

Abstract
The biology of Escherichia coli in its primary niche, the animal intestinal tract, is remarkably unexplored. Studies with the streptomycin-treated mouse model have produced important insights into the metabolic requirements for Escherichia coli to colonize mice. However, we still know relatively little about the physiology of this bacterium growing in the complex environment of an intestine that is permissive for the growth of competing flora. We have developed a system for studying colonization using an E. coli strain, MP1, isolated from a mouse. MP1 is genetically tractable and does not require continuous antibiotic treatment for stable colonization. As an application of this system, we separately knocked out each two-component system response regulator in MP1 and performed competitions against the wild-type strain. We found that only three response regulators, ArcA, CpxR, and RcsB, produce strong colonization defects, suggesting that in addition to anaerobiosis, adaptation to cell envelope stress is a critical requirement for E. coli colonization of the mouse intestine. We also show that the response regulator OmpR, which had previously been hypothesized to be important for adaptation between in vivo and ex vivo environments, is not required for MP1 colonization due to the presence of a third major porin.
INTRODUCTION
Escherichia coli is one of the most extensively studied and best-characterized organisms. Its high growth rate, facile genetics, and simple nutritional requirements have made this bacterium an excellent model system for studying basic aspects of molecular biology and bacteriology and the primary host for DNA and protein engineering. The physiology of E. coli growth and survival under diverse conditions has been intensively studied, and a significant fraction of E. coli gene products and regulatory networks have been characterized. However, for such a well-studied organism, we know remarkably little about the biology of E. coli in its primary niche: the animal gastrointestinal tract.
E. coli is generally the most abundant aerobe in the intestines of warm-blooded vertebrates, although its numbers vary considerably with animal host and geography (1–3). As a species, this bacterium has a remarkable genetic diversity; the number of genes in common among fully sequenced isolates is less than half the number of genes in any individual strain (4–6). Some E. coli strains are pathogenic, depending on the host and site of infection (3, 7–9), and have been intensively studied to understand the factors controlling their virulence. However, the majority of E. coli strains associated with animals are believed to be part of the normal flora of the intestine, growing asymptomatically as commensals.
Most of our knowledge about E. coli colonization of the animal intestine comes from studies with streptomycin-resistant strains colonizing mice fed streptomycin continuously in their drinking water (10, 11). This streptomycin-treated mouse model has played a key role in the characterization of the growth of E. coli in the intestine and the identification of nutritional and metabolic requirements for colonization (10, 12–16). The model has been particularly effective because it not only overcomes colonization resistance—the barrier to establishing an infection in an animal whose microbial flora is unperturbed—but also enables colonization with strains that would otherwise be unable to compete with bacteria that are well adapted to the host (10, 11, 17). Streptomycin eliminates a significant portion of the microbial diversity in the mouse intestine (17) and enables E. coli to greatly expand the niches that it occupies. Indeed, the number of E. coli CFU in mouse feces in the streptomycin-treated mouse is 3 to 4 orders of magnitude higher than the number typically found in untreated mice (17). Thus, while this model system remains an invaluable tool for studying colonization, continuous streptomycin treatment may eliminate stresses or other environmental factors that emerge from increased competition with other bacteria or from attendant host responses.
We have developed a system for stable E. coli colonization of mice that does not require continuous antibiotic treatment, enabling the study of colonization without preventing growth of competing bacteria from the environment. The system is based on a genetically manipulable E. coli strain (designated MP1) that we isolated from a mouse. Streptomycin pretreatment is still necessary for MP1 to gain an initial hold in the intestine, but no subsequent treatment with antibiotics is needed for stable colonization.
We used this system to test the requirements for individual two-component signaling systems in intestinal colonization. Of the 32 histidine kinase-response regulator pairs identified in the MP1 genome, we found that only three (Cpx, Rcs, and Arc) are essential for colonization in competitions out to at least 20 days. Other two-component systems, many of which have been identified as important for infection by pathogenic E. coli or related bacteria, did not show strong colonization defects. We further demonstrate that MP1 expresses a third major porin, in addition to the classical porins OmpF and OmpC, which renders the EnvZ/OmpR two-component system nonessential for colonization. This work is the first systematic study of the role of two-component signaling in E. coli colonization of the intestine and introduces a valuable resource for studies of E. coli physiology and colonization as a commensal.
MATERIALS AND METHODS
E. coli HS was obtained from James Nataro, and Nissle 1917 was obtained from Dean Hamer.
Isolation of MP1.
Fecal pellets from CD-1 mice (Charles River Laboratories) were resuspended in 10 mM MgSO4, and dilutions were spread on MacConkey lactose agar plates (BD-Difco). Red colonies were picked and streaked on LB agar plates containing 40 μg/ml X-Gluc (5-bromo-4-chloro-3-indolyl-β-d- glucuronic acid) to identify cells expressing beta-glucuronidase. Dark blue colonies were picked and tested in a spot titer assay with the phage P1vir to screen for E. coli strains that are susceptible to P1 (18). Two of six colonies tested produced plaques. One of these was selected and named MP1.
Genome sequencing and analysis.
The MP1 genome was sequenced by 454 pyrosequencing at the University of Florida ICBR Genomic Core facility. De novo assembly was performed with Newbler and produced 84 contigs with approximately 20× coverage. The phylogenetic group of MP1 was derived by the reconstruction of a maximum-likelihood tree from concatenated sequences of complete coding regions of 7 housekeeping genes frequently used for multilocus sequence typing (MLST) analysis of E. coli: adk, fumC, gyrB, icd, mdh, purA, and recA. The sequence type (ST) of each strain was based on the internal fragments of these 7 genes, as described in the MLST online database (http://mlst.ucc.ie/mlst/dbs/Ecoli) (19).
Construction of marked strains.
The plasmid pAS07 (A. Siryaporn and M. Goulian, unpublished data) consists of the integration vector pCAH63 (20) with the uidA gene replaced with a sequence consisting of tetR and an operon fusion of tetA with gfpmut3.1 (Clontech). The tetR tetA cassette was isolated by PCR from the transposon Tn10 in the E. coli strain XL1-Blue (Stratagene). pML8 is pAS07 with mcherry (taken from pRSETb-mCherry [21]) in place of gfpmut3.1. The plasmids pAS07 and pML8 were integrated into the MP1 genome at the phage lambda attachment site and verified to be present in single copy by PCR as described in reference 20, resulting in MP13 and MP7, respectively. Both pAS07 and pML8 carry the genes cat and tetA, conferring resistance to chloramphenicol and tetracycline. However, for reasons that we do not understand, the cat gene does not provide high-level resistance to chloramphenicol in MP1.
Construction of deletion strains.
