

Abstract
Human monocytic ehrlichiosis (HME) is caused by a tick-borne obligate intracellular pathogen of the order Rickettsiales. HME disease can range from mild to a fatal, toxic shock-like syndrome, yet the mechanisms regulating pathogenesis are not well understood. We define a central role for type I interferons (alpha interferon [IFN-α] and IFN-β) in severe disease in a mouse model of fatal ehrlichiosis caused by Ixodes ovatus Ehrlichia (IOE). IFN-α and IFN-β were induced by IOE infection but not in response to a less virulent strain, Ehrlichia muris. The major sources of type I IFNs during IOE infection were plasmacytoid dendritic cells and monocytes. Mice lacking the receptor for type I IFNs (Ifnar deficient) or neutralization of IFN-α and IFN-β resulted in a reduced bacterial burden. Ifnar-deficient mice exhibited significantly increased survival after IOE infection, relative to that of wild-type (WT) mice, that correlated with increased type II IFN (IFN-γ) production. Pathogen-specific antibody responses were also elevated in Ifnar-deficient mice, and this required IFN-γ. Remarkably, increased IFN-γ and IgM were not essential for protection in the absence of type I IFN signaling. The direct effect of type I IFNs on hematopoietic and nonhematopoietic cells was evaluated in bone marrow chimeric mice. We observed that chimeric mice containing Ifnar-deficient hematopoietic cells succumbed to infection early, whereas Ifnar-deficient mice containing WT hematopoietic cells exhibited increased survival, despite having a higher bacterial burden. These data demonstrate that IFN-α receptor signaling in nonhematopoietic cells is important for pathogenesis. Thus, type I IFNs are induced during a rickettsial infection in vivo and promote severe disease.
INTRODUCTION
Alpha interferon (IFN-α) and IFN-β, which belong to the type I IFN family, bind to the heterodimeric IFN-α receptor (IFN-αR) (1). At homeostasis, IFN-β is constitutively expressed (2) and provides immune regulatory functions that include maintaining hematopoietic stem cells in the bone marrow (3), phagocytic potential of macrophages (4), and normal NK cell numbers and function (5). In response to viral infections, induction of type I IFNs regulates expression of antiviral genes, including IFN-stimulated genes (ISGs) and IFN-regulated factors (IRFs), thus controlling the antiviral state (1, 6, 7).
Although well recognized for supporting antiviral immunity, the impact of type I IFNs during bacterial infections is complex and, in some cases, can be pathogenic (8). Mice lacking the IFN-αR are more resistant to infection with Listeria monocytogenes (9, 10). Type I IFNs also render macrophages more susceptible to necroptosis, thus contributing to severe disease in Salmonella infection (11). In contrast, type I IFNs are protective during certain bacterial infections. For instance, type I IFN signaling is crucial for host resistance to group B streptococcal infection, which correlated with increased tumor necrosis factor alpha (TNF-α) and nitric oxide production by macrophages in the presence of type I IFN signaling (12). Recently, the significance of type I IFNs during fungal infection was also observed, in that renal dendritic cell (DC)-derived IFN-β is necessary for host defense against Candida albicans infection (13). Thus, it appears that type I IFNs can be protective during extracellular bacterial and fungal infections but contribute to pathogenesis during intracellular bacterial infections (7); however, the mechanisms for the dual effects of type I IFNs during bacterial infection are not well defined.
One possible explanation for the pathogenic role of type I IFNs during bacterial infection may be due to the ability of type I IFNs to suppress IFN-γ expression and signaling. IFN-γ is essential for clearance of many intracellular bacterial infections, including L. monocytogenes, Mycobacterium tuberculosis, and Ehrlichia muris (14–16), and the ability of type I IFNs to suppress IFN-γ signaling may contribute to bacterial pathogenesis. This idea is supported by observations in humans. It was shown in human leprosy patients that type I IFNs correlated with disseminated and progressive disease, whereas IFN-γ was expressed in self-healing lesions (17). The authors further demonstrated that IFN-γ-induced signaling and induction of antimicrobial pathways were inhibited by IFN-β. In addition, type I IFNs correlate with active disease in M. tuberculosis patients (18). Highly virulent strains of M. tuberculosis upregulate type I IFNs, impair Th1 responses (19), and are less pathogenic in the absence of IFN-αR signaling (20). Impaired IFN-γ-mediated signaling by type I IFNs in these contexts may rely on downregulation of the IFN-γ receptor, as observed in L. monocytogenes infection (9), thus allowing increased pathogen growth in macrophages, or may interfere with downstream signaling pathways induced by IFN-γ (17).
The rickettsiae are a group of highly pathogenic emerging and reemerging bacteria transmitted by insect and tick vectors, and our understanding of immunity and pathogenesis during these infections is still incomplete. Misdiagnosis is very common for rickettsial infections, as presentation often occurs with nonspecific symptoms, and delayed treatment correlates with a poor outcome (21, 22). There are currently no therapeutic treatments for severe rickettsial infections that are unresponsive to antibiotics. Human monocytic ehrlichiosis (HME) is caused by the obligate intracellular pathogen Ehrlichia chaffeensis. In recent years, additional isolates have been identified (23), and it is not yet clear whether strain differences contribute to disease outcome. HME can be mild in some patients, whereas in others it progresses rapidly to a toxic shock-like syndrome or even death (24). HME can be modeled in mice by using two genetically related pathogens, Ehrlichia muris, which causes mild disease (25), and Ixodes ovatus Ehrlichia (IOE), which causes severe, fatal ehrlichiosis (16, 25–28). IOE-infected mice exhibit severe liver injury and leukocyte necrosis, similar to what is observed in severely ill HME patients; thus, IOE is an ideal model of severe ehrlichiosis and infection-induced shock (24).
Current understanding of IOE virulence centers on the overproduction of TNF-α by CD8+ T cells (28, 29). In addition, fatal recall responses after low-dose infection of IOE were due to CD8 T cells and significant production of TNF-α (30). TNF-α may promote shock-like disease during IOE by driving apoptosis and necrosis, as has been shown in mouse models of lethal inflammation (31) and in humans receiving high doses of TNF-α (32). Despite the pathogenic inflammatory response induced by TNF-α, mice deficient in TNF receptors still succumbed to primary IOE infection, suggesting that TNF-α-mediated signaling may also be important for control of bacterial growth. In addition, this finding suggests that additional mechanisms are likely involved in promoting severe disease.
Protection against lethal IOE challenge can be achieved by prior infection with the less virulent strain, E. muris (33). One correlate of protection appears to be IFN-γ-producing CD4 T cells (34), and relative to E. muris infection, IOE infection elicits very little IFN-γ expression (28). A strong pathogen-specific IgM response is also elicited during E. muris infection, which is sufficient to protect against IOE challenge (35, 36), and it has been speculated that IOE infection does not elicit a strong pathogen-specific IgM response (35, 36). Thus, CD4 T cells and antibodies are protective, whereas CD8 T cells, NKT cells, and neutrophils are detrimental during IOE infection and contribute to severe inflammation and shock-like disease (30, 37–39).
The impact of type I IFNs on ehrlichial pathogenesis has not been investigated. Here, we first wanted to determine if type I IFNs were induced during ehrlichial infection, and then we sought to address their impact on disease. We demonstrate that induction of type I IFNs from plasmacytoid DC (pDC) and monocytes occurs in response to the highly virulent ehrlichial strain IOE but not in response to less virulent E. muris. We demonstrate that type I IFNs contribute significantly to disease and modulate host immune responses. We made the novel observation that IFN-γ maintains the humoral immune response to IOE infection in the absence of type I IFN signaling. Surprisingly, however, IFN-γ was not required for host defense to IOE infection in Ifnar-deficient mice. Moreover, we found that the host death during IOE infection was not dependent upon uncontrolled bacterial growth but was promoted by type I IFN signaling in nonhematopoietic cells. Our finding reveals a previously unrecognized role for type I IFNs in mediating pathogenesis during severe ehrlichial infection in vivo.
MATERIALS AND METHODS
Mice.
