

Abstract
The silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) is an established histone deacetylase 3 (HDAC3)-dependent transcriptional corepressor. Microarray analyses of MCF-7 cells transfected with control or SMRT small interfering RNA revealed SMRT regulation of genes involved in DNA damage responses, and the levels of the DNA damage marker γH2AX as well as poly(ADP-ribose) polymerase cleavage were elevated in SMRT-depleted cells treated with doxorubicin. A number of these genes are established p53 targets. SMRT knockdown decreased the activity of two p53-dependent reporter genes as well as the expression of p53 target genes, such as CDKN1A (which encodes p21). SMRT bound directly to p53 and was recruited to p53 binding sites within the p21 promoter. Depletion of GPS2 and TBL1, components of the SMRT corepressor complex, but not histone deacetylase 3 (HDAC3) decreased p21-luciferase activity. p53 bound to the SMRT deacetylase activation domain (DAD), which mediates HDAC3 binding and activation, and HDAC3 could attenuate p53 binding to the DAD region of SMRT. Moreover, an HDAC3 binding-deficient SMRT DAD mutant coactivated p53 transcriptional activity. Collectively, these data highlight a biological role for SMRT in mediating DNA damage responses and suggest a model where p53 binding to the DAD limits HDAC3 interaction with this coregulator, thereby facilitating SMRT coactivation of p53-dependent gene expression.
INTRODUCTION
The tumor suppressor protein p53 is a master regulator of cellular responses to genotoxic and other cellular stress signals that functions to maintain genomic integrity. In response to stress, p53 regulates the transcription of a number of specific target genes, such as CDKN1A (which encodes p21), BRCA1, BRCA2, CHEK1, and CHEK2, in order to control various cellular processes, including cell cycle progression, DNA repair, apoptosis, and autophagy (1, 2). Several posttranslational modifications (e.g., phosphorylation, acetylation, methylation, ubiquitination, sumoylation, and neddylation) affect p53 protein stability, sequence-specific DNA binding properties, and transcriptional activity (3). Transcriptional coregulators also play an integral role in the signaling mechanisms by which transcription factors such as p53 elicit their physiological functions. They can either activate (coactivator) or repress (corepressor) gene transcription, at least in part by altering the acetylation of lysine residues in histone tails, thereby favoring an open or closed chromatin structure and thereby regulating the accessibility of DNA to the general transcriptional machinery. Among p53 coactivators, p300, CREB-binding protein (CBP), and CBP-associated factor (p/CAF) are well characterized for their histone acetyltransferase activity and ability to interact with and stimulate p53 transactivation functions (4–6). In contrast, corepressors such as mSin3a play a role in p53-mediated transcriptional repression, likely through p53 tethering a repressor complex containing histone deacetylase 1 (HDAC1) to target genes (7). Moreover, as acetylation of p53 promotes its transcriptional activity (8), a number of HDACs (e.g., HDACs 1 to 4) can also downregulate p53-dependent gene expression by deacetylation of the p53 protein (9).
The silencing mediator of retinoic acid and thyroid hormone receptors (SMRT; also known as NCoR2) and its paralog, nuclear receptor corepressor (NCoR), are transcriptional corepressors that are best characterized relative to their abilities to bind to unliganded or antagonist-bound nuclear receptors and repress their transcriptional activity (10, 11). These large (∼270-kDa) proteins share a molecular architecture and an overall sequence identity of ∼36% (12). Nonetheless, each of them can also perform distinct functions in neurogenesis, development, and hormonal regulation pathways (13–15), and their repression activities can be regulated by distinct regulatory pathways (16). Moreover, deletion of either gene in knockout experiments is embryonically lethal, demonstrating that these corepressors cannot compensate for one another during development (11). While the repressive activity of SMRT resides largely within the N-terminal portion of the molecule, the interaction domains for binding to nuclear receptors, referred to as CoRNR box motifs, are located in the C terminus (11, 17). In addition, the C-terminal and central regions of SMRT can interact with several other DNA binding transcription factors, including BCL6, ETO, NF-κB, and CBF1, and repress the expression of their target genes (18).
The inhibitory chromatin remodeling elicited by SMRT is mediated by its recruitment of deacetylases, particularly HDAC3, to SMRT-associated transcription factors and, hence, their target genes (11, 19). In spite of a high degree of disordered structure (20), there are several well-structured regions within the N-terminal region of SMRT (11), including two highly conserved Swi3-Ada2-NCoR-TFIIIB (SANT)-like domains, as well as repression domain 1 (RD1). These regions provide binding sites for other components of the core SMRT repression complex, namely, HDAC3, G-protein suppressor 2 (GPS2), and the exchange factors TBL1/TBL1R, and consequently are critical for mediating SMRT's repressor activity (19, 21, 22). The RD1 domain of SMRT, TBL1, and GPS2 make contacts with each other, leading to the formation of a stable complex (20, 22). These proteins, in turn, enable interaction of SMRT with HDAC3 through the first of the corepressor's two extended SANT domains, termed the deacetylase activation domain (DAD); the second mediates SMRT interactions with histones and is referred to as the histone interaction domain (23). Inositol tetraphosphate (IP4) binds to the DAD region of SMRT and is an important mediator of the corepressor's interaction with HDAC3 (24). Binding of SMRT to HDAC3 specifically activates the latent activity of the latter to form an enzymatically active deacetylase complex (23). Thus, SMRT molecules with mutations in the DAD or deletion of the DAD are unable to stimulate the deacetylase activity of HDAC3 (19, 24). Moreover, in mice carrying point mutations in the DADs of SMRT and NCoR, HDAC3 activity was undetectable, despite normal expression of the HDAC3 protein, and there was an increase in nucleosomal histone acetylation and gene expression (25). Overall, these studies suggest that HDAC3 interaction with SMRT is indispensable for the repressive activity of this coregulator.
