

ABSTRACT
Hepatitis C virus (HCV)-mediated liver diseases are one of the major health issues in the United States and worldwide. HCV infection has been reported to modulate microRNAs (miRNAs) that control various cell surface receptors and gene-regulatory complexes involved in hepatic inflammation and liver diseases. We report here that specific downregulation of miRNA-107 and miRNA-449a following HCV infection in patients with HCV-mediated liver diseases modulates expression of CCL2, an inflammatory chemokine upregulated in patients with chronic liver diseases, by targeting components of the interleukin-6 receptor (IL-6R) complex. Computational analysis for DNA-bound transcription factors in the CCL2 promoter identified adjacent binding sites for CCAAT/CEBPα, spleen focus-forming virus, proviral integration oncogene (SPI1/PU.1), and STAT3. We demonstrate that CEBPα, PU.1, and STAT3 interacted with each other physically to cooperatively bind to the promoter and activate CCL2 expression. Analysis of IL-6R and JAK1 expression in HCV patients by quantitative PCR showed significant upregulation when there was impaired miRNA-107 and miRNA-449a expression, along with upregulation of PU.1 and STAT3, but not CEBPα. miRNA-449a and miRNA-107 target expression of IL-6R and JAK1, respectively, in vitro and also inhibit IL-6 signaling and impair STAT3 activation in human hepatocytes. Taken together, our results demonstrate a novel gene-regulatory mechanism in which HCV-induced changes in miRNAs (miRNA-449a and miRNA-107) regulate CCL2 expression by activation of the IL-6-mediated signaling cascade, which we propose will result in HCV-mediated induction of inflammatory responses and fibrosis.
IMPORTANCE Hepatitis C virus (HCV)-induced hepatitis is a major health concern worldwide. HCV infection results in modulation of noncoding microRNAs affecting major cellular pathways, including inflammatory responses. In this study, we have identified a microRNA-regulated pathway for the chemokine CCL2 in HCV-induced hepatitis. Understanding microRNA-mediated transcriptional-regulatory pathways will result in development of noninvasive biomarkers for better disease prediction and development of effective therapeutics.
INTRODUCTION
Hepatitis C virus (HCV)-mediated liver diseases are major health issues, with an estimated 300 million people worldwide and 4 million in the United States affected (1). Most often, HCV infection leads to gradual development of fibrosis and cirrhosis—end-stage liver diseases—and poses significant risk for developing hepatocellular carcinoma (HCC) (2).
CCL2, or monocyte chemotactic protein 1, is a cytokine (11 kDa) of the CC chemokine family secreted by a variety of cell types, including fibroblasts; peripheral blood mononuclear cells; monocytes; macrophages; and epithelial, endothelial, smooth muscle, and dendritic and microglial cells (3–6). CCL2 is upregulated in patients with chronic liver diseases, such as alcoholic hepatitis, nonalcoholic steatohepatitis (NASH), and HCV infection, which are accompanied by inflammation (7–9). In mice, removal of the cytokine interleukin-6 (IL-6) and components of the IL-6 receptor (IL-6R) complex and STAT3 impairs CCL2 expression (10). IL-6 transmits a signal through components of the IL-6R complex, through dimerization of the ligand binding IL-6R and non-ligand binding gp130 (11). Upon ligand binding, JAK1, JAK2, and TYK2 are activated by phosphorylation, which in turn phosphorylates gp130 (12). Phosphorylated gp130 provides a docking site for the DNA binding STAT1 and STAT3 (12). The STATs, including STAT3, are then phosphorylated by activated JAKs in the receptor complex, leading to dimerization, followed by nuclear translocation and activation of target genes (12, 13). The DNA binding transcription factor proviral integration oncogene (SPI1/PU.1) regulates gene expression during hematopoietic differentiation and development, and its knockdown results in the absence of B-lymphoid and myeloid lineage cells (14). PU.1 is required for the development and maintenance of lymphoid and myeloid lineages of fetal liver hematopoietic stem cells (15). The homodimeric DNA binding bZIP transcription factor CCAAT/CEBPα controls cell proliferation and differentiation (16). In a subset of patients diagnosed with HCC, CEBPα is upregulated and controls the expression of genes, including hepatocyte-specific microRNA-122 (miRNA-122), involved in hepatocarcinogenesis (17, 18).
In recent years, miRNAs have been shown to play an important role in immune evasion, allograft rejection, cell cycle regulation, and cancer progression (19–21). HCV infection has been shown to result in aberrant miRNA expression that regulates HCV particle entry, propagation, and gene expression mechanisms, thus playing a pivotal role in host immune evasion and inflammatory responses (22, 23). In this report, we demonstrate that HCV-mediated regulation of miRNA-449a and miRNA-107 results in upregulation of CCL2 and modulates components of the IL-6R complex, which we propose will result in HCV-mediated induction of inflammatory responses and fibrosis.
MATERIALS AND METHODS
Patients.
Liver biopsy specimens were obtained from 10 healthy-donor livers (control), 10 alcoholic-hepatitis patients, 10 NASH patients, and 10 chronic-HCV patients (see Table S1 in the supplemental material). Patients with HIV and/or hepatitis B virus infection were not included in this study. The collection of human samples and the studies were approved by the Human Research Protection Committee at Washington University (protocol 201104075), and the patients were enrolled after obtaining written informed consent.
Plasmids and siRNA constructs.
For the reporter constructs, the CCL2 promoter regions were amplified from human genomic DNA (Zyagen, CA) by PCR using iProof High-Fidelity DNA Polymerase (Bio-Rad, CA). The PCR products were subcloned into the pGL4.11 vector (Promega, WI) upstream of a luciferase gene using the KpnI/EcoRV restriction sites. Mutation in the STAT3 binding site in the base pair (bp) −300 reporter construct was carried out using PCR. pCMV-XL4-PU.1 (SC315715) and pCMV-XL4-STAT3 (SC124165), Hsa-miRNA-449a (SC400399), Hsa-miRNA-107 (SC400023), and controls were obtained from Origene, MD. The construction of pcDNA-CEBPα is described elsewhere (23). Small interfering RNAs (siRNAs) specific for PU.1 (sc-36330), STAT3 (sc-29493), CEBPα (sc-37047), IL-6R (sc-35663), JAK1 (sc-35719), and a control (sc-37007) were purchased from Santa Cruz Biotechnology, CA. 3′-untranslated-region (UTR) clones of IL-6R (HmiT009673-MT01) and JAK1 (HmiT009849-MT01) were purchased from Genecopoeia, MD. Computational analysis to identify promoter-bound transcription factors was done using the Transcription Element Search System (TESS) (http://www.cbil.upenn.edu/cgi-bin/tess/tess). miRNA target gene analysis was done using the website http://www.targetscan.org.
miRNA and mRNA expression analysis.
