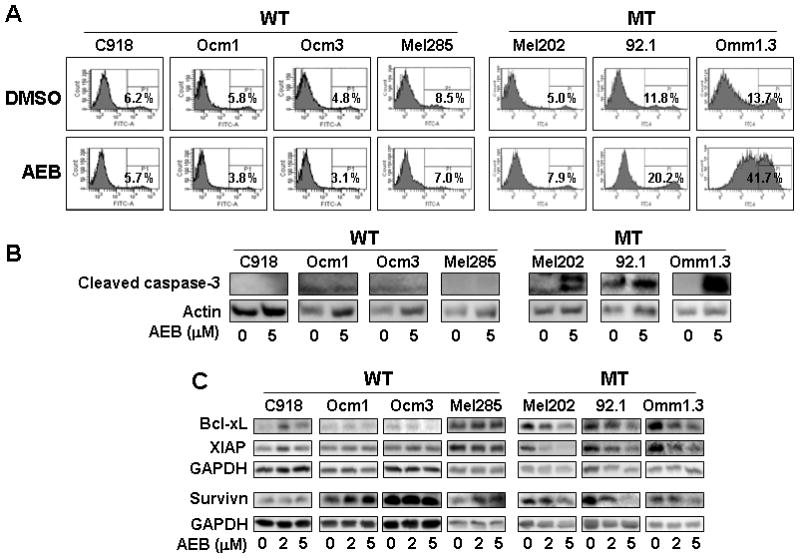
AEB071 induced apoptosis in UM cells harboring GNAQ mutations. (A) Cells were treated with DMSO or 5 μM AEB071 for 72 hours. Apoptotic cells were detected using Annexin V-FITC staining and FACS analysis. (B) Cells were treated with DMSO or 5 μM AEB071 for 72 hours and subjected to immunoblot analysis of cleaved casapase-3. Increased caspase-3 cleavage was observed in GNAQ mutated UM cells. (C) AEB071 decreased the expression of antiapoptotic proteins Bcl-xL, XIAP, and survivin in GNAQ mutated cells. WT = GNAQ wild type; MT = GNAQ mutation.
AEB071 inhibits expression and phosphorylation of PKC isoforms in UM cells
To better understand differential responses of UM cells to AEB071 based on GNAQ mutational status, we examined inhibition of PKC isoforms by AEB071 in UM cells. Immunoblotting demonstrated that treatment with AEB071 for 6 hours in the absence of serum resulted in decreased PKCδ/θSer643/676 phosphorylation in Mel202, 92.1 and C918 cells, and PKCθThr538 phosphorylation in Mel202 cells (Figure 4A). It has been reported that expression of PKC isoforms can be downregulated by PKC inhibition (21). We thus examined the expression of PKC isoforms after prolonged AEB071 treatment (5 μM for 24 hours) in the presence of 10% FBS. UM cell lines showed varying reduction in PKC expression after AEB071 treatment (Figure 4B). PKCα was reduced in some wild type (C918 and Ocm1) and in GNAQ mutated (92.1 and Omm1.3) cells, while PKCβ was reduced only in mutated cells (Mel202 and 92.1). PKCθ was decreased in all three GNAQ mutated but only in one wild type (Ocm3) cells. PKCε was downregulated in all three GNAQ wild type cell lines and two mutated cell lines (Mel202 and 92.1). These findings suggest that AEB071 may have greater overall inhibitory effect on multiple PKC isoforms in UM cells with mutated GNAQ: PKCα, β, δ, ε and/or θ expression was suppressed by AEB071 in GNAQ mutated cells while PKCα, and PKCδ and PKCε expression was affected in GNAQ wild type cells.
Figure 4.

