

Abstract
The upper respiratory tract (URT) is a distinct microbial niche of low-density bacterial communities and, also, a portal of entry for many potential pathogens, including Streptococcus pneumoniae. Thus far, animal models have been used to study the dynamics of and interactions between limited numbers of different species in the URT. Here, we applied a deep sequencing approach to explore, for the first time, the impact of S. pneumoniae acquisition on URT microbiota in a mouse model, as well as potential age-dependent effects. Young-adult and elderly mice were inoculated intranasally with S. pneumoniae, and nasal lavage samples were collected for up to 28 days postcolonization. Bacterial DNA extracted from lavage samples was subjected to barcoded pyrosequencing of the V5-to-V7 hypervariable region of the small-subunit rRNA gene. We observed highly diverse microbial profiles, with the presence overall of 15 phyla and approximately 645 operational taxonomic units (OTUs). We noted differences in the composition of microbiota between young and elderly mice, with a significantly higher abundance of Bacteroidetes in the young mice. The introduction of S. pneumoniae into the URT led to a temporary dominance of pneumococci in the microbiota of all mice, accompanied by a significant decrease in microbial diversity. As mice gradually cleared the colonization, the diversity returned to baseline levels. Diversification was accompanied by an early expansion of Bacteroidetes, Staphylococcus spp., and Lachnospiraceae. Moreover, the Bacteroidetes expansion was significantly greater in young-adult than in elderly mice. In conclusion, we observed differences in URT microbiota composition between naive young-adult and elderly mice that were associated with differences in pneumococcal clearance over time.
INTRODUCTION
The bacteria indigenous to the mammalian body are a diverse and important component of a healthy existence, even outnumbering the cells of the host. New techniques in sequencing have allowed a greater understanding of the breadth and diversity of these microbial populations, while experimental animal models have elucidated mechanisms of commensal-host-pathogen interactions, including beneficial immune stimulation and species-specific pathogen protection (1, 2). The upper respiratory tract (URT) is a distinct microbial niche of low-density bacterial communities (3) and, also, a portal of entry for many pathogens, including Streptococcus pneumoniae, which is a frequent cause of pneumonia worldwide and a major cause of death, particularly in the elderly population (4). Furthermore, pneumococcal colonization of the URT is a prerequisite state before disease develops (5, 6). Due to the key role that colonization plays in both the survival of the bacteria in the upper respiratory tract of the human host and the development of disease, studies on the relationship between the bacterial community composition of the URT and pneumococcal acquisition and clearance are of great importance (3).
Despite being a frequent asymptomatic component of the URT microbiota, pneumococci can cause serious diseases, such as pneumonia, septicemia, and meningitis (5). Young children and the elderly are especially at risk for pneumococcal disease, and while immature immunity causes the increased risk in children, age-related defects in the immune system, termed immunosenescence, are thought to contribute to the increased disease risk in the elderly (7). In a previous study, we determined that baseline inflammation and delayed cellular responses were correlated with a delay in pneumococcal colonization clearance in elderly mice compared to the time for clearance in young-adult mice (8). We observed that a higher proinflammatory status in the nasal-associated lymphoid tissue (NALT) of elderly mice occurred simultaneously with the increased expression of tolerance pathways in the nasal epithelium, suggesting that homeostasis in the URT of elderly mice is altered. We hypothesized that the URT microbiota of elderly mice might be different from that in younger mice and that the difference might contribute to this phenomenon.
Thus far, animal models have been used to study interactions in the URT for a limited number of prokaryotic species at a time. Here, we applied a deep sequencing approach to explore the correlation between URT microbiota and the dynamics of S. pneumoniae acquisition in a mouse model of pneumococcal colonization. We used the model to investigate age-dependent differences in microbiota responses to pneumococcal carriage in the context of immunosenescence.
MATERIALS AND METHODS
Ethics statement.
Animal experiments were performed in accordance with the Dutch Animal Experimentation Act and EU directives 86/609/CEE and 2010/63/EU related to the protection of vertebrate animals used for experimental or other scientific purposes. The experimental protocols were approved by the Committee on Animal Experiments of the University of Utrecht (DEC 2009.II.10.107, 2010.II.07.125, and 2011.II.11.175). C57BL/6 female specific-pathogen-free mice were purchased from Harlan (Venray, Netherlands). In line with previous studies (9, 10), we studied 3- to 4-month-old mice as representative for young-adult and 18- to 23-month-old mice as representative for elderly mice. Animals with noticeable tumors were excluded from the experiments, and animals with neoplasms detected in postmortem examinations were excluded from analysis. Mice were euthanized with 120 mg/kg of body weight pentobarbital (Veterinary Department Pharmacy) administered intraperitoneally to induce respiratory arrest.
