

Abstract
Persistent Helicobacter pylori infection induces chronic inflammation in the human gastric mucosa, which is associated with development of peptic ulceration, gastric atrophy, and gastric adenocarcinoma. It has been postulated that secretion of immunomodulatory molecules by H. pylori facilitates bacterial persistence, and membrane vesicles (MV), which have the potential to cross the gastric epithelial barrier, may mediate delivery of these molecules to host immune cells. However, bacterial MV effects on human immune cells remain largely uncharacterized to date. In the present study, we investigated the immunomodulatory effects of H. pylori MV with and without the vacuolating cytotoxin, VacA, which inhibits human T cell activity. We show a high degree of variability in the toxin content of vesicles between two H. pylori strains (SS1 and 60190). Vesicles from the more toxigenic 60190 strain contain more VacA (s1i1 type) than vesicles from the SS1 strain (s2i2 VacA), but engineering the SS1 strain to produce s1i1 VacA did not increase the toxin content of its vesicles. Vesicles from all strains tested, including a 60190 isogenic mutant null for VacA, strongly induced interleukin-10 (IL-10) and IL-6 production by human peripheral blood mononuclear cells independently of the infection status of the donor. Finally, we show that H. pylori MV induce T cell apoptosis and that this is enhanced by, but not completely dependent on, the carriage of VacA. Together, these findings suggest a role for H. pylori MV in the stimulation of innate pro- and anti-inflammatory responses and in the suppression of T cell immunity.
INTRODUCTION
Helicobacter pylori is a Gram-negative microaerophilic bacterium that colonizes the stomachs of around half the world's population. Infection persists lifelong if untreated, stimulating chronic inflammation of the gastric mucosa (1, 2). Host, environmental, and bacterial factors affect disease risk, with carriage of more virulent bacterial strains associated with a higher incidence of ulceration and cancer (1, 2).
H. pylori produces multiple virulence factors, including the vacuolating cytotoxin VacA, which is polymorphic in its signal (s), intermediate (i), and middle (m) regions. The s1, i1, and m1 alleles confer higher toxin activity and broader cell specificity, while the s2, i2, and m2 alleles are less toxigenic (3–5). Carriage of strains producing more toxigenic forms of VacA is associated with a higher disease incidence (5, 6).
H. pylori is known to have profound effects on gastric epithelial cells, including stimulation of interleukin-8 (IL-8) production (7, 8), and toxic effects of VacA on epithelial cells are now well characterized (9). In vitro, the toxin induces vacuolation (10) and apoptosis (11–14) in epithelial cell lines, and oral administration of purified VacA causes epithelial damage in experimental animals (15–17).
During persistent colonization, H. pylori exerts both pro- and anti-inflammatory effects on the human immune system (18–21). However, since the majority of bacteria remain in the mucus overlying the gastric epithelial cell layer in the stomach lumen (22), it is unclear how bacteria or bacterial products cross this barrier to access cells of the host immune system and stimulate these responses. Secreted proteins might be readily broken down in the harsh gastric environment and might access host cells in relatively small quantities. VacA is now known to inhibit T cell proliferation and IL-2 production (23–27), but while these effects are striking in vitro, the secreted toxin would likely be present in the stomach lumen at low concentrations, and whole bacteria (carrying VacA on their surfaces) are not generally thought to penetrate the epithelial barrier.
In common with other Gram-negative bacteria, H. pylori constitutively produces membrane vesicles (MV) (28, 29). These 20- to 200-nm diameter “blebs” from the outer surface of the bacterium contain mainly outer membrane and periplasmic components, including lipopolysaccharide (LPS), peptidoglycan, and proteins (30, 31). H. pylori MV are known to carry VacA and can deliver active toxin to epithelial cells, although the toxin is also secreted conventionally (29). Independently of VacA status, and in common with MV from other bacteria, H. pylori MV also stimulate proliferation, IL-8 secretion, and apoptosis in epithelial cells (32, 33). There is growing interest in the role of membrane vesicles in bacterial pathogenesis, both in H. pylori and more widely. However, MV-mediated bacterial effects on cells of the host immune system are poorly understood to date. H. pylori is known to secrete multiple immune-modulatory proteins, and membrane vesicle-mediated delivery of these might represent a possible route by which H. pylori can exert long-range effects on host immune cells, delivering discrete concentrated packages of a “cocktail” of H. pylori molecules, including VacA and other virulence factors.
In the present study, we set out to characterize the effects of H. pylori MV on human immune cells and to determine which, if any, are due to the carriage of VacA. We show that there are substantial differences in the quantity of VacA associated with MV between the more toxigenic 60190 strain and less toxigenic SS1. H. pylori MV from both strains strongly stimulate release of pro- and anti-inflammatory cytokines from human peripheral blood mononuclear cells (PBMCs), independently of VacA, and induce apoptosis in T cells. Toxic effects on T cells are enhanced by, but not dependent on, the presence of VacA.
MATERIALS AND METHODS
H. pylori strains.
The strains used in this study were 60190 (ATCC 49503, VacA s1i1m1), SS1WT (VacA s2i2m2) (34), 60190 ΔvacA (35), and SS1s1i1. The SS1s1i1 mutant was generated by replacing the s2i2-carrying region of SS1 wild-type vacA with the s1i1-carrying region from strain 60190 by natural transformation with plasmid pJR100 and allelic exchange by homologous recombination and chloramphenicol resistance marker rescue following established methods (5). The SS1 vacA open reading frame was replaced with the 60190 sequence (GenBank accession no. U05676) up to nucleotide 1684.
MV purification.
H. pylori strains from frozen stocks were grown for 24 h on blood agar base 2 plates containing 7% (vol/vol) defibrinated horse blood (Oxoid Ltd., Basingstoke, United Kingdom) at 37°C in a microaerobic cabinet and then passaged once onto fresh blood plates and grown for a further 24 h. Bacteria were then inoculated into 500 ml brain heart infusion broth (Oxoid Ltd., Basingstoke, United Kingdom) containing 0.2% β-cyclodextrin and shaken at 150 rpm under microaerophilic conditions for 48 h. Bacterial cells were removed by centrifugation at 6,000 × g for 10 min and confirmed helical by Gram staining and microscopy. The culture supernatants were passaged sequentially through 0.45-μm and then 0.20-μm syringe filters. MV were precipitated at room temperature using 40% (wt/vol) ammonium sulfate for 1 h and then harvested by centrifugation at 10,000 × g for 15 min. The pellet containing MV was resuspended in 20 ml low-endotoxin Dulbecco's phosphate-buffered saline (PBS) (Sigma-Aldrich Co. Ltd., Gillingham, Dorset, United Kingdom) and MV harvested by ultracentrifugation at 100,000 × g for 2 h at 4°C. The pellet containing purified MV was resuspended in 500 μl PBS, adjusted to a 500-μg/ml total protein concentration following quantification using the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL, USA), and stored at −20°C until use.
TEM.
All MV preparations were confirmed to be free of bacterial cells and flagella by transmission electron microscopy (TEM). MV samples on 200-mesh Formvar-copper grids (Agar Scientific) were negatively stained using 0.5% uranyl acetate and visualized using a Fei Tecnai Biotwin electron microscope.
Preparation of bacterial water extracts.
H. pylori strains were grown for 24 h on blood agar base 2 plates as described above, and then bacteria were harvested into 2 ml PBS, pelleted by centrifugation, and resuspended in 200 μl sterile distilled water (dH2O). After vigorous vortexing, bacterial suspensions were incubated at room temperature for 30 min and then centrifuged at 10,000 × g for 5 min. Supernatants were stored at −20°C until use.
SDS-PAGE and Western blotting.