C57BL/6 mice and a CD45 congenic strain (B6.SJL-Ptprca/BoyAiTac) were purchased from Taconic (Petersburgh, NY). IFN-α/β receptor gene (Ifnar1; referred to here as Ifnar)-knockout mice were provided by Jacob Kohlmeier and David Woodland (Trudeau Institute, Saranac Lake, NY). β-Actin (β-act) enhanced green fluorescent protein (EGFP) [B6-Tg(CAG-EGFP)131Osb/LeySopJ] mice express EGFP, driven by the β-act promoter, and thus all cells of the hematopoietic compartment, aside from red blood cells, express EGFP. IFN-β reporter mice (B6.129-ifnb1tm1Lky/J) and MyD88-deficient [B6.129P2(SJL)-MyD88tm1Defr/J] mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Toll-like receptor 2 (TLR2)-deficient mice with a C57BL/6 background were provided by Timothy Sellati (Albany Medical College). All mice were bred in the Animal Resources Facility at Albany Medical College under specific-pathogen-free (SPF) conditions.
Bacteria.
Mice between 6 and 12 weeks of age were infected, via intraperitoneal injection, with 50,000 E. muris bacteria or 1,200 IOE bacteria in sucrose-phosphate-glutamate (SPG) buffer. Bacteria were obtained from infected mouse splenocytes, as previously described (16, 34). “Mock” or mock-infected mice are our uninfected control mice.
PCR quantification for bacterial burden.
DNA from 2 × 106 cells derived from spleen, lung, or liver was extracted using DNAzol (Molecular Research Center, Cincinnati, OH). The number of bacterial copies was determined using a real-time quantitative probe-based PCR for the disulfide bond formation protein gene (dsb), as previously described (38, 40).
Flow cytometry and antibodies.
Spleen mononuclear cells were harvested and prepared as previously described (40). The antibodies used for flow cytometry include the following: phycoerythrin (PE)-CD45.1 (A20), PB-CD4 (RM4-5), allophycocyanin (APC)-CD4 (GK1.5), fluorescein isothiocyanate (FITC)-CD4 (RM4-5), FITC-CD3 (17A2), APC-Cy7-CD3 (145-2C1), PerCP-Cy5.5-CD8 (53-6.7), PE-CD8 (53-6.7), FITC-B220 (RA3-6B2), biotin-B220 (RA3-6B2), PB-CD19 (6D5), NK1.1-PE-Cy7 (PK136), APC–IFN-γ (XMG1.2), FITC-CD11b (M1/70), PE-Cy7-CD11b (M1/70), APC-CD11c (N418), APC-Cy7-CD11c (N418), PB-Ly6C (HK1.4), APC-Ly6C (HK1.4), PerCP-Cy5.5-Ly6G (1A8), PE-Ly6G (1A8), PE-Siglec H (551), PE-CD115 (AFS98), CD317-PB (927), CD317-PE (927), APC-Cy7-F4/80 (CI:A3-1), PB-F4/80 (BM8), and APC-Cy7-streptavidin (all from BioLegend, San Diego, CA), V500-CD45.2 (104) and FITC–TNF-α (MP6-XT22) (from BD Biosciences), and FITC–IFN-α (RMMA-1) (from PBL Interferon Source, Piscataway, NJ). Fc block (2.4G2; BD Biosciences) was used if necessary. Unstained cells were used as negative controls to establish the flow cytometer voltage settings, and single-color positive controls were used to adjust the instrument compensation. The flow cytometric data were acquired using an LSR II flow cytometer (BD Biosciences), and data analysis was performed using FlowJo software (TreeStar Inc., Ashland, OR).
Intracellular cytokine staining.
For IFN-α staining, C57BL/6 mice were treated with brefeldin A (BFA) intravenously (250 μg/200 μl phosphate-buffered saline [PBS]/mouse) 6 h prior to sacrifice (41). For IFN-γ and TNF-α staining, direct ex vivo staining was performed without BFA application. Splenic single-cell suspensions were prepared, and erythrocytes were removed by a brief hypotonic lysis. A total of 2 × 106 cells were plated in a 96-well plate, and Fc receptors were blocked by incubation with Fc block for 20 min on ice. Cells were then incubated on ice for 30 min with specific antibodies to stain for surface proteins. Cells were then stained with a fixable viability dye (BD Bioscience). Cells were washed and then fixed and permeabilized in Fix/Perm buffer (BD Biosciences). Thereafter, cells were incubated in Wash/Perm buffer (BD Biosciences) with fluorescence-conjugated anti-IFN-α antibody, anti-IFN-γ antibody, or anti-TNF-α antibody for 30 min on ice. Cells were washed two times in Wash/Perm buffer, resuspended in simple wash buffer, and analyzed on an LSR II (BD Biosciences). To verify that staining was specific for intracellular protein, nonpermeabilized cells were also analyzed. Data were analyzed using FlowJo software (TreeStar Inc., Ashland, OR).
Cytokine measurements.
Liver and lung were perfused with PBS before harvest. Spleen, liver, and lung homogenates were made in the presence of IGEPAL CA630 and proteinase inhibitors (Sigma, St. Louis, MO). Total protein concentration in homogenate was measured using a BCA kit (Pierce, Rockford, IL). Enzyme-linked immunosorbent assay (ELISA) kits for IFN-α and IFN-β protein were purchased from eBioscience (San Diego, CA) and PBL interferon (Piscataway, NJ), respectively. Protocols followed the manufactures' instructions. For IFN-γ detection, capture antibody (XMG1.2) and biotin-conjugated detection antibody (R4-6A2) were purchased from Bio X cell (West Lebanon, NH) and BioLegend (San Diego, CA), respectively. Streptavidin-horseradish peroxidase (HRP), and 3,3′,5,5′-tetramethylbenzidine (TMB) liquid ELISA substrate were purchased from BioLegend and Sigma, respectively. Cytokines in homogenates and sera were calculated as pg/mg of total protein and pg/ml, respectively.
OMP-19-reactive IgM measurement.
OMP-19 proteins (generously provided by Gary Winslow, Upstate Medical University, Syracuse, NY) were used as antigens (42), and peroxide-conjugated goat anti-mouse IgM was used as detection antibody (Sigma).
Generation of bone marrow chimeric mice.
To generate mixed WT and Ifnar-deficient bone marrow (BM) chimeric mice, CD45 congenic mice were lethally irradiated (950 rad, administered in 2 doses, 4 h apart). Irradiated mice received a total of 5 × 106 BM cells derived from WT mice expressing GFP (β-act EGFP mice; GFP+ CD45.2+) and Ifnar-deficient mice (GFP− CD45.2+) at a 1:1 ratio. This allowed distinction between WT and Ifnar-deficient donor cells based on GFP expression; radioresistant host cells were identified and excluded based on CD45.1 expression. To generate reverse chimeric mice, CD45 congenic mice were lethally irradiated as described above and then received 5 × 106 BM cells from Ifnar-deficient mice and vice versa. Mice were screened for chimerism at 6 weeks and infected with IOE at 8 to 10 weeks postreconstitution.
Neutralization of type I and II IFNs.
Neutralizing antibodies to mouse IFN-α, IFN-β, or both were administered into WT mice intravenously from day 4 to day 7 post-IOE infection (rat anti-IFN-α [RMMA-1], 2 μg per day except 5 μg at day 7; rabbit anti-IFN-β, 80 μg per day except 200 μg at day 7; all from PBL Interferon Source). Mice were sacrificed at day 8 postinfection. Neutralizing antibodies to mouse IFN-γ (XMG1.2; Bio X cell) were administered into Ifnar-deficient mice since day 2 post-IOE infection every other day through intraperitoneal injection (400 μg/500 μl PBS each time).
Statistics.
Analysis of data was performed using a two-way analysis of variance (ANOVA) (strain and treatment) or one-way ANOVA (treatment), if necessary using Prism. P values of <0.05 were considered a significant difference.
RESULTS
Induction of interferons during IOE infection.
