

SYNOPSIS
Nitric oxide (NO) binds to the receptor, soluble guanylyl cyclase (sGC), stimulating cGMP production. The NO-sGC-cGMP pathway is a key component in the cardiovascular system. Discrepancies in sGC activation and deactivation in vitro vs. in vivo have led to a search for endogenous factors that regulate sGC or assist in cellular localization. In our previous work, which identified Hsp70 as a modulator of sGC, we determined that protein disulfide isomerase (PDI) bound to an sGC-affinity matrix. We here establish and characterize this interaction. Incubation of purified PDI with semi-purified sGC, both reduced and oxidized, resulted in different migration patterns on non-reducing Western blots indicating a redox component to the interaction. In sGC-infected COS-7 cells, transfected Flag-tagged PDI and PDI-CXXS (redox active site “trap-mutant”) pulled down sGC. This PDI-sGC complex was resolved by reductant, confirming a redox interaction. PDI inhibited NO-stimulated sGC activity in COS-7 lysates; however, a PDI redox inactive mutant, PDI-SXXS, did not. Together, these data unveil a novel mechanism of sGC redox modulation via thiol-disulfide exchange. Finally, in smooth muscle cells, endogenous PDI and sGC co-localize by in situ proximity ligation assay, suggesting biological relevance. PDI-dependent redox regulation of sGC NO-sensitivity may provide a secondary control over vascular homeostasis.
Keywords: soluble guanylyl cyclase, protein disulfide isomerase, redox regulation, nitric oxide, smooth muscle cells
INTRODUCTION
Soluble guanylyl cyclase (sGC) is an important enzyme in the cardiovascular system where it mediates vasodilation and inhibits platelet aggregation. Active as a heterodimer, the most abundant form of the enzyme is composed of α1 and β1 subunits, although an active α2/β1 heterodimer exists and is predominantly expressed in brain and placenta [1, 2]. Each subunit contains three major domains: the N-terminal HNOX domain, a central domain with a PAS fold and coiled-coil region, and the C-terminal catalytic domain. The β subunit HNOX domain binds the heme. Binding of nitric oxide (NO) to heme stimulates the enzyme to convert GTP to cGMP at a rate up to 200-fold over the basal level of activity. The formation of a functional catalytic site requires that the α and β C-terminal catalytic domains are associated [3]. The NO-sGC-cGMP pathway is key in the regulation of blood pressure and sGC is activated by glycerin trinitrate (GTN has been used for over a century to treat angina pectoris; [4]). Yet, sGC regulation and activation mechanisms remain poorly understood.
Previous work determined potential endogenous modulators of sGC activity. Our lab found that the soluble fraction of COS-7 cell lysates contained an endogenous “factor” able to stimulate sGC in vitro, increasing NO-stimulated over basal cGMP production by about 1.6 fold. This factor, identified as Hsp70, showed co-localization with sGC in rat lung and smooth muscle cells [5]. In addition, several groups have shown that the molecular chaperone Hsp90 complexes with sGC to stabilize and protect the enzyme from proteosomal degradation, as well as promoting heme insertion [6–9]. Other sGC interacting proteins include post-synaptic density protein 95 (PSD95), the ArfGAP protein AGAP-1, a suit of chaperonin containing t-complex polypeptide (CCTη) and LGN [10–13]. Mass spectrometry analysis used to identify Hsp70 uncovered another potential sGC interacting protein, protein disulfide isomerase (PDI) [5]. PDI is so named because of its ability to isomerase mis-paired disulfides of client proteins as they undergo folding in the endoplasmic reticulum (ER) and is a well-studied member of the thioredoxin oxidoreductase super family. This versatile enzyme is also found outside the ER in other cellular compartments and has defined roles beyond disulfide isomerization [14–18]. Recently, de A. Paes and coworkers have shown that through its active site cysteines, PDI associates with p47phox in a manner that depends on cellular conditions and that occurs in the cytosolic fraction of leukocytes [17].
Recent work on the mechanisms of sGC activation suggests that sGC may be redox regulated [19–22]. The human form of the sGC α1β1 heterodimer contains 37 cysteines (Cys), 2.8% of total residues, an unusually high percentage for a protein thought to reside in the cytosol. Current and past research suggests that certain sGC Cys may be necessary for such functions as enzyme activation, dimerization, and desensitization by S-nitrosation [20–23]. However, sGC is historically purified with millimolar amounts of reductant, most commonly dithiothreitol (DTT), which can mask thiol-redox properties.
We hypothesized that the interaction of sGC with PDI, if confirmed, could be a result of either the chaperone or reduction/oxidation actions of PDI. The interaction of sGC and PDI and the effect on sGC activity by transient expression of sGC, PDI WT and PDI redox null mutant, along with endogenous co-localization in rat aortic smooth muscle cells is investigated.
EXPERIMENTAL
Materials
Talon cobalt resin from Clontech. Superdex 200 10/300 GL column from GE Healthcare. Recombinant human sGC from Axxora. Anti-PDI antibody from Abcam. SDS-PAGE 7.5% TGX gels, transfer/running buffers, and nitrocellulose from Biorad. Amicon centricon filtration device YM-30 from Millipore. Dithiothreitol (DTT) and Expressfect reagent from Denville Scientific. 2-mercaptoethanol, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), anti-β-actin antibody, anti-sGC α antibody, anti-Flag M2 affinity resin, bovine serum albumin (BSA), Bradford reagent, diamide, guanosine triphosphate (GTP), HEPES, leupeptin HC1, N-ethylmaleimide (NEM), MgCl2, phenylmethylsulfonyl fluoride (PMSF), lima bean trypsin inhibitor, Triton X-100 from Sigma Aldrich. All primers from Sigma Genosys. Anti-sGC β antibody and diethylamine NONOate (DEA-NO) from Cayman Chemical. Anti-Flag antibody from Cell Signaling Technology. Phusion® High-Fidelity PCR kit from New England Biolabs. COS-7 / CRL-2018 cells, Dulbecco’s Modified Eagles Medium (DMEM) from ATCC. Fetal bovine serum (FBS) for CRL-2018 cells from GIBCO. Antibiotic-antimycotic, 1X and 10X phosphate buffered saline (PBS) from Cellgro. FBS for COS-7 cells from Atlanta Biologics. Aprotinin and tris(2-carboxyethyl)phosphine HCl (TCEP) resin from Thermo Fisher Scientific. Geneticin, Lipofectamine 2000, PDI clone in pCMV5-Sport6, Opti-MEM I, and Measure iT thiol kit from Invitrogen. Radiolabeled guanosine triphosphate (GTP32) and radiolabled cyclic guanosine monophosphate (cGMP-3H) obtained from Perkin Elmer. Paraformaldehyde (PFA) from Electron Microsopy Sciences. Duolink II assay kit with Plus and Minus PLA probes from Olink Bioscience. PDI in the pET-28A plasmid was a gift from Dr. Colin Thorpe (University of Delaware). Adenoviral vectors (pAdTrack) containing sGC α1 or sGC β1 wild type were from Drs. Andreas Papapetropoulos and Zongmin Zhou (University of Patras, Greece).
