

Abstract
Transcriptional networks regulate cell fate decisions, which occur at the level of individual cells. However, much of what we know about their structure and function comes from studies averaging measurements over large populations of cells, many of which are functionally heterogeneous. Such studies conceal the variability between cells and so prevent us from determining the nature of heterogeneity at the molecular level. In recent years, many protocols and platforms have been developed that allow the high throughput analysis of gene expression in single cells, opening the door to a new era of biology. Here, we discuss the need for single cell gene expression analysis to gain deeper insights into the transcriptional control of cell fate decisions, and consider the insights it has provided so far into transcriptional regulatory networks in development.
Keywords: cell fate control, single cell analysis, transcriptional networks
Introduction
Cell and molecular biology have long been studies of averages. Many of our most commonly used techniques report data based on population averages, yet they are widely used to draw conclusions about how individual cells behave. While the study of populations, at least at the molecular level, has until recently been a necessity, it masks a great deal of information about cellular systems, and as a result it influences our interpretation of data. As some of biology's fundamental questions, such as how fate decisions are made by cells, take place at the level of individuals within populations that we know are heterogeneous 1–8, studies of those individuals are long overdue. Furthermore, some populations are so rare – such as those present in the earliest stages of development – that studies of these cells are only possible when techniques are scaled down to a few or single cells.
It has been possible to isolate and culture single cells for some time. Yet, attempts at molecular analysis of mammalian cells have typically been limited in the number of cells and either genes or proteins analysed 7,9–11. Individual mammalian cells are estimated to contain 1–26 pg of RNA, of which mRNA comprises only a few percent of the total 12,13, hence making the analysis of multiple targets challenging. However, recent technological advances have made single cell transcriptomics feasible and affordable, as well as amenable to high-throughput approaches. This enables the analysis of tens to hundreds of genes in hundreds to thousands of cells simultaneously 14–19. New methods vary from microscopy-based, such as single molecule RNA-FISH (sm mRNA-FISH) that can measure a limited number of targets but in potentially large numbers of cells, through RT-qPCR techniques that balance larger numbers of targets with large numbers of cells 12,15,17,20, to microarray and sequencing-based methods. Whereas the latter are able to sample the entire transcriptome, they are currently limited in cell numbers by cost, making RT-qPCR the current method of choice. These methods, their technical aspects and their relative merits have been reviewed in detail previously 12,14–21.
Single cell studies are already revealing a large amount of gene expression heterogeneity between individual cells of specific populations, and as a result, the approaching single cell era may well force us to reconsider much of what we think we know from population studies in light of such molecular heterogeneity. Here, we discuss the need for single cell studies, referring to recent studies of single cells using high-throughput techniques, and their utility for understanding the transcriptional basis of cell fate choices. Transcription factors (TFs) are major drivers of cellular identity and differentiation, as highlighted by their roles in malignancy and as reprogramming factors 22–25. Transcriptional regulatory networks describe the interactions of TFs with cis-regulatory elements on the DNA, and understanding their structure helps us to understand how they function in regulating cellular decision-making 4,26–30. While much has been learned from population studies, detailed analyses of TF interactions in individual cells will revolutionise our understanding of transcriptional regulation in development and also in malignancy.
Single cell analysis enhances our understanding of transcriptional networks
Population-average gene expression measurements conceal inter-cellular variation
Transcription occurs as bursts, where a gene is transcribed for a short period followed by a period of inactivity 31,32, and individual genes can exhibit differing burst kinetics. This can result in heterogeneity in expression between individual cells of a population, both in terms of which genes they express and the level of expression. However, the extent to which this variation is stochastic or regulated, and whether it drives or facilitates biological changes, is still open for debate, and is already becoming a major focus of single cell analysis 3,4,33,34. In order to understand which TFs are co-regulated or regulate each other, and how this relates to cell fate, it is important to know whether they are expressed in the same individual cells. Beyond masking functional and molecular heterogeneity, population studies also force us to make assumptions about how individuals behave when extrapolating the data to the single cell level, such that we will assume that all cells of a population express similar levels of a given gene (Fig. 1A). Similarly, when multiple genes are analysed we assume that co-expression at the population level corresponds to co-expression in the individual cells (Fig. 1B). However, studies to date have indicated that often neither of these assumptions hold true at the level of individual cells 12,35,36.
Figure 1.
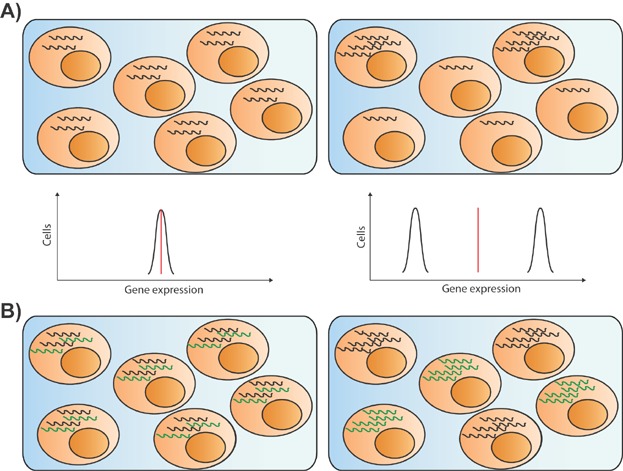
Single cell analysis reveals heterogeneity. A: Single cell analysis can distinguish whether all cells of a population express a similar level of a transcript (top left) or whether a small number of cells account for most of the expression (top right), which cannot be determined from population studies. In single cell studies, a homogeneous population would give a single expression distribution (bottom left) while a heterogeneous population would give a broader distribution, or multiple distributions (bottom right). In population studies, both sets of cells would seem to have the same level of expression (red lines). B: Single cell analysis can reveal whether co-expression observed at the population level actually occurs within the same single cells (left) or not (right).
