

Abstract
Although many microbial infections elicit an adaptive immune response that can protect against reinfection, it is generally thought that Staphylococcus aureus infections fail to generate protective immunity despite detectable T and B cell responses. No vaccine is yet proven to prevent S. aureus infections in humans, and efforts to develop one have been hampered by a lack of animal models in which protective immunity occurs. Our results describe a novel mouse model of protective immunity against recurrent infection, in which S. aureus skin and soft tissue infection (SSTI) strongly protected against secondary SSTI in BALB/c mice but much less so in C57BL/6 mice. This protection was dependent on antibody, because adoptive transfer of immune BALB/c serum or purified antibody into either BALB/c or C57BL/6 mice resulted in smaller skin lesions. We also identified an antibody-independent mechanism, because B cell-deficient mice were partially protected against secondary S. aureus SSTI and adoptive transfer of T cells from immune BALB/c mice resulted in smaller lesions upon primary infection. Furthermore, neutralization of interleukin-17A (IL-17A) abolished T cell-mediated protection in BALB/c mice, whereas neutralization of gamma interferon (IFN-γ) enhanced protection in C57BL/6 mice. Therefore, protective immunity against recurrent S. aureus SSTI was advanced by antibody and the Th17/IL-17A pathway and prevented by the Th1/IFN-γ pathway, suggesting that targeting both cell-mediated and humoral immunity might optimally protect against secondary S. aureus SSTI. These findings also highlight the importance of the mouse genetic background in the development of protective immunity against S. aureus SSTI.
INTRODUCTION
Methicillin-resistant Staphylococcus aureus (MRSA) infections have become epidemic in the United States (1). An increasing percentage of MRSA infections occurs among previously healthy people without identified health care-associated risk factors, so called community-associated MRSA (CA-MRSA) infections (2, 3). CA-MRSA is now the leading cause of skin and soft tissue infections (SSTIs) in the United States, accounting for millions of patient visits per year (4–6). These SSTIs are frequently associated with superficial dermonecrosis and abscess formation in subcutaneous tissues.
The CA-MRSA epidemic has provided an impetus to understand the immunopathogenesis of SSTIs in order to support the development of novel strategies to prevent and treat them. Innate immunity is the first line of defense against S. aureus SSTIs, including neutrophils, interleukin-1β (IL-1β), and pattern recognition receptors (7). Recurrent infections, particularly SSTIs, are common, and the role of adaptive immunity against S. aureus infections is poorly understood. Furthermore, vaccines against S. aureus infection have been unsuccessful; several phase 3 clinical trials have failed despite encouraging preclinical results (8–11). Interestingly, these vaccines elicited high antibody titers among vaccine recipients, raising the possibility that humoral immunity alone may be insufficient to fully protect against S. aureus infections (9, 10).
Evidence supporting a role for cell-mediated immunity in the host defense against S. aureus infections is emerging. For example, patients with poorly controlled HIV and low CD4+ T cell counts have high rates of S. aureus SSTIs (reviewed in reference 12). In addition, patients with the hyper-IgE syndrome, in which Th17 function is impaired, are highly susceptible to S. aureus skin and lung infections (13), as are mice that are deficient in IL-17 (14, 15). Therefore, targeting T cell responses against S. aureus may also be critical in developing protection against infection.
Investigation of the mechanisms of adaptive immunity against recurrent S. aureus infection has been hampered by a lack of an animal model in which “natural” immunity is elicited after primary infection. In this study, we compared the memory response to S. aureus SSTI in two genetic backgrounds and found that S. aureus SSTI strongly protected against secondary SSTI in BALB/c mice but much less so in C57BL/6 mice. Protection against dermonecrosis was mediated by antibody and IL-17A in BALB/c mice and inhibited by IFN-γ in C57BL/6 mice. Passive transfer of BALB/c immune serum into C57BL/6 mice was sufficient to limit lesion size upon infection, demonstrating a potential prophylactic or therapeutic avenue.
MATERIALS AND METHODS
Mouse model of S. aureus SSTI.
All animal experiments were approved by and performed in accordance with the regulations of the Institutional Committee on the Care and Use of Animals at the University of Chicago. Our established model of S. aureus SSTI has been described (16). Six-week-old female C57BL/6, BALB/c, T cell receptor (TCR) βδ−/− (B6.129P2-Tcrbtm1Mom Tcrdtm1Mom/J), CD4+ T cell-deficient (B6.129S2-Cd4tm1Mak/J), and CD8+ T-cell deficient (B6.129S2-Cd8atm1Mak/J) mice were purchased from Jackson Laboratory. BALB/c and B cell-deficient μMT (Igh-Jtm1Dhu) mice were purchased from Taconic.
Bacterial preparation.
The USA300 clinical isolate SF8300 (provided by Henry Chambers, University of California, San Francisco [UCSF]) was used for all infections. Its virulence has been described (16). On the day of inoculation, an overnight culture of SF8300 was diluted 1:100 in fresh tryptic soy broth. The cultures were harvested 3 h later (approximate optical density at 600 nm [OD600] of 1.8). The bacterial cells were pelleted by centrifugation, washed, and resuspended in sterile phosphate-buffered saline to achieve a concentration of 1.5 × 107 CFU/50 μl. The inocula were confirmed by plating serial dilutions on tryptic soy agar.
Inoculation and measurement of skin lesions.
The mice were sedated, and the flank was shaved and cleaned with ethanol, after which 50 μl of S. aureus (or phosphate-buffered saline [PBS control]) was inoculated subcutaneously. Mice were observed to awaken and given access to food and water throughout the experiment. The first inoculation was performed on the right flank, and the second was performed on the left flank. For reinfection experiments, mice were first infected with PBS or S. aureus, and all mice were reinfected with S. aureus 8 weeks later; therefore, the mice were age matched. Mice were observed and lesions were photographed daily. The raw edge of the lesions was measured using Adobe Photoshop software, and the lesion size was calculated digitally compared with a 100-mm2 standard. All measurements were performed by an observer blinded to the experimental groups.
Quantification of bacterial burden and local inflammatory response.
