

Abstract
We previously showed that the Aggregatibacter actinomycetemcomitans lsrACDBFG and lsrRK operons are regulated by LsrR and cyclic AMP receptor protein (CRP) and that proper regulation of the lsr locus is required for optimal biofilm growth by A. actinomycetemcomitans. Here, we identified sequences that reside immediately upstream from both the lsrA and lsrR start codons that closely resemble the consensus recognition sequence of Escherichia coli integration host factor (IHF) protein. A. actinomycetemcomitans IHFα and IHFβ were expressed and purified as hexahistidine fusion proteins, and using electrophoretic mobility shift assays (EMSAs), the IHFα-IHFβ protein complex was shown to bind to probes containing the putative IHF recognition sequences. In addition, single-copy chromosomal insertions of lsrR promoter-lacZ and lsrA promoter-lacZ transcriptional fusions in wild-type A. actinomycetemcomitans and ΔihfA and ΔihfB mutant strains showed that IHF differentially regulates the lsr locus and functions as a negative regulator of lsrRK and a positive regulator of lsrACDBFG. Deletion of ihfA or ihfB also reduced biofilm formation and altered biofilm architecture relative to the wild-type strain, and these phenotypes were partially complemented by a plasmid-borne copy of ihfA or ihfB. Finally, using 5′ rapid amplification of cDNA ends (RACE), two transcriptional start sites (TSSs) and two putative promoters were identified for lsrRK and three TSSs and putative promoters were identified for lsrACDBFG. The function of the two lsrRK promoters and the positive regulatory role of IHF in regulating lsrACDBFG expression were confirmed with a series of lacZ transcriptional fusion constructs. Together, our results highlight the complex transcriptional regulation of the lsrACDBFG and lsrRK operons and suggest that multiple promoters and the architecture of the lsrACDBFG-lsrRK intergenic region may control the expression of these operons.
INTRODUCTION
The dental biofilm is a complex and dynamic microbial community that comprises up to 700 different species of bacteria (1, 2, 58, 60). This biofilm is the prime etiological agent of three common oral diseases in humans, dental caries, gingivitis, and periodontal disease (3–5, 59). The progression of these diseases is associated with major shifts in microbial populations in the oral biofilm, and diseased sites often exhibit increased populations of pathogenic species relative to healthy sites in the oral cavity (3, 4, 6). The stimuli that contribute to these populational shifts have not been well characterized, but the oral cavity is subject to continual environmental flux, including changes in pH, temperature, osmolarity, and nutrient supply. Oral bacteria rapidly detect and respond to these environmental fluctuations, allowing them to successfully coexist and thrive in the oral cavity (4, 7, 8). Both intra- and interspecies communication is known to occur among oral bacteria, and it is likely that these signaling processes enable the organisms to coordinate their behavior and function by regulating gene expression as a community. One mechanism of communication, termed “quorum sensing,” is a cell density-dependent response (9–12), which in Gram-negative bacteria is mediated by the production, release, and detection of soluble signal molecules called “autoinducers.”
Aggregatibacter actinomycetemcomitans is a Gram-negative organism that is associated with aggressive forms of periodontitis and other systemic infections (13–16). This organism expresses LuxS and secretes autoinducer 2 (AI-2), and AI-2-dependent quorum sensing has been shown to regulate the expression of virulence factors, iron acquisition systems, and biofilm formation (17–20). The complex regulatory network involved in A. actinomycetemcomitans biofilm growth and the role of quorum sensing in this process have begun to be explored in recent years. A. actinomycetemcomitans expresses two periplasmic proteins, LsrB and RbsB, that function as receptors for AI-2 (21, 22), and inactivation of either or both of the genes encoding these proteins results in reduced biofilm growth and virulence (18, 20, 21). Like Escherichia coli and Salmonella, A. actinomycetemcomitans LsrB is encoded by an operon consisting of lsrACDBFG (the Salmonella enterica serovar Typhimurium operon also contains an additional gene designated lsrE), where the lsrACD genes encode the AI-2 transporter, lsrF encodes an aldolase-like protein that cleaves AI-2 (23), and lsrG codes for an isomerase of phospho-AI-2 (24). Upstream of and divergently transcribed from the lsrACDBFG operon resides lsrRK, encoding a repressor of lsrACDBFG (LsrR) and an AI-2 kinase (LsrK), which in E. coli regulate the expression of lsrACDBFG operon in an AI-2-dependent manner (25, 26). In E. coli and S. Typhimurium, AI-2 is internalized and phosphorylated at high density by LsrK, and AI-2-PO4 binds to LsrR, resulting in derepression of lsrACDBFG. The structure of both operons is conserved in A. actinomycetemcomitans, but in contrast to what occurs in E. coli, deletion of lsrK had no effect on the transcriptional activity of the A. actinomycetemcomitans lsrA or lsrR promoters (27). However, deletion of lsrR, lsrRK, or the gene encoding the cyclic AMP (cAMP) receptor protein (CRP) results in a significant reduction of biofilm formation by A. actinomycetemcomitans (27). Thus, the proper regulation of the lsr locus has a significant impact on the ability of A. actinomycetemcomitans to thrive in biofilms.
The integration host factor (IHF) is a DNA-binding and -bending protein that consists of two subunits, HimA (IHFα) and HimD (IHFβ), which show 30% identity in their amino acids in E. coli. IHF recognizes and binds to the asymmetric consensus sequence YAANNNNTTGATW, where Y = T or C, N = any base, and W = A or T (28, 29), which usually is found in A/T-rich regions (30). The surrounding regions flanking the IHF consensus site are also important in the IHF binding (31). IHF was originally discovered as being essential for the integration of the bacteriophage λ DNA into the chromosome (32). However, it is now known to play a role in bacterial chromatin organization, recombination, DNA replication, and transcriptional regulation in Gram-negative bacteria, including the regulation of genetic loci associated with virulence in many pathogenic bacteria. IHF has a critical role in the coparticipation with RpoS for the transcriptional activation of genes required for the transition from exponential growth to the stationary phase, and in Salmonella, IHF coordinates the regulation of the major virulence gene clusters. Specifically, IHF has a positive regulatory role in the expression of the pathogenicity island 1 (SP-1) in early and late exponential phases of growth, while at the onset of the stationary phase, IHF inhibits the expression of the genes encoding the type III secretion system (TTSS) and has a positive effect on the expression of the genes coding for secreted effector proteins (33). IHF also has a positive role in the transcriptional control of the che genes that encode the chemotaxis proteins and genes coding for the flagellum in Salmonella (33), E. coli K-12 (34), and Caulobacter (35). IHF contributes to flagellin protein phase variation in Salmonella (33, 36, 37), and in E. coli, IHF participates in the control of phase variation of type 1 fimbria (38). Furthermore, the foundation of the biofilm structure is the extracellular polymeric substance (EPS) that acts as a barrier to protect the bacteria in the biofilm community from host defenses and antibiotics. Extracellular IHF appears to play a role in the integrity of the EPS matrix that contains extracellular DNA, and treatment with IHF antibodies rapidly disrupts the biofilm EPS formed by several human pathogens in vitro (39–41).
In this report, we show that the intergenic region (IGR) separating the A. actinomycetemcomitans lsrACDBFG and lsrRK operons contains two motifs that resemble the consensus IHF binding site and that both interact with purified IHFα-IHFβ complex. We also show that IHF differentially regulates the lsrACDBFG and lsrRK operons, and deletion of either ihfA or ihfB reduces biofilm growth by A. actinomycetemcomitans. These results highlight the complex transcriptional regulation of the lsrACDBFG and lsrRK operons and suggest that the architecture of the lsrACDBFG-lsrRK intergenic region may contribute to the regulation of these operons.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. Luria-Bertani (LB) broth, LB agar (LB broth plus 1.5% agar), TYE broth (1% tryptone, 0.5% yeast extract), brain heart infusion (BHI) broth, and BHI agar (all from Difco) were routinely used for the propagation and plating of bacteria. Aggregatibacter actinomycetemcomitans (afimbriated, smooth-colony morphotype strain 652) was grown at 37°C under microaerophilic conditions in a candle jar and was also used for biofilm experiments (see below). When required, the medium was supplemented with 25 μg/ml kanamycin (Km), 12.5 μg/ml tetracycline (Tc), 50 μg/ml spectinomycin (Sp), or 100 μg/ml ampicillin (Amp).