Protein Expression and Purification
Expression and purification of recombinant sGC-Histag was carried out as described previously [24] with replacement of the second chromatographic step by a size exclusion / gel filtration column. Proteins eluted from Talon cobalt resin (Clontech) in 0.25-0.5 mL fractions were pooled to 1 mL and applied to a Superdex 200 10/300 GL (GE Healthcare) gel filtration column equilibrated in elution buffer (20 mM HEPES, pH 8.0, 2 mM MgCl2, 1 mM 2-mercaptoethanol). Chromatographic separation was performed at a flow rate of 0.5 mL/min using an AKTA Explorer FPLC (GE Healthcare). sGC (150 kDa, heterodimer) was eluted at 23 min. The fractions containing sGC were aliquoted, snap frozen in liquid nitrogen and stored at −80°C until use. Analysis of protein purity by silver staining indicated approximately 80–85% homogeneity of the sGC peak fraction using this method (not shown). In contrast with previous protocols, millimolar DTT was omitted during the preparation of lysates and storage. Expression and purification of PDI was performed as described [25].
Redox Status by electrophoresis
Determination of protein concentration was by UV-visible spectroscopy (Shimadzu 2450) using extinction coefficients (ε280) of 56400 M−1cm−1 and 72,000 M−1cm−1 for PDI and sGC, respectively [25, 26]. sGC (Axxora) and PDI (as purified) were reduced by incubation with at least 5-fold molar excess tris(2-carboxyethyl)phosphine HCl (TCEP) resin or oxidized by incubation with 100 μM diamide on ice in the dark for 15 min. TCEP resin was prewashed with 50 mM HEPES, pH 8.0, this functionalized resin ensures reductant does not carry through. The final diamide concentration in the steps following was at most 35 μM. Denatured sGC was prepared by thermal denaturation in a thermocycler from 25°C – 65°C in a stepwise gradient of 1°C every 30 seconds and held at 65°C for two minutes then placed on ice. Thiol-blocking of PDI was performed by first reducing with 10-fold molar excess TCEP, then incubating with 3 mM N-ethylmaledime (NEM) on ice for 15 min. The reaction was quenched with 0.5 mM DTT. PDI was recovered by Centricon filtration with a YM-30 (30,000 kDa cutoff) filter device. The following samples were prepared: reduced, oxidized, and denatured sGC; reduced, oxidized, and NEM-PDI; and their various combinations (where sGC and PDI are mixed, the ratio of sGC:PDI is 1:3). All mixed samples were incubated on ice for 15 min in the dark. For SDS-PAGE, reducing (10% SDS, 1.47 M 2-mercaptoethanol) and non-reducing (10% SDS) sample buffers were used. All samples were heated at 100°C for 5 min. All SDS-PAGE electrophoresis gels were run at 150V using Biorad 7.5% TGX gels for 45 min. Antibodies for Western blot: anti-PDI (Abcam), anti-sGC β (Cayman Chemical), anti-sGC α and anti-β actin (Sigma).
Thiol Titer Analysis
Determination of free thiols using the canonical DTNB assay was attempted; however, due to the very low concentrations of sGC used, this method was not feasible. To determine more precisely the number of thiols in the various redox samples, the Measure-iT thiol assay kit (Invitrogen) was used according to the manufacturer’s protocol with a reduced glutathione (GSH) standard curve. Recombinant human sGC (Axxora) at 0.4 mg/mL in 50 mM TEA pH 7.4, 2 mM DTT, 1 mM MgCl2, 30% glycerol was diluted to 0.27 μM and divided into two aliquots: 30 μL sGC was treated with either diamide (final concentration 91 μM) or incubated with 5 μL TCEP resin (final concentration ~ 1.1 mM) for 15 minutes on ice, in the dark. The two samples were incubated either with 50 mM potassium phosphate, pH 7.5 (assay buffer) or with 3-fold purified PDI to the same volume with assay buffer; each had the same final concentration of sGC. Additionally, sGC and PDI alone were incubated with 50 mM potassium phosphate, pH 7.5. All samples were incubated in the dark, on ice for 15 min. After this second incubation, samples were measured in triplicate according to the manufacturer’s instruction in a 96-well plate. Negative controls of assay buffer alone and supernatant from TCEP resin with assay buffer alone (no protein) were also measured in triplicate. A set of at least 8 reduced glutathione standards were run in triplicate for each assay. Fluorescence was measured with a Cary Eclipse fluorometer (Varian) equipped with a microplate reader (excitation at 494 nm and emission at 517 nm).
Transfection with PDI thiol mutants
PDI Inactive mutant
The four redox active cysteines of mouse PDI cDNA (Invitrogen Clone ID IOH9865) in the pCMV5-Sport6 vector were mutated to serine (Ser), referred to as PDI SxxS, by site-directed mutagenesis (QuikChange, Stratagene) using the following primers: CxxC site one (Cys55,58) forward – 5′-CGGGGCACCAGACCGGTGAGGTTTCGAGAC-3′, reverse – 5′-GCCCCGTGGTCTGGCCACTCCAAAGCTCTG-3′; CxxC site two (Cys399,402) forward – 5′-CGGGGAACCAGACCAGTGAGGTTCGTCGATCGG-3′, reverse – 5′-GCCCCTTGGTCTGGTCACTCCAAGCAGCTAGCC-3′. COS-7 cells (ATCC), which have endogenous PDI but no detectable sGC, were cultured in DMEM supplemented with 1% antiobiotic-antimyocotic and 10% FBS. Cells at 80 – 90% confluency were transfected with sGC α1 and β1 wild type (WT) in pCMV5, PDI-WT, PDI-SxxS, sGC-WT/PDI-WT, and sGC-WT/PDI-SxxS. After 48 hours, cells were harvested in homogenization buffer (50 mM HEPES, pH 8.0; 150 mM NaCl) containing protease inhibitors: PMSF (35 μg/mL), leupeptin (10 μg/mL), lima bean trypsin inhibitor (10 μg/mL) and aprotinin (40 μg/mL). Cells were lysed on ice by sonication and centrifuged (14000 RPM, 10 min, 4°C) to remove debris. Lysate was collected as supernatant (soluble fraction). A 30 μL aliquot of lysate was reserved for total protein concentration determination by Bradford assay. The remaining lysate was aliquoted, flash frozen and stored at −80°C until use in Western blot analysis or activity assay.
