

Abstract
Chlamydial infection in the lower genital tract can lead to hydrosalpinx, which is accompanied by activation of both pattern recognition receptor TLR2- and inflammatory cytokine receptor TNFR1-mediated signaling pathways. In the current study, we compared the relative contributions of these two receptors to chlamydial induction of hydrosalpinx in mice. We found that mice with or without deficiencies in TLR2 or TNFR1 displayed similar time courses of live organism shedding from vaginal swabs, suggesting that these receptor-mediated signaling pathways are not required for controlling chlamydial lower genital infection. However, mice deficient in TNFR1 but not TLR2 developed significantly reduced hydrosalpinx. The decreased pathogenicity correlated with a significant reduction in interleukin-17 by in vitro-restimulated splenocytes of TNFR1-deficient mice. Although TLR2-deficient mice developed hydrosalpinx as severe as that of wild-type mice, peritoneal macrophages from mice deficient in TLR2 but not TNFR1 produced significantly reduced cytokines upon chlamydial stimulation, suggesting that reduced macrophage responses to chlamydial infection do not always lead to a reduction in hydrosalpinx. Thus, we have demonstrated that the signaling pathways triggered by the cytokine receptor TNFR1 play a more significant role in chlamydial induction of hydrosalpinx than those mediated by the pattern recognition receptor TLR2, which has laid a foundation for further revealing the chlamydial pathogenic mechanisms.
INTRODUCTION
Chlamydia trachomatis infection in the lower genital tract, if not treated, can ascend to the upper genital tract, causing pathologies such as hydrosalpinx, leading to complications such as tubal factor infertility (1). The precise pathogenic mechanisms of C. trachomatis-induced diseases in humans remain unknown. Intravaginal inoculation with C. muridarum in mice has been extensively used to study mechanisms of C. trachomatis pathogenesis and immunity (2–6), although C. muridarum causes no known human diseases. This is because intravaginal infection in some strains of mice with C. muridarum can lead to hydrosalpinx that mimics the upper genital tract pathology in C. trachomatis-infected women. Using this model in combination with antibody depletion and gene knockout (KO), a CD4+ T cell-dependent and gamma interferon (IFN-γ)-mediated immunity has been identified as a major protective mechanism for mice to control chlamydial infection (7). However, the precise inflammatory mechanism of C. muridarum-induced hydrosalpinx is still unclear, although activation of many inflammatory signaling pathways has been detected during infection (8–18).
Hydrosalpinx is a visible tubal pathology observed under laparoscopy and is defined as a long-lasting clear fluid accumulation in the lumen of fallopian tubes or oviducts as a result of fibrotic blockage. Mice intravaginally infected with C. muridarum can develop pyosalpinx during the first 3 weeks and gradually transition into hydrosalpinx as early as 4 weeks after infection, which is accompanied by significantly reduced neutrophil infiltration (9, 12, 19). Some of these early hydrosalpinges are transient, while others are long lasting depending on the nature of the tubal blockage (19). Although it is not clear at what time point hydrosalpinx becomes irreversible, what is certain is that hydrosalpinx observed in mice long after intravaginal infection with C. muridarum may be more relevant to hydrosalpinx observed in women. Mouse hydrosalpinx can be detected reproducibly between days 60 and 80 after intravaginal infection with C. muridarum (14, 15, 20–24), suggesting that mouse hydrosalpinx detected 60 days after infection represents long-lasting upper genital tract pathology.
During chlamydial infection, both pattern recognition receptor TLR2- and inflammatory cytokine receptor TNFR1-mediated signaling pathways are activated (8–13). TLR2 is a MyD88-dependent pattern recognition receptor responsible for detecting microbial components such as peptidoglycan (25). TLR2-mediated signals can lead to activation of both NF-κB and mitogen-activated protein (MAP) kinase signaling pathways (26). Tumor necrosis factor (TNF) signals through two distinct receptors, designated TNFR1 or p55 and TNFR2 or p75, respectively. TNFR1 is the primary signaling receptor that initiates the majority of inflammatory responses classically attributed to TNF. It activates both NF-κB and MAP kinase pathways in addition to apoptosis induction (27–29). Although in general inflammatory pathways activated by either TLR2 or TNFR1 can contribute to both eradication of infectious agents and tissue injury (27, 29), their precise roles in chlamydial infection and pathogenesis, especially the development of long-lasting visible hydrosalpinx, have not been carefully evaluated. For example, although a statistically significant reduction in oviduct acute but not chronic inflammation and decrease in lumenal dilation score from ∼3 to ∼2 were found in TLR2 knockout (TLR2−/−) mice (9), these observations were made under microscopy and on day 35 after infection. This acute inflammation and oviduct dilation, observable only by microscopice on day 35, may not always lead to long-lasting hydrosalpinx observable by the naked eye on day 60 or 80 after infection. The role of TNFR1 in chlamydial infection and pathogenesis also needs further investigation. Adoptive transfer of TNF-α reduced chlamydial lung infection (30), but chlamydial resolution in the genital tract appeared to be independent of TNF-α (31). The role of TNF-α in chlamydial pathogenesis seems to be more certain. First, TNF-α polymorphism has been linked to increased risk of severe tubal damage in women with infertility associated with C. trachomatis (32). Second, mice genetically deficient in TNF-α production displayed significantly reduced hydrosalpinx (33). Since TNF-α can signal via both TNFR1 and TNFR2, it remains unknown which receptor-mediated pathway is more important in chlamydial pathogenesis.