Gene expression patterns reveal TF regulatory interactions
While lineage commitment may to some extent be a stochastic process, interactions between genes and proteins, as well as the kinetics of expression and degradation, enforce rules on expression that reinforce transcriptional programs once they are activated. Single cell studies offer the potential to identify regulatory links between TFs to generate networks on a larger scale than previously achieved.
Much of the work to date has used correlation analysis across large numbers of individual cells to identify interactions. A positive correlation, where two genes are consistently expressed in the same cells, could indicate that either they share a regulatory mechanism or that one activates the expression of the other (Fig. 2A). Conversely, a negative correlation could indicate that they are independently regulated or antagonistic. Bengtsson et al. 36 analysed the expression of three genes, Ins1, Ins2 and Actb at the single cell level after finding that all three were up-regulated in pancreatic beta cells in response to glucose stimulation. While Ins1 and Ins2 were up-regulated in the same cells and so had correlated expression, Actb was expressed in a separate subset of cells and so was not correlated with the other two genes. This indicates that while Ins1 and Ins2 likely share regulatory mechanisms, Actb is independently activated in response to the same stimulus. This information is obscured at the population level, resulting in problems in interpretation, and highlighting how putative regulatory interactions determined using population studies may not really occur in individual cells. Furthermore, robust calculation of correlations requires large sample sizes, which single cell RT-qPCR analysis is uniquely able to provide.
Figure 2.
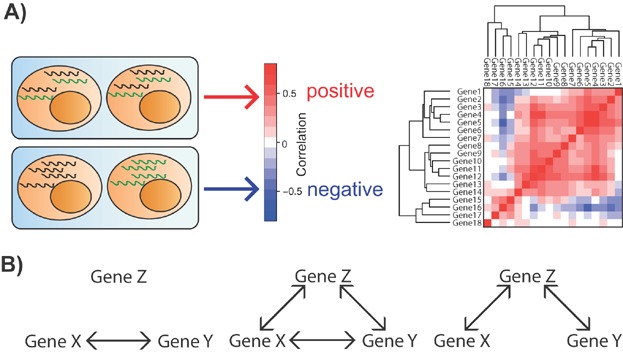
Transcriptional network analysis from single cell gene expression data. A: Single cell expression data can be used to calculate correlations, which describe the likelihood of two genes being expressed at the same time in the same cell. Positive correlations are shown in red and negative correlations in blue. These data can be shown as heatmaps and used to develop hypotheses about transcriptional regulation. B: Partial correlations can be calculated to determine whether the correlation between two factors, X and Y, is direct (left); due to both being regulated by a third factor, Y (right); or a combination of both (middle). These interactions can be validated experimentally using ChIP-seq to identify TF binding to target loci, and reporter assays to show that binding has an effect on gene expression, as well as using perturbation studies to demonstrate that changing the expression of the direct interactor affects expression of the target gene.
Many correlations are generated for even small sets of genes, and not all will represent real regulatory events. ChIP-seq data has been useful in narrowing down the number of correlations that represent true direct regulatory interactions by identifying direct targets of TFs. However, this method is dependent on the existence of data in appropriate cell types, and validation of the function of TF binding events can be time consuming and expensive. The correlations between factors also vary in different cell types due to changes in expression and binding partners. As a result, more efficient computational methods are needed to narrow down the targets for validation and to build networks.
Partial correlations 37 consider whether other genes may interact with the genes of interest and to what extent the correlation between them is the result of interactions with the additional genes rather than a direct interaction (Fig. 2B), as shown in astrocytes for the identification of an interaction network centred around Vim 38. Crucially, however, this method can only consider the genes for which measurements were obtained, and networks are generated iteratively from multiple calculations. Other methods are emerging to distinguish direct and indirect interactions more easily and efficiently to generate networks in a single step, and where interactions occur within complex feedback loops. However, these methods still struggle to delineate direct and indirect interactions where the indirect node is not included in the experimental data 39,40. While this is a problem for RT-qPCR data where the number of genes that can be measured simultaneously is limited to hundreds (with TFs numbering into the thousands in mammals) it will become less of an issue as single cell mRNA-seq matures, giving us access to the entire transcriptome 41,42 of individual cells.
Together these studies indicate that in the short period of time in which high-throughput single cell gene expression analysis has been available it has already facilitated significant insights into the biology of single cells that may have implications for disease processes and regenerative medicine. The data so far indicate that cell fate decisions may be at least partly stochastic processes, but that they occur within a defined transcriptional framework governed by transcription factor networks.