Bacterial strains.
Streptococcus pneumoniae serotype 6B strain 603 is a clinical isolate that has been used previously in murine models of pneumococcal colonization (11). This strain was mouse passaged and grown to mid-log phase in brain heart infusion broth (BHI), aliquoted, and stored frozen in 10% glycerol at −80°C. Prior to animal inoculation, bacterial cells were thawed, washed twice with saline, and resuspended to 5 × 108 CFU/ml, and the titers of cell suspensions were determined by plating 10-fold dilutions of inoculum on blood agar plates supplemented with gentamicin (SB7-Gent; Oxoid).
Mouse colonization experiments.
All samples analyzed for this work were randomly selected from samples collected in a study on the effects of immunosenescence on colonization with S. pneumoniae in the mouse model of pneumococcal carriage (8). Briefly, mice were inoculated intranasally with 10 μl of bacterial suspension containing approximately 5 × 106 CFU. At day 1 pre- and days 3, 7, and 14 postinoculation, 5 mice of each age were sacrificed, and at day 28 postinoculation, 3 mice of each age were sacrificed. From each animal, 500 μl of retrotracheal nasal lavage fluid were collected with calcium- and magnesium-free phosphate-buffered saline supplemented with 1% bovine serum albumin (PBS+BSA), followed by nasal tissue harvest. Tissue samples were crushed in 1 ml of PBS, stored on ice, and processed within 3 h. Samples were vortexed vigorously, and 60 μl of lavage fluid and all volumes of tissue wash fluid were plated in serial dilutions onto SB7-Gent medium in order to quantify the S. pneumoniae bacteria in the URT. The CFU counts from the lavage and nasal tissue samples were summed for each mouse. The remaining volume of the lavage fluid was centrifuged at 1,200 rpm (∼300 × g) for 10 min at 4°C to remove mouse cells. The supernatant (bacterial fraction) was removed and frozen at −80°C until DNA extraction.
Bacterial DNA isolation.
DNA was isolated from 100 μl of the URT lavage fluid with phenol plus bead beating and magnetic bead separation using Agowa reagents as described by Biesbroek et al. (12). The samples chosen to go through to 454 pyrosequencing were randomly selected and representative of the overall population of mice used for all experiments (8).
Deep sequencing analysis of URT samples.
The composition of microbiota was determined by using 454 barcoded pyrosequencing of the 16S rRNA genes as previously described (12). The total bacterial load was analyzed by quantitative real-time PCR (qPCR), using a universal primer-probe set targeting this gene (3). Amplicon library preparation and sequence processing were performed as described by Biesbroek et al. (12). In short, we amplified the V5-to-V7 hypervariable region of the 16S rRNA genes. Amplicons were size checked, quantified, and pooled in equimolar amounts, after which the library was unidirectionally sequenced in the 454 GS-FLX-Titanium sequencer (Life Sciences [Roche]; Branford, CT). Sequences were processed using the modules implemented in the Mothur version 1.20.0 software platform (13); they were first denoised and checked for quality and chimers (using Chimera Slayer) (14), resulting in 80,739 sequences being available for downstream analysis. The remaining high-quality aligned sequences were classified using the RDP-II naive Bayesian Classifier and clustered into operational taxonomic units (OTUs; defined by 97% similarity). For all samples, rarefaction curves were plotted and community diversity indices (Shannon diversity and Simpson's index) calculated. Sequence data were subjected to unweighted UniFrac analysis using the UniFrac module implemented in Mothur (15). The UniFrac algorithm calculates the distance between microbial communities based on the phylogenetic lineages in each sample.
Prior to analysis, OTU tables were filtered: any OTU accounting for 3 or fewer sequences in the entire data set was removed. Relative abundance was calculated as the proportion of sequences assigned to a specific OTU divided by the overall number of sequences obtained per sample (3), and absolute abundance was calculated by multiplying relative abundance by the bacterial load obtained per sample, measured by qPCR.