DNA procedures.
DNA manipulations were carried out as described previously (42). Transformation of E. coli and A. actinomycetemcomitans was done by electroporation (Bio-Rad, Hercules, CA). Transformants containing plasmids were selected on LB agar plates supplemented with the appropriate antibiotics. Plasmid DNA was isolated using the QIAprep Spin miniprep kit (Qiagen, Valencia, CA). Restriction enzymes were used as recommended by the manufacturer (New England BioLabs, Ipswich, MA). All primers used in this study (Integrated DNA Technology, Coralville, IA) are shown in Table S2 in the supplemental material, and those flanked with restriction enzyme sites are underlined in the primer sequences. Primer sequences were designed based on the genome information of A. actinomycetemcomitans strain D11S-1 available from the Pathosystems Resource Integration Center (http://patricbrc.vbi.vt.edu). All constructs were verified by DNA sequencing (University of Louisville Core Sequencing Facilities).
Identification of the lsrR and lsrA transcriptional start sites.
The transcriptional start sites of lsrR and lsrA were determined using a Gene Racer kit (Invitrogen, Grand Island, NY). A. actinomycetemcomitans 652 was grown in LB to the late exponential phase (optical density at 600 nm [OD600] of 0.4). This culture was treated with Qiagen RNAprotect bacterial reagent, and total RNA was extracted using the RNeasy lipid tissue minikit (Qiagen, Valencia, CA) as specified by the manufacturer. RNA was treated with RNase-free DNase I (New England BioLabs, Ipswich, MA) to eliminate contaminating DNA. RNA was quantified by spectrophotometry using a Nanodrop ND-1000 (Thermo Fisher Scientific, Pittsburgh, PA). A 44-base 5′ RNA adaptor oligonucleotide was ligated to the 5′ ends of the total RNA (5 μg) using T4 RNA ligase. Reverse transcription (RT) was performed with avian myeloblastosis virus (AMV) reverse transcriptase and random hexamers that were included in the Gene Racer kit. PCR was performed using the Gene Racer 5′ primer and the lsrA- and lsrR-specific primers ATE-210 and ATE-208, respectively. The PCR products were subsequently cloned into a pCR4-TOPO vector and transformed into E. coli TOP10 (Invitrogen). Thirty-five positive independent clones were sequenced for the lsrR promoter, and 28 clones were analyzed for the lsrACDBFG promoter.
Construction of lsrR and lsrA promoter-lacZ fusion plasmids.
Various fragments containing portions or the entire intergenic region between lsrACDBFG and lsrRK region were amplified by PCR as follows using A. actinomycetemcomitans 652 genomic DNA as a template. The typical amplification profile used was 94°C for 2 min for 1 cycle and then 94°C for 30s, 60°C for 1 min, and 72°C for 2 min for 25 cycles. For the lsrR promoter fusion constructs pATE69, pATE70, and pATE100, portions of the intergenic region were amplified using the primer sets ATE-172F and ATE-6R, ATE-173F and ATE-6R, and ATE-242F and ATE-6R, respectively (see Table S2 in the supplemental material). The 193-, 143-, and 414-bp PCR products were then digested with KpnI-BamHI, and each was cloned into KpnI-BamH-digested pJT3 (43) to create pATE69, pATE70, and pATE100, respectively. A similar approach was used to construct the lsrA promoter fusion plasmids pATE73, pATE74, and pATE75. The primer sets used to amplify the appropriate promoter fragment for these constructs were ATE-165F and ATE-7R, ATE-166F and ATE-7R, and ATE-167F and ATE-7R, respectively.
Construction of pATE92 and pATE93 plasmids.
The promoter region and structural ihfA gene were amplified from A. actinomycetemcomitans genomic DNA using primer sets ATE-216F and ATE-218R, and the resulting 813-bp fragment was digested with BamHI-XbaI and cloned into BamHI-XbaI-digested pJT7 (A. Torres-Escobar, M. D. Juarez-Rodriguez, and D. R. Demuth, submitted for publication) (see Table S2 in the supplemental material) to create pATE92. Similarly, the promoter region and structural ihfB gene were amplified from A. actinomycetemcomitans genomic DNA using primer sets ATE-219F and ATE-221R, and the resulting 714-bp product was digested with KpnI-XbaI and cloned into KpnI-XbaI-digested pJT7 (see Table S2) to create pATE93.
Construction of ihfA and ihfB expression plasmids.
The structural ihfA and ihfB genes were PCR amplified from A. actinomycetemcomitans genomic DNA using primer sets ATE-188F and ATE-189R and ATE-190F and ATE-191R, respectively. The 302-bp product containing the ihfA gene and the 285-bp product containing the ihfB gene were digested with NcoI-ApaI and cloned individually into the NcoI-ApaI-digested pYA3883 (44) expression vector to create pATE76 and pATE77, respectively. However, IHFβ was only moderately expressed from pATE77; thus, a fragment containing the structural ihfB flanked by the sequence encoding the AU1 (44) from pATE77 was amplified using the primer set ATE-192F and ATE-193R in order to increase its expression. The resulting 317-bp PCR product was digested with NcoI-XhoI and cloned into NcoI-XhoI-digested pET28A+ vector (Novagen) to obtain pATE80.
Generation of A. actinomycetemcomitans markerless deletion mutants.
The construction of A. actinomycetemcomitans harboring a markerless deletion mutation of the ihfA gene was carried out by amplifying the upstream and downstream flanking regions of ihfA with primer sets ATE-157F and ATE-134R and ATE-135F and ATE-158R (see Table S2 in the supplemental material). The respective 646-bp and 546-bp PCR products were digested with NheI-XhoI and XhoI-SacI and cloned adjacently (joined by the XhoI restriction site) into NheI-SacI-digested pJT1 suicide vector (43) to create pATE78. A similar approach was used to generate the markerless deletion mutant in ihfB. The upstream and downstream flanking regions of ihfB were amplified by PCR with primer sets ATE-159F and ATE-142R and ATE-143F and ATE-160R (see Table S2). The respective 652-bp and 1,035-bp PCR products were digested with NheI-XhoI, and XhoI-SacI and cloned adjacently (joined by the XhoI restriction site) into the NheI-SacI-digested pJT1 suicide vector to create pATE79. Each recombinant suicide plasmid (20 μg) was introduced individually into A. actinomycetemcomitans by electroporation. Electroporated cells were incubated in 0.5 ml of SOC broth (Super Optimal broth with glucose added for catabolite repression) standing for 5 h at 37°C under microaerophilic conditions (anaerobic jar). Bacterial cells with single recombinant event were selected onto BHI agar containing 50 μg/ml Sp at 37°C under microaerophilic conditions. Ten spectinomycin-resistant (Spr) colonies were randomly selected and subcultured daily for 24 h at 37°C for 3 consecutive days, except that the final cultures were grown in the presence of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) (IPTG-induced Ptrc-sacB). To select for bacteria that had undergone a second recombination event, the culture was diluted 10-fold, spread onto TYE agar supplemented with 1 mM IPTG and 10% sucrose, and grown at 37°C under microaerophilic conditions. One thousand sucrose-resistant (Sucr) colonies were replica plated onto TYE agar supplemented with sucrose and onto BHI agar supplemented with spectinomycin. Spectinomycin-sensitive (Sps) colonies were selected to perform PCR for the deletion mutation of the target genes, using the primer set ATE-137F and ATE-140R for ihfA deletion or ATE-145F and ATE-148R for ihfB deletion. Sps colonies that were PCR positive were selected for further analysis.
Integration of lsrR-lacZ or lsrA-lacZ transcriptional fusion in single copy into the A. actinomycetemcomitans genome.