In addition to the established corepressor role of SMRT-HDAC3 complexes in the regulation of metabolism (26), evidence demonstrating a correlation between high levels of SMRT in breast cancer and a poor prognosis (27, 28) suggested a potential role of this coregulator in breast tumorigenesis. Although the mechanism(s) by which this occurs remains undefined, depletion of SMRT expression reduced the proliferation of estrogen receptor α (ERα)-positive MCF-7 breast cancer cells (15). Unlike for other members of the nuclear receptor superfamily, SMRT can promote estrogen-induced ERα transcriptional activity (15). Since estrogen and ERα signaling are important regulators in breast cancer, this raises the possibility that SMRT promotes breast tumorigenesis, at least in part through amplifying ERα target gene expression. Furthermore, overexpression of SMRT stimulated the intrinsic transcriptional activity of the oncogene steroid receptor coactivator 3 (SRC-3), itself an ERα coactivator and positive regulator of breast cancer cell proliferation, and together these coregulators cooperatively promoted estrogen-dependent expression of some but not all ERα target genes, including the cyclin D1 and progesterone receptor genes (29–31). Collectively, these candidate gene studies demonstrated that SMRT can coactivate ERα transcriptional activity in a gene-selective manner and indicated that SMRT has both coactivator and corepressor activities in breast cancer cells.
In order to obtain a more global perspective of the relative ability of SMRT to coactivate versus corepress gene expression, a microarray analysis of control and SMRT-depleted MCF-7 breast cancer cells was performed. Gene ontological analyses revealed an association between SMRT depletion in these cells and a decrease in the expression of genes involved in DNA damage responses. Interestingly, several of the genes identified in these analyses are targets of p53, suggesting a possible role for SMRT in the regulation of gene expression by this tumor suppressor protein, and we therefore investigated the role of SMRT in modulating p53 transcriptional activity and cellular responses to DNA damage. Silencing of SMRT expression in MCF-7 cells enhanced the DNA damage response, abrogated p53 transcriptional activity, and decreased the doxorubicin-dependent induction of the p53 target genes CDKN1A (which encodes p21), BRAC1, CHEK1, and RAD51. SMRT directly interacted with p53 and was recruited to p53 binding sites in the CDKN1A gene promoter in response to genotoxic stress. Moreover, the minimal interaction region was mapped to the N-terminal region of SMRT that encompasses the DAD, the region essential for HDAC3 enzyme activation, and therefore reveals an HDAC3-independent role for SMRT as a coactivator of p53-dependent gene expression and promoter of DNA damage repair functions.
MATERIALS AND METHODS
Chemicals.
Doxorubicin hydrochloride and 17β-estradiol (E2) were obtained from Fisher Bioreagents (Pittsburgh, PA) and Sigma Chemical Company (St. Louis, MO), respectively. Easytag 35S-labeled l-methionine (specific activity, 1,175 Ci/mmol) was from New England Nuclear/PerkinElmer (Boston, MA). The Lipofectamine RNAiMax, Lipofectamine 2000, and Oligofectamine reagents were from Invitrogen (Carlsbad, CA).
Plasmid constructs.
The mammalian expression constructs for full-length human SMRTτ (pCR3.1-SMRTτ), the SMRTβ splice variant (pCR3.1-SMRTΔ36-254), and N- and C-terminally truncated SMRT mutants (SMRT-NT and SMRT-CT, respectively) in the pCR3.1 vector have been described previously (30). The luciferase (Luc) reporter vectors p21-Luc and p2-mdm2-Luc, as well as the wild-type and mutant 14-3-3σ luciferase reporter genes (32), were kind gifts of Larry Donehower (Baylor College of Medicine), and the SMRT DAD mutant (DADm; Y470A) was obtained from Mitch Lazar (19). Wild-type p53 was obtained from Addgene (Cambridge, MA) and cloned into pGEX-4T, while pGEX-4T-p53(1-300) and pGEX-4T-p53(300-393) were kinds gifts of Mengtao Li (33). The expression vectors for deletions in the SMRT N terminus, namely, SMRT-NTΔ36-312, SMRT-NTΔ36-388, and SMRT-NTΔ36-480, were generated by overlap extension PCR following a standard protocol. Briefly, mutagenesis was achieved by performing the first set of PCRs with specially designed oligonucleotide primers that included the desired deletions in their sequences. The two overlapping PCR-amplified fragments were fused together in a subsequent PCR using the 5′ and 3′ outside primers and the two PCR fragments as the template. The final PCR products were cloned between BamHI and NotI sites of the vector to yield plasmids pCR3.1-SMRT-NTΔ36-312, pCR3.1-SMRT-NTΔ36-388, and pCR3.1-SMRT-NTΔ36-480. The final clones were verified for the desired mutations by sequencing of both DNA strands.