PKCα and PKCδ are functionally important for UM cells harboring GNAQ mutations. (A) Effect of AEB071 on phosphorylation of PKC isoforms in UM cells. Cells were treated with 5 μM AEB071 in the absence of serum and analyzed for PKC phosphorylation. (B) AEB071 inhibited PKC expression in UM cells. Cells were treated with 5 μM AEB071 for 24 hours in the presence of 10% FBS and subjected to immunoblot analysis. PKCδ, ε and θ levels in AEB treated cells relative to control (DMSO treated) cells are also shown. Relative levels (fold changes) are presented as mean ± SD of 3–4 experiments. Except PKCθ in C918 and Ocm1 cells and PKCε in Omm1.3 cells the difference between AEB treated and control cells was statistically significant (Student’s t-test, P < 0.05 or 0.01). (C) PKCα and PKCδ were downregulated by shRNA-mediated knockdown. C918 and Mel202 cells were infected with lentivirus expressing shRNA for PKCα or PKCδ for 4 days and protein levels of these isoforms were determined by immunoblot analyses. (D) Knockdown of PKCα and PKCδ significantly reduced viability of Mel202 and Omm1.3 cells. Cell viability was determined 4 days after infection with lentivirus expressing PKC shRNA using MTS assay. Results are presented as mean ± SD of percent viability from 3 independent experiments. # P < 0.01 versus cells expressing control GFP shRNA.
PKC isoforms are functionally important for UM cells harboring GNAQ mutations
As the expression of PKCα and δ was suppressed by AEB071 in both GNAQ wild type and mutated UM cells, we next investigated if these isoforms were of functional importance for UM cells by shRNA mediated knockdown to better understand the mechanisms for the differential AEB071 response in GNAQ wild type and mutated cells. PKCα and PKCδ expression was significantly downregulated by their shRNA in C918 and Mel202 cells (Figure 4C). Interestingly, knockdown of PKCα or PKCδ significantly decreased viability of Mel202 and Omm1.3 cells, but failed to significantly decrease the viability of C918 cells (Figure 4D), indicating that PKCα and PKCδ are functionally important in UM cells harboring GNAQ mutations. We have previously found that PKC isoforms β, ε, and θ are functionally critical for GNAQ mutated UM cells (17). These findings together suggest that AEB071 suppressed growth of GNAQ mutated UM cells via inhibition of multiple PKC isoforms.
AEB071 selectively decreases Erk1/2 phosphorylation in UM cells harboring GNAQ mutations
To further define the molecular mechanisms underlying the anti-proliferative action of AEB071, we next assessed downstream effectors of PKC mediated pathways that were potentially affected by AEB071. It has been previously reported that PKC can activate the PI3K/Akt and MAPK pathways as well as GSK3βSer9 phosphorylation. The PKC inhibitor enzastaurin inhibits Akt and GSK3βSer9 phosphorylation in some types of cancer cells (21–26). Similarly, AEB071 decreased GSK3βSer9 phsophorylation in all UM cell lines studied here (Supplemental Figure S2). However, AEB071 only significantly inhibited Erk1/2 phosphorylation in GNAQ mutant cells while it had minimal effect on Akt phosphorylation in both GNAQ wild type and mutated cells (Figure 5). Total Akt and Erk1/2 levels were not significantly altered by AEB071 in any of the cell lines examined. These findings demonstrate AEB071-induced selective inhibition of the PKC/MAPK pathway in UM cells carrying GNAQ mutations. We have found that Erk1/2 inhibition decreased UM cell viability (19). Therefore, AEB071 may exert its anti-proliferative effects in part through suppression of Erk1/2 activation in GNAQ mutated UM cells.
Figure 5.

AEB071 selectively inhibited Erk1/2 phosphorylation in GNAQ mutated UM cells. Cells were treated with AEB071 for 72 hours and analyzed for the expression of Erk1/2 and Akt and their phosphorylation by immunoblot analyses. WT = GNAQ wild type; MT = GNAQ mutation.