To integrate a single copy of the lsrR-promoter- or lsrA-promoter-lacZ transcriptional fusions into the chromosome of A. actinomycetemcomitans by homologous recombination, the lsrR-lacZ (in a 3,000-bp KpnI-XbaI fragment) and lsrA-lacZ (in a 3,969-bp KpnI-XbaI fragment) constructs were released from pATE23 and pATE75, respectively, and were subcloned into KpnI-XbaI-digested pJT10 suicide vector (Torres-Escobar et al., submitted) to create pATE94 and pATE95, respectively. Each recombinant suicide plasmid (20 μg) was introduced individually into A. actinomycetemcomitans 652 and isogenic ΔihfA, and ΔihfB mutants by electroporation. A similar approach used to generate the markerless deletion mutants described above was employed, but with the selection of Spr colonies, to obtain A. actinomycetemcomitans with a single copy of the transcriptional fusion inserted in the chromosome by one homologous recombinant event. Ten Spr colonies were selected to verify by PCR the single-copy chromosomal insertion of the lsrR-lacZ or lsrA-lacZ fusion with primer sets ATE-210R and MDJR-123R and MDJR148F and ATE208R and ATE-208R and MDJR-123R and MDJR148F and ATE210R, respectively. The Spr colonies that were PCR positive were selected for further analysis.
Growth kinetics.
A single colony of A. actinomycetemcomitans harboring each recombinant plasmid was independently inoculated into 10 ml of BHI medium supplemented with 25 μg/ml Km and was grown standing for 24 h at 37°C. The next day, the overnight culture (OD600 of 0.6) was diluted at a 1:30 ratio to inoculate 10 ml of BHI with 25 μg/ml Km and grown standing at 37°C. For the first 12 h of growth, an aliquot was removed each hour, and culture density was determined by measuring the OD600. Additional aliquots were taken from each culture for analysis at the 24-, 48-, and 72-h time points. β-Galactosidase (β-Gal) activity was also determined for each aliquot as described below.
β-Gal assays.
β-Galactosidase (β-Gal) activity was qualitatively assessed on LB agar plates that were supplemented with 50 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). Quantitative evaluation of β-Gal activity was carried out using permeabilized cells incubated with o-nitrophenyl-β-d-galactopyranoside (ONPG) substrate (Sigma, St. Louis, MO) as previously described by Miller (45). Average values ± standard deviations) for activity units were routinely calculated from three independent assays, each carried out in triplicate.
Expression and purification of IHF protein.
E. coli LMG194 harboring pATE76 and E. coli BL21(DE3) transformed with the pATE80 plasmid were used for the synthesis of the hexahistidine fusion proteins' IHF alpha (IHFα) and beta (IHFβ) subunits, respectively. The expression and detection of the recombinant proteins were performed essentially as described previously by Torres-Escobar et al. (44). Purification was carried out by cobalt-based immobilized metal affinity chromatography under denaturing conditions. Eluted fractions containing the purified IHFα and IHFβ subunits were selected based on sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis (SDS-PAGE). The IHFα and IHFβ selected pools were mixed in equimolar quantities and dialyzed in a Slyde-A-Lyzer 3K cassette (Pierce) at 4°C against refolding buffer containing 50 mM Tris-HCl (pH 8.5), 4 M urea, 0.5 M l-arginine, 264 mM NaCl, 11 mM KCl, 8 mM MgCl2, 0.1% Triton X-100, and 20% (vol/vol) glycerol for 24 h. Subsequently, samples were dialyzed for 24 h against the buffer described above but containing 2 M urea and then sequentially dialyzed for 12 h each in buffer consisting of 50 mM Tris-HCl (pH 8.5), 100 mM NaCl, 0.1 mM KCl, 4 mM MgCl2, 2 mM Mg(CH3COO)2, 0.1 mM EDTA, 0.1 mM dithiothreitol (DTT), 50% (vol/vol) glycerol, and 2, 1, or 0.5 M urea. A final dialysis was then carried out for 6 h against the buffer described above without urea. Aliquots of the IHF heterodimer reconstituted from purified subunits IHFα and IHFβ was stored at −70°C. The protein concentration was determined by the Bradford assay, using bovine serum albumin (BSA) as a standard.
EMSA.
The DNA fragments used for the nonradioactive electrophoretic mobility shift assay (EMSA) were obtained by PCR using sets of primers described in Table S2 in the supplemental material. A biotin 3′-end labeling kit (Fisher Scientific, Pittsburgh, PA) was used for labeling of DNA fragments according to the manufacturer's instructions. Binding reactions were performed with a total of 50 fmol of each probe mixed with various amounts of the IHF heterodimer reconstituted from purified subunits IHFα and IHFβ (1, 2, and 4 μM) in 20 μl of binding buffer consisting of 10 mM Tris-HCl [pH 7.5], 50 mM KCl, 1 mM dithiothreitol, 1 μg poly(dI-dC), and 100 μg/ml bovine serum albumin (BSA). The reaction mixtures were incubated for 20 min at room temperature. Afterward, 5 μl of gel loading buffer (0.25× Tris-borate-EDTA [TBE], 60%; glycerol, 40%; bromophenol, 0.2% [wt/vol]) was added, and the mixtures were electrophoresed in a 6% native polyacrylamide gel in 0.5× TBE buffer (45 mM Tris-borate, 1 mM EDTA [pH 8.0]) and immunoblotted. DNA bands were detected using the LightShift chemiluminescent EMSA kit according to the manufacturer's instructions.
Biofilm formation and analysis.
A. actinomycetemcomitans biofilms were grown in an FC81 polycarbonate flow chamber (Biosurface Technologies Corp., Bozeman, MT) (chamber dimensions are 50.5 mm by 12.7 mm by 2.54 mm) at a flow rate of 10 ml per hour at 25°C, essentially as described by Shao et al. (20). The resulting biofilm was stained with 0.2 mg/ml fluorescein isothiocyanate (FITC) (Sigma-Aldrich) for 1 h in the dark and then washed with phosphate-buffered saline (PBS) for 2 h. Biofilms were visualized using an Olympus Fluoview FV500 confocal scanning laser microscope (Olympus, Pittsburgh, PA) at a magnification of ×600, using an argon laser. Confocal images were captured from 20 randomly chosen frames from each flow chamber, and z-plane scans from 0 to 100 μm at 1-μm intervals were performed above the glass surface for each frame. The images were analyzed using Volocity image analysis software (PerkinElmer, Inc., Waltham, MA) to reconstruct three-dimensional images. Biofilm depth, biofilm biomass, and total surface area of biofilm were determined using the Volocity software package. The values reported are means of data from the different frames obtained. Biofilm assays were repeated independently three times with each strain. Differences among biofilms were determined by one-way analysis of variance (ANOVA) followed by Tukey's multiple-comparison test. Differences with P values of <0.05 were considered significant. Data were analyzed with GraphPad Prism v5 software.
RESULTS
Identification of putative integration host factor binding sites in the lsrACBFG and lsrRK operon promoters.
In our previous work, we identified key promoter elements in a 414-bp sequence that encompasses the intergenic region between the divergent lsrACBFG and lsrRK operons in A. actinomycetemcomitans and showed that the expression of these operons was controlled by both the LsrR repressor and cAMP receptor protein (CRP) (27). We have now identified two additional sequences in the 414-bp intergenic region that closely resemble the E. coli consensus recognition sequence of the integration host factor (IHF) protein (YAANNNNTTGATW, where Y = T or C, N = any base, and W = A or T) (29). The first putative IHF binding sequence is located at nucleotides −65 to −77 relative to the lsrR start codon. The second sequence comprises nucleotides −66 to −78 relative to the putative lsrA start codon (27). These sequences have been designated IHFs1 and IHFs2, respectively (for details, see Fig. 2). Furthermore, examination of the A. actinomycetemcomitans genome sequence identified two open reading frames designated D11S_1364 and D11S_1626, which are annotated ihfA and ihfB, respectively (see Fig. S1A in the supplemental material). These genes encode proteins that exhibit 82% and 78% amino acid sequence similarity with E. coli IHFα and IHFβ, respectively (not shown) and 40% sequence identity compared to each other (see Fig. S1B).