The pCR3.1-hSMRT(255-480) expression plasmid was generated by PCR amplification of the SMRT region between amino acids (aa) 255 and 480 with forward (5′-CCCAAGCTTAAGCTCCCCGCCGACCCCCACCACCATGCCGCTGTACAACCAGCCCTCCG-3′) and reverse (5′-TTTTGCGGCCGCCTTATAGTTCTCATTCTTCTTAGTCAGG-3′) primers using pCR3.1-SMRTτ as a template. The resulting PCR product was cloned into the pCR3.1 vector between HindIII and NotI restriction sites. The mammalian expression plasmid pBIND-Gal4-DBD-hSMRT(255-480) encoding the SMRT DAD (amino acids 255 to 480) fused to the Gal4 DNA binding domain (Gal4-DBD) was generated by PCR amplification of the DAD using forward (5′-CGCGGATCCGTCCGCTGTACAACCAGCCCTCCG-3′) and reverse (5′-TTTTGCGGCCGCCTTATAGTTCTCATTCTTCTTAGTCAGG-3′) primers and pCR3.1-SMRTτ as the template. The resulting PCR product was ligated into the pBIND vector (Promega, Madison, WI) between BamHI and NotI restriction sites in frame with and downstream of the Gal4-DBD. All clones were verified by sequencing of both DNA strands.
Cell culture.
MCF-7 human breast cancer cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The HCT116 human colon carcinoma and Saos-2 human osteosarcoma cell lines were maintained in McCoy's 5a medium supplemented with 10% and 15% FBS, respectively. ZR-75-1 cells were maintained in RPMI medium supplemented with 10% FBS.
Gene silencing by siRNA.
Small interfering RNA (siRNA) target sequences for SMRT (panSMRT), NCoR, and HDAC3, which were published previously (34), as were the target sequences (siRNAs 1 and 2) for TBL1 (35), were chemically synthesized by Ambion (Grand Island, NY) as oligonucleotide duplexes. For silencing GPS2, an ON-TARGETplus SMARTpool siRNA from Dharmacon (Thermo Scientific, Rockford, IL) was used. MCF-7 cells (4.5 × 105 cells/well) were plated in six-well culture dishes. After 24 h, cells were transfected with 20 or 30 pmol/well of the indicated siRNA or Ambion's Silencer 2 as a negative control using the Oligofectamine reagent (Life Technologies, Grand Island, NY) following the manufacturer's protocol. At 6 h after transfection, the medium was changed to medium containing stripped FBS (sFBS), and 48 h thereafter, cells were treated with vehicle (phosphate-buffered saline [PBS]) or doxorubicin (0.5 or 1.0 μM) for 24 h. Cells from duplicate wells were harvested in the TRIzol reagent (Life Technologies) for RNA extraction for real-time quantitative PCR (RT-qPCR) assays. For RNA analyses, RNA was isolated by an RNeasy kit (Qiagen, Valencia, CA). To assess protein knockdown, cells from 4 wells were harvested in PBS containing protease inhibitors, and cell pellets were kept at −80°C until further analysis by Western blotting. SMRT knockdown by panSMRT siRNA in Saos-2 cells (4 × 105 cells/well) and ZR75-1 cells (6 × 105 cells/well) was performed with the Oligofectamine reagent and the Lipofectamine RNAiMax reagent, respectively, following the above-described protocol.
Microarray and gene ontology analyses.
The robust multiarray average (RMA) algorithm was applied to the Affymetrix human gene (version 1.0) ST microarray (36). Briefly, the raw intensity values were background corrected, log2 transformed, and then quantile normalized, and then a linear model was fit to the normalized data to obtain an expression measure for each probe set on each array. The significance analysis of microarrays (SAM) algorithm was used to detect differentially expressed genes (DGEs) between treatments with the statistical cutoff (q value = 0.1) and effect cutoff (fold change = 1.2) (37). Pathway analyses were performed using Ingenuity Pathway Analysis (IPA) software. Gene ontology terms enriched in DGEs were analyzed using DAVID (http://david.abcc.ncifcrf.gov).
Analyses of endogenous gene expression.
The effects of SMRT depletion on the endogenous expression of the CDKN1A (p21), BRCA1, CHEK1, and RAD51 genes were assessed by reverse transcription and RT-qPCR in an ABI Prism 7500 sequence analyzer (PE Applied Biosystems, Foster City, CA) in conjunction with SYBR green chemistry. Total RNA was isolated by use of the TRIzol reagent (Life Technologies) according to the manufacturer's instructions, and cDNA was prepared from 1 μg of total RNA by using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). The mRNA levels of the indicated genes were measured by RT-qPCR using gene-specific primers (Table 1) and were normalized against the 18S rRNA level. Assays were performed with SYBR green PCR master mix reagents in MicroAmp 96-well plates (PE Applied Biosystems). Cycling conditions were 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Data are presented as the means ± standard errors of the means (SEMs) of at least three independent experiments.