In the current study, we compared the relative contributions of TLR2 and TNFR1 to chlamydial induction of hydrosalpinx in mice. We found that regardless of their deficiencies in TLR2 or TNFR1, mice displayed similar time courses of live organism shedding from vaginal swabs, suggesting that these receptor-mediated signaling pathways are not required for controlling chlamydial lower genital infection. However, mice genetically deficient in TNFR1 but not TLR2 displayed significantly reduced hydrosalpinx on day 60 or 80 after C. muridarum infection. The reduced pathogenicity correlated with a significant reduction in interleukin-17 (IL-17) production in TNFR1-deficient mice. Thus, signaling pathways triggered by the cytokine receptor TNFR1 play a more significant role than those mediated by the pattern recognition receptor TLR2 in chlamydial induction of long-lasting hydrosalpinx, which has provided a foundation for further revealing the chlamydial pathogenic mechanisms.
MATERIALS AND METHODS
Chlamydial organisms and infection.
The C. muridarum organisms (Nigg strain) used in the current study were propagated in HeLa cells (human cervical carcinoma epithelial cells; ATCC CCL2), purified, aliquoted, and stored as described previously (16, 34). Female wild-type C57BL/6J mice (stock number 000664) or mice with gene deficiency in TLR2 (B6.129-Tlr2tm1Kir/J; stock number 004650) or TNFR1 (B6.129-Tnfrsf1atm1Mak/J; stock number 002818) were purchased at the age of 5 to 6 weeks old from Jackson Laboratories (Bar Harbor, ME). Each mouse was inoculated intravaginally with 2 × 105 inclusion-forming units (IFUs) of live C. muridarum organisms as described previously (16). For in vitro infection of HeLa cells, HeLa cells grown on coverslips in 24-well plates containing Dulbecco's modified Eagle medium (DMEM; GIBCO BRL, Rockville, MD) with 10% fetal calf serum (FCS; GIBCO BRL) at 37°C in an incubator supplied with 5% CO2 were inoculated with C. muridarum organisms as described previously (16, 34). The infected cultures were processed for immunofluorescence assay as described below.
Monitoring mouse shedding of live chlamydial organisms.
To monitor live organism shedding, vaginal swabs were taken on different days after the intravaginal infection. Each swab was suspended in 500 μl of SPG followed by sonication on ice, and the released organisms were titrated on HeLa cell monolayers in duplicates as described previously (14, 16, 35, 36). The total number of IFUs per swab was calculated based on the number of IFUs per field, number of fields per coverslip, dilution factors, and inoculation and total sample volumes. An average was taken from the serially diluted and duplicate samples for any given swab. The calculated total number of IFUs/swab was converted into log10 values, and the log10 IFUs were used to calculate means and standard deviations for each group at each time point.
Evaluating mouse genital tract tissue pathology and histological scoring.
Mice were sacrificed on days 60 to 80 after infection, and the mouse urogenital tract tissues were isolated. Before the tissues were removed from the mouse body, an in situ gross examination was performed for evidence of oviduct hydrosalpinx or any other related abnormalities of oviducts. The severity of oviduct hydrosalpinx was scored based on the following criteria: no hydrosalpinx (0), hydrosalpinx detectable only after amplification (1), hydrosalpinx clearly visible with the naked eye but smaller than (2), equal to (3), or larger than (4) the size of the ovary on the same side. The oviducts from left and right sides of the same mouse were scored separately, and the average of these two scores was the score assigned to the mouse. The excised tissues, after photographing, were fixed in 10% neutral formalin, embedded in paraffin, and serially sectioned longitudinally (5 μm/section). Efforts were made to include cervix, both uterine horns, and oviducts, as well as lumenal structures of each tissue in each section. The sections were stained with hematoxylin and eosin (H&E) as described elsewhere (19). The H&E-stained sections were scored for severity of inflammation and pathologies based on the modified schemes established previously (14, 19). Scoring for dilation of oviduct used the following scheme: 0, no significant dilation; 1, mild dilation of a single cross section; 2, one to three dilated cross sections; 3, more than three dilated cross sections; and 4, confluent pronounced dilation. Scoring for inflammatory cell infiltrates (at the chronic stage of infection, the infiltrates mainly contain mononuclear cells) used the following scheme: 0, no significant infiltration; 1, infiltration at a single focus; 2, infiltration at two to four foci; 3, infiltration at more than four foci; and 4, confluent infiltration. Scores assigned to individual mice were calculated as means ± standard errors for each group of animals.
Immunofluorescence assay.
HeLa cells grown on coverslips with or without chlamydial infection were fixed and permeabilized for immunostaining as described previously (36–38). Hoechst (blue; Sigma) was used to visualize nuclear DNA. For titrating IFUs from mouse vaginal swab and kidney tissue homogenate samples, a mouse antichlamydial lipopolysaccharide (LPS) antibody (clone MB5H9; unpublished observation) plus a goat anti-mouse IgG conjugated with Cy3 (red; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used to visualize chlamydial inclusions. All immunofluorescence-labeled samples were observed under an Olympus AX-70 fluorescence microscope equipped with multiple filter sets (Olympus, Melville, NY).
ELISA.
