

Abstract
Caspase recruitment domain-containing protein 9 (CARD9) is an adaptor molecule signal that is critical for NF-κB activation and is triggered through C-type lectin receptors (CLRs), which are pattern recognition receptors that recognize carbohydrate structures. Previous studies have reported that Cryptococcus neoformans, a fungal pathogen that causes meningoencephalitis in AIDS patients, is recognized through some CLRs, such as mannose receptors or DC-SIGN. However, the role of CARD9 in the host defense against cryptococcal infection remains to be elucidated. In the present study, we analyzed the role of CARD9 in the host defense against pulmonary infection with C. neoformans. CARD9 gene-disrupted (knockout [KO]) mice were highly susceptible to this infection, as shown by the reduced fungal clearance in the infected lungs of CARD9 KO mice, compared to that in wild-type (WT) mice. Gamma interferon (IFN-γ) production was strongly reduced in CARD9 KO mice during the innate-immunity phase of infection. Reduced IFN-γ synthesis was due to impaired accumulation of NK and memory phenotype T cells, which are major sources of IFN-γ innate-immunity-phase production; a reduction in the accumulation of these cells was correlated with reduced CCL4, CCL5, CXCL9, and CXCL10 synthesis. However, differentiation of Th17 cells, but not of Th1 cells, was impaired at the adaptive-immunity phase in CARD9 KO mice compared to WT mice, although there was no significant difference in the infection susceptibility between interleukin 17A (IL-17A) KO and WT mice. These results suggest that CARD9 KO mice are susceptible to C. neoformans infection probably due to the reduced accumulation of IFN-γ-expressing NK and memory phenotype T cells at the early stage of infection.
INTRODUCTION
Cryptococcus neoformans, an opportunistic fungal pathogen, causes life-threatening meningoencephalitis in hosts with compromised cellular immune responses, such as AIDS patients (1, 2). The host defense against cryptococcal infection is mediated mainly by the cellular immune response and critically regulated by the balance between type 1 helper T (Th1) and Th2 cytokine responses (3–5). A defect in the synthesis of Th1-related cytokines, such as gamma interferon (IFN-γ), interleukin 12 (IL-12), IL-18, and tumor necrosis factor alpha (TNF-α), exacerbates cryptococcal infection (6–9), while mice lacking Th2 cytokines, including IL-4, IL-10, and IL-13, have less severe infections than control mice (10–12). Recently, many investigators have shown that IL-17A is involved in the host defense mechanism, especially against extracellular microorganisms, including Candida albicans (13–15). This cytokine is produced by Th17 cells and makes a profound contribution to the neutrophilic inflammatory response (15). However, the role of IL-17A in the host defense against C. neoformans is not fully understood. Besides these Th responses, innate immune lymphocytes, such as natural killer (NK) cells, NKT cells, and γδ T cells, are also reported to play important roles in the host defense against cryptococcal infection (16–18).
The C-type lectin receptor (CLR) is a pattern recognition receptor that possesses a carbohydrate recognition domain (CRD), which binds polysaccharide ligands in a calcium-dependent manner (19, 20). Dectin-1 recognizes β-1,3-d-glucan, and Dectin-2 binds high-mannose oligosaccharides (21–23). A large amount of carbohydrates is contained in the fungal capsule and cell wall, and therefore, CLR is a critical recognition system for fungal pathogens. In earlier studies, the mannose receptor (MR) and dendritic-cell-specific ICAM-3-grabbing nonintegrin (DC-SIGN) are reported to recognize C. neoformans (24, 25). Conversely, our previous studies revealed that both Dectin-1 and Dectin-2 are not required for host resistance to C. neoformans infection (23, 26).
Caspase recruitment domain-containing protein 9 (CARD9) is an essential adaptor molecule in the signal delivery that is triggered by CLRs, and high levels are expressed in myeloid cells, such as macrophages and dendritic cells (27). Signaling via some CLRs is dependent on the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) and tyrosine kinase Syk, followed by activation of three adaptor molecule complexes, which consist of CARD9, B-cell lymphoma 10 (Bcl10), and mucosa-associated lymphoid tissue 1 (MALT1). These processes lead to induction of cytokine production and expression of costimulatory molecules via NF-κB activation, which is involved in the host defense against some fungi and bacteria (28–30). However, the role of CARD9 in the host defense against cryptococcal infection has not been elucidated.
In the present study, we examined the effect of CARD9 deficiency on the clearance of C. neoformans and the host immune response using a mouse model of pulmonary infection. Here, we demonstrate that CARD9-mediated signaling is necessary for the early production of IFN-γ at the innate-immunity stage and for the differentiation of Th17 cells, but not of Th1 cells, at the adaptive-immunity stage in the lungs after C. neoformans infection. We also demonstrate that the Th17 response is not required for the host defense against this infection.
MATERIALS AND METHODS
Ethics statement.
This study was performed in strict accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science, and Technology in Japan, 2006. All experimental procedures involving animals followed the Regulations for Animal Experiments and Related Activities at Tohoku University, Sendai, Japan, and were approved by the Institutional Animal Care and Use Committee at Tohoku University (approval numbers 22 IDOU-108, 2011 IDOU-86, 2012 IDOU-124, and 2013 IDOU-257). All experiments were performed under anesthesia, and all efforts were made to minimize suffering of the animals.
Mice.
CARD9 gene-disrupted (knockout [KO]) mice and IL-17A KO mice were generated and established as described previously (27, 31) and backcrossed to C57BL/6 mice for more than 8 generations. Wild-type (WT) C57BL/6 mice, purchased from CLEA Japan (Tokyo, Japan), were used as controls. Male or female mice at 6 to 8 weeks of age were used in the experiments. All mice were kept under specific-pathogen-free conditions at the Institute for Animal Experimentation, Tohoku University Graduate School of Medicine.
Cryptococcus neoformans.
A serotype D strain of C. neoformans, designated B3501 (a kind gift from Kwong Chung, National Institutes of Health, Bethesda, MD, USA), was used. The yeast cells were cultured on potato dextrose agar (PDA; Eiken, Tokyo, Japan) plates for 2 to 3 days before use. Mice were anesthetized by an intraperitoneal injection of 70 mg/kg of pentobarbital (Abbott Laboratory, North Chicago, IL, USA) and restrained on a small board. Live C. neoformans (1 × 106 cells) was inoculated in a 50-μl volume into the trachea of each mouse using a 24-gauge catheter (Terumo, Tokyo, Japan).
Treatment with anti-IFN-γ MAb.
Neutralizing anti-IFN-γ monoclonal antibody (MAb) and control rat IgG were purchased from BioLegend (San Diego, CA, USA) and MP Biomedicals, LLC, Santa Ana, CA, USA), respectively). Mice were injected intraperitoneally with either Ab at 100 μg/mouse 2 h before infection and intratracheally with the same Ab at 25 μg/mouse when they were infected.