Single cell analysis reveals mechanisms of cellular decision-making
Cell fate decisions occur in individual cells
Single cell analysis reveals key regulators of lineage segregation in early mouse embryos
One of the first single cell studies using microfluidics platforms analysed cells from the mouse zygote through to the 64-cell stage blastocyst 43. Principal components analysis (PCA), a mathematical technique that defines new axes in multidimensional space to capture the variation in data, was able to identify three cell populations based on gene expression, corresponding to the trophectoderm, primitive endoderm and epiblast present in the embryo at the 64-cell stage. From this analysis, two TFs, Id2 and Sox2, were identified as early markers discriminating the different cell types. A negative correlation between Fgf4 and its receptor was also identified early in the inner cell mass and preceded changes in the transcriptional program 43, providing some insight into the role of signalling in cell fate choices and changes in transcriptional state. When applied to the same data, Gaussian process latent variable model (GPLVM) analysis – an extension of PCA that accounts for nonlinear changes in gene expression – was able to distinguish the primitive endoderm and epiblast at an earlier stage than conventional PCA 44. This indicates how single cell studies are driving the design of better analysis tools.
Loss of pluripotency and cell reprogramming involve stochastic and hierarchical phases
In ES cells, heterogeneity in the expression of the pluripotency protein Nanog has been suggested to play a role in the balance between self-renewal and differentiation 5. The effect of loss of Nanog on known pluripotency regulatory networks was investigated using a doxycycline-inducible Nanog knockdown 45. While removal of Nanog resulted in transient up-regulation of differentiation-associated transcripts, there was substantial heterogeneity between cells in the expression of the genes analysed and no subpopulations of similar cells were identified, hence indicating that the earliest stages of differentiation are stochastic 45. This confirms previous assumptions, and is consistent with data from the haematopoietic system 46,47. The pluripotency network is relatively stable if unperturbed by external stimuli, as ground state pluripotency can be tightly maintained in culture with inhibitors of Erk signalling and the beta-catenin/Wnt pathway 48. However, feedback loops between Nanog and other pluripotency factors were compromised when Nanog was down-regulated, a situation in which different sub-networks activate transiently to push cells toward differentiation. Interestingly, Nanog has recently been reported to be biallelically expressed based on both single-cell gene expression analysis and sm mRNA-FISH – heterogeneity in Nanog mRNA expression being no greater than for other genes such as Oct4 49. This indicates that the heterogeneity previously observed at the protein level may be due to gene-targeting strategies and monoallelic expression or culture conditions. While it is clear that Nanog is down-regulated during the differentiation of the epiblast, what role gene expression heterogeneity in ES cells plays in the down-regulation of Nanog and commitment of stem cells is therefore still uncertain.
Reprogramming represents the opposite of this differentiation process, where the ectopic expression of TFs can force cells to revert to a previous state or transdifferentiate to an alternative lineage 22,25,50,51. The production of induced pluripotent stem (iPS) cells by reprogramming, however, is inefficient. A greater understanding of this process at the single cell level could therefore inform the development of better reprogramming strategies. As in the case of differentiation, reprogramming has been viewed as a stochastic process because of its inefficiency 52, but these observations originate from studies of functionally heterogeneous populations in which reprogramming is a rare event. Buganim et al. analysed single cells at multiple time points during reprogramming using either single cell gene expression analysis or sm mRNA-FISH 53 to try to understand the molecular acquisition of pluripotency. PCA identified three groups of cells corresponding to the differentiated cells before reprogramming, iPS cells and an intermediate population that is more heterogeneous both in terms of gene expression and the stage of reprogramming. Early stochastic changes in gene expression were observed that were followed by a more hierarchical stage beginning with the activation of Sox2. Bayesian network analysis was then employed to identify linkages between TFs that regulated this hierarchical phase. This was used successfully to predict TFs that could act as reprogramming factors in place of existing protocols.
These single cell studies, in combination with existing literature from population studies, necessarily require changes in how we view cell states such as pluripotency. It has become clear that no single transcriptional state can be associated with, for example the state of pluripotency, even though individual cells may be functionally pluripotent 54. Transcriptional heterogeneity of the pluripotent state may allow the population to respond to a large number of signals, as well as protecting it from perturbations: it has previously been suggested that temporal fluctuations in the levels of key regulators such as Nanog, Stella and Rex1 5–7 influence the ability of ES cells to self renew or differentiate. This has resulted in pluripotency being described in terms of statistical mechanics, a branch of physics that relates the properties of a substance – for example the pressure of a gas – to the stochastic kinetics of its component molecules 54. In the case of cells, the property in question is their transcriptional status. As a population is the average of its components, many configurations or transcriptional states can produce the same functionality.