Cytokines from supernatants of mouse spleen cells restimulated with UV-inactivated chlamydial organisms or peritoneal cavity macrophages infected with live chlamydial organisms or vaginal swabs collected on day 10 after infection were measured using commercially available enzyme-linked immunosorbent assay (ELISA) kits. The kits for mouse IFN-γ (catalog number DY485), IL-17 (DY420), IL-4 (DY404), IL-1α (DY400), IL-6 (DY406), and MIP2 (IL-8; DY452) all were obtained from R&D Systems, Inc. (Minneapolis, MN). The ELISA was performed by following the instructions provided by the manufacturer or as described elsewhere (39–41). A soluble colorimetric substrate [2-2′-azino-di-(3-ethylbenzthiazoline) sulfonic acid; ABTS] was used to detect the secondary antibody conjugate binding. The absorbance taken at 405 nm was used to calculate cytokine concentrations (pg per ml). To prepare mouse spleen cell in vitro restimulation cultures, splenocytes were harvested from mice infected with C. muridarum on days 60 to 80 after infection and cultured in 96-well plates at a density of 5 × 106 cells/well with or without stimulation with UV-inactivated C. muridarum elementary bodies at a final concentration of 1 × 106 IFUs/ml. After culturing for 3 days, the culture supernatants were collected for cytokine measurements. Peritoneal cavity macrophages were harvested by washing the cavity with 3 ml cold phosphate-buffered saline (PBS), and the cell suspensions from each mouse were washed with DMEM containing 10% FCS before seeding to 24-well plates with 1 × 105/well. The overnight cultures were washed once to remove nonadherent cells and were infected with C. muridarum at an multiplicity of infection (MOI) of 1 for 24 h before the supernatants were taken for cytokine measurements.
Statistical analyses.
The Kruskal-Wallis test was used to analyze the differences in IFUs recovered from mouse swabs. The pathology score data were analyzed with the Mann-Whitney rank-sum test. Fisher's exact test was used to analyze category data, including the percentage of mice with oviduct hydrosalpinx. The cytokine concentrations were analyzed using a two-tailed Student t test.
RESULTS
Neither the TNFR1 nor TLR2 signaling pathway is required for controlling chlamydial lower genital tract infection.
First, we compared mice deficient in TLR2 (n = 19 mice) or TNFR1 (n = 20) to wild-type mice (n = 31) in their susceptibility to C. muridarum infection in the urogenital tract. The three groups of mice were intravaginally infected with C. muridarum organisms, and the vaginal shedding of live organisms was monitored postinfection (Fig. 1). All mice, regardless of genotype, shed similar levels of live organisms up to 4 weeks after infection. By day 31, most mice cleared infection. These observations suggest that neither the TLR2- nor TNFR1-mediated signaling pathway is necessary for controlling C. muridarum infection in the mouse lower genital tract.
FIG 1.

Effect of TLR2 or TNFR1 deficiency on live organism recovery from mouse vaginal swabs following C. muridarum infection. Mice without (C57BL/6J wild type; n = 31) or with deficiency in TLR2 (TLR2−/−; n = 19) or TNFR1 (TNFR1−/−; n = 20) were infected intravaginally with C. muridarum. Vaginal swabs were taken on different days after infection, as shown along the x axis, for monitoring live C. muridarum organisms. Shown along the y axis are the recovered live organisms expressed as log10 IFUs (inclusion-forming units) per swab (A) or the percentage of mice positive for shedding live organisms (B). Note that all three groups of mice displayed similar levels of live organism shedding from the lower genital tract. The data came from 3 to 4 independent experiments.
Mice deficient in TNFR1 but not TLR2 developed significantly reduced hydrosalpinx following C. muridarum infection.
All mice described above were sacrificed for observing upper genital tract pathology between days 60 and 80 after infection (Fig. 2). We found that 71% of C57BL/6J wild-type mice developed hydrosalpinx with a severity score of 3.45 ± 3.00. Mice deficient in the inflammatory cytokine receptor TNFR1 developed significantly reduced hydrosalpinx, with only 25% of these knockout mice being positive for hydrosalpinx (P < 0.01), and a severity score of 0.61 ± 1.18 (P < 0.01). This observation is consistent with a previous finding that mice deficient in TNF-α failed to develop severe hydrosalpinx (33). However, 58% of mice deficient in TLR2 also developed hydrosalpinx with a severity score of 2.32 ± 2.03. Although both the absolute number of mice with hydrosalpinx and the hydrosalpinx score were lower in the TLR2 knockout group, these parameters were not significantly different between the TLR2 knockout and wild-type groups (P = 0.37 and P = 0.16, respectively). This observation seemed to contradict a previous claim that the TLR2-mediated signaling pathway is critical for C. muridarum induction of hydrosalpinx (9). The gross pathology was confirmed by the microscopic observation that the oviduct lumen of mice deficient in TNFR1 but not TLR2 was significantly dilated compared to that of the wild-type mice (Fig. 3). However, there was no significant difference in chronic inflammatory infiltration scores among the 3 groups. This is probably due to remission of inflammatory responses after the irreversible hydrosalpinx has already established 60 to 80 days after infection.
FIG 2.

Effect of TLR2 or TNFR1 deficiency on hydrosalpinx development following C. muridarum infection. The same three groups of mice described in the legend to Fig. 1 were sacrificed between days 60 and 80 after infection for harvesting genital tract tissues. One representative tissue image is shown for each group, with the left image for the C57BL/6J wild-type (WT), the middle for TLR2−/−, and the right for TNFR1−/− groups. V, vagina; C, cervix; U, uterus; UH, uterine horn; O, ovary. The genital tract tissues were carefully examined by naked eye or under a stereoscope for hydrosalpinx, as indicated by red arrows in the overall genital tract image and red arrowheads in the magnified images for oviduct/ovary sections. For each group, the total number of mice (N) and percentage of mice with positive hydrosalpinx, along with hydrosalpinx severity scores (determined as described in Materials and Methods), all were listed under the corresponding images. Note that both the hydrosalpinx incidence rates and severity scores were significantly lower in TNFR1−/− (P < 0.01 for both) but not TLR2−/− groups compared to the C57BL/6J wild-type group.
FIG 3.