Enumeration of viable C. neoformans.
Mice were sacrificed 2 weeks after infection, and lungs were dissected carefully, excised, and then homogenized separately in 5 ml of distilled water by teasing with a stainless mesh at room temperature. The homogenates, diluted appropriately with distilled water, were inoculated at 100 μl on PDA plates and cultured for 2 to 3 days, and the resulting colonies were counted.
Histological examination.
The lung specimens obtained from mice were fixed in 10% buffer formalin, dehydrated, and embedded in paraffin. Sections were cut and stained with hematoxylin-eosin (H-E) or periodic acid-Schiff (PAS) stain, using standard staining procedures, at the Biomedical Research Core, Animal Pathology Platform of Tohoku University Graduate School of Medicine.
In vitro stimulation of lymph node cells.
Paratracheal lymph node (LN) cells were prepared on day 7 after infection with C. neoformans and cultured at a concentration of 2 × 106/ml with various doses of viable yeast cells or concanavalin A (ConA; Sigma-Aldrich, St. Louis, MO, USA) in RPMI 1640 medium (Nipro, Osaka, Japan) supplemented with 10% fetal calf serum (FCS) (BioWest, Nuaillé, France), 100 U/ml penicillin G, 100 μg/ml streptomycin, and 50 μM 2-mercaptoethanol (Sigma-Aldrich) in a 5% CO2 incubator for 48 h. The culture supernatants were collected and stored at −70°C before use.
Preparation and culture of dendritic cells.
Bone marrow (BM) cells from WT and CARD9 KO mice were cultured at 2 × 105/ml in 10 ml RPMI 1640 medium supplemented with 10% FCS, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 50 μM 2-mercaptoethanol containing 20 ng/ml murine granulocyte-macrophage colony-stimulating factor (GM-CSF; Wako Pure Chemical Industries, Ltd., Osaka, Japan). On day 3, another 10 ml of the same medium was added, and on day 6, a half change was performed using the GM-CSF-containing culture medium. On day 8, nonadherent cells were collected and used as BM-derived dendritic cells (BM-DCs). The obtained cells were cultured at 1 × 105/ml with various doses of viable yeast cells or lipopolysaccharide (LPS) (Sigma-Aldrich) for 24 h at 37°C.
Extraction of RNA and quantitative real-time RT-PCR.
Total RNA was extracted from the infected lungs or BM-DCs using Isogen (Wako Pure Chemical, Osaka, Japan), and the first-strand cDNA was synthesized using PrimeScript first-strand cDNA synthesis kit (TaKaRa Bio Inc., Otsu, Japan), according to the manufacturer's instructions. Quantitative real-time PCR was performed in a volume of 20 μl using gene-specific primers and FastStart essential DNA green master mix (Roche Applied Science) in a LightCycler nanosystem (Roche Applied Science, Branford, CT, USA). The primer sequences used for amplification are shown in Table 1. Reaction efficiency with each primer set was determined using standard amplifications. Target gene expression levels and that of the hypoxanthine phosphoribosyltransferase (HPRT) gene as a reference were calculated for each sample using the reaction efficiency. The results were analyzed using a relative quantification procedure and are presented as expression relative to HPRT expression.
TABLE 1.
Primer sequences for real-time PCR
| Gene product | Forward primer | Reverse primer |
|---|---|---|
| IFN-γ | ACTGCCACGGCACAGTCATT | TCACCATCCTTTTGCCAGTTCCT |
| TGF-β | TACGCCTGAGTGGCTGTCTTTT | CGTGGAGTTTGTTATCTTTGCTGT |
| IL-6 | CACGGCCTTCCCTACTTCACAA | TTGCACAACTCTTTTCTCATTTCCACG |
| IL-12p35 | GAGTTCCAGGCCATCAACGCA | GCTTCTCCCACAGGAGGTTTCTG |
| IL-17A | AACCGTTCCAC GTCACCCTG | GTCCAGCTTTCCCTCCGCAT |
| IL-23p19 | CTCAGCCAACTCCTCCAGCCAG | CTGCTCCGTGGGCAAAGACC |
| CCL3 | AACCAGCAGCCTTTGCTCCC | GGTCTCTTTGGAGTCAGCGCA |
| CCL4 | AAACCTAACCCCGAGCAACACC | GAAACAGCAGGAAGTGGGAGGG |
| CCL5 | ACTCCCTGCTGCTTTGCCTAC | GGCGGTTCCTTCGAGTGACAA |
| CXCL9 | GGCACGATCCACTACAAATCCCT | AGGCAGGTTTGATCTCCGTTCTTC |
| CXCL10 | GTGCTGCCGTCATTTTCTGCC | AGGATAGGCTCGCAGGGATGA |
| iNOS | AGGGAATCTTGGAGCGAGTGGT | GCAGCCTCTTGTCTTTGACCC |
| T-bet | CACTAAGCAAGGACGGCGAATG | TCCACCAAGACCACATCCACAA |
| ROR-γt | CTTCCTCAGCGCCCTGTGTT | CCCAGGACGGTTGGCATTGA |
| HPRT | GCTTCCTCCTCAGACCGCTT | TCGCTAATCACGACGCTGGG |
Cytokine assay.
IFN-γ and IL-17A concentrations in the lung homogenates and culture supernatants were measured using the appropriate ELISA kit (Biolegend for IFN-γ and eBioscience [San Diego, CA, USA] for IL-17A). The detection limits were 16 pg/ml for IFN-γ and 8 pg/ml for IL-17A.
Preparation of lung leukocytes.
Pulmonary intraparenchymal leukocytes were prepared as previously described (32). Briefly, the chest of the mouse was opened, and the lung vascular bed was flushed by injecting 3 ml of chilled physiological saline into the right ventricle. The lungs were then excised and washed in physiological saline. The lungs were teased with a 40-μm cell strainer (BD Falcon, Bedford, MA, USA) and incubated in RPMI 1640 medium with 5% fetal calf serum, 100 U/ml penicillin G, 100 μg/ml streptomycin, 10 mM HEPES, 50 μM 2-mercaptoethanol, and 2 mM l-glutamine, containing 20 U/ml collagenase and 1 μg/ml DNase I (Sigma-Aldrich). After incubation for 60 min at 37°C with vigorous shaking, the tissue fragments and the majority of dead cells were removed by passing them through the 40-μm cell strainer. After centrifugation, the cell pellet was resuspended in 4 ml of 40% (vol/vol) Percoll (Pharmacia, Uppsala, Sweden) and layered onto 4 ml of 80% (vol/vol) Percoll. After centrifugation at 600 × g for 20 min at 15°C, the cells at the interface were collected, washed three times, and counted using a hemocytometer. The cells were centrifuged onto a glass slide at 110 × g for 3 min using Cytofuge-2 (Statspin Inc., Norwood, MA, USA), stained with Diff-Quick (Sysmex, Kobe, Japan), and observed under a microscope. The number of leukocyte fractions was estimated by multiplying the total leukocyte number by the proportion of each fraction in 200 cells.