TABLE 1.
Sequences of primers used in RT-qPCR analyses
| Gene | Sequence |
|
|---|---|---|
| Forward primer | Reverse primer | |
| CDKN1 | 5′-GGAGACTCTCAGGGTCGAA-3′ | 5′-GGATTAGGGCTTCCTCTTGG-3′ |
| BRCA1 | 5′-GCTCTTCGCGTTGAAGAAGTA-3′ | 5′-CTCCAGACAGATGGGACACT-3′ |
| CHEK1 | 5′-CAAAGGACAGTCCGCCGAG-3′ | 5′-TCCATAGGCACCTTCTCCCA-3′ |
| RAD51 | 5′-CCCGCGGGGTGAAGTCG-3′ | 5′-TGCATTGCCATTACTCGGTCC-3′ |
Transactivation assays for p21, MDM2, and 14-3-3σ promoter activity.
MCF-7 and HCT116 cells (4.5 × 105 cells/well) were grown in six-well culture dishes for 24 h in phenol red-free DMEM containing 10% FBS. The indicated amount of either the p21-Luc, p2-mdm2-Luc, or 14-3-3σ–Luc (wild type or mutant) reporter plasmid and, where indicated, 0.25 to 0.5 μg/well of SMRT expression plasmids were transfected with the X-tremeGene HP DNA transfection reagent (Roche Diagnostics, Indianapolis, IN) per the manufacturer's instruction. At 4 to 6 h after transfection, the medium was replaced by DMEM supplemented with 10% charcoal-stripped FBS, and cells were allowed to grow for 24 to 36 h. Where indicated, cells were treated with 1 μM doxorubicin for 24 h. The cells were harvested, and the luciferase activity determinations were performed in triplicate using a luciferase assay system kit (Promega) and a Luminoskan Ascent Thermo Labsystems apparatus (Thermo Electron Corporation, Milford, MA). Relative light units were normalized to the total cellular protein level measured in a Bio-Rad protein assay. Data are presented as the means ± SEMs of four independent experiments.
ChIP assay.
Chromatin immunoprecipitation (ChIP) was performed using a CHIP-It enzymatic kit (Active Motif, Carlsbad, CA) following the manufacturer's instructions, with some modifications. MCF-7 cells (15 × 106 cells/150-mm plate) maintained in DMEM–10% FCS were treated for 6 h with 1 μM doxorubicin or vehicle (PBS). Cross-linking was performed with 1% formaldehyde for 10 min at 37°C in a CO2 incubator and terminated by adding glycine at a 0.125 M final concentration for 5 min at room temperature with rotation. Cells were harvested by scraping in PBS into 15-ml conical tubes and centrifuged at 3,500 rpm for 7 min. Cell pellets were incubated on ice in 1 ml of lysis buffer provided in the kit. Nuclei were isolated by disruption in a Dounce homogenizer (40 strokes). Nuclear pellets were collected by centrifugation at 2,500 × g for 10 min and resuspended in 350 μl of the provided enzymatic buffer containing protease inhibitors. Nuclear suspensions were sonicated (10-s pulses repeated 4 times) and then subjected to nuclease digestion for 20 min following the manufacturer's instructions. Chromatin was collected after centrifugation at 16,000 × g for 20 min. Washes and reverse cross-linking procedures during immunoprecipitation were performed per the manufacturer's protocol. The following antibodies were used for immunoprecipitation: p53 (DO-1), SMRT (H-300) from Santa Cruz Biotechnology (Santa Cruz, CA), as well as rabbit and mouse normal IgG, as appropriate, for nonspecific controls. DNA was then purified by using Qiagen spin columns and analyzed by quantitative PCR (qPCR) using SYBR green chemistry, and the amount was normalized against the amount of input chromatin. Primer sequences for the p53 binding site at position −2200 of the p21 gene were 5′-GTGGCTCTGATTGGCTTTCTG-3′ and 5′-CTGAAAACAGGCAGCCCAAG-3′ (38), primer sequences for the p53 binding site at position −1200 were 5′-CATCCCCCACAGCAGAGGAGAA-3′ and 5′-ACCCAGGCTTFFAGCAGCTA-3′, and primer sequences for the Sp1 binding site at position −282 (39) were 5′-AGTGCCAACTCATTCTCCAAG-3′ and 5′-GACACATTTCCCCACGAAGT-3′. Nonspecific primers for the p21 gene were 5′-GAGTCCTGTTTGCTTCTGGGCA-3′ and 5′-CTGCATTGGGGCTGCCTATGTA-3′ (40). Data are presented as the means ± SEMs of at least three independent experiments.
Western blot analyses.