Data analysis.
Culture data, qPCR, and diversity indices were graphed and analyzed using GraphPad Prism software. Kruskal-Wallis with Dunn's posttest was used for nonparametric data, and otherwise, one-way analysis of variance (ANOVA) with Bonferroni posttests were used to detect statistical differences. The relative abundances of sequence data were graphed using Adobe Illustrator CS6. Significance analysis of microarrays (SAM) (16) on sequence data was performed with TIGR MeV software (www.tm4.org) (17) using Pearson correlation with complete linkage clustering and a false discovery rate (FDR) of 19% to study differences between groups on the OTU and family levels.
RESULTS
Upper respiratory tract bacterial density is correlated with the introduction of a pathogen.
In a previously published study, we showed that elderly mice have a prolonged duration of pneumococcal colonization compared with the time of colonization in young-adult mice and that this was associated with aberrant innate immune responses. For the microbiota analyses presented here, a subset of mice was selected randomly from the previous study, and we confirmed that the pneumococcal density in this subset did not differ significantly from the mean density dynamics presented in the original study. We isolated the total bacterial DNA from upper respiratory tract lavage fluids of young-adult and elderly mice and quantified the DNA with 16S qPCR. As seen from the data in Fig. 1, the introduction of S. pneumoniae into the URT niche does have a significant effect on the total bacterial density at 3 days postinoculation, as shown by a 3-fold increase in bacterial DNA recovered from young-adult mice and a 4-fold increase in bacterial DNA recovered from elderly mice (Kruskal-Wallis with Dunn's posttest, P < 0.001).
FIG 1.

Quantities of DNA in nasal lavage samples following pneumococcal challenge. DNA was isolated from nasal washes from the mice (mean of 5 mice per group per time point, except for day 28, when there were 3 mice per group). No differences were found between the quantities of DNA isolated from young-adult and elderly mice. Introduction of S. pneumoniae into the niche increased the quantities of DNA isolated at day 3 postinoculation (P < 0.001, Kruskall-Wallis with Dunn's posttest). Shown are means and standard deviations.
Sequence characteristics.
A total of 80,739 sequences were obtained from all mice (n = 46); on average, this corresponded to 1,793 sequences per sample with a standard deviation of 946 sequences, which corresponded to 1,259 unique OTUs. After filtering out OTUs that occurred ≤3 times in the complete data set, a total of 79,757 sequences corresponding to 645 unique OTUs remained. The overwhelming majority of the sequences came from 15 bacterial phyla. Only a very small proportion of the sequences (n = 189, 0.2%) were unclassifiable at the phylum level. The sequence coverage calculated from the rarefaction curves for all samples was high, with a mean of 0.9779, median of 0.9845, and range of 0.9081 to 0.9982.
Bacterial diversity.
Before the introduction of S. pneumoniae into the URT, we observed high levels of bacterial diversity in both age groups, with mean Simpson index values of 0.059 for young and 0.051 for elderly mice (Fig. 2). Although young-adult mice had slightly higher Shannon indices (mean of 3.83) than elderly mice (mean of 3.52), these differences were not significant. After the introduction of S. pneumoniae into the URT niche, diversity was temporarily diminished, with higher interindividual variety in index scores than before inoculation. The decrease in diversity at day 3 postinoculation was significant when compared to the diversity in naive mice (P < 0.001 for both Simpson index and Shannon index, ANOVA with Bonferroni posttest). Starting from 7 days postinoculation, the diversity gradually returned to baseline levels in both young-adult and elderly mice, as indicated in their Simpson index and Shannon index scores (Fig. 2). Although young-adult mice generally clear pneumococcal colonization significantly more rapidly than elderly mice (8), there was no significant difference in diversity dynamics in young-adult and elderly mice over time.
FIG 2.