Mice were sacrificed 3 days after infection, and the skin lesions were removed and homogenized. For bacterial quantification, serial dilutions of the homogenate were plated on mannitol salt agar, and colonies were enumerated 24 h later. The homogenized lesions were centrifuged, and enzyme-linked immunosorbent assay (ELISA) was performed with the supernatants to quantify CXCL-1 (R & D Biosystems), IL-17A (R & D Biosystems), and myeloperoxidase (Hycult Biotechnology). For some mice, skin lesions were removed and fixed in 10% neutral buffered formalin, following which they were paraffin embedded. Sections were stained with hematoxylin and eosin and then were analyzed and photographed using a Zeiss Axioskop microscope (Integrated Microscopy Facility at the University of Chicago).
CD4+ T cell depletion.
Neutralizing antibody against CD4 (rat IgG2b, clone YTS191) and isotype control rat IgG2b (clone LTF2) were purchased from BioXcell. For in vivo CD4+ T cell depletion, mice received antibody (500 μg) via intraperitoneal injection 1 day prior to primary infection with S. aureus. An additional dose was given 7 days after infection. To confirm depletion, the mice were sedated and blood was obtained by retroorbital puncture. CD4+ (anti-CD4–fluorescein isothiocyanate [FITC], clone RM4-5; eBioscience) and CD8+ (anti-CD8–peridinin chlorophyll protein [PerCP], clone 53-6.7; BD Biosciences) T cells were quantified using a BD LSR II flow cytometer (BD Biosciences).
Antibody quantification.
Enzyme immunoassay/radioimmunoassay (EIA/RIA) 96-well plates (Costar; Corning Inc.) were coated with 5 μg/ml alpha-hemolysin (Hla) (Sigma-Aldrich) or 25 μg/ml iron-regulated surface determinant B (IsdB) (Merck). Mouse serum was prepared from whole blood using serum separator tubes (BD Biosciences). The serum was diluted 1:200 in PBS and added to the antibody-containing wells. Detection was performed using alkaline phosphatase (AP)-conjugated goat anti-mouse IgG, IgG1, IgG2a, IgG2b, and IgG3 (1:5,000; AffiniPure, Jackson ImmunoResearch) and AP substrate p-nitrophenylphosphate (pNPP) (Sigma-Aldrich), following the manufacturer's recommendations. Absorbance was measured using a GENios spectrophotometer (Tecan).
Serum transfer and antibody purification.
Mice were sacrificed 14 days after secondary infection with S. aureus (or PBS) by CO2 inhalation. Blood was obtained by cardiac puncture, and serum was isolated using serum separator tubes (Becton, Dickinson). Antibody was purified from immune BALB/c serum using protein A/G columns (Pierce). In order to fully remove antibody, 3 treatments were performed. Adoptive transfer of serum or antibody was performed by retroorbital injection (100 μl) on each of the 2 days prior to infection.
T cell transfer.
Mice were sacrificed 8 weeks after secondary infection with S. aureus (or PBS) by CO2 inhalation, and the spleens were harvested and placed in sterile medium. T lymphocytes were isolated by negative selection using the Pan T cell isolation Kit II or the CD8+ T cell isolation kit II (Miltenyi Biotec), according to the manufacturer's instructions. One day prior to infection, each recipient mouse received 8 × 106 T cells or PBS in a volume of 200 μl by retroorbital injection.
ELISpot.
Enzyme-linked immunosorbent spot (ELISpot) plates (Millipore) were coated with anti-IFN-γ or anti-IL-17 antibody (BD Biosciences) and incubated at 4°C overnight. Wild-type BALB/c or C57BL/6 splenocytes depleted of T cells (rabbit serum complement; Sigma) were used as stimulator cells and incubated with heat-killed S. aureus (HTKL-SA) overnight at 37°C with 5% CO2. The plates were blocked with proliferation medium for 1 h. Responder splenocytes were harvested from BALB/c and C57BL/6 mice and plated at 5 × 105/well with 2.5 × 105 stimulators. The plates were then incubated at 37°C, 5% CO2, for 24 h. Biotinylated anti-IFN-γ and anti-IL17A detection antibodies (BD Biosciences) and horseradish peroxidase (HRP)-conjugated antibiotin were used as the primary and secondary antibodies, respectively (eBioscience). The plates were washed, and substrate solution was added (BD Biosciences); the reaction was terminated, and the plates were read using an ImmunoSpot series 1 analyzer (Cellular Technology).
Intracellular cytokine staining.
HTKL-SA-specific stimulators and responders were prepared as described for the ELISPOT assays and plated in 96-well round-bottom plates with lids at concentrations of 5 × 105 and 1 × 106 per well, respectively, and were incubated at 37°C, 5% CO2, for 18 h, followed by incubation with brefeldin A for 6 h. Cells were collected from plates and transferred to FACS tubes for antibody viability staining. After viability staining (LIVE/DEAD fixable violet dead cell stain, Invitrogen) and blocking nonspecific binding with anti-FcγR (2.4G2; University of Chicago Immunology Facility Core), the following antibodies were used to stain the cell surface at a concentration of 1 μg antibody per 106 cells for flow cytometry analysis: anti-CD90.2–phycoerythrin (PE)–Cy7 (clone 53-2.1; eBioscience), anti-CD4–FITC (clone RM4-5; eBioscience), anti-CD8–PerCP (clone 53-6.7; BD Biosciences), and anti-CD44–allophycocyanin (APC)–Cy7 (clone IM7; BD Biosciences). The cells were washed, and BD fix/perm solution (250 μl per 106 cells) was added. The cells were stained with anti-IFN-γ antibody (eBioscience) and anti-IL-17A antibody (eBioscience) (1 μl per 106 cells), washed, and analyzed using a BD LSR II flow cytometer (BD Biosciences).
Cytokine neutralization.
Neutralizing antibodies against IL-17A (mouse IgG1, clone 17F3) and IFN-γ (rat IgG1, clone XMG1.2), as well as the isotype controls mouse IgG1 (clone MOPC-21) and rat IgG1 (clone HRPN), were purchased from BioXcell. For in vivo neutralization, mice received antibody (500 μg) via intraperitoneal injection 1 day prior to infection with S. aureus.
Data analysis.
Data were compared using Student's t test or one-way analysis of variance (ANOVA) with the Tukey posttest, where appropriate. Differences were considered significant when P values were <0.05. All data were analyzed using GraphPad Prism software.