For Western blot analyses, treated cells were harvested and incubated for 20 min at 4°C in lysis buffer (50 mM Tris-HCl [pH 7.4] containing 150 mM NaCl, 5 mM EDTA, and 0.5% Nonidet P-40 supplemented with Complete protease inhibitor minitablets [Roche]). The protein concentration of the cell lysates was determined using Bio-Rad protein assay reagent, and equal amounts of protein were resolved by SDS-PAGE using precast 3 to 8% NuPAGE Novex gels for detection of SMRT and NCoR or 10% bis-Tris precast gels (Life Technologies) for detection of other proteins and immunoblotted. Actin was used as a loading control. The following antibodies were used for immunoblotting: SMRT, p21, and HDAC3 (BD Biosciences; San Jose, CA); γ-H2AX and poly(ADP-ribose) polymerase (PARP; Cell Signaling; Danvers, MA); p53 and BRCA1 (Santa Cruz Biotechnology); actin (MAB1501R; Millipore, MA); and Flag (Agilent Technologies, Santa Clara CA).
Coimmunoprecipitation.
Cells were washed twice with ice-cold PBS and harvested with PBS containing protease inhibitors, followed by incubation with lysis buffer (20 mM Tris-HCl [pH 7.5], 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 5% glycerol) containing 1× Halt protease and phosphatase inhibitor cocktail (Thermo Scientific) for 30 min at 4°C under rotation. The cell lysates were centrifuged at 13,000 rpm for 10 min at 4°C, the supernatant was collected, and the protein concentration was measured by the Bio-Rad protein assay. Equal amounts of total protein (500 or 1,000 μg) were precleared with 20 μl of protein A/G plus agarose beads (Santa Cruz Biotechnology), followed by incubation with either SMRT (BD Biosciences) or p53 (FL-393 or DO-1; Santa Cruz Biotechnology) antibodies or normal rabbit or mouse IgG on a rotary shaker overnight at 4°C. Immune complexes were further incubated with 50 μl of protein A/G plus agarose beads (Santa Cruz Biotechnology) for 1 h at 4°C on a rotary shaker. For Flag immunoprecipitation, precleared lysates were incubated with Flag-agarose beads (Sigma). The beads were washed 3 times with lysis buffer, and immunoprecipitated proteins were eluted by resuspending the beads in 40 μl of 2× SDS sample buffer and incubating at 75°C for 10 min. The eluted proteins were resolved by SDS-PAGE and transferred onto nitrocellulose membranes for Western blot analysis.
GST pulldown assay.
Glutathione S-transferase (GST) and GST-p53 proteins were expressed in the Escherichia coli BL21(DE3) strain and purified using a glutathione-Sepharose column (Thermo Scientific) following the manufacturer's protocol. The full-length SMRT isoforms, SMRT fragments, and the deletion mutant proteins were expressed in vitro as 35S-labeled proteins using a TNT T7 Quick Coupled transcription/translation system (Promega) per the manufacturer's instruction. GST or GST-tagged p53 wild type or its truncated mutants (5 μg/reaction) were allowed to bind to glutathione agarose beads (Thermo Scientific) for 4 h at 4°C under rotation, followed by 2 washes with binding buffer (20 mM Tris-HCl [pH 8.0], 1 mM EDTA, 100 mM NaCl, 0.5% NP-40, 5% glycerol). Five microliters of TNT reaction lysates was added to the glutathione agarose beads, and the mixture was incubated overnight at 4°C. On the next day, proteins complexed to beads were recovered by centrifuging samples at 1,000 × g for 5 min at 4°C, washed 4 times with wash buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% Triton X-100, 15 mM EGTA, 1 mM dithiothreitol [DTT] supplemented with Halt protease, phosphatase inhibitor cocktail), and suspended in 40 μl of 2× SDS sample buffer. Samples were heat denatured at 75°C for 10 min, and the supernatants were resolved by SDS-PAGE. The gels were dried for 3 h in a gel dryer, and proteins were visualized by autoradiography. Protein band intensities were analyzed semiquantitatively by densitometry using ImageJ software (NIH Image).
Competition assay.
The binding of p53 and HDAC3 to SMRT at the DAD region was assessed in a competition experiment. The SMRT DAD (amino acids 255 to 480) fused to the Gal4-DBD was expressed in HCT116 cells and extracted using a lysis buffer containing 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, and 50 mM KCl supplemented with Complete protease inhibitor cocktail (Roche). The total cell lysate was incubated with anti-Gal4 agarose beads (RK5C1; Santa Cruz Biotechnology) that had previously been blocked with 5% bovine serum albumin (BSA), followed by incubation with 600 μl of in vitro-translated 35S-labeled p53, with or without 30 μl of in vitro-translated 35S-labeled HDAC3 for a further period of 16 h at 4°C in the binding buffer (20 mM Tris-HCl [pH 8.0], 2 mM EDTA, 1% Triton X-100, 1 mM DTT, 10% glycerol, 50 mM KCl, 0.2% BSA). Subsequently, the proteins complexed to agarose beads were recovered by centrifugation, washed 3 times with binding buffer, and finally, suspended in 40 μl of 2× SDS sample buffer. Samples were heat denatured, and the proteins in the supernatants were resolved in 10% bis-Tris SDS-polyacrylamide gels. The gels were subsequently dried for 3 h, and resolved proteins were visualized by autoradiography.