Single cell analysis unravels the haematopoietic hierarchy and identifies commitment events
It is unlikely that these concepts are restricted to ES cells. Nearly 20 years ago, single cell analysis of a limited number of genes in blood cells showed not only that haematopoietic stem cells (HSCs) are heterogeneous in the expression of key regulators – which was subsequently confirmed by us and others 10,46,55,56 – but that they also exhibit promiscuous expression of lineage-affiliated genes 9, which might prime these cells for differentiation. A recent study by Glotzbach et al. 55 aimed to elucidate the relationship between transcriptional and phenotypic variation in HSCs, where subpopulations are known to exist with differing lineage potential. The authors aimed to quantify gene expression heterogeneity and establish whether it constitutes noise around a fixed point or the presence of multiple subpopulations that could correspond to functional states. This is technically challenging, as there is no baseline for gene expression heterogeneity against which to compare expression data. The authors compared CD34hi and CD34lo subsets of HSCs 55, where CD34hi cells have been shown to have a much lower long-term stem cell capacity, and identified nine genes with expression distributions that differed between the two populations and may be important in their differing potentials. The authors used fuzzy c-means clustering 57 to group CD34lo cells on the basis of the expression of these nine genes. This method identifies cells with similar expression and allocates them to clusters to allow their common properties to be identified, but allows each cell to be a member of multiple clusters if it shares properties with several clusters. This identified three clusters of similar sizes. Repeating the analysis on the CD34hi cells that have lower stem cell potential revealed that one cluster was starkly underrepresented compared to CD34lo cells, but this cluster included a similar proportion of HSCs sorted using a different strategy, indicating that this may represent the transcriptional program of true long-term HSCs. Interestingly, expression of some of the nine genes was associated with specific clusters, while others varied between clusters, demonstrating how this analysis can help to discriminate meaningful variation from background noise.
A recent study has also provided insights into how HSCs undergo erythroid commitment. Previous work indicated that the multipotent haematopoietic cell line EML consists of a mixture of cells with differing levels of the surface marker Sca-1, which correspond to differing erythroid and myeloid potentials. Over time, populations sorted for different levels of Sca-1 expression regenerated the original mixed population 8. However, whether individual cells were able to re-establish these mixed cultures had not been shown. Pina et al. 46 used this model system to investigate the transcriptional basis of the erythroid potential of Sca-1lo cells and found that self-renewal and lineage commitment were independent events with correspondingly different transcriptomes. The CD34− compartment of the Sca-1lo cells contained virtually all expression of the erythroid TF Gata1 and had accelerated erythroid differentiation but no culture reconstitution potential, while CD34+ cells were multipotent. This insight facilitated transcriptional analysis of cells either side of the commitment boundary. Significant cell-to-cell variation in the expression of a set of erythroid-associated genes was observed around commitment, which resolved to a more homogeneous expression state upon commitment, similar to expression patterns for the same gene set in differentiated erythroid cells 46. While most of the work was performed in cell lines –as it would be difficult to capture cells at the commitment boundary in vivo – similar heterogeneity in the erythroid program was found in primary megakaryocyte-erythroid progenitors compared to more committed erythroid cells. These results indicate that commitment occurs through the independent activation of key regulators in the absence of a coordinated lineage program, with a low probability of transitioning to a committed state due to the requirement for the activation of additional regulators within the same cell. This also suggests that commitment can occur through multiple pathways and that the sequence of events is not entirely fixed, which may have implications for the design of directed differentiation strategies.
Modelling of this data set identified cells close to the commitment boundary and inferred a time course of commitment from static single-cell gene expression measurements 47. Monitoring gene expression changes during commitment again indicated that commitment was mediated by stochastic and independent modulation of key regulators. However, several genes were identified as key in discriminating between self-renewing and committed cells. These included Gata2, Mpl and to a lesser extent, Gata1, with multiple combinations of expression patterns permissive for commitment in modelling experiments and in vivo. In silico perturbation studies indicated that changes in Gata2 at the mRNA level had the strongest impact on commitment frequency, and permanent activation of Gata1 increased the likelihood of commitment twofold, which was validated experimentally. However, there was little correlation between the expression patterns of the genes studied, and so the network that regulates commitment is not yet understood.
Single cell analysis is also being employed to delineate pathways of differentiation. While the haematopoietic system is well characterised, there is some disagreement about the ontogeny of the adult system. Analysis of 280 genes, including all commonly used cell surface markers and some important TFs, in multiple cell types of the haematopoietic system 58 showed that in the stem cell compartment levels of the marker CD150 (E-Slam) – which has already been shown to enrich for long-term stem cell capacity compared to CD150− cells 59 – were correlated with the expression of a megakaryocyte-erythroid module of TFs. Furthermore, CD150+ cells produced more megakaryocyte-erythroid cells in colony-forming assays. In combination with previous functional studies, this single cell analysis supports the suggestion that megakaryocytic and erythroid cells emerge directly from the HSC, while myelolymphoid cells arise at a later stage 60. This is in contrast to the original model of differentiation in which the HSC gives rise to the CMP, which produces megakaryocytic, erythroid and myeloid cells, and the CLP, which gives rise to the lymphoid lineages 61,62. This study therefore indicates how gene expression analysis can relate transcriptional and cell surface programs, and shows how single cell analysis could be useful in other, less well-defined systems to identify novel surface markers by which to isolate stem cells from contaminating cell types.