MV proteins (5 μg/lane) were reduced by boiling in buffer containing SDS and dithiothreitol (DTT) and then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto nitrocellulose membrane, and probed with rabbit polyclonal anti-VacA p33 and anti-VacA p55 sera (each at 1:10,000) (generated in-house at The University of Nottingham Biomedical Services Unit) followed by horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:10,000). Bands were visualized using Amersham ECL Prime Western blotting reagent (GE Healthcare).
Recombinant VacA purification.
Sequences encoding the p33 and p55 subunits of VacA were cloned from 60190 genomic DNA, recombinantly expressed in Escherichia coli, and purified essentially following the methods of González-Rivera et al. (2010) (36) except that both p33 and p55 domains were purified under denaturing conditions and refolded by dialysis prior to reconstitution of the toxin. The reconstituted toxin was highly active, inducing profound vacuolation of AGS cells and inhibition of Jurkat proliferation and IL-2 secretion, consistent with previously published reports (36).
Vacuolation assay.
RK13 cells (ATCC CCL-37, a rabbit kidney cell line) were maintained in Ham's F-12 medium supplemented with 2 mM l-glutamine and 10% fetal bovine serum (Sigma-Aldrich Co. Ltd., Gillingham, Dorset, United Kingdom). Cells were seeded into 96-well tissue culture-treated flat-bottom plates (Nalge Nunc International, Thermo Fisher Scientific Inc.) at 1 × 104/well and treated with 0 to 4 μg/ml purified recombinant VacA, 50 μg/ml MV, or appropriate buffer controls in the presence of 10 mM NH4Cl for 4 h. Vacuolation extent was assessed by light microscopy, with the mean proportion of vacuolated cells from 4 or 5 randomly selected microscopy fields across duplicate wells calculated for a minimum of 100 cells per condition. Representative data from two independent replicate experiments are shown.
Stimulation of PBMCs by MV.
Blood was collected from patients into EDTA Vacutainers (Greiner Bio-One, Stonehouse, United Kingdom). Patients were attending the Queen's Medical Centre, Nottingham, for routine upper gastrointestinal endoscopy, usually for investigation of dyspepsia, but were otherwise healthy. Written informed consent was obtained from all patients under the approval of the Nottingham Research Ethics Committee 2. Male and female patients with an age range of 17 to 72 (mean age, 45) were included. H. pylori infection status was determined by rapid urease test and bacterial culture from gastric biopsy specimens taken during endoscopy. For healthy donor studies, blood was taken from healthy volunteers of unknown H. pylori infection status, with written informed consent, also under approval from the University of Nottingham Medical School Ethics Committee. Blood cells were separated from plasma by centrifugation (725 × g, 10 min) and resuspended in washing medium (15 ml RPMI 1640 supplemented with 2% fetal bovine serum, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 2 mM l-glutamine [all from Sigma]). The cell suspension was carefully layered on 5 ml Histopaque 1077 (Sigma) and centrifuged at 725 × g for 20 min. PBMCs were collected from the interface between the Histopaque and RPMI layers and washed twice by suspension in washing medium and centrifugation at 400 × g for 5 min. Purified PBMCs were suspended in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 2 mM l-glutamine, and cell viability was confirmed to be >99% by trypan blue staining and microscopy. PBMCs at 1 × 106/ml were coincubated in sterile 96-well plates with 0 to 25 μg/ml MV or medium/buffer negative controls at 37°C in 5% CO2 for 24 or 48 h as indicated. The T cell mitogen concanavalin A was used at 5 μg/ml as a positive control. Cytokine concentrations in culture supernatants were determined by enzyme-linked immunosorbent assay (ELISA) (eBioscience) following the manufacturer's instructions. ELISA sensitivity limits were calculated as means from triplicate wells containing no cytokine plus 3 times the standard deviation (SD) and were <3.2 pg/ml (IL-10), <5 pg/ml (IL-6 and IL-2), <1 pg/ml (IL-4 and IL-12p70), and <4 pg/ml (gamma interferon [IFN-γ]).
Purification of CD4+ cells from human blood.
CD4+ cells were purified from healthy volunteer PBMCs using an EasySep Negative Selection human CD4+ T cell enrichment kit (Stemcell Technologies) following the manufacturer's instructions.
T cell inhibition and apoptosis assays.
Jurkat T cells (a generous gift from R. McIntosh, Academic Clinical Oncology Department, University of Nottingham) were maintained in RPMI 1640 supplemented with 2 mM l-glutamine and 10% fetal bovine serum at 37°C in 5% CO2.
For inhibition assays, Jurkat or CD4+ T cells enriched from PBMCs at 1 × 106/ml and 200 μl per well in a sterile 96-well plate were pretreated for 1 h with MV, recombinant VacA, or appropriate buffer controls at the indicated concentrations and then stimulated with 50 ng/ml phorbol myristate acetate (PMA) and 1 μM ionomycin for 48 h at 37°C in 5% CO2. To determine the extent of proliferation, CellTiter 96 Aqueous One reagent (Promega) was used following the manufacturer's instructions. This reagent, based on the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, causes a color change directly proportional to the number of metabolically active cells in the solution. The IL-2 concentration in each culture supernatant was quantified by ELISA (eBioScience). For apoptosis, viability, and cytotoxicity assays, Jurkat or CD4+ T cells enriched from PBMCs at 1 × 105/ml and 100 μl per well in a sterile 96-well plate were incubated for 24 h at 37°C in 5% CO2 in the presence of MV, recombinant VacA, or appropriate buffer controls at the indicated concentrations. Proteinase K-treated MV (and an equivalent buffer-only control) were preincubated with 100 μg/ml proteinase K for 60 min at 37°C following the methods of Bomberger et al. (37), and then proteinase K was inactivated using P1860 protease inhibitor cocktail (Sigma) before adding the MV to the T cells. An ApoTox-Glo triplex assay (Promega) was used following the manufacturer's instructions to simultaneously determine viability (live-cell protease activity, resulting in a fluorescent signal proportional to the number of living cells per well), cytotoxicity (dead-cell protease activity, resulting in a fluorescent signal proportional to the number of cells that have lost membrane integrity per well), and apoptosis (a luminescent signal proportional to caspase 3/7 activity per well). Data for the first two parameters were combined to give a dead/live cell, or cytotoxicity/viability, ratio for each well.
Statistical analysis.
Data were analyzed by one-way analysis of variance and Tukey's posttest unless otherwise stated. Graphs show representatives of two to four independently repeated experiments, carried out in triplicate unless otherwise stated, and error bars show standard deviations for replicates.
RESULTS
H. pylori MV were harvested from filtered culture supernatants by ultracentrifugation. Before determining their effects on immune cells, we characterized them by TEM, SDS-PAGE, and Western blotting to quantify VacA.
MV with or without VacA have similar morphologies and are produced in similar quantities.
Strains 60190 (expressing s1i1m1-type VacA) and an isogenic mutant null for VacA (69010 ΔvacA) produced similar quantities of MV in liquid culture. The yield was approximately 3.8 mg MV protein per liter culture after 48 h of culture (final optical density at 600 nm [OD600], 0.32 to 0.39). Strains SS1WT (expressing s2i2m2 VacA) and SS1s1i1 (an isogenic mutant engineered to express s1i1m2 VacA) produced 3.6 to 4.4 mg MV protein per liter culture after 48 h (final OD600, 0.14 to 0.20). All four strains were confirmed to be >99% helical after 48 h of liquid culture. The purified MV from all four strains had similar dimensions and morphologies, and all preparations were free of bacteria, bacterial cell wall debris, and flagella when visualized by transmission electron microscopy (a representative image is shown in Fig. 1).
FIG 1.