Total RNA was isolated from the human hepatocytes or liver biopsy specimens using the RNA aqueous kit (Ambion, NY). miRNA-449a and miRNA-107 levels were determined using TaqMan miRNA assays and TaqMan Universal Master Mix-II (Life Technologies, NY) with predesigned primers. Quantitative PCR (qPCR) to analyze CCL2, PU.1, STAT3, CEBPα, JAK1, and IL-6R was performed as described previously (23). Each TaqMan assay was run in triplicate. The ΔΔCT values were calculated by normalizing the threshold cycle (CT) values with GAPDH (glyceraldehyde-3-phosphate dehydrogenase gene) or RNU6B expression and the respective gene expression in control samples.
Primary hepatocytes and HEPG2 cell line transfection.
Primary human hepatocytes (Life Technologies) and HEPG2 (ATCC) cells were grown and gene/siRNA transfections were carried out as described previously (23). The cytokine IL-6 (50 ng/ml; Sigma, MO) was added for 6 h wherever indicated. The optimal amount of IL-6 required for induction was determined by dose-dependent analyses. The knockdown efficiency was measured by qPCR.
Immunofluorescence microscopy and Western blotting.
For localization of PU.1, STAT3, and CEBPα, 50,000 HEPG2 cells were grown on coverslips in 24-well plates. Immunostaining was done as described previously (23). The antibodies (Abs) used for Western blotting and immunofluorescence were goat anti-CCL2 (sc-1304), mouse anti-JAK1 (sc-376996), rabbit anti-IL-6R (sc-661), mouse anti-pSTAT3 (sc-8059), goat anti-CEBPα (sc-9315), and rabbit anti-PU.1 (sc-352). Fluorescein isothiocyanate (FITC)-conjugated rabbit anti-mouse IgG (sc-358916), rhodamine-conjugated rabbit anti-goat IgG (sc-3945), FITC-conjugated goat anti-rabbit IgG (sc-2012), rhodamine-conjugated goat anti-mouse IgG (sc-2092), FITC-conjugated mouse anti-rabbit IgG (sc-2359), and rhodamine-conjugated mouse anti-goat IgG (sc-2490) were used as secondary reagents to demonstrate specific binding. The images were captured using an Eclipse 80i fluorescence microscope (Nikon, NY) and processed using Metamorph version 6.3r2 software (Molecular Devices, CA).
Luciferase reporter assay.
Hepatocytes (1 × 105) were transfected in 24-well plates as described previously (23) with 1 μg pGL4.11 luciferase reporter vector driven by the CCL2 promoters, along with 2 μg control vector or vector expressing miRNA-449a, miRNA-107, PU.1, STAT3, or CEBPα or 80 pmol of either control siRNA or siRNA specific for IL-6R, JAK1, PU.1, STAT3, or CEBPα. To control for efficiency of transfection, 0.1 μg of pRL-TK (Promega, WI), which expresses Renilla luciferase, was included. For miRNA target analysis, hepatocytes were transfected with 2 μg of a luciferase reporter construct containing IL-6R 3′ UTR or JAK1 3′ UTR, along with either control vector or vector expressing miRNA-449a or miRNA-107. Luciferase activity was measured 48 h after transfection using the Dual Luciferase Reporter Assay System (Promega, WI), and the results were normalized to Renilla luciferase.
Coimmunoprecipitation.
For immunoprecipitation of PU.1 with CEBPα, of PU.1 with pSTAT3, or of pSTAT3 with CEBPα, IL-6-treated hepatocytes (1 × 106) were used as described by Sarma and Yaseen (24). One microgram of normal goat IgG, normal mouse IgG, goat anti-CEBPα, or mouse anti-pSTAT3 was used to immunoprecipitate the protein complexes. PU.1 or pSTAT3 was detected with rabbit anti-PU.1 or mouse anti-pSTAT3, respectively.
ChIP.
Chromatin immunoprecipitation (ChIP) was carried out with ChIP-IT Express (Active Motif, CA) according to the manufacturer's instructions, as described previously (23). One microgram of Abs for control IgG, PU.1, pSTAT3, or CEBPα was used to immunoprecipitate DNA-protein complexes. CCL2 promoter regions were amplified using PCR and resolved on 2% agarose gels. The images were acquired with the Chemidoc XRS System (Bio-Rad).
RESULTS
Specific downregulation of miRNA-107 and miRNA-449a and upregulation of CCL2 in HCV livers with fibrosis.
We previously reported differential modulation of miRNAs, including miRNA-107 and miRNA-449a, in HCV patients (23, 25). To determine the expression levels of miRNA-107 and miRNA-449a following HCV infection, biopsy specimens were collected from 10 normal livers from cadaveric donors, 10 alcoholic-hepatitis patients, 10 NASH patients, and 10 chronic-HCV patients. Expression analysis using qPCR demonstrated that miRNA-107 is specifically downregulated by more than 2-fold (2.2-fold) in HCV-infected patients (Fig. 1A). In contrast, no significant difference in the expression of miRNA-107 was observed in alcoholic-hepatitis, NASH, and normal livers. Similarly, expression of miRNA-449a was also found to be specifically downregulated more than 2-fold (2.8-fold) only in the HCV-infected livers, but not in other disease conditions (Fig. 1B). RNU6B was used as the internal control.
FIG 1.

miRNA-107 and miRNA-449a are downregulated and CCL2 is upregulated in HCV patients with fibrosis. RNA was isolated from liver biopsy specimens collected from 10 chronic-HCV patients, 10 alcoholic-hepatitis patients, 10 NASH patients, and 10 healthy-donor livers (control). Expression of miRNA-107 (A), miRNA-449a (B), and CCL2 (C) was determined by qPCR. The ΔΔCT values were calculated by normalizing the CT values with RNU6B or GAPDH and miRNA-107 (A), miRNA-449a (B), or CCL2 (C) levels in controls. *, P < 0.05; two-tailed t test. The error bars represent standard deviations (SD) obtained from three independent replicates.
Expression analysis showed upregulation of CCL2 in alcoholic hepatitis (6.2-fold), NASH (5.8-fold), and HCV (10.0-fold) compared to normal livers (Fig. 1C). Expression of CCL2 in HCV livers was significantly higher than in those of patients with alcoholic hepatitis and NASH (P < 0.001).
IL-6 regulates CCL2 activation through the IL-6R complex.
