

ABSTRACT
Persistent infection is a key feature of hepatitis C virus (HCV). However, chimpanzee infections with cell culture-derived viruses (JFH1 or related chimeric viruses that replicate efficiently in cell culture) have been limited to acute-transient infections with no pathogenicity. Here, we report persistent infection with chronic hepatitis in a chimpanzee challenged with cell culture-derived genotype 1a virus (H77S.2) containing 6 cell culture-adaptive mutations. Following acute-transient infection with a chimeric H77/JFH1 virus (HJ3-5), intravenous (i.v.) challenge with 106 FFU H77S.2 virus resulted in immediate seroconversion and, following an unusual 4- to 6-week delay, persistent viremia accompanied by alanine aminotransferase (ALT) elevation, intrahepatic innate immune responses, and diffuse hepatopathy. This first persistent infection with cell culture-produced HCV provided a unique opportunity to assess evolution of cell culture-adapted virus in vivo. Synonymous and nonsynonymous nucleotide substitution rates were greatest during the first 8 weeks of infection. Of 6 cell culture-adaptive mutations in H77S.2, Q1067R (NS3) had reverted to Q1067 and S2204I (NS5A) was replaced by T2204 within 8 weeks of infection. By 62 weeks, 4 of 6 mutations had reverted to the wild-type sequence, and all reverted to the wild-type sequence by 194 weeks. The data suggest H77S.2 virus has greater potential for persistence and pathogenicity than JFH1 and demonstrate both the capacity of a nonfit virus to persist for weeks in the liver in the absence of detectable viremia as well as strong selective pressure against cell culture-adaptive mutations in vivo.
IMPORTANCE This study shows that mutations promoting the production of infectious genotype 1a HCV in cell culture have the opposite effect and attenuate replication in the liver of the only fully permissive animal species other than humans. It provides the only example to date of persistent infection in a chimpanzee challenged with cell culture-produced virus and provides novel insight into the forces shaping molecular evolution of that virus during 5 years of persistent infection. It demonstrates that a poorly fit virus can replicate for weeks within the liver in the absence of detectable viremia, an observation that expands current concepts of HCV pathogenesis and that is relevant to relapses observed with direct-acting antiviral therapies.
INTRODUCTION
Persistent infections with human hepaciviruses are associated with chronic hepatic inflammation, progressive liver fibrosis, cirrhosis, and hepatocellular carcinoma (1, 2). Worldwide, as many as 200 million persons may be chronically infected with the prototypical hepacivirus, hepatitis C virus (HCV). In the United States, approximately 3.2 million persons are infected (3). Mortality associated with HCV infections has increased significantly in recent years, and it has exceeded that due to human immunodeficiency virus (HIV)/AIDS in the United States for almost a decade (4). The high prevalence of HCV infection and strong association with liver disease has engendered an intense effort to develop specific antiviral therapeutics targeting the virus, and recent results from clinical trials suggest that many HCV infections can be completely cured with relatively brief therapy with all oral combinations of small-molecule, direct-acting antiviral agents (DAAs) (5, 6). However, reinfection with reestablishment of persistent infection is clearly possible, and efforts to develop an effective HCV vaccine have not met with similar success. Despite numerous studies of both innate and adaptive immune responses and multiple hypotheses (7, 8), there is no clear understanding of the primary mechanism underlying the unique capacity of HCV to persist in adult humans who are otherwise immunologically competent. In large part, these gaps in current understanding of hepacivirus biology can be attributed to the lack of a readily available, immunocompetent small-animal model for hepatitis C, compounded by limitations in the capacity of most human HCV strains to be propagated in cell culture.
HCV is a positive-strand RNA virus classified within the family Flaviviridae (genus Hepacivirus). It possesses a genome approximately 9.6 kb in length that encodes a total of 10 mature structural and nonstructural proteins. Only a small number of cloned HCV genomes (RNA transcribed from full-length cDNA molecular clones) replicate efficiently in cell culture, and most of these contain adaptive mutations that were selected in replicons under antibiotic pressure (9–11). JFH1, a genotype 2a genome cloned from the serum of a Japanese patient with fulminant hepatitis C, is relatively unique in that it replicates with high efficiency in Huh-7 cells without the need for adaptive mutations (12, 13). HCV-N, a genotype 1b genome cloned from a Japanese patient with chronic hepatitis C, also replicates efficiently without adaptation to cell culture (14, 15). However, it has an unusual 4-amino-acid insertion in NS5A in proximity to cell culture-adaptive mutations documented in other cloned genomes. JFH1 and some chimeric genomes encoding JFH1 nonstructural proteins are also unique, in that they produce substantial yields of infectious virus particles when transfected into Huh-7 cells (12). These particles (JFH1) are infectious for both naive Huh-7 cells as well as chimpanzees (Pan troglodytes), the only animal species other than humans that is fully permissive for HCV replication (12, 16). We have also documented the release of cell culture-infectious virus from Huh-7 cells transfected with a highly adapted genotype 1a viral genome containing 5 cell culture-adaptive mutations (H77S) (17). The titer of infectious virus released into supernatant fluids of H77S-transfected cells was low compared to that of JFH1, but with modification of the adaptive mutations and optimized cell culture conditions, it approaches or exceeds 104 focus-forming units (FFU) of virus per ml (H77S.2 and H77S.3) (18). More recently, another highly adapted genotype 1a genome, HCV-TNcc, has been shown to produce cell culture-infectious virus with an efficiency rivaling that of JFH1 (19). However, although JFH1 virus produced in cell culture has been shown to be infectious in chimpanzees, this has not been documented for either the H77S or HCV-TNcc virus.
Here, we report the serial challenge of a previously HCV-naive chimpanzee with virus released into supernatant fluids by cells transfected with H77S, an intergenotypic H77/JFH1 chimeric genome (HJ3-5) (20, 21), and H77S.2. As with all previously reported chimpanzee infections with cell culture-produced viruses containing the JFH1 replicase (Table 1), HJ3-5 infection was brief, self-limited, and without demonstrable pathogenic effect. In contrast, subsequent challenge of this animal with H77S.2 virus led to long-term, persistent infection with chronic hepatitis. We describe the evolution of the H77S.2 genome in this animal over a period of 5 years, during which both the level of HCV viremia and intrahepatic expression of interferon-stimulated genes (ISGs) increased substantially and in parallel with the reversion of all 6 H77S.2 cell culture-adaptive mutations to their original wild-type sequence. Thus, this study documents the induction of chronic hepatitis C in a chimpanzee following inoculation of a cell culture-derived virus. We also show that mutations that reduce replication fitness can prevent detectable viremia for weeks despite ongoing intrahepatic replication, a concept that is relevant to some antiviral resistance-associated mutations and relapses after apparently successful DAA therapy.
TABLE 1.
Chimpanzee infections with JFH1 replicase-containing HCV produced in cell culture
| Animal | Virus | Inoculum titer | Viremia time frame (wk) | Time of maximum viremia (wk) | Maximum viremia (GE/ml) | ALT elevation | Time of anti-HCV ELISA (wk) | Reference |
|---|---|---|---|---|---|---|---|---|
| x0483 | FL-J6/JFH1 | 106 TCID50 | 1–15 | 2 | ∼2 × 104 | No | 10 | 50 |
| x0495 | FL-J6/JFH1 | 106 TCID50 | 1–6, 15 | 2 | ∼7 × 104 | No | 13 | 50 |
| x0215 | JFH1 | 8.0 × 106 GE | 2–5 | 4 | 2 ×103 | No | No | 12 |
| 10274 | JFH1 | 9.6 × 106 GE | 2–8 | 4 | ∼1 ×103 | No | No | 16 |
| 4x0193 | HJ3-5 | 106 FFU | 1–3 | 1 | 1 ×104 | No | Noa | This report |
The increase in OD did not reach the threshold for a positive result.
MATERIALS AND METHODS
Viruses and plasmids.
The plasmid pH77S contains the full-length genotype 1a H77c virus sequence (22) with the addition of 5 cell culture-adaptive mutations and has been described previously (17). pH77S.2 is derived from it and includes a sixth cell culture-adaptive mutation, N476D (AAC→GAC). pH77S.3 is derived from pH77S.2 and was created by removal of the Q1067R adaptive mutation in NS3 (18). The chimeric infectious molecular clone, pHJ3-5, is comprised of the structural protein-coding region of H77c virus placed within the background of the genotype 2a JFH1 virus, with adaptive mutations in E1 (Y361H) and NS3 (Q1251L) (20). Viruses were produced by transfection of Huh-7 or Huh-7.5 (23) cells by electroporation of synthetic RNA transcribed from these plasmids, as described previously (17, 18).
Inoculum preparation.
Supernatant fluids from RNA-transfected cell cultures were passed through a 0.45-μm-pore-sized filter and then concentrated approximately 10-fold using a Centricon Plus-70 centrifugal filter unit with an Ultracel-PL membrane (100-kDa exclusion; UFC710008; Millipore). Bacterial sterility was confirmed prior to inoculation. The final cell culture-infectious virus titer was determined by an infectious fluorescent focus (FFU) assay (17).
Chimpanzee infections.
A single, adult, female, HCV- and human immunodeficiency virus-naive chimpanzee (4x0193) was challenged sequentially with 3 successive cell culture-produced virus preparations, each inoculated intravenously as described in Results. This animal expressed Patr class I A and B alleles A*0201, A*0301, B*0101, and B*1301, determined by sequence-based genotyping as described previously (24). The chimpanzee was housed and cared for at the Southwest National Primate Research Center (SNPRC) of the Texas Biomedical Research Institute in accordance with the Guide for the Care and Use of Laboratory Animals. The study protocol was approved by the Institutional Animal Care and Use Committee of the Texas Biomedical Research Institute (formerly the Southwest Foundation for Biomedical Research). The SNPRC is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International and operates in accordance with the NIH and U.S. Department of Agriculture guidelines and the Animal Welfare Act. All experimental manipulations of this animal were completed prior to the release of a report from the Institute of Medicine on the use of chimpanzees in biomedical research (25) and its acceptance by the National Institutes of Health on 15 December 2011.
HCV RNA detection.
HCV RNA levels were monitored in chimpanzee serum and liver biopsy specimens by real-time, quantitative reverse transcription-PCR (qRT-PCR) (TaqMan), as described previously (26). More sensitive, nonquantitative detection of HCV RNA was performed using a transcription-mediated amplification assay (TMA). The Versant HCV RNA qualitative TMA assay amplifies conserved regions within the 5′-untranslated region (UTR) of the hepatitis C viral genome and has a sensitivity of 7.5 IU/ml (Siemens Clinical Laboratory, Berkley CA).
Anti-HCV ELISA.
Serum samples were monitored for antibody response to HCV proteins by ELISA (enzyme-linked immunosorbent assay) using the Ortho 3.0 HCV test (Ortho Diagnostic Systems, Raritan, NJ).