FIG 2.

Binding of reconstituted IHF heterodimer to the lsrA and lsrR promoters. (A) Schematic representation of the lsrA-lsrR promoter region showing the putative binding regions for LsrR (black boxes), CRP (gray boxes), and IHF. PCR fragments used for EMSA reactions encompassing the lsrA and lsrR coding regions and the promoter sequence are numbered from I to XII, and the nucleotides contained by each fragment are indicated to the right. The numbering of the nucleotides comprising the 414-bp promoter sequence is relative to the start codon of lsrR. (B). PCR probes were incubated in the presence (+) or absence (−) of 4 μM reconstituted IHF heterodimer for 20 min at room temperature, and DNA-protein complexes (indicated by asterisks) were resolved in 6% polyacrylamide gels. A 200-bp DNA fragment from the psaA gene of Yersinia pseudotuberculosis (probe Yp) was used as the negative control.
IHF interacts with the lsrACDBFG-lsrRK intergenic region of A. actinomycetemcomitans.
To determine if the A. actinomycetemcomitans IHF protein interacts with the putative binding sites identified in the lsrA and lsrR promoters (lsrAP and lsrRP), A. actinomycetemcomitans IHFα and IHFβ were expressed as hexahistidine fusion proteins and purified as described in Materials and Methods. The purified individual subunits and a heterodimer made by incubating equimolar concentrations of the individual subunits are shown in Fig. 1. Using the purified heterodimer preparation, EMSA reactions were performed with a family of DNA fragments that spanned the 414-bp promoter sequence (Fig. 2A). Preliminary experiments showed that the protein and probe concentrations required for optimal formation of DNA-protein complexes were 4 μM and 50 fmol, respectively (data not shown). As shown in Fig. 2B, IHF bound only to probes that contained the putative binding sites described above (i.e., probes III, IV, V, VI, VIII, IX, X, and XI), whereas probes comprising other regions of the IGR or coding sequences of lsrA or lsrR did not form DNA-IHF complexes (i.e., probes I, II, VII, and XII). Interestingly, upon interaction with IHFs2, the IHF protein produced a single shifted band when incubated with probes III, IV, and VI and two shifted bands with probe V. Similarly, when binding to IHFs1, a single shifted band was detected for probe XI, but multiple shifted bands were observed using probes VIII, IX, and X. There are several possible explanations for this observation: IHF binding may be influenced by sequences flanking the core consensus sequence, IHF binding may be altered when the binding site resides close to the 5′ end of the probe, or alternatively, IHF may induce different DNA-protein conformations in the various probes. IHF binding was not detected using a 200-bp DNA fragment derived from the Yersinia pseudotuberculosis psaA gene, which represented a negative control. Together, these results indicate that IHF interacts with both the lsrRK and lsrACDBFG promoters.
FIG 1.
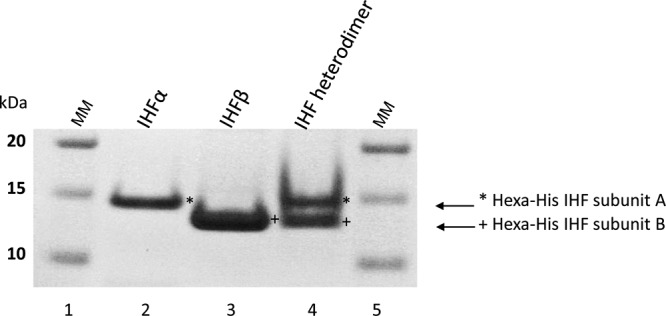
SDS-PAGE analysis of hexahistidine IHF α and β fusion subunits expressed in the cytoplasm of the E. coli LMG109 and BL21(DE3) strains. Lane 1, molecular mass marker (MM); lanes 2 and 3, hexahistidine IHFα and IHFβ subunits, respectively, purified by cobalt-based immobilized metal affinity chromatography; lane 4, IHFα and -β subunits from reconstituted IHF heterodimer (at a 1:1 molar ratio) as described in Materials and Methods.
IHF regulates the expression of the lsrACDBFG and lsrRK operons in A. actinomycetemcomitans.
Single-copy chromosomal insertions of the lsrR promoter-lacZ or lsrA promoter-lacZ transcriptional fusion constructs were introduced into A. actinomycetemcomitans 652 (to generate strains 652-TE-23 and 652-TE-75) and its isogenic ΔihfA and ΔihfB mutant strains (producing stains 652-TE78-23, 652-TE78-75, 652-TE79-23, and 652-TE79-75), as described in Materials and Methods. β-Galactosidase (β-Gal) activity expressed by these strains was measured to determine if the IHF protein influences the expression of the lsrACDBFG and/or lsrRK operon. As shown in Table 1, β-Gal expression directed by the lsrR promoter was induced by 4-fold in both the ΔihfA and ΔihfB mutant strains. In contrast, β-Gal expression directed from the lsrA promoter decreased by 4- to 6-fold in the ΔihfA or ΔihfB strain. Thus, IHF interacts with each promoter, but the binding of IHF differentially regulates the expression of the lsrRK and lsrACDBFG operons, reducing the expression of lsrRK and inducing lsrACDBFG.
TABLE 1.
Differential regulation of the lsrRK and lsrACDBFG promoters by IHF
| Strain | Genotype | β-Galactosidase activity (Miller units) |
|---|---|---|
| 652-TE-23 | Wild type-lsrR::lsrR23-lacZ | 33.3 ± 1.1 |
| 652-TE78-23 | ΔihfA-lsrR::lsrR23-lacZ | 123.5 ± 0.9 |
| 652-TE79-23 | ΔihfB-lsrR::lsrR23-lacZ | 133.0 ± 3.4 |
| 652-TE-75 | Wild type-lsrA::lsrA75-lacZ | 96.6 ± 3.7 |
| 652-TE78-75 | ΔihfA-lsrA::lsrA75-lacZ | 17.1 ± 0.8 |
| 652-TE79-75 | ΔihfB-lsrA::lsrA75-lacZ | 23.8 ± 0.6 |
Transcription of the lsrRK and lsrACDBFG operons initiates from multiple sites.
5′ rapid amplification of cDNA ends (5′-RACE) was employed to map the transcriptional start sites (TSSs) for both the lsrACDBFG and lsrRK promoters. As shown in Table 2, two TSSs (referred to as TSSR1 and TSSR2) were mapped upstream from the lsrR start codon. The main start site (TSSR1) mapped to nucleotide −38 and represented 86% of the positive clones that were identified by 5′-RACE. A secondary TSS (TSSR2) mapped to nucleotide −138. In support of these results, putative −10 and −35 elements are present upstream from both TSSs, at nucleotides −41 to −46 and −66 to −71 for TSS1 and −144 to −149 and −168 to −173 for TSS2. As shown in Fig. 3, the putative −35 element associated with TSS1 overlaps the IHFs1 binding site and the putative −35 element for TSS2 overlaps the CRP2 binding site that was previously identified (27). These results suggest that two promoters, RP1 and RP2, may drive the expression of lsrRK.
TABLE 2.
Transcriptional start sites identified by 5′-RACE
| TSS | Position (nucleotide)a | No. (%) of 5′-RACE clones identified |
|---|---|---|
| lsrR | ||
| TSS1 | −38 (A) | 30 (86) |
| TSS2 | −138 (A) | 5 (14) |
| lsrA | ||
| TSS1 | −184 (G) | 8 (29) |
| TSS2 | −210 (A) | 17 (60) |
| TSS3 | −262 (A) | 3 (11) |
The letter in parentheses indicates the nucleotide determined for that 5′ end.
FIG 3.