Effect of TLR2 or TNFR1 deficiency on microscopic pathology following C. muridarum infection. The genital tract tissues depicted in Fig. 2 from the groups of mice were sectioned for microscopic observation of oviduct inflammatory infiltration and lumenal dilation as described in Materials and Methods. (A) A representative image taken under a 10× objective lens, along with two areas indicated by rectangles taken under a 100× objective lens as indicated at the top of each image, were presented for each mouse group, including wild type (WT) (a), TLR2−/− (b), an TNFR1−/− (c). (B) Both the oviduct inflammatory infiltration and lumenal dilation were semiquantitatively scored as described in Materials and Methods. Note that mice deficient in TNFR1 but not TLR2 developed significantly reduced lumenal dilation (**, P < 0.01 by Mann-Whitney rank-sum test).
The reduced hydrosalpinx development correlates with a decreased production of IL-17 by splenocytes but not IL-1α, IL-6, or IL-8 by macrophages.
We monitored T cell cytokine production between three groups of mice using an in vitro spleen cell restimulation assay (Fig. 4). Splenocytes from all three groups of mice produced high levels of IFN-γ upon restimulation with chlamydial antigens, which may explain why all three groups displayed similar live organism shedding time courses, as shown in Fig. 1, since IFN-γ has been considered a major host defense cytokine for controlling C. muridarum infection in mice. There was no significant difference in IL-4 production between the three groups of mice. However, splenocytes from mice deficient in TNFR1 but not TLR2 produced a significantly reduced level of IL-17 (P < 0.01), which correlated with the significantly reduced hydrosalpinx in TNFR1 knockout mice. We also compared mouse macrophages for inflammatory cytokine production upon chlamydial stimulation in cell cultures (Fig. 5). We found that mice deficient in TLR2 but not TNFR1 produced significantly reduced levels of cytokines IL-1α, IL-6, and MIP2 (mouse IL-8), which is consistent with previous findings that macrophages deficient in either TLR2 or MyD88 were less capable of producing cytokines upon chlamydial stimulation (9, 14). We further monitored these inflammatory cytokines in vaginal swabs collected from mice 10 days after infection and found a similar trend, i.e., the TLR2-deficient mice produced lower levels of these cytokines (Fig. 6). This observation confirmed the in vitro macrophage cytokine profile result described above. However, the inadequate lower genital tract or macrophage responses to chlamydial stimulation in terms of cytokine production may not negatively affect hydrosalpinx development in the upper genital tract, since mice deficient in either TLR2 (Fig. 2) or MyD88 (14) still developed significant hydrosalpinx.
FIG 4.
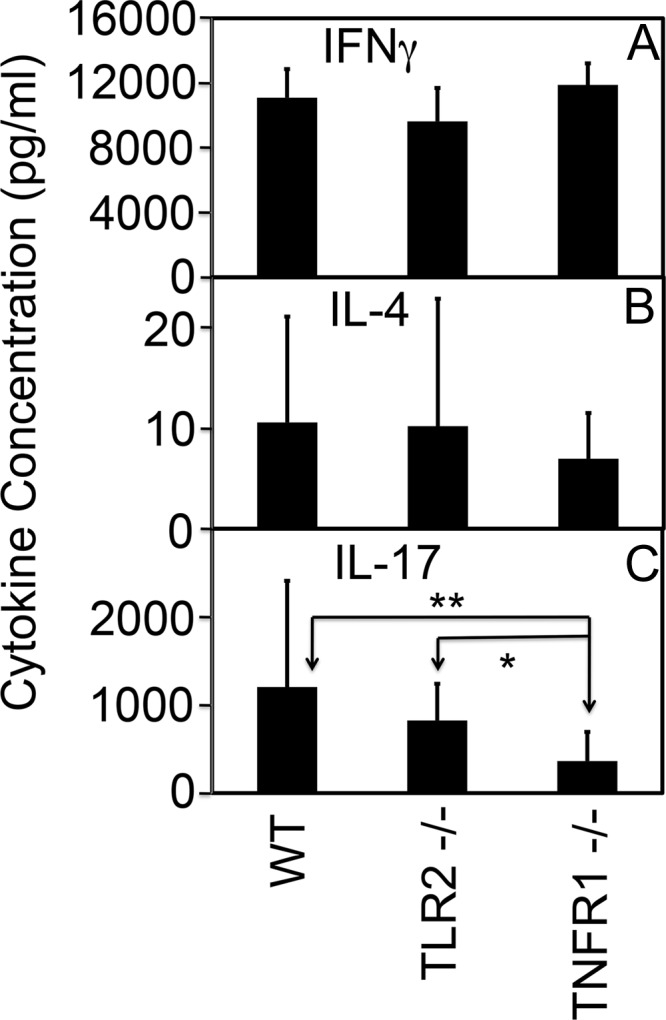
Effect of TLR2 or TNFR1 deficiency on mouse T cell responses following C. muridarum infection. Splenocytes were harvested from TLR2−/−, TNFR1−/−, and wild-type mice between days 60 and 80 after infection, when the mice were sacrificed for examining genital tract pathology as described in the legend to Fig. 2. The splenocytes were restimulated in vitro with UV-inactivated C. muridarum organisms for 72 h. The culture supernatants were measured for IFN-γ (a), IL-4 (b), and IL-17 (c) using ELISA, and the results were expressed as picograms per ml as shown along the y axis. Note that the level of IL-17 produced by splenocytes from TNFR1−/− but not TLR2−/− mice was significantly lower than that from wild-type mice (**, P < 0.01 by Student t test). The TLR2−/− mice produced significantly more IL-17 than the TNFR1−/− mice (*, P < 0.05 by Student t test).