Components of the IL-6R complex were shown to be essential for CCL2 expression (10). To determine the role of IL-6 in CCL2 expression, the reporter construct containing the −2,000-bp region of the human CCL2 promoter was transfected into human hepatocytes and treated or not with IL-6, and luciferase activity was measured. More than a 4-fold (4.7-fold) increase in the CCL2 reporter activity was observed in IL-6-treated cells compared to the untreated cells, which was further confirmed by immunoblotting (Fig. 2A). This strongly suggests that IL-6 activates CCL2 through modulation of upstream transcription factors. Further, knockdown of the IL-6 cell surface receptor gene, IL-6R, resulted in a 2-fold decrease in IL-6-mediated CCL2 activation and protein expression compared to controls (Fig. 2A). Knockdown of JAK1 also resulted in significant impairment (1.8-fold) of IL-6-mediated CCL2 reporter activity and protein expression (Fig. 2B). Quantitative gene expression analysis showed 88% and 86% knockdown of IL-6R and JAK1, respectively, in siRNA-treated cells (see Fig. S1 in the supplemental material).
FIG 2.

IL-6 regulates CCL2 activation through the IL-6 receptor complex components IL-6R and JAK1. (A) (Top) Hepatocytes were transfected with a reporter construct driven by −2,000 bp of the CCL2 promoter with (+) or without (−) IL-6 and nonspecific (control) siRNA or siRNAs specific for IL-6R. Firefly luciferase activity in the cells was measured 48 h after transfection and normalized to a Renilla luciferase internal control. The numbers represent the fold change over the untreated control (average of three independent replicates); the error bars represent SD. *, P < 0.05; two-tailed t test. (Bottom) The hepatocytes were immunoblotted with anti-CCL2. Actin was used as the loading control. (B) (Top) Hepatocytes were transfected with the CCL2 reporter construct with either nonspecific (control) siRNA or siRNAs specific for JAK1. The cells were treated with IL-6, and luciferase activity was measured as described for panel A. (Bottom) The hepatocytes were immunoblotted with anti-CCL2 and anti-actin.
Identification of putative DNA binding transcription factors in the CCL2 promoter regions.
To identify DNA binding transcription factors that regulate the expression of CCL2, a computational analysis of −2,000 bp of the promoter region was performed using TESS. This bioinformatics analysis identified two putative DNA binding sites for CEBPα (GCAAT), four sites for PU.1 (GAGGAA), and one site for STAT3 (TTCCTGGAA) at the bp −138 position (Fig. 3A) in close proximity to each other, suggesting that these three transcription factors may be part of a transcriptional-regulatory complex that mediates CCL2 expression.
FIG 3.

Transcription factors CEBPα, PU.1, and STAT3 cooperate to regulate CCL2 expression. (A) Schematic representation of transcription factor binding sites in the CCL2 promoter identified by computational analysis of −2,000 bp upstream of the open reading frame using TESS. The vertical bars represent the predicted consensus binding sites in the DNA for transcription factors CEBPα, PU.1, and STAT3. (B) (Top) Hepatocytes were transfected with the −2,000-bp CCL2 promoter-driven luciferase construct, in addition to a control vector or vectors expressing CEBPα, PU.1, and STAT3 or any two of these factors. Luciferase activity was measured as described for Fig. 2A. (Bottom) Overexpression of CEBPα, PU.1, or STAT3 was verified by immunoblotting with anti-CEBPα, anti-PU.1, or anti-STAT3. (C) Hepatocytes were transfected with the CCL2 promoter-driven luciferase construct, in addition to either nonspecific (control) siRNA or siRNAs specific for CEBPα, PU.1, STAT3, or any two of these factors. Luciferase activity was measured as described for Fig. 2A. *, P value obtained by a two-tailed t test between the control and the individual transcription factors; **, P value between cotransfected transcription factors and either factor alone. (D) Essential regions in the CCL2 promoter required for IL-6-mediated expression. Hepatocytes were transfected with luciferase reporters driven by deletion constructs of CCL2 promoter constructs (bp −2000, −8000, −300, or −300 with STAT3 binding site mutated [*]) (black bars) with (+) or without (−) IL-6. Luciferase activity was measured as described for Fig. 2A. *, P value obtained by a two-tailed t test between conditions with or without IL-6.
To demonstrate that these transcription factors regulate CCL2 expression, human hepatocytes were transfected with the CCL2 reporter construct, along with either empty vector or vector expressing PU.1, STAT3, or CEBPα, and treated with IL-6 (Fig. 3B). A significant increase in reporter activity was observed compared to the empty vector with overexpression of PU.1 (3.3-fold), STAT3 (7.0-fold), and CEBPα (5.4-fold) (Fig. 3B). To determine whether these transcription factors cooperate in activating CCL2, two of the transcription factors (PU.1/STAT3, PU.1/CEBPα, or STAT3/CEBPα) were cotransfected into hepatocytes, along with the CCL2 reporter construct. Cotransfection resulted in a significant increase in reporter activity compared to either factor alone, suggesting cooperation between the factors in regulating CCL2 expression (Fig. 3B, top). Overexpression of the transcription factors in the hepatocytes was confirmed by immunoblotting (Fig. 3B).
To further demonstrate that these transcription factors are required for IL-6-mediated expression of CCL2, human hepatocytes were transfected with the CCL2 reporter construct, along with either control siRNAs or siRNAs specific for PU.1, STAT3, or CEBPα, and treated with IL-6. Knockdown of PU.1 resulted in 28%, that of STAT3 resulted in 37%, and that of CEBPα resulted in 25% decrease in the CCL2 reporter activity compared to the control (Fig. 3C). Knockdown of any two transcription factors (PU.1/STAT3, PU.1/CEBPα, or STAT3/CEBPα) resulted in a significant decrease in reporter activity compared to knockdown of either factor alone, suggesting cooperation between these factors in the transcriptional activation of CCL2 (Fig. 3C). Quantitative gene expression analysis showed 77%, 73%, and 75% knockdown of PU.1, STAT3, and CEBPα in siRNA-treated cells (see Fig. S1 in the supplemental material). However, coexpression or coknockdown of PU.1, STAT3, and CEBPα together impaired cell viability.