Gamma interferon (IFN-γ) production provides protection during ehrlichial infection (16, 34) and mediates hematopoietic changes, including increased monocyte production (43). Whether type I IFNs are produced in vivo and if they play a protective or pathogenic role in disease outcome has not yet been investigated for any rickettsial pathogen. Here, we sought to determine the kinetics of IFN-α and -β production in models of mild and lethal ehrlichial disease. As expected, E. muris infection elicited robust IFN-γ production, with peak concentrations detected in the sera at day 11 postinfection (43) (Fig. 1A). IOE infection, however, elicited very little IFN-γ production, consistent with what has been previously reported (28). In contrast to IFN-γ, production of IFN-α and IFN-β was found to be high in the sera of IOE-infected animals, with peak concentrations detected on day 7 postinfection (Fig. 1B and C). Type I IFNs were undetectable in E. muris-infected mice. To determine if type I IFNs were also produced in tissues, type I IFN protein was measured in the spleen, lung, and liver. We found that IFN-α was increased in spleen and liver (Fig. 1D and E) but not in the lung (Fig. 1F) of IOE-infected mice, while IFN-β was detected at increased concentrations in all three tissues. These data demonstrate that mild ehrlichial disease correlated with a strong IFN-γ response, whereas severe disease caused by IOE infection correlated with increased type I IFNs. We observed that mice deficient in TLR2 had elevated type I IFNs in the sera, relative to wild-type (WT) and MyD88-deficient mice (Fig. 1G and H). As TLR2-deficient mice are more susceptible to IOE infection (44), these data raised the possibility that type I IFNs mediate more severe disease.
FIG 1.

Reduced IFN-γ production and elevated type I IFNs in a mouse model of fatal ehrlichiosis. Mice were infected with E. muris or IOE, and levels of IFN-γ and type I IFNs (α and β) were measured by ELISA at different times postinfection as indicated. Serum IFN-γ (A), IFN-α (B), and IFN-β (C) concentrations in WT C57BL/6 mice infected with E. muris (closed circles) or IOE (open circles) are shown. IFN-α (closed bars) and IFN-β (open bars) in the spleens (D), livers (E), and lungs (F) of IOE-infected WT mice at day 7 postinfection are shown. (G) IFN-α concentration in the sera of IOE-infected WT (closed bars), MyD88−/− (open bars), and TLR2−/− (striped bars) mice at day 7 postinfection is shown. (H) IFN-β in the sera of IOE-infected WT, MyD88−/−, and TLR2−/− mice at day 7 postinfection is shown. Asterisks indicate a significant difference compared to the counterpart mock controls (P < 0.05). “n.s.” indicates no significant difference (P > 0.05) and “n.d.” indicates not detectable. Number symbols (#) indicate a significant difference between groups as indicated (P < 0.05). At least three animals were used per group.
Type I IFNs are detrimental to the host during IOE infection.
Type I IFNs contribute to pathogenesis in some bacterial infections (11, 17, 45); thus, we next sought to investigate whether type I IFNs were protective or pathogenic during ehrlichial infection. Using mice deficient in the alpha subunit of the IFN receptor (Ifnar deficient), blocking signaling for all type I IFNs (α, β, and ε), we first tested whether mice deficient in type I IFN-mediated signaling exhibited increased survival relative to WT mice after IOE infection. Whereas IOE infection was uniformly fatal in WT mice, 70% of Ifnar-deficient mice survived (Fig. 2A). As expected, IFN-αR-mediated signaling did not affect survival of E. muris-infected mice. We next measured the bacterial burden in different organs of WT and Ifnar-deficient mice during IOE infection. Relative to WT mice, Ifnar-deficient mice exhibited significantly reduced bacterial burdens in the spleen and lung (Fig. 2B). To investigate whether both IFN-α and IFN-β were contributing to the pathogenesis of IOE infection, we neutralized IFN-α or IFN-β, or both, in WT mice. Relative to vehicle control, IFN-α- or IFN-β-neutralizing antibody-treated mice exhibited slightly reduced bacterial burdens in the spleen and lung, whereas mice treated with both anti-IFN-α and anti-IFN-β had significantly reduced bacterial burdens (Fig. 2C and D), indicating that both IFN-α and IFN-β contributed to increased bacterial growth during IOE infection.
FIG 2.
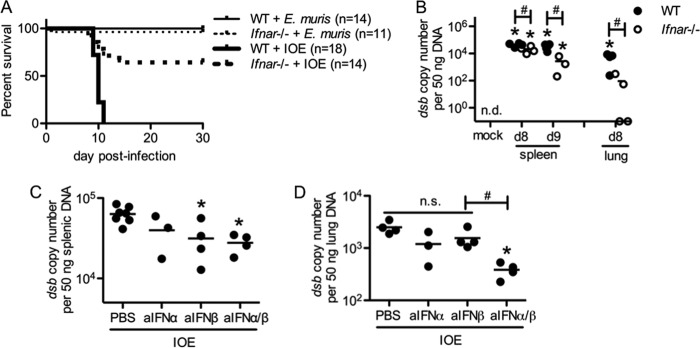
Type I IFNs are detrimental during IOE infection. Survival was assessed and bacterial burden was measured in mice where type I IFN signaling was disrupted. (A) Survival curves of E. muris or IOE-infected WT and Ifnar−/− mice are shown (numbers of mice per group are shown in parentheses). (B) Bacterial burden in the spleens and lungs of IOE-infected WT and Ifnar−/− mice at day 8 and/or day 9 postinfection is shown. Bacterial burden at day 8 postinfection in the spleens (C) and lungs (D) of IOE-infected WT mice that received neutralizing antibodies to IFN-α, IFN-β, or both is shown. Asterisks indicate a significant difference compared to the counterpart mock controls (P < 0.05). Number symbols (#) indicate a significant difference between the groups as indicated (P < 0.05). “n.s.” indicates no significant difference (P > 0.05). “n.d.” indicates not detectable. For the data shown in panels B to D, at least three mice were used for each group.
Monocytes and pDCs are the main source of type I IFNs during IOE infection.
Multiple cell types can produce type I IFNs (6, 45–47); thus, we sought to identify the origin of type I IFNs during ehrlichial infection. We performed intracellular staining to detect cellular sources of IFN-α during IOE infection at day 7 postinfection, as this was the peak of the IFN-α response. Relative to mock-infected mice, IOE-infected WT mice exhibited two populations of IFN-α-positive cells in the spleen: Ly6C+ CD11b− and Ly6C+ CD11b+ cells (Fig. 3A). The Ly6C+ CD11b− cells expressed CD45, CD317 (PDCA-1), Siglec H, and B220 but did not express CD3, CD19, NK1.1, or Ly6G (data not shown), indicating they are likely pDCs (48–50). The Ly6C+ CD11b+ cells expressed CD115 and were F4/80+/−, characteristic of monocytes. Thus, we concluded that the two populations of IFN-α+ cells were pDCs and monocytes, respectively, both of which were significantly increased (Fig. 3C). To identify IFN-β-producing cells, we used an IFN-β reporter mouse (51). During IOE infection at day 7 postinfection, IOE-infected IFN-β–yellow fluorescent protein (YFP) mice exhibited an increased number of splenic IFN-β+ pDCs, relative to that of mock-infected mice (Fig. 3B and D). Taken together, our data demonstrate that pDCs and monocytes are the primary hematopoietic cells that produce IFN-α and pDCs are producers of IFN-β during IOE infection.
FIG 3.

pDCs and monocytes produce type I IFNs during IOE infection. IOE-infected mice were analyzed for production of type I IFNs by intracellular staining for IFN-α and by using IFN-β–YFP reporter mice on day 7 postinfection. (A) Representative flow cytometric plots of intracellular staining for IFN-α+ cells are shown. Monocytes were characterized as Ly6C+ CD11b+ CD115+ F4/80+/− (top). pDCs were characterized as Ly6C+ CD11b− Siglec H+ CD11clo CD317+ B220lo (bottom). (B) Representative flow cytometric plots of the gating strategy to identify YFP+ cells (IFN-β+ cells) are shown. Monocytes were characterized as Ly6C+ CD11b+ CD115+ F4/80+/− (top). pDCs were characterized as Ly6C+ CD11b− Siglec H+ CD11clo CD317+ B220lo (bottom). The numbers shown in the plots indicate the frequency (%) of cells within the gated region. (C) Absolute numbers of IFN-α+ pDCs and monocytes are shown. (D) Absolute numbers of YFP (IFN-β)+ pDCs and monocytes are shown. Asterisks indicate a significant difference compared to the counterpart mock controls (P < 0.05). Data are from two separate experiments, and at least four animals were used for each group.