RESULTS
S. aureus SSTI strongly protected against secondary dermonecrosis in BALB/c mice but much less so in C57BL/6 mice.
We modified an established model of S. aureus SSTI with dermonecrosis (16) to assess the efficacy of a primary S. aureus SSTI in protecting against reinfection in two commonly used strains of mice, C57BL/6 and BALB/c. All BALB/c and C57BL/6 mice developed dermonecrotic lesions after primary infection with S. aureus, and there were no significant differences in the size of skin lesions observed in the mouse strains (data not shown). In contrast, while some BALB/c mice developed small raised abscesses within 3 to 5 days after the secondary S. aureus SSTI (8 weeks after the primary infection), none developed a dermonecrotic lesion, demonstrating that S. aureus SSTI induced protective immunity in BALB/c mice (Fig. 1A to C). In support of this, there were fewer bacteria recovered from the lesions of mice 3 days after secondary infection than after primary infection (P = 0.02) (Fig. 1D). There were also lower levels of myeloperoxidase (MPO), a marker of neutrophil and macrophage activity (P < 0.01) (Fig. 1G) and of the inflammatory chemokine CXCL-1 (P < 0.001) (Fig. 1E) but no difference in IL-17A (P = 0.4) (Fig. 1F) in the lesions of mice 3 days after secondary infection. The different nature of the skin lesions was evident with histologic analysis; skin lesions of BALB/c mice 3 days after primary infection were characterized by dermonecrosis (Fig. 1H). In contrast, there was evidence of subcutaneous abscess formation with intact epidermis after secondary infection (Fig. 1I). Therefore, the absence of dermonecrosis observed after secondary infection of BALB/c mice was associated with fewer bacteria recovered from the lesions and a less vigorous local inflammatory response.
FIG 1.

S. aureus SSTI strongly protected against recurrent infection in BALB/c mice. (A) BALB/c mice had dermonecrotic lesions after primary S. aureus SSTI but not after secondary infection (n = 8 to 10 mice/group). (B and C) Photograph of representative lesions after primary (B) or secondary (C) infection. There were fewer bacteria recovered from the lesions (D) and lower levels of CXCL-1 (E) and myeloperoxidase (MPO) (G) but no difference in IL-17A (F) 3 days after secondary infection compared with findings after primary infection. (H and I) Hemotoxylin-and-eosin-stained skin lesions from mice 3 days after primary (H) or secondary (I) SSTI. Data are presented as means ± SEM. *, P < 0.05 by Student's t test. Bars in photographs represent 1 cm. The results of one representative experiment are presented; each was repeated at least twice.
In contrast, all C57BL/6 mice developed dermonecrotic lesions after secondary infection. Although the skin lesions observed were slightly smaller than those of C57BL/6 mice after primary infection (P < 0.05 on days 3 and 6; P > 0.1 thereafter) (Fig. 2A to C), there were no significant differences in numbers of bacterial CFU or levels of CXCL-1, IL-17A, or MPO recovered from the skin lesions of C57BL/6 mice 3 days after primary or secondary infection (P = 0.2) (Fig. 2D to G). Collectively, we found that S. aureus SSTI induced vastly superior protective immunity against secondary SSTI in BALB/c mice compared with C57BL/6 mice.
FIG 2.

S. aureus SSTI protected minimally against recurrent infection in C57BL/6 mice. (A) C57BL/6 mice had smaller lesions 3 and 6 days after secondary SSTI than C57BL/6 mice had after primary infection (n = 8 to 10 mice/group). (B and C) Photographs of representative lesions after primary (B) or secondary (C) infection. There were no significant differences in the numbers of bacteria recovered from the lesions of C57BL/6 mice (D) or levels of CXCL-1 (E), IL-17 (F), or myeloperoxidase (MPO) (G) after secondary SSTI, compared with results after primary infection. Data are presented as means ± SEM. *, P < 0.05 by Student's t test. Bars in photographs represent 1 cm. For each experiment, the results of one representative experiment are presented; each was repeated at least twice.
Adaptive immunity was necessary for protection against secondary skin infection.
To determine if T cell responses contributed to protective immunity against secondary SSTI, BALB/c mice were treated with anti-CD4 neutralizing antibody prior to primary infection, resulting in depletion of CD4+ T cells from the spleen, blood, and draining lymph nodes for up to 8 weeks (see Fig. S1A in the supplemental material). This depletion of CD4+ T cells had no effect on primary SSTI (data not shown) but resulted in abrogation of protection against secondary SSTI; there was no significant difference in the size of skin lesions between CD4-depleted mice after secondary SSTI and control mice after primary infection (P > 0.2) (Fig. 3A). Because antibody responses require CD4+ T cells, we quantified antibodies against alpha-hemolysin (Hla) and iron-regulated surface determinant B (IsdB), two antigens reported to be important in the immune response against S. aureus and included in prospective vaccines (10, 17). While anti-Hla and anti-IsdB IgG were present in control mice after primary infection, both were undetectable in CD4-depleted mice (see Fig. S1B). We also found that the modest protection observed in C57BL/6 mice was not present in TCR βδ−/− mice (P = 0.8, Fig. 3B). Therefore, protective immunity in both mouse backgrounds required T cells. However, because antibody responses were dependent on CD4+ T cells, the relative contributions of antibody and T cells to protective immunity were examined.
FIG 3.
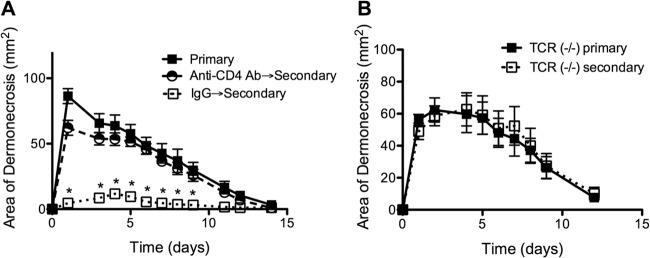
Adaptive immunity was necessary for protection against secondary S. aureus SSTI. (A) CD4+ T cell depletion prior to primary infection of BALB/c mice abrogated protection after secondary SSTI compared with treatment with an isotype control antibody (IgG) (n = 8 mice/group). (B) There was no difference in lesion size between TCR βδ−/− mice after primary or secondary infection (C57BL/6 background; n = 8 mice/group). Data are presented as means ± SEM. *, P < 0.05 by one-way ANOVA with Tukey's posttest. For each experiment, the results of one representative experiment are presented; each was repeated once.