AEB071 selectively inhibits NF-κB activity in GNAQ mutated UM cells
PKC isoforms are involved in GPCR-mediated NF-κB activation (27–30). In most cells, NF-κB is present as a latent, inactive, IκB-bound complex in the cytoplasm. On activation, IκB is phosphorylated and targeted to degradation. The released NF-κB translocates into the nucleus where it drives the expression of its target genes (29). It is well known that aberrant NF-κB activity promotes tumorigenesis and metastasis (31). We next examined whether AEB071 altered NF-κB activity in UM cells. In the absence of AEB071, nuclear p65 (RelA) levels are more or less similar among wild type and mutant cell lines (Figure 6A). In contrast, in the presence of AEB071, nuclear p65 levels were significantly decreased in GNAQ mutated UM cells while they were minimally altered in wild type cells (Figure 6A). Consistent with this, reduced DNA binding activity of NF-κB was detected only in the nuclear extracts of GNAQ mutated cells exposed to AEB071 (Figure 6B). In agreement with decreased NF-κB activity, elevated IκBα levels were detected in AEB071-treated GNAQ mutated cells (Figure 6C). Besides, the secretion of IL-6, one of the target genes of NF-κB, was substantially inhibited by AEB071 in GNAQ mutated Omm1.3 cells but not GNAQ wild type C918 cells (Figure 6D). These findings together indicate that AEB071 selectively inhibited NF-κB activation in UM cells harboring GNAQ mutations. This notion is also in agreement with the selective downregulation of NF-κB target genes including survivin, Bcl-xL, XIAP and cyclin D1, by AEB071 in GNAQ mutated cells (Figures 2C and 3C).
Figure 6.

AEB071 selectively inhibited NF-κB activation in UM cells harboring GNAQ mutations. (A) Immunoblot analysis of nuclear RelA (p65) levels of UM cells treated with 0 or 5 μM AEB071 for 40 hours. HDAC1 (histone deacetylase 1) was used as loading control for nuclear extracts (56). (B) EMSA analysis of NF-κB DNA binding activity in the same nuclear extracts used in (A). NF-κB/DNA complexes were determined as described in Materials and Methods. (C) AEB071 increased IκBα protein levels in UM cells with mutated GNAQ. Cells were treated with DMSO or 5 μM AEB071 for 20 hours and subjected to immunoblot analysis of IκBα. (D) AEB071 reduced IL-6 secretion from Omm1.3 cells. Cells were treated with DMSO or 5 μM AEB071 for 72 hours. Medium was collected for ELISA analysis of IL-6. Data presented as mean ± SD of % secretion of 3 independent experiments. * P < 0.05.
NF-κB activity is functionally important for UM cells
To determine whether NF-κB activity is functionally important for UM cells, both GNAQ wild type and mutated UM cells were treated with an IKK1/2 inhibitor that possesses potent NF-κB inhibition activity (32). This treatment resulted in the dramatic decrease in viability of both GNAQ wild type and mutant UM cell lines (Supplemental Figure S3). These findings suggest that NF-κB activity is critically important for UM cells and its suppression contributes to AEB071 induced growth inhibition of UM cells harboring GNAQ mutations.
DISCUSSION
There are currently no drugs available targeting the oncogenic GNAQ mutations that occur frequently in primary and metastatic UM. In the present study we describe the first small molecule inhibitor that selectively exhibits antiproliferative activity of UM cells harboring GNAQ mutations: the novel PKC inhibitor AEB071 reduced viability of GNAQ mutated UM cell lines, but had little effect on those carrying wild type GNAQ. AEB071-induced growth inhibition is associated with reduced expression of PKC isoforms α, β, δ, ε and/or θ, accompanied by inhibition of Erk1/2 phosphorylation, and NF-κB activation. We have previously shown that PKCθ, PKCβ and PKCε are functionally important for GNAQ mutated UM cells and that inhibition of Erk1/2 by MEK1/2 inhibitors reduced UM cell viability (17). Here, we demonstrate that PKCα, PKCδ and NF-κB are also functionally important for GNAQ mutated UM cells. Together, our findings suggest that AEB071 may selectively exert antiproliferative activity on GNAQ mutated UM cells via targeting the PKC/Erk1/2 and PKC/NF-κB pathways.
AEB071 induced growth suppression of GNAQ mutant cells is associated with pronounced G1 arrest and induction of apoptosis. The molecular mechanisms for AEB071 induced G1 arrest involve altered expression of positive and negative regulators of transition through G1 phase of cell cycle, including cyclin D1 and p27Kip1(33, 34). AEB071 induced apoptosis is associated with decreased expression of antiapoptotic proteins, yet the underlying molecular mechanisms remain to be revealed.