To determine whether the transcription factor binding sites are essential, deletion mutants were designed based on the locations of the DNA binding sites for CEBPα, PU.1, and STAT3 on the CCL2 promoter. These deletion mutants (Fig. 3D, black bars) were introduced into hepatocytes, treated or not with IL-6, and the luciferase activity was measured. Sequential deletion of the CCL2 promoter impaired IL-6-mediated transcriptional induction compared to the bp −2000 promoter (Fig. 3D). No significant differences in reporter activity were observed in untreated cells, indicating that IL-6 is required for transcriptional regulators to activate CCL2 (Fig. 3D). For reporter constructs (bp −2000, −800, and −300), IL-6 treatment resulted in more than 2.5-fold increase in reporter activity compared to untreated constructs. However, a point mutation in the STAT3 binding site on the CCL2 promoter completely abolished the IL-6 response (Fig. 3D). This indicates that STAT3, along with CEBPα and PU.1, plays a major role in IL-6-mediated CCL2 induction.
CEBPα, PU.1, and STAT3 form a transcriptional-activation complex and bind to the CCL2 promoter.
To determine whether CEBPα, PU.1, and STAT3 bind to adjacent DNA binding sites on the CCL2 promoter, DNA-protein complexes from IL-6-treated hepatocytes were immunoprecipitated with anti-CEBPα,r anti-PU.1, anti-STAT3, or control IgGs. ChIP and PCR using primers specific for CCL2 promoter regions (Fig. 4A, black bars) demonstrated that CEBPα, PU.1, and STAT3 bind to the promoter (Fig. 4B). CEBPα binds strongly to the −800/−1200 and −400/−800 regions and weakly to the −1/−400 region; PU.1 binds strongly to the −800/−1200 and −1/−400 regions and weakly to the −400/−800 region of the CCL2 promoter. The observed binding of CEBPα in the −800/−1200 region is likely due to the presence of binding sites within ±250 bp of the PCR amplicon. The DNA binding pattern of CEBPα and PU.1 strongly correlates with the locations of the consensus binding sites on the CCL2 promoter. STAT3 binds strongly to the −1/−400 region, encompassing the binding site (bp −138). ChIP with the control IgGs did not enrich CCL2 promoter regions, demonstrating the specificity for these transcription factors. No binding was observed in PCRs carried out with primers specific for the −300/−700 region of the ACTIN (ACTA1) promoter that lacks these binding sites (Fig. 4B).
FIG 4.

CEBPα, PU.1, and STAT3 interact to bind to the CCL2 promoter. (A) Schematic representation of the CCL2 promoter showing binding sites for CEBPα, PU.1, and STAT3. The black horizontal bars represent regions specific for the PCR primers. (B) ChIP showing transcription factors binding to the CCL2 promoter. DNA-protein complexes were immunoprecipitated with anti-CEBPα, anti-PU.1, anti-STAT3, or isotype control IgG from hepatocytes. Specific CCL2 promoter regions (shown in panel A) were detected by PCR. The top four rows (CEBPα, PU.1, STAT3, IgG) represent immunoprecipitated chromatin, and the bottom row represents total chromatin (Input). ACTIN promoter amplification is shown as the negative control. (C) STAT3 is required for PU.1 binding to the CCL2 promoter. Hepatocytes were transfected with either nonspecific (control) siRNA or siRNAs specific for STAT3. DNA-protein complexes were immunoprecipitated with anti-PU.1. The top row represents CCL2 promoter regions immunoprecipitated by anti-PU.1 in hepatocytes transfected with nonspecific siRNA, the middle row represents hepatocytes transfected with siRNAs specific for STAT3, and the bottom row shows input chromatin (Input). (D) Coimmunoprecipitation showing interaction between CEBPα, PU.1, and STAT3 in hepatocytes. Whole-cell lysates (Input) from IL-6-treated hepatocytes were subjected to immunoprecipitation with either isotype control IgG, anti-CEBPα, or anti-STAT3, and the immunoprecipitated complexes (IP) were detected by immunoblotting with anti-PU.1 or anti-STAT3. (E) Nuclear colocalization of CEBPα, PU.1, and STAT3. HEPG2 cells were treated with IL-6 and coimmunostained with anti-STAT3/anti-CEBPα (top row), anti-PU.1/anti-STAT3 (middle row), or anti PU.1/anti-CEBPα (bottom row).
To demonstrate that these transcription factors are interdependent, human hepatocytes were transfected with either control siRNA or siRNA specific for STAT3 and treated with IL-6, and ChIP was carried out with either control IgG or anti-PU. Knockdown of STAT3 impaired PU.1 DNA binding to its consensus site in the CCL2 promoter (Fig. 4D). PU.1 binding to the −800/−1200 and −1/−400 regions in the STAT3 knockout cells was weaker than to the cells with nonspecific siRNA. No DNA binding was observed in the −400/−800 region in the STAT3 knockout cells.
To determine that CEBPα, PU.1, and STAT3 form a transcriptional-regulatory complex, lysates from IL-6-treated hepatocytes were subjected to immunoprecipitation with either isotype control IgG, anti-CEBPα, or anti-STAT3, followed by immunoblotting with anti-PU.1 or anti-STAT3. Both PU.1 and STAT3 were coimmunoprecipitated with endogenous CEBPα, whereas no binding was observed with the control IgG (Fig. 4D, left). PU.1 was coimmunoprecipitated with endogenous STAT3, and no binding was observed with the control IgG (Fig. 4D, right).
To further define the interaction between CEBPα, PU.1, and STAT3, HEPG2 cells were treated with IL-6 and coimmunostained with anti-STAT3/anti-CEBPα, anti-PU.1/anti-STAT3, or anti-PU.1/anti-CEBPα. All three transcription factors primarily colocalized to the nucleus (Fig. 4E; for high-resolution images and controls, see Fig. S2 and S3 in the supplemental material). The nuclear colocalization of STAT3 with CEBPα (Fig. 4E, top), PU.1 with STAT3 (4E, middle), and PU.1 with CEBPα (4E, bottom) and coimmunoprecipitation (Fig. 4D) demonstrate that these transcription factors physically interact and are part of a larger transcription-regulatory complex.
miRNA-449a and miRNA-107 regulate IL-6-mediated CCL2 expression and STAT3 phosphorylation by targeting IL-6R and JAK1.
To define the role of STAT3 in IL-6-mediated CCL2 expression, IL-6R or JAK1 knockout hepatocytes were treated or not with IL-6 and subjected to immunoblotting using anti-pSTAT3, which is specific for the phosphorylated form of STAT3. IL-6 treatment resulted in increased accumulation of phosphorylated STAT3, and knockdown of IL-6R and JAK1 resulted in impairment of IL-6-mediated STAT3 phosphorylation (Fig. 5A).
FIG 5.