Gene regulatory networks can be characterised using single-cell data
TFs regulate gene expression through interactions with the chromatin at regulatory elements such as promoters and enhancers. Much is known about individual TFs, but while it is clear from functional studies that they act together as part of larger gene regulatory networks 4,27,29,30, less is known about how these networks function to regulate cell fate. Networks are assembled from interlinked motifs, such as positive and negative feedback loops, which perform particular functions. For example auto-regulatory loops can act to reinforce and maintain a factor's expression once activated, while negative auto-regulation results in the repression of a gene by its own product, which can reduce cell-to-cell variation or ‘noise’ in expression 63,64. The connectivity or ‘logic’ of a network determines which factors will be expressed together, so understanding network structure helps us to understand how particular cell states arise, how cells move forward through differentiation and how they decide between alternative fates.
Much of the early work on gene regulatory network construction came from Eric Davidson's studies of the sea urchin 65. Regulatory elements can be defined by examination of conserved regions of the genome, and through analysis of regions bound by TFs in chromatin immunoprecipitation analysis. Perturbation studies, on the other hand, can be used to infer the regulatory relationships or ‘logic’ between factors 28. Our laboratory identified a small network model in haematopoietic cells, in which Gata2, Scl and Fli1 are connected to one another through three enhancers 66. Modelling of this triad has shown that it can function as a bistable switch, being either on or off, hence allowing the network to filter noise when responding to external cues 67. A similar triad has been identified between Oct4, Sox2 and Nanog in ES cells 68, but, as each factor may have many targets, it can be difficult to identify and validate large networks this way. Networks have also been generated from microarray studies by identifying statistical dependencies between gene products 69,70. However, many systems have inherent heterogeneity, both functionally and in terms of gene expression between individual cells, that is not taken into account using these approaches, and that can only be examined through analysis of individual cells.
Regulatory interactions revealed from hundreds of single haematopoietic cells
Several studies have specifically used single cell analysis to characterise gene regulatory networks. We calculated pairwise correlations within a set of 18 TFs in 597 single primary haematopoietic stem and progenitor cells analysed on the Fluidigm BioMark platform 56. This revealed many strong correlations, among which were several known interactions including antagonisms between Gata1 and PU.1, and Gfi1 and Gfi1b. Calculating the significance of correlations and displaying this data as a network allowed us to identify a putative regulatory triad in which Gata2 is involved in the Gfi1-Gfi1b antagonism. Importantly, we were able to validate these correlations as direct interactions with an impact on gene expression using ChIP-sequencing and transcriptional assays, suggesting that single cell gene expression analysis is most powerful in combination with existing experimental techniques. Analysis of expression patterns suggested that this network may be important in regulating the exit of cells from the stem cell compartment toward the myelolymphoid lineages. As loss of function of Gata2 has been implicated in acute myeloid leukaemias 71,72, while inhibition of Gfi1 could prolong survival in some mouse models of leukaemia 73, the regulatory triad identified from single cell analysis may also be important in the balance between normal differentiation and malignancy. Interestingly, while Gfi1−/− HSCs have defects in long-term haematopoiesis due to elevated proliferation and stem cell exhaustion, we identified relatively few HSCs with strong Gfi1 expression, with the majority expressing the related but antagonistic TF Gfi1b. This pattern was also observed in two other recent studies 74,75, indicating that PCR-based single cell studies are highly reproducible, and may require us to revisit and reinterpret existing data. In order to provide robust new insights into developmental and disease processes, this would ideally include non-PCR based assessments of gene expression levels.
Guo et al. 58 also calculated covariances between genes in individual cells from multiple haematopoietic stem and progenitor populations to discover potential regulatory linkages, and integrated their data with existing ChIP-seq studies to exclude interactions where there is no direct TF binding event. An interaction network was generated that highlighted Gata2 as a core stem cell regulator, and examination of Gata2 heterozygotes indicated that the stem cell network is sensitive to modulations in the expression of individual TFs: transcriptional changes were identified consistent with the known expression pattern and function of Gata2 in regulating megakaryocyte-erythroid development. However, this study did not experimentally validate the function of binding events. We found that while TF binding events occur at the Gfi1b promoter, this region is not sufficient to drive expression in haematopoietic cells without binding at additional regulatory elements 56. This indicates that not all TF binding events have a functional consequence and so they cannot alone be used to validate regulatory interactions without functional studies.
Perspectives
The ultimate power of single cell analysis depends to a large extent on the number of cells that can be analysed in parallel. Analysing large numbers of cells simultaneously provides statistical power when calculating correlations and covariances in expression between pairs of genes. It also allows for the capture of rare events that would be hidden in large population studies, such as the commitment of stem cells. This should allow researchers to delineate the steps involved and the molecular mechanisms that underlie them in a way that is not possible when taking average measurements of populations. For example work in multiple systems has indicated that the early stages of lineage commitment are stochastic: some multipotent cells express lineage-affiliated genes that reversibly sway the balance between self-renewal and differentiation 5–7. Furthermore, whether all cells take the same route to a particular committed state, activating a suite of lineage-affiliated TFs, or whether alternative routes exist is also an important question – both in normal development and disease. Studies in this area have the potential to aid the development of directed differentiation strategies for regenerative medicine. While much has yet to be determined, a number of studies, including those discussed above, have begun to make advances towards answering these questions.