Deletions were constructed by recombineering essentially as described in reference 22, except that electrocompetent cells were prepared by washing with an ice cold solution containing 20% glycerol and 1 mM unbuffered 3-(N-morpholino)propanesulfonic acid (MOPS) (23). For those response regulator genes in MP1 with at least 50 bp of flanking sequence identical to the sequence in E. coli K-12, we used the corresponding response regulator deletion from the Keio collection (24) as the template for the PCR. This made it possible in some cases to use long regions of homology on one or both sides of the response regulator gene without exceptionally long primers or extra PCR steps. In the few cases where the flanking sequence in MP1 was significantly different from the sequence in K-12 for at least one side of the gene, as well as for the genes that were not present in K-12, PCR primers with 40 to 50 bases of homology to the MP1 genome sequence were used with the template pKD13 (22). For these cases, gene knockouts were constructed in the same manner as for the Keio collection—using the same segment of pKD13 for the insertion and replacing all but the start codon and the last six codons of the targeted gene (24). Deletions were constructed in MP1 with selection on 35 μg/ml kanamycin, verified by PCR, and then moved into other strain backgrounds (e.g., MP13) by transduction with P1vir. In cases where kanamycin sensitivity was required to move in a second deletion, the kanamycin resistance gene was removed with Flp recombinase by transformation with pCP20 as described in reference 22.
Construction of complementation strains.
To complement the arcA, rcsB, and cpxR deletions, the corresponding genes with their promoters were isolated from MP1 genomic DNA by PCR with primers that added flanking BamHI and SacI restriction sites upstream and downstream of the gene, respectively. Following digestion with BamHI and SacI, the DNA segments were ligated into the (similarly digested) chromosomal integration plasmid pAH70 (20). The kanamycin resistance gene in the corresponding recipient strain (ΔarcA::kan, ΔrcsB::kan, or ΔcpxR::kan) was removed with pCP20 (22), and the complementing gene was inserted at the HK022 attachment site by transient expression of the phage integrase from plasmid pAH69 and verified to be in single copy as described in reference 20.
Inoculation of mice.
All animal studies were carried out in accordance with animal protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Five-week-old CD-1 mice (Charles River Laboratories) were fed streptomycin and glucose in their drinking water, both at a concentration of 5 mg/ml, for 72 h. The drinking water was then replaced with water without antibiotic and glucose, and the mice were not given any further antibiotic treatment. After an additional 24 h, the mice were orally inoculated by gavage with ∼109 cells in 100 μl phosphate-buffered saline (PBS), resuspended from an overnight culture as described below. For each strain, a single colony was picked from an LB agar plate containing kanamycin (35 μg/ml) and resuspended in 1 ml of LB (LB Miller medium; BD Difco). Eight microliters of this suspension was inoculated into 8 ml of LB and grown overnight (∼16 h) at 37°C on a roller drum at ∼40 rpm. A volume of this culture equal to 4.8 ml per unit of optical density at 600 nm (OD600) of a 1:10 dilution of the overnight culture was spun down at 3,800 × g and 4°C, resuspended in 2 ml of ice-cold PBS, spun down again as described above, and resuspended in a volume of ice-cold PBS equal to 1/50 of the culture volume used for the first spin. The mutant and wild-type cell suspensions were mixed 1:1, and mice were orally inoculated with 100 μl. A portion of the remaining cell suspension was then serially diluted and plated on LB agar containing 15 μg/ml tetracycline to determine the input CFU. Mice were raised on standard laboratory rodent diet (LabDiet 5001).
Determination of E. coli CFU.
Fresh feces were weighed and resuspended as a slurry in PBS, and serial dilutions were plated on LB containing 15 μg/ml tetracycline. Fluorescence images of plates were obtained using a homemade system as described previously (25). The competitive index (CI) was determined as [(GFP fluorescent CFU)/(mCherry fluorescent CFU)]/[(input GFP fluorescent CFU)/(input mCherry fluorescent CFU)], where the input CFU was determined from the inoculum.
Analysis of outer membrane proteins.
Outer membranes were isolated using a modification of the Sarkosyl extraction protocol described in reference 26. Cells from 1 ml of an overnight culture grown in LB at 37°C were resuspended in 1 ml 10 mM HEPES (pH 7.4). The cells were then lysed by sonication and centrifuged at 6,000 × g for 4 min. The supernatant was then centrifuged at 50,000 × g for 1 h at 4°C. The pellet was resuspended in 400 μl 1% (wt/vol) N-lauroylsarcosine (Sarkosyl) in 10 mM HEPES (pH 7.4), incubated at 37°C for 30 min with shaking, and then centrifuged at 50,000 × g for 1 h at 4°C. The pellet was resuspended in 20 μl 10 mM HEPES (pH 7.4). Outer membranes were analyzed by SDS-PAGE with gels containing 6 M urea.
Nucleotide sequence accession numbers.
The whole-genome shotgun project for E. coli MP1 has been deposited at DDBJ/EMBL/GenBank under accession no. JEMI00000000. The version described in this paper is version JEMI01000000. The MP1 genome sequencing project is registered at NCBI under Bioproject ID PRJNA196008.
RESULTS
Isolation and characterization of MP1.
We initially attempted to colonize mice with two human commensal E. coli strains, Nissle 1917 and HS (4, 27–29), without continuous selection with an antibiotic. However, in both cases, we found that colonization was often relatively short-lived and showed significant mouse-to-mouse variability (data not shown). Furthermore, while we could move selectable markers from E. coli K-12 into Nissle 1917 and HS by transduction with bacteriophage P1vir, we were unable to produce transducing particles, or infectious P1 virions, from either of these strains. Therefore, to find E. coli strains that are robust colonizers of the mouse gastrointestinal (GI) tract and are also convenient to manipulate genetically, we isolated E. coli from the feces of healthy mice and screened for strains that produce plaques with bacteriophage P1vir in a soft-agar assay. After testing two isolates for their colonization potential, we settled on one strain, which we named MP1.
To follow colonization, we constructed fluorescently marked derivatives of MP1 using a tightly regulated expression system based on the tetracycline repressor and resistance genes tetR and tetA from the transposon Tn10. Operon fusions tetA-mcherry or tetA-gfp, together with tetR, were integrated at the lambda phage attachment site. (MP1 is λ− and has the attBλ sequence found in E. coli K-12.) In the absence of tetracycline, mCherry and GFP expression are tightly off. In the presence of tetracycline, however, fluorescence is easily detectable in colonies growing on agar plates (Fig. 1C) (30).
FIG 1.

Colonization of mice with E. coli strain MP1. (A) Schematic of the colonization protocol. Mice were fed streptomycin (Strep.) in their drinking water for 72 h. The water with streptomycin was then removed and replaced with antibiotic-free water, and there was no further antibiotic treatment. After an additional 24 h, mice were orally inoculated with approximately 109 CFU of E. coli cells by gavage. (B) Circles show data for 16 mice colonized with MP7, an mcherry-marked MP1 derivative (MP1 attλ::pML8). CFU were measured on the indicated days following inoculation. Triangles show measurements of total E. coli (Lac+ colonies on MacConkey plates) for three mice that were not treated with streptomycin and were not exposed to MP1. (C) Colonies of MP7 and MP13, a gfp-marked MP1 derivative (MP1 attλ::pAS07). The image is an overlay of red and green fluorescence images of colonies growing on LB agar containing 15 μg/ml tetracycline.