Double-stranded DNA sequence of the 414-bp lsrA-lsrR promoter region. The numbers on the top and bottom strands are relative to the lsrR and lsrA start codons, respectively. Inverted repeat sequences that represent LsrR binding sites are labeled “O1” to “O4.” Regions that resemble the consensus CRP binding site are shown in green boxes and are labeled “CRP1” and “CRP2.” Regions that resemble the consensus IHF binding site are shown in blue boxes and are labeled “IHFs1” and “IHFs2.” The transcriptional start sites are indicated with bent arrows. For lsrR, start sites are labeled “TSSRX”; for lsrA, start sites are labeled “TSSAX.” Predicted −35 and −10 sequences are shown in shown in red text. Potential Shine-Dalgarno sequences are labeled “SD.”
Three TSSs (designated TSSA1, TSSA2, and TSSA3) were mapped for the lsrACDBFG operon. The main TSS (TSSA2) was located 210 nucleotides upstream from the lsrA start codon and represented 60% of the clones that were identified by 5′-RACE. Two secondary TSSs mapped to nucleotides −184 (TSSA1) and −262 (TSSA3) relative to the putative lsrA start codon (27), and these sites represented 28% and 11% of the 5′-RACE clones identified, respectively (Table 2). Consistent with these assignments, putative −10 and −35 elements were associated with each TSS, located at nucleotides −191 to −196 and −212 to −217 for TSSA1, −215 to 220 and −238 to 243 for TSSA2, and −268 to 273 and −293 to 298 for TSSA3 (Fig. 3). Similar to the results for the lsrRK promoter, the −10 and −35 elements associated with TSSA1 overlap the LsrR binding sites O4 and O3, which were previously characterized (27). In addition, the −10 element associated with TSSA2 overlaps the LsrR binding site, O3, and its corresponding −35 element overlaps a previously identified CRP2 binding site. Finally, the −35 element associated with TSSA3 overlaps the LsrR binding site O2. Thus, similar to the lsrRK operon, multiple promoters (AP1, AP2, and AP3) may control the expression of lsrACDBFG. Overall, these results suggest that several regulatory proteins, including IHF, LsrR, and CRP, interact with multiple overlapping promoters to regulate the expression of the lsrACDBFG and lsrRK operons in A. actinomycetemcomitans.
Functional characterization of the lsrACDBFG and lsrRK promoters.
Our previous results suggested that the lsrRK promoter resides within nucleotides −1 to −255 upstream of the lsrR start codon (27). As shown in Fig. 4A, there was no significant difference in lacZ expression produced from constructs pATE23 and pATE100, confirming that no additional regulatory elements required for expression of lsrRK exist between nucleotides −256 and −414. In contrast, our previous results showed that the 255-bp fragment did not promote the expression of lsrA, but the entire 414-bp sequence was necessary for expression of lsrACDBFG (27). The results presented above now suggest that both the lsrRK and lsrACDBFG promoter regions contain multiple transcriptional initiation sites, and to demonstrate the function of the putative promoters associated with these TSSs, a series of DNA fragments encompassing portions of the 414-bp IGR were amplified by PCR and cloned individually into the low-copy-number promoterless lacZ plasmid pJT3 (Fig. 4). The recombinant plasmids and a control vector without a promoter were introduced into A. actinomycetemcomitans by electroporation, and β-galactosidase (β-Gal) activity was determined. As shown in Fig. 4A, a 5-fold reduction in β-Gal expression occurred when nucleotides −1 to −81 upstream from lsrR were deleted (see pATE68). This suggests that RP1 is the main promoter driving lsrRK expression under the growth conditions tested and is consistent with our results indicating that TSSR1 is the major TSS of the lsrRK operon (Table 1). However, even in the absence of RP1, significant β-Gal expression was observed, indicating that RP2 is also functional and contributes to lsrRK expression. Deletion of the LsrR binding sites O3 and O4 (pATE69 in Fig. 4A) resulted in a modest increase in lacZ expression, suggesting that O1 and O2 may be the primary sites for the LsrR-mediated repression of lsrRK that was previously reported (27).
FIG 4.

Schematic diagrams of the lsrR (A) and lsrA (B) transcriptional fusion constructs showing the putative binding regions for LsrR (black boxes labeled “O1” to “O4”), CRP (gray boxes), IHF (solid ovals), and transcriptional start sites (bent arrows). Fragments derived from the 414-bp promoter fragment are represented by thin lines, and the lsrA and lsrR coding sequences are shown by the large open arrows. The numbering of the nucleotides is relative to the lsrR start codon in panel A and from the putative lsrA start codon in panel B. The lacZ gene is indicated by a solid black arrow. β-Galactosidase activity for each construct is expressed as Miller units and was measured in A. actinomycetemcomitans (Aa) strains transformed individually with each plasmid and grown in BHI medium as described in Materials and Methods. Measurements were made at the mid-exponential phase of growth, and values are means of results from three independent experiments ± standard deviations.
As shown in Fig. 4B, little lacZ expression occurred in the strain containing pATE71, in which the putative promoter elements associated with the three TSSs identified above were deleted. In addition, similar low levels of lacZ expression were observed with pATE73, where IHFs2 was deleted but the upstream promoter elements were left intact. This suggests that IHF is a positive regulator of lsrA expression, consistent with the results shown in Table 1. This was further confirmed by restoring IHFs2 in construct pATE74, which produced significantly higher levels of lacZ expression. Interestingly, lacZ expression from pATE74 was approximately 5-fold higher than that observed with pATE75, which contains the entire 414-bp promoter fragment, suggesting that an additional lsrA negative regulatory element may exist in the region comprising nucleotides −1 to −44 relative to the putative lsrA start codon. At present, it is not known if this putative element functions at the transcriptional or posttranscriptional level.
IHF influences biofilm formation by A. actinomycetemcomitans.
We previously showed that deletion of lsrR or crp reduced biofilm growth of A. actinomycetemcomitans, suggesting that proper regulation of lsrACDBFG and lsrRK expression is important for the formation of A. actinomycetemcomitans biofilms (27). To determine if deletion of IHF affected biofilm formation, strains 652-TE-78 and 652-TE-79 (ΔihfA and ΔihfB, respectively) were cultured in flow cells, and the resulting biofilms were analyzed by confocal laser scanning microscopy using Volocity software. Representative reconstructed three-dimensional images of the biofilms formed by each strain are shown in Fig. 5, and measurements of biofilm biomass, depth, and surface coverage are presented in Table 3. Deletion of ihfA or ihfB resulted in a significant decrease in biofilm depth and an increase in total biomass relative to the wild type. Biofilms produced by the mutant strains appeared less structured than the wild type and more closely resembled a confluent layer of cells (and thus a higher total surface coverage) rather than the distinct microcolonies observed with wild-type strain 652. Consistent with this, average biomass per microcolony was significantly increased in the mutant strains since the Volocity software delineated fewer distinct microcolonies (Table 3). Complementation of the ΔihfA and ΔihfB strains with the low-copy-number plasmids pATE92 and pATE93 (copy number of approximately 5 to 10 per cell) containing the promoter and the structural ihfA and ihfB genes, respectively, resulted in a biofilm phenotype that was similar to the that of the wild type (Fig. 5D and E). However, total biomass of the complemented cultures was less than that of the parent strain and could be due to the effect of multiple copies of the ihfA and ihfB genes in these strains. Together, these results suggest that ihfA and ihfB are required for biofilm formation by A. actinomycetemcomitans and may act at least in part through the regulation of the lsrRK and lsrACDBFG loci.
FIG 5.

Three-dimensional reconstructions of biofilms formed by wild-type A. actinomycetemcomitans 652 (A) and isogenic ΔihfA (B) and ΔihfB (C) mutants. Mutations were complemented by a plasmid-borne copy of ihfA or ihfB in plasmids pATE92 and pATE93 (D and E), respectively. Biofilms were cultured in open flow cells as described in Materials and Methods and were visualized by confocal laser scanning microscopy. Image stacks were assembled and analyzed using Volocity image analysis software. The yellow and blue scale bars in the x-y, x-z, and y-z sections represent 50 μm and 10 μm, respectively.
TABLE 3.