Further technological advances are providing more opportunities for studying single cells. Index sorting during FACS allows us to identify the cell surface marker profiles of individual sorted cells. If this information could be correlated with gene expression signatures, it would become possible to isolate particular gene expression states based on surface markers, which may be useful for isolating tissue-specific stem cells 76,77. Protein analysis is also possible at the single cell level, although it has not yet become as popular as gene expression analysis. Mass cytometry uses isotope-labelled antibodies and mass spectrometry to detect over 50 different proteins simultaneously within individual cells 78–80. Analysis pipelines are already in place to visualise relationships between cells and the proteins that drive them 81,82. This methodology has the potential to analyse tens of thousands of cells in a single run, and has been used to study immune signalling in the haematopoietic system 79,80. Microfluidic gene expression platforms are also being utilised for protein analysis, through the use of proximity ligation assays 83. When two different oligonucleotide-tagged antibodies bind to the same protein, the nucleotides are brought into proximity, facilitating a PCR reaction 84 that can be analysed, for example on Fluidigm's BioMark platform.
In the era of ‘big data’, single cell studies are likely to take centre stage, particularly as single cell mRNA and genome sequencing technologies mature. The growing interest in and necessity for studying individual cells is highlighted by the meetings, conferences and courses now dedicated to single cell biology. Many challenges lie ahead, not least for the optimisation of protocols to limit variation in sample collection and processing, but also for the analysis and visualisation of multidimensional data and the production of novel hypotheses. Studies to date have highlighted the insights that can be gained by studying single types of biomolecules, but coupling genomics, transcriptomics and proteomics in individual cells will take systems biology to a whole new level. While this is a rapidly growing field in its own right, the power of single cell studies is in complimenting existing population studies rather than completely supplanting them as it is still necessary to validate findings, and many techniques may remain impractical at the single cell level. However, potentially the biggest challenge for single cell biology will be the inevitable requirement for researchers to forsake established paradigms based on population data in the light of new evidence from the single cell analysis.
Glossary
- ChIP-seq
chromatin immunoprecipitation with deep sequencing
- FACS
fluorescence activated cell sorting
- mRNA-seq
sequencing of messenger RNA
- RT-qPCR
reverse transcription quantitative polymerase chain reaction
- sm mRNA-FISH
single molecule RNA in situ hybridisation
- TF
transcription factor
References
- Copley MR, Beer PA, Eaves CJ. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell. 2012;10:690–7. doi: 10.1016/j.stem.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Graf T, Stadtfeld M. Heterogeneity of embryonic and adult stem cells. Cell Stem Cell. 2008;3:480–3. doi: 10.1016/j.stem.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development. 2009;136:3853–62. doi: 10.1242/dev.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enver T, Pera M, Peterson C, Andrews PW. Stem cell states, fates, and the rules of attraction. Cell Stem Cell. 2009;4:387–97. doi: 10.1016/j.stem.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Chambers I, Silva J, Colby D, Nichols J, et al. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–4. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- Toyooka Y, Shimosato D, Murakami K, Takahashi K, et al. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development. 2008;135:909–18. doi: 10.1242/dev.017400. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Lopes SM, Tang F, Surani MA. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell. 2008;3:391–401. doi: 10.1016/j.stem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HH, Hemberg M, Barahona M, Ingber DE, et al. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–7. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Krause D, Greaves M, Sharkis S, et al. Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 1997;11:774–85. doi: 10.1101/gad.11.6.774. [DOI] [PubMed] [Google Scholar]
- Ramos CA, Bowman TA, Boles NC, Merchant AA, et al. Evidence for diversity in transcriptional profiles of single hematopoietic stem cells. PLoS Genet. 2006;2:e159. doi: 10.1371/journal.pgen.0020159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady G, Barbara M, Iscove NN. Representative in vitro cDNA amplification from individual hemopoietic cells and colonies. Methods Mol Cell Biol. 1990;2:17–25. [Google Scholar]
- Stahlberg A, Bengtsson M. Single-cell gene expression profiling using reverse transcription quantitative real-time PCR. Methods. 2010;50:282–8. doi: 10.1016/j.ymeth.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Kawasaki ES. Microarrays and the gene expression profile of a single cell. Ann N Y Acad Sci. 2004;1020:92–100. doi: 10.1196/annals.1310.010. [DOI] [PubMed] [Google Scholar]
- Lecault V, White AK, Singhal A, Hansen CL. Microfluidic single cell analysis: from promise to practice. Curr Opin Chem Biol. 2012;16:381–90. doi: 10.1016/j.cbpa.2012.03.022. [DOI] [PubMed] [Google Scholar]