To overcome colonization resistance, mice were pretreated with streptomycin in their drinking water (5 mg/ml) for 72 h but received no additional streptomycin following this treatment. After an additional 24 h to allow clearance of antibiotic, mice were orally inoculated (intragastrically) with ∼109 CFU of E. coli cells (Fig. 1A). With this protocol, we found that MP1 colonizes mice to at least 71 days—the longest interval that we tested—with a colonization level in the range of 105 to 106 CFU per gram of feces (Fig. 1B). We also verified that MP1 is significantly better than Nissle or HS at colonizing mice (Fig. 2). In competition experiments with MP1, Nissle was below the detection limit by 6 days postinfection. HS was a better colonizer than Nissle but was below the detection limit in 3 of 4 mice by 14 days. We note that the colonization levels measured for MP1 (105 to 106 CFU/gram feces) are comparable to the levels we measured for E. coli in the normal flora of untreated mice (Fig. 1) and are consistent with previous reports (for example, see reference 17).
FIG 2.

Mouse colonization competitions between MP1 and HS or Nissle. Each symbol represents CFU measurements for a single mouse, taken at the indicated days postinoculation. The dashed lines indicate the detection limit. The strains used for colonization were MP7 (MP1 attλ::pML8), HS attλ::pAS07, and Nissle attλ::pAS07.
MP1 genome.
We assembled 84 contigs from 454 pyrosequencing of the MP1 genome. Multilocus sequence typing (MLST) (19) assigns MP1 to the sequence type ST491. At present, two other strains in the E. coli MLST database (http://mlst.ucc.ie/mlst/dbs/Ecoli) belong to ST491, and both are human isolates: ROAR372, a B2 commensal isolated in France (31), and G110b, a Lac− enteroaggregative E. coli (EAEC) strain isolated in Nigeria (Iruka Okeke, personal communication). A phylogenetic tree for 67 genomes based on the sequences of the 7 housekeeping genes used for MLST places MP1 squarely within the B2 phylogenetic group and closest to the aggregative and invasive E. coli (AIEC) strains LF82 (32) and NRG 857C (33), the urinary pathogenic E. coli (UPEC) strains CFT073 (34) and D i14 and D i2 (35), and the human commensal strain ED1a (5) (Fig. 3).
FIG 3.

MLST-based tree of 67 E. coli strains. The gray boxes denote each phylogenetic group, and the ST of each strain is indicated.
The B2 phylogenetic group has a high representation of extraintestinal pathogenic E. coli (ExPEC). More generally, the genomes of B2 strains, whether or not they are ExPEC, often encode significant numbers of ExPEC virulence factors (36, 37). MP1 is no exception; its genome encodes many proteins that are potentially associated with extraintestinal virulence, especially adhesins and iron acquisition systems (Table 1).
TABLE 1.
List of potential ExPEC-associated virulence factors and their presence or absence in MP1a
| Factor type or function | Gene | Resultb |
|---|---|---|
| Fimbriae/adhesins | fimH | + |
| fmlA (c1936) | + | |
| c2395 | + | |
| yadN | + | |
| aufA | + | |
| ygiL | + | |
| yfcV | + | |
| pixC | + | |
| ppdD | + | |
| yehA | + | |
| csgA | + | |
| sfaS | − | |
| papG | − | |
| focH | − | |
| draC | − | |
| iha | − | |
| Toxins | picU | ±c |
| hlyA | − | |
| cnf1 | − | |
| cdtA | − | |
| sat | − | |
| astA (EAST1) | − | |
| Iron acquisition | irp1 | + |
| fyuA | + | |
| chuA | + | |
| entF | + | |
| iutA | − | |
| iucA | − | |
| iroN | − | |
| ireA | − | |
| cvaC | − | |
| Protectins | kpsMT group III | − |
| kpsMT group II | − | |
| traT | − | |
| ibeA | − | |
| Miscellaneous | ompT | + |
| iss | + | |
| aslA | + | |
| usp | − |
+, the gene is present and the coding sequence is intact; −, the gene is absent; ±, the gene is present but not functional.
Based on the current MP1 genome sequence, the picU gene has a frameshift caused by a single base deletion.
MP1 also harbors an 8.5-kb plasmid that carries the colicin gene colY (38). Consistent with colicin production, MP1 colonies produced zones of clearing in a soft-agar overlay with E. coli K-12 strain MG1655, and this bactericidal activity was lost when MP1 was cured of the plasmid (data not shown).
Two-component system knockouts.
We applied this new model system to study the effects of disrupting individual two-component signaling response regulators on MP1 colonization of mice. The E. coli K-12 strain MG1655 has 30 histidine kinase-response regulator pairs that have been shown or predicted to function as two-component signaling systems (39–41). From the MP1 genome sequence, we identified 32 two-component systems (31 histidine kinase-response regulator pairs plus the CheA/CheB/CheR chemotaxis proteins), 30 of which are shared with MG1655. Of the two that are not found in MG1655, one is the KguS/KguR two-component system, which was recently identified as a regulator of α-ketoglutarate utilization in E. coli CFT073 and other UPEC strains (42), and the other is orthologous to the PgtB/PgtA system from Salmonella, which is associated with regulation of phosphoglycerate transport (43, 44).
Response regulator gene deletions were constructed by recombineering and moved into an MP1 derivative marked with gfp (MP13) by P1 transduction. Competitions were performed against an MP1 derivative marked with mcherry (MP7). Competitive indices determined from mouse feces after at least 20 days following inoculation are shown in Fig. 4. Of the 32 deletions, three (arcA, cpxR, and rcsB) resulted in severe colonization defects, with competitive indices that were below our detection limit of 10−4 for all or virtually all of the mice. To verify that the observed colonization defects were due to the absence of the targeted response regulators, we tested whether integration of the genes at an ectopic site complemented the deletions. The arcA, cpxR, and rcsB genes with their native promoters were integrated at the bacteriophage HK022 attachment site in the chromosome in the respective deletion strains. Integration at this site, which is between torS and torT, may disrupt TorS/TorR signal transduction. However, a torR deletion did not show a detectable colonization defect (Fig. 4). For all three cases, restoration of the response regulator gene complemented the deletion, indicating that the observed colonization defects from the deletions are due to loss of the ArcA, CpxR, and RcsB response regulators (Fig. 5).
FIG 4.

Mouse colonization competitions between response regulator deletion strains and wild type. The competitive index (CI) is shown for competitions with the indicated response regulator deletion. Response regulator deletions were in the strain MP13 (MP1 attλ::pAS07) and competed against the marked wild-type strain MP7 (MP1 attλ::pML8). Each symbol represents measurements for a single mouse, taken at least 20 days postinoculation, and the horizontal bars indicate the geometric means. The lower dashed line is the detection limit (10−4).
FIG 5.
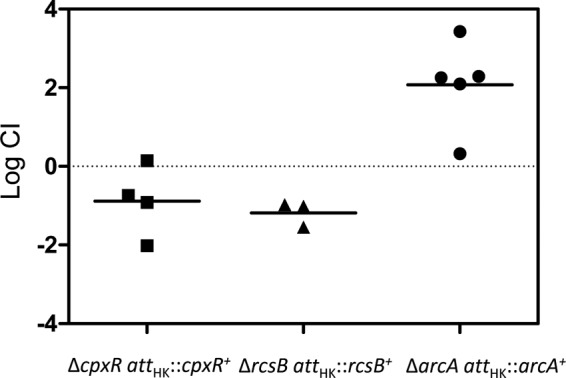
Complementation of arcA, rcsB, and arcA deletions in mouse colonization competition assays. The complemented genes were inserted at the phage HK022 attachment site, as described in Materials and Methods. Each symbol represents an individual mouse, and the bars indicate the geometric means. The competitive indices were determined from the numbers of CFU in feces at 21 days (cpxR and arcA) or 14 days (rcsB) postinoculation.