PDI Thiol Trap Mutant
Mouse PDI cDNA (Invitrogen Clone ID IOH9865) in the pCMV5-Sport6 vector was used. First, a single base pair was changed (no change in amino acid coding) to remove an internal EcoRI site using the following primers: forward – 5′-CGTGTTTGTTGAGTTCTATGCCCCTTGG-3′; reverse – 5′-CCAAGGGGCATAGAACTCAACAAACACG-3′ by site-directed mutagenesis (QuikChange, Stratagene). Then, two new restriction sites were inserted via PCR amplification of the PDI cDNA and a Flag sequence was inserted at the 3′ end using the following primers: 5′ EcoRI – 5′-GATCGAATTCGCCGCCACCATGCTGAGCCGTGCTTTG-3′ and 3′ BamHI/Flag – 5′-GCAACGGGATCCTCACTACTTGTCATCGTCGTCCTTGTAGTCGCCCAGTTCATCCTTCACAGC-3′ and Phusion® High-Fidelity PCR Kit as per the manufacturer’s instructions (New England Biolabs). The newly created PDI-Flag insert was cut and ligated into a pCMV5 vector previously digested with EcoRI and BamHI. To create the PDI thiol-trap mutant, the second Cys of both redox active CxxC sites was mutated to serine (referred to as PDI-Flag CxxS) by site-directed mutagenesis (QuikChange, Stratagene) using the following primers: CxxC site one (Cys58) forward – 5′-GGCCACTCCAAAGCTCTGGCTCCCGAG-3′, reverse – 5′-CTCGGGAGCCAGAGCTTTGGAGTGGCC-3′; CxxC site two (Cys402) forward – 5′-GGTGTGGTCACTCCAAGCAGCTAGCC-3′, reverse – 5′-GGCTAGCTGCTTGGAGTGACCACACC-3′. The molecular cloning steps resulted in two pCMV5 vectors: PDI-Flag WT and PDI-Flag CxxS. COS-7 cells (ATCC) were cultured as described above. Cells at 80 – 90% confluency were transfected with PDI-Flag WT, PDI-Flag CxxS, or left untransfected using Lipofectamine 2000 (Invitrogen). After 24 hours, all cells were infected with sGC α1 and β1 WT adenovirus at an multiplicity of infection (MOI) of 1, with the exception of a set of PDI-Flag WT and PDI-Flag CxxS dishes left un-infected as control. Twenty-four hours post-infection, cells were harvested in lysis buffer (50 mM Tris-HCl, pH 7.4; 150 mM NaCl; 1 mM EDTA; 1% Triton X-100) containing the same protease inhibitors as above. for Flag immunoprecipitation (IP) as per the Flag-affinity resin manufacturer’s instructions (Sigma-Aldrich). Cells were lysed and stored as described above. 100μL of each lysate (sGC WT; sGC WT / PDI-Flag WT; sGC WT / PDI-Flag CxxS; PDI-Flag WT; PDI-Flag CxxS) was incubated with 50 μL of anti-Flag M2 affinity resin (Sigma Aldrich) for either 2h at room temperature or overnight at 4°C with end-over-end mixing. The lysate-resin mixture was then centrifuged at 9000RPM in a benchtop microcentrifuge for 2 min. Supernatant was removed and collected as the unbound fraction, stored at −20°C. The resin was washed 3 times with 10 volumes of TBS. The bound sample was eluted from the Flag affinity resin with 20 μL of 2X non-reducing Laemmli buffer by boiling for 3m. The samples were centrifuged and supernatant collected as the bound fraction. The bound fraction was split into two aliquots, 10 μL was used for non-reducing gels and 1 μL of 2-mercaptoethanol was added to 10 μL of sample for reducing gels. SDS-PAGE gels were run as described above and subsequently transferred to nitrocellulose for Western Blot analysis, anti-β-actin as loading control.
Assay of sGC activity
Activity of sGC was assayed as described [27] under both basal and NO-stimulated conditions (100 μM DEA-NO). The reaction was allowed to proceed for 5 min at 30°C.
In situ proximity ligation assay
CRL-2018 rat aortic smooth muscle cells (ATCC) containing endogenous PDI and sGC were cultured in DMEM supplemented with 1% antibiotic-antimycotic and 0.2 mg/mL G-418 for in situ proximity ligation assay (PLA) with the Duolink II fluorescence kit (Olink bioscience). CRL-2018 cells at 30–40% confluency were plated on coverslips in 6-well plates washed with 1X PBS, then fixed and permeabilized with 3% PFA and 0.2% Triton X-100 for 10 min. After washing, coverslips were incubated in blocking solution (1% BSA in 1X PBS) for 30 min at room temperature and incubated overnight at 4°C with following primary antibodies: anti-sGC α1 (1:300, rabbit) or anti-sGC β1 (1:200, rabbit), with anti-PDI (1:300, mouse). Duolink II PLA probes anti-rabbit Plus and Duolink II PLA probes anti-mouse Minus were used as per the manufacturer’s instruction. Cells were mounted onto glass slides using the provided mounting medium with DAPI. Fluorescence signals were imaged using a 200M Axiovert fluorescence microscope (Zeiss), 40X objective and analysed with Axiovision software. Cells incubated with only anti-PDI or without primary antibody were used as negative controls.
Statistical Analysis
Data were plotted using Sigma Plot and statistical analysis was performed using the Student’s t-test function in Sigma Plot or Microsoft Excel to determine the significance of difference (p<0.05 was considered significant) and standard error mean (SEM).