FIG 5.

Effect of TLR2 or TNFR1 deficiency on mouse macrophage cytokine production. Macrophages harvested from the peritoneal cavity of wild-type C57 mice (WT; n = 5) and TLR2−/− (n = 5) or TNFR1−/− (n = 5) mice were infected with C. muridarum organisms for 24 h. The culture supernatants were used for cytokine measurements, and the cytokine concentrations were expressed in picograms per milliliter (shown along the y axis). Macrophages from TLR2−/− mice produced significantly lower levels of cytokines (**, P < 0.01 by Student t test).
FIG 6.

Effect of TLR2 or TNFR1 deficiency on cytokine production in the lower genital tract. Swabs were collected from mouse vaginas 10 days after infection as described in the legend to Fig. 1. Swabs from 10 mice randomly selected from each group (WT, TLR2−/−, and TNFR1−/−) were used for measuring cytokines, and the cytokine concentrations were expressed in picograms per milliliter (shown along the y axis). The TLR2−/− mice produced the lowest levels of cytokines.
DISCUSSION
Chlamydia muridarum infection induces long-lasting hydrosalpinx in mice, which is accompanied by activation of both pattern recognition receptor TLR2- and inflammatory cytokine receptor TNFR1-mediated signaling pathways. However, the relative roles of these two receptor-mediated pathways in chlamydial induction of long-lasting hydrosalpinx have not been carefully compared side by side. The current study is aimed at clarifying the relative contributions of these receptor-mediated signaling events to C. muridarum infection and induction of hydrosalpinx. The hydrosalpinx was visually examined 60 days or later after infection, since long-lasting hydrosalpinx is more relevant to hydrosalpinx observed in women and may be used as a hallmark for tubal factor infertility. We found that in 3 to 4 independent experiments, mice with or without deficiencies in TLR2 or TNFR1 displayed similar time courses of live organism shedding from the lower genital tract, suggesting that these receptor-mediated signaling pathways are not required for controlling chlamydial lower genital infection. More importantly, in these same experiments, mice deficient in TNFR1 but not TLR2 consistently developed significantly reduced hydrosalpinx. These experiments have convincingly demonstrated that TLR2 signaling pathways are not required for C. muridarum induction of long-lasting hydrosalpinx, since there were no significant differences in either the incidence rates or severity scores of hydrosalpinx between TLR2−/− and wild-type mice. On the contrary, TNFR1-mediated pathways are far more important for the development of hydrosalpinx during C. muridarum infection. Both the incidence rates and severity scores of hydrosalpinx were significantly reduced in the TNFR1−/− mice.
Darville et al. reported a statistically significant reduction in oviduct acute but not chronic inflammation in TLR2−/− mice and a decrease in the oviduct lumenal dilation score from ∼3 in wild-type mice to ∼2 in TLR2−/− mice (9). These observations have led the field to believe that TLR2-mediated pathways are critical for C. muridarum induction of hydrosalpinx. However, these observations were made under microscopy and on day 35 after infection. Acute inflammation and oviduct dilation observable only at the microscopic level on day 35 may not always lead to the long-lasting hydrosalpinx observable by the naked eye on day 60 or 80 after infection. Although transient hydrosalpinx may also affect fertility, it is the long-lasting hydrosalpinx that is more clinically relevant to tubal factor infertility. This is because most tubal factor infertility patients with high titers of anti-C. trachomatis antibodies developed tubal blockage, as revealed under hysterosalpingography, and hydrosalpinx observable under a laparoscope (42, 43). Thus, we have used the visual observation of hydrosalpinx on day 60 or longer after infection as a primary parameter for measuring C. muridarum pathogenesis in mice (15, 20, 24). Using this criterion in the current study, we found that the TLR2−/− mice developed hydrosalpinx as severe as that of the wild-type mice. Although both the incidence rates and severity scores of the TLR2−/− mice were slightly lower, they were not significantly lower than those in the wild-type mice. Given the large sample size (n = 31 for wild-type mice and n = 19 for TLR2−/− mice) in the current study, we are confident that these observations are reproducible. On the contrary, the previous study that claimed a role of TLR2 in upper genital pathology was based on only 10 mice per group (9). Our observation is supported by a previous finding that mice deficient in MyD88, a critical adaptor molecule required for TLR2-mediated signaling, developed more severe hydrosalpinx (14). This suggested that MyD88-independent inflammatory pathways are sufficient for causing hydrosalpinx. However, it is worth noting that in the absence of MyD88, mice experienced more severe infection (14), and the severe infection might be responsible for activating MyD88-independent signaling pathways for exacerbating hydrosalpinx. In the absence of TLR2, mice did not develop more severe infection, suggesting that redundant pathways involving MyD88 are sufficient for keeping chlamydial infection at a level similar to that of the wild-type mice. However, it is not clear at the moment whether the same redundant pathways were also responsible for mediating hydrosalpinx-causing inflammation in the TLR2 KO mice. Since the current study has also revealed that TNFR1 KO mice displayed significantly reduced hydrosalpinx, we hypothesize that TNFR1 pathways play a significant role in chlamydial induction of hydrosalpinx in the TLR2 KO mice. It will be interesting to determine how the TNFR1 signaling pathway is activated during chlamydial infection in TLR2 KO mice.