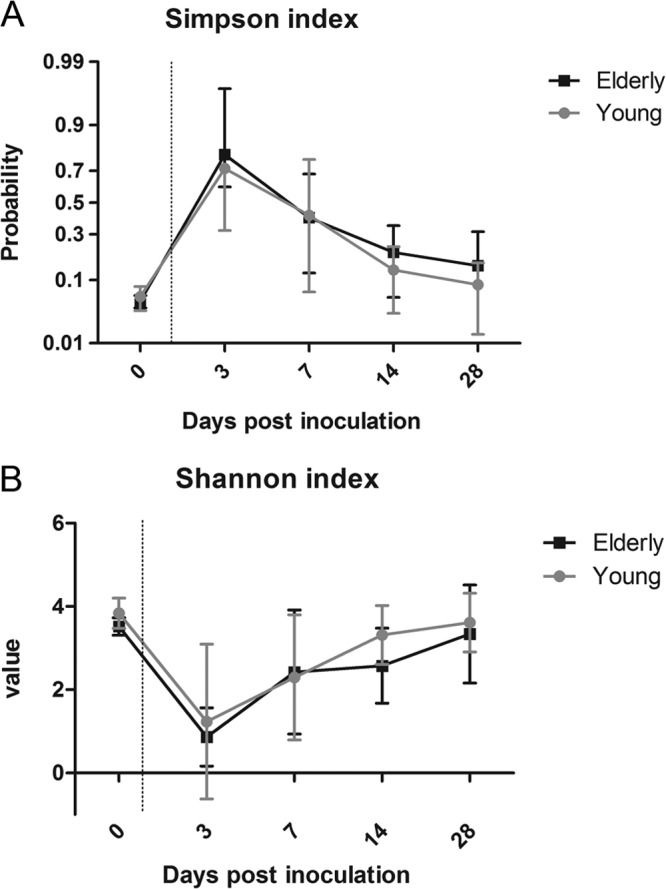
Alpha diversity of bacterial communities in respiratory lavage samples following pneumococcal challenge. Both the Simpson (A) and Shannon (B) indices show a large amount of diversity before the introduction of S. pneumoniae into the URT niche and a subsequent loss of diversity during pneumococcal colonization. The mean value of each index and the standard deviation are shown. As pneumococcal colonization is cleared, diversity returns to baseline. Five mice per group per time point were sequenced, except for day 28 when 3 mice per group were sequenced. Introduction of S. pneumoniae significantly reduced diversity (P < 0.0001 at day 3 for the Simpson index and P < 0.001 at day 3 for the Shannon index, ANOVA with Bonferroni posttest).
Respiratory microbiota composition in young and elderly naive mice.
Sequences resulting from 454 pyrosequencing of URT samples were classified in Mothur according to 97% similarity and analyzed taxonomically at the phylum, family, and OTU levels. At the phylum level, the microbiota of both young-adult and elderly mice contained approximately 50% Gram-positive and 50% Gram-negative bacteria, with Firmicutes, Bacteroidetes, and Proteobacteria being the most predominant phyla in the URT of both age groups (Fig. 3). However, in young-adult mice, there were consistently more Bacteroidetes observed than were found in elderly mice. Furthermore, the young-adult and elderly naive mice cluster in different quadrants when plotted using principle coordinate analysis (PCoA) of unweighted UniFac distances (Fig. 4), suggesting that the phylogenetic composition at baseline is different.
FIG 3.

Microbiota profiles of the URT after introduction of S. pneumoniae into the niche, showing mean relative abundances of bacteria observed in the upper respiratory tract of 5 mice per group (days 0, 3, 7, and 14) or 3 mice per group (day 28), represented at the family level (only the top 30 most abundant families are shown). Data for young mice are represented in the top 5 bars, and data for elderly mice in the bottom bars. Gram-positive phyla are represented by warm colors (orange bracket, Firmicutes, and yellow bracket, Actinobacteria), and Gram-negative phyla by cool colors (teal bracket, Bacteroidetes, and dark-blue bracket, Proteobacteria). The OTU correlating with S. pneumoniae observed by culture is indicated by the pink boxes outlined in black denoted F. Spn. Bacteroidetes OTUs, especially Porphyromonas, were significantly higher in young-adult than in elderly mice at baseline. Clearance of S. pneumoniae was accompanied by expansion of Bacteroidetes, Staphylococcaceae, and Lachnospiraceae in both groups. Bacteria that were significantly different between young and elderly mice are indicated at the family level (*F) and OTU level (*O). Initials of family names refer back to phyla, as follows: P, Proteobacteria; F, Firmicutes; A, Actinobacteria; B, Bacteroidetes; V, Verrucomicrobia.
FIG 4.