Protective antibody was induced in BALB/c mice but not in C57BL/6 mice.
To determine if differences in the antibody response were sufficient to explain the superior protection observed in BALB/c mice, naive and immune BALB/c serum was adoptively transferred to naive BALB/c mice prior to primary infection with S. aureus. Mice that received immune serum developed smaller dermonecrotic lesions than those that received PBS or naive serum prior to infection (P < 0.01) (Fig. 4A). Adoptive transfer of antibody purified from immune BALB/c serum but not antibody-depleted immune serum conferred protection on naive mice, confirming that antibody mediated the protective effects of immune serum (P < 0.01) (Fig. 4B). In contrast to BALB/c mice, adoptive transfer of immune C57BL/6 serum to naive C57BL/6 mice had no effect on lesion size (P > 0.3) (Fig. 4C). This observation suggested that protective antibody was not elicited in C57BL/6 mice after S. aureus SSTI or that C57BL/6 mice were unable to respond effectively to passively administered antibodies.
FIG 4.

S. aureus SSTI elicited protective antibody in BALB/c mice but not in C57BL/6 mice. (A) Adoptive transfer of immune BALB/c serum into naive BALB/c mice prior to primary infection with S. aureus resulted in smaller lesions than were seen with naive serum or PBS. (B) Adoptive transfer of antibody purified from immune BALB/c serum but not antibody-depleted serum prior to primary infection resulted in significantly smaller lesions than were seen with BALB/c mice that received PBS. (C) Adoptive transfer of immune C57BL/6 serum into C57BL/6 mice prior to primary infection with S. aureus did not result in significantly smaller lesions than were seen with mice that received naive serum or PBS. (D) There were no significant differences in anti-IsdB IgG levels between C57BL/6 and BALB/c mice 8 weeks after primary (“1”) or secondary (“2”) infection. (E) There were no significant differences in anti-IsdB IgG1, IgG2a, IgG2b, or IgG3 after secondary infection between the groups. (F) Anti-Hla IgG levels were higher for BALB/c mice than for C57BL/6 mice after secondary but not primary infection. (G) There were higher levels of anti-Hla IgG1, IgG2a, and IgG3 but not IgG2b after secondary infection in BALB/c mice. (H) Adoptive transfer of immune BALB/c serum into C57BL/6 mice prior to primary infection resulted in smaller lesions than were seen with mice that received naive BALB/c serum or PBS. (I) In contrast, adoptive transfer of immune C57BL/6 serum into BALB/c mice prior to primary infection did not result in smaller lesions. Each experiment used 5 to 10 mice/group. Data are presented as means ± SEM. *, P < 0.05; **, P < 0.01 by one-way ANOVA with Tukey's posttest. The results of one representative experiment are presented; each was repeated at least twice.
To distinguish between these possibilities, we compared antibody responses against IsdB and Hla in BALB/c and C57BL/6 mice 8 weeks after primary and secondary infection. Antibody levels against both antigens increased after the primary infection and increased further after secondary infection in both mouse backgrounds (Fig. 4D and F). There were no significant differences in anti-IsdB total IgG levels between BALB/c and C57BL/6 mice after primary infection or in levels of anti-IsdB total IgG, IgG1, IgG2a, IgG2b, or IgG3 after secondary infection (Fig. 4D and E). Similarly, total anti-Hla IgG levels did not differ between the two backgrounds after primary infection (P = 0.4) (Fig. 4F). In contrast, there were higher levels of anti-Hla total IgG, IgG1, IgG2a, and IgG3 (P < 0.01) but not IgG2b after secondary infection of BALB/c mice, compared with results for C57BL/6 mice (Fig. 4F and G). These data suggested that a selective difference in the antibody response in BALB/c mice from that in C57BL/6 mice might be the basis for differential antibody-mediated protection. To test this possibility, crossover serum transfer was performed, in which immune BALB/c serum was transferred into C57BL/6 recipients or vice versa. Immune BALB/c sera conferred protection to naive C57BL/6 recipients (P < 0.01) (Fig. 4H), while immune C57BL/6 sera did not confer protection to naive BALB/c recipients (P > 0.3) (Fig. 4I). Collectively, these results suggested that the superior protection observed in the BALB/c mice was mediated, at least partly, by the quality of the protective antibody response and not by differences in antibody-mediated effector mechanisms that confer protection.
Protective T cell responses were induced in BALB/c mice but not in C57BL/6 mice.
To test whether there also was a role for B cell/antibody-independent protective responses, we infected B cell-deficient (μMT) BALB/c mice. μMT mice had smaller lesions after secondary infection than after primary infection (P < 0.05) (Fig. 5A). This confirmed that some protection occurred even in the absence of B cells and protective antibodies, and we hypothesized that this protection was mediated by T cells.
FIG 5.

T lymphocytes mediated B cell/antibody-independent protective immunity in BALB/c mice but not in C57BL/6 mice. (A) Secondary infection of B cell-deficient BALB/c mice resulted in smaller skin lesions than those seen after primary SSTI (n = 8 mice/group). (B) Adoptive transfer of immune BALB/c T cells into BALB/c mice prior to primary infection resulted in smaller lesions than were seen with mice that received naive T cells (n = 8 mice/group). (C) In contrast, adoptive transfer of immune C57BL/6 T cells into C57BL/6 mice prior to primary SSTI resulted in minimal protection; lesions were significantly smaller 2 and 4 days after infection but not thereafter (n = 8 mice/group). (D) There were no differences in the size of skin lesions of CD4-deficient mice after primary or secondary SSTI (n = 8 mice/group). (E) There was a trend toward smaller lesions in CD8-deficient mice after secondary SSTI than after primary SSTI. Data are presented as means ± SEM. *, P < 0.05 by Student's t test. The results of one representative experiment are presented; each was repeated at least once.