Flow cytometry.
The lung leukocytes were cultured at 1 × 106/ml with 5 ng/ml phorbol 12-myristate 13-acetate (PMA), 500 ng/ml ionomycin, and 2 nM monensin (Sigma-Aldrich) in RPMI 1640 medium supplemented with 10% FCS for 4 h. The cells were washed three times in phosphate-buffered saline (PBS) containing 1% FCS and 0.1% sodium azide and then stained with allophycocyanin (APC)-conjugated anti-CD3ε MAb (clone 145-2C11; Biolegend) and phycoerythrin (PE)-conjugated anti-γδ T cell receptor (γδTCR) or PE-NK1.1 MAb (clone eBioGL3 [eBioscience] for γδTCR and clone PK136 [eBioscience] for NK1.1). To identify CD4+ or CD8+ memory phenotype T cells, the cells were stained with PE–anti-CD4, APC–Cy7–anti-CD8, and Pacific blue-conjugated anti-CD44 MAb (clones GK1.5, 53-6.7, and IM7, respectively; Biolegend). CD44dull+ and CD44bright+ cells were identified as naive T and memory phenotype T cells, respectively (33). After being washed twice, the cells were incubated in the presence of Cytofix/Cytoperm (BD Bioscience), washed twice in BD perm/wash solution (BD Bioscience), and stained with fluorescein isothiocyanate (FITC)-conjugated anti-IFN-γ (clone XMG1.2, Biolegend). Isotype-matched IgG was used for control staining. The stained cells were analyzed using a BD FACS Canto II flow cytometer (BD Bioscience). Data were collected from 20,000 to 30,000 individual cells using forward-scatter and side-scatter parameters to set a gate on the lymphocyte population. The number of particular lymphocyte subsets was estimated by multiplying the lymphocyte number, calculated as mentioned above, by the proportion of each subset.
Statistical analysis.
Data were analyzed using JMP Pro 10.0.2 software (SAS Institute Japan, Tokyo, Japan). Data are expressed as means ± standard deviations (SD). Differences between groups were examined for statistical significance using Welch's t test. A P value less than 0.05 was considered significant.
RESULTS
Increased susceptibility of CARD9 KO mice to cryptococcal infection.
To elucidate the effect of CARD9 deficiency on the clinical course of cryptococcal infection, CARD9 KO and WT mice were infected with C. neoformans, and the lung weights and numbers of live microorganisms in lungs were measured on day 14 after infection. As shown in Fig. 1, the lung burdens of C. neoformans were significantly higher in CARD9 KO mice than in WT mice, and the lung weights evaluated relative to body weights were significantly increased in CARD9 KO mice compared to WT mice. We then conducted a histological analysis of the lungs on day 14 after infection. Massive multiplication of yeast cells with poor granulomatous responses was observed in the alveolar spaces in CARD9 KO mice, whereas WT mice showed a marked reduction in the number of yeast cells that were not found in the alveolar spaces and mostly encapsulated in the granulomatous tissues (Fig. 2A). When lungs were observed at a higher magnification, macrophages and lymphocytes were found to have accumulated around the yeast cells in WT mice, whereas accumulation of neutrophils was markedly increased in CARD9 KO mice (Fig. 2B). Consistent with this finding, neutrophils were significantly increased in the lung leukocytes from CARD9 KO mice compared with those in WT mice (Fig. 2C). These results indicate that CARD9-mediated signaling is involved in the elimination of cryptococcal infection and induction of a protective inflammatory response.
FIG 1.
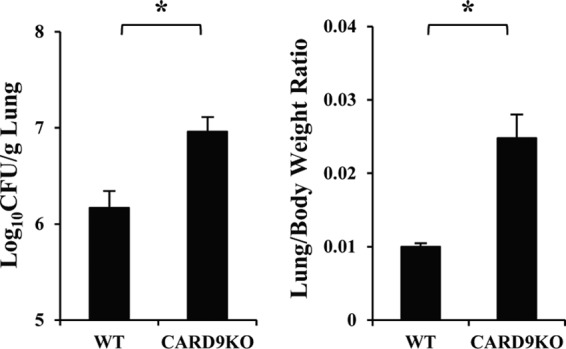
C. neoformans infection in CARD9 KO mice. WT and CARD9 KO mice were infected intratracheally with C. neoformans. The lung/body weight ratio was measured and the number of live colonies in lungs was counted on day 14 after infection. Each column represents the mean and SD for eight mice. Experiments were repeated twice with similar results. *, P < 0.05.
FIG 2.

Effect of CARD9 deficiency on the histological changes after infection with C. neoformans. WT and CARD9 KO mice were infected intratracheally with C. neoformans. Sections of lungs on day 14 postinfection were stained with H-E or PAS and observed under a light microscope at magnifications of ×200 (A) and ×1,000 (B). Representative pictures from three mice are shown. (C) The lung leukocytes prepared on day 14 postinfection were stained with Diff-Quick and observed under a light microscope. Representative pictures from three mice are shown. Magnification, ×200. The number of cells in each leukocyte fraction was counted. Each column represents the mean and SD for three mice. NS, not significant; *, P < 0.05.
Effect of CARD9 deficiency on IFN-γ production.
IFN-γ is a critical cytokine in the host defense against cryptococcal infection (6), and therefore, we investigated the effect of CARD9 deficiency on the production of this cytokine in the infected lungs. As shown in Fig. 3A, IFN-γ was quickly produced with a peak level on day 3 and then gradually decreased on days 7 and 14 in WT mice. In contrast, early production of this cytokine on day 3 was significantly reduced in CARD9 KO mice. In agreement with this finding, expression of inducible nitric oxide synthase (iNOS), a downstream effector molecule induced by IFN-γ, was almost abolished in CARD9 KO mice on day 3, whereas this expression was equivalent between these mouse strains on day 7 (Fig. 3B). These results indicate that CARD9 is a signaling molecule that is critical in the production of IFN-γ during the innate-immunity phase of infection.
FIG 3.