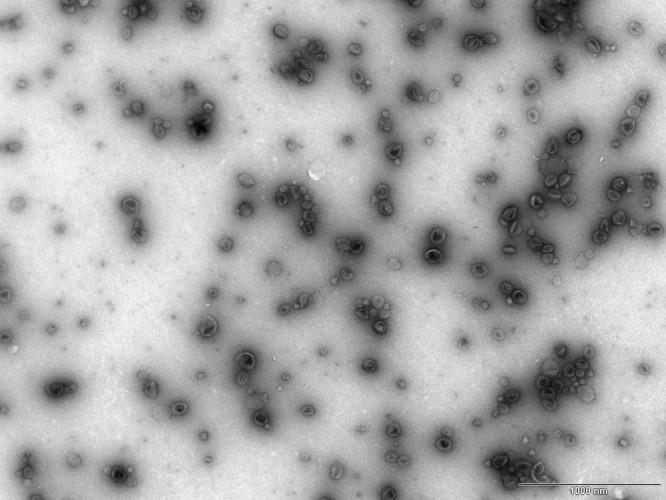
Electron micrograph showing the size distribution, morphology, and purity of 60190 MV. Purified MV were diluted negatively stained using 0.5% uranyl acetate and visualized using a Tecnai FEI electron microscope to confirm vesicle purity. All MV were free from bacteria, flagella, and other bacterial debris. A representative image of 60190 MV at a magnification of ×20,500 is shown. Scale bar, 1,000 nm.
There is substantial strain variation in the quantity of VacA associated with MV.
Equal concentrations of 60190 MV with and without VacA were analyzed by SDS-PAGE and Western blotting for VacA (Fig. 2). Although secreted p88 VacA sometimes separates into p33 and p55 domains, all MV-associated VacA migrated at a position consistent with intact p88 on SDS-PAGE. VacA protein was completely absent from 60190 ΔvacA MV, as expected. Protein profiles of MV from the two strains were otherwise indistinguishable by SDS-PAGE, consistent with the isogenic nature of the ΔvacA mutant.
FIG 2.
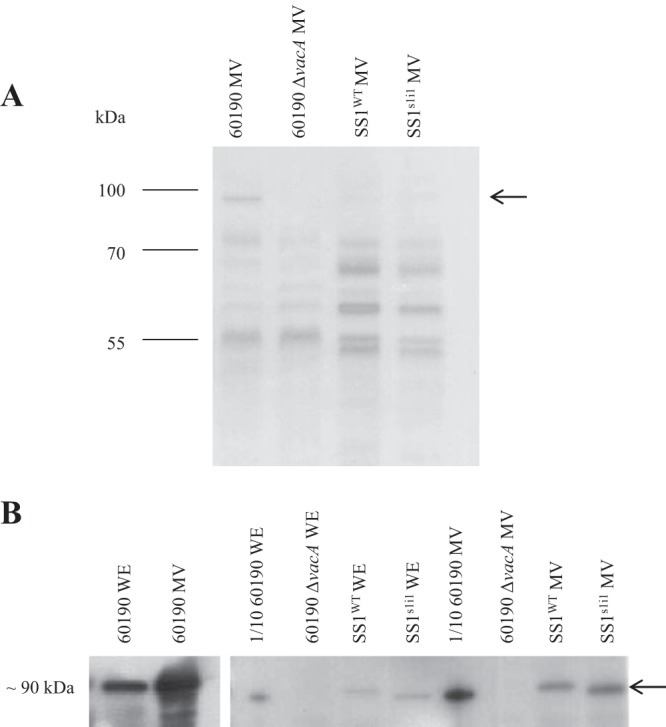
Comparison of protein profiles and VacA contents in membrane vesicles (MV) from 60190 and SS1 strains. Reduced proteins (5 μg/lane) were separated on 12% SDS-polyacrylamide gels and Coomassie blue stained (A) or transferred onto nitrocellulose and immunoblotted for VacA (B). Isogenic mutant MV contained proteins similar to those in MV purified from their respective parental strains, but MV from the 60190 and SS1 strain backgrounds had different protein profiles. VacA (arrow) was barely detectable in MV from the SS1 strains but present in much larger quantities in 60190 MV. For comparison, water extracts (WE) from whole bacteria are shown. High VacA quantities in 60190 samples caused overexposure of blots, so 0.5-μg/lane (1/10) 60190 samples were analyzed directly alongside SS1 samples, and 5-μg/lane 60190 samples developed in parallel on separate film are also shown (left).
MV from SS1WT and SS1s1i1 had protein profiles markedly different from that of the 60190 MV, reflecting the high level of strain-to-strain variability in H. pylori. When crude water extracts from whole bacteria were compared by Western blotting, the 60190 extracts contained substantially more VacA than the SS1 extracts. MV from strain 60190 contained very high concentrations of VacA, but in MV from strains SS1WT and SS1s1i1 VacA was barely detectable on the same blot. In order to compare VacA contents in 60190 and SS1 preparations on the same blot without overexposure, 60190 samples had to be diluted 10-fold, indicating marked strain-to-strain variation in the quantities of VacA associated with MV (Fig. 2).
VacA remains active when vesicle associated.
As expected, RK13 cells were heavily vacuolated (49% of cells were vacuolated) following 4 h of incubation in the presence of 50 μg/ml 60190 MV (Fig. 3A), but application of 60190 ΔvacA MV did not induce significant vacuolation above background levels (3% vacuolated cells with 60190 ΔvacA MV versus 4% with PBS only) (Fig. 3B and E). SS1WT MV induced minimal vacuolation (13%), and SS1s1i1 MV induced vacuolation at an intermediate level (27%) (Fig. 3C to E), consistent with the delivery of small quantities of s2i2 and s1i1 VacA, respectively.
FIG 3.
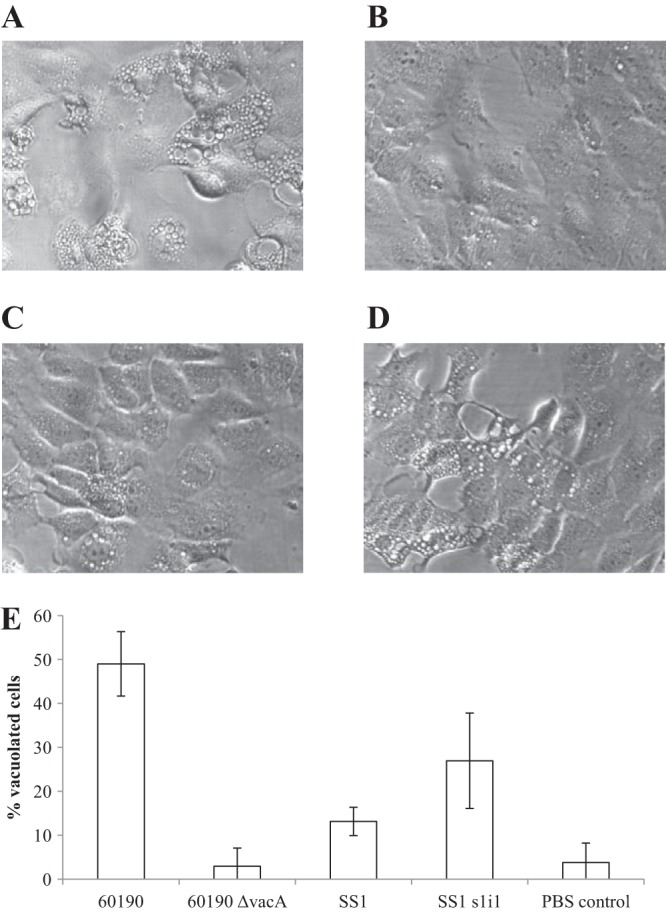
MV containing active VacA cause vacuolation in RK13 cells. (A to D) RK13 cells were incubated for 4 h in the presence of 50 μg/ml MV purified from strain 60190 (carrying active s1i1m1 VacA) (A), 60190 ΔvacA (B), SS1WT (carrying s2i2m2 VacA with impaired vacuolating activity) (C), or SS1s1i1 (carrying relatively small quantities of active s1i1m2 VacA) (D). (E) Mean proportion of heavily vacuolated cells (±SD) across a minimum of four randomly selected microscopy fields (>100 cells) per condition.