- Tischler J, Surani MA. Investigating transcriptional states at single-cell-resolution. Curr Opin Biotechnol. 2013;24:69–78. doi: 10.1016/j.copbio.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Plessy C, Desbois L, Fujii T, Carninci P. Population transcriptomics with single-cell resolution: a new field made possible by microfluidics: a technology for high throughput transcript counting and data-driven definition of cell types. BioEssays. 2013;35:131–40. doi: 10.1002/bies.201200093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Freire V, Ebert AD, Kalisky T, Quake SR, et al. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat Protoc. 2012;7:829–38. doi: 10.1038/nprot.2012.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AK, VanInsberghe M, Petriv OI, Hamidi M, et al. High-throughput microfluidic single-cell RT-qPCR. Proc Natl Acad Sci USA. 2011;108:13999–4004. doi: 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Pang ZP, Sudhof TC, Wernig M, et al. Comprehensive qPCR profiling of gene expression in single neuronal cells. Nat Protoc. 2012;7:118–27. doi: 10.1038/nprot.2011.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Lao K, Surani MA. Development and applications of single-cell transcriptome analysis. Nat Methods. 2011;8:S6–11. doi: 10.1038/nmeth.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahlberg A, Rusnakova V, Forootan A, Anderova M, et al. RT-qPCR work-flow for single-cell data analysis. Methods. 2013;59:80–8. doi: 10.1016/j.ymeth.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol. 2007;7:105–17. doi: 10.1038/nri2024. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH. Chromosomal translocations in human cancer. Nature. 1994;372:143–9. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Schutte J, Moignard V, Gottgens B. Establishing the stem cell state: insights from regulatory network analysis of blood stem cell development. Wiley Interdiscip Rev Syst Biol Med. 2012;4:285–95. doi: 10.1002/wsbm.1163. [DOI] [PubMed] [Google Scholar]
- Pimanda JE, Gottgens B. Gene regulatory networks governing haematopoietic stem cell development and identity. Int J Dev Biol. 2010;54:1201–11. doi: 10.1387/ijdb.093038jp. [DOI] [PubMed] [Google Scholar]
- Foster SD, Oram SH, Wilson NK, Gottgens B. From genes to cells to tissues – modelling the haematopoietic system. Mol Biosyst. 2009;5:1413–20. doi: 10.1039/B907225j. [DOI] [PubMed] [Google Scholar]
- Petricka JJ, Benfey PN. Reconstructing regulatory network transitions. Trends Cell Biol. 2011;21:442–51. doi: 10.1016/j.tcb.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonzanni N, Garg A, Feenstra KA, Schutte J, et al. Hard-wired heterogeneity in blood stem cells revealed using a dynamic regulatory network model. Bioinformatics. 2013;29:i80–8. doi: 10.1093/bioinformatics/btt243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK. Noise in gene expression: origins, consequences, and control. Science. 2005;309:2010–3. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–26. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder T. Hematopoietic stem cell heterogeneity: subtypes, not unpredictable behavior. Cell Stem Cell. 2010;6:203–7. doi: 10.1016/j.stem.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Eldar A, Elowitz MB. Functional roles for noise in genetic circuits. Nature. 2010;467:167–73. doi: 10.1038/nature09326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriello NM, Douglas ES, Thaitrong N, Hsiao SC, et al. Integrated microfluidic bioprocessor for single-cell gene expression analysis. Proc Natl Acad Sci USA. 2008;105:20173–8. doi: 10.1073/pnas.0806355106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson M, Stahlberg A, Rorsman P, Kubista M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005;15:1388–92. doi: 10.1101/gr.3820805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente A, Bing N, Hoeschele I, Mendes P. Discovery of meaningful associations in genomic data using partial correlation coefficients. Bioinformatics. 2004;20:3565–74. doi: 10.1093/bioinformatics/bth445. [DOI] [PubMed] [Google Scholar]
- Stahlberg A, Andersson D, Aurelius J, Faiz M, et al. Defining cell populations with single-cell gene expression profiling: correlations and identification of astrocyte subpopulations. Nucleic Acids Res. 2011;39:e24. doi: 10.1093/nar/gkq1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzel B, Barabasi AL. Network link prediction by global silencing of indirect correlations. Nat Biotechnol. 2013;31:720–5. doi: 10.1038/nbt.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feizi S, Marbach D, Medard M, Kellis M. Network deconvolution as a general method to distinguish direct dependencies in networks. Nat Biotechnol. 2013;31:726–33. doi: 10.1038/nbt.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Yang M, Guo H, Yang L, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20:1131–9. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–82. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Guo G, Huss M, Tong GQ, Wang C, et al. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell. 2010;18:675–85. doi: 10.1016/j.devcel.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Buettner F, Theis FJ. A novel approach for resolving differences in single-cell gene expression patterns from zygote to blastocyst. Bioinformatics. 2012;28:I626–32. doi: 10.1093/bioinformatics/bts385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur BD, Sevilla A, Lenz M, Muller FJ, et al. Nanog-dependent feedback loops regulate murine embryonic stem cell heterogeneity. Nat Cell Biol. 2012;14:1139–47. doi: 10.1038/ncb2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina C, Fugazza C, Tipping AJ, Brown J, et al. Inferring rules of lineage commitment in haematopoiesis. Nat Cell Biol. 2012;14:287–94. doi: 10.1038/ncb2442. [DOI] [PubMed] [Google Scholar]
- Teles J, Pina C, Eden P, Ohlsson M, et al. Transcriptional regulation of lineage commitment – a stochastic model of cell fate decisions. PLoS Comput Biol. 2013;9:e1003197. doi: 10.1371/journal.pcbi.1003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–23. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faddah DA, Wang H, Cheng AW, Katz Y, et al. Single-cell analysis reveals that expression of nanog is biallelic and equally variable as that of other pluripotency factors in mouse ESCs. Cell Stem Cell. 2013;13:23–9. doi: 10.1016/j.stem.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–32. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Melton DA. Extreme makeover: converting one cell into another. Cell Stem Cell. 2008;3:382–8. doi: 10.1016/j.stem.2008.09.015. [DOI] [PubMed] [Google Scholar]