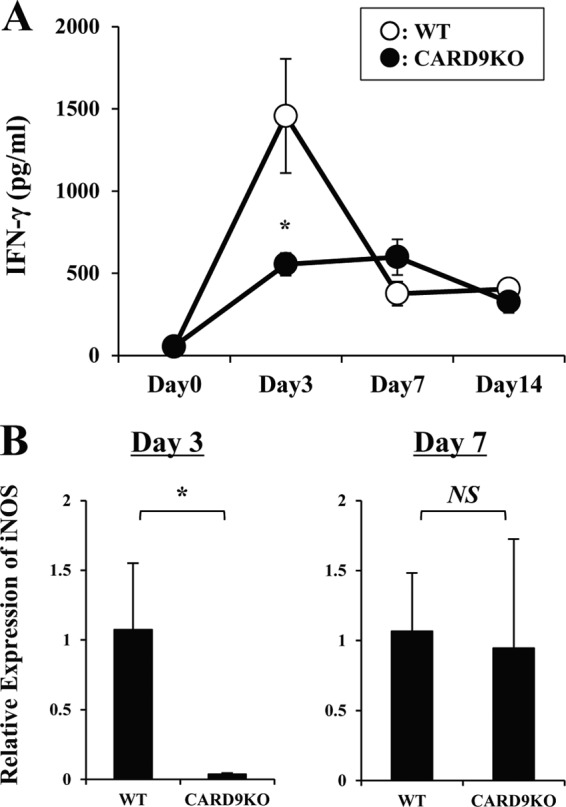
Production of IFN-γ and iNOS after infection with C. neoformans. WT and CARD9 KO mice were infected intratracheally with C. neoformans. (A) IFN-γ production in lungs was measured at various time points. Data are means and SD for five or six mice. Experiments were repeated twice with similar results. *, P < 0.05. (B) Expression of iNOS mRNA in lungs was measured on days 3 and 7. Data are means and SD for five mice. Experiments were repeated twice with similar results. NS, not significant; *, P < 0.05.
Role of innate-immunity-phase IFN-γ production on the host defense against cryptococcal infection.
To define the role of IFN-γ in the early host defense, we initially examined the clearance of C. neoformans in lungs on day 5 postinfection. As shown in Fig. 4A, the number of live colonies was significantly higher in CARD9 KO mice than in WT mice. Next, we tested the effect of neutralizing anti-IFN-γ MAb administered 2 h before and at the time of infection on the clearance of this fungal pathogen. Administration of anti-IFN-γ MAb led to significantly increased fungal burdens on day 14 postinfection compared with that of PBS or control rat IgG (Fig. 4B). These results suggested that the reduced IFN-γ production during the innate-immunity phase may be related to the impaired host defense against cryptococcal infection in CARD9 KO mice.
FIG 4.
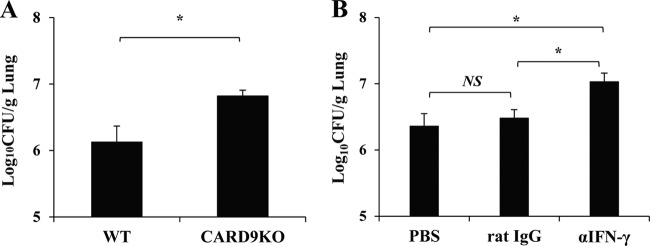
Role of early produced IFN-γ on the host defense against cryptococcal infection. (A) WT and CARD9 KO mice were infected intratracheally with C. neoformans, and the number of live colonies in lungs was counted on day 5 after infection. Data are means and SD for five mice. Experiments were repeated twice with similar results. *, P < 0.05. (B) WT mice were infected intratracheally with C. neoformans and injected intraperitoneally with anti-IFN-γ MAb or control rat IgG at 200 μg/mouse 2 h before infection and intratracheally with the same Ab at 25 μg/mouse when they were infected. The number of live colonies in lungs was counted on day 14 after infection. Data are means and SD for five mice. Experiments were repeated twice with similar results. *, P < 0.05; NS, not significant.
Early producers of IFN-γ after infection with C. neoformans.
To define the cellular source of IFN-γ production, we analyzed the intracellular expression of this cytokine in NK, NKT, γδ T, and T cells in the lungs on day 3 postinfection. As shown in Fig. 5, in WT mice, the major producers of this cytokine were NK cells and T cells, identified as NK1.1+ CD3− and NK1.1− CD3+ populations, respectively. The number of IFN-γ-expressing cells was strikingly reduced in both populations in CARD9 KO mice. Among T cells, the CD44bright+ subset (memory phenotype T cells), but not the CD44dull+ subset (naive T cells), expressed IFN-γ (Fig. 6A). IFN-γ-expressing CD8+ memory phenotype T cells were found at a higher level in lungs than CD4+ memory phenotype T cells expressing this cytokine, and the number of these cells was significantly reduced in CARD9 KO mice compared to WT mice (Fig. 6B). These results suggest that NK cells and memory phenotype T cells expressing IFN-γ accumulate in lungs after infection with C. neoformans, which is impaired in CARD9 KO mice.
FIG 5.

Early producers of IFN-γ after infection with C. neoformans. WT and CARD9 KO mice were infected intratracheally with C. neoformans, and the lung leukocytes were prepared on day 3 postinfection. (A) IFN-γ expression in NK, NKT, γδ T, and T cells was analyzed using flow cytometry. Gray areas show isotype-matched IgG; solid lines correspond to anti-IFN-γ MAb. Representative histograms from five WT and four CARD9 KO mice are shown. (B) The number of each subset expressing IFN-γ was calculated. Data are means and SD for five WT and four CARD9 KO mice. Experiments were repeated twice with similar results. NS, not significant; *, P < 0.05.
FIG 6.

IFN-γ production by memory phenotype T cells. WT and CARD9 KO mice were infected intratracheally with C. neoformans, and the lung leukocytes were prepared on day 3 postinfection. (A) Expression of IFN-γ in CD4+ or CD8+ naive and memory phenotype T cells, identified as CD44dull+ and CD44bright+ cells, was analyzed in a using flow cytometry. Representative histograms from three WT mice are shown. (B) The number of each subset expressing IFN-γ was calculated. Data are means and SD for three mice. Experiments were repeated twice with similar results. MPT cells, memory phenotype T cells. NS, not significant; *, P < 0.05.
Effect of CARD9 deficiency on the expression of chemokines.
Chemokines play a central role in the accumulation of leukocytes at the infected sites. Therefore, we next examined the lung expression of CCL3, -4, and -5 and of CXCL9 and -10, which are chemokines for CCR5 and CXCR3, respectively, and which attract NK cells and memory T cells (34–37). As shown in Fig. 7A, expression of these chemokines, except for CCL3, on day 3 postinfection was significantly reduced in CARD9 KO mice compared to WT mice. In further experiments, we examined the in vitro expression of these chemokines in WT and CARD9 KO mice using BM-DCs that were stimulated with C. neoformans. As shown in Fig. 7B, expression of CCL3 and CCL4 was strikingly reduced in CARD9 KO mice, although CCL5 was not detected even in WT mice. Conversely, CXCL9 and CXCL10 were not produced by BM-DCs that were stimulated with C. neoformans (data not shown).