Although we have presented convincing data that TLR2 is redundant in chlamydial induction of hydrosalpinx in our mouse model system, the precise role of TLR2 in chlamydial infection and pathogenesis in other models and humans remains unclear. TLR2 signaling pathways were activated during chlamydial infection, and TLR2-deficient mice did display a slight reduction in hydrosalpinx rate and severity (although there was no statistical significance). These observations suggested that TLR2 still plays some role in chlamydial infection and pathogenesis, especially under different infection conditions, for example, during less severe infection or in hosts with different genetic backgrounds. It is worth noting that the C. muridarum genital tract infection in C57BL/6J mice is a relatively severe infection model, since a single infection in the lower genital tract is sufficient to induce hydrosalpinx in the upper genital tract. However, during natural infection in humans, multiple C. trachomatis infections often correlate with tubal factor infertility. Thus, each single infection with C. trachomatis in humans may be less severe than the experimental C. muridarum infection in our mouse model. It is possible that TLR2 signaling pathways play a more significant role in C. trachomatis infection in humans. However, the TLR2 genotype was not associated with tubal infertility in women (44). Thus, more investigations are required to address the precise role of TLR2 signaling pathways in human chlamydial infection and pathogenesis.
The cytokine receptor TNFR1 plays a significant role in hydrosalpinx development during C. muridarum infection. Both the incidence rates and severity scores of hydrosalpinx were highly significantly reduced in TNFR1−/− mice compared to those of either wild-type or TLR2−/− mice. The question is why these two receptors, both leading to activation of NF-κB and MAP kinase pathways, played distinct roles in chlamydial pathogenesis. When macrophages from these mice were compared, we found that TLR2−/− but not TNFR1−/− macrophages displayed significantly reduced cytokine production after C. muridarum infection. Similarly, MyD88−/− macrophages were also deficient in cytokine production (14). Clearly, lack of cytokine production by macrophages deficient in innate immunity receptor-mediated signaling did not result in reduced hydrosalpinx. Thus, we can conclude that MyD88-dependent signaling pathways are not essential for chlamydial induction of hydrosalpinx. On the contrary, the cytokine receptor TNFR1-mediated signaling contributes significantly to hydrosalpinx development during C. muridarum infection, which is consistent with a previously demonstrated role of TNF-α in chlamydial pathogenesis (33). The reduced hydrosalpinx was further correlated with a significant reduction in IL-17 production by splenocytes from TNFR1−/− mice. This finding is supported by a recent observation that mice deficient in IL-17 developed significantly reduced chronic pathology (45) and also is consistent with our previous study showing that although the IL-23/TH17 axis was not required for host resistance to C. muridarum infection, it contributed significantly to Chlamydia-induced pathology (20). These observations together have suggested that IL-17-mediated responses are critical for hydrosalpinx development during C. muridarum infection. The next question is how TNFR1 signaling may affect IL-17 production. During fungal infection in the mouse airway, TNF-α produced by inflammatory dendritic cells was found to promote IL-17A production by CD4+ T cells, and a lack of TNF-α decreased IL-17A levels (46). Thus, it is likely that in TNFR1-deficient mice, inadequate IL-17 production leads to the reduced development of hydrosalpinx. We can now hypothesize that TNFR1 signaling-promoted Th17 cells contribute significantly to chlamydial induction of hydrosalpinx.
In all, we have demonstrated that TLR2 signaling is not required for chlamydial induction of hydrosalpinx despite the observation that TLR2-deficient macrophages produced significantly reduced levels of cytokines upon chlamydial infection. In contrast, the TNFR1 signaling is essential for hydrosalpinx development during chlamydial infection, which correlates with IL-17 production by splenocytes. These observations have set a correct direction for further understanding chlamydial pathogenic mechanisms.
ACKNOWLEDGMENT
This work was supported in part by grants from the U.S. National Institutes of Health (G.Z.).
Footnotes
Published ahead of print 18 February 2014
REFERENCES
- 1.Sherman KJ, Daling JR, Stergachis A, Weiss NS, Foy HM, Wang SP, Grayston JT. 1990. Sexually transmitted diseases and tubal pregnancy. Sex Transm. Dis. 17:115–121. 10.1097/00007435-199007000-00001 [DOI] [PubMed] [Google Scholar]
- 2.Cotter TW, Meng Q, Shen ZL, Zhang YX, Su H, Caldwell HD. 1995. Protective efficacy of major outer membrane protein-specific immunoglobulin A (IgA) and IgG monoclonal antibodies in a murine model of Chlamydia trachomatis genital tract infection. Infect. Immun. 63:4704–4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pal S, Peterson EM, de la Maza LM. 2005. Vaccination with the Chlamydia trachomatis major outer membrane protein can elicit an immune response as protective as that resulting from inoculation with live bacteria. Infect. Immun. 73:8153–8160. 10.1128/IAI.73.12.8153-8160.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu H, Zhong G. 1999. Interleukin-12 production is required for chlamydial antigen-pulsed dendritic cells to induce protection against live Chlamydia trachomatis infection. Infect. Immun. 67:1763–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murthy AK, Sharma J, Coalson JJ, Zhong G, Arulanandam BP. 2004. Chlamydia trachomatis pulmonary infection induces greater inflammatory pathology in immunoglobulin A deficient mice. Cell Immunol. 230:56–64. 10.1016/j.cellimm.2004.09.002 [DOI] [PubMed] [Google Scholar]
