

ABSTRACT
Barrier dysfunction of airway epithelium may increase the risk for acquiring secondary infections or allergen sensitization. Both rhinovirus (RV) and polyinosinic-polycytidilic acid [poly(I·C)], a double-stranded RNA (dsRNA) mimetic, cause airway epithelial barrier dysfunction, which is reactive oxygen species (ROS) dependent, implying that dsRNA generated during RV replication is sufficient for disrupting barrier function. We also demonstrated that RV or poly(I·C)-stimulated NADPH oxidase 1 (NOX-1) partially accounts for RV-induced ROS generation. In this study, we identified a dsRNA receptor(s) contributing to RV-induced maximal ROS generation and thus barrier disruption. We demonstrate that genetic silencing of the newly discovered dsRNA receptor Nod-like receptor X-1 (NLRX-1), but not other previously described dsRNA receptors, abrogated RV-induced ROS generation and reduction of transepithelial resistance (RT) in polarized airway epithelial cells. In addition, both RV and poly(I·C) stimulated mitochondrial ROS, the generation of which was dependent on NLRX-1. Treatment with Mito-Tempo, an antioxidant targeted to mitochondria, abolished RV-induced mitochondrial ROS generation, reduction in RT, and bacterial transmigration. Furthermore, RV infection increased NLRX-1 localization to the mitochondria. Additionally, NLRX-1 interacts with RV RNA and poly(I·C) in polarized airway epithelial cells. Finally, we show that NLRX-1 is also required for RV-stimulated NOX-1 expression. These findings suggest a novel mechanism by which RV stimulates generation of ROS, which is required for disruption of airway epithelial barrier function.
IMPORTANCE Rhinovirus (RV), a virus responsible for a majority of common colds, disrupts the barrier function of the airway epithelium by increasing reactive oxygen species (ROS). Poly(I·C), a double-stranded RNA (dsRNA) mimetic, also causes ROS-dependent barrier disruption, implying that the dsRNA intermediate generated during RV replication is sufficient for this process. Here, we demonstrate that both RV RNA and poly(I·C) interact with NLRX-1 (a newly discovered dsRNA receptor) and stimulate mitochondrial ROS. We show for the first time that NLRX-1 is primarily expressed in the cytoplasm and at the apical surface rather than in the mitochondria and that NLRX-1 translocates to mitochondria following RV infection. Together, our results suggest a novel mechanism for RV-induced barrier disruption involving NLRX-1 and mitochondrial ROS. Although ROS is necessary for optimal viral clearance, if not neutralized efficiently, it may increase susceptibility to secondary infections and alter innate immune responses to subsequently inhaled pathogens, allergens, and other environmental factors.
INTRODUCTION
The airway mucosa is the body's largest mucosal surface. It is prone to microbial encounters due to the high volume of inspired gas that crosses it. The polarized airway epithelium lining the respiratory tract regulates the movement of solutes and ions and prevents inhaled pathogens, allergens, and particulate material from gaining access to the submucosal compartment and activating immune cells. Apical junction complexes located at the apicolateral borders of adjacent cells contribute to epithelial barrier function by regulating paracellular permeability. Since the barrier function is a fundamental innate defense mechanism of the airway mucosa, disruption of apical junctional complexes could increase susceptibility to secondary infections and also alter cytokine responses to allergens, infection, and other environmental factors.
Viruses have been shown to disrupt barrier function by binding to integral membrane proteins of the tight-junction complex, thereby interfering with the interactions between junctional proteins. For instance, coxsackievirus and adenovirus bind to coxsackievirus adenovirus receptor (CAR) and induce disassembly of the tight junctions and a reduction in transepithelial resistance (RT) (1, 2). Claudins 1, 6, and 9 are coreceptors for hepatitis C virus, and binding of hepatitis C virus to claudins is required at the late stages of viral entry into epithelial cells (3, 4). Binding of reovirus to junctional adherence molecule A (JAM-A) is required for dissemination of virus (5). We and others have demonstrated that rhinovirus (RV) dissociates ZO-1, occludin, and claudin 4 from the tight junctions and increases bacterial association and translocation across the polarized airway epithelial cells (6–8). Unlike other viruses, barrier dysfunction caused by RV is dependent on viral replication and reactive oxygen species (ROS) generation. In addition, we and others have demonstrated that the double-stranded RNA (dsRNA) mimetic polyinosinic-polycytidilic acid [poly(I·C)] also causes barrier dysfunction (6, 9). These observations imply that dsRNA generated during RV replication is sufficient to cause ROS-dependent barrier dysfunction.
dsRNA generated during viral replication induces host responses via pattern recognition molecules, which include Toll-like receptor 3 (TLR3), protein kinase R (PKR) (10), and the retinoic acid-inducible gene I (RIG-I)-like receptors RIG-I and myeloma differentiation antigen 5 (MDA-5) (reviewed in references 11 and 12). Recently, Nod-like receptor X-1 (NLRX-1) has also been shown to recognize dsRNA and to inhibit RIG-I like receptor-stimulated interferon responses (13–15). RV has been shown to induce antiviral interferon responses in airway epithelial cells via TLR3, PKR, RIG-I, and MDA-5 (16–19). RV has also been shown to stimulate chemokine expression and mucin gene expression by activating TLR3 (20–22). However, recently, we showed that TLR3 is not required for RV-induced barrier dysfunction (6). Moreover, we have demonstrated that the expression of TLR3 is minimal and is confined to intracellular locations in both polarized immortalized cells and mucociliary differentiated primary airway epithelial cells (21). This study was undertaken to identify the dsRNA recognition receptor(s) that plays a role in RV-induced barrier dysfunction.
Membrane-bound NADPH oxidases (NOX) and mitochondria are the two major sources of intracellular ROS. Both RV and poly(I·C) have been shown to stimulate dual oxidase 2, a membrane-bound NOX in airway epithelial cells (23) that regulates production of extracellular ROS (24). Recently, we showed that RV and poly(I·C) also stimulate expression of NOX-1, which partially contributes to RV-induced intracellular ROS and barrier dysfunction, suggesting that intracellular ROS from sources other than NOX-1 may also contribute to barrier disruption caused by RV or poly(I·C) (6). Mitochondria are another major source of intracellular ROS (25, 26). The cytoplasmic dsRNA receptors RIG-I and MDA5, upon ligation to dsRNA, translocate and interact with mitochondrial antiviral signaling protein (MAVS) (27–29), and this interaction could potentially stimulate mitochondrial ROS generation. Also, NLRX-1, which localizes to the mitochondrial outer membrane, translocates to the mitochondrial inner membrane upon treatment with poly(I·C) and stimulates mitochondrial ROS generation (30, 31). Based on these observations, we hypothesized that RV may activate dsRNA receptors other than TLR3 that stimulate mitochondrial ROS generation. In addition, we hypothesized that mitochondrial ROS generation, in combination with NOX-1-dependent ROS, is critical for RV-induced barrier dysfunction in airway epithelial cells. Here, we demonstrate that NLRX-1, but not other dsRNA receptors, plays a role in RV-induced maximal ROS generation and thus in the barrier function of airway epithelial cells.