The PI3K/Akt and MAPK pathways are frequently activated in malignant tumors and are critical for cancer cell survival and proliferation (35–37). Erk1/2 activation is common in UM and has been reported to play a crucial role in UM development (38, 39). AEB071 inhibited Erk1/2 phosphorylation in GNAQ mutated but not GNAQ wild type UM cells, and had minimal impact on Akt phophorylation. Similarly, we have recently found that PKC inhibitor enzastaurin inhibits phosphorylation of Erk1/2 but not Akt in GNAQ mutated cell lines (17), although enzastaurin has been reported to inhibit Akt phosphorylation but not Erk1/2 phosphorylation in other types of cancer cells (24, 26, 40). The inhibition of Erk1/2 phosphorylation is therefore likely a common mechanism for the anti-proliferative action of PKC inhibitors in GNAQ mutated UM cells. Further studies are needed to identify the PKC isoform(s) which are most crucial for Erk1/2 phosphorylation in GNAQ mutant UM, and whose inhibition by AEB071 leads to decreased Erk1/2 phosphorylation. PKCβII and PKCθ are among the candidates, because activation of these isoforms triggers several signaling pathways including MAPK (9, 13, 17, 41), and siRNA downregulation of PKCβII decreased Erk1/2 phosphorylation in metastatic hepatocellular carcinoma cells (42).
In addition to the PKC/Erk1/2 pathway, activation of many GPCRs can trigger the PKC/IKK/NF-κB pathway through G proteins, including GNAQ (27–30, 43). Overexpression of mutant GNAQ(Q209L) leads to constitutive activation of NF-κB which is mediated by PKCδ and to a lesser extent PKCα and PKCε in HUVEC (27, 28). It is not known whether the same PKC isoforms have comparable roles in UM cells. Importantly, we demonstrate that AEB071 selectively repressed NF-κB activation in UM cells harboring GNAQ mutations and this is accompanied by downregulation of multiple PKC isoforms, in particular PKCα, PKCδ, and PKCε. The downregulation of these PKC isoforms may contribute to AEB071 suppression of NF-κB activity. Comparison studies using an IKK inhibitor corroborate that the NF-κB pathway has critical functional roles in UM cells. NF-κB inhibition might therefore be another mechanism for the antiproliferative action of AEB071 on UM cells harboring GNAQ mutations. The association between PKC inhibition and decreased NF-κB activity in GNAQ mutated UM cells suggests that these cells might rely on GNAQ-PKC-NF-κB pathways for NF-κB activation. Other pathways presumably regulate NF-κB activation in GNAQ wild type cells. For example, Ocm1 and Ocm3 cells have been shown to carry the common V600E BRAF mutation that constitutively activates the MAPK and NF-κB pathways (19, 20, 44–46). High c-Met expression has been found in C918 and Mel285 cells (47) and it has been reported that NF-κB can be activated by HGF/c-Met signaling (48, 49).
Our data indicate that multiple PKC isoforms including α, β, δ, ε, and θ are suppressed by AEB071 in GNAQ mutated UM cells while only PKCα and PKCδ were affected in GNAQ wild type cells. The findings from shRNA knockdown studies confirmed the functional importance of PKCα and PKCδ in GNAQ-mutated UM cells. Knockdown of these two PKC isoforms had no (PKCα) or little (PKCδ) affect on viability of GNAQ wild type cells, suggesting that these two PKC isoforms are less important in GNAQ wild type cells compared to GNAQ mutated cells. Similarly, our previous shRNA-mediated knockdown studies have found that PKCβ, PKCε and PKCθ are functionally more important for GNAQ-mutated than wild type UM cells (17). Together, these findings suggest that GNAQ mutated UM cells are more dependent on these PKC isoforms than GNAQ wild type cells, and that AEB071 exerts anti-proliferative action on GNAQ mutated cells via suppression of these PKC isoforms. These findings also provide a plausible explanation for the differential response/sensitivity of GNAQ wild type and mutated UM cells to AEB071.