RESULTS
SMRT depletion reduces expression of DNA damage response genes.
Given our previous observation that SMRT can function as a dual coactivator/corepressor in a cell/gene-specific context (15), a microarray analysis was performed to further evaluate the relative ability of SMRT to coactivate versus corepress gene expression using MCF-7 cells transfected with control or SMRT-specific siRNA (Fig. 1A). Inhibition of SMRT expression was associated with a decrease in the expression of a set of genes involved in the DNA damage response (Fig. 1B). The microarray analysis was originally performed with and without estradiol (E2) treatment in order to assess the dual coactivator/corepressor function of SMRT on ERα-regulated gene expression. However, cells exposed to E2 also showed a reduced expression of DNA damage response genes (data not shown), indicating a lack of significant ERα involvement in this altered gene expression profile. The microarray data were validated by RT-qPCR measurements of the mRNAs for two DNA repair-specific genes, namely, BRCA1 and CHEK1, whose expression was significantly decreased in SMRT-depleted cells in comparison to that in nonspecific siRNA-transfected control MCF-7 cells (Fig. 1C); the efficiency of SMRT knockdown is shown in Fig. 1D. Among the genes identified in the microarray analysis, several were previously reported to be regulated by the tumor suppressor protein, p53, including CHEK1, RAD51, BRCA1, BRCA2, and RFC3 (41–43). These data demonstrated that SMRT, in addition to its well-characterized corepressor activity, can also promote the expression of a select group of genes, potentially through regulation of p53 transcriptional activity.
FIG 1.
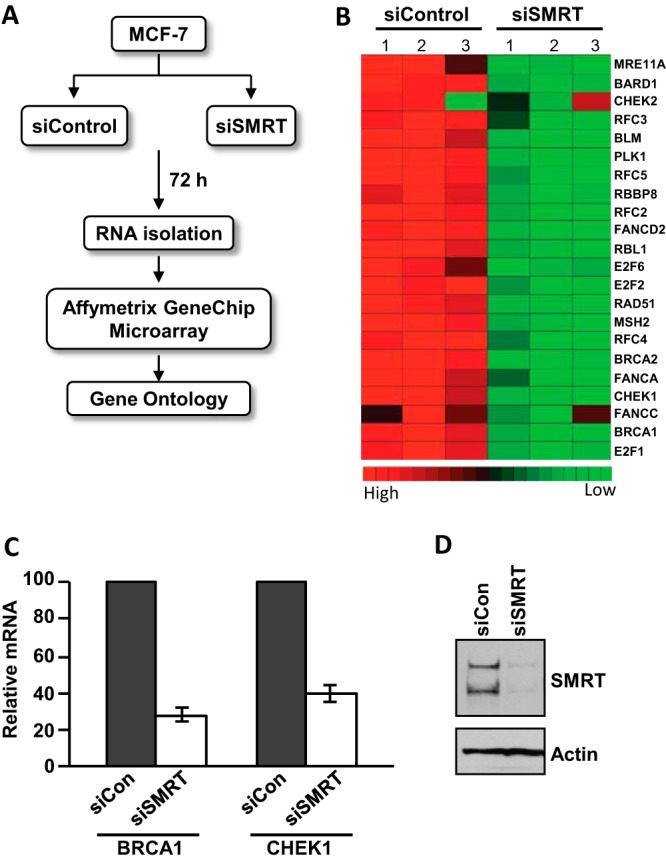
Profiling of SMRT-regulated gene expression in MCF-7 cells. (A) Scheme of siRNA-mediated depletion of SMRT and subsequent analysis of mRNA by microarray. MCF-7 cells were transfected with 20 pmol of control siRNA (siControl) or SMRT-specific siRNA (siSMRT). Cells were harvested 72 h after transfection, and the RNA was isolated and analyzed by an Affymetrix GeneChip microarray, followed by gene ontological analysis. (B) Heat map showing that SMRT depletion leads to decreased expression of genes involved in the DNA damage response. (C) Depletion of SMRT expression in MCF-7 cells leads to a decrease in BRCA1 and CHEK1 mRNAs. MCF-7 cells were transfected with 20 pmol of control siRNA (siCon) or SMRT-specific siRNA (siSMRT), and at 48 h thereafter, RNA was isolated and the levels of BRCA1 and CHEK1 mRNAs were quantitated by RT-qPCR and normalized to the signals obtained for 18S RNA. Values are the averages of four experiments ± SEMs. (D) Representative Western blot showing the efficiency of SMRT depletion by SMRT-specific siRNA in MCF-7 cells.
Impairment of DNA damage response in SMRT-depleted cells.