HCV neutralization assays.
Neutralizing antibodies to genotype 1a H77 virus were quantified using an FFU reduction assay as described previously (27).
qRT-PCR quantitation of ISG transcripts.
Interferon and interferon-stimulated gene (ISG) transcript abundance in liver biopsy specimens was determined by qRT-PCR (TaqMan). Total RNA was prepared using RNA Bee extraction (AMS Biotechnology, Ltd.), a guanidinium HCl phenol-based extraction procedure, with isopropanol precipitation. Total RNA was quantified using a Nanodrop spectrophotometer, and RNA quality was verified by the ratio of the optical density at 260 nm (OD260) to that at 280 nm (i.e., the OD260/280 ratio) or by an Experion automated microfluidics system for analysis of RNA (Bio-Rad). The high homology existing between chimpanzee and human genomes (28) enabled use of human RT-PCR Assays-on-Demand primers (Applied Biosystems).
Immunohistochemistry.
Antigen retrieval and immunohistochemical staining of liver biopsy sections for ISG-15, activated caspase 3, and the proliferation marker Ki-67 were carried out as described previously (29).
Cytokine and chemokine analysis.
Monitoring of 16 cytokines and chemokines was performed by the SNPRC Immunology Core using Luminex100 with the xMAP multianalyte platform and a customized panel optimized for cross-reactivity to primate samples.
HCV RNA sequence determination.
HCV RNA was extracted from serum with the ViraRNA Minikit (Qiagen) and then reverse transcribed to cDNA with the SuperScript III first-strand synthesis system for RT-PCR kit (Invitrogen) and random hexamers by following the manufacturer's suggested protocol. Overlapping segments of the HCV genome were amplified by nested PCR, using 2 μl undiluted cDNA as the template for the first round of PCR in a 10-μl reaction mix and 1 μl undiluted first-round PCR product as the template for the second round of PCR in a 50-μl reaction mix and reagents provided with the PrimeSTAR HS (Premix) kit (TaKaRa) with a two-step PCR protocol (98°C for 10 s, 68°C for 2 min). PCR products were purified from a 1% agarose gel using a gel extraction kit (Qiagen) and sequenced directly with HCV-specific primers. Rates of nonsynonymous substitutions per nonsynonymous site (dN) and synonymous substitutions per synonymous site (dS) were calculated by the method of Nei and Gojobori (30) using the Codon Suite server (http://www.cbrg.ethz.ch/services/CodonSuite).
Reverse molecular genetics experiments.
Selected nucleotide substitutions identified in virus from serum collected 8 weeks after inoculation of H77S.2 were introduced into pH77S.2 using the QuikChange II XL site-directed mutagenesis kit (Stratagene). The correct insertion of the substitution and absence of other changes within the manipulated segments of the plasmid were verified by DNA sequencing. RNA was transcribed from the plasmid and electroporated into FT3-7 cells as described previously (20). Replication was assessed by real-time quantitative RT-PCR measurement of HCV RNA and FFU assay for release of cell culture-infectious virus (17).
Informatics analysis of amino acid substitutions in HCV nonstructural proteins.
Protein structures were retrieved from the RCSB Protein Databank (PDB) (http://www.rcsb.org/pdb/home/home.do): PDB entry 1CU1 (31) for the NS3 protease-helicase, PDB entries 3FQQ (32) and 1ZH1 (33) for NS5A domain I, and PDB entry 2XI2 (34) for the NS5B polymerase. ProMate, a structure-based prediction program running on the ProMate server (http://bioinfo.weizmann.ac.il/promate/) (35), was used to calculate the probability that protein residues altered by nonsynonymous nucleotide substitutions contribute to protein-protein interaction sites. Predictions were carried out with the standard configuration. The algorithm predicts unknown protein-protein interaction sites based on knowledge of the properties of known protein-binding sites, including physicochemical properties, bound versus unbound water molecules, and the B-factor reflecting the relative dynamic motion, all derived from a database of proteins with known protein-protein interactions. The PyMOL Molecular Graphics System (http://www.pymol.org; DeLano Scientific, San Carlos, CA, USA) with Color B script (http://pldserver1.biochem.queensu.ca/∼rlc/work/pymol/) was used to project ProMate output onto the surfaces of the NS3, NS5A, and NS5B structures, respectively. WHAT IF was used to compute the solvent accessibilities (in Å2) of residues in the structural models of NS3, NS5A domain I, and NS5B (described above). The solvent accessibilities of residues affected by nonsynonymous substitutions were then compared to those of all residues in each structural model. P values were calculated using the nonparametric Mann-Whitney U test.
Alb-uPa/SCID mouse studies.
Mice were transplanted as previously described with human hepatocytes purchased from Invitrogen (36, 37). Mice with human albumin levels greater than 2 mg/ml were infected with either 1.5 × 105 FFU of H77S.3 virus injected intraperitoneally or 240 μg of genomic RNA encoding H77c, H77S, H77S.2, H77S.2/I2204T, H77S.3, and H77S.3/I2204T viruses injected intrahepatically. Blood was drawn weekly, and serum HCV RNA levels were determined as described previously (37). All mice were housed and treated according the Canadian Council on Animal Care guidelines, with experimental approval from the University of Alberta Animal Welfare Committee.
RESULTS
H77S virus challenge in chimpanzee 4x0193.
The original cell culture-adapted genotype 1a H77S clone contains 5 cell culture-adaptive mutations located in NS3, NS4A, and NS5A (17). Cells transfected with synthetic H77S RNA release virus into supernatant culture fluid that has a buoyant density of ∼1.13 gm/cm3 and is capable of infecting naive Huh-7.5 cells (102 to 103 FFU/ml). To determine whether this virus was infectious in the chimpanzee, we concentrated H77S virus from supernatant fluids by centrifugal filtration. A sterile 10-ml suspension containing 104 FFU H77S virus was inoculated intravenously into a 25-year-old, previously HCV-naive female chimpanzee (4x0193). Subsequent monitoring of the animal over a period of 28 weeks revealed an isolated increase in serum alanine aminotransferase (ALT) from a baseline of 30 ± 5.9 U/liter (means ± standard deviations [SD]) to 88 U/liter 3 weeks postinoculation but no evidence of HCV viremia or anti-HCV seroconversion (ELISA or anti-HCV neutralization) (Fig. 1A). The results led us to conclude that the H77S inoculum did not contain sufficient chimpanzee-infectious virus to initiate an infection.
FIG 1.

Serial challenge of chimpanzee 4x0193 with cell culture-derived HCVs. (A) Virologic markers (serum and intrahepatic HCV RNA and ELISA detection of anti-HCV) and serum ALT activity over a 290-week period. Viruses were inoculated intravenously at the time shown from the nominal week 0 for each; intervening gaps in time are not to scale (see the text for details). Horizontal dotted lines indicate baseline ALT activity (means ± 2 SD). (B) Expanded view of the 40 weeks following inoculation of HJ3-5 virus (second infectious challenge, ∼106 FFU). The results of transcription-mediated amplification (TMA) assays for detection of HCV RNA in serum and FFU reduction assays for detection of neutralizing antibody (nAb) to HCV (H77 envelope) are shown at the top. (C) Similar expanded view of the 40 weeks following challenge with H77S.2 virus (third infectious challenge, ∼106 FFU). Note that the liver biopsy specimen taken at 2 weeks after H77S.2 inoculation contained low levels of HCV RNA.
HJ3-5 virus challenge.
To document the susceptibility of 4x0193 to HCV infection and also assess the infectivity of a chimeric H77/JFH1 virus produced in cell culture, the animal was inoculated intravenously with 106 FFU HJ3-5 virus 32 weeks after the initial H77S virus challenge. The HJ3-5 genome is comprised of H77S RNA encoding the core-to-NS2 segment of the polyprotein placed within the JFH1 background (21). It contains adaptive mutations in E1 (Y361H) and NS3 (Q1251L) that promote its replication and facilitate the production of infectious virus in cell culture at yields that are approximately 100-fold higher than those of the original JFH1 cDNA clone (20). The animal became viremic within 1 week of inoculation (1.1 ×104 GE/ml) and remained so for the ensuing 3 weeks (Fig. 1A and B). HJ3-5 virus was isolated from week 1 and week 3 postinoculation sera in Huh-7.5 cells, with discrete foci of replication detected by immunofluorescence for HCV core antigen within 4 days of inoculation with week 1 serum and 11 days with week 3 serum (data not shown). By 4 weeks after inoculation, viremia had declined to a level no longer detectable by conventional real-time qRT-PCR but was still evident in a sensitive, qualitative transcription-mediated amplification (TMA) assay (sensitivity, 7.5 IU/ml). Repeat serum TMA assays were negative 16 and 51 weeks after inoculation of HJ3-5 virus. Reactivity in the anti-HCV ELISA rose from a preinoculation baseline OD of 0.039 ± 0.019 (means ± SD) to a peak of 0.247 at week 16 and then declined to baseline, never reaching the threshold for seroconversion (Fig. 1B). Neutralizing antibodies were not detected by an FFU reduction assay at 6 and 34 weeks after challenge, despite the brief period of viremia. Maximum serum ALT was 43, just 2 standard deviations above baseline, at 3 weeks (Fig. 1B). Collectively, these results indicate that HJ3-5 virus produced in cell culture is infectious in the chimpanzee, but that it engenders little serological response. Like other cell culture-derived HCVs containing the JFH1 replicase that have been studied in chimpanzees (Table 1), HJ3-5 virus caused only an acute transient infection without persistence of virus or evidence of significant liver injury.
Viral RNA present in sera collected from 4x0193 at 1 and 3 weeks after inoculation of HJ3-5 virus was extracted, converted to cDNA, amplified by RT-PCR, and sequenced to confirm that the circulating virus was HJ3-5. Both cell culture-adaptive mutations were present in the week 1 virus, and the Q1251L mutation remained present at week 3. Sequence could not be obtained to confirm the continuing presence of Y361H in week 3 sera, but it was present in virus isolated in cell culture from this sample (data not shown). Thus, there appeared to be no selective pressure against these adaptive mutations in HJ3-5 during replication in the chimpanzee. Several new substitutions were identified in the NS2 and NS3 regions of the circulating virus, including nonsynonymous substitutions in NS2 and the protease domain of NS3 (Table 2).
TABLE 2.