G-protein coupled receptors (GPCRs) have a pivotal role in many physiological functions and in multiple diseases, including tumorigenesis and metastasis of cancers (50). Frequent somatic mutations in GPCRs have been found in melanoma and mutations in GRM3, which is a glutamate receptor and a member of the metabolic GPCRs, activate the MAPK/ERK pathway and promote growth and migration of melanoma cells (51). Furthermore, overexpression of diverse GPCRs were found in number of primary and metastatic cancers including melanoma, breast, prostate, non-small cell lung cancer, gastric tumors, head and neck squamous cell carcinoma (HNSCC), and diffused large B cell lymphoma (52, 53). Overexpression of G protein-coupled receptors in cancer cells: involvement in tumor progression. GPCRs can also influence cancer progression through crosstalk with growth factor receptors such as EGFR and IGF-1R, G-proteins (such as Gaq/11 and Ga12/13), chemokines, Hedgehog and WNT signaling pathways, regulation of the apoptotic response and viral factors. Various GPCRs can be involved in cancer through activating NF-κB (50). As more data linking GPCRs to cancer emerge, these receptors are attractive potential targets for tumor therapy. In particular, the GPCR-ERK and GPCR-NF-κB pathways could be valuable targets for innovative anticancer drug discovery.
We demonstrate that PKC inhibitor AEB071 (sotrastaurin) at low micromolar concentrations exerts significant antiproliferative effect on GNAQ mutated UM cells through targeting the PKC/MAPK and PKC/NF-κB pathways. Our findings support PKCs as important targets for therapeutic intervention of UM harboring GNAQ mutations. Clinical studies have shown that a blood concentration of 4 μM was achieved within five hours after an oral dose of 500 mg AEB071(18). AEB071 could thus be of therapeutic potential for UM with GNAQ mutations. A clinical trial with AEB071 in UM patients is currently enrolling patients. AEB071 has primarily been studied as an anti-inflammatory agent for the treatment of diseases such as psoriasis and in preventing solid organ rejection following transplant (54, 55). How the immunosuppressive effects of this drug may influence the clinical efficacy will have to be determined, and monitoring of changes in anti-tumor immune responses as a function of treatment should be helpful to discern such changes. Given that preclinical models for uveal melanoma are difficult and mostly comprise xenograft studies, much of these questions will likely be answered by clinical investigation. There may be future opportunities to combine AEB071 or other PKC inhibitors with other rational small molecule inhibitors (e.g. MEK or PI3K) or immune therapy such as ipilimumab to improve the efficacy and durability of any clinical activity. It would be also of great interest to investigate antitumor effects of other specific PKC inhibitors in melanoma and other cancers with GPCR mutations that may activate MAPK/ERK and/or NF-kB pathways.
Supplementary Material
Acknowledgments
This work was supported in part by Sharon Crowley Martin Memorial Fund for Melanoma Research (F.S. Hodi), the Malcolm and Emily Mac Naught Fund for Melanoma Research (F.S. Hodi) at Dana-Farber Cancer Institute, NIH 1P50CA127003-04 (J.A. Fletcher) and the Virginia and Daniel K. Ludwig Trust for Cancer Research (J.A. Fletcher).
Footnotes
F.S.H. has served as a non-paid consultant to Novartis and received clinical trial support from Novartis.