miRNA-449a and miRNA-107 regulate IL-6-mediated CCL2 expression and STAT3 phosphorylation by targeting IL-6R and JAK. (A) Hepatocytes were transfected with either nonspecific (control) siRNA or siRNAs specific for IL-6R (left) or JAK1 (right) and treated (+) or not (−) with IL-6. Phosphorylated STAT3 was detected by immunoblotting with anti-pSTAT3. (B) Hepatocytes were transfected with a CCL2 luciferase reporter construct, in addition to the control vector or vector expressing miRNA-449a, miRNA-107, or both. The cells were treated (+) or not (−) with IL-6, and luciferase activity was measured as described for Fig. 2A. (C and D) Detection of CCL2, pSTAT3, and actin in hepatocytes from panel A using anti-CCL2, anti-pSTAT3, or anti-actin.
To test whether HCV-mediated upregulation of CCL2 is regulated by miRNAs, human hepatocytes were transfected with the CCL2 reporter construct, along with a control vector or vector expressing miRNA-449a and/or miRNA-107, and treated or not with IL-6. Expression of either miRNA-449a or miRNA-107 resulted in significant impairment (>2.5-fold) of IL-6-mediated expression of CCL2 (Fig. 5B). Expression of both miRNA-449a and miRNA-107 resulted in further impairment (6.1-fold) of CCL2 activation compared to either miRNA alone, indicating cooperation between the two miRNAs (Fig. 5B). Expression of either of these miRNAs also resulted in decreased CCL2 protein and STAT3 phosphorylation (Fig. 5C), which was further accentuated by expression of both miRNAs together, indicating cooperation in mediating transcriptional signaling (Fig. 5D).
Computational target prediction of miRNA-449a and miRNA-107 using Targetscan identified the 3′-UTR regions of IL-6R and JAK1, respectively, as putative targets for translational silencing (Fig. 6A and E). To test the specificity of these miRNAs, hepatocytes were transfected with a luciferase reporter containing the 3′-UTR region of IL-6R or JAK1, along with either control vector or vector expressing miRNA-449a or miRNA-107, and luciferase activity was measured. Expression of miRNA-449a and miRNA-107 resulted in significant reduction in the IL-6R (3.8-fold) and JAK1 (3-fold) UTR reporter activity, indicating direct interaction between the miRNAs and the 3′-UTR regions (Fig. 6B and F). To further demonstrate that these miRNAs impair gene expression, hepatocytes were transfected with either control vector or vector expressing miRNA-449a or miRNA-107, and expression of IL-6R and/or JAK1 was determined by qPCR and immunoblotting. Increased expression of miRNA-449a or miRNA-107 resulted in more than 2-fold downregulation of IL-6R (55%) and JAK1 (67%) RNA and protein levels (Fig. 6C, D, G, and H).
FIG 6.

miRNA-449a and miRNA-107 target IL-6R and JAK1 for silencing. Schematic representation of the miRNA-449a (A) and miRNA-107 (E) target sites in the IL-6R and JAK1 3′ UTRs, respectively. (B and F) Hepatocytes were transfected with a luciferase reporter construct containing the IL-6R UTR (B) or the JAK1 UTR (F), along with either a control vector or a vector expressing miRNA-449a or miRNA-107, and luciferase activity was measured. (C, D, G, and H) Hepatocytes were transfected with an empty vector (−) or a vector expressing (+) miRNA-449a (C and D) or miRNA-107 (G and H), and expression of IL-6R (C) and JAK1 (G) was determined by qPCR. The ΔΔCT values were calculated by normalizing the CT values with GAPDH and expression of IL-6R and JAK1, respectively, in controls. *, P < 0.005; two-tailed t test. IL-6R (D) and JAK1 (H) protein levels in hepatocytes from panels C and G were detected using anti-IL-6R and anti-JAK1, respectively. Actin is shown as the loading control.
IL-6R, JAK1, PU.1, and STAT3 are upregulated in HCV patients.
To determine whether downregulation of miRNA-449a and miRNA-107 in HCV livers is accompanied by upregulation of their respective targets, IL-6R and JAK1, RNA was isolated from 10 normal, 10 alcoholic-hepatitis, 10 NASH, and 10 chronic-HCV livers. qPCR analysis showed that IL-6R and JAK1 are upregulated more than 2-fold (2.7- and 2.2-fold, respectively) in HCV livers (Fig. 7A and B). In contrast, no significant difference in the expression of IL-6R and JAK1 was noted in alcoholic-hepatitis, NASH, and normal livers. These results, along with our results from in vitro studies using human hepatocytes, clearly indicate that upregulation of IL-6R, JAK1, and CCL2 (Fig. 1C) in HCV livers can be attributed to downregulation of miRNA-449a and miRNA-107.
FIG 7.

IL-6R, JAK1, PU.1, and STAT3 are upregulated in HCV patients. Total RNA was isolated from liver biopsy specimens obtained from 10 chronic-HCV patients, 10 alcoholic-hepatitis patients, 10 NASH patients, and 10 healthy-donor livers (control). Expression levels of IL-6R (A), JAK1 (B), PU.1 (C), STAT3, and CEBPα (E) were determined by qPCR. The ΔΔCT values were calculated by normalizing the CT values with GAPDH expression and IL-6R, JAK1, PU.1, STAT3, and CEBPα expression in controls. *, P < 0.05; two-tailed t test. The error bars represent SD.
Expression analysis of the transcription factors also showed that PU.1 is upregulated (3.6-fold) only in HCV livers compared to non-HCV and normal livers (Fig. 7C). However, expression of STAT3 is upregulated 2.4-fold in alcoholic-hepatitis patients, 3.1-fold in NASH patients, and 6.6-fold in HCV livers compared to normal livers (Fig. 7D). We did not detect any differences in the expression of CEBPα in any of these patient populations (Fig. 7E). GAPDH was used as the internal control.
DISCUSSION
HCV infection has been shown to induce inflammatory cytokines/chemokines, leading to activation of hepatic stellate cells (HSCs) and development of fibrosis (26). We have previously identified differential regulation of inflammatory cytokines, such as IL-17, IL-1β, IL-6, IL-8, monocyte chemoattractant protein 1 (MCP-1), gamma interferon (IFN-γ), IL-4, IL-5, IL-10, and YKL40 in HCV patients with fibrosis (23, 27). In this study, we demonstrate that HCV-mediated specific regulation of miRNA-449a and miRNA-107 alters expression of CCL2, a major inflammatory chemokine that is regulated by the IL-6R complex components IL-6R and JAK1. It is known that CCL2 is expressed in the HSCs, regulates HSC chemotaxis to the site of injury, and promotes HCV-induced hepatic fibrosis (28–30). Increased expression of CCL2 is associated with chronic HCV infection, and IFN-α therapy has been shown to reduce its expression (9). Further, inhibition of CCL2 results in reduced cellular infiltration, steatohepatitis, inflammatory responses, and fibrosis during liver injury (31, 32). IL-6 has also been shown to be upregulated in HCV patients through NS5A-mediated induction of Toll-like receptor 4 (TLR4) (33), and IL-6 can induce CCL2, leading to increased inflammatory responses (34). The results presented in this report indicate that HCV-induced induction of IL-6 can also lead to induction of CCL2, resulting in HSC chemotaxis and hepatic fibrosis (Fig. 2).