Changes in HJ3-5 virus sequence during acute transient infection in chimpanzee 4x0193e
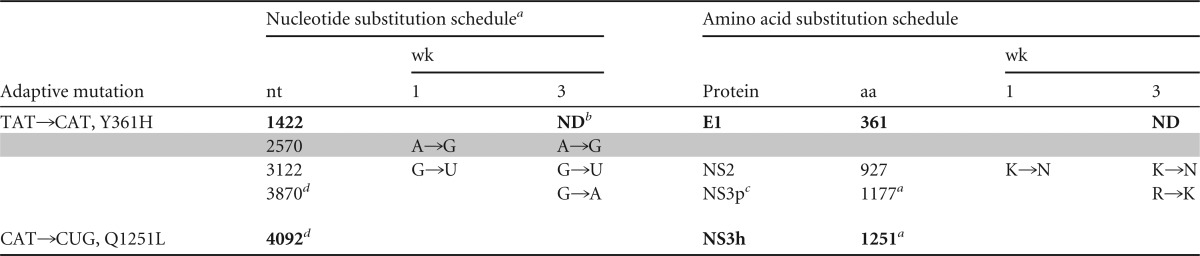
Virus in week 1 serum was sequenced between nt 250 and 5113, and virus from week 3 serum was sequenced between nt 1880 and 5113 only.
ND, not determined.
NS3p, protease domain; NS3h, helicase domain.
Numbered according to the JFH1 sequence.
Cell culture-adaptive mutations and reversions are shown in boldface; synonymous nucleotide substitutions are shaded. nt, nucleotide; aa, amino acid.
H77S.2 challenge.
Two years (108 weeks) after the initial challenge of 4x0193 with 104 FFU H77S virus (76 weeks after HJ3-5 challenge), 4x0193 was rechallenged intravenously with 106 FFU of H77S.2 virus, also produced in cell culture. H77S.2 virus differs from H77S by the presence of an Asn-to-Asp substitution at residue 476 (E2 protein) that results in a reproducible 2- to 4-fold increase (means, 3.1; standard errors of the means [SEM], 0.41) in the titer of infectious virus released by RNA-transfected Huh-7 cells (Fig. 2A) without influencing the kinetics of RNA replication (Fig. 2B). The mutation was identified originally in an intergenotypic chimeric HCV RNA, H77-(NS2)-JFH1 (21). Despite probable immunologic priming of 4x0193 by the preceding HJ3-5 infection, this third infectious challenge with a high-titer H77S.2 inoculum resulted in a sustained, persistent infection associated with evidence of significant liver injury. However, the course of the infection was unusual in several respects.
FIG 2.

Impact of cell culture-adaptive mutations and reversions on viral replication in Huh-7.5 cells. (A) Infectious virus released by cells 2 days after transfection with the indicated HCV RNA. Values shown represent the means ± SD of 2 (H77S) or 4 (all others) FFU assays from two independent experiments. An asterisk indicates P < 0.03 for the difference in yield from H77S.2 by two-sided Mann-Whitney test. (B) qRT-PCR analysis of the fold increase in H77S versus H77S.2 RNA relative to H77S/AAG RNA (lethal NS5B mutant) following transfection of Huh-7.5 cells. (C) Fold increase of H77S.2 mutant RNAs, as described for panel B. (D) Immunofluorescence detection of HCV core protein in cells transfected with H77S.2 versus H77S.2/12204T RNA.
First, although both ELISA and neutralizing antibody (FFU reduction) assays were negative for anti-HCV at the time of H77S.2 challenge, the ELISA was positive 1 week later (Fig. 1C). Consistent with an anamnestic response, the serum neutralizing antibody titer rose from nondetectable at the time of inoculation to >1:80 at week 2. Despite this, there was no detectable viremia until week 4, when the very sensitive TMA assay first detected the presence of HCV RNA; the standard qRT-PCR assay became positive at 1.36 ×103 GE/ml 1 week later (Fig. 1C). Notably, however, a very low level of HCV RNA, resulting in erratic detection in repeat assays, was present in a liver biopsy specimen taken at week 2, when the serum TMA was negative. Following its first appearance in the serum at week 4, the magnitude of viremia increased rapidly, peaking at 1.39 × 104 GE/ml at week 12 and then dipping to 1.85 × 103 GE/ml at week 26 before beginning a slow, sustained rise to 105 to 106 GE/ml by 250 to 288 weeks (5.5 years) (Fig. 1A). Thus, the course of H77S.2 infection in 4x0193 was marked by an initial 4-week period of very low-level replication in the liver in the absence of detectable viremia, followed by the apparent emergence of viral variants with greater replicative fitness (described below).
Despite the presence of intrahepatic viral RNA and slight increases in serum ALT (54 and 48 U/liter at 1 and 2 weeks postinoculation, respectively) (Fig. 1C), histopathologic changes in the liver were confined to minimal hepatocellular cytoplasmic swelling (Fig. 3A, compare preinfection to week 2). Following the appearance of viremia, however, the serum ALT activity became substantially increased (peaking at 114 U/liter) and remained well above the baseline subsequently (Fig. 1A). This was associated with a minimal increase in periportal lymphoid infiltrates by week 8 that were notably increased on subsequent liver biopsy specimens at week 194 (Fig. 3A). Remarkably, Masson trichrome stains of liver tissue demonstrated significant hepatic fibrosis at 194 weeks, particularly in and around portal triads (Fig. 3B). However, while collagen deposition was clearly increased from the preinfection biopsy specimen, there was no bridging fibrosis. There was also an increase in caspase 3-positive cells, many observed within inflammatory infiltrates, and greater numbers of Ki67-positive cells undergoing proliferation (Fig. 4). Interferon-stimulated gene 15 (ISG-15) expression was evident within hepatocytes by immunohistochemistry as early as 8 weeks postinfection (Fig. 5A, top), although mRNA transcripts for it and other ISGs (IP-10, CXCL11, CXCL9, and IFIT1) were not detectably elevated within the liver until week 26 (Fig. 5B). Despite hepatocellular ISG-15 expression being markedly increased by 194 weeks (Fig. 5A and B), only minimal increases in type I and III interferon (IFN-β and IFN-λ2/3) and IFN-γ transcripts were detected at the latter time point (Fig. 5C). Proinflammatory cytokine expression also became prominent following the appearance of viremia. Serum interleukin-12 (IL-12), IP-10, and MIP-1β protein levels, all of which became markedly elevated at the termination of HJ3-5 viremia, became reelevated with the appearance of H77S.2 RNA in the serum, consistent with a resurgent cellular immune response (Fig. 5A, top). In contrast, serum levels of the inflammasome-related cytokines IL-1β and IL-18 increased immediately after H77S.2 inoculation but did not track closely with HCV viremia or ALT activity (Fig. 5A, bottom). They may have represented a nonspecific response to contaminating cellular antigens in the inoculum.
FIG 3.
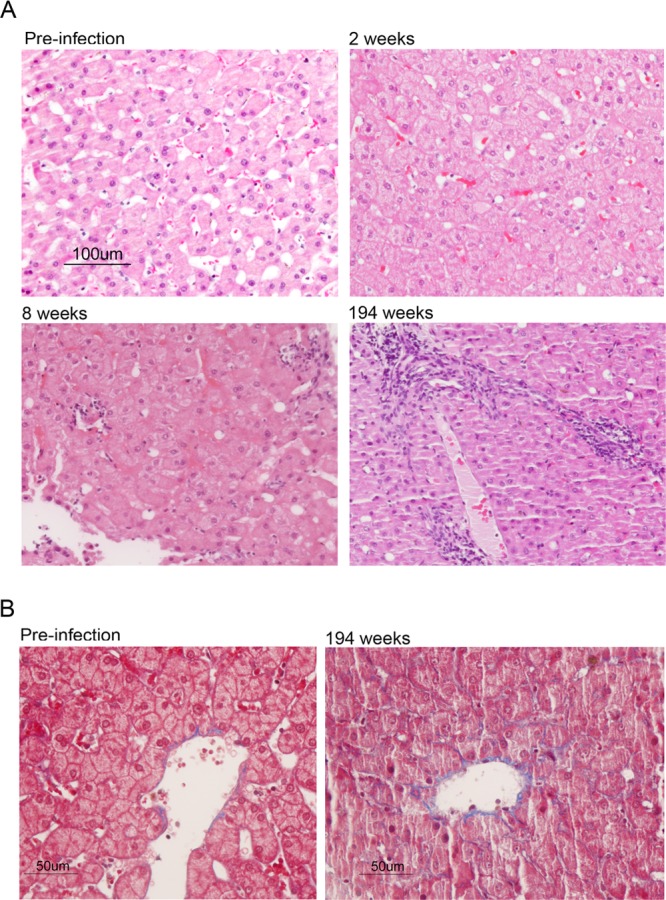
Liver histopathology in chimpanzee 4x0193. (A) Hematoxylin and eosin-stained sections of tissue collected prior to and following infection with H77S.2 virus. Mild hepatocellular swelling with minimal impingement of hepatic sinuses and disruption of cords is evident at 2 weeks postinoculation of virus. Following the appearance of viremia, 8 weeks after inoculation, there was moderate hepatocellular swelling with increased impingement of sinuses. Minimal lymphocyte infiltration was evident in portal regions. By 194 weeks, the hepatic cords had become disorganized and the sinusoidal spaces markedly narrowed. Periportal mononuclear cell infiltrates were increased, and there was diffuse moderate hepatocellular swelling with increased amounts of pale, glassy cytoplasm, mild anisocytosis, occasional binucleate hepatocytes, and rare single-cell necrosis. (B) Masson trichrome stains of preinfection (left) and week 194 (right) liver tissue showing evidence of fibrosis (collagen, blue) with septae lining hepatic sinusoids in tissue collected at 194 weeks.
FIG 4.
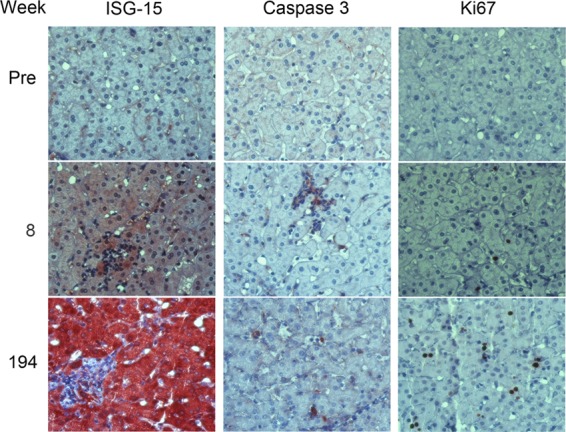
Immunohistochemical staining of 4x0193 liver tissue for markers of innate immune response (ISG-15), apoptosis (caspase 3), and cellular proliferation (nuclear Ki67 expression). A marked increase in hepatocellular ISG-15 expression is evident between preinfection liver and tissue collected at 8 and especially 194 weeks. Caspase 3 expression is also increased, especially in association with collections of lymphoid cells. Multiple, single, and paired Ki-67-positive nuclei are evident at 194 weeks, consistent with increased hepatocellular proliferation.
FIG 5.