References
- 1.Bakalian S, Marshall JC, Logan P, Faingold D, Maloney S, Di Cesare S, et al. Molecular pathways mediating liver metastasis in patients with uveal melanoma. Clin Cancer Res. 2008;14:951–6. doi: 10.1158/1078-0432.CCR-06-2630. [DOI] [PubMed] [Google Scholar]
- 2.Singh A, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18:75–84. doi: 10.1016/j.ohc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Onken MD, Worley LA, Long MD, Duan S, Council ML, Bowcock AM, et al. Oncogenic mutations in GNAQ occur early in uveal melanoma. Investigative ophthalmology & visual science. 2008;49:5230–4. doi: 10.1167/iovs.08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 363:2191–9. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hubbard KB, Hepler JR. Cell signalling diversity of the Gqalpha family of heterotrimeric G proteins. Cellular signalling. 2006;18:135–50. doi: 10.1016/j.cellsig.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Koivunen J, Aaltonen V, Peltonen J. Protein kinase C (PKC) family in cancer progression. Cancer Lett. 2006;235:1–10. doi: 10.1016/j.canlet.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 8.Martiny-Baron G, Fabbro D. Classical PKC isoforms in cancer. Pharmacol Res. 2007;55:477–86. doi: 10.1016/j.phrs.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003;63:780–6. [PubMed] [Google Scholar]
- 10.Koren R, Ben Meir D, Langzam L, Dekel Y, Konichezky M, Baniel J, et al. Expression of protein kinase C isoenzymes in benign hyperplasia and carcinoma of prostate. Oncology reports. 2004;11:321–6. [PubMed] [Google Scholar]
- 11.da Rocha AB, Mans DR, Regner A, Schwartsmann G. Targeting protein kinase C: new therapeutic opportunities against high-grade malignant gliomas? Oncologist. 2002;7:17–33. doi: 10.1634/theoncologist.7-1-17. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Anastasiadis PZ, Liu Y, Thompson EA, Fields AP. Protein kinase C (PKC) betaII induces cell invasion through a Ras/Mek-, PKC iota/Rac 1-dependent signaling pathway. J Biol Chem. 2004;279:22118–23. doi: 10.1074/jbc.M400774200. [DOI] [PubMed] [Google Scholar]
- 13.Yoshiji H, Kuriyama S, Ways DK, Yoshii J, Miyamoto Y, Kawata M, et al. Protein kinase C lies on the signaling pathway for vascular endothelial growth factor-mediated tumor development and angiogenesis. Cancer Res. 1999;59:4413–8. [PubMed] [Google Scholar]
- 14.Herbst RS, Oh Y, Wagle A, Lahn M. Enzastaurin, a protein kinase Cbeta-selective inhibitor, and its potential application as an anticancer agent in lung cancer. Clin Cancer Res. 2007;13:s4641–6. doi: 10.1158/1078-0432.CCR-07-0538. [DOI] [PubMed] [Google Scholar]
- 15.Goekjian PG, Jirousek MR. Protein kinase C inhibitors as novel anticancer drugs. Expert opinion on investigational drugs. 2001;10:2117–40. doi: 10.1517/13543784.10.12.2117. [DOI] [PubMed] [Google Scholar]
- 16.Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends in endocrinology and metabolism: TEM. 2000;11:91–9. doi: 10.1016/s1043-2760(99)00232-5. [DOI] [PubMed] [Google Scholar]
- 17.Wu X, Zhu M, Fletcher JA, Giobbie-Hurder A, Hodi FS. The Protein Kinase C Inhibitor Enzastaurin Exhibits Antitumor Activity against Uveal Melanoma. PloS one. 7:e29622. doi: 10.1371/journal.pone.0029622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skvara H, Dawid M, Kleyn E, Wolff B, Meingassner JG, Knight H, et al. The PKC inhibitor AEB071 may be a therapeutic option for psoriasis. J Clin Invest. 2008;118:3151–9. doi: 10.1172/JCI35636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calipel A, Lefevre G, Pouponnot C, Mouriaux F, Eychene A, Mascarelli F. Mutation of B-Raf in human choroidal melanoma cells mediates cell proliferation and transformation through the MEK/ERK pathway. J Biol Chem. 2003;278:42409–18. doi: 10.1074/jbc.M308709200. [DOI] [PubMed] [Google Scholar]