EnvZ/OmpR and porins in MP1.
We were surprised that an ompR deletion did not have a strong effect on colonization. In the absence of OmpR, the classical porins OmpF and OmpC are not expressed, which greatly reduces outer membrane permeability and the rate of nutrient uptake (45–49). We compared the high abundance outer membrane proteins of MG1655 and MP1 by urea–SDS-PAGE and found that the two strains have different outer membrane protein profiles. In particular, MP1 and MP1 ΔompR show a strong band not present in MG1655 (Fig. 6), suggesting at least one additional MP1 outer membrane protein is highly expressed in cells growing in LB at 37°C. We hypothesized that this protein could be a major porin in MP1 not present in MG1655 that provides a significant component of the outer membrane permeability in ompR-null strains. To test this hypothesis, we compared the sensitivity of MG1655 and MP1 to the beta-lactam antibiotic cefoxitin. Deletion of ompR in K-12 strains confers cefoxitin resistance due to decreased outer membrane permeability from the loss of OmpC and OmpF expression (50), (Table 2). In contrast, MP1 ΔompR is cefoxitin sensitive (Table 2), which is consistent with additional porins contributing to outer membrane permeability. To look for a porin that provides cefoxitin access to the periplasm in MP1 ΔompR, i.e., in the absence of the porins OmpF and OmpC, we moved selected kanamycin resistance insertions from the E. coli K-12 Keio collection (24) into MP1 ΔompR by P1 transduction and screened for resistance to 15 μg/ml cefoxitin. We chose kanamycin insertions in genes that are near candidate porin genes in MP1 (identified by sequence similarity to OmpF). This strategy was predicated on the assumption that only a single porin locus was responsible for cefoxitin permeation across the outer membrane in MP1 ΔompR and that there would be sufficient flanking homology for P1 transduction to replace the locus with a K-12 chromosomal segment with reasonable efficiency. Two transductants that we tested were cefoxitin resistant, and the chromosomal regions for both were linked to two candidate porin genes that are within 25 kb of each other. Based on sequence similarity and chromosome location, one of the genes is yedS, a pseudogene in MG1655 that is intact in MP1 and in some other E. coli isolates, including CFT073. The second gene encodes a protein that is 90% identical to NmpC, a porin in MG1655 that is encoded by a gene within the cryptic prophage DLP12 that is disrupted by an insertion sequence (51, 52). Since this porin gene in MP1 is located in a different region of the chromosome, we have named it nmpD, to avoid confusion with E. coli K-12 nmpC. Several strains closely related to MP1, including CFT073, LF82, and NRG 857C, have both nmpC (without the insertion sequence) and nmpD (32–34). In particular, NmpD from MP1 has a predicted amino acid sequence that is identical to that of the protein C2348, encoded in the CFT073 genome. We deleted nmpD and yedS in MP1 ΔompR and tested the sensitivity of these strains to cefoxitin. MP1 ΔompR ΔyedS failed to grow on 15 μg/ml cefoxitin, but MP1 ΔompR ΔnmpD was resistant (Table 2). In addition, comparison of urea–SDS-PAGE gels of MP1 and MP1 nmpD outer membranes indicates that NmpD is an abundant protein in the outer membrane (Fig. 6). In mouse competitions we found that MP1 ΔnmpD has a relatively mild colonization defect, similar to MP1 ΔompR, but the double-deletion strain MP1 ΔompR ΔnmpD is severely compromised in its ability to colonize mice (Fig. 7). Taken together, these results suggest that that NmpD is a major porin in the MP1 outer membrane that makes a significant contribution to outer membrane permeability and does not require OmpR for expression.
FIG 6.
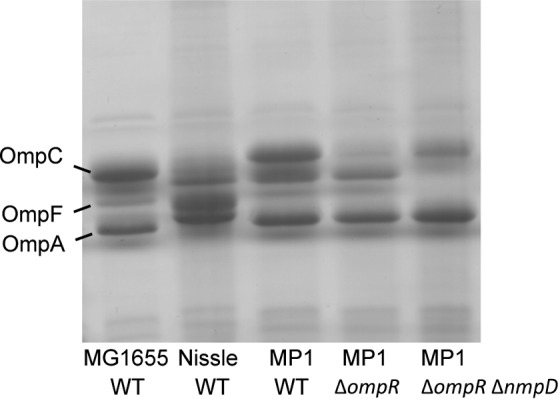
Outer membrane profiles for wild-type (WT) strains and the indicated mutants. Outer membranes were resolved by urea–SDS-PAGE followed by staining with Coomassie brilliant blue.
TABLE 2.
Cefoxitin susceptibility
| Strain | Growth on cefoxitin (15 μg/ml) |
|---|---|
| MG1655 | No |
| MG1655 ompR | Yes |
| MP1 | No |
| MP1 ΔompR | No |
| MP1 ΔompR ΔyedS | No |
| MP1 ΔompR ΔnmpD | Yes |
| MP1 ΔnmpC | No |
FIG 7.
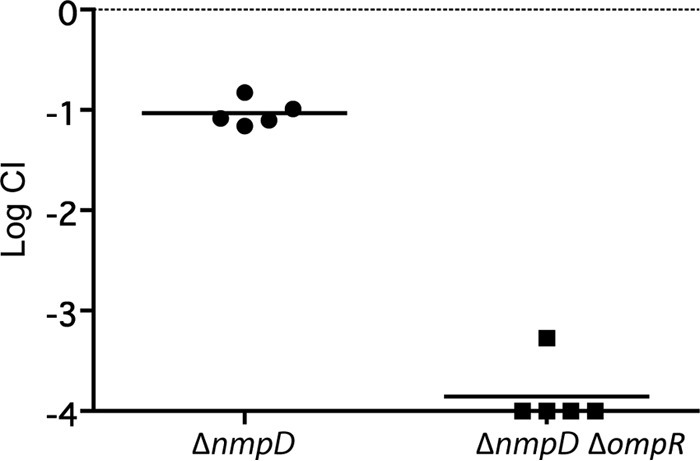
Mouse colonization competitions between WT MP1 and the ΔnmpD and ΔnmpD ΔompR strains. Each symbol denotes an individual mouse, and the bars indicate the geometric means. The competitive indices were determined from numbers of CFU in feces at 21 days postinoculation.
DISCUSSION
MP1 provides a new model system for studying E. coli colonization of the mouse gastrointestinal tract. This strain does not require continuous antibiotic treatment or the use of germfree mice, enabling studies of growth and survival in complex polymicrobial environments. MP1 can be manipulated with the standard genetic techniques that have proved so valuable for working with laboratory E. coli strains, including P1 transduction and recombineering. Therefore, this system can be used to engineer strains not only to study E. coli physiology but also to monitor physical, chemical, and biological properties of the mouse large intestine and to create defined perturbations of this environment.