The importance of T cells in protection was tested by the adoptive transfer of T cells from naive or immune BALB/c mice into naive BALB/c mice prior to primary infection. Indeed, BALB/c mice that received immune BALB/c T cells had smaller lesions than mice that received T cells from naive mice (P < 0.01) (Fig. 5B). In contrast, adoptive transfer of immune C57BL/6 T cells into naive C57BL/6 mice was minimally protective; skin lesions were smaller early after infection (P < 0.05 at 2 and 4 days), but these differences did not persist (P > 0.1) (Fig. 5C). There were higher numbers of IFN-γ and IL-17A staining cells among the transferred immune BALB/c T cells than among nonprotective naive T cells, but there were no differences in the numbers of CD8+ T cells, suggesting that CD4+ T cells mediated protective immunity (see Fig. S2A in the supplemental material). In further support of a role for CD4+ T cells, adoptive transfer of immune CD8+ T cells had no significant effects on lesion size (P > 0.1) (see Fig. S2B and C). Also, CD4−/− mice were not protected against secondary SSTI, while there was modest protection observed in CD8−/− mice (Fig. 5D and E). Thus, CD4+ T cells contributed to the protective responses observed in BALB/c mice, and the protection conferred by immune T cells from BALB/c mice was greater than that of immune T cells from C57BL/6 mice.
To define differences in T cell responses in the two mouse backgrounds, IFN-γ and IL-17A ELISpot was performed with splenocytes isolated from C57BL/6 and BALB/c mice 8 weeks after primary and secondary SSTI. S. aureus-specific IFN-γ and IL-17A responses for both mouse backgrounds increased after primary and secondary infection compared with those for naive mice (P < 0.01) (Fig. 6A and B). The IFN-γ response was higher in C57BL/6 mice after primary infection than in BALB/c mice (P < 0.01) (Fig. 6A), whereas the IL-17A response was higher in BALB/c mice (P < 0.001) (Fig. 6B). The IFN-γ/IL-17A ratio was therefore higher in C57BL/6 mice than in BALB/c mice (P < 0.05) (Fig. 6C). Similar responses were observed after secondary infection, with a stronger IFN-γ response in C57BL/6 mice (P < 0.001) and a higher IFN-γ/IL-17A ratio in C57BL/6 mice (P < 0.001) (see Fig. S3A to C in the supplemental material).
FIG 6.
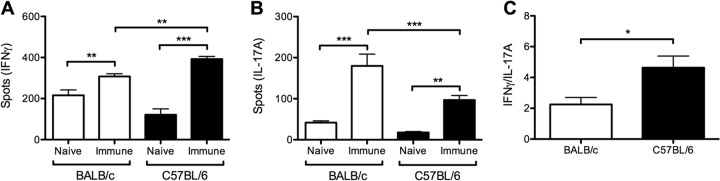
Protective immunity in BALB/c mice was associated with a Th17/IL-17A-polarized response, compared with a Th1/IFN-γ-polarized response in C57BL/6 mice. S. aureus-specific T cell responses were assessed 8 weeks after primary infection with S. aureus by ELISpot (n = 3 to 5 mice/group). IFN-γ (A) or IL-17A (B) produced by S. aureus-stimulated splenocytes was increased in C57BL/6 and BALB/c mice after primary infection compared with results for naive controls. (A) IFN-γ responses were higher for C57BL/6 mice than for BALB/c mice. (B) In contrast, IL-17A responses were higher for BALB/c mice than for C57BL/6 mice. (C) IFN-γ/IL-17A ratios were higher for C57BL/6 mice than for BALB/c mice. Data are presented as means ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; by one-way ANOVA with Tukey's posttest. The results of one representative experiment are presented; each was repeated at least once.
To confirm that IFN-γ and IL-17A were produced by CD4+ T cells, intracellular cytokine staining was performed after secondary infection. Consistent with the ELISpot assay, we observed a higher percentage of CD4+ CD44+ IFN-γ+ cells in C57BL/6 mice (P < 0.001) (see Fig. S3D in the supplemental material), a trend toward a higher percentage of CD4+ CD44+ IL-17A+ cells in BALB/c mice (P = 0.08) (see Fig. S3E), and a higher IFN-γ/IL-17A ratio in C57BL/6 mice (P < 0.05) (see Fig. S3F). Therefore, the superior protection in BALB/c mice, compared with that in C57BL/6 mice, was associated with a higher ratio of IL-17A-/IFN-γ-producing CD4+ T cells after infection with S. aureus. Based on these observations, we hypothesized that IL-17A production by T cells was protective, whereas IFN-γ production was not.
IL-17A-polarized responses contributed to protection in BALB/c mice, whereas IFN-γ-polarized T cell responses inhibited protection in C57BL/6 mice.
We tested whether IL-17A was necessary for protection against secondary SSTI in BALB/c mice by treatment with neutralizing antibody against IL-17A prior to reinfection, with the expectation that this would abrogate protection. However, neutralization of IL-17A had no effect on protection in BALB/c mice (data not shown), which could be explained by the contribution of antibody-mediated protective immunity. Therefore, we performed the same experiment with BALB/c μMT mice. Consistent with a role for IL-17A in protection, neutralization of IL-17A prior to secondary SSTI in μMT mice abrogated protection (P < 0.01) (Fig. 7A). Furthermore, the protection mediated by adoptive transfer of immune BALB/c T cells into naive BALB/c mice was abolished by the administration of anti-IL-17A antibody at the time of T cell transfer (P < 0.05) (Fig. 7B). The importance of IL-17A was limited to adaptive immune responses, because treatment with anti-IL-17A antibody had no effect on lesion size in primary infection (P > 0.4) (see Fig. S4A in the supplemental material). These data collectively demonstrate that IL-17A mediated the protection conferred by transfer of immune T cells.
FIG 7.