Impact of type I IFNs on TNF-α and IFN-γ production.
During IOE infection, CD8 T cell production of TNF-α is significantly increased (28), and abrogation of TNF-αR-mediated signaling delays death after IOE infection (29). Thus, we next compared TNF-α production between WT and Ifnar-deficient mice during IOE infection. As T cells are the major cells that produced TNF-α during IOE infection (28), we performed intracellular staining for TNF-α during IOE infection. Both CD4 and CD8 T cells produced more TNF-α in the absence of Ifnar-mediated signaling during IOE infection (Fig. 4A and B). TNF-α can induce apoptosis during infection (52); thus, we compared apoptosis of leukocyte populations between WT and Ifnar-deficient mice during IOE infection using flow cytometric analysis of annexin V and 7-aminoactinomycin D. WT and Ifnar-deficient mice had similar frequencies of apoptotic and necrotic leukocytes, including T cells, B cells, NK cells, NKT cells, monocytes, macrophages, and neutrophils (data not shown). Thus, in the absence of type I IFNs, increased TNF-α does not contribute significantly to lethal inflammation, consistent with a mouse model of lethal shock caused by injection of TNF-α, where type I IFNs are required for morbidity (53).
FIG 4.
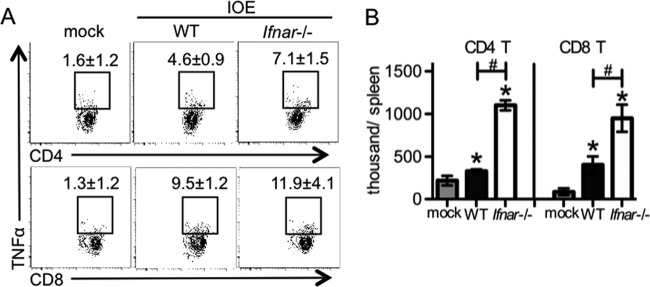
TNF-α-producing T cells were increased in the absence of IFN-αR signaling during IOE infection. WT and Ifnar−/− mice were infected with IOE, and TNF-α was measured by intracellular staining at day 9 postinfection. (A) Representative flow cytometric plots of intracellular staining of TNF-α in splenic CD4 and CD8 T cells (CD3+) are shown. The numbers (means ± standard deviation [SD]) above the square gates represent the percentage of TNF-α+ cells among all CD4 or CD8 T cells. (B) Absolute numbers of TNF-α+ CD4 T cells and CD8 T cells are shown. Asterisks indicate a significant difference compared to the counterpart mock controls (P < 0.05). Number symbols (#) indicate a significant difference between WT and Ifnar−/− mice (P < 0.05). At least four animals were used for each group.
IFN-γ has been shown to be protective during Ehrlichia infection (16, 34). IFN-γ production and IFN-γ signaling can be suppressed by type I IFNs during infection (17, 54); thus, we next measured IFN-γ in the absence of type I IFNs. Relative to WT mice, Ifnar-deficient mice had increased concentrations of IFN-γ in both serum and spleen during IOE infection (Fig. 5A and B). CD4 T cells are the major source of IFN-γ during ehrlichial infection (34, 43); thus, we performed intracellular staining for IFN-γ and confirmed that CD4 T cells were the major IFN-γ-producing cells in both WT mice and Ifnar-deficient mice during IOE infection. Moreover, relative to WT mice, Ifnar-deficient mice had significantly more IFN-γ-producing CD4 T cells in the spleen (Fig. 5C and D). As IFN-γ is protective during ehrlichial infection, we reasoned that increased IFN-γ in Ifnar-deficient mice was in part responsible for their increased survival.
FIG 5.
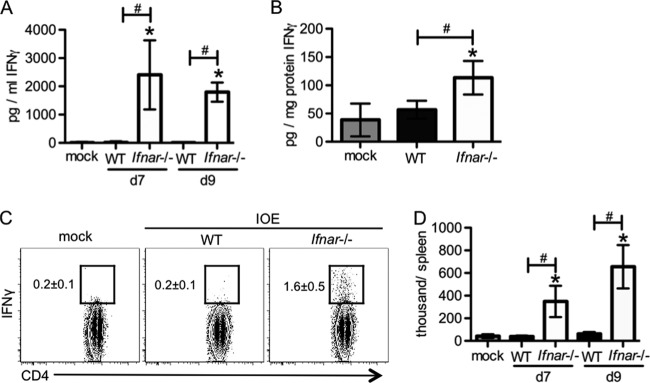
Suppression of IFN-γ production by type I IFNs during IOE infection. WT mice and Ifnar−/− mice were infected with IOE. IFN-γ expression was measured by ELISA and intracellular staining. (A) Serum IFN-γ concentrations at days 7 and 9 postinfection are shown. (B) Spleen IFN-γ concentrations at day 9 postinfection are shown. (C) Representative flow cytometric plots of intracellular staining for IFN-γ by CD4 T cells (gated on CD3+ CD4+ cells) in the spleens at day 7 postinfection are shown. The numbers (means ± SD) next to the square gates represent the percentage of IFN-γ+ cells among all the CD4 T cells. (D) Absolute numbers of CD4+ IFN-γ+ T cells for each spleen are shown. Asterisks indicate a significant difference compared to mock controls (P < 0.05). Number symbols (#) indicate a significant difference between the groups as indicated (P < 0.05). Data are from two separate experiments, and at least four animals were used for each group.
IFN-γ-dependent increase in Ehrlichia-specific serum IgM.
Infection with the pathogen E. muris elicits a robust expansion of B220loCD11clo plasmablasts that produce a majority of pathogen-specific IgM (35). The Ehrlichia-specific IgM is also sufficient to protect against IOE infection in a challenge model (36). Compared to WT mice, we found that Ifnar-deficient mice had increased plasmablasts in the spleen (Fig. 6A and B). In addition, pathogen-reactive IgM in serum was also increased in the absence of Ifnar-mediated signaling (Fig. 6C). These data suggest that type I IFNs were able to suppress the generation of plasmablasts and pathogen-specific IgM, resulting in increased susceptibility to IOE infection. Next, to test whether the impaired generation of plasmablasts during IOE infection was due to Ifnar-mediated signaling in B cells, we generated mixed BM chimeric mice that contained an equal mixture of WT and Ifnar-deficient BM. After IOE infection, we observed equal frequencies of splenic plasmablasts derived from Ifnar-deficient and WT cells (Fig. 6D and E), indicating that intrinsic Ifnar-mediated signaling was not suppressive for plasmablast generation. Because the higher pathogen-specific IgM production in Ifnar-deficient mice correlated with increased IFN-γ production, we next tested whether increased plasmablasts and pathogen-reactive IgM production in Ifnar-deficient mice required IFN-γ signaling. Thus, we treated Ifnar-deficient mice with a neutralizing antibody against IFN-γ. Neutralization of IFN-γ resulted in significantly reduced splenic plasmablasts (Fig. 6F and G) and serum IgM (Fig. 6H), which was comparable to the levels observed in infected WT mice. These findings demonstrate an important role for IFN-γ in driving expansion of the CD11clo B220lo plasmablast population and production of IgM during ehrlichial infection.
FIG 6.