FIG 7.

Effect of CARD9 deficiency on the chemokine production. (A) WT and CARD9 KO mice were infected intratracheally with C. neoformans. Expression of CCL3, CCL4, CCL5, CXCL9, and CXCL10 in lungs was measured on day 3. Each column represents the mean ± SD of five mice. (B) BM-DCs were cultured with C. neoformans (MOI, 1) or LPS (1 μg/ml) for 24 h, and expression of CCL3, CCL4, and CCL5 was measured. Data are means and SD from triplicate cultures. Experiments were repeated twice with similar results. NS, not significant; *, P < 0.05.
Effect of CARD9 deficiency on the Th1/Th17-related response.
Th1 cells play a critical role in the host defense against cryptococcal infection at an adaptive-immunity stage (5). In the next series of experiments, we investigated the effect of CARD9 deficiency on the Th1-related response. As shown in Fig. 8A, IL-12p35, a key cytokine for Th1 cell differentiation, was expressed on day 3 at an equivalent level in the infected lungs in the WT and CARD9 KO mice. In contrast, among cytokines critical for Th17 cell differentiation, transforming growth factor β1 (TGF-β1) showed a significantly reduced expression in CARD9 KO mice compared to WT mice, though IL-6 expression was not significantly different between these mouse strains. In addition, expression of IL-23p19, an important cytokine for expansion of Th17 cells, was significantly lower in CARD9 KO mice than that in WT mice. These results suggest that differentiation of Th1 cells is not affected whereas that of Th17 cells is attenuated in the absence of CARD9. In agreement with this possibility, there was no difference in T-bet and IFN-γ expression between the two mouse strains(Fig. 8A), and regional LN cells produced a comparable level of IFN-γ upon restimulation with C. neoformans in CARD9 KO mice compared to that in WT mice (Fig. 8B). In contrast, expression of ROR-γt and IL-17A in the lungs and production of IL-17A by the restimulated LN cells were significantly reduced in CARD9 KO mice (Fig. 8).
FIG 8.

Effect of CARD9 deficiency on the Th1/Th17-related response. WT and CARD9 KO mice were infected intratracheally with C. neoformans. (A) Expression of IL-12p35, IL-6, TGF-β, and IL-23p19 on day 3 and of T-bet, ROR-γt, IFN-γ, and IL-17A on day 7 was measured in the lung homogenates by real-time PCR. Data are means and SD for five mice. (B) LN cells obtained on day 7 postinfection were stimulated with indicated doses of C. neoformans or ConA (1 μg/ml) for 48 h, and production of IFN-γ and IL-17A was measured. Data are means and SD of triplicate cultures. Experiments were repeated twice with similar results. NS, not significant; *, P < 0.05.
Effect of IL-17A deficiency on the host defense against cryptococcal infection.
To define the role of IL-17A, a cytokine produced by Th17 cells, we examined the effect of IL-17A deficiency on the host defense against cryptococcal infection. As shown in Fig. 9, the C. neoformans lung burdens were not increased but rather decreased on day 14 postinfection in IL-17A KO mice compared to those in WT mice. Histological analysis of the infected lungs did not show a difference between the two mouse strains (data not shown). These results indicate that the reduced Th17 response does not directly contribute to the attenuated host defense against this infection in CARD9 KO mice.
FIG 9.
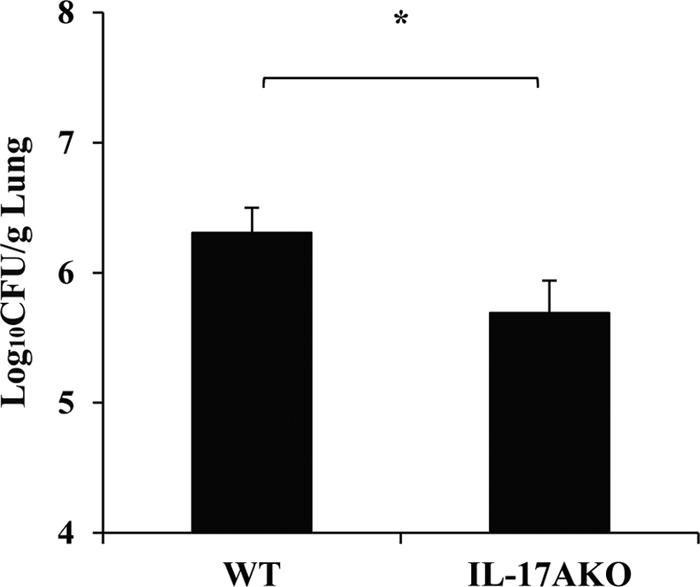
Effect of IL-17A deficiency on the clearance of C. neoformans in lungs. WT and IL-17A KO mice were infected intratracheally with C. neoformans. The number of live colonies in lungs was counted on day 14 after infection. Data are means and SD for six WT and seven IL-17A KO mice. Experiments were repeated three times with similar results. *, P < 0.05.
DISCUSSION
In the present study, CARD9 KO mice were more susceptible to pulmonary C. neoformans infection than were WT mice. In CARD9 KO mice, IFN-γ production and iNOS expression were strongly reduced at the innate-immunity phase of infection compared to those in WT mice. Reduced IFN-γ synthesis partially explained the impaired accumulation of NK cells and CD4+ or CD8+ memory phenotype T cells, the major sources of the innate-immunity-phase IFN-γ production in the infected lungs, which was correlated with reduced synthesis of CCL4, CCL5, CXCL9, and CXCL10, which are reported to recruit NK cells and memory T cells (34–38). In contrast, Th1 cell differentiation, a critical process in the host defense against cryptococcal infection (5), was almost equivalent between WT and CARD9 KO mice.
A defect in CARD9 or Dectin-1 and Dectin-2, upstream receptors of CARD9-mediated signaling (39), compromises host defenses against infection with Candida albicans, an extracellularly growing fungal pathogen (21, 23). In intracellular pathogens, Listeria monocytogenes and Mycobacterium tuberculosis were reported to cause severe infection in CARD9-deficient mice (29, 30). Macrophages derived from CARD9 KO mice had lower phagocytosis and killing activities against L. monocytogenes (40). Thus, CARD9-mediated signaling is involved in the host defense against both types of microbial pathogens. In the present study, CARD9 KO mice were susceptible to infection with C. neoformans, an intracellular fungal pathogen (41), as shown by the increased fungal burdens in the infected lungs. These mice also showed marked multiplication of the yeast cells in the alveolar spaces with poor granulomatous responses and increased accumulation of neutrophils, which are typical histopathological findings as this infection gets worse. Thus, CARD9-mediated signaling was found to play a critical role in rendering the hosts resistant to infection with C. neoformans, similar to what has been reported for other pathogenic microorganisms.