We found that pretreating mice with streptomycin prior to inoculation was necessary to ensure reproducibly stable colonization from a single inoculation. This pretreatment presumably renders specific niches in the intestinal tract accessible to MP1 by eliminating pre-existing bacteria, especially facultative anaerobes (10, 17, 53). Previous studies suggest that the normal flora returns by 5 to 6 days following streptomycin treatment (54, 55). Our observation that within a week MP1 colonization stabilizes at 105 to 106 CFU/gram of feces—a colonization level comparable to E. coli counts in untreated mice (Fig. 1) (17)—is consistent with a rapid return of competing flora.
MP1 is in the B2 E. coli phylogenetic group (reviewed in reference 3), and is closely related to several ExPEC and human commensal strains (Fig. 3). Furthermore, as with many other B2 group E. coli strains, MP1 contains a considerable number of genes associated with ExPEC virulence (Table 1). We cannot rule out the possibility that MP1 is a bona fide ExPEC strain, capable of causing infection at extraintestinal sites in addition to colonizing the GI tract as a commensal (8, 9, 37). Interestingly, Nissle 1917, a B2 commensal strain closely related to MP1, colonized mice more poorly than E. coli HS, a member of phylogenetic group A that is quite distant from MP1 (Fig. 2–3). This suggests that fitness in the mouse GI tract depends on elements of the pangenome that are not necessarily shared by otherwise closely related strains (4, 5) and is consistent with previous studies demonstrating that individual species are often colonized by E. coli from multiple phylogenetic groups (2, 56).
To study the role of specific signal transduction systems in colonization, we constructed individual knockouts of the response regulators from histidine kinase-response regulator pairs identified in the MP1 genome and performed competitions against a marked derivative of wild-type MP1 in mouse colonization experiments. Interestingly, severe defects were observed only for three response regulator deletions: arcA, cpxR, and rcsB. The defect for ΔarcA is the most easily interpreted of the three, as the ArcB/ArcA two-component system is important for controlling the transition from aerobic to microaerobic or anaerobic growth (57). Thus, the requirement for arcA likely reflects the low-oxygen environment of the large intestine, where E. coli proliferates. Previous studies using the streptomycin-treated mouse model also identified a requirement for arcA and demonstrated that proper respiratory control is critical for efficient colonization (14, 16).
The colonization defects observed for the Cpx-null and Rcs-null strains suggests that E. coli cells experience significant envelope stress in the GI tract. The CpxA/CpxR two-component system is stimulated by various perturbations to the cell envelope, and many of the genes regulated by CpxR encode secreted proteins that repair or degrade misfolded proteins in the periplasm (58–60). This envelope stress-responsive system is also important for the assembly of pili and other surface structures that are required for infection by pathogenic E. coli and for host colonization by other bacteria (61–67). The RcsC/RcsD/RcsB signal transduction system is also associated with envelope stress (68, 69). The specific inducing signals for this system are not well characterized. However, many of the conditions associated with Rcs stimulation perturb the cell envelope. In addition, RcsB-regulated genes affect cell surface properties such as capsule and other surface structures and confer resistance to envelope perturbants such as lysozyme and some antimicrobial peptides (68–73). The colonization defects associated with cpxR and rcsB strains may indicate chemical or physical properties of the GI tract that damage the E. coli cell envelope and require the Cpx and Rcs systems for protection. It is also possible that colonization requires specific macromolecules on the cell surface that depend on the Cpx or Rcs systems for regulation or efficient assembly. Dysregulation of any of these pathways may impair E. coli growth and survival in the mouse intestine.
It is striking that 30 of the 33 response regulator deletion strains that we tested did not exhibit severe colonization defects. Many of these response regulators have been associated with colonization or virulence of various pathogens (for example, see references 74, 75, 76, 77, 78, and 79). It is particularly noteworthy that an ompR deletion does not show a strong colonization defect. The EnvZ/OmpR system is a key component of the complex network that differentially regulates the major porins OmpF and OmpC (80, 81). It has been hypothesized that this network enables E. coli cells to sense and adapt to in vivo and ex vivo environments (49). This model posits that OmpF, the higher-permeability porin, will be expressed in low-nutrient, low-toxin environments that are likely to be encountered outside the host, whereas OmpC, the lower permeability porin, will be expressed in the intestine, where levels of nutrients as well as toxins are likely to be high. Deletion of ompR abrogates expression of both OmpF and OmpC (82), significantly reducing outer membrane permeability and nutrient uptake (47–49). However, studies of outer membrane permeability in ΔompR strains have mainly focused on E. coli K-12 and B/r backgrounds. We found that MP1 expresses a third major porin, NmpD, that is not regulated by OmpR and that accounts for the ability of MP1 ΔompR to colonize mice. Thus, whether or not EnvZ/OmpR plays a role in distinguishing between environments inside and outside animal hosts, this signaling system is not essential for colonization. Furthermore, the results suggest the differential regulation of OmpF and OmpC may have other physiological consequences in addition to modulating outer membrane permeability.
Interestingly, studies of MG1655 colonization in both germfree and streptomycin-treated mouse models have found that adaptation of this strain to the mouse intestine selects for envZ missense mutations that produce high levels of OmpR phosphorylation (83–85). We have not looked for the emergence of such mutants in our mouse colonization experiments. However, since MP1 was isolated from mice, it would be surprising if such a mutation increased the fitness of this strain in the mouse intestine under our colonization conditions.
Several response regulator deletion strains showed average competitive indices between 10−1 and 10−2 (Fig. 4), which may indicate mild colonization defects. In addition, results for one deletion (ΔntrC) suggest a possible fitness advantage. However, further studies with more mice and more time points will be required to determine whether these effects are significant. Of course, laboratory competition experiments will always have limited sensitivity and will be unable to detect small but evolutionarily significant fitness defects. It is also likely that many two-component systems do not confer any selective advantage in the colonization experiments described here because these systems are not activated. For example, the robust colonization by cusR and phoB deletions suggests that the concentration of copper ions is relatively low and that there is ample inorganic phosphate in the large intestine (86, 87).
More generally, if the primary environment where MP1 proliferates is the gastrointestinal tract, then this bacterium may have evolved so that its natural set point is adapted to the average conditions it encounters in this niche. Hence, many of the two-component systems in MP1 may not be required for colonizing the intestine of a healthy well-fed mouse but are instead critical for adaptation to fluctuations in this environment—e.g., changes in diet, host microbial flora, or physiological state of the mouse—as well as transitions to other environments within or outside the animal. Further studies of E. coli colonization of the mouse gastrointestinal tract, and the response to perturbations associated with entrance, persistence in, and exit from the animal, or expansion to extraintestinal niches, promise to provide new insights into the physiology and evolution of signal transduction systems in E. coli and the role of these systems in interactions with the host.
ACKNOWLEDGMENTS
This work was supported by NIH grants GM080279 (M.G.), AI072479 (J.Z.), and RC4 AI092828 (E.S.) and Bacteriology Training Grant T32 AI060516 (M.L.).