IFN-γ induces pathogen-specific IgM production during IOE infection in the absence of Ifnar signaling. Plasmablasts (B220lo CD11clo) and serum Ehrlichia-specific IgM were measured at different time points post-IOE infection as indicated. (A) Representative flow cytometric plots of plasmablasts in the spleens of WT and Ifnar−/− mice at day 8 postinfection are shown. The numbers (means ± SD) above the circle gates represent the frequency of plasmablasts. (B) The absolute numbers of splenic plasmablasts are shown. (C) Titers of serum OMP-19-specific IgM in WT and Ifnar−/− mice at days 9 and 10 postinfection are shown. (D) Representative flow cytometric plots of WT (CD45.1− CD45.2+ GFP+) and Ifnar−/− (CD45.1− CD45.2+ GFP−) plasmablasts in spleens of mixed BM chimeric mice at day 7 postinfection are shown. The numbers in the quadrants represent the percentage of WT or Ifnar−/− cells among total splenic plasmablasts. (E) Quantification of percentage of WT and Ifnar−/− plasmablasts in panel D is shown. (F) Representative flow cytometric plots of splenic plasmablasts on day 8 postinfection in Ifnar−/− mice with or without anti-IFN-γ treatment are shown. The numbers above the gate represent the percentage of plasmablasts among all splenocytes. (G) Quantification of plasmablasts in panel F is shown. (H) Titers of serum OMP-19-specific IgM in Ifnar−/− mice with or without anti-IFN-γ treatment at day 8 postinfection are shown. Asterisks indicate a significant difference compared to uninfected controls (P < 0.05). Number symbols (#) indicate a significant difference between the groups as indicated (P < 0.05). “n.s.” indicates no significant difference (P > 0.05). “n.d.” indicates not detectable. At least four animals were used for each group.
Increased IFN-γ and IFN-γ-dependent IgM is dispensable for increased survival.
IFN-γ and IgM are important for protection against primary infection with the less virulent strain, E. muris, and are involved in protection against lethal challenge with IOE (33, 34, 36, 43). To determine whether type I IFN-mediated suppression of IFN-γ and the IFN-γ-dependent plasmablast response was responsible for IOE-induced death in WT animals, we neutralized IFN-γ during the course of infection in Ifnar-deficient mice. Unexpectedly, we observed that neutralization of IFN-γ had no impact on bacterial burden in the spleen or lung and did not impair survival (Fig. 7A and B). Neutralization of IFN-γ was effective as determined by ELISA (Fig. 7C). In addition, we found that the IFN-γ-dependent splenomegaly observed in Ifnar-deficient mice was abrogated by IFN-γ neutralization (data not shown). Thus, increased survival in the absence of type I IFNs does not require increased IFN-γ or IgM.
FIG 7.
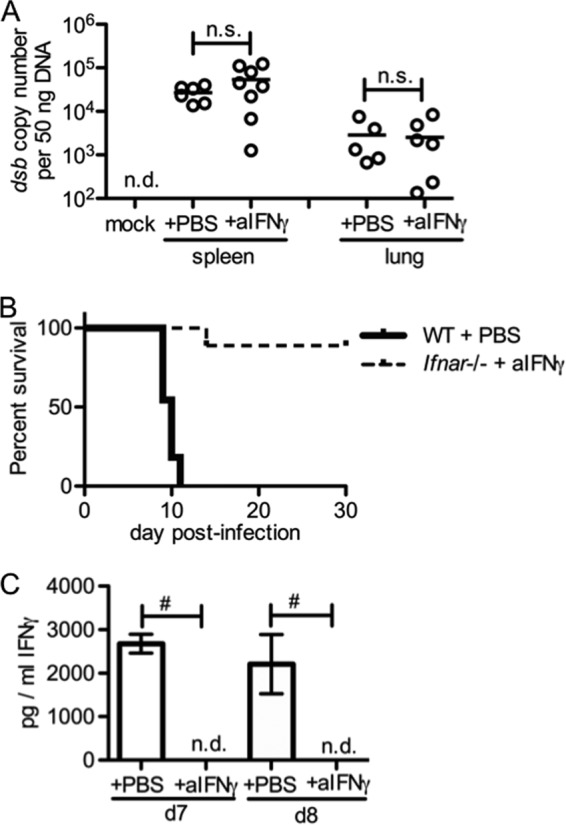
IFN-γ and pathogen-specific IgM are not essential for host defense against IOE infection in the absence of IFN-αR signaling. Ifnar−/− mice were treated with anti-IFN-γ or PBS every other day, beginning on day 2 post-IOE infection. (A) Bacterial burden in the spleens and lungs was measured in Ifnar−/− mice treated with or without anti-IFN-γ antibodies (day 8 postinfection). (B) Survival curves of WT mice treated with PBS and Ifnar−/− mice that received anti-IFN-γ are shown. (C) Serum IFN-γ concentrations in Ifnar−/− mice that received anti-IFN-γ treatment are shown. Number symbols (#) indicate a significant difference between the groups as indicated (P < 0.05). “n.s.” indicates no significant difference (P > 0.05). “n.d.” indicates not detectable. For data shown in panels A and C, at least five animals for each group were used. For panel B, at least nine animals for each group were used.
Critical role of type I IFN signaling in nonhematopoietic cells in lethality of IOE infection.
As increased IFN-γ and IgM were dispensable for survival in Ifnar-deficient mice, we sought to address whether type I IFN signaling in hematopoietic or nonhematopoietic cells was critical for IOE-induced death. In order to examine this, we generated reverse BM chimeric mice (WT BM to Ifnar-deficient hosts and Ifnar-deficient BM to WT hosts). After IOE infection, Ifnar-deficient hosts with a WT hematopoietic system survived significantly longer than WT hosts with an Ifnar-deficient hematopoietic system (Fig. 8A). WT hosts with an Ifnar-deficient hematopoietic system also had a slightly, but not significantly, higher concentration of serum IFN-γ (Fig. 8B). We found no difference in serum concentration of IFN-α between the reverse chimeric mice (Fig. 8C). However, we noted that IFN-β concentrations were significantly reduced in chimeric mice where the nonhematopoietic system was WT and the hematopoietic compartment lacked Ifnar. Thus, improved survival is associated with a nonhematopoietic compartment that cannot respond to type I IFNs, despite increased IFN-β in these mice, and a presumably greater impact of IFN-β on the nonhematopoietic system. It also suggests that IFN-αR signaling in hematopoietic cells is required for increase IFN-β production. Although mice with a WT hematopoietic system and Ifnar-deficient nonhematopoietic cells exhibited increased survival, they had a higher bacterial burden in both the spleen and liver (Fig. 8D). Our data demonstrate that IFN-αR signaling in nonhematopoietic cells is more important in mediating host death during IOE infection than the effects of type I IFNs on the hematopoietic system. Thus, type I IFNs promote shock-like disease during severe ehrlichial disease by targeting nonhematopoietic tissues.
FIG 8.

IFN-αR signaling in the nonhematopoietic system contributes significantly to host death during IOE infection. Reverse chimeric mice (WT BM to Ifnar−/− recipients and Ifnar−/− BM to WT recipients, respectively) were generated and subsequently infected with IOE. (A) Survival curves for reverse chimeric mice during IOE infection are shown. P value shown (log-rank [Mantel-Cox] test) indicates the comparison of survival curves between the two reverse chimeric mice. (B) Serum IFN-γ concentrations in reverse chimeric mice at day 8 postinfection are shown. (C) Serum IFN-α and IFN-β in reverse chimeric mice at day 8 postinfection are shown. (D) IOE burden in the spleens and livers of reverse chimeric mice at day 8 postinfection is shown. Number symbols (#) indicate a significant difference between the groups as indicated (P < 0.05). “n.s.” indicates no significant difference (P > 0.05). For data shown in panel A, at least eight animals for each group were used. For panels B and C, three animals for each group were used.