IFN-γ is a required cytokine for elimination of C. neoformans, with a mechanism that works by activating the NO-dependent killing activity of macrophages (6, 42). In our model, IFN-γ production was markedly reduced on day 3 and clearance of C. neoformans was attenuated on day 5 postinfection in CARD9 KO mice. In addition, neutralization of IFN-γ activity only at the early phase led to the impaired clearance of C. neoformans. These results suggest that reduction in innate-immunity-phase IFN-γ production may be a reason for the compromised host resistance to C. neoformans. Our results support this hypothesis, because iNOS expression was abrogated on day 3 but not on day 7 postinfection in these mice. The major sources of IFN-γ production at this time point were NK cells and CD4+ or CD8+ memory phenotype T cells, but not NKT cells or γδ T cells. Previous investigations reported that CD8+ memory phenotype T cells existed in naive mice and played an important role in the phase of innate-immunity resistance to L. monocytogenes infection by rapidly producing IFN-γ in response to cytokine stimulation, such as that by IL-12 and IL-18, in an antigen (Ag)-independent manner (43, 44). In contrast, in the present study, 20% to 40% of CD4+ and CD8+ memory phenotype T cells expressed IFN-γ in the peripheral blood even before infection, which was similar between WT and CARD9 KO mice (data not shown). After infection with C. neoformans, IFN-γ-expressing CD4+ and CD8+ memory phenotype T cells had accumulated in the lungs, probably from the circulatory system, with almost no change in the rate of positivity for IFN-γ expression. In CARD9 KO mice, accumulation of CD4+ and CD8+ memory phenotype T cells expressing IFN-γ in the lungs was impaired, compared with that in WT mice. Similarly, IFN-γ-expressing NK cells were markedly reduced in the lungs of CARD9 KO mice. Thus, these findings may account for the reduction in IFN-γ production during the innate-immunity phase in CARD9 KO mice.
Accumulation of inflammatory cells is critically regulated by interaction between chemokines and their particular receptors. NK cells and memory T cells are known to express CCR5 and CXCR3, receptors for CCL3, -4, and -5 and for CXCL9 and -10, respectively (45). In an earlier study by Kohlmeier and coworkers (34), CCR5 was found to be essential for the recruitment of memory CD8+ T cells in the lungs during viral infection. In addition, CXCR3 ligands play an important role in the recruitment of central memory T cells during antiviral recall response (35). Similarly, recruitment of NK cells is reported to require CCR5 and CXCR3 during viral infection (36, 37). Consistent with these ideas, the innate-immunity-phase production of CCL3, CCL4, CCL5, CXCL9, and CXCL10 in the lungs of CARD9 KO mice was reduced during infection with C. neoformans, although significant reduction was not observed in CCL3 expression. Also, in vitro experiments showed that in mice lacking CARD9, C. neoformans-stimulated synthesis of CCL3 and -4 by BM-DCs was decreased, although CCL5, CXCL9, and CXCL10 expression was not detected (data not shown for CXCL9 and -10). Because CXCL9 and -10 are known to be induced by IFN-γ (45), the observed reduction of IFN-γ synthesis in vivo may account for the downregulation of these chemokines.
Although the innate-immunity-phase IFN-γ production was markedly reduced, there was no reduction during the adaptive-immunity phase in CARD9 KO mice. Consistent with these data, expression of Th1-related molecules, including IL-12p35 on day 3 and T-bet, IFN-γ, and iNOS on day 7, was detected at equivalent levels in these mouse strains, although expression of Th17-related molecules such as IL-23p19, ROR-γt, and IL-17A was lower in CARD9 KO mice than in WT mice. In addition, Th1 response in the Ag-restimulated LN cells was detected at similar levels in WT and CARD9 KO mice, whereas the Th17 response was abrogated in CARD9 KO mice. The reduced Th17 response is consistent with previous investigations, indicating that the critical role of CARD9 and its upstream receptors, such as Dectin-1 and Dectin-2, in the host defense against pathogenic microorganisms, such as C. albicans, is mediated by a Th17 response (21, 23, 28). In contrast, in the present study, clearance of C. neoformans in the lungs was not impaired but rather promoted in IL-17A KO mice, which was consistent with previous studies showing that depletion of IL-17A had no effect on the resolution of pulmonary infection with C. neoformans (46, 47). These findings suggest that Th17 response may be dispensable for the local host defense against cryptococcal infection in lungs, although the possible involvement of IL-17F (48) and IL-22 (49), which are produced by Th17 cells, remains to be elucidated. In addition, CARD9 KO mice were less susceptible to infection with C. neoformans than IFN-γ KO mice that showed severe impairment in the lung clearance of this fungal pathogen (see Fig. S1 in the supplemental material). These results are not incompatible with the notion that the impaired host defense in CARD9 KO mice is due to the reduced production of IFN-γ but not of IL-17A. Thus, CARD9-mediated signaling is likely to have an impact on the host defense against cryptococcal infection at the innate-immunity stage rather than at the adaptive-immunity stage, similar to what has been reported for L. monocytogenes infection (30).
In conclusion, the present study demonstrates that the signaling mediated by CARD9 makes a critical contribution to the host defense against pulmonary infection with C. neoformans and suggests that this pathway may promote the innate-immunity-phase recruitment of IFN-γ-producing cells at the infection site rather than inducing the differentiation of Th1 cells. In contrast, MyD88, an adapter molecule operating downstream of TLRs, is reported to play a critical role in the differentiation of Th1 cells during this infection (50, 51), suggesting that CARD9 and MyD88 may play distinct roles in the different stages of infection. Because the precise role of the CARD9-mediated signaling pathway in the host defense against C. neoformans remains to be defined, the present study provides an important implication for understanding the pathogenic mechanism of this infection. Further investigations are necessary to identify the upstream receptors contributing to the recognition of this fungal microorganism.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a Grant-in-Aid for Scientific Research (B) (23390263) and a Grant-in-Aid for Challenging Exploratory Research (24659477) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by a grant (Research on Emerging and Re-emerging Infectious Diseases [H22-SHINKOU-IPPAN-008 and H25-SHINKOU-IPPAN-006]) from the Ministry of Health, Labor and Welfare of Japan.
We have no financial conflict of interest.
Footnotes
Published ahead of print 27 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01089-13.