Footnotes
Published ahead of print 21 February 2014
REFERENCES
- 1.Gordon DM, FitzGibbon F. 1999. The distribution of enteric bacteria from Australian mammals: host and geographical effects. Microbiology 145:2663–2671 [DOI] [PubMed] [Google Scholar]
- 2.Gordon DM, Cowling A. 2003. The distribution and genetic structure of Escherichia coli in Australian vertebrates: host and geographic effects. Microbiology 149:3575–3586. 10.1099/mic.0.26486-0 [DOI] [PubMed] [Google Scholar]
- 3.Tenaillon O, Skurnik D, Picard B, Denamur E. 2010. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 8:207–217. 10.1038/nrmicro2298 [DOI] [PubMed] [Google Scholar]
- 4.Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sebaihia M, Thomson NR, Chaudhuri R, Henderson IR, Sperandio V, Ravel J. 2008. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 190:6881–6893. 10.1128/JB.00619-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, Bingen E, Bonacorsi S, Bouchier C, Bouvet O, Calteau A, Chiapello H, Clermont O, Cruveiller S, Danchin A, Diard M, Dossat C, Karoui ME, Frapy E, Garry L, Ghigo JM, Gilles AM, Johnson J, Le Bouguenec C, Lescat M, Mangenot S, Martinez-Jehanne V, Matic I, Nassif X, Oztas S, Petit MA, Pichon C, Rouy Z, Ruf CS, Schneider D, Tourret J, Vacherie B, Vallenet D, Medigue C, Rocha EP, Denamur E. 2009. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 5:e1000344. 10.1371/journal.pgen.1000344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukjancenko O, Wassenaar TM, Ussery DW. 2010. Comparison of 61 sequenced Escherichia coli genomes. Microb. Ecol. 60:708–720. 10.1007/s00248-010-9717-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donnenberg MS. 2002. Escherichia coli: virulence mechanisms of a versatile pathogen. Academic Press, Amsterdam, The Netherlands [Google Scholar]
- 8.Le Gall T, Clermont O, Gouriou S, Picard B, Nassif X, Denamur E, Tenaillon O. 2007. Extraintestinal virulence is a coincidental by-product of commensalism in B2 phylogenetic group Escherichia coli strains. Mol. Biol. Evol. 24:2373–2384. 10.1093/molbev/msm172 [DOI] [PubMed] [Google Scholar]
- 9.Leimbach A, Hacker J, Dobrindt U. 2013. E. coli as an all-rounder: the thin line between commensalism and pathogenicity. Curr. Top. Microbiol. Immunol. 358:3–32. 10.1007/82_2012_303 [DOI] [PubMed] [Google Scholar]
- 10.Conway T, Krogfelt KA, Cohen PS. 29 December 2004. Chapter 8.3.1.2, The life of commensal Escherichia coli in the mammalian intestine. In EcoSal Plus, ASM Press, Washington, DC. 10.1128/ecosalplus.8.3.1.2 [DOI] [PubMed] [Google Scholar]
- 11.Myhal ML, Laux DC, Cohen PS. 1982. Relative colonizing abilities of human fecal and K 12 strains of Escherichia coli in the large intestines of streptomycin-treated mice. Eur. J. Clin. Microbiol. 1:186–192. 10.1007/BF02019621 [DOI] [PubMed] [Google Scholar]
- 12.Chang DE, Smalley DJ, Tucker DL, Leatham MP, Norris WE, Stevenson SJ, Anderson AB, Grissom JE, Laux DC, Cohen PS, Conway T. 2004. Carbon nutrition of Escherichia coli in the mouse intestine. Proc. Natl. Acad. Sci. U. S. A. 101:7427–7432. 10.1073/pnas.0307888101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miranda RL, Conway T, Leatham MP, Chang DE, Norris WE, Allen JH, Stevenson SJ, Laux DC, Cohen PS. 2004. Glycolytic and gluconeogenic growth of Escherichia coli O157:H7 (EDL933) and E. coli K-12 (MG1655) in the mouse intestine. Infect. Immun. 72:1666–1676. 10.1128/IAI.72.3.1666-1676.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones SA, Chowdhury FZ, Fabich AJ, Anderson A, Schreiner DM, House AL, Autieri SM, Leatham MP, Lins JJ, Jorgensen M, Cohen PS, Conway T. 2007. Respiration of Escherichia coli in the mouse intestine. Infect. Immun. 75:4891–4899. 10.1128/IAI.00484-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fabich AJ, Jones SA, Chowdhury FZ, Cernosek A, Anderson A, Smalley D, McHargue JW, Hightower GA, Smith JT, Autieri SM, Leatham MP, Lins JJ, Allen RL, Laux DC, Cohen PS, Conway T. 2008. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect. Immun. 76:1143–1152. 10.1128/IAI.01386-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones SA, Gibson T, Maltby RC, Chowdhury FZ, Stewart V, Cohen PS, Conway T. 2011. Anaerobic respiration of Escherichia coli in the mouse intestine. Infect. Immun. 79:4218–4226. 10.1128/IAI.05395-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hentges DJ, Que JQ, Casey SW, Stein AJ. 1984. The influence of streptomycin on colonization resistance in mice. Microecol. Ther. 14:53–62 [Google Scholar]
- 18.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 19.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M. 2006. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60:1136–1151. 10.1111/j.1365-2958.2006.05172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haldimann A, Wanner BL. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J. Bacteriol. 183:6384–6393. 10.1128/JB.183.21.6384-6393.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22:1567–1572. 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]
- 22.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy KC, Campellone KG. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4:11. 10.1186/1471-2199-4-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siryaporn A, Goulian M. 2008. Cross-talk suppression between the CpxA-CpxR and EnvZ-OmpR two-component systems in E. coli. Mol. Microbiol. 70:494–506. 10.1111/j.1365-2958.2008.06426.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hobb RI, Fields JA, Burns CM, Thompson SA. 2009. Evaluation of procedures for outer membrane isolation from Campylobacter jejuni. Microbiology 155:979–988. 10.1099/mic.0.024539-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grozdanov L, Raasch C, Schulze J, Sonnenborn U, Gottschalk G, Hacker J, Dobrindt U. 2004. Analysis of the genome structure of the nonpathogenic probiotic Escherichia coli strain Nissle 1917. J. Bacteriol. 186:5432–5441. 10.1128/JB.186.16.5432-5441.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cress BF, Linhardt RJ, Koffas MA. 2013. Draft genome sequence of Escherichia coli strain Nissle 1917 (serovar O6:K5:H1). Genome Announc. 1:e0004713. 10.1128/genomeA.00047-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine MM, Bergquist EJ, Nalin DR, Waterman DH, Hornick RB, Young CR, Sotman S. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet i:1119–1122 [DOI] [PubMed] [Google Scholar]