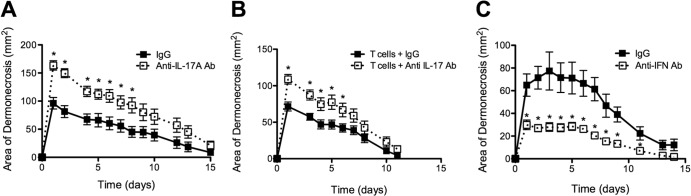
Th17/IL-17A mediated B cell/antibody-independent protection in BALB/c mice, whereas Th1/IFN-γ inhibited protection in C57BL/6 mice. (A) BALB/c B cell-deficient mice treated with IL-17A neutralizing antibody prior to secondary SSTI had larger lesions than mice treated with an isotype control antibody (IgG) (n = 9 to 12 mice/group). (B) Treatment with IL-17A neutralizing antibody after immune T cell transfer resulted in larger lesions than were seen with mice that received an isotype control antibody (n = 8 mice/group). (C) C57BL/6 mice treated with IFN-γ neutralizing antibody prior to secondary SSTI had smaller lesions than mice treated with an isotype control antibody (IgG) (n = 6 to 8 mice/group). Data are presented as means ± SEM. *, P < 0.05 by Student's t test. The results of one representative experiment are presented; each was repeated at least once.
Because the poorer protection in C57BL/6 mice was associated with an IFN-γ-polarized (versus IL-17A) response, we tested whether neutralization of IFN-γ prior to secondary SSTI in C57BL/6 mice would enhance protection and reduce lesion severity. Consistent with this hypothesis, treatment of C57BL/6 mice with neutralizing anti-IFN-γ antibody prior to secondary infection resulted in smaller lesions than those in control mice (P < 0.01) (Fig. 7C). As we observed with IL-17A, the effects of IFN-γ neutralization were specific for memory responses, because treatment with anti-IFN-γ antibody prior to primary SSTI had no effect on lesion size (P > 0.4) (see Fig. S4B in the supplemental material). Taken together, these findings demonstrate that the IFN-γ-skewed responses in C57BL/6 mice, in combination with a poor antibody response, resulted in poor protection against secondary SSTI.
DISCUSSION
We have described herein a novel mouse model in which S. aureus SSTI successfully elicited protective immunity against dermonecrosis during secondary S. aureus SSTI. Furthermore, this protective immunity was mediated by antibody and IL-17A and prevented by IFN-γ. These observations demonstrate that, contrary to popular belief, S. aureus SSTIs are in fact capable of eliciting memory responses that protect against subsequent infection, but the development of protective immunity was critically dependent on the genetic background of the host. Although several previous studies have suggested that S. aureus infections elicit protective immunity, there are important differences between those and our findings. For example, Agarwal described the protective effects of S. aureus SSTI against recurrent infection in Wright-Fleming Institute mice, but the model required the coadministerion of cotton dust to enhance lesion formation (18). Others have found that mutant S. aureus strains but not wild-type isolates elicit adaptive immunity against S. aureus sepsis in single strains of mice (19–22). Therefore, our model, in which a natural SSTI with a wild-type S. aureus clinical isolate elicited protective immunity in BALB/c but not C57BL/6 mice, afforded us a unique opportunity to dissect the determinants of protection against recurrent infection.
The vastly superior protection against dermonecrosis observed in BALB/c mice after secondary SSTI, compared with results for C57BL/6 mice, was characterized by the absence of dermonecrotic lesions, fewer bacteria recovered from the skin lesions, and a less vigorous local inflammatory response. It is noteworthy that despite the absence of dermonecrosis, the decrease in bacterial recovery was less than 1 log, suggesting that limiting local inflammation may also be important in determining the severity of infection. Protection from dermonecrosis was abrogated in BALB/c mice when CD4+ T cells were depleted prior to primary infection and in C57BL/6 TCR βδ−/− mice, demonstrating that a CD4+ T cell-dependent adaptive immune response mediated protection. Humoral immunity, dependent on CD4+ T cells, was critical to the development of protective immunity in BALB/c mice, because adoptive transfer of immune serum into naive mice prior to primary infection resulted in smaller lesions. We confirmed that antibody mediated protective immunity in BALB/c mice by demonstrating that adoptive transfer of antibody purified from immune serum but not antibody-depleted immune serum conferred protection.
Whereas BALB/c serum was protective against dermonecrosis when transferred to either C57BL/6 or BALB/c mice, C57BL/6 serum did not confer protection to either mouse strain. These observations suggest important differences in the serologic response in these two strains of mice but not in their ability to respond to protective antibodies by limiting the severity of S. aureus SSTI. In support of this hypothesis, we observed higher levels of anti-Hla total IgG, IgG1, IgG2a, and IgG3 but not IgG2b after secondary infection of BALB/c mice than were seen for C57BL/6 mice. Anti-Hla antibodies have been reported to be protective (17, 23), but whether differences in the anti-Hla IgG response are the basis for the difference in protection or hint at a broader difference in the quality of antibody response between C57BL/6 and BALB/c mice is under investigation.
The findings that S. aureus SSTI elicited antibody responses in both C57BL/6 and BALB/c mice, but only BALB/c responses were protective, may have important implications in understanding the mechanisms of protective immunity against S. aureus. In particular, the observation that naturally occurring antibodies against many S. aureus antigens develop shortly after birth but have not been demonstrated to be protective led to the conclusion that antibodies are not protective (24, 25). Our data suggest an alternative hypothesis that these antibodies may resemble the polyclonal antibody response elicited by S. aureus SSTI in C57BL/6 mice, where there is no protection. Our data further suggest that S. aureus antigens specifically responsible for protection can be identified by comparing the specificity of the antibody response after S. aureus infection in BALB/c and C57BL/6 mice.
Recent findings support a role for specific T cells in defense against S. aureus infections, particularly SSTI, primarily by enhancing recruitment of phagocytes. For example, patients with the hyper-IgE syndrome, in whom Th17 pathways are defective, have increased risk of S. aureus skin and lung infection (13). These clinical observations are supported by experimental evidence; for example, IL-17-deficient mice develop spontaneous S. aureus mucocutaneous infections (15). IL-17 deficiency in mice also specifically inhibited skin and lung defenses against S. aureus by limiting the production of antimicrobial peptides and neutrophil-recruiting chemokines in keratinocytes and bronchial epithelial cells (26). Our data also support a role for T cells and IL-17 in protective immunity against recurrent S. aureus SSTI. B cell-deficient BALB/c μMT mice were protected against secondary SSTI, and adoptive transfer of immune BALB/c T cells conferred protection to naive mice. As we observed with antibody-mediated protection, S. aureus SSTI elicited protective T cell responses in BALB/c mice but not C57BL/6 mice. The S. aureus-specific T cell response was strongly skewed to Th1 in C57BL/6 mice, whereas the Th1 and Th17 responses in BALB/c mice were more balanced. Neutralization of IFN-γ improved protection in C57BL/6 mice, whereas neutralization of IL-17A abrogated protection in BALB/c mice. Therefore, our observations are consistent with recent reports that the Th17/IL-17 pathway is protective and that Th1/IFN-γ responses are deleterious (14, 27–31). However, our observations suggest that the ratio of the Th17 responses to Th1 responses rather than the magnitude of the Th17 response per se determines protection and that this ratio of T cell responses is determined by the genetic background of the infected host.