Transcription factors play a significant role in regulating inflammatory responses, development of fibrosis, and HCC. Our results demonstrate that CEBPα, PU.1, and STAT3 form a functional transcriptional-activation complex to regulate CCL2 expression in hepatocytes (Fig. 3 and 4). CEBPα has been shown to regulate HSC activation, which plays a major role in hepatic fibrosis (35, 36). PU.1 knockout mice lack neutrophils, macrophages, and mast cells and do not elicit inflammatory responses or fibrosis (37). Both CEBPα and PU.1 bind and cooperate to activate expression of myeloid-specific genes, such as murine neutrophil elastase promoter and granulocyte colony-stimulating factor (G-CSF) receptor, which is necessary for neutrophil differentiation (38, 39). Further, STAT3 binding to the PU.1 promoter is required for its expression and subsequent hematological differentiation (40). STAT3 has also been shown to bind to the promoter and activate CCL2 expression in rat smooth muscle cells (41). Activated STAT3 is a proviral host factor and is required for HCV replication (42). Therefore, the CEBPα-PU.1-STAT3 transcription-regulatory complex plays a significant role in cellular differentiation, inflammatory gene expression, and development of hepatic fibrosis.
miRNAs are regulators of HCV infection, fibrosis, cirrhosis, and development of HCC following liver transplantation (21, 43). miRNAs regulate cytokine signaling on HSCs and expression of inflammatory cytokines and interferons, leading to fibrosis (44, 45). We have shown that miRNA-449a and miRNA-107 are specifically downregulated in HCV patients (Fig. 1A and B) and that miRNA-449a regulates expression of the inflammatory cytokine YKL40 through components of the NOTCH signaling pathway (23). miRNA-125b and miRNA-124, which are downregulated in hepatocarcinoma patients, target IL-6R to modulate the IL-6/STAT3 pathway and IL-6 production and induce tumorigenicity (46–48). We demonstrate that expression of miRNA targets IL-6R and JAK1 and that transcription factors PU.1 and STAT3 are upregulated specifically in HCV patients compared to non-HCV patients and healthy individuals (Fig. 7). Although STAT3 was upregulated in alcoholic-hepatitis and NASH patients compared to healthy donors, the expression was 2-fold higher in HCV patients. We propose that in cases of HCV infection, the viral protein NS5A or the core protein may increase its expression and promote phosphorylation and nuclear translocation of STAT3 (49, 50). Therefore, HCV-induced downregulation of miRNA-449a and miRNA-107 in human livers can activate transcriptional-regulatory complexes (CEBPα-PU.1-STAT3) through modulation of cell surface receptor (IL-6R/JAK1) expression, thereby promoting inflammatory responses that lead to development of fibrosis and HCC (Fig. 8). Since we and others have found that CCL2 is also elevated in patients with alcoholic hepatitis and NASH (Fig. 1C) (7–9), although significantly lower than in HCV infection, it is possible that parallel gene-regulatory mechanisms may operate in non-HCV-mediated liver diseases.
FIG 8.
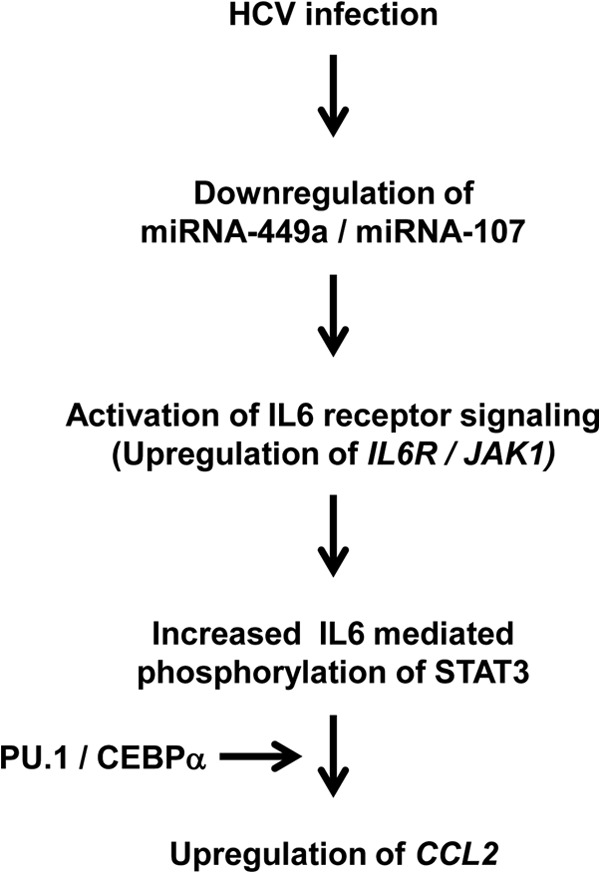
Schematic representation of HCV-mediated modulation of miRNA-449a and miRNA-107 leading to CCL2 expression. HCV infection results in downregulation of miRNA-449a and miRNA-107, which leads to upregulation of IL-6 receptor complex components IL-6R and JAK1. This results in increased IL-6-mediated activation and nuclear translocation of phosphorylated STAT3. Activated STAT3, in cooperation with PU.1 and CEBPα, regulates CCL2 expression through DNA binding.
Taken together, the results of this study provide novel insights into the miRNA-mediated gene regulation mechanisms that play a role in HCV-mediated inflammatory responses and development of liver fibrosis. The HCV-specific modulation of miRNA, transcription factors, and cell surface receptors identified in this report can be utilized for the potential development of noninvasive biomarkers that can identify HCV patients with higher risk for developing fibrosis, cirrhosis, and HCC, as well as in developing methods for altering miRNA expression levels (miRNA mimics or antagomirs) as a therapeutic strategy.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH DK065982 and the Barnes-Jewish Foundation (T.M.).
We thank Billie Glasscock for her help in preparing the manuscript.
We have no financial conflicts of interest.
Footnotes
Published ahead of print 15 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03060-13.