- 30.Lasaro MA, Salinger N, Zhang J, Wang Y, Zhong Z, Goulian M, Zhu J. 2009. F1C fimbriae play an important role in biofilm formation and intestinal colonization by the Escherichia coli commensal strain Nissle 1917. Appl. Environ. Microbiol. 75:246–251. 10.1128/AEM.01144-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rendueles O, Travier L, Latour-Lambert P, Fontaine T, Magnus J, Denamur E, Ghigo JM. 2011. Screening of Escherichia coli species biodiversity reveals new biofilm-associated antiadhesion polysaccharides. mBio 2:e00043–11. 10.1128/mBio.00043-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miquel S, Peyretaillade E, Claret L, de Vallee A, Dossat C, Vacherie B, Zineb EH, Segurens B, Barbe V, Sauvanet P, Neut C, Colombel JF, Medigue C, Mojica FJ, Peyret P, Bonnet R, Darfeuille-Michaud A. 2010. Complete genome sequence of Crohn's disease-associated adherent-invasive E. coli strain LF82. PLoS One 5:e12714. 10.1371/journal.pone.0012714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nash JH, Villegas A, Kropinski AM, Aguilar-Valenzuela R, Konczy P, Mascarenhas M, Ziebell K, Torres AG, Karmali MA, Coombes BK. 2010. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics 11:667. 10.1186/1471-2164-11-667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welch RA, Burland V, Plunkett G, III, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HL, Donnenberg MS, Blattner FR. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 99:17020–17024. 10.1073/pnas.252529799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reeves PR, Liu B, Zhou Z, Li D, Guo D, Ren Y, Clabots C, Lan R, Johnson JR, Wang L. 2011. Rates of mutation and host transmission for an Escherichia coli clone over 3 years. PLoS One 6:e26907. 10.1371/journal.pone.0026907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picard B, Garcia JS, Gouriou S, Duriez P, Brahimi N, Bingen E, Elion J, Denamur E. 1999. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 67:546–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson JR, Delavari P, Kuskowski M, Stell AL. 2001. Phylogenetic distribution of extraintestinal virulence-associated traits in Escherichia coli. J. Infect. Dis. 183:78–88. 10.1086/317656 [DOI] [PubMed] [Google Scholar]
- 38.Riley MA, Cadavid L, Collett MS, Neely MN, Adams MD, Phillips CM, Neel JV, Friedman D. 2000. The newly characterized colicin Y provides evidence of positive selection in pore-former colicin diversification. Microbiology 146:1671–1677 [DOI] [PubMed] [Google Scholar]
- 39.Mizuno T. 1997. Compilation of all genes encoding two-component phosphotransfer signal transducers in the genome of Escherichia coli. DNA Res. 4:161–168. 10.1093/dnares/4.2.161 [DOI] [PubMed] [Google Scholar]
- 40.Ulrich LE, Zhulin IB. 2010. The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 38:D401–D407. 10.1093/nar/gkp940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barakat M, Ortet P, Whitworth DE. 2011. P2CS: a database of prokaryotic two-component systems. Nucleic Acids Res. 39:D771–D776. 10.1093/nar/gkq1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai W, Wannemuehler Y, Dell'anna G, Nicholson B, Barbieri NL, Kariyawasam S, Feng Y, Logue CM, Nolan LK, Li G. 2013. A novel two-component signaling system facilitates uropathogenic Escherichia coli's ability to exploit abundant host metabolites. PLoS Pathog. 9:e1003428. 10.1371/journal.ppat.1003428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Timme TL, Lawrence CB, Moses RE. 1989. Two new members of the OmpR superfamily detected by homology to a sensor-binding core domain. J. Mol. Evol. 28:545–552. 10.1007/BF02602935 [DOI] [PubMed] [Google Scholar]
- 44.Jiang SQ, Yu GQ, Li ZG, Hong JS. 1988. Genetic evidence for modulation of the activator by two regulatory proteins involved in the exogenous induction of phosphoglycerate transport in Salmonella typhimurium. J. Bacteriol. 170:4304–4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall MN, Silhavy TJ. 1981. Genetic analysis of the major outer membrane proteins of Escherichia coli. Annu. Rev. Genet. 15:91–142. 10.1146/annurev.ge.15.120181.000515 [DOI] [PubMed] [Google Scholar]
- 46.Nara F, Matsuyama S, Mizuno T, Mizushima S. 1986. Molecular analysis of mutant ompR genes exhibiting different phenotypes as to osmoregulation of the ompF and ompC genes of Escherichia coli. Mol. Gen. Genet. 202:194–199. 10.1007/BF00331636 [DOI] [PubMed] [Google Scholar]
- 47.Von Meyenburg K. 1971. Transport-limited growth rates in a mutant of Escherichia coli. J. Bacteriol. 107:878–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lutkenhaus JF. 1977. Role of a major outer membrane protein in Escherichia coli. J. Bacteriol. 131:631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nikaido H, Vaara M. 1985. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 49:1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaffe A, Chabbert YA, Semonin O. 1982. Role of porin proteins OmpF and OmpC in the permeation of beta-lactams. Antimicrob. Agents Chemother. 22:942–948. 10.1128/AAC.22.6.942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pugsley AP, Schnaitman CA. 1978. Identification of three genes controlling production of new outer membrane pore proteins in Escherichia coli K-12. J. Bacteriol. 135:1118–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hindahl MS, Crockford GW, Hancock RE. 1984. Outer membrane protein NmpC of Escherichia coli: pore-forming properties in black lipid bilayers. J. Bacteriol. 159:1053–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vollaard EJ, Clasener HA. 1994. Colonization resistance. Antimicrob. Agents Chemother. 38:409–414. 10.1128/AAC.38.3.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, Dougan G, von Mering C, Hardt WD. 2007. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 5:2177–2189. 10.1371/journal.pbio.0050244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Antunes LC, Han J, Ferreira RB, Lolic P, Borchers CH, Finlay BB. 2011. Effect of antibiotic treatment on the intestinal metabolome. Antimicrob. Agents Chemother. 55:1494–1503. 10.1128/AAC.01664-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Escobar-Paramo P, Le Menac'h A, Le Gall T, Amorin C, Gouriou S, Picard B, Skurnik D, Denamur E. 2006. Identification of forces shaping the commensal Escherichia coli genetic structure by comparing animal and human isolates. Environ. Microbiol. 8:1975–1984. 10.1111/j.1462-2920.2006.01077.x [DOI] [PubMed] [Google Scholar]
- 57.Malpica R, Sandoval GR, Rodriguez C, Franco B, Georgellis D. 2006. Signaling by the arc two-component system provides a link between the redox state of the quinone pool and gene expression. Antioxid. Redox Signal. 8:781–795. 10.1089/ars.2006.8.781 [DOI] [PubMed] [Google Scholar]
- 58.Ruiz N, Silhavy TJ. 2005. Sensing external stress: watchdogs of the Escherichia coli cell envelope. Curr. Opin. Microbiol. 8:122–126. 10.1016/j.mib.2005.02.013 [DOI] [PubMed] [Google Scholar]
- 59.Vogt SL, Raivio TL. 2012. Just scratching the surface: an expanding view of the Cpx envelope stress response. FEMS Microbiol. Lett. 326:2–11. 10.1111/j.1574-6968.2011.02406.x [DOI] [PubMed] [Google Scholar]
- 60.Hunke S, Keller R, Muller VS. 2012. Signal integration by the Cpx-envelope stress system. FEMS Microbiol. Lett. 326:12–22. 10.1111/j.1574-6968.2011.02436.x [DOI] [PubMed] [Google Scholar]