MV stimulate release of IL-6 and IL-10 (but not IL-2, IL-4, IL-12p70, or IFN-γ) from PBMCs in a dose-dependent manner.
Having characterized the VacA protein contents and vacuolating activities of 60190, 60190 ΔvacA, SS1WT, and SS1s1i1 MV, we aimed to investigate effects of MV on human immune cells. First, peripheral blood mononuclear cells (PBMCs) were purified from healthy donors of unknown H. pylori infection status and cultured in the presence of 0 to 12.5 μg/ml SS1or 60190 MV for 48 h. Cytokines secreted into the culture medium were quantified by ELISA. H. pylori MV strongly stimulated the release of IL-10 and IL-6 from PBMCs, in a dose-dependent manner (Fig. 4A). Incubation with up to 25 μg/ml MV did not stimulate IL-2, IL-4, IL-12p70, or IFN-γ secretion at concentrations above the limit of sensitivity of the assays (data not shown), indicating that the cytokine response in PBMCs from healthy donors was largely innate rather than T cell derived. H. pylori MV also stimulated a dose-dependent proliferative response in human PBMCs, which was maximal at approximately 3 μg/ml (see Fig. S1 in the supplemental material).
FIG 4.

MV stimulate IL-10 and IL-6 production from PBMCs. (A) PBMCs from healthy donors of unknown H. pylori infection status were incubated for 48 h in the presence of 0 to 12.5 μg/ml SS1WT or 60190 MV, and then IL-10 (left) and IL-6 (right) concentrations in culture supernatants were measured by ELISA. Similar data were obtained from independent experiments using PBMCs from three different donors; representative data from one donor are shown. (B) PBMCs from H. pylori-infected (closed circles) and uninfected (open circles) patients were incubated for 24 h in the presence of 10 μg/ml MV, 5 μg/ml concanavalin A, or an equivalent volume of PBS, then IL-10 (left) and IL-6 (right) concentrations in culture supernatants were measured by ELISA. NS, not significant.
Next, we compared the effects of MV at 10 μg/ml from the two wild-type H. pylori strains, 60190 (VacA s1i1m1) and SS1 (VacA s2i2m2), on PBMCs from a panel of patients presenting at the Queens Medical Centre, Nottingham, with gastric symptoms for routine endoscopy. PBMCs were incubated for 24 h in the presence of 5 μg/ml concanavalin A (a T cell mitogen), 10 μg/ml MV, or a buffer-only control, and then IL-10 and IL-6 levels in culture medium were determined by ELISA. MV from both strains stimulated high levels of IL-6 and IL-10 production by PBMCs from all patients tested (P < 0.001 compared with PBS-treated control cells) (Fig. 4B).
We hypothesized that MV might activate a memory T cell response in PBMCs from H. pylori-infected patients. In order to determine whether the strong cytokine response to H. pylori MV was innate or adaptive, we stratified the data according to the infection status of the patients (Fig. 4B). There was no significant difference in the levels of cytokines produced by PBMCs from infected and uninfected patients in response to H. pylori MV (P = 0.26 for IL-10 and P = 0.34 for IL-6 by unpaired Student t tests comparing infected-group against uninfected-group responses to 60190 MV). This indicates that human PBMCs mount a strong, innate cytokine response to H. pylori MV, likely due to the delivery of concentrated pathogen-associated molecular patterns (PAMPs) such as LPS and peptidoglycan.
Since VacA can directly activate mast cells to produce IL-6 and IL-10 (38), we hypothesized that MV containing active VacA might provoke more pronounced cytokine responses. In order to examine the contribution of VacA to MV-induced IL-10 and IL-6 secretion from PBMCs, the effects of MV from 60190 ΔvacA and from the isogenic SS1 mutant engineered to produce more toxigenic s1i1 VacA (SS1s1i1) were also measured (Fig. 4B). All four MV types stimulated the production of similar levels of IL-10 and IL-6 (P > 0.05), indicating that VacA is not required at the concentration of MV tested.
H. pylori MV are toxic to Jurkat T cells.
We expected MV to stimulate IL-2 production by T cells present in the PBMC cultures, but this was not the case. PBMCs stimulated with MV produced IL-10 and IL-6, as described above, but the anticipated T cell-derived IL-2 response was absent. Since free soluble VacA inhibits T cell proliferation and IL-2 secretion (23–25, 27, 39), we set out to determine whether or not MV-associated VacA exerts inhibitory effects on T cells. Jurkat cells were pretreated for 1 h with 25 μg/ml MV and then stimulated for 48 h using PMA and ionomycin, following methods described by Gebert et al. (23). PMA-ionomycin-stimulated cells proliferated (i.e., the number of metabolically active cells per well increased) compared with unstimulated control cells (Fig. 5A, PBS controls with or without PMA-ionomycin). Pretreatment with MV caused a loss of metabolically active cells rather than proliferation, indicating a toxic effect on MV on Jurkat cells (P < 0.001) (Fig. 5A). Consistent with this, PMA-ionomycin-driven IL-2 production was markedly inhibited by pretreatment with MV (P < 0.001) (Fig. 5B). These cytotoxic effects were dose dependent, with MV-mediated cytotoxicity detectable at 60190 MV doses of 6.25 μg/ml and above (data not shown).
FIG 5.
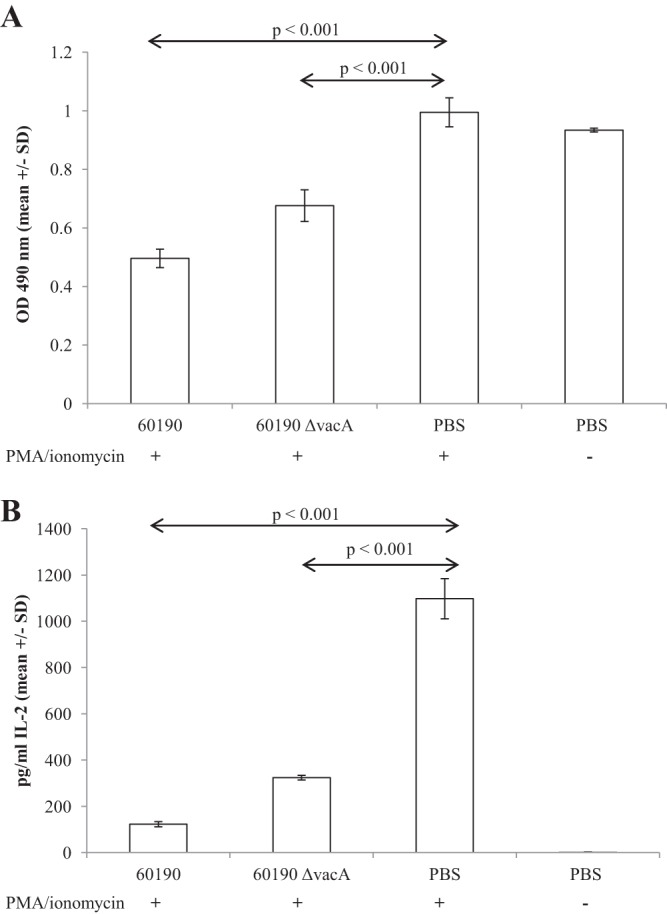
Pretreatment with MV reduces Jurkat cell viability and IL-2 production in response to PMA and ionomycin stimulation. Jurkat cells were pretreated with 25 μg/ml MV for 1 h and then stimulated with PMA and ionomycin. After 48 h, the concentration of metabolically active cells per well was measured using a CellTiter colorimetric assay (A), and IL-2 concentrations in culture supernatants were quantified by ELISA (B).