RESULTS
Purified sGC and PDI interact via thiol-disulfide exchange
PDI was identified by mass spectrometry as a potential sGC interacting protein in our study that uncovered Hsp70 [5]. In the original study, the His-tagged end of the β subunit of sGC bound to cobalt resin created an sGC-affinity matrix through which COS-7 cell lysates were passed, helping to confirm that Hsp70 interacts with sGC. In the present study, we initially confirmed that PDI interacts, either directly or indirectly, with sGC using the same sGC-affinity matrix (Supplemental Fig. 1). Next, we investigated the nature of the interaction between sGC and PDI in vitro using purified enzyme. TCEP is a phosphine reagent that reduces protein disulfides to free thiols [28]. PDI disulfides were reduced with TCEP, or free thiols were oxidized to disulfides with diamide [17]. Similarly, diamide is used to oxidized free thiols to disulfides in sGC [20]. Reduced, oxidized or denatured sGC was incubated with either reduced, oxidized, as purified, or free thiol-blocked PDI (alkylated using NEM, Alk PDI), as described in Materials and Methods. Following incubation, samples were run on non-reducing and reducing gels; subsequently transferred to nitrocellulose and probed with anti-sGC to determine whether complexes were formed under the various conditions and if disulfide bonds were involved. Representative sGC reducing and non-reducing blots are shown in Figure 1 (PDI probed blots are provided as Supplemental Figure 2).
Figure 1. Redox dependent interaction between sGC and purified PDI.
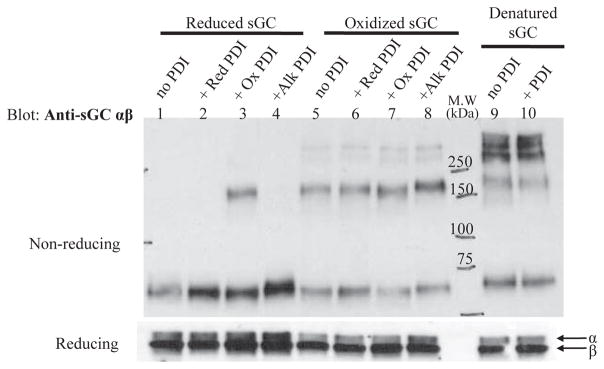
A. Representative non-reducing (upper panel) and reducing (lower panel) Western blots probed for sGC α and β of mixed redox samples. Lanes 1–4 are reduced sGC alone and with PDI at different redox states as labeled. Lanes 4–8 are the same with oxidized sGC as labeled. Lanes 9 and 10 are heat-denatured sGC alone and with PDI as purified (partially oxidized). The different redox states of sGC and PDI and experimental conditions are detailed in Materials and Methods. sGC is from Axxora, PDI was purified as described. The assay was independently repeated three times with sGC, both supplied and purified, and purified PDI with similar results. (Red: reduced; Ox: oxidized, Alk: PDI alkylated with NEM)
The non-reducing blot, probed for sGC is shown in Figure 1 (upper panel), lanes 1–4 contain reduced sGC. Only reduced sGC incubated with oxidized PDI (lane 3) shows a significant shift in migration pattern, indicating an interaction between sGC and PDI that is not resistant to reduction (reducing blots lower panel vs. non-reducing blots upper panel) suggesting the involvement of a mixed disulfide between the two proteins. Lanes 5–8 contain oxidized sGC, all of which show no significant change in redox pattern in the presence of reduced, oxidized, or alkylated PDI (Red, Ox, Alk PDI). Thus, under the conditions of this in vitro redox Western blot assay, reduced sGC interacts with oxidized PDI; but the opposite: oxidized sGC and reduced PDI do not interact. If PDI was acting as a molecular chaperone, it is expected that denatured sGC would also be bound by PDI. However, lanes 9 and 10 show that heat-denatured sGC does not interact with purified PDI as there is no change in the migration pattern of sGC with or without PDI. In addition, neither reduced or oxidized sGC when incubated with PDI that was blocked with NEM, showed a change in sGC migration pattern (lanes 4 and 8), again suggesting a thiol-dependent interaction. All together, these observations point to an interaction between sGC and PDI mediated via thiol-disulfide exchange, dependent on the redox state of the two proteins. A reduced Western blot probed for sGC, Figure 1 lower panel, shows similar protein levels for the reduced sample group and amongst the oxidized sample group indicating that the different pattern was not due to a difference in loaded amount. Reduced and non-reduced gels probed by Western blot for PDI are available in supplemental information.
Thiol titer of sGC is modified in the presence of PDI
The canonical DTNB thiol titer assay was initially used to estimate the free thiols in samples as prepared for the redox Western blots, but the method was not sensitive enough to reliably determine the thiol titer of redox samples at the low concentrations of sGC used (not shown). Therefore, we evaluated the thiol titer of samples prepared in the same way as for the redox Western blots using a more sensitive fluorometric plate reader assay, as described in Materials and Methods. In Figure 2A, the average thiols per mole of purified PDI is 1.5 ± 0.4, in good agreement with the literature where PDI has two free thiols as purified [29]. Recombinant sGC (Axxora) has 21 ± 5 free thiols per mole of sGC, while fully reduced sGC (Red sGC) is slightly higher at 24 ± 4 per mole. Oxidized sGC (Ox sGC) has a much lower value at 5.3 ± 2 thiols per mole as expected. The total thiol titer for reduced and oxidized sGC with and without purified PDI (two free thiols, partially oxidized) is shown in Figure 2B. There is a slight increase in total thiols for oxidized sGC with PDI as compared to oxidized sGC alone (0.61 ± 0.2 μM versus 0.51 ± 0.3 μM). However, for reduced sGC, addition of PDI almost doubles the total thiol titer, 2.3 ± 0.3 μM versus 4.0 ± 0.6 μM (p<0.05). The supernatant from TCEP resin with assay buffer was also measured and showed no increase in total thiols over assay buffer alone (not shown), ensuring that there was no reductant carryover between incubation steps. The data from this thiol titer also supports that the interaction of sGC and PDI results in thiol-disulfide exchange in vitro.
Figure 2. Purified PDI increases thiol titer of reduced sGC.