Principle coordinate analysis of the unweighted UniFrac distances between sample profiles. Shown are coordinates 1 and 2 for the microbiota of all mice. Profiles of young mice are depicted in red, and profiles of elderly mice are depicted in blue. Profiles at days 0, 3, and 7 (5 mice each) are plotted separately, whereas profiles from days 14 and 28 are merged (8 mice total). Young and elderly naive mice (day 0) cluster separately, whereas mice colonized with pneumococci cluster away from both groups of naive mice. Over time, the profiles gradually return to precolonization levels, i.e., those of naive mice.
At the family level, the profiles of young-adult mice showed a significantly higher relative abundance of symbionts, such as the Bacteroidetes family Porphyromonadaceae and the Firmicutes family Lachnospiraceae. In contrast, elderly mice had a significantly higher abundance of Staphylococcaceae. At the OTU level, three Barnesiella OTUs, three OTUs belonging to the Porphyromonadaceae, and a single Lachnospiraceae OTU were significantly more abundant in young-adult than in elderly mice before nasal inoculation with S. pneumoniae.
Niche composition during S. pneumoniae colonization.
At the phylum level and the family level (Fig. 3), the most abundant bacteria in the URT niche during pneumococcal colonization were Firmicutes and Streptococcaceae, respectively; specifically, the streptococcal OTU that matched our pneumococcal culture data was most abundant (the median relative abundance at day 3 postchallenge was 0.97 in young-adult mice and 0.90 in elderly mice). This was also apparent in the PCoA, where colonized mice grouped at a distance from their naive counterparts (Fig. 4). Although greatly reduced, all other major phyla (Bacteroidetes, Proteobacteria, and Actinobacteria) were still present in the niche in both age groups during the temporary dominance of pneumococci. Although both the culture and the sequence data indicate that the young-adult mice cleared pneumococci faster than elderly mice, the microbiota composition and diversity shift between days 7 and 14 were similar in both age groups, and the distance between colonized mice and naive counterparts became less at these time points. However, the microbiota composition did not return to the profiles present before the introduction of pneumococci in the URT niche within the 28 days of observation. For example, Staphylococcaceae were the first bacteria to reestablish in both age groups, with a tendency to grow out to greater abundance than before the pneumococcal challenge (Fig. 5). Two commensal bacterial families that maintained a relatively high presence throughout pneumococcal colonization were the Porphyromonadaceae and Lachnospiraceae (Fig. 3). Furthermore, Porphyromonadaceae (Barnesiella in particular) were the first commensals to reestablish their abundance to preinoculation levels, even gaining slightly higher abundance levels at day 28 in young-adult inoculated mice than in naive mice (Fig. 5).
FIG 5.
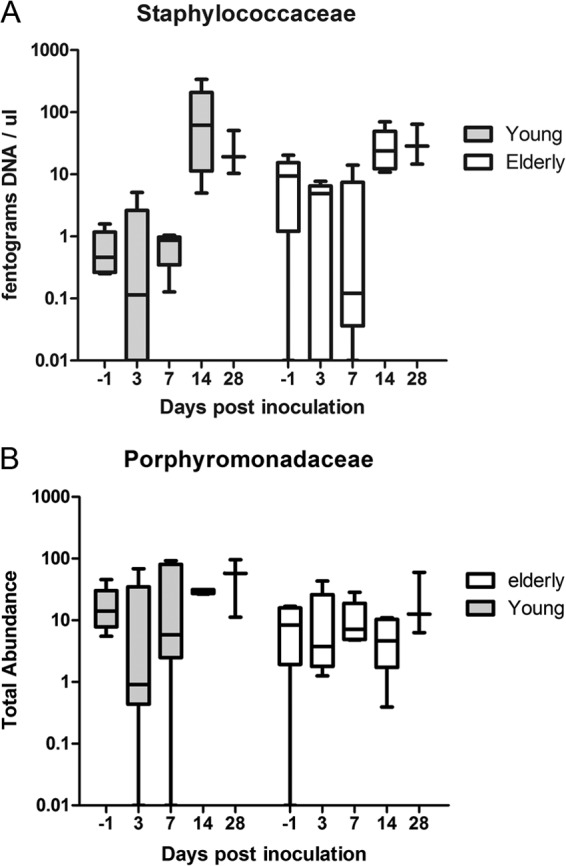
Absolute abundances of Staphylococcaceae (A) and Porphyromonadaceae (B) over time in young-adult and elderly mice pre- and postcolonization with S. pneumoniae. Shown are the 5th to 95th percentiles (whiskers) and the 25th to 75th percentiles (boxes), with the medians marked by horizontal bars. Where Porphyromonadaceae colonization stayed fairly stable over time, Staphylococcaceae tended to grow out simultaneously with pneumococcal clearance, especially in young mice.