REFERENCES
- 1.Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, Kaslow RA, Margolis HS. 1999. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N. Engl. J. Med. 341:556–562. 10.1056/NEJM199908193410802 [DOI] [PubMed] [Google Scholar]
- 2.Seeff LB. 2002. Natural history of chronic hepatitis C. Hepatology 36:S35–S46. 10.1053/jhep.2002.36806 [DOI] [PubMed] [Google Scholar]
- 3.Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. 1994. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc. Natl. Acad. Sci. U. S. A. 91:3652–3656. 10.1073/pnas.91.9.3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, Gerrity R, Schwartz CJ, Fogelman AM. 1990. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. U. S. A. 87:5134–5138. 10.1073/pnas.87.13.5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM. 1991. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J. Biol. Chem. 266:9912–9918 [PubMed] [Google Scholar]
- 6.Xu LL, Warren MK, Rose WL, Gong W, Wang JM. 1996. Human recombinant monocyte chemotactic protein and other C-C chemokines bind and induce directional migration of dendritic cells in vitro. J. Leukoc. Biol. 60:365–371 [DOI] [PubMed] [Google Scholar]
- 7.Bertola A, Bonnafous S, Anty R, Patouraux S, Saint-Paul MC, Iannelli A, Gugenheim J, Barr J, Mato JM, Le Marchand-Brustel Y, Tran A, Gual P. 2010. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One 5:e13577. 10.1371/journal.pone.0013577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Degre D, Lemmers A, Gustot T, Ouziel R, Trepo E, Demetter P, Verset L, Quertinmont E, Vercruysse V, Le Moine O, Deviere J, Moreno C. 2012. Hepatic expression of CCL2 in alcoholic liver disease is associated with disease severity and neutrophil infiltrates. Clin. Exp. Immunol. 169:302–310. 10.1111/j.1365-2249.2012.04609.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narumi S, Tominaga Y, Tamaru M, Shimai S, Okumura H, Nishioji K, Itoh Y, Okanoue T. 1997. Expression of IFN-inducible protein-10 in chronic hepatitis. J. Immunol. 158:5536–5544 [PubMed] [Google Scholar]
- 10.Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J. 2013. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J. Biol. Chem. 288:1489–1499. 10.1074/jbc.M112.419788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. 1990. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 63:1149–1157. 10.1016/0092-8674(90)90411-7 [DOI] [PubMed] [Google Scholar]
- 12.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. 1998. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 334:297–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang S. 2007. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin. Cancer Res. 13:1362–1366. 10.1158/1078-0432.CCR-06-2313 [DOI] [PubMed] [Google Scholar]
- 14.Scott EW, Simon MC, Anastasi J, Singh H. 1994. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265:1573–1577. 10.1126/science.8079170 [DOI] [PubMed] [Google Scholar]
- 15.Kim HG, de Guzman CG, Swindle CS, Cotta CV, Gartland L, Scott EW, Klug CA. 2004. The ETS family transcription factor PU.1 is necessary for the maintenance of fetal liver hematopoietic stem cells. Blood 104:3894–3900. 10.1182/blood-2002-08-2425 [DOI] [PubMed] [Google Scholar]
- 16.Johnson PF. 2005. Molecular stop signs: regulation of cell-cycle arrest by C/EBP transcription factors. J. Cell Sci. 118:2545–2555. 10.1242/jcs.02459 [DOI] [PubMed] [Google Scholar]
- 17.Lu GD, Leung CH, Yan B, Tan CM, Low SY, Aung MO, Salto-Tellez M, Lim SG, Hooi SC. 2010. C/EBPalpha is up-regulated in a subset of hepatocellular carcinomas and plays a role in cell growth and proliferation. Gastroenterology 139:632–643. 10.1053/j.gastro.2010.03.051 [DOI] [PubMed] [Google Scholar]
- 18.Zeng C, Wang R, Li D, Lin XJ, Wei QK, Yuan Y, Wang Q, Chen W, Zhuang SM. 2010. A novel GSK-3 beta-C/EBP alpha-miR-122-insulin-like growth factor 1 receptor regulatory circuitry in human hepatocellular carcinoma. Hepatology 52:1702–1712. 10.1002/hep.23875 [DOI] [PubMed] [Google Scholar]
- 19.Gracias DT, Katsikis PD. 2011. MicroRNAs: key components of immune regulation. Adv. Exp. Med. Biol. 780:15–26. 10.1007/978-1-4419-5632-3_2 [DOI] [PubMed] [Google Scholar]
- 20.McManus MT. 2003. MicroRNAs and cancer. Semin. Cancer Biol. 13:253–258. 10.1016/S1044-579X(03)00038-5 [DOI] [PubMed] [Google Scholar]
- 21.Sarma NJ, Tiriveedhi V, Ramachandran S, Crippin J, Chapman W, Mohanakumar T. 2012. Modulation of immune responses following solid organ transplantation by microRNA. Exp. Mol. Pathol. 93:378–385. 10.1016/j.yexmp.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girard M, Jacquemin E, Munnich A, Lyonnet S, Henrion-Caude A. 2008. miR-122, a paradigm for the role of microRNAs in the liver. J. Hepatol. 48:648–656. 10.1016/j.jhep.2008.01.019 [DOI] [PubMed] [Google Scholar]
- 23.Sarma NJ, Tiriveedhi V, Subramanian V, Shenoy S, Crippin JS, Chapman WC, Mohanakumar T. 2012. Hepatitis C virus mediated changes in miRNA-449a modulates inflammatory biomarker YKL40 through components of the NOTCH signaling pathway. PLoS One 7:e50826. 10.1371/journal.pone.0050826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarma NJ, Yaseen NR. 2011. Amino-terminal enhancer of split (AES) interacts with the oncoprotein NUP98-HOXA9 and enhances its transforming ability. J. Biol. Chem. 286:38989–39001. 10.1074/jbc.M111.297952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramachandran S, Ilias Basha H, Sarma NJ, Lin Y, Crippin JS, Chapman WC, Mohanakumar T. 2013. Hepatitis C virus induced miR200c down modulates FAP-1, a negative regulator of Src signaling and promotes hepatic fibrosis. PLoS One 8:e70744. 10.1371/journal.pone.0070744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishitsuji H, Funami K, Shimizu Y, Ujino S, Sugiyama K, Seya T, Takaku H, Shimotohno K. 2013. HCV infection induces inflammatory cytokines and chemokines mediated by the cross-talk between hepatocytes and stellate cells. J. Virol. 10.1128/JVI.00974-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basha HI, Subramanian V, Seetharam A, Nath DS, Ramachandran S, Anderson CD, Shenoy S, Chapman WC, Crippin JS, Mohanakumar T. 2011. Characterization of HCV-specific CD4+Th17 immunity in recurrent hepatitis C-induced liver allograft fibrosis. Am. J. Transplant. 11:775–785. 10.1111/j.1600-6143.2011.03458.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marra F, Romanelli RG, Giannini C, Failli P, Pastacaldi S, Arrighi MC, Pinzani M, Laffi G, Montalto P, Gentilini P. 1999. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology 29:140–148. 10.1002/hep.510290107 [DOI] [PubMed] [Google Scholar]