MV induce apoptosis in Jurkat T cells, which is enhanced by, but not dependent on, the presence of VacA.
Since Jurkat cells pretreated with MV died in proliferation assays, we decided to characterize the toxic effect of MV on T cells further by measuring apoptosis. Free soluble VacA is known to induce apoptosis in gastric epithelial cells (11–14) but not in Jurkat cells (23, 27, 39). H. pylori MV effects on gastric epithelial cells are complex, with induction of proliferation at low doses and of apoptosis at higher doses (33), but their effects on immune cells are unknown.
We cultured Jurkat cells in the presence of 25 μg/ml MV for 24 h and then measured intracellular and extracellular protease and cleaved caspase 3/7 activities using an ApoTox-Glo triplex assay (Promega) to determine levels of cell viability, cytotoxicity, and apoptosis, respectively.
H. pylori MV markedly decreased Jurkat cell viability and increased cytotoxicity compared with those for cells treated with an equivalent volume of PBS without MV (P < 0.001 for 60190 and SS1s1i1 MV, P < 0.01 for 60190 ΔvacA MV, and P < 0.05 for SS1 MV) (Fig. 6A). MV carrying more active forms of VacA had a more profound cytotoxic effect than MV with s2i2m2-type VacA or no VacA, although this difference did not reach statistical significance in the SS1 background (P < 0.01 for 60190 versus 60190 ΔvacA), but 60190 ΔvacA MV remained cytotoxic in the absence of VacA.
FIG 6.
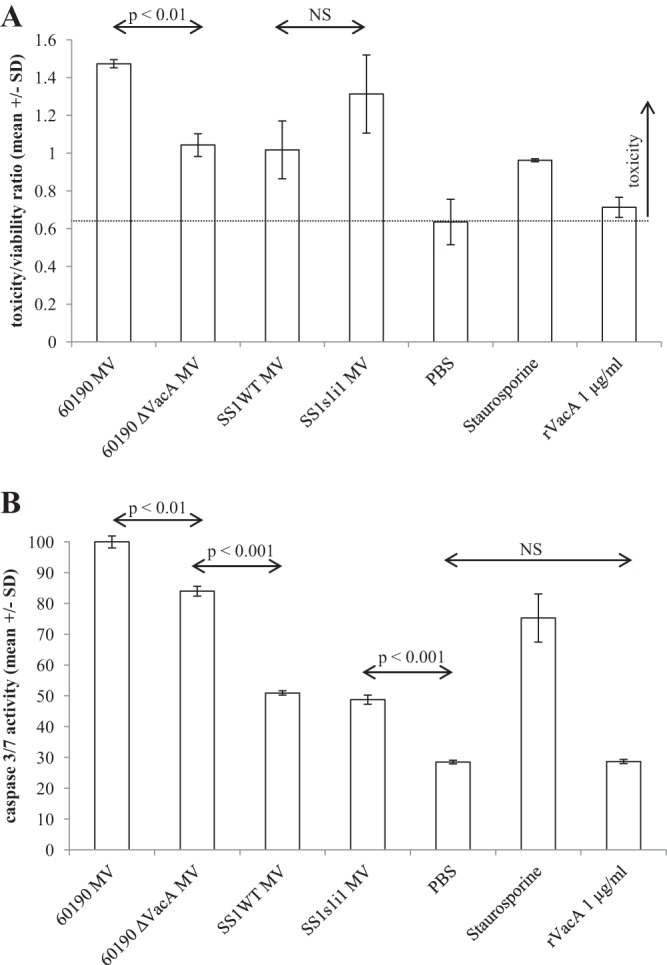
H. pylori MV induce apoptosis in Jurkat cells. Jurkat cells were incubated in the presence of 25 μg/ml MV for 24 h, and then cytotoxicity, viability, and caspase 3/7 activity were quantified using an ApoTox-Glo triplex assay. Mean values from triplicate wells are shown (±SD). (A) Overall cytotoxicity expressed as a toxicity/viability ratio with reference to healthy cells treated with a PBS control. (B) Caspase 3/7 activity normalized to the maximum level seen across all conditions tested.
H. pylori MV induced apoptosis in Jurkat cells (Fig. 6B). This was enhanced by, but not completely dependent on, the presence of VacA: Jurkat cells treated with 60190 MV had significantly more caspase 3/7 activity than those treated with 60190 ΔvacA MV (P < 0.001). 60190 MV at 25 μg/ml were more potent than the apoptosis-inducing chemical staurosporine at 1 μM (P < 0.001). SS1 and SS1s1i1 MV also induced apoptosis (P < 0.001) but to a lesser extent than MV from either of the 60190 background strains (P < 0.001). An MV dose of 25 μg/ml was the minimum required to induce apoptosis in Jurkat cells.
In order to confirm that VacA enhanced MV-induced Jurkat apoptosis but did not itself induce apoptosis when free and soluble, we treated Jurkat cells with purified recombinant s1i1m1 VacA expressed in E. coli using the 60190 vacA p33 and p55 genes and prepared following the method of González-Rivera et al. (36) (Fig. 7A). The activity of the recombinant toxin was confirmed by vacuolation assay on RK13 cells (Fig. 7B), and, as expected, recombinant VacA inhibited IL-2 secretion from PMA-ionomycin-stimulated Jurkat cells (Fig. 7C) but did not induce apoptosis (Fig. 6B), consistent with the published reports of other researchers (23, 27, 39).
FIG 7.
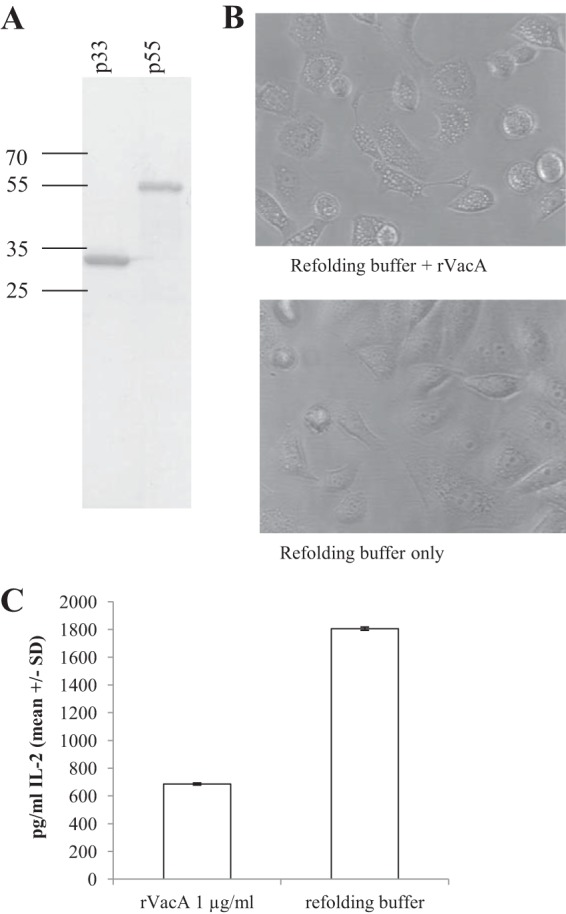
Recombinant VacA is active on epithelial and Jurkat cells. (A) Recombinant s1i1m1-type VacA (rVacA) was reconstituted from purified, refolded p33 and p55 subunits. Purity was confirmed by SDS-PAGE. (B) RK13 cells were incubated overnight in the presence of 1 μg/ml rVacA and vacuolation confirmed by light microscopy. (C) Jurkat cells were pretreated with rVacA or buffer control for 1 h and then stimulated with PMA and ionomycin. After 48 h, IL-2 secretion was quantified by ELISA.