DISCUSSION
Type I IFNs can be protective or detrimental during infection, depending on the infectious organism (6). A possible role for type I IFNs in immunity to ehrlichial infection has not been previously investigated. We demonstrate here that infection with IOE, which causes fatal disease, elicits a robust type I IFN response that contributes to lethality. Type I IFNs exert a variety of immunomodulatory effects on host immune responses, and we found that type I IFNs impaired the production of IFN-γ. However, increased IFN-γ was not essential for reduced bacterial burden or increased survival in the Ifnar-deficient mice. This was surprising, as impaired IFN-γ has previously been thought to contribute to severity of disease during IOE infection. In fact, IFN-αR-mediated signaling in the nonhematopoietic system contributed significantly to death during IOE infection. IOE-infected mice die of a toxic shock-like syndrome characterized by extensive liver damage and interstitial pneumonitis (27). Vascular dysfunction is evident in extremely ill mice, and although the precise mechanism causing death is not clear, lung pathology and liver damage are likely involved. Our data demonstrate for the first time that direct IFN-αR signaling in nonhematopoietic cells, which may include endothelial cells, epithelial cells, or hepatocytes, is critical in causing death in a model of lethal ehrlichiosis.
Type I IFNs have been associated with endothelial activation and apoptosis, but the mechanisms underlying these effects, particularly during rickettsial infections, are unclear (55). Vascular permeability is regulated, in part, by the opening and closing of endothelial tight junctions. It was recently shown that phosphorylation of vascular endothelial (VE) cadherin, a key protein in maintenance of adherens junctions, precedes the increase in endothelial cell permeability in an in vitro model of Rickettsia montanensis infection (56), supporting the idea that endothelial tight junctions may be dysfunctional during severe ehrlichiosis. Vascular endothelial cell growth factor (VEGF) and the angiopoietin-Tie2 ligand receptor system function to regulate both angiogenesis and microvascular permeability (57). Angiopoietin 2 (Ang2) contributes to increased expression of intercellular and vascular cell adhesion molecules (ICAM-1 and VCAM-1) on endothelial cells (58), which promotes recruitment of monocytes and neutrophils. Interestingly, increased Ang2 is observed in anthrax (59), dengue hemorrhagic fever (60), and severe malaria, all diseases associated with vascular dysfunction and induction of type I IFNs. Future studies are required to address the precise downstream mechanisms by which type I IFN signaling in nonhematopoietic cells contributes to lethal infection and vascular dysfunction during rickettsial infections.
As TNF-α has previously been shown to be an important driver of disease, we were surprised by our observation that Ifnar-deficient mice exhibited slightly increased TNF-α and significantly more TNF-α-producing CD4 and CD8 T cells. We believe this finding illustrates the requirement of type I IFNs in driving shock-like disease. Whereas TNF-α may initiate disease during IOE infection, type I IFNs appear to be necessary for development of the severe toxic shock-like syndrome. This is consistent with a mouse model of shock caused by TNF-α administration. In comparison to WT mice, TNF-α administration to Ifnar-deficient mice results in reduced levels of proinflammatory cytokines, including interleukin 6 (IL-6), and less apoptosis in the liver (53). Further supporting this idea is the observation that TNF-R2-mediated signaling in endothelial cells induced the expression of IFN-β, which acted in an autocrine fashion to regulate chemokine expression, thus controlling recruitment of monocytes and macrophages to the endothelium (61). This proinflammatory circuit was shown to drive severe kidney disease in vivo. Thus, type I IFNs may mediate severe disease only in a context where TNF-α is already present. This experimental finding is supported by the clinical observation that type I IFNs are used frequently for treating hepatitis C virus and some types of cancer, such as melanoma, without causing shock (62, 63), whereas systemic administration of TNF-α causes significant toxicity, including liver damage and hypotension (32, 64). Understanding the relationship between TNF-α and type I IFNs in driving shock-like disease is likely relevant to other inflammatory diseases, such as sepsis.
Type I IFNs are produced constitutively at low levels (2), and their expression can be triggered during infection by pathogen-associated molecular patterns (PAMPs). Current dogma suggests bacterial pathogens elicit type I IFN production via bacterial ligands, such as lipopolysaccharide (LPS) and peptidoglycan (PDG), that can stimulate TLRs. Ehrlichia lacks the genes encoding LPS and PDG; thus, the mechanism driving IFN-α and IFN-β production may involve sensing of bacterial nucleic acids. Recognition of nucleic acids depends on membrane-bound receptors, including TLR3, TLR7, and TLR9. We observed production of type I IFNs in the absence of MyD88 and TLR2, suggesting that TLR-mediated signaling is not required for induction of type I IFNs in response to IOE infection. However, it is possible that TLR3 is involved, as it can signal independently of MyD88. Cytosolic receptors, including stimulator of IFN genes (STING), can also sense nucleic acids and appear to be particularly important for type I IFN induction in response to intracellular bacterial infections (65). It is possible that STING may be involved in the induction of type I IFNs during IOE infection, and future experiments will address this possibility.
Plasmacytoid dendritic cells (pDCs) are a potent source of type I IFNs during viral infections, whereas monocytes and macrophages appear to be the primary source of type I IFNs during infection with the intracellular bacterial pathogens Brucella abortus (66), Listeria monocytogenes (67), and Mycobacterium tuberculosis (68). Here, we demonstrate that pDCs and monocytes produce both IFN-α and IFN-β during IOE infection. It is not yet clear whether type I IFNs are elicited during IOE infection due to direct pathogen recognition or due to infection-induced damage. Production of type I IFNs by pDCs and monocytes during IOE infection may also involve distinct mechanisms. Identifying the mechanism of IFN induction in response to IOE may reveal novel therapeutic targets for severe rickettsial infections that cause shock-like disease.
We observed improved bacterial clearance in the absence of IFN-αR-mediated signaling, but the precise mechanisms by which type I IFNs impair bacterial clearance are not yet clear. We observed an enhanced IFN-γ response and increased IgM responses in Ifnar-decifient mice, yet these were not required for effective control of bacteria, as a similar bacterial burden was observed upon IFN-γ neutralization. These data were surprising and indicate that a unique IFN-αβ-driven pathway impairs ehrlichial clearance. Increased TNF-α was also observed in the absence of type I IFNs, and it is possible that TNF-α is essential to control bacterial growth.
We made the surprising observation that mortality did not appear to correlate with bacterial burden. In WT mice that contained an Ifnar-deficient hematopoietic system, we observed reduced bacterial burden in both the spleen and liver. However, mice died early, with kinetics similar to those of WT mice. On the other hand, Ifnar-deficient hosts containing WT hematopoietic cells exhibited higher bacterial burdens but prolonged survival. These data suggest that improved survival in the absence of type I IFNs is due, in part, to a reduced damage or a diminished pathogenic inflammatory response. Underscoring the role of type I IFNs in promoting a detrimental inflammatory response, we found that mice lacking TLR2 exhibited significantly increased type I IFNs, relative to WT mice, and TLR2-deficient mice are highly susceptible to IOE (44). The precise mechanisms by which type I IFNs participate in severe disease require further study and are likely part of a more complex story. Herein, we demonstrate for the first time, to our knowledge, the critical role for type I IFNs in driving severe disease during a rickettsial infection. These findings provide rationale to examine the contribution of type I IFNs to disease in other rickettsial infections and to address the precise mechanisms by which type I IFN signaling in nonhematopoietic cells contribute to severe shock-like disease.
ACKNOWLEDGMENTS
We gratefully acknowledge Gary Winslow for kindly providing reagents. We also thank Yili Lin in the Flow Cytometry Core at Albany Medical College. We thank Lei Jin for helpful comments and suggestions regarding this project and in preparing the manuscript.