- 6.Morrison SG, Morrison RP. 2005. A predominant role for antibody in acquired immunity to chlamydial genital tract reinfection. J. Immunol. 175:7536–7542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison RP, Caldwell HD. 2002. Immunity to murine chlamydial genital infection. Infect. Immun. 70:2741–2751. 10.1128/IAI.70.6.2741-2751.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perfettini JL, Darville T, Gachelin G, Souque P, Huerre M, Dautry-Varsat A, Ojcius DM. 2000. Effect of Chlamydia trachomatis infection and subsequent tumor necrosis factor alpha secretion on apoptosis in the murine genital tract. Infect. Immun. 68:2237–2244. 10.1128/IAI.68.4.2237-2244.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darville T, O'Neill JM, Andrews CW, Jr, Nagarajan UM, Stahl L, Ojcius DM. 2003. Toll-like receptor-2, but not Toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J. Immunol. 171:6187–6197 [DOI] [PubMed] [Google Scholar]
- 10.Yang X, Gartner J, Zhu L, Wang S, Brunham RC. 1999. IL-10 gene knockout mice show enhanced Th1-like protective immunity and absent granuloma formation following Chlamydia trachomatis lung infection. J. Immunol. 162:1010–1017 [PubMed] [Google Scholar]
- 11.Reddy BS, Rastogi S, Das B, Salhan S, Verma S, Mittal A. 2004. Cytokine expression pattern in the genital tract of Chlamydia trachomatis positive infertile women–implication for T-cell responses. Clin. Exp. Immunol. 137:552–558. 10.1111/j.1365-2249.2004.02564.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darville T, Andrews CW, Jr, Sikes JD, Fraley PL, Rank RG. 2001. Early local cytokine profiles in strains of mice with different outcomes from chlamydial genital tract infection. Infect. Immun. 69:3556–3561. 10.1128/IAI.69.6.3556-3561.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bas S, Neff L, Vuillet M, Spenato U, Seya T, Matsumoto M, Gabay C. 2008. The proinflammatory cytokine response to Chlamydia trachomatis elementary bodies in human macrophages is partly mediated by a lipoprotein, the macrophage infectivity potentiator, through TLR2/TLR1/TLR6 and CD14. J. Immunol. 180:1158–1168 [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Lei L, Chang X, Li Z, Lu C, Zhang X, Wu Y, Yeh IT, Zhong G. 2010. Mice deficient in MyD88 develop a Th2-dominant response and severe pathology in the upper genital tract following Chlamydia muridarum infection. J. Immunol. 184:2602–2610. 10.4049/jimmunol.0901593 [DOI] [PubMed] [Google Scholar]
- 15.Lu C, Peng B, Li Z, Lei L, Chen L, He Q, Zhong G, Wu Y. 2013. Induction of protective immunity against Chlamydia muridarum intravaginal infection with the chlamydial immunodominant antigen macrophage infectivity potentiator. Microbes Infect. 15:329–338. 10.1016/j.micinf.2013.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng W, Shivshankar P, Li Z, Chen L, Yeh IT, Zhong G. 2008. Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection. Infect. Immun. 76:515–522. 10.1128/IAI.01064-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng W, Shivshankar P, Zhong Y, Chen D, Li Z, Zhong G. 2008. Intracellular interleukin-1alpha mediates interleukin-8 production induced by Chlamydia trachomatis infection via a mechanism independent of type I interleukin-1 receptor. Infect. Immun. 76:942–951. 10.1128/IAI.01313-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su H, McClarty G, Dong F, Hatch GM, Pan ZK, Zhong G. 2004. Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J. Biol. Chem. 279:9409–9416. 10.1074/jbc.M312008200 [DOI] [PubMed] [Google Scholar]
- 19.Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, Ramsey KH. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex Transm. Dis. 32:49–56. 10.1097/01.olq.0000148299.14513.11 [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Lei L, Zhou Z, He J, Xu S, Lu C, Chen J, Yang Z, Wu G, Yeh IT, Zhong G, Wu Y. 2013. Contribution of interleukin-12 p35 (IL-12p35) and IL-12p40 to protective immunity and pathology in mice infected with Chlamydia muridarum. Infect. Immun. 81:2962–2971. 10.1128/IAI.00161-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu C, Zeng H, Li Z, Lei L, Yeh IT, Wu Y, Zhong G. 2012. Protective immunity against mouse upper genital tract pathology correlates with high IFNgamma but low IL-17 T cell and anti-secretion protein antibody responses induced by replicating chlamydial organisms in the airway. Vaccine 30:475–485. 10.1016/j.vaccine.2011.10.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lei L, Chen J, Hou S, Ding Y, Yang Z, Zeng H, Baseman J, Zhong G. 2013. Reduced live organism recovery and lack of hydrosalpinx in mice infected with plasmid-free Chlamydia muridarum. Infect. Immun. 82:983–992. 10.1128/IAI.01543-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang L, Yang Z, Zhang H, Zhou Z, Arulanandam B, Baseman J, Zhong G. 1 November 2013. Induction of protective immunity against Chlamydia muridarum intracervical infection in DBA/1j mice. Vaccine 10.1016/j.vaccine.2013.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang L, Zhang H, Lei L, Gong S, Zhou Z, Baseman J, Zhong G. 2013. Oviduct infection and hydrosalpinx in DBA1/j mice is induced by intracervical but not intravaginal inoculation with Chlamydia muridarum. PLoS One 8:e71649. 10.1371/journal.pone.0071649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar S, Ingle H, Prasad DV, Kumar H. 2013. Recognition of bacterial infection by innate immune sensors. Crit. Rev. Microbiol. 39:229–246. 10.3109/1040841X.2012.706249 [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto M, Takeda K. 2010. Current views of toll-like receptor signaling pathways. Gastroenterol. Res. Pract. 2010:240365. 10.1155/2010/240365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waters JP, Pober JS, Bradley JR. 2013. Tumour necrosis factor in infectious disease. J. Pathol. 230:132–147. 10.1002/path.4187 [DOI] [PubMed] [Google Scholar]