Analysis of A. actinomycetemcomitans biofilmsa
| Strain | Maximal biofilm depth (μm) | Biomass (μm3 × 103) |
Total surface area (μm2 × 103) | |
|---|---|---|---|---|
| Avg of microcolony | Total | |||
| 652 | 12.19 ± 1.6 | 4.7 ± 1.5 | 594.9 ± 47.7 | 808.1 ± 25.1 |
| ΔihfA mutant | 7.59 ± 1.4§ | 67.6 ± 40† | 690.3 ± 58.8† | 784.0 ± 19.2 |
| ΔihfB mutant | 8.35 ± 1.1* | 95.7 ± 70‡ | 745.9 ± 21.8‡ | 891.5 ± 75.8§ |
| ΔihfA/pATE92 mutant | 14.89 ± 3.8 | 0.78 ± 0.3 | 376.6 ± 41.2‡ | 643.8 ± 30.3‡ |
| ΔihfB/pATE93 mutant | 12.11 ± 4.3 | 0.19 ± 0.05 | 283.6 ± 43.6‡ | 515.6 ± 61.9‡ |
Significance: *, P < 0.05; §, P < 0.01; †, P < 0.001; ‡, P < 0.0001.
DISCUSSION
In E. coli and S. Typhimurium, the products of the divergent lsrACDBFG-lsrRK operons are involved in a regulatory network that controls the uptake and processing of autoinducer 2 (AI-2). The structure of both operons is conserved in A. actinomycetemcomitans, and recent results from our laboratory showed that the LsrR repressor protein and cyclic AMP receptor protein (CRP) regulate the expression of both the lsrACDBFG and lsrRK operons in A. actinomycetemcomitans (27). As currently annotated, the A. actinomycetemcomitans D11S genome indicates that a 255-bp intergenic region resides between lsrACDBFG and lsrRK, and our present results indicate that all of the regulatory elements required to control the expression of lsrRK reside within this sequence. However, the 255-bp fragment did not function as the lsrA promoter, but instead, expression of lsrACDBFG required an additional 159 nucleotides comprising nucleotides +1 to +158 of the annotated lsrA sequence. This led us to speculate that the lsrA-lsrR intergenic region might comprise 414 bp and the actual lsrA start codon may be the ATG at nucleotides +159 to +161 of the annotated lsrA gene (27). Examination of the region from −256 to −414 identified a sequence (IHFs2) that resembled the E. coli consensus IHF binding site at nucleotides −66 to −78 upstream from the putative lsrA start codon, and EMSA showed that purified IHFα-IHFβ complex bound to probes containing this sequence. IHF functions as a positive regulator of lsrACDBFG expression, since deletion of IHFs2 and expression of a single-copy lsrA promoter-lacZ reporter in the ΔihfA and ΔihfB backgrounds both resulted in a significant reduction in lacZ expression. The mechanism of IHF-mediated induction lsrA operon expression is currently not known. However, one possibility is that DNA bending arising from IHF binding to IHFs2 stimulates transcription by effecting a conformational change that alters the structure of the DNA helix and facilitates open complex formation at one or more of the lsrA promoters (see below), similar to that described for PilvG by Parekh and Hatfield (46) or for systems such as phage lambda pL promoter (47) and the Caulobacter crescentus flbG promoter (48). 5′-RACE showed that the lsrA transcriptional start sites (TSSs) reside a considerable distance upstream from the putative lsrA start codon and IHFs2, with the most prominent TSS mapping to nucleotide −210 and two additional sites mapping to nucleotides −184 and −262 at lower frequency. This suggests that lsrACDBFG expression may be directed from three promoters, and putative −10 and −35 elements were associated with each of the TSSs identified. Interestingly, elements from each of the putative promoters overlap binding sites for the LsrR repressor, suggesting that a mechanism of LsrR-mediated repression of lsrACDBFG may be through restriction of RNA polymerase (RNAP) access to these promoters when LsrR is bound. In addition, the lsrAP2 promoter (TSS at −210) is similar to the lsrA promoter of E. coli (49) and resembles a class III CRP-dependent promoter, possessing two or more CRP binding sites that function to increase binding of RNAP and initiation of transcription (50). Similar to some class III CRP-dependent promoters, lsrAP2 contains one CRP site (CRP1) centered approximately 99 nucleotides upstream from the TSS and a second CRP site (CRP2) located around nucleotide −39 and overlapping the −35 element of lsrAP2. Indeed, two CRP binding sites located in similar positions in a semisynthetic CRP-dependent promoter resulted in a 2- to 4-fold increase in transcription compared to a semisynthetic promoter with a single CRP site overlapping the −35 element (51). Thus, the two CRP sites in lsrAP2 may act cooperatively where one α subunit C-terminal domain (αCTD) of RNAP interacts with the upstream CRP dimer subunit bound at CRP2, the second αCTD interacts with the downstream CRP dimer subunit bound at CRP1, and the downstream subunit of CRP at CRP2 interacts with RNAP α subunit N-terminal domain (αNTD) and σ subunit, consistent with known class III CRP-dependent promoters (Fig. 6).
FIG 6.
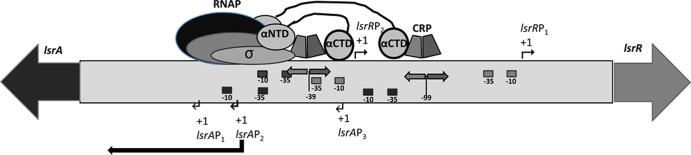
Model showing similarity of lsrAP2 to the class III CRP-dependent promoter, in which regulation of lsrAP2 may require the interaction of RNA polymerase (RNAP) α subunit C-terminal domains (αCTD) with bound CRP. The locations of the three putative lsrA promoters (lsrAP1, lsrAP2, and lsrAP3) are shown along the bottom of the figure, and the putative −10 and −35 sites for each are represented by black boxes. The two lsrR promoters (lsrRP1 and lsrRP2) are shown at the top of the figure, and the putative −10 and −35 sites for each are represented by gray boxes. CRP binding sites are indicated by opposing arrows.
Or current model is that the regulation of lsrACDBFG expression is highly complex and is directed by regulatory elements in a 414-bp region that extends upstream from the ATG codon that is currently annotated as nucleotides +159 to +161 of lsrA. Transcription is primarily driven by lsrAP2, which produces a transcript containing a 5′-untranslated region of 210 nucleotides. Transcription of the operon may be induced by DNA bending mediated by IHF binding to IHFs2 and by CRP, but it is also negatively controlled by LsrR and an unknown regulatory element in the −1 to −42 region relative to the putative start codon (compare plasmids pATE74 and pATE75 in Fig. 5). Currently it is not known if this additional regulation occurs at the level of transcription or by posttranscriptional mechanisms, such as the activity of small RNAs involved in regulation of polycistronic mRNA (52). Finally, we cannot conclusively exclude the possibility that IHFs2 and other regulatory elements reside within the coding sequence of the gene and that the lsrA start codon as currently annotated in the A. actinomycetemcomitans genome sequence is correct. Determination of the N-terminal protein sequence of native LsrA would address this possibility.
A second IHF binding site (IHFs1) that interacts with purified IHF complex was located at nucleotides −65 to −77 relative to the lsrR start codon. Interestingly, IHF differentially regulates the lsrRK and lsrACDBFG operons, since a single-copy genomic lsrRK promoter-lacZ fusion expressed in ΔihfA or ΔihfB backgrounds indicated that IHF negatively regulates lsrRK expression. Two TSS were identified by 5′-RACE for the lsrRK promoter: the most prevalent TSS mapped to nucleotide −26, and a second site mapped to residue −126 at much lower frequency, and putative −10 and −35 elements were associated with each TSS. Thus, lsrRK may be expressed by two promoters. Deletion of lsrRP1 (TSS at nucleotide −26) resulted in a significant reduction in lsrRK expression, consistent with the RACE results, suggesting that the main TSS occurred at residue −26. In addition, the putative −35 element of lsrRP1 overlaps IHFs1, suggesting that downregulation of lsrRK by IHF may arise from IHF blocking of RNAP at this promoter. Repression of transcription by IHF binding to sites that overlap core promoter elements has also been described for the ompB promoter of E. coli (53) and for bacteriophage λ PL2 and PR, bacterial ilvGMEDA PG1 and promoters of ihfA and ihfB genes (54–57). In addition, the downregulation of lsrRK by IHF may also indirectly contribute to the induction of lsrACDBFG by reducing the levels of the LsrR repressor protein that is produced.