Our findings underscore the importance of considering the contribution of the genetic background to protective immunity. Although different mouse strains have been found to have variable resistance against S. aureus bacteremia (32, 33), we did not find any difference in lesion size after primary infection between C57BL/6 and BALB/c mice. In contrast, we found that adaptive T cell and antibody responses differed markedly between infected C57BL/6 and BALB/c mice. We speculate that these differences in mice may reflect the potential spectrum of immune responses against S. aureus infection in humans. Indeed, the genetic background of the host is increasingly appreciated as being important in defining the immune response. For example, the genomic responses in C57BL/6 mouse models of burn injury, endotoxemia, and trauma poorly reflected those observed in human disease (34), although it is possible that other mouse genetic backgrounds may have responses that are more similar to those of humans. Therefore, it is critical to assess immunity against S. aureus infection in multiple mouse backgrounds in order to fully appreciate the translational impact. An alternative approach would be to use outbred mice to more fully define the spectrum of immune responses that are elicited by S. aureus SSTI.
The failure of human vaccines against specific S. aureus antigens despite high vaccine-specific antibody titers among vaccine recipients has led to a reconsideration of vaccine design (reviewed in reference 8). Our findings support the idea of a multimechanistic (i.e., targeting both humoral and cell-mediated immunity) approach by demonstrating important roles for both antibody and T cells in protecting against secondary SSTI. This notion is further supported by experimental findings that both antibody (17, 19, 23) and T cell/Th17 (35–37) vaccination strategies have shown promise in preclinical studies.
In summary, we observed that primary S. aureus SSTI strongly protected against dermonecrosis during secondary SSTI in BALB/c mice, but protection was greatly inferior in C57BL/6 mice. This protection was mediated by antibody and also by B cell/antibody-independent mechanisms dependent on IL-17A and curtailed by IFN-γ. These findings advance our understanding of the fundamental mechanisms of adaptive immunity against S. aureus SSTI, provide insight into why protection is sometimes not observed despite evidence of a strong adaptive immune response, and suggest that vaccine strategies aimed at eliciting the appropriate T cell-mediated and humoral immunity should be prioritized.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Child Health and Human Development (Pediatric Critical Care Scholar Development Program, HD047349, to C.P.M.), the National Institute of Allergy and Infectious Diseases (AI076596, to C.P.M.; AI072630, to M.D. and A.S.C.; AI97113, to A.S.C.; and AI040481, to R.S.D.), the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR059414, to R.S.D. and M.-L.A.), and the National Heart, Lung, and Blood Institute (T32 HL07605, to M.D.).
R.S.D. reports serving as a paid consultant for Pfizer and receiving a research grant from Pfizer. C.P.M., M.D., F.Z., M.-L.A., and A.S.C. have no conflicts of interest to declare.
Footnotes
Published ahead of print 10 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01491-14.
REFERENCES
- 1.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. 10.1001/jama.298.15.1763 [DOI] [PubMed] [Google Scholar]
- 2.David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23:616–687. 10.1128/CMR.00081-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herold BC, Immergluck LC, Maranan MC, Lauderdale DS, Gaskin RE, Boyle-Vavra S, Leitch CD, Daum RS. 1998. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 279:593–598. 10.1001/jama.279.8.593 [DOI] [PubMed] [Google Scholar]
- 4.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. 2006. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 355:666–674. 10.1056/NEJMoa055356 [DOI] [PubMed] [Google Scholar]
- 5.McCaig LF, McDonald LC, Mandal S, Jernigan DB. 2006. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg. Infect. Dis. 12:1715–1723. 10.3201/eid1211.060190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hersh AL, Chambers HF, Maselli JH, Gonzales R. 2008. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch. Intern. Med. 168:1585–1591. 10.1001/archinte.168.14.1585 [DOI] [PubMed] [Google Scholar]
- 7.Miller LS, Cho JS. 2011. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 11:505–518. 10.1038/nri3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daum RS, Spellberg B. 2012. Progress toward a Staphylococcus aureus vaccine. Clin. Infect. Dis. 54:560–567. 10.1093/cid/cir828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinefield H, Black S, Fattom A, Horwith G, Rasgon S, Ordonez J, Yeoh H, Law D, Robbins JB, Schneerson R, Muenz L, Fuller S, Johnson J, Fireman B, Alcorn H, Naso R. 2002. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N. Engl. J. Med. 346:491–496. 10.1056/NEJMoa011297 [DOI] [PubMed] [Google Scholar]
- 10.Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, Corey GR, Carmeli Y, Betts R, Hartzel JS, Chan IS, McNeely TB, Kartsonis NA, Guris D, Onorato MT, Smugar SS, DiNubile MJ, Sobanjo-ter Meulen A. 2013. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA 309:1368–1378. 10.1001/jama.2013.3010 [DOI] [PubMed] [Google Scholar]
- 11.Matalon AM, Buerkert J, Block G, Hohenboken M, Fattom A, Horwirth G, Rasmussen H, Damasco S, Bourtriau D. 2012. Efficacy profile of a bivalent Staphylococcus aureus glycoconjugate investigational vaccine in adults on hemodialysis: phase III randomized study, p 109–114 Fifteenth International Symposium on Staphylococcal Infections, Lyon, France [Google Scholar]
- 12.Shadyab AH, Crum-Cianflone NF. 2012. Methicillin-resistant Staphylococcus aureus (MRSA) infections among HIV-infected persons in the era of highly active antiretroviral therapy: a review of the literature. HIV Med. 13:319–332. 10.1111/j.1468-1293.2011.00978.x [DOI] [PubMed] [Google Scholar]