A. Thiols calculated per mole of protein for PDI as purified, oxidized sGC, reduced sGC and sGC from the supplier (± SEM; n = 4 for PDI, oxidized sGC and supplied sGC; n = 5 for reduced sGC). B. Total thiol titer for oxidized sGC and reduced sGC in the presence or absence of purified PDI shown as total protein thiols in μM (±SEM; n = 4 for oxidized sGC/PDI; n = 5 for reduced sGC/PDI); *: p<0.05 for reduced sGC alone vs. reduced sGC with PDI. Experimental details are described in Materials and Methods.
sGC associates with PDI in COS-7 cells
The in vitro PDI-sGC association was dependent on the redox state of both proteins, where sGC is reduced and PDI is oxidized, suggesting thiol-disulfide exchange. We next sought to determine whether the PDI-sGC interaction would take place in cells, using a Flag-tagged PDI construct (see Materials and Methods). The Flag-tag allows for robust pull-down of a PDI-sGC complex using an anti-Flag affinity resin. Because of the transient nature of a potential thiol-disulfide interaction, we also constructed Flag-tagged PDI-CxxS or PDI “trap-mutant” in which the second, resolving Cys in both redox active sites is replaced with Ser allowing it to “trap” a transient PDI-sGC complex, as in Jessop, et al. [30]. To test the interaction in a cellular context, COS-7 cells were transfected with PDI constructs and infected with sGC adenoviruses in order to provide enough protein for analysis. Neither PDI nor sGC were drastically over-expressed (see lysate blots probed for sGC α and β and Flag compared to β-actin loading control, Figure 3B). The bound eluant was divided into non-reducing and reducing samples and Western blots probed with anti-sGC are shown in Figure 3 A. In lane 1, as expected, no sGC is present (band at ~175 kDa is non-specific, and was present in COS-7 cells not infected with sGC, not shown). In the left panel of Fig. 3 A, lanes 2 & 3 show that both Flag-tagged PDI-WT and PDI-CxxS pulled down sGC in COS-7 lysates, as a complex of about 200 kDa (which could correspond to sGC 150 kDa + PDI 57 kDa) and of higher molecular weight (potential multimers). Addition of reductant (2-mercaptoethanol) to the bound samples resolves the high molecular weight complexes, to the expected sGC α and β subunit size (~ 80 and 75 kDa), right panel Fig. 3 A (the same blots probed with anti-Flag are shown in Supplemental Figure 3). Both Flag-PDI-WT and Flag-PDI-CxxS pulled down sGC. Thus, PDI-GC is not as transient as expected for a mixed disulfide intermediate (potential reasons are addressed in the discussion), it could be that the interaction was due to PDI chaperone activity, rather than a mixed-disulfide. However, this is in contradiction with the data in Fig. 1 and with the fact that both sGC-PDI and sGC-PDI-trap complexes were resolved under reducing conditions (Fig 3A, right panel), confirming that the interaction was due to a reductant-sensitive mixed disulfide in cells.
Figure 3. Redox-based immunoprecipitation of sGC by Flag-tagged PDI-WT/PDI-CxxS.

COS-7 cells were transiently transfected with Flag-tagged PDI-WT or PDI-CxxS and infected with sGC α and β WT adenovirus; immunoprecipitation of lysates was performed using anti-Flag affinity resin as described in Material and Methods. A. Representative Western blots of bound eluant from anti-Flag affinity resin probed with anti-α1 and β1; gels were run under non-reducing (left panel) and -reducing conditions (right panel). B. Lysates probed for sGC α1 and β1, Flag, PDI and β-actin (loading control).
sGC activity is inhibited by wild-type but not thiol mutant PDI in transfected COS-7 cells
By redox Western blot, thiol titer, and Flag-PDI-WT/CxxS sGC pull-down, we established that the interaction between sGC and PDI was redox-mediated in vitro and in cells. To determine if thiol-disulfide exchange between sGC and PDI in a cellular setting modulates sGC activity, COS-7 cells were transiently transfected to determine the effect of PDI on cGMP production. PDI-WT or PDI-SxxS, in which all four Cys in the two redox active CxxC motifs were replaced with Ser, were co-transfected with sGC α and β. sGC activity under basal and NO-stimulated conditions was measured in cell lysates to determine the effect of PDI-WT or PDI-SxxS co-transfection. Basal activity was not significantly affected by PDI-SxxS and only slightly inhibited by PDI-WT, 8 ± 3% (Figure 4A). The difference in sGC inhibition between PDI-WT and PDI-SxxS was not significant (p=0.28). However, under NO-stimulation (100 μM DEA-NO), co-transfection of sGC with PDI-WT led to 41 ± 6% inhibition of sGC activity (Fig 4A). In contrast, the presence of PDI-SxxS led to 13 ± 7% decrease in cGMP production over sGC alone. Under NO-stimulation, the inhibitory effect on sGC of PDI-WT as compared to PDI-SxxS was significant (p<0.005). Additionally, for sGC/PDI-WT the difference of inhibition under basal vs. NO-stimulation was significant (p<0.002), while basal vs. NO-stimulated for sGC/PDI-SxxS was not (p=0.18). In Figure 4B, Western blot indicates similar expression levels for sGC, PDI, and β-actin These results posit that NO-stimulated sGC activity is inhibited by PDI in cells only when the CxxC active sites are functional.
Figure 4. NO-stimulated sGC activity was inhibited by PDI-WT but not PDI-SxxS.

A. COS-7 cells were transiently co-transfected with sGC and PDI WT or PDI-SxxS as described in Material and Methods. Data is represented as percent inhibition of sGC activity in the presence of PDI-WT or PDI-SxxS (four redox active Cys changed to Ser), under basal and NO-stimulated (100 μM DEA-NO) conditions (± SEM; n = 7 from four separate transfections). *: p<0.05 with student’s t-test sGC/PDI-WT vs. sGC/PDI-SxxS. B. A representative Western blot, top panel probed for sGC α1 and β1, bottom panel probed for PDI and β-actin. COS-7 cells do not contain detectable sGC as confirmed in Western blot, right panel.
In situ proximity ligation assay shows close proximity of sGC and PDI
Thus far, the interaction between sGC and PDI was shown to be the result of thiol-disulfide exchange by Western blot, thiol titer and PDI-Flag IP. In addition, PDI inhibited sGC NO-stimulated activity Although PDI is typically found in the ER, other subcellular locations are known [14–18]; and, sGC while “soluble,” is also found in non-cytosolic subcellular locations [10, 31, 32]. In order for the redox interaction to be physiologically relevant, we sought to confirm this interaction in smooth muscle cells (SMC). The sensitive in situ PLA assay [33, 34] was performed in CRL-2018 rat aortic SMC, which contain both endogenous sGC and PDI. In Figure 5, the top left-hand panel shows PDI with sGC α1, the bottom left-hand panel shows PDI with sGC β1. The positive co-localization of sGC and PDI is visualized as red dots (rhodamine) fluorescence. The fluorescence indicates a perinuclear and cytosolic localization when compared with the same cells in brightfield (inset). Negative controls of the two PLA probes alone and anti-PDI with PLA probes are in the right-hand panels, top and bottom, respectively. Notably, both α1 and β1 sGC subunits showed co-localization with PDI. Therefore, PDI most likely undergoes thiol-disulfide exchange with either individual subunits or with the intact heterodimer (i.e. sGC active form). The signal intensity in the PDI/sGC β1 panel is higher than PDI/sGC α1; thus we speculate that the β1 subunit contains the sGC Cys that mediate thiol-disulfide exchange with PDI.