MATERIALS AND METHODS
Airway epithelial cell cultures.
Immortalized 16HBE14o− bronchial epithelial cells (provided by Dieter Gruenert, California Pacific Medical Center, San Francisco, CA) were cultured in transwells (Corning, Tewksbury, MA) as described previously (7) to promote polarization. The cells displaying RT values between 800 and 1,000 Ω · cm2 were used in this study. Primary airway epithelial cells were cultured at the air-liquid interface to promote mucociliary differentiation (7).
Rhinovirus and infection.
Rhinovirus 39 (RV39) was purchased from the American Type Culture Collection (Manassas, VA), propagated in H1 HeLa cells, and partially purified by ultrafiltration, and the viral titer was determined by measurement as the 50% tissue culture infective dose (TCID50) (7). Supernatants from uninfected H1 HeLa cells purified similarly to RV were used as a sham control.
Polarized cells were infected apically with RV39 at a multiplicity of infection (MOI) of 1 or with a similar volume of sham control and incubated for 90 min at 33°C (6, 7). The infection media were replaced with fresh media, and incubation was continued for an additional 24 h at 33°C. Mucociliary differentiated primary airway epithelial cells were infected apically at an MOI of 1 with RV suspended in 10 μl of phosphate-buffered saline (PBS) and incubated for 24 h at 33°C. In some experiments, polarized 16HBE14o− cells were treated with 2-aminopurine (Invivogen, San Diego, CA) or Mito-Tempo (Enzo Life Sciences Inc., Farmingdale, NY) 1 h prior to and during RV infection.
Treatment with poly(I·C).
Polarized 16HBE14o− cells were treated apically with 300 μl of medium containing 1, 5, or 10 μg/ml of high-molecular-weight poly(I·C) (1.5 to 8 kb; Invivogen) and incubated at 37°C for up to 6 h.
RT measurement.
The RT of polarized epithelial cell cultures or mucociliary differentiated airway epithelial cells was measured with an Evom voltmeter equipped with an EndOhm 6 tissue resistance measurement chamber (World Precision Instruments, Sarasota, FL) (6, 7).
Determination of transmigration of NTHI across polarized airway epithelial cell cultures.
Nontypeable Haemophilus influenzae (NTHI) organisms cultured on chocolate agar plates were suspended in cell culture medium to a density of 1 × 108 CFU/ml. The polarized monolayer of 16HBE14o− cells was either infected with RV or sham infected and incubated for 24 h. One hundred microliters of NTHI suspension was added to the apical surface and incubated for 4 h, and the bacterial load in the basolateral chamber was determined to assess the bacterial transmigration from the apical to the basolateral chambers (6, 21). The cells were lysed in 0.1% Triton X-100 and plated to determine the total number of bacteria associated with cells.
Assessment of RV RNA binding to NLRX-1.
The 16HBE14o− cells cultured in collagen-coated six-well plates were sham infected or infected with RV as described above and incubated for 16 h. Cell lysates were prepared as described previously (32). Briefly, the cells were washed with cold PBS and lysed in 100 μl of 10 mM Tris buffer, pH 7.4, containing 100 mM sodium chloride, 2.5 mM magnesium chloride, 0.5% NP-40, 2 mM dithiothreitol, 1 mM EDTA, 0.5 mM phenylmethanesulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml pepstatin, and 0.2 U/ml RNasin (Promega Corporation, Madison, WI) for 30 min. The lysates were left on ice for 30 min and then centrifuged at 16,000 × g for 15 min. The lysates from 3 wells (equivalent to 1 × 107 cells) were combined and incubated with 10 μg of NLRX-1 antibody or normal goat IgG conjugated to agarose beads at 4°C overnight. The immunoprecipitate complexes were washed with lysis buffer two times and then with cold PBS and finally solubilized in TRIzol, total RNA was isolated by using a miRNeasy kit (Qiagen), and the viral RNA copy number was estimated by quantitative PCR (qPCR), as described previously (33). An aliquot of the immunoprecipitate was subjected to Western blot analysis to confirm the pulldown of NLRX-1 by anti-NLRX-1 antibody.
Western blot analysis.
After relevant treatment, total proteins or cytoskeletal proteins (NP-40-insoluble proteins) were isolated as described previously (6, 7). Mitochondrial proteins were extracted using a mitochondrial isolation kit (Thermo Fisher Scientific Inc., Rockford, IL). An equal amount of protein (for total protein or mitochondrial proteins) or an equal volume of extracts (for cytoskeletal proteins) was subjected to Western blot analysis with antibodies to NLRX-1 (Santa Cruz Biotechonology Inc., Santa Cruz, CA), occludin (BD Biosciences, San Jose, CA), mitochondrially encoded cytochrome c oxidase 1 (MT-CO1) (Abcam, Cambridge, MA), or β-actin (Sigma-Aldrich, St. Louis, MO). Specific bands were quantified by densitometry using NIH Image J and expressed as the fold change over β-actin.
Indirect immunofluorescence confocal microscopy.
The localization of NLRX-1, MT-CO1, and occludin was determined by indirect immunofluorescence confocal microscopy as described previously (6, 7). Briefly, after appropriate treatment, cells were fixed in methanol, blocked with bovine serum albumin (BSA), and incubated with primary antibody at 4°C overnight. Bound antibody was detected by using an appropriate secondary antibody conjugated with Alexa Fluor 488 or Alexa Fluor 598. Cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole) to visualize nuclei.
The polarized 16HBE14o− cells were incubated with rhodamine-labeled poly(I·C) (Invivogen) for 3 h, fixed in methanol, incubated with NLRX-1 antibody conjugated with Alexa Fluor 488 for 1 h at room temperature, counterstained with DAPI, and visualized by confocal microscopy.
Measurement of mitochondrial ROS.
16HBE14o− cells were grown in collagen-coated cover glass chamber slides (Thermo Fisher Scientific Inc.) as polarized monolayers. After relevant treatment, the cells were briefly washed with Hanks balanced salt solution (HBSS) and incubated with 5 μM MitoSox Red (Life Technologies, Grand Island, NY) for 15 min. The cells were washed and counterstained with Hoechst stain, and live cells were immediately imaged using confocal microscopy.
To quantify mitochondrial ROS, 16HBE14o− cells were grown as polarized monolayers in collagen-coated 6-well plates or mucociliary differentiated primary airway epithelial cells were used. After relevant treatment, the cells were loaded with MitoSox Red, incubated for 30 min, and washed with warm HBSS. Then, the cells were detached from the wells using trypsin-EDTA, neutralized with serum, centrifuged, suspended in warm HBSS, and immediately analyzed by flow cytometry.
Transfection of 16HBE14o− cells growing in transwells.