- Hanna J, Saha K, Pando B, van Zon J, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–22. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur BD, Lemischka IR. Statistical mechanics of pluripotency. Cell. 2013;154:484–9. doi: 10.1016/j.cell.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Glotzbach JP, Januszyk M, Vial IN, Wong VW, et al. An information theoretic, microfluidic-based single cell analysis permits identification of subpopulations among putatively homogeneous stem cells. PLoS One. 2011;6:e21211. doi: 10.1371/journal.pone.0021211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moignard V, Macaulay IC, Swiers G, Buettner F, et al. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat Cell Biol. 2013;15:544. doi: 10.1038/ncb2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr G, Ruskin HJ, Crane M, Doolan P. Techniques for clustering gene expression data. Comput Biol Med. 2008;38:283–93. doi: 10.1016/j.compbiomed.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Guo G, Luc S, Marco E, Lin TW, et al. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell. 2013;13:492–505. doi: 10.1016/j.stem.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent DG, Copley MR, Benz C, Wohrer S, et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood. 2009;113:6342–50. doi: 10.1182/blood-2008-12-192054. [DOI] [PubMed] [Google Scholar]
- Adolfsson J, Mansson R, Buza-Vidas N, Hultquist A, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121:295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–7. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Kondo M, Scherer DC, Miyamoto T, King AG, et al. Cell-fate conversion of lymphoid-committed progenitors by instructive actions of cytokines. Nature. 2000;407:383–6. doi: 10.1038/35030112. [DOI] [PubMed] [Google Scholar]
- Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–61. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- Swiers G, Patient R, Loose M. Genetic regulatory networks programming hematopoietic stem cells and erythroid lineage specification. Dev Biol. 2006;294:525–40. doi: 10.1016/j.ydbio.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Davidson EH, Rast JP, Oliveri P, Ransick A, et al. A genomic regulatory network for development. Science. 2002;295:1669–78. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- Pimanda JE, Ottersbach K, Knezevic K, Kinston S, et al. Gata2, Fli1, and Scl form a recursively wired gene-regulatory circuit during early hematopoietic development. Proc Natl Acad Sci USA. 2007;104:17692–7. doi: 10.1073/pnas.0707045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula J, Smith AM, Gottgens B, Igoshin OA. Modeling reveals bistability and low-pass filtering in the network module determining blood stem cell fate. PLoS Comput Biol. 2010;6:e1000771. doi: 10.1371/journal.pcbi.1000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Wang K, Lim WK, Kustagi M, et al. Reverse engineering cellular networks. Nat Protoc. 2006;1:662–71. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, et al. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–90. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- Bonadies N, Foster SD, Chan WI, Kvinlaug BT, et al. Genome-wide analysis of transcriptional reprogramming in mouse models of acute myeloid leukaemia. PLoS One. 2011;6:e16330. doi: 10.1371/journal.pone.0016330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–7. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandanpour C, Phelan JD, Vassen L, Schutte J, et al. Growth factor independence 1 antagonizes a p53-induced DNA damage response pathway in lymphoblastic leukemia. Cancer Cell. 2013;23:200–14. doi: 10.1016/j.ccr.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock H, Hamblen MJ, Rooke HM, Schindler JW, et al. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. 2004;431:1002–7. doi: 10.1038/nature02994. [DOI] [PubMed] [Google Scholar]
- Zeng H, Yucel R, Kosan C, Klein-Hitpass L, et al. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 2004;23:4116–25. doi: 10.1038/sj.emboj.7600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne GW. Recent advances in flow cytometric cell sorting. Methods Cell Biol. 2011;102:533–56. doi: 10.1016/B978-0-12-374912-3.00021-3. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Shibata N, Okumura R, Kudome T, et al. Single-cell gene profiling of planarian stem cells using fluorescent activated cell sorting and its “index sorting” function for stem cell research. Dev Growth Differ. 2010;52:131–44. doi: 10.1111/j.1440-169X.2009.01157.x. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler's guide to cytometry. Trends Immunol. 2012;33:323–32. doi: 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir EAD, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–96. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW, Sigal N, Bendall SC, Nolan GP, et al. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–52. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir ED, Davis KL, Tadmor MD, Simonds EF, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–52. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Simonds EF, Bendall SC, Gibbs KD, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–91. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, et al. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–7. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- Stahlberg A, Thomsen C, Ruff D, Aman P. Quantitative PCR analysis of DNA, RNAs, and proteins in the same single cell. Clin Chem. 2012;58:1682–91. doi: 10.1373/clinchem.2012.191445. [DOI] [PubMed] [Google Scholar]