Since 60190 ΔvacA MV were capable of inducing Jurkat cell apoptosis but the effect was enhanced by the presence of VacA in the vesicles, we hypothesized that VacA on the vesicle surface promoted binding to and/or uptake into Jurkat cells. To test this hypothesis, we used proteinase K to remove surface-bound proteins, including VacA from 60190 MV, following the methods of Bomberger et al. (37). As predicted, proteinase K-treated 60190 MV still induced Jurkat cell apoptosis, but only to 60190 ΔvacA MV levels (Fig. 8).
FIG 8.
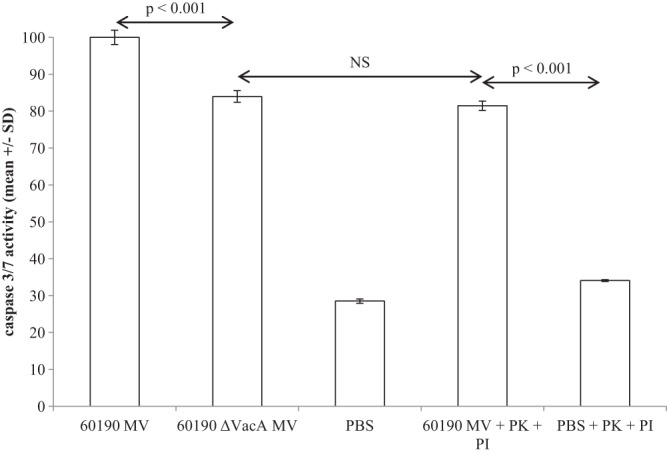
Induction of Jurkat cell apoptosis by MV is enhanced by, but not dependent on, carriage of VacA. 60190 MV were pretreated with proteinase K (PK) (inactivated using a protease inhibitor [PI] prior to cell culture). Treated and untreated MV were added to Jurkat cells at 25 μg/ml and caspase 3/7 activity measured after 24 h of incubation using an ApoTox-Glo triplex assay. Data are expressed as the percentage of maximal apoptosis induction and are means from triplicate wells.
MV effects on CD4+ T cells purified from human blood.
The Jurkat cell line is a widely used IL-2-producing leukemic T cell line, but experimental results using such cell lines might not always be representative of host-pathogen interactions in vivo. To confirm H. pylori MV effects on native human T cells, we purified CD4+ T cells from human blood and repeated the proliferation and apoptosis assays described above.
The Jurkat cell line proliferates rapidly even in the absence of stimulation, so addition of PMA and ionomycin did not markedly increase the number of metabolically active cells per well in Jurkat cell assays (Fig. 5). Native CD4+ cells grow more slowly in vitro, and proliferation was markedly increased in response to PMA and ionomycin. Pretreatment with 25 μg/ml H. pylori 60190 MV (with or without VacA) significantly inhibited this proliferative response (P < 0.001) (Fig. 9A).Similar to the results obtained with Jurkat cells (Fig. 6), incubation with 25 μg/ml 60190 MV induced apoptosis in native CD4+ cells at levels comparable to those with the chemical apoptosis inducer staurosporine at 1 μM (P < 0.05) (Fig. 9B). However, MV from the other H. pylori strains tested did not induce significant levels of apoptosis, indicating that MV-associated H. pylori components, including VacA and other, as-yet-unidentified factors may contribute to T cell inhibition during infection.
FIG 9.
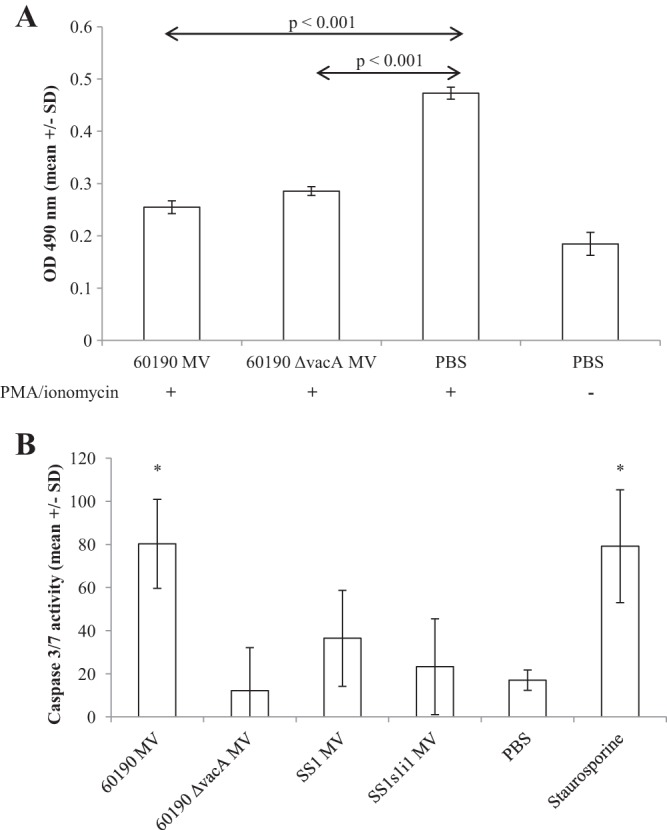
H. pylori MV effects on CD4+ T cells purified from human blood. (A) CD4+ T cells purified from human blood were pretreated with 25 μg/ml MV for 1 h and then stimulated with PMA and ionomycin (results for an unstimulated control are also shown). After 48 h, the concentration of metabolically active cells per well was measured using a CellTiter colorimetric assay. (B) Alternatively, CD4+ T cells were incubated in the presence of 25 μg/ml MV, PBS, or the chemical apoptosis-inducer staurosporine for 24 h, and then levels of apoptosis were determined by measuring caspase 3/7 activity using an ApoTox-Glo triplex assay. Data are expressed as the percentage of maximal apoptosis induction and are means from triplicate wells.
DISCUSSION
H. pylori infects humans lifelong, inducing profound immunological changes, including chronic inflammation of the stomach lining (gastritis) and elevated levels of IL-10-producing regulatory T cells (19). Coevolution of bacterium and host has likely resulted in a balance between induction of an inflammatory response in the host gastric mucosa, perhaps to promote release of host nutrients into the stomach lumen, and an anti-inflammatory response to prevent clearance of the bacteria by the host immune system (18).
Gram-negative bacteria constitutively produce MV, and multiple functions have been attributed to them, including envelope stress response (40) and toxin delivery (reviewed by Deatherage and Cookson [41]). There is growing interest in MV because they are now believed to play a role in bacterial pathogenesis for many species. Since MV resemble the bacterial surface, they can act as decoys for host factors such as antibodies and antimicrobial peptides (42–44) to promote bacterial survival and persistence. MV from a number of bacterial species have been proven to deliver virulence factors and PAMPs to host epithelial cells, inducing production of the proinflammatory cytokine IL-8, but direct effects of MV on host immune cells remain largely uncharacterized to date. Consequently, although we have focused on H. pylori, the findings described here may also be of interest to researchers studying mechanisms of immune modulation and pathogenesis in other bacterial infections.
H. pylori clearance is T cell dependent in a mouse infection model (45), and hence there has been great interest in the discovery of two proteins secreted by H. pylori, VacA and γ-glutamyl transferase (GGT), which inhibit human T cells via distinct mechanisms (23, 46). However, it is not yet clear how, and in what quantities, these factors access T cells in the lamina propria underlying the gastric epithelial barrier when the infecting bacteria remain on or close to the apical surface of the epithelium.