16HBE14o− cells were transfected with small interfering RNA (siRNA) of interest as described previously (6). Briefly, partially polarized confluent monolayers of 16HBE14o− cells were transfected with MDA-5, RIG-I, NLRX-1, or nontargeting (NT) siRNA using Lipofectamine siRNA max according to the manufacturer's instructions. A pool of each gene-specific siRNA or NT siRNA was purchased from Thermo Fisher Scientific Inc. and used at 10 pmol/well. The cells were incubated for another 2 days to promote polarization, at which time the cells showed RT values ranging between 800 and 1,000 Ω · cm2. The efficiency of siRNA transfection was confirmed by Western blotting.
Real-time PCR.
After relevant treatment, total RNA was isolated from airway epithelial cells, and 0.2 μg of total RNA was used to quantify viral RNA, as described previously, by qPCR (33). NOX-1 expression was determined using gene-specific primers as described previously and expressed as the fold change over that of the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) housekeeping gene (6).
Statistical analysis.
Statistical significance was assessed by unpaired Student t test, analysis of variance (ANOVA), or ANOVA on ranks, as appropriate. Differences identified by ANOVA or ANOVA on ranks were pinpointed by Tukey-Kramer or Dunn's test, respectively.
RESULTS
Roles of MDA-5, RIG-I, and PKR in RV-induced reduction in RT.
Previously, dsRNA receptors, MDA5 and RIG-I, have been shown to contribute to RV-induced interferon responses in airway epithelial cells (16–18). To determine the roles of MDA5 and RIG-I in RV-induced tight-junction disruption, 16HBE14o− cells transfected with NT, MDA-5, or RIG-I siRNA were infected with either RV or an equal volume of sham control, and the RT was measured after 24 h. Compared to sham-infected controls, RV-infected cultures showed reduced RT, as observed previously in NT siRNA-transfected cells (Fig. 1A). Knockdown of either MDA-5 or RIG-I had no effect on RV-induced reduction in RT. Knockdown of MDA5 and RIG-I in cells transfected with gene-specific siRNA was confirmed by Western blotting (Fig. 1B to D).
FIG 1.

Knockdown of MDA5 or RIG-I or inhibition of PKR does not affect RV-induced changes in RT. 16HBE14o− cells were transfected with NT, MDA5, or RIG-I siRNA, and the cells were allowed to polarize. (A) Cells were infected apically with RV or sham control, and RT was measured after 24 h. (B) Total proteins from cells transfected with NT, MDA5, or RIG-I siRNA were subjected to Western blot analysis with antibodies to MDA5, RIG-I, or β-actin. (C and D) Band intensities were quantified by NIH Image, and levels of MDA5 or RIG-I were normalized to the respective β-actin controls. (E) Cells were pretreated with 2-AP for 1 h, infected with RV or sham control, and incubated for 24 h in the presence or absence of 2-AP, and then RT was measured. The data in panels A, C, D, and E represent means and standard deviations (SD) calculated from 3 to 6 independent experiments (*, P ≤ 0.05; ANOVA). The percentages in panels C and D represent percent inhibition of MDA5 and RIG-I expression, respectively, in gene-specific siRNA-transfected cells compared to NT siRNA-transfected cells. The images in panel B are representative of 3 independent experiments.
Next, we examined the role of another dsRNA receptor, PKR, in RV-induced reduction in RT. Polarized monolayers of 16HBE14o− cells were pretreated with the PKR inhibitor 2-aminopurine (2-AP) for 1 h and then infected with sham control or RV in the presence or absence of 2-AP, and the RT was measured after 24 h. RV infection reduced the RT in cells treated with 2-AP similarly to that observed in untreated controls (Fig. 1E), indicating that PKR may not contribute to this process. Since RIG-I, MDA5, and PKR were not required for RV-induced reduction in RT, we examined the role of NLRX-1 in RV-induced reduction in RT because of its capacity to recognize dsRNA (15).
NLRX-1 is essential for RV-induced reduction in RT.
Cells were transfected with NT or NLRX-1 siRNA and infected with RV or sham control, and the RT was measured after 24 h. Interestingly, cells transfected with NLRX-1 siRNA were found to be resistant to RV-induced reduction in RT (Fig. 2A) despite showing similar viral loads, as assessed by viral RNA copy numbers (Fig. 2B), implying a requirement for NLRX-1 in RV-induced reduction in RT.
FIG 2.

NLRX-1 is required for RV-induced tight-junction breakdown. 16HBE14o− cells were transfected with NT or NLRX-1 siRNA and allowed to polarize for 48 h. (A) Cells were infected with either RV or sham control and incubated for 24 h, and then RT was measured. (B) Total RNA isolated from sham- or RV-infected cells was used to determine viral RNA copy numbers. (C and D) NP-40-insoluble fractions from cells infected with sham control or RV were subjected to Western blot analysis using antibodies to occludin or β-actin, and band intensities were quantified and expressed as fold change over β-actin. (E) NT or NLRX-1 siRNA-transfected cell cultures were infected with RV or sham control and incubated for 16 h. NTHI was added to the apical surface and incubated for an additional 4 h, and the number of bacteria in the basolateral chamber was determined by plating. (F) Total proteins from cells transfected with NT or NLRX-1 siRNA and infected with sham control or RV were subjected to Western blot analysis with antibody to NLRX-1 or β-actin to confirm knockdown of NLRX-1. The data in panels A, B, and D represent means and SD calculated from 3 or 4 independent experiments (*, P ≤ 0.05; ANOVA). The data in panel D represent medians with ranges from 3 independent experiments (*, P ≤ 0.05; ANOVA on ranks). The images in panels B and E are representatives of 3 or 4 independent experiments.
Previously, we have shown that RV-induced reduction in RT correlates with dissociation of occludin from tight junctions (6). To examine whether NLRX-1-dependent reduction in RT induced by RV causes dissociation of occludin from the tight-junction complex, we subjected the NP-40-insoluble fraction enriched in tight-junction complex proteins to Western blot analysis. Reduction in occludin levels in the NP-40-insoluble fraction indicates dissociation of the protein from the tight junctions (6). As expected, NT siRNA-transfected cells showed reduction in occludin levels following RV infection (Fig. 2C and D). In contrast, NLRX-1 siRNA-transfected cells did not show significant reduction in occludin levels in the NP-40-insoluble fraction after RV infection.
To determine whether NLRX-1-dependent tight-junction disruption induced by RV also causes barrier dysfunction, NTHI was added to the apical surfaces of cell cultures infected with either RV or sham control, and the total number of bacteria in the basolateral chamber was assessed to determine transmigration of bacteria across the polarized cells (6, 7). Cell lysates were plated to determine the total number of bacteria associated with cells. Compared to sham infection, RV-infected NT siRNA-transfected cells showed increased transmigration of apically added bacteria to the basolateral compartment (Fig. 2E). In contrast, RV-infected NLRX-1 siRNA-transfected cells showed significantly less transmigration of bacteria than similarly infected NT siRNA-transfected cells. However, there was no difference in the total bacterial counts recovered from the cells (data not shown), suggesting that decreased transmigration in NLRX-1 siRNA-transfected cells infected with RV is not due to decreased bacterial association with cells or bacterial survival. Together, these results suggest that NLRX-1 is required for RV-induced barrier dysfunction. Western blot analysis of whole-cell lysate confirmed efficient knockdown of NLRX-1 expression by gene-specific siRNA and not by NT siRNA (Fig. 2F).