Innate immune response to sequential HJ3-5 and H77S.2 infections in chimpanzee 4x0193. (A) Serum cytokine levels measured by Luminex assay during the period of HJ3-5 infection and the first 26 weeks of persistent H77S.2 infection: (top) IL-12, IP-10, and MIP-1β serum levels; (bottom) inflammasome-related serum cytokine levels of IL-1β and IL-18. (B) Intrahepatic ISG mRNA transcript levels measured by real-time qRT-PCR 2, 26, 79, and 194 weeks after intravenous inoculation of cell culture-derived H77S.2 virus. (C) Intrahepatic IFN transcripts 2 and 194 weeks after infection with H77S.2 virus: IFN-β, IL-28 (IFN-λ2 and -λ3), IL-29 (IFN-λ1), and IFN-γ.
Evolution of the H77S.2 genome during persistence in 4x0193.
The persistence of H77S.2 virus in animal 4x0193 provided a unique opportunity to characterize the evolution of a cell culture-adapted HCV genome in vivo. We determined the sequence of all but the extreme 5′ end of the H77S.2 genome from serum samples collected at weeks 8, 62, 194, and 250. Numerous synonymous and nonsynonymous nucleotide substitutions were evident as early as 8 weeks (Fig. 6), including changes in 2 of the 6 cell culture-adaptive mutations: reversion of Q1067R (NS3 protease) to the wild-type Gln and a change in S2204I (NS5A) to Thr (Table 3). This S2204T mutation was also present in a small fragment of RNA amplified from the liver at week 2. These changes were accompanied by several other nonsynonymous substitutions in the NS3 sequence of circulating virus at week 8 and multiple additional substitutions throughout the genome by week 62. Rates for both synonymous and nonsynonymous substitutions, estimated according to the method of Nei and Gojobori (30), declined progressively over time (Fig. 6B); the ratio of nonsynonymous substitutions per site to synonymous substitutions per site (dN/dS) fell from ∼0.50 during the first 62 weeks of infection to ∼0.18 by week 194 (Fig. 6C). By week 62, the original S2204I adaptive mutation had fully reverted to the wild-type Ser, and two other adaptive mutations, N476D and K1691R, had also reverted to the wild-type H77c sequence. By week 194, all 6 of the cell culture-adaptive mutations had reverted to the original amino acid sequence (Table 4). The reversion of adaptive mutations and decreasing rates of both synonymous and nonsynonymous substitutions elsewhere in the genome correlated with progressive increases in the level of viremia (Fig. 6B).
FIG 6.

Substitutions arising in the H77S.2 genome during persistent infection of 4x0193. (A) The viral genome with nucleotide and amino acid coordinates is shown in the middle of the figure with synonymous (above) and nonsynonymous (below) substitutions identified at 8, 62, and 194 weeks after infection. Each triangular symbol represents a change from the base or amino acid residue present at the preceding time point; the sequence present at each position is shown at week 194, with intermediate changes shown in a single-letter entry next to the relevant symbol. Changes in the 6 cell culture-adaptive mutations are shown in boldface; all had reverted to the wild-type H77c sequence by week 194. Table 3 provides a list of additional substitutions identified at week 250. (B) Rates of nonsynonymous and synonymous nucleotide substitutions within the polyprotein coding region of H77S.2 virus shown by period following infection. Rates were calculated as the number of substitutions per nonsynonymous and synonymous sites by the method of Nei and Gojobori (30). The overall rate of nucleotide substitutions (shown as all substitutions per base per year) declined over the 250-week period. (C) Ratio of the rates of nonsynonymous to synonymous substitutions (dN/dS) by period following infection.
TABLE 3.
Changes in H77S.2 virus sequence during persistent infection in chimpanzee 4x0193h
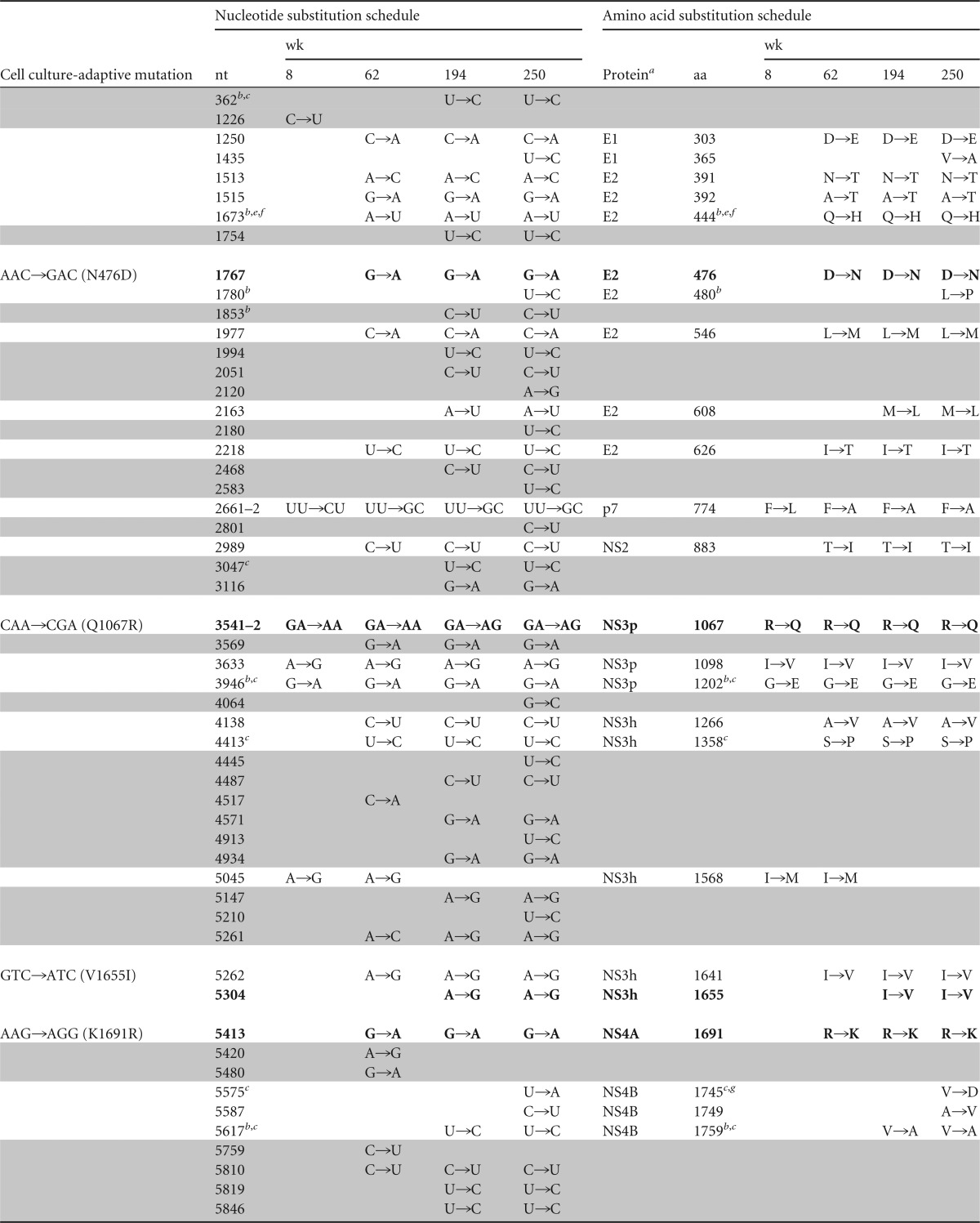
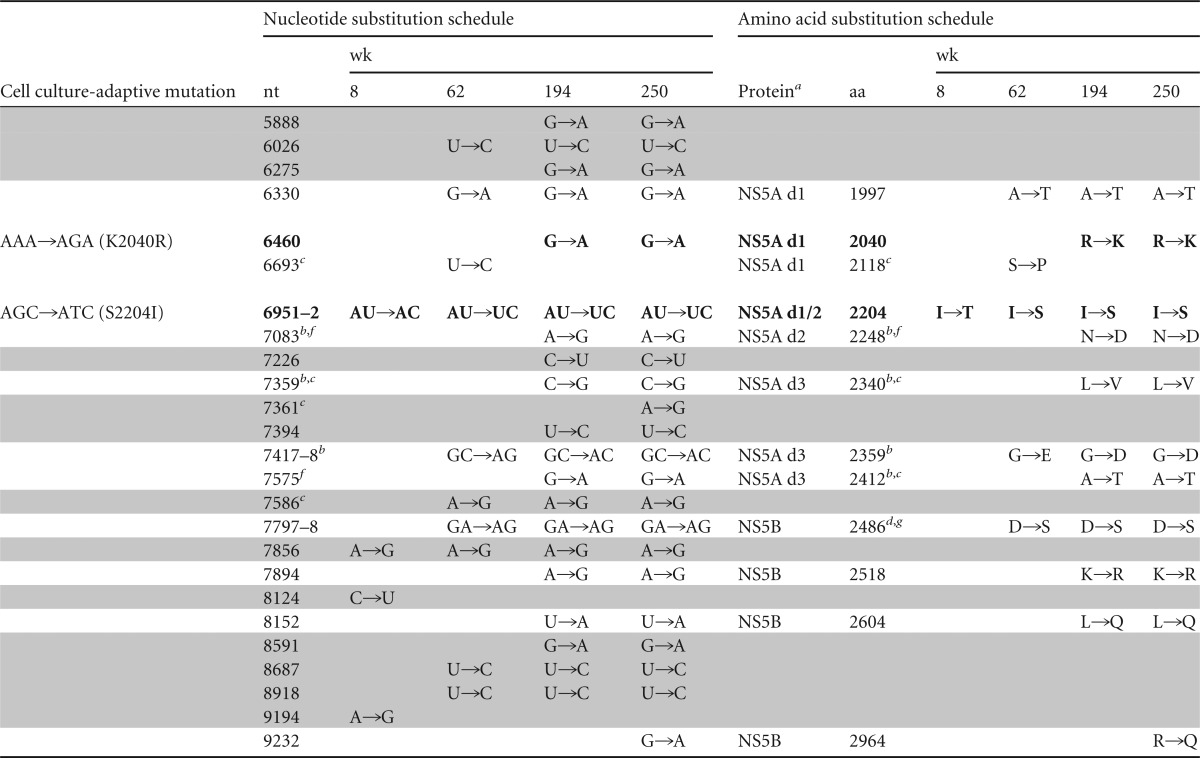
NS3p, protease domain; NS3h, helicase domain; NS5a domains (d1, d2, and d3) are according to Tellinghuisen et al. (68).
Identical substitution at this position in chimpanzee 1530 (39).
Identical substitution at this position in chimpanzee 1564 (39).
Alternative amino acid substitutions (A2412D, D2486E) are at this position in chimpanzee 1564 (39).
Identical substitution at this position in chimpanzee 1535 (59).
Identical substitution at this position in chimpanzee 1536 (59).
Alternative amino acid substitution (V1745A, D2486E) at this position in chimpanzee 1535 (59).