REFERENCES
- 1.Cunha BA. 2001. Central nervous system infections in the compromised host: a diagnostic approach. Infect. Dis. Clin. North Am. 15:567–590. 10.1016/S0891-5520(05)70160-4 [DOI] [PubMed] [Google Scholar]
- 2.Jarvis JN, Harrison TS. 2007. HIV-associated cryptococcal meningitis. AIDS 21:2119–2129. 10.1097/QAD.0b013e3282a4a64d [DOI] [PubMed] [Google Scholar]
- 3.Lim TS, Murphy JW. 1980. Transfer of immunity to cryptococcosis by T-enriched splenic lymphocytes from Cryptococcus neoformans-sensitized mice. Infect. Immun. 30:5–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perfect JR, Casadevall A. 2002. Cryptococcosis. Infect. Dis. Clin. North Am. 16:837–874. 10.1016/S0891-5520(02)00036-3 [DOI] [PubMed] [Google Scholar]
- 5.Koguchi Y, Kawakami K. 2002. Cryptococcal infection and Th1-Th2 cytokine balance. Int. Rev. Immunol. 21:423–438. 10.1080/08830180213274 [DOI] [PubMed] [Google Scholar]
- 6.Yuan RR, Casadevall A, Oh J, Scharff MD. 1997. T cells cooperate with passive antibody to modify Cryptococcus neoformans infection in mice. Proc. Natl. Acad. Sci. U. S. A. 94:2483–2488. 10.1073/pnas.94.6.2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawakami K, Koguchi Y, Qureshi MH, Miyazato A, Yara S, Kinjo Y, Iwakura Y, Takeda K, Akira S, Kurimoto M, Saito A. 2000. IL-18 contributes to host resistance against infection with Cryptococcus neoformans in mice with defective IL-12 synthesis through induction of IFN-γ production by NK cells. J. Immunol. 165:941–947 [DOI] [PubMed] [Google Scholar]
- 8.Decken K, Köhler G, Palmer-Lehmann K, Wunderlin A, Mattner F, Magram J, Gately MK, Alber G. 1998. Interleukin-12 is essential for a protective Th1 response in mice infected with Cryptococcus neoformans. Infect. Immun. 66:4994–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rayhane N, Lortholary O, Fitting C, Callebert J, Huerre M, Dromer F, Cavaillon JM. 1999. Enhanced sensitivity of tumor necrosis factor/lymphotoxin-α-deficient mice to Cryptococcus neoformans infection despite increased levels of nitrite/nitrate, interferon-γ, and interleukin-12. J. Infect. Dis. 180:1637–1647. 10.1086/315061 [DOI] [PubMed] [Google Scholar]
- 10.Blackstock R, Buchanan KL, Adesina AM, Murphy JW. 1999. Differential regulation of immune responses by highly and weakly virulent Cryptococcus neoformans isolates. Infect. Immun. 67:3601–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blackstock R, Murphy JW. 2004. Role of interleukin-4 in resistance to Cryptococcus neoformans infection. Am. J. Respir. Cell Mol. Biol. 30:109–117. 10.1165/rcmb.2003-0156OC [DOI] [PubMed] [Google Scholar]
- 12.Müller U, Stenzel W, Köhler G, Werner C, Polte T, Hansen G, Schutzen N, Straubinger RK, Blessing M, Ackenzie AN, Brombacher F, Alber G. 2007. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J. Immunol. 179:5367–5377 http://www.jimmunol.org/content/179/8/5367.long [DOI] [PubMed] [Google Scholar]
- 13.Huang W, Na L, Fidel PL, Schwarzenberger P. 2004. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 190:624–631. 10.1086/422329 [DOI] [PubMed] [Google Scholar]
- 14.Conti HR, Gaffen SL. 2010. Host responses to Candida albicans: Th17 cells and mucosal candidiasis. Microbes Infect. 12:518–527. 10.1016/j.micinf.2010.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korn T, Bettelli E, Oukka M, Kuchroo VK. 2009. IL-17 and Th17 cells. Annu. Rev. Immunol. 27:485–517. 10.1146/annurev.immunol.021908.132710 [DOI] [PubMed] [Google Scholar]
- 16.Lipscomb MF, Alvarellos T, Toews GB, Tompkins R, Evans Z, Koo G, Kumar V. 1987. Role of natural killer cells in resistance to Cryptococcus neoformans infections in mice. Am. J. Pathol. 128:354–361 [PMC free article] [PubMed] [Google Scholar]
- 17.Kawakami K, Kinjo Y, Uezu K, Yara S, Miyagi K, Koguchi Y, Nakayama T, Taniguchi M, Saito A. 2001. Monocyte chemoattractant protein-1-dependent increase of Vα14 NKT cells in lungs and their roles in Th1 response and host defense in cryptococcal infection. J. Immunol. 167:6525–6532 http://www.jimmunol.org/content/167/11/6525.long [DOI] [PubMed] [Google Scholar]
- 18.Uezu K, Kawakami K, Miyagi K, Kinjo Y, Kinjo T, Ishikawa H, Saito A. 2004. Accumulation of γδ T cells in the lungs and their regulatory roles in Th1 response and host defense against pulmonary infection with Cryptococcus neoformans. J. Immunol. 172:7629–7634 http://www.jimmunol.org/content/172/12/7629.long [DOI] [PubMed] [Google Scholar]
- 19.Figdor G, van Kooyk Y, Adema GJ. 2002. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol. 2:77–84. 10.1038/nri723 [DOI] [PubMed] [Google Scholar]
- 20.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis e Sousa C. 2007. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 8:630–638. 10.1038/ni1460 [DOI] [PubMed] [Google Scholar]
- 21.Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 8:31–38. 10.1038/ni1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, Kinjo T, Nakamura K, Kawakami K, Iwakura Y. 2007. Dectin-1 is required for the host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 8:39–46. 10.1038/ni1425 [DOI] [PubMed] [Google Scholar]
- 23.Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH, Komatsu H, Miura N, Adachi Y, Ohno N, Shibuya K, Yamamoto N, Kawakami K, Yamasaki S, Saito T, Akira S, Iwakura Y. 2010. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for the host defense against Candida albicans. Immunity 32:681–691. 10.1016/j.immuni.2010.05.001 [DOI] [PubMed] [Google Scholar]
- 24.Mansour MK, Schlesinger LS, Levitz SM. 2002. Optimal T cell responses to Cryptococcus neoformans mannoprotein are dependent on recognition of conjugated carbohydrates by mannose receptors. J. Immunol. 168:2872–2879 http://www.jimmunol.org/content/168/6/2872.long [DOI] [PubMed] [Google Scholar]
- 25.Mansour MK, Latz E, Levitz SM. 2006. Cryptococcus neoformans glycoantigens are captured by multiple lectin receptors and presented by dendritic cells. J. Immunol. 176:3053–3061 http://www.jimmunol.org/content/176/5/3053.long [DOI] [PubMed] [Google Scholar]