Figure 5. Duolink PLA assay shows co-localization of sGC and PDI in smooth muscle cells.

CRL-2018 smooth muscle cells containing endogenous sGC and PDI were probed with the Duolink PLA assay as described under Materials and Methods. Top left panel shows positive staining of PDI with sGC α1. Bottom left panel shows positive staining of PDI with sGC β1. Top right panel is the negative control of PLA probes with no primary antibody. Bottom right panel is the negative control of PDI primary antibody with both PLA probes. Nuclei are stained with DAPI; brightfield images shown in inset.
DISCUSSION
NO signaling is a key modulator of cardiovascular homeostasis. NO stimulates sGC several hundred fold to produce cGMP, which in turn relaxes smooth muscle cells, inhibits platelet aggregation, and protects from cardiac hypertrophy. Considering that dysfunction in the NO-sGC-cGMP pathway contributes to development of atherosclerosis, hypertension and cardiac hypertrophy, understanding the molecular mechanisms of sGC modulation is crucial.
An earlier study uncovered Hsp70 as a sGC-interacting protein as part of a purified sGC-activating fraction [5]. Other sGC-interacting candidates were identified by mass spectrometry analysis including PDI, a member of the thiol oxidoreductase family. PDI has several cellular roles, the most well known as a molecular chaperone in oxidative protein folding in the ER. Yet, PDI is also found at other cellular locations and under certain conditions, including a recently described thiol-dependent interaction with the subunit p47 of NADPH oxidase in the cytosol, ([14, 17]; reviewed in [16, 18]). Of note, sGC, which is described as a “soluble” protein, has subcellular locations other than the cytosol including the plasma membrane and nucleus [10, 31, 32]. Interestingly, mammalian form sGC α1β1 has a high number of conserved Cys (2.8% total amino acid content). This is an unexpectedly high number for a cytosolic/soluble enzyme (typical intracellular proteins contain ~ 1.4% Cys [35]). In fact, several lines of evidence now suggest that sGC Cys are post-translationally modified and these modifications whether they are thiol oxidation [36], disulfide bond formation [21] or S-nitrosation [23] affect sGC activity Thus, we hypothesize that the oxidoreductase PDI would not only associate with sGC but affect sGC Cys, including the possible conversion between monomeric and dimeric forms via disulfide bond formation or reduction, thereby impacting sGC activity. Following confirmation that sGC and PDI interact using an sGC affinity matrix (Supplemental Fig 1), we first focused on determining the mode of interaction between these two proteins. Redox experiments looked at the change in SDS-PAGE migration patterns under non-reducing and reducing conditions. These experiments showed that adding oxidized PDI to reduced sGC results in distinct changes in the migration of sGC. Addition of PDI to denatured sGC showed no difference from denatured sGC alone, indicating that PDI does not bind denatured sGC, i.e. does not act as a molecular chaperone. Furthermore, NEM thiol-blocked PDI also had no effect on reduced or oxidized sGC migration pattern; confirming that the interaction is dependent on the availability of PDI Cys. Non-reducing SDS-PAGE gels probed with PDI (see supplemental information) showed no significant change in PDI migration pattern across all samples. Together, the redox Western blots show that in vitro, the sGC/PDI interaction is dependent on the redox state of the two enzymes.
For this study, we also found that the addition of PDI to reduced sGC resulted in an approximate two-fold increase in overall thiol titer. It is not possible from this approach to determine which protein cysteines are reduced. Nonetheless, the large increase in thiol content cannot be attributed solely to the known amount of PDI present; sGC free thiols must increase to obtain an overall thiol concentration of ~ 4 μM. This is further evidence that not only do sGC and PDI interact in vitro but they do so in a thiol-dependent manner, i.e. via thiol-disulfide exchange.
Both Flag-tagged PDI-WT and PDI-CxxS were able to associate with sGC in COS-7 cells. The high molecular weight complex seen under non-reducing conditions is resolved by reduction (Fig. 3B), similar to the in vitro experiment (Fig. 1). There appears to be no difference in the amount of sGC in the complex formed or fully reduced, when comparing the results of Flag IP between PDI-WT and thiol-trap mutant PDI-CxxS. Replacing the second, resolving Cys in both CxxC active sites of PDI with Ser creates a thiol-trapping mutant, PDI-CxxS [30], which should not be able to resolve an sGC-PDI-CxxS mixed disulfide as expected (Fig. 3A). However, a longer-lived intermediate is also seen in sGC-PDI-WT; a typical PDI-protein mixed disulfide is expected to be transient and thus not detected as a complex (Fig. 3A). This is intriguing and has several possible explanations. The first is that PDI forms an intramolecular sGC disulfide and the two remain complexed through a secondary interaction, perhaps with the hydrophobic region of PDI. However, this is unlikely as the complex is resolved by reductant and denatured sGC does not complex with PDI (Fig. 1). A second explanation is that the mixed disulfide between PDI and sGC does not involve the resolving Cys in the PDI redox active site; hence, no difference seen by Western blot between sGC-PDI WT and sGC-PDI CxxS. The formation of this sGC-PDI mixed disulfide, between the first Cys of the PDI redox active site and a reactive accessible sGC Cys, may result in a conformational change in sGC that moves the newly formed disulfide out of reach of the second resolving Cys in the PDI active site. The third explanation is that, given the high sGC Cys content, the Cys resolving the mixed disulfide instead depends on a nearby reactive, solvent exposed sGC Cys. This Cys would not be as efficient as that of a thiol oxidoreductase, which would account for the long-lived intermediate seen under non-reducing conditions (Fig. 3A, left panel) but that can be resolved (reducing, Fig. 3A, right panel). The presence of a reactive, accessible sGC Cys is supported by redox Western blot (Fig. 1), only reduced and oxidized PDI show an interaction.