- 29.Muhlbauer M, Bosserhoff AK, Hartmann A, Thasler WE, Weiss TS, Herfarth H, Lock G, Scholmerich J, Hellerbrand C. 2003. A novel MCP-1 gene polymorphism is associated with hepatic MCP-1 expression and severity of HCV-related liver disease. Gastroenterology 125:1085–1093. 10.1016/S0016-5085(03)01213-7 [DOI] [PubMed] [Google Scholar]
- 30.Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, Schwabe RF, Brenner DA. 2009. CCR2 promotes hepatic fibrosis in mice. Hepatology 50:185–197. 10.1002/hep.22952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T, Trautwein C, Tacke F. 2012. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 61:416–426. 10.1136/gutjnl-2011-300304 [DOI] [PubMed] [Google Scholar]
- 32.Galastri S, Zamara E, Milani S, Novo E, Provenzano A, Delogu W, Vizzutti F, Sutti S, Locatelli I, Navari N, Vivoli E, Caligiuri A, Pinzani M, Albano E, Parola M, Marra F. 2012. Lack of CC chemokine ligand 2 differentially affects inflammation and fibrosis according to the genetic background in a murine model of steatohepatitis. Clin. Sci. 123:459–471. 10.1042/CS20110515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Machida K, Cheng KT, Sung VM, Levine AM, Foung S, Lai MM. 2006. Hepatitis C virus induces Toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J. Virol. 80:866–874. 10.1128/JVI.80.2.866-874.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biswas P, Delfanti F, Bernasconi S, Mengozzi M, Cota M, Polentarutti N, Mantovani A, Lazzarin A, Sozzani S, Poli G. 1998. Interleukin-6 induces monocyte chemotactic protein-1 in peripheral blood mononuclear cells and in the U937 cell line. Blood 91:258–265 [PubMed] [Google Scholar]
- 35.Huang J, Zhang JS, Huang GC, Tang QQ, Chen C, Zhang XR, Chen Q. 2004. Expression of CCAAT/enhancer-binding protein in cultured rat hepatic stellate cells and its significance. Zhonghua Gan Zang Bing Za Zhi 12:259–262 [PubMed] [Google Scholar]
- 36.Tao LL, Cheng YY, Ding D, Mei S, Xu JW, Yu J, Ou-Yang Q, Deng L, Chen Q, Li QQ, Xu ZD, Liu XP. 2012. C/EBP-alpha ameliorates CCl(4)-induced liver fibrosis in mice through promoting apoptosis of hepatic stellate cells with little apoptotic effect on hepatocytes in vitro and in vivo. Apoptosis 17:492–502. 10.1007/s10495-012-0700-y [DOI] [PubMed] [Google Scholar]
- 37.Martin P, D'Souza D, Martin J, Grose R, Cooper L, Maki R, McKercher SR. 2003. Wound healing in the PU.1 null mouse: tissue repair is not dependent on inflammatory cells. Curr. Biol. 13:1122–1128. 10.1016/S0960-9822(03)00396-8 [DOI] [PubMed] [Google Scholar]
- 38.Oelgeschlager M, Nuchprayoon I, Luscher B, Friedman AD. 1996. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol. Cell. Biol. 16:4717–4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith LT, Hohaus S, Gonzalez DA, Dziennis SE, Tenen DG. 1996. PU.1 (Spi-1) and C/EBP alpha regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood 88:1234–1247 [PubMed] [Google Scholar]
- 40.Hegde S, Ni S, He S, Yoon D, Feng GS, Watowich SS, Paulson RF, Hankey PA. 2009. Stat3 promotes the development of erythroleukemia by inducing Pu.1 expression and inhibiting erythroid differentiation. Oncogene 28:3349–3359. 10.1038/onc.2009.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Potula HS, Wang D, Quyen DV, Singh NK, Kundumani-Sridharan V, Karpurapu M, Park EA, Glasgow WC, Rao GN. 2009. Src-dependent STAT-3-mediated expression of monocyte chemoattractant protein-1 is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. J. Biol. Chem. 284:31142–31155. 10.1074/jbc.M109.012526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCartney EM, Helbig KJ, Narayana SK, Eyre NS, Aloia AL, Beard MR. 22 May 2013. Signal transducer and activator of transcription 3 (STAT3) is a pro-viral host factor for hepatitis C virus. Hepatology 10.1002/hep.26496 [DOI] [PubMed] [Google Scholar]
- 43.He Y, Huang C, Zhang SP, Sun X, Long XR, Li J. 2012. The potential of microRNAs in liver fibrosis. Cell Signal. 24:2268–2272. 10.1016/j.cellsig.2012.07.023 [DOI] [PubMed] [Google Scholar]
- 44.He Y, Huang C, Sun X, Long XR, Lv XW, Li J. 2012. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell Signal. 24:1923–1930. 10.1016/j.cellsig.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 45.McCoy CE. 2012. The role of miRNAs in cytokine signaling. Front. Biosci. 17:2161–2171 [DOI] [PubMed] [Google Scholar]
- 46.Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. 2010. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 31:766–776. 10.1093/carcin/bgp250 [DOI] [PubMed] [Google Scholar]
- 47.Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K, Hadzopoulou-Cladaras M, Iliopoulos D. 2011. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 147:1233–1247. 10.1016/j.cell.2011.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jia HY, Wang YX, Yan WT, Li HY, Tian YZ, Wang SM, Zhao HL. 2012. MicroRNA-125b functions as a tumor suppressor in hepatocellular carcinoma cells. Int. J. Mol. Sci. 13:8762–8774. 10.3390/ijms13078762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gong G, Waris G, Tanveer R, Siddiqui A. 2001. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. U. S. A. 98:9599–9604. 10.1073/pnas.171311298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida T, Hanada T, Tokuhisa T, Kosai K, Sata M, Kohara M, Yoshimura A. 2002. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J. Exp. Med. 196:641–653. 10.1084/jem.20012127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.