- 61.Jones CH, Danese PN, Pinkner JS, Silhavy TJ, Hultgren SJ. 1997. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J. 16:6394–6406. 10.1093/emboj/16.21.6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hung DL, Raivio TL, Jones CH, Silhavy TJ, Hultgren SJ. 2001. Cpx signaling pathway monitors biogenesis and affects assembly and expression of P pili. EMBO J. 20:1508–1518. 10.1093/emboj/20.7.1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nevesinjac AZ, Raivio TL. 2005. The Cpx envelope stress response affects expression of the type IV bundle-forming pili of enteropathogenic Escherichia coli. J. Bacteriol. 187:672–686. 10.1128/JB.187.2.672-686.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raivio TL. 2005. Envelope stress responses and Gram-negative bacterial pathogenesis. Mol. Microbiol. 56:1119–1128. 10.1111/j.1365-2958.2005.04625.x [DOI] [PubMed] [Google Scholar]
- 65.Vogt SL, Nevesinjac AZ, Humphries RM, Donnenberg MS, Armstrong GD, Raivio TL. 2010. The Cpx envelope stress response both facilitates and inhibits elaboration of the enteropathogenic Escherichia coli bundle-forming pilus. Mol. Microbiol. 76:1095–1110. 10.1111/j.1365-2958.2010.07145.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herbert EE, Cowles KN, Goodrich-Blair H. 2007. CpxRA regulates mutualism and pathogenesis in Xenorhabdus nematophila. Appl. Environ. Microbiol. 73:7826–7836. 10.1128/AEM.01586-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Herbert Tran EE, Andersen AW, Goodrich-Blair H. 2009. CpxRA influences Xenorhabdus nematophila colonization initiation and outgrowth in Steinernema carpocapsae nematodes through regulation of the nil locus. Appl. Environ. Microbiol. 75:4007–4014. 10.1128/AEM.02658-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59:379–405. 10.1146/annurev.micro.59.050405.101230 [DOI] [PubMed] [Google Scholar]
- 69.Clarke DJ. 2010. The Rcs phosphorelay: more than just a two-component pathway. Future Microbiol. 5:1173–1184. 10.2217/fmb.10.83 [DOI] [PubMed] [Google Scholar]
- 70.Laubacher ME, Ades SE. 2008. The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J. Bacteriol. 190:2065–2074. 10.1128/JB.01740-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Callewaert L, Vanoirbeek KG, Lurquin I, Michiels CW, Aertsen A. 2009. The Rcs two-component system regulates expression of lysozyme inhibitors and is induced by exposure to lysozyme. J. Bacteriol. 191:1979–1981. 10.1128/JB.01549-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farris C, Sanowar S, Bader MW, Pfuetzner R, Miller SI. 2010. Antimicrobial peptides activate the Rcs regulon through the outer membrane lipoprotein RcsF. J. Bacteriol. 192:4894–4903. 10.1128/JB.00505-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lehti TA, Heikkinen J, Korhonen TK, Westerlund-Wikstrom B. 2012. The response regulator RcsB activates expression of Mat fimbriae in meningitic Escherichia coli. J. Bacteriol. 194:3475–3485. 10.1128/JB.06596-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gunn JS. 2008. The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 16:284–290. 10.1016/j.tim.2008.03.007 [DOI] [PubMed] [Google Scholar]
- 75.Fass E, Groisman EA. 2009. Control of Salmonella pathogenicity island-2 gene expression. Curr. Opin. Microbiol. 12:199–204. 10.1016/j.mib.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moreira CG, Weinshenker D, Sperandio V. 2010. QseC mediates Salmonella enterica serovar typhimurium virulence in vitro and in vivo. Infect. Immun. 78:914–926. 10.1128/IAI.01038-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alteri CJ, Lindner JR, Reiss DJ, Smith SN, Mobley HL. 2011. The broadly conserved regulator PhoP links pathogen virulence and membrane potential in Escherichia coli. Mol. Microbiol. 82:145–163. 10.1111/j.1365-2958.2011.07804.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Njoroge JW, Nguyen Y, Curtis MM, Moreira CG, Sperandio V. 2012. Virulence meets metabolism: Cra and KdpE gene regulation in enterohemorrhagic Escherichia coli. mBio 3:e00280–12. 10.1128/mBio.00280-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Freeman ZN, Dorus S, Waterfield NR. 2013. The KdpD/KdpE two-component system: integrating K(+) homeostasis and virulence. PLoS Pathog. 9:e1003201. 10.1371/journal.ppat.1003201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pratt LA, Hsing W, Gibson KE, Silhavy TJ. 1996. From acids to osmZ: multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Mol. Microbiol. 20:911–917. 10.1111/j.1365-2958.1996.tb02532.x [DOI] [PubMed] [Google Scholar]
- 81.Batchelor E, Walthers D, Kenney LJ, Goulian M. 2005. The Escherichia coli CpxA-CpxR envelope stress response system regulates expression of the porins ompF and ompC. J. Bacteriol. 187:5723–5731. 10.1128/JB.187.16.5723-5731.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Slauch JM, Silhavy TJ. 1996. The porin regulon: a paradigm for the two-component regulatory systems, p 383–417 In Lin ECC, Lynch AS. (ed), Regulation of gene expression in Escherichia coli. Chapman & Hall, New York, NY [Google Scholar]
- 83.Giraud A, Arous S, De Paepe M, Gaboriau-Routhiau V, Bambou JC, Rakotobe S, Lindner AB, Taddei F, Cerf-Bensussan N. 2008. Dissecting the genetic components of adaptation of Escherichia coli to the mouse gut. PLoS Genet. 4:e2. 10.1371/journal.pgen.0040002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leatham-Jensen MP, Frimodt-Moller J, Adediran J, Mokszycki ME, Banner ME, Caughron JE, Krogfelt KA, Conway T, Cohen PS. 2012. The streptomycin-treated mouse intestine selects Escherichia coli envZ missense mutants that interact with dense and diverse intestinal microbiota. Infect. Immun. 80:1716–1727. 10.1128/IAI.06193-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Paepe M, Gaboriau-Routhiau V, Rainteau D, Rakotobe S, Taddei F, Cerf-Bensussan N. 2011. Trade-off between bile resistance and nutritional competence drives Escherichia coli diversification in the mouse gut. PLoS Genet. 7:e1002107. 10.1371/journal.pgen.1002107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wanner BL. 1996. Phosphorus assimilation and control of the phosphate regulon, p 1357–1381 In Neidhardt FC, Curtis R, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC [Google Scholar]
- 87.Munson GP, Lam DL, Outten FW, O'Halloran TV. 2000. Identification of a copper-responsive two-component system on the chromosome of Escherichia coli K-12. J. Bacteriol. 182:5864–5871. 10.1128/JB.182.20.5864-5871.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Johnson JR, Russo TA. 2005. Molecular epidemiology of extraintestinal pathogenic (uropathogenic) Escherichia coli. Int. J. Med. Microbiol. 295:383–404. 10.1016/j.ijmm.2005.07.005 [DOI] [PubMed] [Google Scholar]
- 89.Spurbeck RR, Stapleton AE, Johnson JR, Walk ST, Hooton TM, Mobley HL. 2011. Fimbrial profiles predict virulence of uropathogenic Escherichia coli strains: contribution of ygi and yad fimbriae. Infect. Immun. 79:4753–4763. 10.1128/IAI.05621-11 [DOI] [PMC free article] [PubMed] [Google Scholar]