DISCUSSION
To the best of our knowledge, this is the first study to (i) describe the URT microbiota composition in mice, (ii) show the community dynamics in response to the acquisition of a potential pathogen, i.e., S. pneumoniae, and (iii) study differences between young-adult and elderly mice. We found differences between young-adult and elderly mice in the microbiota composition already present in naive mice, with consistently more Bacteroidetes in young than in elderly mice. The introduction of S. pneumoniae into the niche led to a temporary dominance of pneumococci and a loss of diversity. During the dominance of pneumococci in the niche, all four major phyla observed before were still detected in the mice, albeit at very low abundance. The mice gradually cleared the pneumococci, and during this process, the microbial diversity gradually returned to baseline, with Porphyromonadaceae and Lachnospiraceae among the first to reestablish. However, return to baseline was not complete within 28 days. We observed that certain families of bacteria that were in low abundance prior to the introduction of the pneumococcus, for example, staphylococci, were able to grow out in parallel to the clearance of pneumococci.
Before the introduction of the pathobiont S. pneumoniae into the URT niche, the diversity in the mouse URT was high, with Simpson index scores comparable to the nostril and oropharyngeal scores in humans (18) and the mouse gut (19), indicating that although the microbiota of the URT are less dense than those of the gut (3, 12), a large variety of species are present. The Shannon index scores for the URT of the mice in this study, however, are about half of those found in the mouse gut (19), which is probably caused by the general predominance of certain bacterial species in the URT, a phenomenon which is also observed in humans (3, 12). In the URT of naive mice, young-adult mice showed a high abundance of normal commensals, such as Streptococcaceae, Porphyromonadaceae, and Lachnospiraceae, which are generally considered beneficial for the host (1, 20–22). Interestingly, elderly mice had consistently lower abundances of anaerobic bacteria like the Porphyromonadaceae, and specifically, of Barnesiella bacteria. This bacterium has previously been found to contribute to clearance of intestinal pathogens, such as antibiotic-resistant enterococci, in mice (22) and was therefore suggested to contribute to microbiota homeostasis.
In line with culture data from previous mouse colonization studies (23), the relative and absolute abundance of pneumococci peaked at day 3 in both age groups. This correlated with an increase in total bacterial DNA, suggesting a temporary overgrowth of the pathobiont. Furthermore, a significant reduction in diversity, both in richness and evenness, as measured by alpha diversity indices accompanied the dominance of pneumococci in the URT. This is the first study to show that pneumococci rapidly overtake the niche in both young adult and elderly mice.
In the period between pneumococcal dominance and full clearance, the mice appeared to gradually clear pneumococci and return to baseline levels of diversity. During this period of time, the commensal anaerobes of the Porphyromonadaceae and Lachnospiraceae were among the first to reestablish in the niche. Both classes of bacteria, Bacteroidales and Clostridia, have been identified as key modulators of homeostasis in the gut, including immune maturation and modulation (20), and they may therefore be functioning similarly in the URT as well. Since Bacteroidetes especially are present in lower abundance in the URT of elderly mice, we hypothesize that this might relate to the increased baseline inflammation and decreased pneumococcal clearance that we observed previously in the URT of elderly mice (8), and therefore, it deserves further study.
The clearance of pneumococci in the URT of mice over time seems a less homogenous process, with high variability in the amount of time needed for clearance of this potential pathogen. Since the majority of young-adult mice cleared pneumococci by 21 days in our previous studies, we hypothesized that the microbiota should have returned back to baseline by day 28. While the diversity indices did return to the same level as in naive young-adult mice, we were surprised to find that the composition of the microbiota had not completely returned to baseline. Even more surprising was that in both young-adult and elderly mice, bacteria that were low in abundance in naive mice were enriched in challenged mice that were mostly clear of their pneumococci. In our study, the bacteria that grew out to the highest abundance were staphylococci; interestingly, there is epidemiological evidence for a competitive relationship between Streptococcus (S. pneumoniae) and Staphylococcus (S. aureus) in humans as well, suggesting that complex bacterial-bacterial and host-bacterial interactions are involved in microbiota dynamics (24–26). This was observed especially in children, where a temporary increase in S. aureus colonization and disease was observed in individuals vaccinated with a pneumococcal conjugate vaccine versus nonimmunized children, both in a randomized controlled trial setting and in surveillance studies in the population (26, 27).