RV stimulates mitochondrial ROS generation.
NLRX-1 is thought to be a mitochondrial protein and has been shown to induce mitochondrial ROS generation following transfection with poly(I·C) in nonpolarized HeLa cells (31). Since NLRX-1 was required for RV-induced reduction in RT, we determined whether RV stimulates mitochondrial ROS using MitoSox Red, which selectively detects mitochondrial ROS (34). As assessed by confocal microscopy, RV-infected polarized monolayers of 16HBE14o− cells showed more MitoSox Red-positive cells than sham-infected cultures (Fig. 3A). Analysis of cells by flow cytometry indicated that RV-infected cells generate significantly higher levels of mitochondrial ROS than sham-infected cells (Fig. 3B and C). Furthermore, knockdown of NLRX-1 blocked RV-induced mitochondrial ROS (Fig. 3D), suggesting a role for NLRX-1 in this process.
FIG 3.

RV stimulates NLRX-1-dependent mitochondrial ROS in polarized airway epithelial cells. The polarized monolayers of 16HBE14o− cells were infected with RV or sham control and incubated for 90 min at 33°C. The infection medium was replaced with fresh medium, and incubation continued for an additional 16 h. The cells were washed once with HBSS and incubated with 5 μM MitoSox Red for 15 min at 37°C. The cells were washed with warm HBSS. (A) Cells were incubated with HBSS containing Hoechst dye (nuclear stain) for 10 min, which was then replaced with warm HBSS, and the cells were imaged immediately. The images are representative of 3 independent experiments (red, MitoSox Red fluorescence indicative of mitochondrial ROS; blue, nuclei). (B and C) Cells were detached from the plate, suspended in warm HBSS, and analyzed by flow cytometry. MFI, mean fluorescence intensity. (D) NT or NLRX-1 siRNA-transfected cells were infected with sham control or RV, and mitochondrial ROS was quantified by flow cytometry 16 h postinfection. (B) Representative histogram of 3 independent experiments. (C and D) Data represent means and SD calculated from 3 or 4 independent experiments (*, P ≤ 0.05; ANOVA).
Mitochondrial ROS contributes to RV-induced reduction in RT.
To confirm that mitochondrial ROS generation stimulated by RV is required to reduce the RT of polarized 16HBE14o− cells, we infected cells with RV in the absence or presence of Mito-Tempo, an antioxidant targeted to mitochondria (35), and measured the RT 24 h later. Compared to sham infection, RV infection caused significant reduction in RT (Fig. 4A) and Mito-Tempo blocked RV-induced reduction in RT in a dose-dependent manner. Determination of ROS generation by flow cytometry confirmed that Mito-Tempo inhibits RV-stimulated ROS generation (Fig. 4B). Indirect immunoconfocal microscopy indicated that Mito-Tempo also prevents dissociation of occludin from the peripheries of RV-infected cells (Fig. 4C). Quantification of occludin in the NP-40-insoluble fraction (cytoskeletal fraction) by Western blotting suggested that Mito-Tempo significantly blocks RV-induced dissociation of occludin from the cytoskeleton (Fig. 4D and E). The observed effects of Mito-Tempo were not due to changes in the RV load, because there was no difference in viral RNA between untreated and Mito-Tempo-treated groups, as assessed by qPCR (data not shown). Next, we examined the transmigration of apically added bacteria to the basolateral chamber. Mito-Tempo treatment significantly attenuated the transmigration of bacteria in RV-infected cells (Fig. 4F). However, there was no significant difference in cell-associated bacteria between untreated and Mito-Tempo-treated cells, suggesting that Mito-Tempo does not affect bacterial survival or association with cells (data not shown). Together, these results suggest that ROS generated by mitochondria in response to RV infection contributes significantly to disruption of tight junctions in polarized airway epithelial cells.
FIG 4.

Mito-Tempo, an antioxidant targeted to mitochondria, blocks RV-induced reduction in RT and mitochondrial ROS generation. Polarized monolayers of 16HBE14o− cells were treated with 0, 10, or 50 μM Mito-Tempo for 1 h both apically and basolaterally. (A and B) Cells were infected with RV or sham control apically and incubated for 90 min. The infection medium was replaced with fresh medium containing 0, 10, or 50 μM Mito-Tempo and incubated for an additional 24 h. The RT was measured, and then the cells were washed and incubated with MitoSox Red as described for Fig. 3 and analyzed by flow cytometry. (C) Polarized cells were infected with RV or sham control in the presence of 0 or 50 μM Mito-Tempo and incubated for 24 h. The cells were fixed, blocked with 1% BSA in PBS, and incubated with antibody to occludin. Bound antibody was detected with antirabbit IgG conjugated with Alexa Fluor 488, and the cells were counterstained with DAPI and then subjected to indirect immunofluorescence microscopy. The images are representative of 3 or 4 independent experiments (*, dissociation of occludin from the tight-junction complex; green, occludin; blue, nuclei). (D and E) NP-40-insoluble fractions from cells infected with RV or sham control in the presence or absence of Mito-Tempo were subjected to Western blot analysis, and the band intensities were quantified using NIH Image J and expressed as fold change over β-actin. (F) Serial dilutions of basolateral media from cell cultures infected with RV or sham control in the presence or absence of Mito-Tempo were plated to determine the number of bacteria transmigrated from the apical to the basolateral surface. The data in panels A, B, and D represent means and SD calculated from 3 or 4 independent experiments (*, P ≤ 0.05; ANOVA). The data in panel F represent medians with ranges from 3 independent experiments (*, P ≤ 0.05; ANOVA on ranks).
Poly(I·C)-induced reduction in RT also requires mitochondrial ROS generation that is NLRX-1 dependent.
To assess the contribution of mitochondrial ROS in poly(I·C)-induced reduction of RT in polarized 16HBE14o− cells, we examined whether poly(I·C) stimulates mitochondrial ROS and whether treatment with Mito-Tempo protects cells against poly(I·C)-induced reduction in RT. Poly(I·C) stimulated mitochondrial ROS generation in a dose-dependent manner in 16HBE14o− cells (Fig. 5A), and this was inhibited significantly by Mito-Tempo (Fig. 5B). In addition, Mito-Tempo also reduced poly(I·C)-induced mitochondrial ROS (Fig. 5C). These results indicate a role for mitochondrial ROS in poly(I·C)-induced RT reduction in polarized 16HBE14o− cells, as observed with RV.
FIG 5.