Cell culture-adaptive mutations and reversions are shown in boldface; synonymous nucleotide substitutions are shaded.
TABLE 4.
Nonsynonymous substitutions occurring within potential class 1 epitopes in H77S.2 virus during persistent infection in chimpanzee 4x0193
| Protein | Amino acid substitution(s) | Chimpanzee class I | Chimpanzee epitope sequence | Mutation direction |
|---|---|---|---|---|
| E2 | L546M | B1301 | TRPPLGNWFb | Away |
| E2 | M608L | Not mappeda | ITPRCMVDYPYRLWHYPC | Toward |
| NS3h | S1358P | B1301 | VPHPNIEEV | Toward |
| NS3h | I1568M | Not mappeda | FTGLTHIDAHFLSQTKQS | Away |
| NS3h | I1641V, I1655V | Not mappeda | ITKYIMTCMSADLEIV | Away |
| NS5A d1 | A1997T | B0101 | KTWLKAKLM | Away |
| NS5B | K2518R | A0301 | CSLTPPHSAK | Away |
Epitopes were identified using CD8+ T cell lines, but class I molecules responsible for presentation were not mapped.
Residue undergoing substitution is underlined.
The 4- to 6-week delay in appearance of H77S.2 viremia (Fig. 1C) coupled with the onset of viremia only after loss of adaptive mutations at Q1067 and S2204 strongly suggests that these mutations compromise in vivo viral fitness despite promoting replication in Huh-7 cells. This has been reported previously for mutations facilitating replication of a genotype 1b RNA replicon in cell culture (38). To better understand the significance of the change from Ile to Thr at residue 2204 that we documented at 8 weeks in 4x0193 (Table 3), we introduced this mutation into the H77S.2 genome. Interestingly, despite being associated with the appearance of viremia in the chimpanzee, the resulting RNA (H77S.2/I2204T) was substantially handicapped in its ability to replicate in transfected FT3-7 cells compared to H77S.2, with an approximate 10-fold decrease in RNA accumulation at 48 h (Fig. 2C) with reduced expression of HCV core protein (Fig. 2D). Infectious virus yields were also reduced by at least 50-fold (Fig. 2A). Collectively, these and earlier results demonstrating the importance of the S2204I mutation for replication in cell culture (11) identify this residue in NS5A (NS5A residue 232) as pivotal in determining the contrasting abilities of wild-type versus cell culture-adapted genotype 1a HCV to replicate in vivo versus within cultured Huh-7 cells. It is likely that the delayed kinetic of H77.2 viremia in 4x0193 was caused by a severe restriction imposed by S2204I on H77S.2 replication in vivo that was partially reversed by the change at 2204 from Ile to Thr and then more fully reversed by complete reversion to the wild-type Ser residue by week 62 (Table 3).
The reversion of the cell culture adaptation mutation Q1067R (NS3 residue 41) to the wild-type Gln residue by week 8 (Fig. 6A and Table 3) suggests it also impairs fitness in vivo. However, mutagenesis studies demonstrated that it is quite different from S2204I. Loss of the mutation had no effect on RNA replication in transfected cells (H77S.2/R1067Q) (Fig. 2C) but instead significantly enhanced the yield of infectious virus (Fig. 2A). Thus, this mutation is likely to reduce fitness both in vivo and in vitro. Although our early studies showed it to promote replication of subgenomic HCV RNA replicons in Huh-7 cells, this effect appears to be lost or otherwise made redundant within genome-length H77S.2 RNA containing other adaptive mutations (see reference 18 for additional details).
We also studied an additional mutation in NS3, G1202E (NS3 residue 177), identified at week 8 in 4x0193 (Table 3). This residue is Glu in most genotype 1 viruses but is Gly in wild-type H77c as well as H77S.2 virus. Previous studies show that a Glu-to-Gly substitution at this position promotes replication of the genotype 1b Con1 virus in Huh-7 cells while possibly impairing its replication in chimpanzees (38). Altering the wild-type G1202 residue in H77S.2 to Glu resulted in a moderate decrease in RNA replication as well as infectious virus production following electroporation of the RNA into cells (Fig. 2C). Thus, the H77S.2 G1202 residue may serve as a natural adaptive mutation that enhances H77S.2 replication in Huh-7 cells but reduces its fitness in chimpanzees. Consistent with the latter notion, G1202E substitutions have also been noted in two chimpanzees infected by intrahepatic inoculation of synthetic H77c RNA in other studies (Table 3) (39).
Replication phenotype of H77S.2 versus wild-type H77c virus in humanized Alb-uPA/SCID mice.
Although the prolonged interval between intravenous inoculation of H77S.2 virus and the appearance of viremia in 4x0193 (Fig. 1C) likely reflects the need for reversion of cell culture-adaptive mutations as suggested above, it is also possible that prior immunologic priming during the brief, self-limited HJ3-5 infection suppressed the initial replication of the virus. In an effort to distinguish between these possibilities, we assessed replication phenotypes in the absence of an immune response in immunodeficient Alb-uPa/SCID mice with chimeric human livers (36). None of 6 animals became infected after being inoculated intravenously with 1.5 ×105 FFU H77S.2/R1067Q (H77S.3) virus produced in cell culture. We also attempted infection by direct intrahepatic inoculation of synthetic RNA. Of 4 animals challenged with the wild-type H77c RNA (22), only 1 developed an infection marked by significant and sustained viremia, while the other 3 did not show convincing evidence of infection (Fig. 7, left). This lack of infection likely reflects a combination of RNase activity in the liver and failure of the synthetic RNA to enter human hepatocytes in the chimeric liver. Intrahepatic inoculation of HCV RNA has also failed to initiate infection in the chimpanzee in some experiments (38). Similarly, only 1 of 3 animals inoculated intrahepatically with H77S.2 RNA showed definitive evidence of infection (Fig. 7, right). Thus, the infection “take rate” was similar following inoculation of either wild-type or cell culture-adapted H77S.2 RNA. There were marked differences in the course of the infection in the two infected animals, however. Wild-type H77c RNA produced high-grade viremia (up to 1.3 × 106 GE/ml) that persisted for over 80 days, whereas the maximum H77S.2 viremia was 1,000-fold less (1.5 × 103 GE/ml) and present for only 28 days (Fig. 7). These data are consistent with the H77S.2 RNA having a markedly attenuated replication phenotype in vivo compared to the wild-type H77c genome.
FIG 7.

HCV viremia in Alb-uPa/SCID mice inoculated intrahepatically with synthetic wild-type H77c RNA (left) or H77S.2 RNA (right). Despite comparable infection take rates (1 of 4 versus 1 of 3 animals), H77c viremia peaked at 1,000-fold higher levels than H77S.2 RNA.
Structural context of amino acid substitutions occurring during H77S.2 infection in 4x0193.
Experimental high-resolution models exist for the three-dimensional structures of several nonstructural HCV proteins, including the NS2 protease, the NS3 protease-helicase, NS5A domain I, and the NS5B RNA-dependent RNA polymerase (31–34, 40). To gain insight into the selective forces driving evolution of the H77S.2 genome in 4x0193, we assessed the structural context of substitutions occurring within these nonstructural proteins. We considered it likely that amino acid substitutions influencing interactions with host cell proteins would be located at or near the solvent-accessible surface of the nonstructural proteins. In contrast, mutations reflecting escape from T cell recognition of peptide epitopes presented on class I molecules may be more randomly distributed between surface-exposed and nonexposed residues. Surprisingly, each of the nonsynonymous substitutions occurring within domains of the nonstructural proteins for which structural information exists involved amino acid residues that were at least partially surface exposed.
No substitutions were identified within the catalytic domain of the NS2/NS3 protease for which a crystallographic structure exists (40). However, 8 nonsynonymous substitutions occurred in the NS3 protease and helicase domains (Table 3). Each of these 8 substitutions involved a surface-exposed residue in the three-dimensional structure of this bifunctional complex from PDB entry 1CU1 (31). Four substitutions involved residues located within or near a predicted protein-protein interaction site, I1098 (NS3-72), G1202 (NS3-176), I568 (NS3-542), and I1641 (NS3-615), while A1266 (NS3-240) is in close proximity to the ATP-binding site (Fig. 8). The 3 remaining substitutions are not located near predicted protein-protein interaction sites. These include Q1067R (NS3-41), which is close to the protease active site, and I1655V (NS3-629), which is close to the site of scission and part of the natural substrate for NS3 proteolysis. Both are reversions of cell culture-adaptive mutations in the H77S.2 clone. Interestingly, G1202 and I1655, as well as S1358 (NS3-332), which represents a change from the wild-type H77c sequence to the consensus genotype 1a sequence (39), are also located within known T cell epitopes that are potentially relevant in this animal (see the following section).
FIG 8.

Structural context of mutations occurring in the NS3 protein of H77S.2 virus during persistent infection of chimpanzee 4x0193, modeled on the structure of the bifunctional protease-helicase of genotype 1b BK strain of HCV (PDB entry 1CU1). Residues at which mutations were identified (Table 1) are labeled according to the H77S.2 sequence with the authentic BK residue shown in parentheses. Solvent-accessible surfaces of these residues are shaded in yellow, while the overall surface of the structure is colored according to ProMate output, with red shading representing high probability and blue shading a low probability that each surface residue contributes to a protein-protein interaction site (see Materials and Methods). Protein-protein interaction sites predicted by ProMate are shown encircled by a dashed red line. The single mutation identified in NS4A (R1691K, a reversion of a cell culture-adaptive mutation) occurs at residue 34 of NS4A and is located just outside the ordered structure of the NS4A peptide strand in the NS3/4A complex.
Two nonsynonymous substitutions occurred within that part of NS5A domain I for which structural models are available. Both R2040K (NS5A-68, representing a reversion of the K2040R cell culture-adaptive mutation) (Table 3) and S2118 (NS5A-146) involve surface-exposed residues within the proposed homodimer interface (Fig. 9A, left) in PDB entry 3FQQ (32). S2118 is immediately adjacent to E2120 (NS5A-148), a residue that plays a key role in homodimer formation (32). An alternate dimer structure, with the dimerization domain on the opposing side of the monomer, was proposed in an earlier model in PDB entry 1ZH1 (33), but the monomer folds are similar for both (Fig. 9A, right). In both proposed dimeric structures, the membrane association of NS5A is mediated through the N-terminal part of the monomer, and a role for K2040 in modulating membrane association is consistent with both models. All 4 nonsynonymous substitutions occurring in the NS5B polymerase also involve surface-accessible residues in the structural model proposed in PDB entry 2XI2 (34) (Fig. 9B). L2604 (NS5B-184) is located within the putative RNA-binding groove, while K2518 (NS5B-98) is close to the NTP channel. Both are located within predicted protein-protein interaction sites (Fig. 9B). The two remaining residues, 2486D (NS5B-66) and 2964R (NS5B-544), are close to the RNA-binding groove but not associated with predicted sites of protein-protein interactions.