Using PDI WT and a PDI thiol mutant, in which the four redox active Cys of the two CxxC active site motifs were replaced with Ser (PDI-SxxS), we were able to determine the effect of the sGC-PDI interaction on sGC activity. There was no significant effect on the basal level of sGC activity when co-transfected with PDI-SxxS or PDI-WT. However, when lysates were stimulated with DEA-NO, PDI-WT effected a 41% inhibition of sGC activity over sGC alone; while PDI SxxS showed no significant inhibition over sGC alone (COS-7 cells have endogenous PDI and this contributes to the small inhibition seen). Thus, PDI can inhibit NO-stimulated sGC activity in cells, probably via thiol-disulfide exchange. The in vitro data thus far, confirmed that sGC and PDI could interact in cells, albeit through exogenous expression of PDI and sGC (Fig. 3 and 4). A relatively novel method, in situ PLA, confirmed that the interaction between sGC and PDI is biologically relevant in CRL-2018 SMC that contain endogenous sGC and PDI. Notably, both α1 and β1 sGC subunits showed potential co-localization with PDI. Indicating that PDI most likely undergoes thiol-disulfide exchange with either individual subunits or with the intact heterodimer (i.e. sGC active form). Of note, the signal intensity in the PDI-sGC β1 panel is somewhat higher than PDI-sGC α1; thus we speculate that the β subunit contains the sGC Cys that mediate thiol-disulfide exchange with PDI.
The effects of PDI on sGC activity underscore the importance of determining the role of cysteines in the molecular mechanism of modulation of this crucial cardiovascular enzyme. Analysis using mass spectrometry methods is currently underway to determine which sGC Cys are involved in this interaction. While this work focused on aspects of the interaction between sGC and PDI, further work will be required to determine the physiological relevance of this interaction, in particular the inhibitory, thiol-dependent effect of PDI on NO-stimulated sGC activity.
Supplementary Material
Acknowledgments
We would like to thank Drs. Nataliya Balashova and Nazish Sayed for their initial contribution and two anonymous reviewers for experimental suggestions.
FUNDING SOURCE:
This work was supported by the National Institutes of Health GM067640 and the F.M. Kirby foundation.
Abbreviations used
- AGAP-1
ADP-ribosylation factor GTPase activating protein 1
- CCTη
chaperonin containing t-complex polypeptide
- DEA-NO
diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- HNOX domain
heme nitric oxide/oxygen binding domain
- Hsp70
heat shock protein 70
- Hsp90
heat shock protein 90
- LGN
Leu-Gly-Asn repeat-enriched protein
- NEM
n-ethyl maleimide
- NO
nitric oxide
- PAS domain
PER-ARNT-SIM family domain
- PDI
protein disulfide isomerase
- PLA
proximity ligation assay
- PSD95
post-synaptic density protein 95
- sGC
soluble guanylyl cyclase
- TEA
triethanolamine
- TCEP
tris(2-carboxyethyl)phosphine
- WT
wild type
References
- 1.Gibb BJ, Garthwaite J. Subunits of the nitric oxide receptor, soluble guanylyl cyclase, expressed in rat brain. Euro J Neurosci. 2001;13:539. doi: 10.1046/j.1460-9568.2001.01421.x. [DOI] [PubMed] [Google Scholar]
- 2.Russwurm M, Behrends S, Harteneck C, Koesling D. Functional properties of a naturally occurring isoform of soluble guanylyl cyclase. Biochem J. 1998;335:125–130. doi: 10.1042/bj3350125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winger JA, Marletta MA. Expression and characterization of the catalytic domains of soluble guanylate cyclase: interaction with the heme domain. Biochemistry. 2005;44:4083. doi: 10.1021/bi047601d. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto S, Kobayashi A. Clinical pharmacokinetics and pharmacodynamics of glyceryl trinitrate and its metabolites. Clin Pharmacokin. 2003;42:205–221. doi: 10.2165/00003088-200342030-00001. [DOI] [PubMed] [Google Scholar]
- 5.Balashova N, Chang FJ, Lamothe M, Sun Q, Beuve A. Characterization of a novel type of endogenous activator of soluble guanylyl cyclase. J Biol Chem. 2005;280:2186–2196. doi: 10.1074/jbc.M411545200. [DOI] [PubMed] [Google Scholar]
- 6.Nedvetsky PI, Meurer S, Opitz N, Nedvetskaya TY, Müller H, Schmidt HW. Heat shock protein 90 regulates stabilization rather than activation of soluble guanylate cyclase. FEBS Letters. 2008;582:327–331. doi: 10.1016/j.febslet.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 7.Papapetropoulos A, Zhou Z, Gerassimou C, Yetik G, Venema RC, Roussos C, Sessa WC, Catravas JD. Interaction between the 90-kDa heat shock protein and soluble guanylyl cyclase: physiological significance and mapping of the domains mediating binding. Mol Pharmacol. 2005;68:1133–1141. doi: 10.1124/mol.105.012682. [DOI] [PubMed] [Google Scholar]
- 8.Yetik-Anacak G, Xia T, Dimitropoulou C, Venema RC, Catravas JD. Effects of hsp90 binding inhibitors on sGC-mediated vascular relaxation. Am J Physiol - Heart and Circ Physiol. 2006;291:H260–H268. doi: 10.1152/ajpheart.01027.2005. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh A, Stuehr DJ. Soluble guanylyl cyclase requires heat shock protein 90 for heme insertion during maturation of the NO-active enzyme. Proc Natl Acad of Sci. 2012;109:12998–13003. doi: 10.1073/pnas.1205854109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russwurm M, Wittau N, Koesling D. Guanylyl cyclase/PSD-95 interaction: targeting of the nitric oxide-sensitive alpha2/beta1 guanylyl cyclase to synaptic membranes. J Biol Chem. 2001;276:44647–44652. doi: 10.1074/jbc.M105587200. [DOI] [PubMed] [Google Scholar]
- 11.Hanafy KA, Martin E, Murad F. CCTη, a novel soluble guanylyl cyclase-interacting protein. J Biol Chem. 2004;279:46946–46953. doi: 10.1074/jbc.M404134200. [DOI] [PubMed] [Google Scholar]
- 12.Meurer S, Pioch S, Wagner K, Müller-Esterl W, Gross S. AGAP1, a novel binding partner of nitric oxide-sensitive guanylyl cyclase. J Biol Chem. 2004;279:49346–49354. doi: 10.1074/jbc.M410565200. [DOI] [PubMed] [Google Scholar]