Poly(I·C) induces mitochondrial ROS in polarized 16HBE14o− cells in an NLRX-1- dependent manner. (A) Cells were treated with 1, 5, or 10 μg/ml poly(I·C) and incubated for 6 h at 37°C. Mitochondrial ROS was determined by flow cytometry as described for Fig. 3. (B and C) Polarized monolayers of 16HBE14o− cells were incubated with 0, 10, or 50 μM Mito-Tempo for 1 h both apically and basolaterally and then treated with 5 μg/ml poly(I·C) in the presence of 0, 10, or 50 μM Mito-Tempo. After 6 h, RT was measured, and then the cells were treated with MitoSox Red to determine the levels of mitochondrial ROS. (D and E) Cells were transfected with NT or NLRX-1 siRNA, allowed to polarize for 2 days, and then treated with 5 μg/ml poly(I·C). After 6 h, RT was measured, and then the cells were treated with MitoSox Red to determine the levels of mitochondrial ROS. The data represent means and SD calculated from 3 independent experiments (*, P ≤ 0.05; ANOVA).
To determine the role of NLRX-1 in poly(I·C)-stimulated mitochondrial ROS generation and thus reduction in RT, cells transfected with NT or NLRX-1 siRNA were treated with medium or poly(I·C), and the levels of mitochondrial ROS and RT were measured. NLRX-1 siRNA-transfected cells treated with poly(I·C) showed significant reductions in the levels of mitochondrial ROS compared to similarly treated NT siRNA-transfected cells (Fig. 5D). Cells transfected with NLRX-1 siRNA were also protected against poly(I·C)-induced reduction in RT (Fig. 5E). Together, these results imply that, similar to RV, poly(I·C)-induced tight-junction disruption also requires NLRX-1-dependent mitochondrial ROS.
NLRX-1 colocalizes to mitochondria upon infection with RV in airway epithelial cells.
NLRX-1 contains a mitochondrial signature in its C terminus (15) and has been shown to localize to the mitochondrial outer membrane in nonpolarized HeLa cells (a cervical cancer cell line) (13). However, the cellular distribution of NLRX-1 in polarized airway epithelial cells is not known. Therefore, we examined the cellular location of NLRX-1 and possible colocalization with occludin or mitochondria by indirect immunofluorescence microscopy. NLRX-1 was observed in the cytoplasm, at the cell surface, and occasionally at the apicolateral surface colocalizing with occludin in sham-infected cells (Fig. 6A). RV-infected cells showed redistribution of NLRX-1 from the periphery to an intracellular location (Fig. 6B). Z sections of sham-infected cells revealed more NLRX-1 on the cell surface than RV-infected cells (6C and D).
FIG 6.

NLRX-1 redistributes in airway epithelial cells following RV infection. (A to F) Polarized 16HBE14o− cells were infected with RV or sham control. After 16 h, the cells were fixed and incubated with a mixture of antibodies to occludin and NLRX-1 (A to D) or a mixture of antibodies to MT-CO1 (mitochondrial protein) and NLRX-1 (E and F). Bound antibodies were detected by secondary antibodies conjugated with either Alexa Fluor 598 (NLRX-1) or Alexa Fluor 488 (occludin [A to D] and MT-CO1 [E and F]). The cells were counterstained with DAPI and subjected to indirect immunofluorescence microscopy. The arrows in panel A represent colocalization (yellow) of NLRX-1 with occludin. Panels C and D are the Z sections of panels A and B, respectively, showing the distribution of NLRX-1 in relation to occludin, and the apical surface of the cultures is marked by the white line. The arrows in panels E and F represent colocalization (yellow) of NLRX-1 with MT-CO1. (G) Cytoplasmic or mitochondrial proteins isolated from sham- or RV-infected cells were subjected to Western blot analysis with antibodies to NLRX-1, MT-CO1, or GAPDH. The absence of a band corresponding to MT-CO1 in the cytoplasmic fraction and the absence of GAPDH in the mitochondrial fraction indicate efficient separation of cytoplasmic proteins from mitochondrial proteins. The images are representative of 3 or 4 experiments.
Next, we examined colocalization of NLRX-1 with a mitochondrial marker, MT-CO1. Sham-infected cells showed only minimal NLRX-1 colocalization with mitochondria (Fig. 6E). In contrast, RV-infected cells often showed colocalization of NLRX-1 with MT-CO1 (Fig. 6F), implying that RV may promote translocation to mitochondria. To confirm these observations, mitochondrial and cytosolic proteins were isolated from sham- or RV-infected cells and subjected to Western blot analysis with antibodies to either NLRX-1 or MT-CO1. Compared to sham-infected cells, RV-infected cells showed more NLRX-1 in the mitochondrial fraction (Fig. 6G). Successful separation of mitochondrial proteins from cytosolic proteins was confirmed by the presence of MT-CO1 and absence of GAPDH in the mitochondrial fraction, and vice versa in the cytosolic fraction. These results confirmed that RV promotes translocation of NLRX-1 to mitochondria.
NLRX-1 interacts with RV RNA and poly(I·C).
To assess whether NLRX-1 directly interacts with RV RNA, cells infected with RV were immunoprecipitated with NLRX-1 antibody or normal goat IgG, and total RNA isolated from the immunoprecipitates was used to determine viral RNA. Compared to total RNA from immunoprecipitates of normal goat IgG, immunoprecipitates of NLRX-1 antibody showed significantly higher viral RNA levels, indicating that NLRX-1 may interact with RV RNA (Fig. 7A). Western blot analysis of immunoprecipitates confirmed the pulldown of NLRX-1 by NLRX-1 antibody (Fig. 7B).
FIG 7.

NLRX-1 interacts with viral RNA and poly(I·C). The polarized airway epithelial cells were RV or sham infected and incubated for 16 h, and cell lysates were immunoprecipitated with normal IgG or anti-NLRX-1 antibody. (A and B) Total RNA was isolated from the immunoprecipitates, the RV RNA copy number was determined (A), and an aliquot of immunoprecipitates (IP) was subjected to Western blot (IB) analysis with NLRX-1 antibody (B). (C to E) Rhodamine-labeled poly(I·C) was added to the apical surfaces of polarized airway epithelial cells and incubated for 3 h. The cells were fixed in methanol, blocked with BSA, incubated with antibody to NLRX-1 conjugated with Alexa Fluor 488, and counterstained with DAPI. The cells were visualized by confocal microscopy. Panel D is a magnified view of the boxed area in panel C. Panel E shows a Z section of panel C. The white arrows in panels C and D indicate colocalization of poly(I·C) with NLRX-1. The white and black arrows in panel E represent NLRX-1 colocalization with poly(I·C) in the subapical and apical locations, respectively. The data in panel A represent medians and ranges from 5 independent experiments (*, P ≤ 0.05; ANOVA). The images are representative of three or five independent experiments.