In addition to free soluble secretion, both VacA and GGT are packaged into MV (31, 47) by H. pylori. MV are more likely to cross the epithelial barrier than whole bacteria and so might exert long-range effects on host immune cells. They could potentially protect VacA and GGT from degradation/inactivation during transit, and they would facilitate simultaneous delivery of multiple H. pylori molecules to a single host cell in a concentrated “dose,” preventing rapid dilution of bacterial secreted factors in the stomach lumen and potentially contributing to H. pylori-mediated carcinogenesis. With this in mind, we set out to characterize immunomodulatory effects of H. pylori MV on human immune cells, first using PBMCs to broadly assess cytokine responses to the MV and then focusing on T cell-specific effects.
We show that H. pylori MV are strong innate stimulators of human immune cells, inducing proliferation and release of high concentrations of both proinflammatory (IL-6) and anti-inflammatory (IL-10) cytokines. Since PBMCs from people without H. pylori infection produced quantities of IL-6 and IL-10 similar to those produced by PBMCs from patients with a current infection, these cytokines are likely to be produced innately (e.g., by monocytes and NK cells rather than by H. pylori-specific T cells) in response to MV-associated PAMPs such as LPS and peptidoglycan. Despite high levels of strain-to-strain variability in their contents, MV from strains with 60190 or SS1 backgrounds stimulated indistinguishable cytokine responses independent of the presence, absence, or type of VacA. MV, like whole bacteria, are also known to induce IL-8 release from gastric epithelial cells (7, 8), so it is possible that MV are involved in stimulating and maintaining pro- and anti-inflammatory host responses during persistent infection, via direct effects on both epithelial and innate immune cells.
Given that H. pylori MV are enriched for LPS (28, 48), strong innate stimulation of IL-6 and IL-10 from human PBMCs was not unexpected. However, the complete absence of T cell activation, characterized by IL-2 secretion, led us to examine MV effects on T cells more closely. Since MV-associated VacA is active on gastric epithelial cells, we expected to find that MV carrying VacA were capable of delivering the toxin to T cells, resulting in inhibition of proliferation and IL-2 secretion as previously established (23–27) for free toxin. In fact, any such VacA-mediated T cell inhibition was masked by the strong toxic effect of the vesicles themselves. H. pylori culture supernatant has been previously reported to induce apoptosis in T cells (49), and here we show that MV alone are able to induce apoptosis to similar, or higher, levels than the chemical inducer staurosporine used as a positive control in this study. Although active recombinant VacA in free soluble form did not induce T cell apoptosis, MV-mediated apoptosis was enhanced by, but not completely dependent on, carriage of the toxin. MV from the less toxic SS1 strain did not induce T cell apoptosis to the same extent as MV from 60190, even when the SS1 strain was engineered to produce a more active s1i1 VacA toxin form. Comparison of MV contents by SDS-PAGE and Western blotting revealed striking differences in relative VacA concentrations between the 60190 and SS1 strain backgrounds.
Further investigation of strain-strain variability in MV contents and activities is now under way in our laboratory, and we also aim to define the specific components of H. pylori MV that induce T cell apoptosis and the mechanisms by which they do so. We propose that membrane vesicle-mediated delivery of H. pylori immune-modulatory factors to host immune cells may contribute to bacterial pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from Cancer Research UK (grant C8968/A11204), the University of Nottingham Biomedical Research Committee, and the National Institute of Health Research through its Nottingham Digestive Diseases Centre Biomedical Research Unit.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank T. Cover (Vanderbilt University School of Medicine) for the 60190 ΔvacA mutant. PBMC purification was carried out with assistance from R. Ingram, K. Cook, and A. Greenaway, School of Clinical Sciences, University of Nottingham. TEM was carried out at the Advanced Microscopy Unit, School of Biomedical Sciences, University of Nottingham, with technical assistance from D. Christie. Jurkat cells were a generous gift from R. McIntosh, Academic Clinical Oncology Department, University of Nottingham. We thank C. Lambert for assistance with ultracentrifugation carried out at the Institute of Genetics, University of Nottingham.
Footnotes
Published ahead of print 13 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01443-13.
REFERENCES
- 1.Atherton JC, Blaser MJ. 2009. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J. Clin. Invest. 119:2475–2487. 10.1172/JCI38605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peek RM, Jr, Fiske C, Wilson KT. 2010. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 90:831–858. 10.1152/physrev.00039.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atherton JC, Cao P, Peek RM, Jr, Tummuru MK, Blaser MJ, Cover TL. 1995. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 270:17771–17777 [DOI] [PubMed] [Google Scholar]
- 4.Letley DP, Rhead JL, Twells RJ, Dove B, Atherton JC. 2003. Determinants of non-toxicity in the gastric pathogen Helicobacter pylori. J. Biol. Chem. 278:26734–26741. 10.1074/jbc.M304071200 [DOI] [PubMed] [Google Scholar]
- 5.Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, Atherton JC. 2007. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 133:926–936. 10.1053/j.gastro.2007.06.056 [DOI] [PubMed] [Google Scholar]
- 6.Basso D, Zambon CF, Letley DP, Stranges A, Marchet A, Rhead JL, Schiavon S, Guariso G, Ceroti M, Nitti D, Rugge M, Plebani M, Atherton JC. 2008. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology 135:91–99. 10.1053/j.gastro.2008.03.041 [DOI] [PubMed] [Google Scholar]
- 7.Sharma SA, Tummuru MK, Miller GG, Blaser MJ. 1995. Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect. Immun. 63:1681–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim SY, Lee YC, Kim HK, Blaser MJ. 2006. Helicobacter pylori CagA transfection of gastric epithelial cells induces interleukin-8. Cell. Microbiol. 8:97–106. 10.1111/j.1462-5822.2005.00603.x [DOI] [PubMed] [Google Scholar]
- 9.Boquet P, Ricci V. 2012. Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol. 20:165–174. 10.1016/j.tim.2012.01.008 [DOI] [PubMed] [Google Scholar]
- 10.Cover TL, Blaser MJ. 1992. Purification and characterization of the vacuolating toxin from Helicobacter pylori. J. Biol. Chem. 267:10570–10575 [PubMed] [Google Scholar]
- 11.Matsumoto A, Isomoto H, Nakayama M, Hisatsune J, Nishi Y, Nakashima Y, Matsushima K, Kurazono H, Nakao K, Hirayama T, Kohno S. 2011. Helicobacter pylori VacA reduces the cellular expression of STAT3 and pro-survival Bcl-2 family proteins, Bcl-2 and Bcl-XL, leading to apoptosis in gastric epithelial cells. Dig. Dis. Sci. 56:999–1006. 10.1007/s10620-010-1420-1 [DOI] [PubMed] [Google Scholar]
- 12.Galmiche A, Rassow J, Doye A, Cagnol S, Chambard JC, Contamin S, de Thillot V, Just I, Ricci V, Solcia E, Van Obberghen E, Boquet P. 2000. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 19:6361–6370. 10.1093/emboj/19.23.6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cover TL, Krishna US, Israel DA, Peek RM., Jr 2003. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 63:951–957 [PubMed] [Google Scholar]
- 14.Yamasaki E, Wada A, Kumatori A, Nakagawa I, Funao J, Nakayama M, Hisatsune J, Kimura M, Moss J, Hirayama T. 2006. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J. Biol. Chem. 281:11250–11259. 10.1074/jbc.M509404200 [DOI] [PubMed] [Google Scholar]