It is difficult to determine whether the bacteria that reestablish the niche are taking advantage of the immune system clearing out pneumococci (indirect competition) or whether they are directly competing with the pneumococci and thereby aiding in pneumococcal clearance. Further studies should be undertaken to investigate the mechanisms involved in these potential interactions in more detail. A limitation of this study is that colonization dynamics were studied up to 28 days postinoculation, and we did not observe a return of the microbiota back to baseline. However, since the alpha indices had returned to baseline around day 28, we speculate that the microbial community might have developed a new equilibrium with a slightly different composition and balance from the baseline situation. Future studies should consider following up for at least another 1 to 2 weeks to answer the question of whether microbiota will fully return to the prechallenge baseline.
In conclusion, this study was an in-depth, culture-independent survey of microbiota in the URT of young-adult and elderly mice. We show significant changes in microbiota composition in the elderly mice that are correlated with altered mucosal homeostasis as previously observed (8). The decreased abundance of Bacteroidetes observed in the URT of elderly mice is in agreement with human studies of elderly gut dysbiosis, where lower numbers of bacteroidetes were also observed in the elderly (28). Furthermore, we show that acquisition of the pathobiont S. pneumoniae into the URT niche causes temporary pathogen overgrowth, a temporary loss of diversity, and dysbiosis that lasts beyond the point of pathobiont clearance from the URT. The dynamic changes in microbiota composition reported here can serve as the foundation for future research into host-pathogen-commensal interactions in the polymicrobial mucosal surfaces of the upper respiratory tract.
Footnotes
Published ahead of print 10 February 2014
REFERENCES
- 1.Sansonetti PJ. 2011. To be or not to be a pathogen: that is the mucosally relevant question. Mucosal Immunol. 4:8–14. 10.1038/mi.2010.77 [DOI] [PubMed] [Google Scholar]
- 2.Artis D. 2008. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 8:411–420. 10.1038/nri2316 [DOI] [PubMed] [Google Scholar]
- 3.Bogaert D, Keijser B, Huse S, Rossen J, Veenhoven R, van Gils E, Bruin J, Montijn R, Bonten M, Sanders E. 2011. Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS One 6:e17035. 10.1371/journal.pone.0017035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janssens JP, Krause KH. 2004. Pneumonia in the very old. Lancet Infect. Dis. 4:112–124. 10.1016/S1473-3099(04)00931-4 [DOI] [PubMed] [Google Scholar]
- 5.Bogaert D, De Groot R, Hermans PW. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect. Dis. 4:144–154. 10.1016/S1473-3099(04)00938-7 [DOI] [PubMed] [Google Scholar]
- 6.Simell B, Auranen K, Kayhty H, Goldblatt D, Dagan R, O'Brien KL. 2012. The fundamental link between pneumococcal carriage and disease. Exp. Rev. Vaccines 11:841–855. 10.1586/erv.12.53 [DOI] [PubMed] [Google Scholar]
- 7.Krone CL, vande Groep K, Trzcinski K, Sanders E, Bogaert D. 2013. Immunosenescence and pneumococcal disease: an imbalance in host-pathogen interaction. Lancet Resp. Med. 2:141–153. 10.1016/S2213-2600(13)70165-6 [DOI] [PubMed] [Google Scholar]
- 8.Krone CL, Trzcinski K, Zborowski T, Sanders EA, Bogaert D. 2013. Impaired innate mucosal immunity in aged mice permits prolonged Streptococcus pneumoniae colonization. Infect. Immun. 81:4615–4625. 10.1128/IAI.00618-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller RA, Nadon NL. 2000. Principles of animal use for gerontological research. J. Gerontol. A Biol. Sci. Med. Sci. 55:B117–B123. 10.1093/gerona/55.3.B117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinojosa E, Boyd AR, Orihuela CJ. 2009. Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J. Infect. Dis. 200:546–554. 