- 20.Kilic E, Bruggenwirth HT, Verbiest MM, Zwarthoff EC, Mooy NM, Luyten GP, et al. The RAS-BRAF kinase pathway is not involved in uveal melanoma. Melanoma Res. 2004;14:203–5. doi: 10.1097/01.cmr.0000130006.46885.a0. [DOI] [PubMed] [Google Scholar]
- 21.Fields AP, Calcagno SR, Krishna M, Rak S, Leitges M, Murray NR. Protein kinase Cbeta is an effective target for chemoprevention of colon cancer. Cancer Res. 2009;69:1643–50. doi: 10.1158/0008-5472.CAN-08-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem. 1992;267:16878–82. [PubMed] [Google Scholar]
- 23.Fang X, Yu S, Tanyi JL, Lu Y, Woodgett JR, Mills GB. Convergence of multiple signaling cascades at glycogen synthase kinase 3: Edg receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol Cell Biol. 2002;22:2099–110. doi: 10.1128/MCB.22.7.2099-2110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, et al. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615. HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005;65:7462–9. doi: 10.1158/0008-5472.CAN-05-0071. [DOI] [PubMed] [Google Scholar]
- 25.Rizvi MA, Ghias K, Davies KM, Ma C, Weinberg F, Munshi HG, et al. Enzastaurin (LY317615), a protein kinase Cbeta inhibitor, inhibits the AKT pathway and induces apoptosis in multiple myeloma cell lines. Mol Cancer Ther. 2006;5:1783–9. doi: 10.1158/1535-7163.MCT-05-0465. [DOI] [PubMed] [Google Scholar]
- 26.Jane EP, Pollack IF. The heat shock protein antagonist 17-AAG potentiates the activity of enzastaurin against malignant human glioma cells. Cancer Lett. 2008;268:46–55. doi: 10.1016/j.canlet.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie P, Browning DD, Hay N, Mackman N, Ye RD. Activation of NF-kappa B by bradykinin through a Galpha(q)- and Gbeta gamma-dependent pathway that involves phosphoinositide 3-kinase and Akt. J Biol Chem. 2000;275:24907–14. doi: 10.1074/jbc.M001051200. [DOI] [PubMed] [Google Scholar]
- 28.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circulation research. 2002;91:398–405. doi: 10.1161/01.res.0000033520.95242.a2. [DOI] [PubMed] [Google Scholar]
- 29.Blonska M, Lin X. NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell research. 21:55–70. doi: 10.1038/cr.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grabiner BC, Blonska M, Lin PC, You Y, Wang D, Sun J, et al. CARMA3 deficiency abrogates G protein-coupled receptor-induced NF-{kappa}B activation. Genes Dev. 2007;21:984–96. doi: 10.1101/gad.1502507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harbor perspectives in biology. 2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waelchli R, Bollbuck B, Bruns C, Buhl T, Eder J, Feifel R, et al. Design and preparation of 2-benzamido-pyrimidines as inhibitors of IKK. Bioorganic & medicinal chemistry letters. 2006;16:108–12. doi: 10.1016/j.bmcl.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 33.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–65. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 34.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 35.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 36.Cohen C, Zavala-Pompa A, Sequeira JH, Shoji M, Sexton DG, Cotsonis G, et al. Mitogen-actived protein kinase activation is an early event in melanoma progression. Clin Cancer Res. 2002;8:3728–33. [PubMed] [Google Scholar]
- 37.Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–9. [PubMed] [Google Scholar]
- 38.Rimoldi D, Salvi S, Lienard D, Lejeune FJ, Speiser D, Zografos L, et al. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003;63:5712–5. [PubMed] [Google Scholar]
- 39.Zuidervaart W, van Nieuwpoort F, Stark M, Dijkman R, Packer L, Borgstein AM, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005;92:2032–8. doi: 10.1038/sj.bjc.6602598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee KW, Kim SG, Kim HP, Kwon E, You J, Choi HJ, et al. Enzastaurin, a protein kinase C beta inhibitor, suppresses signaling through the ribosomal S6 kinase and bad pathways and induces apoptosis in human gastric cancer cells. Cancer Res. 2008;68:1916–26. doi: 10.1158/0008-5472.CAN-07-3195. [DOI] [PubMed] [Google Scholar]