To examine whether NLRX-1 interacts with apically added dsRNA, polarized 16HBE14o− cells treated with rhodamine-labeled poly(I·C) were fixed and immunolabeled with antibody to NLRX-1. Indirect immunofluorescence confocal microscopy revealed localization of poly(I·C) with NLRX-1 at the apical surface and also in the subapical location (Fig. 7C to E). Together, these results imply that NLRX-1 may directly interact with viral RNA or poly(I·C) in polarized airway epithelial cells and that this interaction may promote translocation of NLRX-1 from the apical surface and/or cytoplasm to mitochondria.
RV infection promotes redistribution of NLRX-1 in primary airway epithelial cells.
Next, we determined the localization of NLRX-1 in primary mucociliary differentiated airway epithelial cells, a model that closely resembles airway epithelium in vivo. Similar to 16HBE14o− cells, mucociliary differentiated airway epithelial cells showed NLRX-1 at the cell surface, occasionally in the apicolateral junctions colocalizing with occludin, and also in the cytoplasm (Fig. 8A and B). Following RV infection, however, these cells showed increased colocalization of NLRX-1 with MT-CO1 (Fig. 8C and D), suggesting that RV promotes translocation of NLRX-1 to mitochondria, as observed in the polarized 16HBE14o− cells.
FIG 8.

RV promotes NLRX-1 translocation to mitochondria in primary airway epithelial cells. (A and B) Mucociliary differentiated primary airway epithelial cells were fixed and immunolabeled with a mixture of antibodies to occludin (green) and NLRX-1 (red). Panel B is a Z section of panel A showing NLRX-1 on the apical surface and in the cytoplasm. The arrows indicate colocalization of NLRX-1 with occludin. (C and D) Mucociliary differentiated primary airway epithelial cells were apically infected with sham control or RV, incubated for 24 h, fixed, immunolabeled with antibodies to NLRX-1 (red) and MT-CO1 (green), counterstained with DAPI, and subjected to confocal microscopy. The images represent optical sections taken at a depth of 9 μm to show both NLRX-1 and MT-CO1, presumably in the middle of the cells. The total thickness of cell cultures was approximately 20 μm. The inset in panel D is a magnified view of the boxed area. All images are representative of three independent experiments.
Mitochondrial ROS induced by RV contributes to RT reduction in mucociliary differentiated primary airway epithelial cells.
First, we determined whether RV induces mitochondrial ROS by flow cytometry. Compared to sham infection, RV infection significantly increased mitochondrial ROS, and this was effectively blocked by treatment with Mito-Tempo (Fig. 9A). Mito-Tempo also inhibited RV-induced reduction of RT in primary mucociliary differentiated cells, indicating that RV-stimulated mitochondrial ROS contribute to this process (Fig. 9B).
FIG 9.
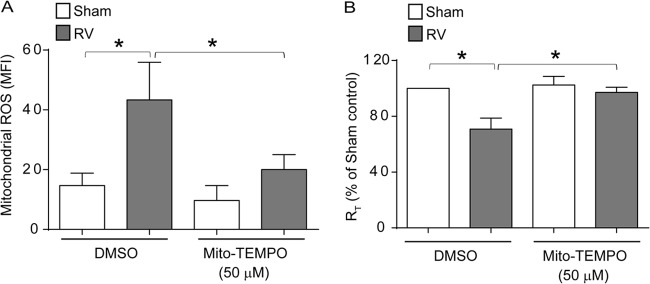
RV-stimulated mitochondrial ROS is required for RT reduction in primary airway epithelial cells. Mucociliary differentiated primary airway epithelial cells were apically infected with sham control or RV in the presence or absence of 50 μM Mito-Tempo and incubated for 24 h. (A) Cells were incubated with MitoSox Red and analyzed by flow cytometry to determine the levels of mitochondrial ROS. (B) RT was measured and expressed as a percentage of sham-infected vehicle-treated controls. The data represent means and SD calculated from three independent experiments (*, P ≤ 0.05; ANOVA).
NLRX-1 and mitochondrial ROS are required for RV-induced NOX-1 expression.
Previously, we demonstrated that RV-induced NOX-1-dependent ROS generation partially contributes to RV-induced barrier disruption. Since mitochondrial ROS has been shown to regulate NOX-1-dependent ROS generation (36, 37), we examined whether NLRX-1 or mitochondrial ROS is required for RV-induced NOX-1 expression in polarized 16HBE14o− cells. Genetic inhibition of NLRX-1, as well as Mito-Tempo, inhibited RV-induced NOX-1 expression completely, (Fig. 10A and B). Similarly, Mito-Tempo also abolished RV-induced NOX-1 expression in primary mucociliary differentiated airway epithelial cells (Fig. 10C). Together, these results indicate that NLRX-1-induced mitochondrial ROS is required for RV-induced NOX-1 expression.
FIG 10.

Inhibition of NLRX-1 or mitochondrial ROS blocks RV-induced NOX-1 expression. (A) 16HBE14o− cells were transfected with NT or NLRX-1 siRNA, infected with sham control or RV, and incubated for 16 h. (B and C) 16HBE14o− cells or mucociliary differentiated cells were infected with RV or sham control in the presence or absence of Mito-Tempo and incubated for 16 h. Total RNA was harvested, and the expression of NOX-1 mRNA was determined by qPCR and expressed as the fold change over the GAPDH housekeeping gene. The data represent means and SD calculated from 3 independent experiments (*, P ≤ 0.05; ANOVA).
DISCUSSION
The present study suggests an essential role for a member of the NLR family, NLRX-1, which has been shown to possess a dsRNA binding site (15), in RV-induced disruption of tight junctions. We demonstrate for the first time that RV stimulates mitochondrial ROS generation and that this requires the participation of NLRX-1. Interestingly, we found that, in both polarized 16HBE14o− cells and primary airway epithelial cells differentiated into a mucociliary phenotype, NLRX-1 is expressed primarily on the apical and apicolateral surfaces, locations in which it may interact with tight-junction proteins, and also in the cytoplasm not colocalized with mitochondria. In addition, we show that following RV infection, the majority of the NLRX-1 colocalizes to mitochondria in both immortalized and primary airway epithelial cells. Further, we show that poly(I·C), a double-stranded RNA mimetic, also stimulates NLRX-1-dependent mitochondrial ROS generation, which contributes to poly(I·C)-induced tight-junction disruption. In addition, we found that NLRX-1 interacts with intracellular viral RNA and also apically added poly(I·C). Finally, we demonstrate that NLRX-1 is also required for NOX-1 expression, which has been previously shown to partially contribute to RV-induced ROS generation and thus to barrier disruption (6). Based on these results, we speculate that dsRNA generated during RV replication may interact directly or indirectly and promote translocation of NLRX-1 to mitochondria, thereby stimulating the generation of mitochondrial ROS and disruption of barrier function.