- 28.Twu YC, Gold MR, Teh HS. 2011. TNFR1 delivers pro-survival signals that are required for limiting TNFR2-dependent activation-induced cell death (AICD) in CD8+ T cells. Eur. J. Immunol. 41:335–344. 10.1002/eji.201040639 [DOI] [PubMed] [Google Scholar]
- 29.Aggarwal BB. 2003. Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 3:745–756. 10.1038/nri1184 [DOI] [PubMed] [Google Scholar]
- 30.Pal S, de la Maza LM. 2013. Mechanism of T-cell mediated protection in newborn mice against a Chlamydia infection. Microbes Infect. 15:607–614. 10.1016/j.micinf.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamalakaran S, Chaganty BK, Gupta R, Guentzel MN, Chambers JP, Murthy AK, Arulanandam BP. 2013. Vaginal chlamydial clearance following primary or secondary infection in mice occurs independently of TNF-alpha. Front. Cell Infect. Microbiol. 3:11. 10.3389/fcimb.2013.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohman H, Tiitinen A, Halttunen M, Lehtinen M, Paavonen J, Surcel HM. 2009. Cytokine polymorphisms and severity of tubal damage in women with Chlamydia-associated infertility. J. Infect. Dis. 199:1353–1359. 10.1086/597620 [DOI] [PubMed] [Google Scholar]
- 33.Murthy AK, Li W, Chaganty BK, Kamalakaran S, Guentzel MN, Seshu J, Forsthuber TG, Zhong G, Arulanandam BP. 2011. Tumor necrosis factor alpha production from CD8+ T cells mediates oviduct pathological sequelae following primary genital Chlamydia muridarum infection. Infect. Immun. 79:2928–2935. 10.1128/IAI.05022-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C, Chen D, Sharma J, Cheng W, Zhong Y, Liu K, Jensen J, Shain R, Arulanandam B, Zhong G. 2006. The hypothetical protein CT813 is localized in the Chlamydia trachomatis inclusion membrane and is immunogenic in women urogenitally infected with C. trachomatis. Infect. Immun. 74:4826–4840. 10.1128/IAI.00081-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murthy AK, Chambers JP, Meier PA, Zhong G, Arulanandam BP. 2007. Intranasal vaccination with a secreted chlamydial protein enhances resolution of genital Chlamydia muridarum infection, protects against oviduct pathology, and is highly dependent upon endogenous gamma interferon production. Infect. Immun. 75:666–676. 10.1128/IAI.01280-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z, Lu C, Peng B, Zeng H, Zhou Z, Wu Y, Zhong G. 2012. Induction of protective immunity against Chlamydia muridarum intravaginal infection with a chlamydial glycogen phosphorylase. PLoS One 7:e32997. 10.1371/journal.pone.0032997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193:935–942. 10.1084/jem.193.8.935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong G, Reis e Sousa C, Germain RN. 1997. Production, specificity, and functionality of monoclonal antibodies to specific peptide-major histocompatibility complex class II complexes formed by processing of exogenous protein. Proc. Natl. Acad. Sci. U. S. A. 94:13856–13861. 10.1073/pnas.94.25.13856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong G, Toth I, Reid R, Brunham RC. 1993. Immunogenicity evaluation of a lipidic amino acid-based synthetic peptide vaccine for Chlamydia trachomatis. J. Immunol. 151:3728–3736 [PubMed] [Google Scholar]
- 40.Zhong GM, Brunham RC. 1990. Immunoaccessible peptide sequences of the major outer membrane protein from Chlamydia trachomatis serovar C. Infect. Immun. 58:3438–3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma J, Bosnic AM, Piper JM, Zhong G. 2004. Human antibody responses to a Chlamydia-secreted protease factor. Infect. Immun. 72:7164–7171. 10.1128/IAI.72.12.7164-7171.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Budrys NM, Gong S, Rodgers AK, Wang J, Louden C, Shain R, Schenken RS, Zhong G. 2012. Chlamydia trachomatis antigens recognized in women with tubal factor infertility, normal fertility, and acute infection. Obstet. Gynecol. 119:1009–1016. 10.1097/AOG.0b013e3182519326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodgers AK, Budrys NM, Gong S, Wang J, Holden A, Schenken RS, Zhong G. 2011. Genome-wide identification of Chlamydia trachomatis antigens associated with tubal factor infertility. Fertil. Steril. 96:715–721. 10.1016/j.fertnstert.2011.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laisk T, Peters M, Saare M, Haller-Kikkatalo K, Karro H, Salumets A. 2010. Association of CCR5, TLR2, TLR4 and MBL genetic variations with genital tract infections and tubal factor infertility. J. Reprod. Immunol. 87:74–81. 10.1016/j.jri.2010.06.001 [DOI] [PubMed] [Google Scholar]
- 45.Andrew DW, Cochrane M, Schripsema JH, Ramsey KH, Dando SJ, O'Meara CP, Timms P, Beagley KW. 2013. The duration of Chlamydia muridarum genital tract infection and associated chronic pathological changes are reduced in IL-17 knockout mice but protection is not increased further by immunization. PLoS One 8:e76664. 10.1371/journal.pone.0076664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fei M, Bhatia S, Oriss TB, Yarlagadda M, Khare A, Akira S, Saijo S, Iwakura Y, Fallert Junecko BA, Reinhart TA, Foreman O, Ray P, Kolls J, Ray A. 2011. TNF-alpha from inflammatory dendritic cells (DCs) regulates lung IL-17A/IL-5 levels and neutrophilia versus eosinophilia during persistent fungal infection. Proc. Natl. Acad. Sci. U. S. A. 108:5360–5365. 10.1073/pnas.1015476108 [DOI] [PMC free article] [PubMed] [Google Scholar]