- 15.Fujikawa A, Shirasaka D, Yamamoto S, Ota H, Yahiro K, Fukada M, Shintani T, Wada A, Aoyama N, Hirayama T, Fukamachi H, Noda M. 2003. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat. Genet. 33:375–381. 10.1038/ng1112 [DOI] [PubMed] [Google Scholar]
- 16.Ghiara P, Marchetti M, Blaser MJ, Tummuru MK, Cover TL, Segal ED, Tompkins LS, Rappuoli R. 1995. Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA, and urease in a mouse model of disease. Infect. Immun. 63:4154–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Telford JL, Ghiara P, Dell'Orco M, Comanducci M, Burroni D, Bugnoli M, Tecce MF, Censini S, Covacci A, Xiang Z, et al. 1994. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 179:1653–1658. 10.1084/jem.179.5.1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blaser MJ, Atherton JC. 2004. Helicobacter pylori persistence: biology and disease. J. Clin. Invest. 113:321–333. 10.1172/JCI20925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, Zaitoun AM, Atherton JC. 2008. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut 57:1375–1385. 10.1136/gut.2007.137539 [DOI] [PubMed] [Google Scholar]
- 20.Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, Edebo A, Johnsson E, Suri-Payer E, Larsson P, Rudin A, Svennerholm AM, Lundin BS. 2005. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect. Immun. 73:523–531. 10.1128/IAI.73.1.523-531.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindholm C, Quiding-Jarbrink M, Lonroth H, Hamlet A, Svennerholm AM. 1998. Local cytokine response in Helicobacter pylori-infected subjects. Infect. Immun. 66:5964–5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warren JR, Marshall B. 1983. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet i:1273–1275 [PubMed] [Google Scholar]
- 23.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. 2003. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099–1102. 10.1126/science.1086871 [DOI] [PubMed] [Google Scholar]
- 24.Sundrud MS, Torres VJ, Unutmaz D, Cover TL. 2004. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. U. S. A. 101:7727–7732. 10.1073/pnas.0401528101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres VJ, VanCompernolle SE, Sundrud MS, Unutmaz D, Cover TL. 2007. Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J. Immunol. 179:5433–5440 [DOI] [PubMed] [Google Scholar]
- 26.Schmees C, Gerhard M, Treptau T, Voland P, Schwendy S, Rad R, Prinz C. 2006. VacA-associated inhibition of T-cell function: reviewed and reconsidered. Helicobacter 11:144–146. 10.1111/j.1523-5378.2006.00393.x [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez-Rivera C, Algood HM, Radin JN, McClain MS, Cover TL. 2012. The intermediate region of Helicobacter pylori VacA is a determinant of toxin potency in a Jurkat T cell assay. Infect. Immun. 80:2578–2588. 10.1128/IAI.00052-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keenan J, Day T, Neal S, Cook B, Perez-Perez G, Allardyce R, Bagshaw P. 2000. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol. Lett. 182:259–264. 10.1111/j.1574-6968.2000.tb08905.x [DOI] [PubMed] [Google Scholar]
- 29.Fiocca R, Necchi V, Sommi P, Ricci V, Telford J, Cover TL, Solcia E. 1999. Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J. Pathol. 188:220–226 [DOI] [PubMed] [Google Scholar]
- 30.McBroom AJ, Kuehn MJ. 12 May 2005. Chapter 2.2.4, Outer membrane vesicles. In Curtiss R, III, et al. (ed), EcoSal—Escherichia coli and Salmonella: cellular and molecular biology ASM Press, Washington, DC [Google Scholar]
- 31.Olofsson A, Vallstrom A, Petzold K, Tegtmeyer N, Schleucher J, Carlsson S, Haas R, Backert S, Wai SN, Grobner G, Arnqvist A. 2010. Biochemical and functional characterization of Helicobacter pylori vesicles. Mol. Microbiol. 77:1539–1555. 10.1111/j.1365-2958.2010.07307.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ismail S, Hampton MB, Keenan JI. 2003. Helicobacter pylori outer membrane vesicles modulate proliferation and interleukin-8 production by gastric epithelial cells. Infect. Immun. 71:5670–5675. 10.1128/IAI.71.10.5670-5675.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayala G, Torres L, Espinosa M, Fierros-Zarate G, Maldonado V, Melendez-Zajgla J. 2006. External membrane vesicles from Helicobacter pylori induce apoptosis in gastric epithelial cells. FEMS Microbiol. Lett. 260:178–185. 10.1111/j.1574-6968.2006.00305.x [DOI] [PubMed] [Google Scholar]
- 34.Lee A, O'Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. 1997. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112:1386–1397. 10.1016/S0016-5085(97)70155-0 [DOI] [PubMed] [Google Scholar]
- 35.Cover TL, Tummuru MK, Cao P, Thompson SA, Blaser MJ. 1994. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem. 269:10566–10573 [PubMed] [Google Scholar]
- 36.Gonzalez-Rivera C, Gangwer KA, McClain MS, Eli IM, Chambers MG, Ohi MD, Lacy DB, Cover TL. 2010. Reconstitution of Helicobacter pylori VacA toxin from purified components. Biochemistry 49:5743–5752. 10.1021/bi100618g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bomberger JM, Maceachran DP, Coutermarsh BA, Ye S, O'Toole GA, Stanton BA. 2009. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 5:e1000382. 10.1371/journal.ppat.1000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Supajatura V, Ushio H, Wada A, Yahiro K, Okumura K, Ogawa H, Hirayama T, Ra C. 2002. VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J. Immunol. 168:2603–2607 [DOI] [PubMed] [Google Scholar]
- 39.Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amedei A, D'Elios MM, Telford JL, Baldari CT. 2003. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 198:1887–1897. 10.1084/jem.20030621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McBroom AJ, Kuehn MJ. 2007. Release of outer membrane vesicles by Gram-negative bacteria is a novel envelope stress response. Mol. Microbiol. 63:545–558. 10.1111/j.1365-2958.2006.05522.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deatherage BL, Cookson BT. 2012. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect. Immun. 80:1948–1957. 10.1128/IAI.06014-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettit RK, Judd RC. 1992. The interaction of naturally elaborated blebs from serum-susceptible and serum-resistant strains of Neisseria gonorrhoeae with normal human serum. Mol. Biol. 6:729–734 [DOI] [PubMed] [Google Scholar]
- 43.Manning AJ, Kuehn MJ. 2011. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 11:258. 10.1186/1471-2180-11-258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaar V, Paulsson M, Morgelin M, Riesbeck K. 2013. Outer membrane vesicles shield Moraxella catarrhalis β-lactamase from neutralization by serum IgG. J. Antimicrob. Chemother. 68:593–600. 10.1093/jac/dks444 [DOI] [PubMed] [Google Scholar]
- 45.Ermak TH, Giannasca PJ, Nichols R, Myers GA, Nedrud J, Weltzin R, Lee CK, Kleanthous H, Monath TP. 1998. Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J. Exp. Med. 188:2277–2288. 10.1084/jem.188.12.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, Bauer S, Brenner L, Schmid RM, Gerhard M. 2007. Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology 132:1820–1833. 10.1053/j.gastro.2007.02.031 [DOI] [PubMed] [Google Scholar]
- 47.Mullaney E, Brown PA, Smith SM, Botting CH, Yamaoka YY, Terres AM, Kelleher DP, Windle HJ. 2009. Proteomic and functional characterization of the outer membrane vesicles from the gastric pathogen Helicobacter pylori. Proteomics Clin. Appl. 3:785–796. 10.1002/prca.200800192 [DOI] [PubMed] [Google Scholar]
- 48.Keenan JI, Allardyce RA, Bagshaw PF. 1997. Dual silver staining to characterise Helicobacter spp. outer membrane components. J. Immunol. Methods 209:17–24. 10.1016/S0022-1759(97)00141-5 [DOI] [PubMed] [Google Scholar]
- 49.Ganten TM, Aravena E, Sykora J, Koschny R, Mohr J, Rudi J, Stremmel W, Walczak H. 2007. Helicobacter pylori-induced apoptosis in T cells is mediated by the mitochondrial pathway independent of death receptors. Eur. J. Clin. Invest. 37:117–125. 10.1111/j.1365-2362.2007.01761.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.