Previously, we demonstrated that both RV and poly(I·C) perturb the barrier function of the airway epithelium and that treatment with the antioxidant diphenylene iodonium (DPI) completely abrogated the disruptive effects of RV and poly(I·C). This indicates that dsRNA generated during RV replication is sufficient and that oxidative stress induced by replicating RV is required for the disruption of barrier function. RV has been shown to activate the dsRNA recognition receptors TLR3, PKR, RIG-I, and MDA5 (10, 16–18, 20, 22). For example, RV was demonstrated to stimulate interferon expression by activating PKR and mucin expression that is dependent on TLR3 (10, 20). We showed that TLR3 and MDA5 are required for maximal interferon production and also for interleukin 8 (IL-8) expression (16, 38). Recently, TLR3 was shown to coordinate with MDA5 and RIG-I in stimulating innate immune responses to RV in vivo and in vitro (18). Despite their important roles in RV-stimulated antiviral responses in several cell types, none of these dsRNA recognition receptors were found to contribute to RV-induced disruption of tight junctions in polarized airway epithelial cells.
Recently, a member of the NLR family, NLRX-1, was shown to interact with poly(I·C) through its C-terminal domain and also to stimulate mitochondrial ROS in nonpolarized HeLa cells (15, 31). Therefore, we examined the contribution of NLRX-1 to RV-induced disruption of tight junctions, as well as ROS. We found that knockdown of NLRX-1 not only protected against RV- or poly(I·C)-induced reductions in RT, but also abrogated RV- or poly(I·C)-stimulated ROS generation. In addition, Mito-Tempo, which inhibits mitochondrial ROS, blocked RV- and poly(I·C)-induced ROS. Collectively, these observations suggest that dsRNA generated during RV may interact with NLRX-1, thereby stimulating mitochondrial ROS and subsequent barrier disruption in airway epithelial cells. However, it raised a question, that is, how does NLRX-1, which has been shown to be expressed only in the mitochondria (13, 30), interact with RV dsRNA that primarily accumulates in the cytoplasm and apically added poly(I·C)? In addition, although, NLRX-1 has been proposed to be one of the receptors for virus-derived dsRNA based on X-ray crystallography or indirect experimental evidence (15, 31), so far, interaction of mitochondrial NLRX-1 with viral RNA or synthetic dsRNA under physiological conditions has not been demonstrated. Our results for the first time demonstrate that, unlike in nonpolarized HeLa cells, NLRX-1 is primarily expressed in the cytoplasm, at the apical surface, and on the apicolateral surface, occasionally colocalizing with occludin, and minimally in mitochondria in polarized or mucociliary differentiated primary airway epithelial cells. However, following RV infection, NLRX-1 increasingly colocalized with mitochondria, in addition to interacting with intracellular viral RNA. Moreover, NLRX-1 colocalized with poly(I·C) at the apical surface, as well as in the subapical location, in polarized airway epithelial cells, implying that NLRX-1 also interacts with apically added poly(I·C). Since NLRX-1 also colocalizes with poly(I·C) in the subapical location, it is plausible that interaction with NLRX-1 may promote internalization of poly(I·C); however, further experiments are required to confirm this notion. Based on these observations, we speculate that NLRX-1 is readily available to interact with apically added poly(I·C) and cytosolic RV dsRNA and that this interaction promotes translocation of NLRX-1 to mitochondria, thereby stimulating mitochondrial ROS and subsequent barrier disruption in airway epithelial cells. Although mechanisms by which NLRX-1 translocate to mitochondria after binding to dsRNA are yet to elucidated, we speculate that binding of dsRNA to NLRX-1 in the cytoplasm or on the apical surface may lead to dissociation of NLRX-1 from yet unknown molecular partners and/or oligomerization, and this may in turn promote NLRX-1 translocation to mitochondria. However, further biochemical and structural studies are required to understand the molecular mechanisms underlying translocation of NLRX-1 to mitochondria upon binding to dsRNA.
In our previous studies, we have demonstrated that NOX-1 is partially required for both RV-induced ROS generation and barrier disruption in airway epithelial cells (6). However, in the present study, we found that Mito-Tempo, which primarily inhibits mitochondrial ROS, almost completely abrogated RV-induced ROS generation and reduction in RT, implying that RV-induced NOX-1 expression may require mitochondrial ROS stimulated by RV. Consistent with this notion, we found that inhibition of mitochondrial ROS by Mito-Tempo blocked RV-induced NOX-1 expression in both polarized and mucociliary differentiated airway epithelial cells. This is not surprising, because previously, mitochondrial ROS has been shown to regulate NOX-1 expression, as well as NOX-1-dependent ROS generation (36, 39). Interestingly, genetic ablation of NLRX-1 also inhibited RV-induced NOX-1 expression in the present study, linking NLRX-1 to NOX-1 expression via mitochondrial ROS.
In conclusion, we propose that (i) NLRX-1, but not other dsRNA recognition receptors, plays a major role in RV-stimulated maximal ROS generation and thus barrier disruption in airway epithelial cells; (ii) NOX-1 expression and activity are regulated by RV-stimulated mitochondrial ROS generation, which is dependent on NLRX-1; and (iii) NLRX-1, which is expressed primarily in the cytoplasm and on the apical surfaces of polarized or mucociliary differentiated airway epithelial cells, readily interacts with dsRNA generated during RV replication and translocates to mitochondria. Although, ROS generation is required for optimal viral clearance following infection, defective neutralization of ROS, as observed in patients with cystic fibrosis or chronic obstructive pulmonary disease (40–42), may lead to persistent airway epithelial barrier dysfunction. This in turn could alter innate immune responses to subsequent environmental or infectious stimuli and also enhance the risk for acquiring secondary infections.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health, HL089772 and AT004793, to U.S.S.
We thank Brenton Kinker for his help in editing the manuscript.