FIG 9.

Structural context of mutations occurring in domain I of NS5A and NS5B protein of H77S.2 virus during persistent infection of chimpanzee 4x0193. (A) NS5A residues involved in mutations are highlighted in yellow on the surface of domain I of the protein from the genotype 1b Con1 strain of HCV (PDB entry 3FQQ) (left). Residues with key roles in homodimer formation are highlighted in green (32). Also shown is an imposition of the monomer structures proposed in alternate structural models (right), with the peptide backbone from PDB entry 2FQQ (32) shown in gray and that from PDB entry 1ZH1 (33) in cyan. The side chain of K68 is highlighted in yellow in both structures. Side chains of residues presumed to be closest to the membrane surface in PDB entry 1ZH1 (33) are shown as stick models. The structural superposition was calculated using the jCE/jFATCAT Structure Alignment Server v2.6 (http://source.rcsb.org/jfatcatserver/index.jsp) with jCE algorithm, PDB entry 3FQQ chain A, and 1ZH1 chain A. (B) Residues involved in nonsynonymous mutations in the NS5B protein are highlighted in a structural model of the polymerase from wild-type H77c virus (PDB entry 2XI2). Residues are labeled and colored as described in the legend to Fig. 8.
It is remarkable that all of the nonsynonymous substitutions identified in protein domains for which structural models are available involved surface-accessible residues. To assess whether this could be due simply to chance, we compared the surface accessibility of the amino acid residues at which substitutions occurred to that of residues included in the model structures (see Materials and Methods). The difference was statistically significant for NS5B alone (P = 0.021 by nonparametric Mann-Whitney U test) and even more significant when all substitutions were considered in the NS3, NS5A, and NS5B structures (P < 0.002).
Sequence evolution and predicted CD8+ T cell escape.
Immune selection pressure has generally been considered to be the dominant force driving increased nonsynonymous substitutions in HCV genomes during the acute phase of infection (7). Nonsynonymous substitutions in nonstructural proteins are concentrated in class I epitopes during the first year of infection and then subside, presumably as CD8+ T cell effector functions fail (39, 41). Because this pattern of substitutions was observed in 4x0193, we sought to determine whether any amino acid changes fell within known class I epitopes. The Immune Epitope Database (http://www.iedb.org) and Los Alamos National Laboratory HCV Database (http://hcv.lanl.gov) were searched for class I epitopes matched to the H77S.2 challenge sequence and presented by the Patr class I A*0201, A*0301, B*0101, and B*1301 of chimpanzee 4x0193. An additional 6 epitopes presented by these chimpanzee class I molecules but not yet deposited in public databases (C. Walker, unpublished data) were also included. Of the 36 nonsynonymous substitutions observed over 5 years of follow-up, 8 (approximately 22% of the total) were located in epitopes known to be presented by class I molecules expressed by this animal (Table 4). Of these 8, 4 (L546M, I1568M, I1641V, and A1997T) appeared before week 62, a time frame associated with the highest rate of nonsynonymous substitution (Fig. 6B) and CD8+ T cell selection pressure (39). S1358P occurred in the same time frame, but this substitution reconstitutes an epitope rather than providing escape from one. As indicated above, Pro is the consensus amino acid at position 1358 in genotype 1a virus. The three remaining nonsynonymous changes (M608L, I1655V, and K2518R) occurred sometime after week 62 but before week 194. Escape mutations in HCV class I epitopes have been documented in chimpanzees 2 years after infection (42), but other selective forces cannot be excluded for changes that appeared well after chronic infection was established. For instance, I1655V represented reversion of a cell culture-adaptive mutation that facilitated replication of H77S.2 virus in cell culture.
DISCUSSION
The chimpanzee, Pan troglodytes, is the only animal species other than Homo sapiens that is naturally fully permissive for HCV infection, and it has served as an exceptionally valuable model of the infection in humans (28). In the past, experimental infections of the chimpanzee established hepatitis C (then known as parenterally transmitted non-A, non-B hepatitis) as a distinct disease entity and also provided unique biological samples that facilitated the identification and molecular cloning of the responsible virus (43, 44). The chimpanzee model has also played a key role in the preclinical evaluation of candidate hepatitis C vaccines (45). However, in recent years the need for the chimpanzee model has lessened given progress in developing improved cell culture systems supporting replication of the virus (12, 17, 46, 47), as well as innovative mouse models that can be infected in vivo (36, 48, 49). Coupled with the ethical implications of the uniquely close relationship of chimpanzees to humans, such advances have led to increasing restrictions on the use of the chimpanzee in hepatitis C research (25). We report here the 5.5-year follow-up of a chimpanzee that developed persistent infection following intravenous challenge with a cell culture-adapted genotype 1a virus produced in Huh-7.5 cells. This study is unique in that, to our knowledge, this is the only chimpanzee to have sustained a persistent infection after challenge with virus produced in cell culture. All other chimpanzee infections with HCV produced in cell culture (HCVcc) were with viruses possessing genomes derived in whole or in part from the genotype 2a JFH1 virus and expressing JFH1 replicase proteins (12, 16, 50). All of these infections were self-limited, with no demonstrable liver injury (Table 1). In contrast, 4x0193, infected with H77S.2 virus, experienced long-term viral persistence (288 weeks at the last health check) associated with continued elevation of serum ALT activity, strong intrahepatic innate immune responses, and histologic evidence of hepatic inflammation and fibrosis, typical features of chronic hepatitis C in humans.
Several aspects of the infection in chimpanzee 4x0193 are particularly notable. First, the animal became persistently infected when challenged with H77S.2 2 years after a brief, transient infection with HJ3-5 virus, an H77/JFH1 chimera expressing structural proteins that differ at only 2 residues (one each in E1 and E2) from H77S.2. Although infection with HJ3-5 virus did not result in seroconversion in the ELISA or detectable levels of serum neutralizing antibodies, the H77S.2 challenge resulted in an immediate, anamnestic antibody response (Fig. 1C). The potential for reinfection of chimpanzees following clearance of an initial HCV infection has been clearly established previously (51). However, prior infection typically results in a robust CD8+ T cell response on rechallenge that limits the extent and duration of secondary infections (52, 53). It is noteworthy that prior HJ3-5 virus infection did not prevent an unfit, cell culture-adapted HCV inoculum (see below) from establishing persistent infection and chronic hepatitis.
A second notable feature of the infection in 4x0193 is the lengthy delay this animal experienced prior to the onset of viremia. This is unusual. All 3 chimpanzees infected by intrahepatic inoculation of clonal wild-type H77 RNA were viremic within a week (54, 55). It is possible that immunologic priming during the preceding HJ3-5 infection suppressed early H77S.2 viremia (52). However, we believe it more likely that the 6 cell culture-adaptive mutations in H77S.2 conferred a strong, negative effect on intrahepatic replication, and that detectable levels of viremia required the evolution of a more fit virus within the liver. The S2204I mutation in NS5A seems particularly likely to have contributed to inefficient initial replication of the virus in vivo, as it plays a key role in the cell culture adaptation of H77S (11). It is also in close proximity (in the linear peptide sequence of NS5A) to a cell culture-adaptive mutation, S2197P, that limited the capacity of genotype 1b Con1 RNA to replicate in chimpanzees in a previous study (38). This earlier study examined a set of mutations that enhance replication of Con1 RNA replicons but that do not provide for significant infectious virus production in cell culture. Thus, chimpanzees were challenged by intrahepatic RNA injection (38) rather than inoculation with infectious, cell culture-produced virus. Nonetheless, despite these methodologic differences, both NS5A mutations (S2197 in Con1 and S2204I in H77S.2) appear to similarly restrict replication in vivo. The S2204I mutation in H77S.2 was partially reverted by 8 weeks after challenge (I2204T mutation) (Table 3), shortly after viremia was first detected (Fig. 1C). The I2204T substitution was also present in a cDNA fragment of HCV RNA recovered from the liver biopsy specimen taken at 2 weeks. Thus, this NS5A mutation appears to have reverted very early in the course of infection, consistent with substantial selective pressure acting at this site in the genome. A second mutation in the NS3 protease of H77S.2, Q1067R, had also reverted by 8 weeks (Table 3). It was originally found to promote replication of a subgenomic replicon, but it is not required for optimal replication of the complete H77S.2 genome in cell culture (11, 17, 18) (Fig. 2). Further evidence for a primary role for S2204I rather than Q1067R in impeding replication in vivo is provided by the absence of infection in 6 SCID-Alb/uPA mice inoculated intravenously with H77S.3 virus. This virus contains all of the cell culture-adaptive mutations in H77S.2 (including S2204I) with the exception of Q1067R. S2204 and Q1067 are both very highly conserved, however, and are present in >99% of genotype 1a viruses. Inefficient replication of H77S.2 virus in vivo may have been due to both of these cell culture-adaptive mutations.
The absence of detectable viremia, even with the very sensitive TMA assay, for a month following infection of 4x0193 with H77S.2 virus (Fig. 1C) is relevant to recent clinical trials of DAAs where some subjects were found to relapse after completing treatment with mericitabine or sofosbuvir, potent nucleosides or nucleotide inhibitors of the NS5B polymerase that effectively suppressed replication (56, 57). S282T is a key mutation that confers resistance to these compounds, and its absence following the reappearance of viremia has been interpreted as indicative of the absence of virologic resistance (56, 57). Importantly, because the S282T mutation impacts the active site of the polymerase, S282T viruses are very impaired in replication. The course of the infection in 4x0193 demonstrates that a nonfit virus can replicate for weeks within the liver in the absence of detectable viremia, and that when the virus does eventually mutate to a more fit phenotype the mutations that had made it unfit may no longer be detectable. Thus, caution must be taken in inferring the absence of virologic resistance from the absence of detectable resistance-associated variants following the reappearance of virus in relapsing patients.
Each of the 6 cell culture-adaptive mutations in H77S.2 virus eventually reverted to the original wild-type sequence during persistent infection in 4x0193 (Table 3). This is strong evidence that mutations conferring enhanced replication in Huh-7 cells reduce fitness in the liver and are negatively selected in vivo. Importantly, the involved nonstructural protein residues are highly conserved among genotype 1a viruses and are present among 216 sequences downloaded from the European database (58) at the following rates: Q1067, 99.1%; V1655, 97.7%; K1691, 98.1%; K2040, 97.7%; and S2204, 100%. The mechanism(s) underlying the conservation of these residues is uncertain. The cell culture-adaptive mutations are likely to promote replication in cell culture by optimizing functionally important interactions between nonstructural viral proteins and host cell factors present in Huh-7 cells but not hepatocytes in vivo. Such a model is consistent with the surface location of the 3 cell culture-adaptive mutations that are positioned within nonstructural protein domains for which structural models exist: Q1067R (NS3 protease), V1655I (NS3 helicase), and K2040R (NS5A domain I) (Fig. 8 and 9). However, there may have been multiple factors driving the selection of the revertants in 4x0193, as reversion of the cell culture-adaptive mutation V1655I (I1655V mutation first noted at week 62) (Table 3) would also provide for escape from a putative T cell epitope (Table 4).