Footnotes
Published ahead of print 3 February 2014
REFERENCES
- 1.Piehler J, Thomas C, Garcia KC, Schreiber G. 2012. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 250:317–334. 10.1111/imr.12001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. 2012. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 36:166–174. 10.1016/j.immuni.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. 2009. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458:904–908. 10.1038/nature07815 [DOI] [PubMed] [Google Scholar]
- 4.Vogel SN, Fertsch D. 1984. Endogenous interferon production by endotoxin-responsive macrophages provides an autostimulatory differentiation signal. Infect. Immun. 45:417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, Hertzog P, Smyth MJ. 2007. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J. Immunol. 178:7540–7549 [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G. 2010. Type I interferon: friend or foe? J. Exp. Med. 207:2053–2063. 10.1084/jem.20101664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrero JA. 2013. Confounding roles for type I interferons during bacterial and viral pathogenesis. Int. Immunol. 25:663–669. 10.1093/intimm/dxt050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Decker T, Muller M, Stockinger S. 2005. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 5:675–687. 10.1038/nri1684 [DOI] [PubMed] [Google Scholar]
- 9.Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. 2010. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J. Exp. Med. 207:327–337. 10.1084/jem.20091746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brzoza-Lewis KL, Hoth JJ, Hiltbold EM. 2012. Type I interferon signaling regulates the composition of inflammatory infiltrates upon infection with Listeria monocytogenes. Cell. Immunol. 273:41–51. 10.1016/j.cellimm.2011.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. 2012. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 13:954–962. 10.1038/ni.2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. 2007. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J. Immunol. 178:3126–3133 [DOI] [PubMed] [Google Scholar]
- 13.Del Fresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, Ruland J, Ardavin C. 2013. Interferon-beta production via dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 38:1176–1186. 10.1016/j.immuni.2013.05.010 [DOI] [PubMed] [Google Scholar]
- 14.Buchmeier NA, Schreiber RD. 1985. Requirement of endogenous interferon-gamma production for resolution of Listeria monocytogenes infection. Proc. Natl. Acad. Sci. U. S. A. 82:7404–7408. 10.1073/pnas.82.21.7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. 1993. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178:2249–2254. 10.1084/jem.178.6.2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacNamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, Choi K, Borjesson DL, Winslow GM. 2011. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-gamma signaling. J. Immunol. 186:1032–1043. 10.4049/jimmunol.1001893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, Modlin RL. 2013. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339:1448–1453. 10.1126/science.1233665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O'Garra A. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977. 10.1038/nature09247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, Kaplan G. 2005. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 25:694–701. 10.1089/jir.2005.25.694 [DOI] [PubMed] [Google Scholar]
- 20.Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE, III, Freedman VH, Kaplan G. 2001. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc. Natl. Acad. Sci. U. S. A. 98:5752–5757. 10.1073/pnas.091096998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamburg BJ, Storch GA, Micek ST, Kollef MH. 2008. The importance of early treatment with doxycycline in human ehrlichiosis. Medicine 87:53–60. 10.1097/MD.0b013e318168da1d [DOI] [PubMed] [Google Scholar]
- 22.Kirkland KB, Wilkinson WE, Sexton DJ. 1995. Therapeutic delay and mortality in cases of Rocky Mountain spotted fever. Clin. Infect. Dis. 20:1118–1121. 10.1093/clinids/20.5.1118 [DOI] [PubMed] [Google Scholar]
- 23.Pritt BS, Sloan LM, Johnson DK, Munderloh UG, Paskewitz SM, McElroy KM, McFadden JD, Binnicker MJ, Neitzel DF, Liu G, Nicholson WL, Nelson CM, Franson JJ, Martin SA, Cunningham SA, Steward CR, Bogumill K, Bjorgaard ME, Davis JP, McQuiston JH, Warshauer DM, Wilhelm MP, Patel R, Trivedi VA, Eremeeva ME. 2011. Emergence of a new pathogenic Ehrlichia species, Wisconsin and Minnesota, 2009. N. Engl. J. Med. 365:422–429. 10.1056/NEJMoa1010493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker DH, Ismail N, Olano JP, McBride JW, Yu XJ, Feng HM. 2004. Ehrlichia chaffeensis: a prevalent, life-threatening, emerging pathogen. Trans. Am. Clin. Climatol. Assoc. 115:375–382; discussion 382–374 [PMC free article] [PubMed] [Google Scholar]
- 25.Feng HM, Walker DH. 2004. Mechanisms of immunity to Ehrlichia muris: a model of monocytotropic ehrlichiosis. Infect. Immun. 72:966–971. 10.1128/IAI.72.2.966-971.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shibata S, Kawahara M, Rikihisa Y, Fujita H, Watanabe Y, Suto C, Ito T. 2000. New Ehrlichia species closely related to Ehrlichia chaffeensis isolated from Ixodes ovatus ticks in Japan. J. Clin. Microbiol. 38:1331–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sotomayor EA, Popov VL, Feng HM, Walker DH, Olano JP. 2001. Animal model of fatal human monocytotropic ehrlichiosis. Am. J. Pathol. 158:757–769. 10.1016/S0002-9440(10)64018-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ismail N, Soong L, McBride JW, Valbuena G, Olano JP, Feng HM, Walker DH. 2004. Overproduction of TNF-alpha by CD8+ type 1 cells and down-regulation of IFN-gamma production by CD4+ Th1 cells contribute to toxic shock-like syndrome in an animal model of fatal monocytotropic ehrlichiosis. J. Immunol. 172:1786–1800 [DOI] [PubMed] [Google Scholar]
- 29.Ismail N, Stevenson HL, Walker DH. 2006. Role of tumor necrosis factor alpha (TNF-alpha) and interleukin-10 in the pathogenesis of severe murine monocytotropic ehrlichiosis: increased resistance of TNF receptor p55- and p75-deficient mice to fatal ehrlichial infection. Infect. Immun. 74:1846–1856. 10.1128/IAI.74.3.1846-1856.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bitsaktsis C, Winslow G. 2006. Fatal recall responses mediated by CD8 T cells during intracellular bacterial challenge infection. J. Immunol. 177:4644–4651 [DOI] [PubMed] [Google Scholar]
- 31.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. 2011. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35:908–918. 10.1016/j.immuni.2011.09.020 [DOI] [PubMed] [Google Scholar]
- 32.Kimura K, Taguchi T, Urushizaki I, Ohno R, Abe O, Furue H, Hattori T, Ichihashi H, Inoguchi K, Majima H, Niitani H, Ota K, Saito T, Suga S, Suzuoki Y, Wakui A, Yamada K, The A-TNF Cooperative Study Group 1987. Phase I study of recombinant human tumor necrosis factor. Cancer Chemother. Pharmacol. 20:223–229. 10.1007/BF00570490 [DOI] [PubMed] [Google Scholar]
- 33.Thirumalapura NR, Stevenson HL, Walker DH, Ismail N. 2008. Protective heterologous immunity against fatal ehrlichiosis and lack of protection following homologous challenge. Infect. Immun. 76:1920–1930. 10.1128/IAI.01293-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bitsaktsis C, Huntington J, Winslow G. 2004. Production of IFN-gamma by CD4 T cells is essential for resolving Ehrlichia infection. J. Immunol. 172:6894–6901 [DOI] [PubMed] [Google Scholar]