Footnotes
Published ahead of print 15 January 2014
REFERENCES
- 1.Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131. 10.1016/j.cell.2005.10.035 [DOI] [PubMed] [Google Scholar]
- 2.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323. 10.1126/science.275.5304.1320 [DOI] [PubMed] [Google Scholar]
- 3.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 4.Zheng A, Yuan F, Li Y, Zhu F, Hou P, Li J, Song X, Ding M, Deng H. 2007. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J. Virol. 81:12465–12471. 10.1128/JVI.01457-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. 10.1016/j.chom.2008.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comstock AT, Ganesan S, Chattoraj A, Faris AN, Margolis BL, Hershenson MB, Sajjan US. 2011. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 85:6795–6808. 10.1128/JVI.02074-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. 2008. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 178:1271–1281. 10.1164/rccm.200801-136OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chattoraj SS, Ganesan S, Jones AM, Helm JM, Comstock AT, Bright-Thomas R, LiPuma JJ, Hershenson MB, Sajjan US. 2011. Rhinovirus infection liberates planktonic bacteria from biofilm and increases chemokine responses in cystic fibrosis airway epithelial cells. Thorax 66:333–339. 10.1136/thx.2010.151431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rezaee F, Meednu N, Emo JA, Saatian B, Chapman TJ, Naydenov NG, De Benedetto A, Beck LA, Ivanov AI, Georas SN. 2011. Polyinosinic:polycytidylic acid induces protein kinase D-dependent disassembly of apical junctions and barrier dysfunction in airway epithelial cells. J. Allergy Clin. Immunol. 128:1216–1224. 10.1016/j.jaci.2011.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Hamati E, Lee PK, Lee WM, Wachi S, Schnurr D, Yagi S, Dolganov G, Boushey H, Avila P, Wu R. 2006. Rhinovirus induces airway epithelial gene expression through double-stranded RNA and IFN-dependent pathways. Am. J. Respir. Cell Mol. Biol. 34:192–203. 10.1165/rcmb.2004-0417OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. 2008. Toll-like receptor and RIG-I-like receptor signaling. Ann. N. Y. Acad. Sci. 1143:1–20. 10.1196/annals.1443.020 [DOI] [PubMed] [Google Scholar]
- 12.Munir M, Berg M. 2013. The multiple faces of protein kinase R in antiviral defense. Virulence 4:85–89. 10.4161/viru.23134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, Lich JD, Heise MT, Chen Z, Ting JP. 2008. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451:573–577. 10.1038/nature06501 [DOI] [PubMed] [Google Scholar]
- 14.Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, Ranjan P, Monroe KM, Pickles RJ, Sambhara S, Ting JP. 2011. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity 34:854–865. 10.1016/j.immuni.2011.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong M, Yoon SI, Wilson IA. 2012. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity 36:337–347. 10.1016/j.immuni.2011.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Nagarkar DR, Bowman ER, Schneider D, Gosangi B, Lei J, Zhao Y, McHenry CL, Burgens RV, Miller DJ, Sajjan U, Hershenson MB. 2009. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol. 183:6989–6997. 10.4049/jimmunol.0901386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Triantafilou K, Vakakis E, Richer EA, Evans GL, Villiers JP, Triantafilou M. 2011. Human rhinovirus recognition in non-immune cells is mediated by Toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence 2:22–29. 10.4161/viru.2.1.13807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slater L, Bartlett NW, Haas JJ, Zhu J, Message SD, Walton RP, Sykes A, Dahdaleh S, Clarke DL, Belvisi MG, Kon OM, Fujita T, Jeffery PK, Johnston SL, Edwards MR. 2010. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 6:e1001178. 10.1371/journal.ppat.1001178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calven J, Yudina Y, Hallgren O, Westergren-Thorsson G, Davies DE, Brandelius A, Uller L. 2012. Viral stimuli trigger exaggerated thymic stromal lymphopoietin expression by chronic obstructive pulmonary disease epithelium: role of endosomal TLR3 and cytosolic RIG-I-like helicases. J. Innate Immun. 4:86–99. 10.1159/000329131 [DOI] [PubMed] [Google Scholar]
- 20.Zhu L, Lee PK, Lee WM, Zhao Y, Yu D, Chen Y. 2009. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR-dependent pathway. Am. J. Respir. Cell Mol. Biol. 40:610–619. 10.1165/rcmb.2008-0223OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sajjan US, Jia Y, Newcomb DC, Bentley JK, Lukacs NW, LiPuma JJ, Hershenson MB. 2006. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 20:2121–2123. 10.1096/fj.06-5806fje [DOI] [PubMed] [Google Scholar]
- 22.Hewson CA, Jardine A, Edwards MR, Laza-Stanca V, Johnston SL. 2005. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J. Virol. 79:12273–12279. 10.1128/JVI.79.19.12273-12279.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harper RW, Xu C, Eiserich JP, Chen Y, Kao CY, Thai P, Setiadi H, Wu R. 2005. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 579:4911–4917. 10.1016/j.febslet.2005.08.002 [DOI] [PubMed] [Google Scholar]
- 24.Forteza R, Salathe M, Miot F, Conner GE. 2005. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 32:462–469. 10.1165/rcmb.2004-0302OC [DOI] [PubMed] [Google Scholar]
- 25.Balaban RS, Nemoto S, Finkel T. 2005. Mitochondria, oxidants, and aging. Cell 120:483–495. 10.1016/j.cell.2005.02.001 [DOI] [PubMed] [Google Scholar]
- 26.Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, Sack MN, Kastner DL, Siegel RM. 2011. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208:519–533. 10.1084/jem.20102049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 28.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. 10.1038/nature04193 [DOI] [PubMed] [Google Scholar]
- 29.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988. 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- 30.Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. 2009. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J. Cell Sci. 122:3161–3168. 10.1242/jcs.051193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, Arnoult D, Girardin SE. 2008. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 9:293–300. 10.1038/sj.embor.7401161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. 10.1016/j.cell.2010.01.020 [DOI] [PubMed] [Google Scholar]
- 33.Schneider D, Ganesan S, Comstock AT, Meldrum CA, Mahidhara R, Goldsmith AM, Curtis JL, Martinez FJ, Hershenson MB, Sajjan U. 2010. Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 182:332–340. 10.1164/rccm.200911-1673OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. 2006. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. U. S. A. 103:15038–15043. 10.1073/pnas.0601945103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trnka J, Blaikie FH, Logan A, Smith RA, Murphy MP. 2009. Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radic. Res. 43:4–12. 10.1080/10715760802582183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee SB, Bae IH, Bae YS, Um HD. 2006. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J. Biol. Chem. 281:36228–36235. 10.1074/jbc.M606702200 [DOI] [PubMed] [Google Scholar]
- 37.Fu Y, Shi G, Wu Y, Kawai Y, Tian Q, Yue L, Xia Q, Miyamori I, Fan C. 2011. The mitochondria mediate the induction of NOX1 gene expression by aldosterone in an ATF-1-dependent manner. Cell Mol. Biol. Lett. 16:226–235. 10.2478/s11658-011-0002-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Miller DJ, Bowman ER, Nagarkar DR, Schneider D, Zhao Y, Linn MJ, Goldsmith AM, Bentley JK, Sajjan US, Hershenson MB. 2011. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 7:e1002070. 10.1371/journal.ppat.1002070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wosniak J, Jr, Santos CX, Kowaltowski AJ, Laurindo FR. 2009. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox Signal. 11:1265–1278. 10.1089/ars.2009.2392 [DOI] [PubMed] [Google Scholar]
- 40.Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P, Tuder RM, Biswal S. 2008. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am. J. Respir. Crit. Care Med. 178:592–604. 10.1164/rccm.200803-380OC [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Chen J, Kinter M, Shank S, Cotton C, Kelley TJ, Ziady AG. 2008. Dysfunction of Nrf-2 in CF epithelia leads to excess intracellular H2O2 and inflammatory cytokine production. PLoS One 3:e3367. 10.1371/journal.pone.0003367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartling TR, Drumm ML. 2009. Oxidative stress causes IL8 promoter hyperacetylation in cystic fibrosis airway cell models. Am. J. Respir. Cell Mol. Biol. 40:58–65. 10.1165/rcmb.2007-0464OC [DOI] [PMC free article] [PubMed] [Google Scholar]