- 13.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O'Shea J, Holland SM, Paul WE, Douek DC. 2008. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773–776. 10.1038/nature06764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, Cheng G, Modlin RL, Miller LS. 2010. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest. 120:1762–1773. 10.1172/JCI40891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. 2009. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30:108–119. 10.1016/j.immuni.2008.11.009 [DOI] [PubMed] [Google Scholar]
- 16.Montgomery CP, Boyle-Vavra S, Daum RS. 2009. The arginine catabolic mobile element is not associated with enhanced virulence in experimental invasive disease caused by the community-associated methicillin-resistant Staphylococcus aureus USA300 genetic background. Infect. Immun. 77:2650–2656. 10.1128/IAI.00256-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bubeck Wardenburg J, Schneewind O. 2008. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205:287–294. 10.1084/jem.20072208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal DS. 1967. Subcutaneous staphylococcal infection in mice. I. The role of cotton-dust in enhancing infection. Br. J. Exp. Pathol. 48:436–449 [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HK, Cheng AG, Kim HY, Missiakas DM, Schneewind O. 2010. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J. Exp. Med. 207:1863–1870. 10.1084/jem.20092514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HK, Kim HY, Schneewind O, Missiakas D. 2011. Identifying protective antigens of Staphylococcus aureus, a pathogen that suppresses host immune responses. FASEB J. 25:3605–3612. 10.1096/fj.11-187963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burnside K, Lembo A, Harrell MI, Klein JA, Lopez-Guisa J, Siegesmund AM, Torgerson TR, Oukka M, Molina DM, Rajagopal L. 2012. Vaccination with a UV-irradiated genetically attenuated mutant of Staphylococcus aureus provides protection against subsequent systemic infection. J. Infect. Dis. 206:1734–1744. 10.1093/infdis/jis579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmaler M, Jann NJ, Ferracin F, Landmann R. 2011. T and B cells are not required for clearing Staphylococcus aureus in systemic infection despite a strong TLR2-MyD88-dependent T cell activation. J. Immunol. 186:443–452. 10.4049/jimmunol.1001407 [DOI] [PubMed] [Google Scholar]
- 23.Kennedy AD, Wardenburg JB, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, Deleo FR. 2010. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202:1050–1058. 10.1086/656043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verkaik NJ, Boelens HA, de Vogel CP, Tavakol M, Bode LG, Verbrugh HA, van Belkum A, van Wamel WJ. 2010. Heterogeneity of the humoral immune response following Staphylococcus aureus bacteremia. Eur. J. Clin. Microbiol. Infect. Dis. 29:509–518. 10.1007/s10096-010-0888-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verkaik NJ, Lebon A, de Vogel CP, Hooijkaas H, Verbrugh HA, Jaddoe VW, Hofman A, Moll HA, van Belkum A, van Wamel WJ. 2010. Induction of antibodies by Staphylococcus aureus nasal colonization in young children. Clin. Microbiol. Infect. 16:1312–1317. 10.1111/j.1469-0691.2009.03073.x [DOI] [PubMed] [Google Scholar]
- 26.Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, Yamada M, Kawamura N, Ariga T, Tsuge I, Karasuyama H. 2009. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J. Exp. Med. 206:1291–1301. 10.1084/jem.20082767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLoughlin RM, Solinga RM, Rich J, Zaleski KJ, Cocchiaro JL, Risley A, Tzianabos AO, Lee JC. 2006. CD4+ T cells and CXC chemokines modulate the pathogenesis of Staphylococcus aureus wound infections. Proc. Natl. Acad. Sci. U. S. A. 103:10408–10413. 10.1073/pnas.0508961103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLoughlin RM, Lee JC, Kasper DL, Tzianabos AO. 2008. IFN-gamma regulated chemokine production determines the outcome of Staphylococcus aureus infection. J. Immunol. 181:1323–1332 [DOI] [PubMed] [Google Scholar]
- 29.Weidenmaier C, McLoughlin RM, Lee JC. 2010. The zwitterionic cell wall teichoic acid of Staphylococcus aureus provokes skin abscesses in mice by a novel CD4+ T-cell-dependent mechanism. PLoS One 5:e13227. 10.1371/journal.pone.0013227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satorres SE, Alcaraz LE, Cargnelutti E, Di Genaro MS. 2009. IFN-gamma plays a detrimental role in murine defense against nasal colonization of Staphylococcus aureus. Immunol. Lett. 123:185–188. 10.1016/j.imlet.2009.03.003 [DOI] [PubMed] [Google Scholar]
- 31.Parker D, Prince A. 2012. Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J. Immunol. 189:4040–4046. 10.4049/jimmunol.1201055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahn SH, Deshmukh H, Johnson N, Cowell LG, Rude TH, Scott WK, Nelson CL, Zaas AK, Marchuk DA, Keum S, Lamlertthon S, Sharma-Kuinkel BK, Sempowski GD, Fowler VG., Jr 2010. Two genes on A/J. chromosome 18 are associated with susceptibility to Staphylococcus aureus infection by combined microarray and QTL analyses. PLoS Pathog. 6:e1001088. 10.1371/journal.ppat.1001088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Kockritz-Blickwede M, Rohde M, Oehmcke S, Miller LS, Cheung AL, Herwald H, Foster S, Medina E. 2008. Immunological mechanisms underlying the genetic predisposition to severe Staphylococcus aureus infection in the mouse model. Am. J. Pathol. 173:1657–1668. 10.2353/ajpath.2008.080337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. U. S. A. 110:3507–3512. 10.1073/pnas.1222878110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spellberg B, Ibrahim AS, Yeaman MR, Lin L, Fu Y, Avanesian V, Bayer AS, Filler SG, Lipke P, Otoo H, Edwards JE., Jr 2008. The antifungal vaccine derived from the recombinant N terminus of Als3p protects mice against the bacterium Staphylococcus aureus. Infect. Immun. 76:4574–4580. 10.1128/IAI.00700-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, Fu Y, French SW, Edwards JE, Jr, Spellberg B. 2009. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 5:e1000703. 10.1371/journal.ppat.1000703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joshi A, Pancari G, Cope L, Bowman EP, Cua D, Proctor RA, McNeely T. 2012. Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model. Hum. Vaccin. Immunother. 8:336–346. 10.4161/hv.18946 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.