The rate with which nonsynonymous substitutions accumulated during the first 8 weeks of infection (5.79 ×10−3 substitutions/site/year) was almost twice that observed previously during the first 26 weeks of infection initiated by intrahepatic inoculation of wild-type H77 RNA in two other chimpanzees (2.59 to 2.88 ×10−3 substitutions/site/year) (59). It is not clear, however, whether the method used to estimate these rates in the earlier study was the same as that used here for 4x0193 (see Materials and Methods). As in previous studies involving long-term follow-up of wild-type H77 infection in chimpanzees (39, 59), the ratio of nonsynonymous to synonymous substitutions fell after the first year of infection, with the rate of all observed substitutions reaching a plateau of 1.26 × 10−3 substitutions/base/year by 250 weeks (Fig. 6). In addition to the reversion of cell culture-adaptive mutations in 4x0193, it is likely that the higher rate of nonsynonymous substitutions during the first year of infection (Fig. 6B and C) reflects the emergence of T cell escape mutations. While we sought to identify substitutions affecting known T cell epitopes of relevance to the Patr type of this particular animal (Table 4), our analysis of potential escape mutations almost certainly underestimated the impact of CD8+ T cell selection pressure on evolution of the H77S.2 virus. The repertoire of known epitopes presented by Patr class I A*0201, A*0301, B*0101, and B*1301 molecules is still quite small. Because of substantial variability in the pattern of epitope dominance among HCV-infected individuals, it is likely that 4x0193 exerted immune pressure against other class I-presented epitopes that have yet to be identified. Our analysis also does not account for compensatory mutations that occur outside epitopes to offset any fitness cost of immune escape to virus replication (60).
B cell pressure may have also contributed to the evolution of H77S.2 virus in this animal. A strong anamnestic antibody response was noted in the anti-HCV ELISA within 1 week of intravenous challenge with H77S.2 virus despite the absence of viremia, and neutralizing antibodies were detected as early as week 2. Two nonsynonymous substitutions in E2 (N391T and A392T) are located within the central part of the hypervariable region I (HVR1) domain of this envelope protein that is known to interact with antibodies (61). Although HVR1 displays a lower rate of nonsynonymous substitutions in infected chimpanzees than in humans (62), it is reasonable to surmise that the changes at N391 and A392 have occurred in response to the selective pressure of antibodies. Q444H is also located within a domain of E2 (residues 434 to 446) that is bound by neutralizing antibodies and also interacts with a cellular receptor for HCV, CD81 (63).
Altogether, of 36 nonsynonymous substitutions occurring during the first 250 weeks of infection in 4x0193, 6 were reversions of engineered cell culture-adaptive mutations, while 8 can be related to potential T cell escape (one of which is also a reversion of a cell culture-adaptive mutation) and 3 may represent B cell escape (Table 3). Interestingly, for 10 of the remaining 20 nonsynonymous substitutions, identical or very similar substitutions have been observed in other chimpanzees that became persistently infected with HCV following intrahepatic inoculation of wild-type H77c RNA (39, 59) (Table 3). In 4 instances, similar or identical substitutions were observed in animals inoculated with RNA from an independently constructed molecular clone (54, 59), arguing against these common changes arising from errors in construction of consensus clones. These common changes could represent reversions of archived T cell escape mutations that were selected for in patient H, the source of H77c virus, or perhaps the source from which patient H acquired the infection. Alternatively, it is possible that they are chimpanzee-adaptive mutations for which no direct evidence yet exists.
Despite the slow start to the infection in 4x0193, this animal developed impressive evidence of liver disease in parallel with an increasing viremia. The serum ALT became persistently elevated above baseline (Fig. 1A), and liver biopsy specimens showed progressively increasing inflammatory infiltrates with fibrosis (Fig. 3). Serum IP-10 (CXCL10), dramatically elevated transiently with the earlier HJ3-5 virus infection, became reelevated and persisted (Fig. 5A). In both infections, IP-10 elevations occurred in parallel but approximately 1 to 2 weeks after the increase in serum HCV RNA. This was matched by substantial upregulation of IP-10 and other ISG transcripts within the liver (Fig. 5B), as observed in many human subjects with chronic hepatitis C (64). Importantly, there was good congruence between the increases in intrahepatic IP-10 and ISG-15 transcript abundance (Fig. 5B), IP-10 protein detected in serum (Fig. 5A), and ISG-15 protein expression in the liver (Fig. 4). This suggests that the translation of these innate immune effector proteins is not significantly blocked by protein kinase R activation, as suggested by Garaigorta et al. (65). While serum IL-1β, recently proposed as a central regulator of hepatic inflammation in chronic hepatitis C (66), was also elevated in H77S.2 infection, the expression of this cytokine did not correlate with viremia, ALT, or hepatic inflammation. It may have reflected a nonspecific response to the cell culture-derived inoculum. Collectively, these data suggest a striking difference in the pathogenic potential of H77S.2 virus and JFH1-related viruses produced in cell culture (Table 1). They also reaffirm the ability of the chimpanzee to closely model many aspects of hepatitis C in humans and suggest that this particular animal should be considered for treatment with direct-acting antiviral therapy.
As with many studies of HCV infection in chimpanzees, the conclusions that can be drawn from this study are tempered by the use of one or only a small number of animals. Nonetheless, as described above, important conclusions can be drawn from the pattern of H77S.2 infection observed in animal 4x0193. Given an increasing variety of systems in which different aspects of HCV can be studied, it is unlikely that a study such as this would be contemplated today. Future efforts to characterize the in vivo replication potential of HCV produced in cell culture will likely depend on the use of genetically humanized or xenotransplanted murine models (49, 67).
ACKNOWLEDGMENTS
This work was supported in part by grants and contracts from the National Institutes of Health (NIH) (N01-AI25488 [N.B.], U19-AI40035, R01-CA164029, RO1-AI095690 [S.M.L.], R01-AI075090 [M.Y.], and R37-AI028433 [C.M.W.]) and the University Cancer Research Fund of the University of North Carolina. F.H. received support from the Chinese National Key Technologies R&D Program for the 12th Five-Year Plan, 2012ZX10001003-003. C.W. was supported by the Deutsche Forschungsgemeinschaft, WE 4388/3-1 and WE 4388/6-1. L.T. and M.J. were supported by the Canadian Institutes for Health Research, MOP126142, and Alberta Advanced Education and Technology, ALSI-10-G2. The Southwest National Primate Research Center is supported by an NIH primate center base grant (previously NCRR grant P51 RR13986; currently Office of Research Infrastructure Programs/OD P51 OD011133) and by Research Facilities Improvement Program Grants C06 RR 12087 and C06 RR016228.
We thank Lucinda Hensley for expert technical assistance.
Footnotes
Published ahead of print 15 January 2014
REFERENCES
- 1.Alter HJ. 2005. HCV natural history: the retrospective and prospective in perspective. J. Hepatol. 43:550–552. 10.1016/j.jhep.2005.07.002 [DOI] [PubMed] [Google Scholar]
- 2.Thomas DL. 2013. Global control of hepatitis C: where challenge meets opportunity. Nat. Med. 19:850–858. 10.1038/nm.3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmberg SD, Spradling PR, Moorman AC, Denniston MM. 2013. Hepatitis C in the United States. N. Engl. J. Med. 368:1859–1861. 10.1056/NEJMp1302973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ly KN, Xing J, Klevens RM, Jiles RB, Ward JW, Holmberg SD. 2012. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann. Intern. Med. 156:271–278. 10.7326/0003-4819-156-4-201202210-00004 [DOI] [PubMed] [Google Scholar]
- 5.Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM. 2013. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N. Engl. J. Med. 368:34–44. 10.1056/NEJMoa1208953 [DOI] [PubMed] [Google Scholar]
- 6.Osinusi A, Meissner EG, Lee YJ, Bon D, Heytens L, Nelson A, Sneller M, Kohli A, Barrett L, Proschan M, Herrmann E, Shivakumar B, Gu W, Kwan R, Teferi G, Talwani R, Silk R, Kotb C, Wroblewski S, Fishbein D, Dewar R, Highbarger H, Zhang X, Kleiner D, Wood BJ, Chavez J, Symonds WT, Subramanian M, McHutchison J, Polis MA, Fauci AS, Masur H, Kottilil S. 2013. Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics: a randomized clinical trial. JAMA 310:804–811. 10.1001/jama.2013.109309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker CM. 2010. Adaptive immunity to the hepatitis C virus. Adv. Virus Res. 78:43–86. 10.1016/B978-0-12-385032-4.00002-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rehermann B. 2013. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat. Med. 19:859–868. 10.1038/nm.3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. 10.1126/science.285.5424.110 [DOI] [PubMed] [Google Scholar]
- 10.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190. 10.1128/JVI.77.5.3181-3190.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi M, Lemon SM. 2004. Adaptive mutations producing efficient replication of genotype 1a hepatitis C virus RNA in normal Huh7 cells. J. Virol. 78:7904–7915. 10.1128/JVI.78.15.7904-7915.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796. 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. 10.1053/j.gastro.2003.09.023 [DOI] [PubMed] [Google Scholar]
- 14.Ikeda M, Yi M, Li K, Lemon SM. 2002. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol. 76:2997–3006. 10.1128/JVI.76.6.2997-3006.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beard MR, Abell G, Honda M, Carroll A, Gartland M, Clarke B, Suzuki K, Lanford R, Sangar DV, Lemon SM. 1999. An infectious molecular clone of a Japanese genotype 1b hepatitis C virus. Hepatology 30:316–324. 10.1002/hep.510300137 [DOI] [PubMed] [Google Scholar]