In summary, we have shown that IHF differently regulates the expression of lsrRK and lsrACDBFG and that each operon may be transcribed by multiple promoters. The essential −10 and/or −35 elements for each of the promoters overlap binding sites for IHF, LsrR, or CRP, suggesting that RNAP competes for binding to the lsrA and lsrR promoters with these regulatory proteins. Finally, lsrAP2 primarily drives expression of lsrACDBFG, and this promoter resembles a class III CRP-dependent promoter in which the two CRP binding sites may function cooperatively to stimulate transcription.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the Public Health Service grant RO1DE14605 from the NIDCR.
Footnotes
Published ahead of print 14 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00006-14.
REFERENCES
- 1.Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. 2005. Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 43:5721–5732. 10.1128/JCM.43.11.5721-5732.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whittaker CJ, Klier CM, Kolenbrander PE. 1996. Mechanisms of adhesion by oral bacteria. Annu. Rev. Microbiol. 50:513–552. 10.1146/annurev.micro.50.1.513 [DOI] [PubMed] [Google Scholar]
- 3.Marsh PD. 2006. Dental plaque as a biofilm and a microbial community—implications for health and disease. BMC Oral Health 6(Suppl 1):S14. 10.1186/1472-6831-6-S1-S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marsh PD, Bradshaw DJ. 1997. Physiological approaches to the control of oral biofilms. Adv. Dent. Res. 11:176–185. 10.1177/08959374970110010901 [DOI] [PubMed] [Google Scholar]
- 5.Socransky SS, Haffajee AD. 1992. The bacterial etiology of destructive periodontal disease: current concepts. J. Periodontol. 63:322–331. 10.1902/jop.1992.63.4s.322 [DOI] [PubMed] [Google Scholar]
- 6.Fong KP, Gao L, Demuth DR. 2003. luxS and arcB control aerobic growth of Actinobacillus actinomycetemcomitans under iron limitation. Infect. Immun. 71:298–308. 10.1128/IAI.71.1.298-308.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Actis LA, Rhodes ER, Tomaras AP. 2003. Genetic and molecular characterization of a dental pathogen using genome-wide approaches. Adv. Dent. Res. 17:95–99. 10.1177/154407370301700122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James CE, Hasegawa Y, Park Y, Yeung V, Tribble GD, Kuboniwa M, Demuth DR, Lamont RJ. 2006. LuxS involvement in the regulation of genes coding for hemin and iron acquisition systems in Porphyromonas gingivalis. Infect. Immun. 74:3834–3844. 10.1128/IAI.01768-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuramitsu HK, He X, Lux R, Anderson MH, Shi W. 2007. Interspecies interactions within oral microbial communities. Microbiol. Mol. Biol. Rev. 71:653–670. 10.1128/MMBR.00024-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.James D, Shao H, Lamont RJ, Demuth DR. 2006. The Actinobacillus actinomycetemcomitans ribose binding protein RbsB interacts with cognate and heterologous autoinducer 2 signals. Infect. Immun. 74:4021–4029. 10.1128/IAI.01741-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schauder S, Shokat K, Surette MG, Bassler BL. 2001. The LuxS family of bacterial autoinducers: biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol. 41:463–476. 10.1046/j.1365-2958.2001.02532.x [DOI] [PubMed] [Google Scholar]
- 12.Zhao G, Wan W, Mansouri S, Alfaro JF, Bassler BL, Cornell KA, Zhou ZS. 2003. Chemical synthesis of S-ribosyl-l-homocysteine and activity assay as a LuxS substrate. Bioorg. Med. Chem. Lett. 13:3897–3900. 10.1016/j.bmcl.2003.09.015 [DOI] [PubMed] [Google Scholar]
- 13.Block PJ, Yoran C, Fox AC, Kaltman AJ. 1973. Actinobacillus actinomycetemcomitans endocarditis: report of a case and review of the literature. Am. J. Med. Sci. 266:387–392. 10.1097/00000441-197311000-00006 [DOI] [PubMed] [Google Scholar]
- 14.Page MI, King EO. 1966. Infection due to Actinobacillus actinomycetemcomitans and Haemophilus aphrophilus. N. Engl. J. Med. 275:181–188. 10.1056/NEJM196607282750403 [DOI] [PubMed] [Google Scholar]
- 15.Slots J, Reynolds HS, Genco RJ. 1980. Actinobacillus actinomycetemcomitans in human periodontal disease: a cross-sectional microbiological investigation. Infect. Immun. 29:1013–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zambon JJ, DeLuca C, Slots J, Genco RJ. 1983. Studies of leukotoxin from Actinobacillus actinomycetemcomitans using the promyelocytic HL-60 cell line. Infect. Immun. 40:205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsh PD. 2003. Are dental diseases examples of ecological catastrophes? Microbiology 149:279–294. 10.1099/mic.0.26082-0 [DOI] [PubMed] [Google Scholar]
- 18.Novak EA, Shao H, Daep CA, Demuth DR. 2010. Autoinducer-2 and QseC control biofilm formation and in vivo virulence of Aggregatibacter actinomycetemcomitans. Infect. Immun. 78:2919–2926. 10.1128/IAI.01376-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaeffer LM, Schmidt ML, Demuth DR. 2008. Induction of Aggregatibacter actinomycetemcomitans leukotoxin expression by IS1301 and orfA. Microbiology 154:528–538. 10.1099/mic.0.2007/012195-0 [DOI] [PubMed] [Google Scholar]
- 20.Shao H, Lamont RJ, Demuth DR. 2007. Autoinducer 2 is required for biofilm growth of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infect. Immun. 75:4211–4218. 10.1128/IAI.00402-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNab R, Lamont RJ. 2003. Microbial dinner-party conversations: the role of LuxS in interspecies communication. J. Med. Microbiol. 52:541–545. 10.1099/jmm.0.05128-0 [DOI] [PubMed] [Google Scholar]
- 22.Shao H, James D, Lamont RJ, Demuth DR. 2007. Differential interaction of Aggregatibacter (Actinobacillus) actinomycetemcomitans LsrB and RbsB proteins with autoinducer 2. J. Bacteriol. 189:5559–5565. 10.1128/JB.00387-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaz Z, Xavier KB, Miller T. 2009. The crystal structure of the Escherichia coli autoinducer-2 processing protein LsrF. PLoS One 28:e6820. 10.1371/journal.pone.0006820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marques JC, Lamosa P, Russell C, Ventura R, Maycock C, Semmelhack MF, Miller ST, Xavier KB. 2011. Processing the interspecies quorum-sensing signal autoinducer-2 (AI-2): characterization of phospho-(S)-4,5-dihydroxy-2,3-pentanedione isomerization by LsrG protein. J. Biol. Chem. 286:18331–18343. 10.1074/jbc.M111.230227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taga ME, Miller ST, Bassler BL. 2003. Lsr-mediated transport and processing of AI-2 in Salmonella Typhimurium. Mol. Microbiol. 50:1411–1427. 10.1046/j.1365-2958.2003.03781.x [DOI] [PubMed] [Google Scholar]
- 26.Xavier KB, Bassler BL. 2005. Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J. Bacteriol. 187:238–248. 10.1128/JB.187.1.238-248.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres-Escobar A, Juárez-Rodríguez MD, Lamont RJ, Demuth DR. 2013. Transcriptional regulation of Aggregatibacter actinomycetemcomitans lsrACDBFG and lsrRK operons and their role in biofilm formation. J. Bacteriol. 195:56–65. 10.1128/JB.01476-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig N, Nash HA. 1984. E. coli integration host factor binds to specific sites in DNA. Cell 39:707–716. 10.1016/0092-8674(84)90478-1 [DOI] [PubMed] [Google Scholar]