- 41.Greco S, Storelli C, Marsigliante S. Protein kinase C (PKC)-delta/-epsilon mediate the PKC/Akt-dependent phosphorylation of extracellular signal-regulated kinases 1 and 2 in MCF-7 cells stimulated by bradykinin. The Journal of endocrinology. 2006;188:79–89. doi: 10.1677/joe.1.06433. [DOI] [PubMed] [Google Scholar]
- 42.Guo K, Liu Y, Zhou H, Dai Z, Zhang J, Sun R, et al. Involvement of protein kinase C beta-extracellular signal-regulating kinase 1/2/p38 mitogen-activated protein kinase-heat shock protein 27 activation in hepatocellular carcinoma cell motility and invasion. Cancer science. 2008;99:486–96. doi: 10.1111/j.1349-7006.2007.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naylor TL, Tang H, Ratsch BA, Enns A, Loo A, Chen L, et al. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 71:2643–53. doi: 10.1158/0008-5472.CAN-10-2525. [DOI] [PubMed] [Google Scholar]
- 44.Dhawan P, Richmond A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J Biol Chem. 2002;277:7920–8. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ikenoue T, Hikiba Y, Kanai F, Aragaki J, Tanaka Y, Imamura J, et al. Different effects of point mutations within the B-Raf glycine-rich loop in colorectal tumors on mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase and nuclear factor kappaB pathway and cellular transformation. Cancer Res. 2004;64:3428–35. doi: 10.1158/0008-5472.CAN-03-3591. [DOI] [PubMed] [Google Scholar]
- 46.Liu J, Suresh Kumar KG, Yu D, Molton SA, McMahon M, Herlyn M, et al. Oncogenic BRAF regulates beta-Trcp expression and NF-kappaB activity in human melanoma cells. Oncogene. 2007;26:1954–8. doi: 10.1038/sj.onc.1209994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdel-Rahman MH, Boru G, Massengill J, Salem MM, Davidorf FH. MET oncogene inhibition as a potential target of therapy for uveal melanomas. Investigative ophthalmology & visual science. 51:3333–9. doi: 10.1167/iovs.09-4801. [DOI] [PubMed] [Google Scholar]
- 48.Damm S, Koefinger P, Stefan M, Wels C, Mehes G, Richtig E, et al. HGF-promoted motility in primary human melanocytes depends on CD44v6 regulated via NF-kappa B, Egr-1, and C/EBP-beta. J Invest Dermatol. 130:1893–903. doi: 10.1038/jid.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muller M, Morotti A, Ponzetto C. Activation of NF-kappaB is essential for hepatocyte growth factor-mediated proliferation and tubulogenesis. Mol Cell Biol. 2002;22:1060–72. doi: 10.1128/MCB.22.4.1060-1072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- 51.Prickett TD, Wei X, Cardenas-Navia I, Teer JK, Lin JC, Walia V, et al. Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nature genetics. 43:1119–26. doi: 10.1038/ng.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li S, Huang S, Peng S. Overexpression of G protein-coupled receptors in cancer cells: involvement in tumor progression. Int J Oncol. 2005;27:1329–39. [PubMed] [Google Scholar]
- 53.Qin Y, Verdegaal EM, Siderius M, Bebelman JP, Smit MJ, Leurs R, et al. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: the constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 24:207–18. doi: 10.1111/j.1755-148X.2010.00781.x. [DOI] [PubMed] [Google Scholar]
- 54.Yamashita K, Todo S. Sotrastaurin, a new selective protein kinase C inhibitor, on the way. Transplantation. 93:146–7. doi: 10.1097/TP.0b013e31823d4b1f. [DOI] [PubMed] [Google Scholar]
- 55.Manicassamy S. Sotrastaurin, a protein kinase C inhibitor for the prevention of transplant rejection and treatment of psoriasis. Curr Opin Investig Drugs. 2009;10:1225–35. [PubMed] [Google Scholar]
- 56.Aguilera C, Fernandez-Majada V, Ingles-Esteve J, Rodilla V, Bigas A, Espinosa L. Efficient nuclear export of p65-IkappaBalpha complexes requires 14-3-3 proteins. Journal of cell science. 2006;119:3695–704. doi: 10.1242/jcs.03086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.