- 26.Nakamura K, Kinjo T, Saijo S, Miyazato A, Adachi Y, Ohno N, Fujita J, Kaku M, Iwakura Y, Kawakami K. 2007. Dectin-1 is not required for the host defense to Cryptococcus neoformans. Microbiol. Immunol. 51:1115–1119. 10.1111/j.1348-0421.2007.tb04007.x [DOI] [PubMed] [Google Scholar]
- 27.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, Iwakura Y, Ohno N, Koseki H, Yoshida H, Penninger JM, Saito T. 2007. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 8:619–629. 10.1038/ni1466 [DOI] [PubMed] [Google Scholar]
- 28.Gross O, Gewies A, Finger F, Schafer M, Sparwasser T, Peschel C, Forster I, Ruland J. 2006. Card9 controls a non-TLR signaling pathway for innate anti-fungal immunity. Nature 442:651–656. 10.1038/nature04926 [DOI] [PubMed] [Google Scholar]
- 29.Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, Hanke K, Gross O, Ruland J, Kaufmann SH. 2010. The adaptor molecule CARD9 is essential for tuberculosis control. J. Exp. Med. 207:777–792. 10.1084/jem.20090067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, Lin X. 2007. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 8:198–205. 10.1038/ni1426 [DOI] [PubMed] [Google Scholar]
- 31.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. 2002. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17:375–387. 10.1016/S1074-7613(02)00391-6 [DOI] [PubMed] [Google Scholar]
- 32.Kawakami K, Kohno S, Morikawa N, Kadota J, Saito A, Hara K. 1994. Activation of macrophages and expansion of specific T lymphocyte in the lungs of mice intratracheally inoculated with Cryptococcus neoformans. Clin. Exp. Immunol. 96:230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yajima T, Nishimura H, Ishimitsu R, Watase T, Busch DH, Pamer ED, Kuwano H, Yoshikai Y. 2002. Overexpression of IL-15 in vivo increases antigen-driven memory CD8+ T cells following a microbe exposure. J. Immunol. 168:1198–1203 [DOI] [PubMed] [Google Scholar]
- 34.Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, Roberts AD, Woodland DL. 2008. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity 29:101–113. 10.1016/j.immuni.2008.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sung JH, Zhang H, Moseman EA, Alvarez D, Iannacone M, Henrickson SE, de la Torre JC, Goom JR, Luster AD, von Andrian UH. 2012. Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 150:1249–1263. 10.1016/j.cell.2012.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grégoire C, Cognet C, Chasson L, Coupet CA, Dalod M, Reboldi A, Marvel J, Sallusto F, Vivier E, Walzer T. 2008. Intrasplenic trafficking of natural killer cells is redirected by chemokines upon inflammation. Eur. J. Immunol. 38:2076–2084. 10.1002/eji.200838550 [DOI] [PubMed] [Google Scholar]
- 37.Pak-Wittel MA, Yang L, Sojka DK, Rivenbark JG, Yokoyama WM. 2013. Interferon-γ mediates chemokine-dependent recruitment of natural killer cells during viral infection. Proc. Natl. Acad. Sci. U. S. A. 110:E50–E59. 10.1073/pnas.1220456110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson MJ. 2002. Role of chemokines in the biology of natural killer cells. J. Leukoc. Biol. 71:173–183 http://www.jleukbio.org/content/71/2/173.long [PubMed] [Google Scholar]
- 39.Drummond RA, Saijo S, Iwakura Y, Brown GD. 2011. The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur. J. Immunol. 41:276–281. 10.1002/eji.201041252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu W, Hsu YM, Bi L, Songyang Z, Lin X. 2009. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nat. Immunol. 10:1208–1214. 10.1038/ni.1788 [DOI] [PubMed] [Google Scholar]
- 41.Feldmesser M, Tucker S, Casadevall A. 2001. Intracellular parasitism of macrophages by Cryptococcus neoformans. Trends Microbiol. 9:273–278. 10.1016/S0966-842X(01)02035-2 [DOI] [PubMed] [Google Scholar]
- 42.Tohyama M, Kawakami K, Futenma M, Saito A. 1996. Enhancing effect of oxygen radical scavengers on murine macrophage anticryptococcal activity through production of nitric oxide. Clin. Exp. Immunol. 103:436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yajima T, Nishimura H, Ishimitsu R, Yamamura K, Watase T, Busch DH, Pamer EG, Kuwano H, Yoshikai Y. 2001. Memory phenotype CD8+ T cells in IL-15 transgenic mice are involved in early protection against a primary infection with Listeria monocytogenes. Eur. J. Immunol. 31:757–766. 10.1002/1521-4141(200103)31:3<757::AID-IMMU757>3.0.CO;2-Q [DOI] [PubMed] [Google Scholar]
- 44.Yajima T, Nishimura H, Sad S, Shen H, Kuwano H, Yoshikai Y. 2005. A novel role of IL-15 in early activation of memory CD8+ CTL after reinfection. J. Immunol. 174:3590–3597 http://www.jimmunol.org/content/174/6/3590.long [DOI] [PubMed] [Google Scholar]
- 45.Rossi D, Zlotnik A. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18:217–242. 10.1146/annurev.immunol.18.1.217 [DOI] [PubMed] [Google Scholar]
- 46.Hardison SE, Wozniak KL, Kolls JK, Wormley FL., Jr 2010. Interleukin-17 is not required for classical macrophage activation in a pulmonary mouse model of Cryptococcus neoformans infection. Infect. Immun. 78:5341–5351. 10.1128/IAI.00845-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wozniak KL, Hardison SE, Kolls JK, Wormley FL. 2011. Role of IL-17A on resolution of pulmonary C. neoformans infection. PLoS One 6:e17204. 10.1371/journal.pone.0017204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu C, Wu L, Li X. 2013. IL-17 family: cytokines, receptors and signaling. Cytokines 64:477–485. 10.1016/j.cyto.2013.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabat R, Ouyang W, Wolk K. 2013. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat. Rev. Drug Discov. 13:21–38. 10.1038/nrd4176 [DOI] [PubMed] [Google Scholar]
- 50.Yauch LE, Mansour MK, Shoham S, Rottman JB, Levitz SM. 2004. Involvement of CD14, toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect. Immun. 72:5373–5382. 10.1128/IAI.72.9.5373-5382.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Wang F, Bhan U, Huffnagle GB, Toews GB, Standiford TJ, Olszewski MA. 2010. TLR9 signaling is required for generation of the adaptive immune protection in Cryptococcus neoformans-infected lungs. Am. J. Pathol. 177:754–765. 10.2353/ajpath.2010.091104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.