In order to ascertain the role of SMRT in the DNA damage response, the level of the DNA damage marker γH2AX (44) was assessed in MCF-7 cells treated with doxorubicin, a DNA-intercalating agent that induces DNA double-strand breaks (45). Western blot analysis revealed higher levels of DNA damage in SMRT-depleted cells than control cells following doxorubicin treatment (Fig. 2A). This increase in γH2AX levels was doxorubicin dose dependent and unaffected by E2 treatment. This is consistent with the microarray results and suggests that SMRT regulation of this cellular response to the DNA-damaging agent is independent of ERα. In contrast, there was no significant difference in γH2AX levels in SMRT-depleted versus control Saos-2 cells, which are p53 deficient (Fig. 2B), consistent with the hypothesis that SMRT impacts DNA damage responses through a p53-dependent mechanism. In addition to γH2AX, cleavage of the apoptotic marker PARP (46), a nuclear protein implicated in DNA repair, was elevated in SMRT-depleted cells compared to control MCF-7 cells in a doxorubicin-induced, dose-dependent manner (Fig. 2C). These data, combined with the microarray finding of lowered expression of multiple p53 target genes in SMRT-depleted cells, suggested that SMRT can promote p53 transcriptional activity as part of the cellular response to DNA damage.
FIG 2.
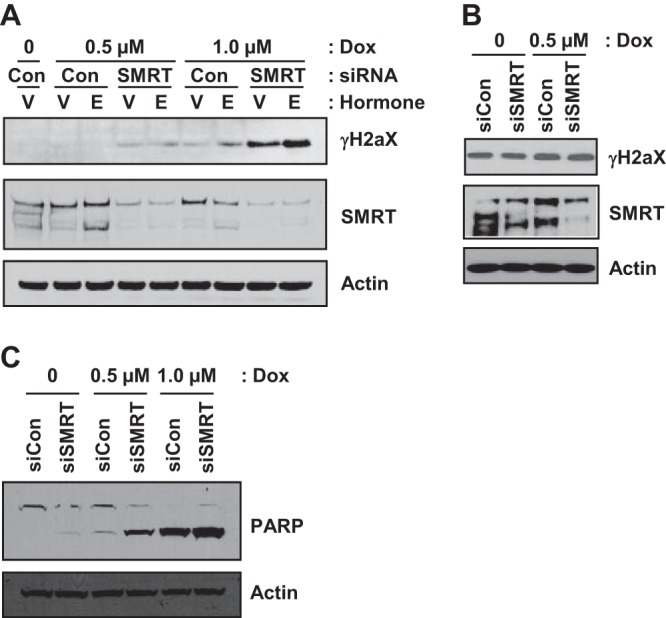
Depletion of SMRT expression compromises the DNA damage response in MCF-7 cells. (A) Western blot analysis showing the impact of SMRT depletion on doxorubicin (Dox)-induced DNA damage. MCF-7 cells were transfected with control (Con) or SMRT-specific siRNA. After 48 h, cells were treated with 0.1% ethanol (lanes V) or 1 nM estradiol (lanes E) for 4 h and subsequently treated with doxorubicin at the indicated concentrations for 16 h. Whole-cell lysates were resolved in SDS-polyacrylamide gels and immunoblotted for γH2AX and SMRT. (B) Saos-2 cells, which are deficient for p53, were analyzed as described for MCF-7 cells in the legend to panel A, except that cells were not treated with hormone. (C) Western blot analysis of PARP cleavage in SMRT-depleted versus control MCF-7 cells with and without doxorubicin treatment. For all blots, actin was used as a loading control.
To further substantiate the role of SMRT in regulating p53 transcriptional activity, the expression of genes involved in the DNA double-strand repair pathway, namely, BRCA1, CHEK1, RAD51, and CDKN1A (which encodes p21), whose levels are upregulated in response to DNA damage and/or regulated by p53 (41, 43), were measured in MCF-7 cells pretreated with control or SMRT-specific siRNA and exposed to doxorubicin for 16 h. As expected, analyses by RT-qPCR demonstrated little effect of doxorubicin on CHEK1 mRNA levels in MCF-7 cells (47); however, knockdown of SMRT significantly reduced the levels of mRNA for all of these genes in doxorubicin-treated cells (Fig. 3). The levels of doxorubicin-induced p21 mRNA following SMRT depletion were 30% lower than those in control cells, while for the other three genes measured (BRCA1, RAD51, and CHEK1), there was no doxorubicin induction of their expression in SMRT-depleted cells, consistent with a role for SMRT in the positive regulation of these p53 target genes.
FIG 3.

SMRT regulates expression of p53 target genes in doxorubicin-treated MCF-7 cells. Cells were transfected with 20 pmol of control siRNA or SMRT-specific siRNA (siSMRT) and after 48 h were treated with vehicle (PBS; filled bars) or doxorubicin (1 μM; white bars) for 16 h. RNA was subsequently isolated, and the levels of mRNA for the p21 gene (A), BRCA1 (B), RAD51 (C), and CHEK1 (D) were quantitated by RT-qPCR and normalized to the signals obtained for 18S RNA. The changes in the mRNA levels of the indicated genes are represented relative to the levels of their respective control siRNAs in doxorubicin-treated cells. Values represent the means ± SEMs of data from five independent experiments. P values were determined by Student's t test. *, P ≤ 0.05; **, P ≤ 0.001; ***, P ≤ 0.0001.
SMRT coactivates p53 transcriptional activity.