- 13.Chauhan S, Jelen F, Sharina I, Martin E. The G-protein regulator LGN modulates the activity of the NO receptor soluble guanylate cyclase. Biochem J. 2012;446:445–453. doi: 10.1042/BJ20111882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koivu J, Myllylä R, Helaakoski T, Pihlajaniemi T, Tasanen K, Kivirikko KI. A single polypeptide acts both as the beta subunit of prolyl 4-hydroxylase and as a protein disulfide-isomerase. J Biol Chem. 1987;262:6447–6449. [PubMed] [Google Scholar]
- 15.Wetterau JR, Combs KA, McLean LR, Spinner SN, Aggerbeck LP. Protein disulfide isomerase appears necessary to maintain the catalytically active structure of the microsomal triglyceride transfer protein. Biochem. 1991;30:9728–9735. doi: 10.1021/bi00104a023. [DOI] [PubMed] [Google Scholar]
- 16.Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: Unpredicted non-ER locations and functions. J Cell Physiol. 2002;193:154–163. doi: 10.1002/jcp.10172. [DOI] [PubMed] [Google Scholar]
- 17.de A, Paes AM, Veríssimo-Filho S, Guimarães LL, Silva ACB, Takiuti JT, Santos CXC, Janiszewski M, Laurindo FRM, Lopes LR. Protein disulfide isomerase redox-dependent association with p47phox: evidence for an organizer role in leukocyte NADPH oxidase activation. J Leukocyte Biol. 2011;90:799–810. doi: 10.1189/jlb.0610324. [DOI] [PubMed] [Google Scholar]
- 18.Laurindo FRM, Pescatore LA, de Castro Fernandes D. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Rad Biol Med. 2012;52:1954–1969. doi: 10.1016/j.freeradbiomed.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 19.Friebe A, Wedel B, Harteneck C, Foerster J, Schultz G, Koesling D. Functions of conserved cysteines of soluble guanylyl cyclase. Biochem. 1997;36:1194–1198. doi: 10.1021/bi962047w. [DOI] [PubMed] [Google Scholar]
- 20.Mingone CJ, Gupte SA, Ali N, Oeckler RA, Wolin MS. Thiol oxidation inhibits nitric oxide-mediated pulmonary artery relaxation and guanylate cyclase stimulation. Am J Physiol - Lung Cell Mol Physiol. 2006;290:L549–557. doi: 10.1152/ajplung.00331.2005. [DOI] [PubMed] [Google Scholar]
- 21.Zheng X, Ying L, Liu J, Dou D, He Q, Leung SWS, Man RYK, Vanhoutte PM, Gao Y. Role of sulfhydryl-dependent dimerization of soluble guanylyl cyclase in relaxation of porcine coronary artery to nitric oxide. Cardiovasc Res. 2011;90:565–572. doi: 10.1093/cvr/cvr016. [DOI] [PubMed] [Google Scholar]
- 22.Kamisaki Y, Waldman SA, Murad F. The involvement of catalytic site thiol groups in the activation of soluble guanylate cyclase by sodium nitroprusside. Arch Biochem Biophys. 1986;251:709–714. doi: 10.1016/0003-9861(86)90380-2. [DOI] [PubMed] [Google Scholar]
- 23.Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Pro Natl Aca Sci. 2007;104:12312–12317. doi: 10.1073/pnas.0703944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamothe M, Chang FJ, Balashova N, Shirokov R, Beuve A. Functional characterization of nitric oxide and YC-1 activation of soluble guanylyl cyclase: structural implication for the YC-1 binding site? Biochem. 2004;43:3039. doi: 10.1021/bi0360051. [DOI] [PubMed] [Google Scholar]
- 25.Rancy PC, Thorpe C. Oxidative protein folding in vitro: a study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochem. 2008;47:12047–12056. doi: 10.1021/bi801604x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gasteiger E, HC, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. Humana Press; 2005. [DOI] [PubMed] [Google Scholar]
- 27.Domino SE, Tubb DJ, Garbers DL. Assay of guanylyl cyclase catalytic activity. Methods Enzymol. 1991;195:345–355. doi: 10.1016/0076-6879(91)95179-n. [DOI] [PubMed] [Google Scholar]
- 28.Cline DJ, Redding SE, Brohawn SG, Psathas JN, Schneider JP, Thorpe C. New water-soluble phosphines as reductants of peptide and protein disulfide bonds: reactivity and membrane permeability. Biochem. 2004;43:15195–15203. doi: 10.1021/bi048329a. [DOI] [PubMed] [Google Scholar]
- 29.Quan H, Fan G, Wang C-c. Independence of the chaperone activity of protein disulfide isomerase from its thioredoxin-like active site. J Biol Chem. 1995;270:17078–17080. doi: 10.1074/jbc.270.29.17078. [DOI] [PubMed] [Google Scholar]
- 30.Jessop CE, Watkins RH, Simmons JJ, Tasab M, Bulleid NJ. Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J Cell Sci. 2009;122:4287–4295. doi: 10.1242/jcs.059154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pifarre P, Baltrons MA, Foldi I, Garcia A. NO-sensitive guanylyl cyclase bete1 subunit is peripherally associated to chromosomes during mitosis: novel role in chromatin condensation and cell cycle progression. Int J Biochem Cell Biol. 2009;41:1719–1730. doi: 10.1016/j.biocel.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 32.Zabel U, Kleinschnitz C, Oh P, Nedvetsky P, Smolenski A, Muller H, Kronich P, Kugler P, Walter U, Schnitzer JE, Schmidt HHHW. Calcium-dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nat Cell Biol. 2002;4:307. doi: 10.1038/ncb775. [DOI] [PubMed] [Google Scholar]
- 33.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Meth. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 34.Weibrecht I, Leuchowius KJ, Clausson CM, Conze T, Jarvius M, Howell WM, Kamali-Moghaddam M, Söderberg O. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev Proteomics. 2010;7:401–409. doi: 10.1586/epr.10.10. [DOI] [PubMed] [Google Scholar]
- 35.Fahey RC, Hunt JS, Windham GC. On the cysteine and cystine content of proteins: differences between intracellular and extracellular proteins. J Mol Evol. 1977;10:155–160. doi: 10.1007/BF01751808. [DOI] [PubMed] [Google Scholar]
- 36.Maron BA, Zhang YY, Handy DE, Beuve A, Tang SS, Loscalzo J, Leopold JA. Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinyl thiol oxidation in vascular smooth muscle cells. J Biol Chem. 2009;284:7665–7672. doi: 10.1074/jbc.M809460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.