10.1086/600870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malley R, Lipsitch M, Stack A, Saladino R, Fleisher G, Pelton S, Thompson C, Briles D, Anderson P. 2001. Intranasal immunization with killed unencapsulated whole cells prevents colonization and invasive disease by capsulated pneumococci. Infect. Immun. 69:4870–4873. 10.1128/IAI.69.8.4870-4873.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biesbroek G, Sanders EA, Roeselers G, Wang X, Caspers MP, Trzcinski K, Bogaert D, Keijser BJ. 2012. Deep sequencing analyses of low density microbial communities: working at the boundary of accurate microbiota detection. PLoS One 7:e32942. 10.1371/journal.pone.0032942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methe B, DeSantis TZ, Petrosino JF, Knight R, Birren BW. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21:494–504. 10.1101/gr.112730.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235. 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tusher VG, Tibshirani R, Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 98:5116–5121. 10.1073/pnas.091062498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378 [DOI] [PubMed] [Google Scholar]
- 18.Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, Kolter R. 2010. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. mBio 1(3):e00129–10. 10.1128/mBio.00129-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke SF, Murphy EF, O'Sullivan O, Ross RP, O'Toole PW, Shanahan F, Cotter PD. 2013. Targeting the microbiota to address diet-induced obesity: a time dependent challenge. PLoS One 8:e65790. 10.1371/journal.pone.0065790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sommer F, Backhed F. 2013. The gut microbiota—masters of host development and physiology. Nat. Rev. Microbiol. 11:227–238. 10.1038/nrmicro2974 [DOI] [PubMed] [Google Scholar]
- 21.Cosseau C, Devine DA, Dullaghan E, Gardy JL, Chikatamarla A, Gellatly S, Yu LL, Pistolic J, Falsafi R, Tagg J, Hancock RE. 2008. The commensal Streptococcus salivarius K12 downregulates the innate immune responses of human epithelial cells and promotes host-microbe homeostasis. Infect. Immun. 76:4163–4175. 10.1128/IAI.00188-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ubeda C, Bucci V, Caballero S, Djukovic A, Toussaint NC, Equinda M, Lipuma L, Ling L, Gobourne A, No D, Taur Y, Jenq RR, van den Brink MR, Xavier JB, Pamer EG. 2013. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infect. Immun. 81:965–973. 10.1128/IAI.01197-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301. 10.1038/nrmicro1871 [DOI] [PubMed] [Google Scholar]
- 24.Bogaert D, van Belkum A, Sluijter M, Luijendijk A, de Groot R, Rumke HC, Verbrugh HA, Hermans PW. 2004. Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363:1871–1872. 10.1016/S0140-6736(04)16357-5 [DOI] [PubMed] [Google Scholar]
- 25.van den Bergh MR, Biesbroek G, Rossen JW, de Steenhuijsen Piters WA, Bosch AA, van Gils EJ, Wang X, Boonacker CW, Veenhoven RH, Bruin JP, Bogaert D, Sanders EA. 2012. Associations between pathogens in the upper respiratory tract of young children: interplay between viruses and bacteria. PLoS One 7:e47711. 10.1371/journal.pone.0047711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veenhoven R, Bogaert D, Uiterwaal C, Brouwer C, Kiezebrink H, Bruin J, Hermans E IJP, de Groot R, Zegers B, Kuis W, Rijkers G, Schilder A, Sanders E. 2003. Effect of conjugate pneumococcal vaccine followed by polysaccharide pneumococcal vaccine on recurrent acute otitis media: a randomised study. Lancet 361:2189–2195. 10.1016/S0140-6736(03)13772-5 [DOI] [PubMed] [Google Scholar]
- 27.Spijkerman J, Prevaes SM, van Gils EJ, Veenhoven RH, Bruin JP, Bogaert D, Wijmenga-Monsuur AJ, van den Dobbelsteen GP, Sanders EA. 2012. Long-term effects of pneumococcal conjugate vaccine on nasopharyngeal carriage of S. pneumoniae, S. aureus, H. influenzae and M. catarrhalis. PLoS One 7:e39730. 10.1371/journal.pone.0039730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woodmansey EJ. 2007. Intestinal bacteria and ageing. J. Appl. Microbiol. 102:1178–1186. 10.1111/j.1365-2672.2007.03400.x [DOI] [PubMed] [Google Scholar]