- 16.Kato T, Choi Y, Elmowalid G, Sapp RK, Barth H, Furusaka A, Mishiro S, Wakita T, Krawczynski K, Liang TJ. 2008. Hepatitis C virus JFH-1 strain infection in chimpanzees is associated with low pathogenicity and emergence of an adaptive mutation. Hepatology 48:732–740. 10.1002/hep.22422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 103:2310–2315. 10.1073/pnas.0510727103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimakami T, Welsch C, Yamane D, McGivern D, Yi M, Zeuzem S, Lemon SM. 2011. Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 140:667–675. 10.1053/j.gastro.2010.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li YP, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J. 2012. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc. Natl. Acad. Sci. U. S. A. 109:19757–19762. 10.1073/pnas.1218260109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi M, Ma Y, Yates J, Lemon SM. 2009. Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog. 5:e1000403. 10.1371/journal.ppat.1000403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi M, Ma Y, Yates J, Lemon SM. 2007. Compensatory mutations in E1, p7, NS2 and NS3 enhance yields of cell culture-infectious inter-genotypic chimeric hepatitis C virus. J. Virol. 81:629–638. 10.1128/JVI.01890-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yanagi M, Purcell RH, Emerson SU, Bukh J. 1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into liver of a chimpanzee. Proc. Natl. Acad. U. S. A. 97:8738–8743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014. 10.1128/JVI.76.24.13001-13014.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kowalski H, Erickson AL, Cooper S, Domena JD, Parham P, Walker CM. 1996. Patr-A and B, the orthologues of HLA-A and B, present hepatitis C virus epitopes to CD8+ cytotoxic T cells from two chronically infected chimpanzees. J. Exp. Med. 183:1761–1775. 10.1084/jem.183.4.1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altevogt BM, Pankevich DE, Shelton-Davenport MK, Kahn JP. (ed). 2011. Chimpanzees in biomedical and behavioral research: assessing the necessity. The National Academies Press, Washington, DC: [PubMed] [Google Scholar]
- 26.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. 2010. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201. 10.1126/science.1178178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gal-Tanamy M, Keck ZY, Yi M, McKeating JA, Patel AH, Foung SK, Lemon SM. 2008. In vitro selection of a neutralization-resistant hepatitis C virus escape mutant. Proc. Natl. Acad. Sci. U. S. A. 105:19450–19455. 10.1073/pnas.0809879105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanford RE, Lemon SM, Walker C. 2011. The chimpanzee model of hepatitis C infections and small animal surrogates, p 99–132 In He Y, Tan T. (ed), Hepatitis C antiviral drug discovery and development. Horizons Scientific Press, Norwich, CT [Google Scholar]
- 29.Lanford RE, Feng Z, Chavez D, Guerra B, Brasky KM, Zhou Y, Yamane D, Perelson AS, Walker CM, Lemon SM. 2011. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc. Natl. Acad. Sci. U. S. A. 108:11223–11228. 10.1073/pnas.1101939108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3:418–426 [DOI] [PubMed] [Google Scholar]
- 31.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. 1999. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure Fold Des. 7:1353–1363. 10.1016/S0969-2126(00)80025-8 [DOI] [PubMed] [Google Scholar]
- 32.Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. 2009. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 83:4395–4403. 10.1128/JVI.02352-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379. 10.1038/nature03580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harrus D, Ahmed-El-Sayed N, Simister PC, Miller S, Triconnet M, Hagedorn CH, Mahias K, Rey FA, Astier-Gin T, Bressanelli S. 2010. Further insights into the roles of GTP and the C terminus of the hepatitis C virus polymerase in the initiation of RNA synthesis. J. Biol. Chem. 285:32906–32918. 10.1074/jbc.M110.151316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neuvirth H, Raz R, Schreiber G. 2004. ProMate: a structure based prediction program to identify the location of protein-protein binding sites. J. Mol. Biol. 338:181–199. 10.1016/j.jmb.2004.02.040 [DOI] [PubMed] [Google Scholar]
- 36.Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, Addison WR, Fischer KP, Churchill TA, Lakey JR, Tyrrell DL, Kneteman NM. 2001. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 7:927–933. 10.1038/90968 [DOI] [PubMed] [Google Scholar]
- 37.Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N, Doyle JS, Katze MG, Tyrrell DL. 2009. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 5:e1000291. 10.1371/journal.ppat.1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bukh J, Pietschmann T, Lohmann V, Krieger N, Faulk K, Engle RE, Govindarajan S, Shapiro M, St Claire M, Bartenschlager R. 2002. Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc. Natl. Acad. Sci. U. S. A. 99:14416–14421. 10.1073/pnas.212532699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callendret B, Bukh J, Eccleston HB, Heksch R, Hasselschwert DL, Purcell RH, Hughes AL, Walker CM. 2011. Transmission of clonal hepatitis C virus genomes reveals the dominant but transitory role of CD8(+) T cells in early viral evolution. J. Virol. 85:11833–11845. 10.1128/JVI.02654-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorenz IC, Marcotrigiano J, Dentzer TG, Rice CM. 2006. Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature 442:831–835. 10.1038/nature04975 [DOI] [PubMed] [Google Scholar]
- 41.Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, Cameron B, Maher L, Dore GJ, White PA, Lloyd AR. 2011. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog. 7:e1002243. 10.1371/journal.ppat.1002243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, McKinney D, Sette A, Hughes AL, Walker CM. 2001. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 15:883–895. 10.1016/S1074-7613(01)00245-X [DOI] [PubMed] [Google Scholar]
- 43.Tabor E, Gerety RJ, Drucker JA, Seeff LB, Hoofnagle JH, Jackson DR, April M, Barker LF, Pineda-Tamondong G. 1978. Transmission of non-A, non-B hepatitis from man to chimpanzee. Lancet i:463–466 [DOI] [PubMed] [Google Scholar]
- 44.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362. 10.1126/science.2523562 [DOI] [PubMed] [Google Scholar]
- 45.Folgori A, Capone S, Ruggeri L, Meola A, Sporeno E, Ercole BB, Pezzanera M, Tafi R, Arcuri M, Fattori E, Lahm A, Luzzago A, Vitelli A, Colloca S, Cortese R, Nicosia A. 2006. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat. Med. 12:190–197. 10.1038/nm1353 [DOI] [PubMed] [Google Scholar]
- 46.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- 47.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299. 10.1073/pnas.0503596102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Washburn ML, Bility MT, Zhang L, Kovalev GI, Buntzman A, Frelinger JA, Barry W, Ploss A, Rice CM, Su L. 2011. A humanized mouse model to study hepatitis C virus infection, immune response, and liver disease. Gastroenterology 140:1334–1344. 10.1053/j.gastro.2011.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, Akira S, Law M, Rice CM, Ploss A. 2013. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 501:237–241. 10.1038/nature12427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, Rice CM. 2006. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. U. S. A. 103:3805–3809. 10.1073/pnas.0511218103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farci P, Alter HJ, Govindarajan S, Wong DC, Engle R, Lesniewski RR, Mushahwar IK, Desai SM, Miller RH, Ogata N, Purcell RH. 1992. Lack of protective immunity against reinfection with hepatitis C virus. Science 258:135–140. 10.1126/science.1279801 [DOI] [PubMed] [Google Scholar]
- 52.Bassett SE, Guerra B, Brasky K, Miskovsky E, Houghton M, Klimpel GR, Lanford RE. 2001. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology 33:1479–1487. 10.1053/jhep.2001.24371 [DOI] [PubMed] [Google Scholar]
- 53.Shoukry NH, Grakoui A, Houghton M, Chien DY, Ghrayeb J, Reimann KA, Walker CM. 2003. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 197:1645–1655. 10.1084/jem.20030239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kolykhalov AA, Agapov AA, Blight KJ, Mihalik K, Feinstone SM, Rice CM. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570–574. 10.1126/science.277.5325.570 [DOI] [PubMed] [Google Scholar]
- 55.Yanagi M, St Claire M, Shapiro M, Emerson SU, Purcell RH, Bukh J. 1998. Transcripts of a chimeric cDNA clone of hepatitis C virus genotype 1b are infectious in vivo. Virology 244:161–172. 10.1006/viro.1998.9092 [DOI] [PubMed] [Google Scholar]
- 56.Tong X, Le Pogam S, Li L, Haines K, Piso K, Baronas V, Yan JM, So SS, Klumpp K, Najera I. 2013. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low-level resistance to the HCV polymerase inhibitors mericitabine and sofosbuvir. J. Infect. Dis. 10.1093/infdis/jit562 [DOI] [PubMed] [Google Scholar]
- 57.Lawitz E, Lalezari JP, Hassanein T, Kowdley KV, Poordad FF, Sheikh AM, Afdhal NH, Bernstein DE, Dejesus E, Freilich B, Nelson DR, Dieterich DT, Jacobson IM, Jensen D, Abrams GA, Darling JM, Rodriguez-Torres M, Reddy KR, Sulkowski MS, Bzowej NH, Hyland RH, Mo H, Lin M, Mader M, Hindes R, Albanis E, Symonds WT, Berrey MM, Muir A. 2013. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect. Dis. 13:401–408. 10.1016/S1473-3099(13)70033-1 [DOI] [PubMed] [Google Scholar]
- 58.Combet C, Garnier N, Charavay C, Grando D, Crisan D, Lopez J, Dehne-Garcia A, Geourjon C, Bettler E, Hulo C, Le Mercier P, Bartenschlager R, Diepolder H, Moradpour D, Pawlotsky JM, Rice CM, Trepo C, Penin F, Deleage G. 2007. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res. 35:D363–D366. 10.1093/nar/gkl970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandez J, Taylor D, Morhardt DR, Mihalik K, Puig M, Rice CM, Feinstone SM, Major ME. 2004. Long-term persistence of infection in chimpanzees inoculated with an infectious hepatitis C virus clone is associated with a decrease in the viral amino acid substitution rate and low levels of heterogeneity. J. Virol. 78:9782–9789. 10.1128/JVI.78.18.9782-9789.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Honegger JR, Kim S, Price AA, Kohout JA, McKnight KL, Prasad MR, Lemon SM, Grakoui A, Walker CM. 2013. Loss of immune escape mutations during persistent HCV infection in pregnancy enhances replication of vertically transmitted viruses. Nat. Med. 19:1529–1533. 10.1038/nm.3351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kato N, Sekiya H, Ootsuyama Y, Nakazawa T, Hijikata M, Ohkoshi S, Shimotohno K. 1993. Humoral immune response to hypervariable region 1 of the putative envelope glycoprotein (gp70) of hepatitis C virus. J. Virol. 67:3923–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ray SC, Mao Q, Lanford RE, Bassett S, Laeyendecker O, Wang YM, Thomas DL. 2000. Hypervariable region 1 sequence stability during hepatitis C virus replication in chimpanzees. J. Virol. 74:3058–3066. 10.1128/JVI.74.7.3058-3066.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krey T, Meola A, Keck ZY, Damier-Piolle L, Foung SK, Rey FA. 2013. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 9:e1003364. 10.1371/journal.ppat.1003364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, Heim MH. 2008. Interferon signaling and treatment outcome in chronic hepatitis C. Proc. Natl. Acad. Sci. U. S. A. 105:7034–7039. 10.1073/pnas.0707882105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garaigorta U, Chisari FV. 2009. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host. Microbe 6:513–522. 10.1016/j.chom.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, Delker DA, Jo J, Bertoletti A, Hagedorn CH, Gale M., Jr 2013. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 9:e1003330. 10.1371/journal.ppat.1003330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Jong YP, Rice CM, Ploss A. 2010. New horizons for studying human hepatotropic infections. J. Clin. Investig. 120:650–653. 10.1172/JCI42338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. 2004. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 279:48567–48587. 10.1074/jbc.M407787200 [DOI] [PubMed] [Google Scholar]