- 29.Friedman DI. 1988. Integration host factor: a protein for all reasons. Cell 55:545–554. 10.1016/0092-8674(88)90213-9 [DOI] [PubMed] [Google Scholar]
- 30.Goodrich JA, Schwartz ML, McClure WR. 1990. Searching for and predicting the activity of sites for DNA binding proteins: compilation and analysis of the binding sites for Escherichia coli integration host factor (IHF). Nucleic Acid Res. 18:4993–5000. 10.1093/nar/18.17.4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Rijn PA, van de Putte P, Goosen N. 1991. Analysis of the IHF binding site in the regulatory region of bacteriophage Mu. Nucleic Acid Res. 19:2825–2834. 10.1093/nar/19.11.2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nash HA. 1981. Integration and excision of bacteriophage lambda: the mechanism of conservation site specific recombination. Annu. Rev. Genet. 15:143–167. 10.1146/annurev.ge.15.120181.001043 [DOI] [PubMed] [Google Scholar]
- 33.Mangan MW, Lucchini S, Danino V, Cróinín TO, Hilton JC, Dorman CJ. 2006. The integration host factor (IHF) integrates stationary-phase and virulence gene expression in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 59:1831–1847. 10.1111/j.1365-2958.2006.05062.x [DOI] [PubMed] [Google Scholar]
- 34.Yona-Nadler C, Umanski T, Aizawa S, Friedberg D, Rosenshine I. 2003. Integration host factor (IHF) mediates repression of flagella in enteropathogenic and enterohaemorrhagic Escherichia coli. Microbiology 149:877–884. 10.1099/mic.0.25970-0 [DOI] [PubMed] [Google Scholar]
- 35.Marques MV, Gober JW. 1995. Activation of a temporally regulated Caulobacter promoter by upstream and downstream sequence elements. Mol. Microbiol. 16:279–289. 10.1111/j.1365-2958.1995.tb02300.x [DOI] [PubMed] [Google Scholar]
- 36.Beach MB, Osuna R. 1998. Identification and characterization of the fis operon in enteric bacteria. J. Bacteriol. 180:5932–5946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goshima N, Kano Y, Tanaka H, Kohno K, Iwaki T, Imamoto F. 1994. IHF suppresses the inhibitory effect of H-NS on HU function in the hin inversion system. Gene 141:17–23. 10.1016/0378-1119(94)90122-8 [DOI] [PubMed] [Google Scholar]
- 38.Blomfield IC, Kulasekara DH, Eisenstein BI. 1997. Integration host factor stimulates both FimB- and FimE-mediated site-specific DNA inversion that controls phase variation of the type 1 fimbria expression in Escherichia coli. Mol. Microbiol. 23:705–717. 10.1046/j.1365-2958.1997.2241615.x [DOI] [PubMed] [Google Scholar]
- 39.Goodman SD, Obergfell KP, Jurcisek JA, Downey JS, Ayala EA, Tjokro N, Li B, Justice SS, Bakaletz LO. 2011. Biofilms can be dispersed by focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal Immunol. 4:625–637. 10.1038/mi.2011.27 [DOI] [PubMed] [Google Scholar]
- 40.Brandstetter KA, Jurcisek JA, Goodman SD, Bakaletz LO, Das S. 2013. Antibodies directed against integration host factor mediated biofilm clearance from Nanospore. Laryngoscope 123:2626–2632. 10.1002/lary.24183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gustave JE, Jurcisek JA, McCoy KS, Goodman SD, Bakaletz LO. 2013. Targeting bacterial integration host factor to disrupt biofilms associated with cystic fibrosis. J. Cyst. Fibros. 12:384–389. 10.1016/j.jcf.2012.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 43.Juárez-Rodríguez MD, Torres-Escobar A, Demuth DR. 2013. Construction of new cloning, lacZ reporter and scarless-markerless suicide vectors for genetic studies in Aggregatibacter actinomycetemcomitans. Plasmid 69:211–222. 10.1016/j.plasmid.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torres-Escobar A, Juárez-Rodríguez MD, Curtiss R. 2010. Biogenesis of Yersinia pestis PsaA in recombinant attenuated Salmonella Typhimurium vaccine (RASV) strain. FEMS Microbiol. Lett. 302:106–113. 10.1111/j.1574-6968.2009.01827.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 46.Parekh BS, Hatfield GW. 1996. Transcriptional activation by protein-induced DNA bending: evidence for a DNA structural transmission model. Proc. Natl. Acad. Sci. U. S. A. 93:1173–1177. 10.1073/pnas.93.3.1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giladi H, Gottesman M, Oppenheim AB. 1990. Integration host factor stimulates the phage lambda pL promoter. J. Mol. Biol. 213:109–121. 10.1016/S0022-2836(05)80124-X [DOI] [PubMed] [Google Scholar]
- 48.Gober JW, Shapiro L. 1990. Integration host factor is required for the activation of developmentally regulated genes in Caulobacter. Genes Dev. 4:1494–1504. 10.1101/gad.4.9.1494 [DOI] [PubMed] [Google Scholar]
- 49.Byrd CM. 2011. Local and global gene regulation analysis of the autoinducer-2 mediated quorum sensing mechanism in Escherichia coli. Ph.D. thesis University of Maryland, College Park, MD [Google Scholar]
- 50.Busby S, Ebright RH. 1999. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 293:199–213. 10.1006/jmbi.1999.3161 [DOI] [PubMed] [Google Scholar]
- 51.Belyaeva TA, Rhodius VA, Webster CL, Busby SJ. 1998. Transcription activation at promoters carrying tandem DNA sites for the Escherichia coli cyclic AMP receptor protein: organisation of the RNA polymerase alpha subunits. J. Mol. Biol. 277:789–804. 10.1006/jmbi.1998.1666 [DOI] [PubMed] [Google Scholar]
- 52.Rice JB, Balasubramanian D, Vanderpool CK. 2012. RNA binding-site multiplicity involved in translational regulation of a polycistronic mRNA. Proc. Natl. Acad. Sci. U. S. A. 109:E2691–E2698. 10.1073/pnas.1207927109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsui P, Huang L, Freundlich M. 1991. Integration host factor binds specifically to multiple sites in the ompB promoter of Escherichia coli and inhibits transcription. J. Bacteriol. 173:5800–5807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giladi H, Murakami K, Ishihama A, Oppenheim AB. 1996. Identification of an UP element within the IHF binding site at the PL1-PL2 tandem promoter of bacteriophage λ. J. Mol. Biol. 260:484–491. 10.1006/jmbi.1996.0416 [DOI] [PubMed] [Google Scholar]
- 55.Kur J, Hasan N, Szybalski W. 1989. Physical and biological consequences of interactions between integration host factor (IHF) and coliphage lambda late P′R promoter and its mutants. Gene 81:1–15. 10.1016/0378-1119(89)90331-4 [DOI] [PubMed] [Google Scholar]
- 56.Tsui P, Freundlich M. 1988. Integration host factor binds specifically to sites in the ilvGMEDA operon in Escherichia coli. J. Mol. Biol. 203:817–820. 10.1016/0022-2836(88)90212-4 [DOI] [PubMed] [Google Scholar]
- 57.Aviv M, Giladi H, Schreiber G, Oppenheim AB, Glaser G. 1994. Expression of the genes coding for the Escherichia coli integration host factor are controlled by growth phase, rpoS, ppGpp and by autoregulation. Mol. Microbiol. 14:1021–1031. 10.1111/j.1365-2958.1994.tb01336.x [DOI] [PubMed] [Google Scholar]
- 58.Kolenbrander PE, London J. 1993. Adhere today, here tomorrow: oral bacterial adherence. J. Bacteriol. 175:3247–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lamont RJ, Jenkinson HF. 1998. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62:1244–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paster BJ, Olsen I, Aas JA, Dewhirst FE. 2006. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol. 2000 42:80–87. 10.1111/j.1600-0757.2006.00174.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.