- 35.Racine R, Chatterjee M, Winslow GM. 2008. CD11c expression identifies a population of extrafollicular antigen-specific splenic plasmablasts responsible for CD4 T-independent antibody responses during intracellular bacterial infection. J. Immunol. 181:1375–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Racine R, McLaughlin M, Jones DD, Wittmer ST, MacNamara KC, Woodland DL, Winslow GM. 2011. IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J. Immunol. 186:1011–1021. 10.4049/jimmunol.1002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ismail N, Crossley EC, Stevenson HL, Walker DH. 2007. Relative importance of T-cell subsets in monocytotropic ehrlichiosis: a novel effector mechanism involved in Ehrlichia-induced immunopathology in murine ehrlichiosis. Infect. Immun. 75:4608–4620. 10.1128/IAI.00198-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stevenson HL, Crossley EC, Thirumalapura N, Walker DH, Ismail N. 2008. Regulatory roles of CD1d-restricted NKT cells in the induction of toxic shock-like syndrome in an animal model of fatal ehrlichiosis. Infect. Immun. 76:1434–1444. 10.1128/IAI.01242-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Q, Ghose P, Ismail N. 2013. Neutrophils mediate immunopathology and negatively regulate protective immune responses during fatal bacterial infection-induced toxic shock. Infect. Immun. 81:1751–1763. 10.1128/IAI.01409-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacNamara KC, Racine R, Chatterjee M, Borjesson D, Winslow GM. 2009. Diminished hematopoietic activity associated with alterations in innate and adaptive immunity in a mouse model of human monocytic ehrlichiosis. Infect. Immun. 77:4061–4069. 10.1128/IAI.01550-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, Whitton JL. 2005. Cutting edge: re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J. Immunol. 174:5936–5940 [DOI] [PubMed] [Google Scholar]
- 42.Nandi B, Hogle K, Vitko N, Winslow GM. 2007. CD4 T-cell epitopes associated with protective immunity induced following vaccination of mice with an ehrlichial variable outer membrane protein. Infect. Immun. 75:5453–5459. 10.1128/IAI.00713-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Jones M, McCabe A, Winslow GM, Avram D, MacNamara KC. 2013. MyD88 signaling in CD4 T cells promotes IFN-gamma production and hematopoietic progenitor cell expansion in response to intracellular bacterial infection. J. Immunol. 190:4725–4735. 10.4049/jimmunol.1203024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chattoraj P, Yang Q, Khandai A, Al-Hendy O, Ismail N. 2013. TLR2 and Nod2 mediate resistance or susceptibility to fatal intracellular Ehrlichia infection in murine models of ehrlichiosis. PLoS One 8:e58514. 10.1371/journal.pone.0058514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwandt T, Schumak B, Gielen GH, Jungerkes F, Schmidbauer P, Klocke K, Staratschek-Jox A, van Rooijen N, Kraal G, Ludwig-Portugall I, Franken L, Wehner S, Kalff JC, Weber O, Kirschning C, Coch C, Kalinke U, Wenzel J, Kurts C, Zawatzky R, Holzmann B, Layland L, Schultze JL, Burgdorf S, den Haan JM, Knolle PA, Limmer A. 2012. Expression of type I interferon by splenic macrophages suppresses adaptive immunity during sepsis. EMBO J. 31:201–213. 10.1038/emboj.2011.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cervantes-Barragan L, Zust R, Weber F, Spiegel M, Lang KS, Akira S, Thiel V, Ludewig B. 2007. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 109:1131–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng D, Gommerman JL. 2013. The regulation of immune responses by DC derived type I IFN. Front. Immunol. 4:94. 10.3389/fimmu.2013.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wendland M, Czeloth N, Mach N, Malissen B, Kremmer E, Pabst O, Forster R. 2007. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc. Natl. Acad. Sci. U. S. A. 104:6347–6352. 10.1073/pnas.0609180104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Raper A, Sugita N, Hingorani R, Salio M, Palmowski MJ, Cerundolo V, Crocker PR. 2006. Characterization of Siglec-H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood 107:3600–3608. 10.1182/blood-2005-09-3842 [DOI] [PubMed] [Google Scholar]
- 50.Barchet W, Cella M, Colonna M. 2005. Plasmacytoid dendritic cells—virus experts of innate immunity. Semin. Immunol. 17:253–261. 10.1016/j.smim.2005.05.008 [DOI] [PubMed] [Google Scholar]
- 51.Scheu S, Dresing P, Locksley RM. 2008. Visualization of IFNbeta production by plasmacytoid versus conventional dendritic cells under specific stimulation conditions in vivo. Proc. Natl. Acad. Sci. U. S. A. 105:20416–20421. 10.1073/pnas.0808537105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wajant H, Pfizenmaier K, Scheurich P. 2003. Tumor necrosis factor signaling. Cell Death Differ. 10:45–65. 10.1038/sj.cdd.4401189 [DOI] [PubMed] [Google Scholar]
- 53.Huys L, Van Hauwermeiren F, Dejager L, Dejonckheere E, Lienenklaus S, Weiss S, Leclercq G, Libert C. 2009. Type I interferon drives tumor necrosis factor-induced lethal shock. J. Exp. Med. 206:1873–1882. 10.1084/jem.20090213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen KB, Cousens LP, Doughty LA, Pien GC, Durbin JE, Biron CA. 2000. Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox. Nat. Immunol. 1:70–76. 10.1038/76940 [DOI] [PubMed] [Google Scholar]
- 55.Nakano M, Fujii T, Hashimoto M, Yukawa N, Yoshifuji H, Ohmura K, Nakaizumi A, Mimori T. 2012. Type I interferon induces CX3CL1 (fractalkine) and CCL5 (RANTES) production in human pulmonary vascular endothelial cells. Clin. Exp. Immunol. 170:94–100. 10.1111/j.1365-2249.2012.04638.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gong B, Ma L, Liu Y, Gong Q, Shelite T, Bouyer D, Boor PJ, Lee YS, Oberhauser A. 2012. Rickettsiae induce microvascular hyperpermeability via phosphorylation of VE-cadherins: evidence from atomic force microscopy and biochemical studies. PLoS Negl. Trop. Dis. 6:e1699. 10.1371/journal.pntd.0001699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y, Pampou S, Fujikawa K, Varticovski L. 2004. Opposing effect of angiopoietin-1 on VEGF-mediated disruption of endothelial cell-cell interactions requires activation of PKC beta. J. Cell. Physiol. 198:53–61. 10.1002/jcp.10386 [DOI] [PubMed] [Google Scholar]
- 58.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, Gale NW, Witzenrath M, Rosseau S, Suttorp N, Sobke A, Herrmann M, Preissner KT, Vajkoczy P, Augustin HG. 2006. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat. Med. 12:235–239. 10.1038/nm1351 [DOI] [PubMed] [Google Scholar]
- 59.Ghosh CC, Mukherjee A, David S, Knaus UG, Stearns-Kurosawa DJ, Kurosawa S, Parikh SM. 2012. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc. Natl. Acad. Sci. U. S. A. 109:10024–10029. 10.1073/pnas.1120755109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michels M, van der Ven AJ, Djamiatun K, Fijnheer R, de Groot PG, Griffioen AW, Sebastian S, Faradz SM, de Mast Q. 2012. Imbalance of angiopoietin-1 and angiopoetin-2 in severe dengue and relationship with thrombocytopenia, endothelial activation, and vascular stability. Am. J. Trop. Med. Hyg. 87:943–946. 10.4269/ajtmh.2012.12-0020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Venkatesh D, Ernandez T, Rosetti F, Batal I, Cullere X, Luscinskas FW, Zhang Y, Stavrakis G, Garcia-Cardena G, Horwitz BH, Mayadas TN. 2013. Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-beta autocrine signaling to promote monocyte recruitment. Immunity 38:1025–1037. 10.1016/j.immuni.2013.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pawlotsky JM. 2013. Treatment of chronic hepatitis C: current and future. Curr. Top. Microbiol. Immunol. 369:321–342 [DOI] [PubMed] [Google Scholar]
- 63.Tarhini AA, Gogas H, Kirkwood JM. 2012. IFN-alpha in the treatment of melanoma. J. Immunol. 189:3789–3793. 10.4049/jimmunol.1290060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blick M, Sherwin SA, Rosenblum M, Gutterman J. 1987. Phase I study of recombinant tumor necrosis factor in cancer patients. Cancer Res. 47:2986–2989 [PubMed] [Google Scholar]
- 65.Barber GN. 2011. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 23:10–20. 10.1016/j.coi.2010.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Almeida LA, Carvalho NB, Oliveira FS, Lacerda TL, Vasconcelos AC, Nogueira L, Bafica A, Silva AM, Oliveira SC. 2011. MyD88 and STING signaling pathways are required for IRF3-mediated IFN-beta induction in response to Brucella abortus infection. PLoS One 6:e23135. 10.1371/journal.pone.0023135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stockinger S, Kastner R, Kernbauer E, Pilz A, Westermayer S, Reutterer B, Soulat D, Stengl G, Vogl C, Frenz T, Waibler Z, Taniguchi T, Rulicke T, Kalinke U, Muller M, Decker T. 2009. Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS Pathog. 5:e1000355. 10.1371/journal.ppat.1000355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stanley SA, Johndrow JE, Manzanillo P, Cox JS. 2007. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 178:3143–3152 [DOI] [PubMed] [Google Scholar]