Given that SMRT depletion decreased expression of multiple DNA damage repair genes known to be p53 targets, it was hypothesized that SMRT promotes p53 transcriptional activity. In order to test this, luciferase reporter assays were conducted using promoters from two distinct p53-responsive genes, namely, CDKN1A (which encodes p21) and MDM2. In addition, since the DNA damage response genes were first identified in ERα-positive MCF-7 cells and p53 can interact with ERα (40), the ability of ERα to impact SMRT coactivation of p53 transcriptional activity was evaluated by measuring p53 transcriptional activity in cells treated or not with E2. The results showed that the SMRT depletion achieved by silencing with siRNA significantly decreased p53 transcriptional activity, as indicated by the strong reduction in the amount of luciferase produced by either reporter gene (Fig. 4A and B). This reduction was unaltered by E2 treatment, indicating an ERα-independent action of SMRT in the regulation of p53 activity in MCF-7 cells. In contrast, transient overexpression of exogenous SMRT in MCF-7 cells resulted in increased p53 transcriptional activity, as revealed by the 3.5-fold higher p21-Luc reporter activity compared to that for the empty-vector (basal) control (Fig. 4C).
FIG 4.

SMRT coactivates p53 transcriptional activity in MCF-7 cells. Cells were transfected with 20 pmol of control siRNA or SMRT-specific siRNA (siSMRT), prior to transfection with 1 μg of either the p21 (A) and MDM2 (B) luciferase reporter genes, and treated with vehicle (Veh; 0.1% ethanol) or 1 nM E2 for 24 h. Luciferase activity data are the means ± SEMs of three (p21) and two (MDM2) experiments. (C) MCF-7 cells were transfected with 500 ng of a Flag-tagged expression vector for SMRT or an empty-vector control (basal) along with 500 ng of the p21-Luc reporter gene. Cells were harvested 48 h after transfection, and luciferase activity was measured. The activity for SMRT is shown relative to that for the empty-vector control. Values represent the averages ± SEMs (n = 4). (D) MCF-7 cells were transfected with control siRNA or SMRT-specific siRNA, and 48 h later, the cells were treated with vehicle (PBS; without doxorubicin [−Dox]) or doxorubicin (+Dox; 1 μM) for 16 h. Whole-cell lysates were resolved in SDS-polyacrylamide gels, and the expression of the p21, p53, SMRT, and actin proteins was analyzed by Western blotting. (E) ZR75-1 cells were transfected with 30 pmol of control siRNA or SMRT-specific siRNA. After 48 h, the cells were treated with vehicle (PBS) or doxorubicin (1 μM) for 16 h, followed by RNA isolation and quantitation by RT-qPCR. The p21 mRNA levels were normalized to the signals obtained for 18S RNA. The changes in the mRNA levels are represented relative to the level of control siRNA in doxorubicin-treated cells. Values represent the means ± SEMs of data from two independent experiments. (F) Saos-2 cells were transfected with control siRNA (siC) or SMRT-specific siRNA (siS), and 48 h later, the cells were treated with vehicle (PBS without doxorubicin) or doxorubicin (+Dox; 0.5 μM) for 16 h. Whole-cell lysates were resolved by SDS-PAGE, and the expression of p21, BRCA1, SMRT, and actin proteins was analyzed by Western blotting. (G) MCF-7 cells were transfected with 500 ng of either the 14-3-3σ wild-type (WT) or mutant (MT) luciferase reporter genes, along with 500 ng pCR3.1 (basal) or full-length human SMRT plasmids. After 24 h, cells were treated with vehicle or 1 μM doxorubicin for 24 h. Luciferase activities are presented relative to those obtained for cells transfected with wild-type 14-3-3σ–Luc and SMRT and treated with doxorubicin. Values represent the averages ± SEMs (n = 3). RLU, relative light units.
To test whether SMRT was regulating the expression of p53, the levels of this transcription factor in doxorubicin-treated MCF-7 cells transfected with control or SMRT-specific siRNA were assessed by Western blotting. As expected, doxorubicin induction of p21 protein expression was reduced in SMRT-depleted versus control MCF-7 cells, but this was not accompanied by any change in p53 protein levels (Fig. 4D), indicating that SMRT is regulating the activity rather than the expression of p53. Regulation of p53-dependent gene expression was not cell type specific, since SMRT knockdown in another p53-positive breast cancer cell line, ZR-75-1, also yielded a significant decrease in doxorubicin-induced p21 mRNA expression (Fig. 4E) similar to that observed for MCF-7 cells, as described above. To address whether SMRT regulation of p53 target genes was dependent upon p53, the impact of SMRT depletion on p21 and BRCA1 expression was evaluated in p53-deficient Saos-2 cells; SMRT knockdown did not impact the levels of either target (Fig. 4F). In addition, transactivation assays revealed that SMRT coactivation of doxorubicin-induced 14-3-3σ luciferase reporter activity was absent in parallel experiments conducted with a mutated 14-3-3σ reporter lacking a binding site for p53 (Fig. 4G). Taken together, these results indicate that SMRT promotes p53 transcriptional activity in a cell-independent manner without altering its protein expression.
SMRT associates with p53 binding sites on the p21 gene promoter.
If SMRT is a p53 coactivator, it should localize to p53 binding sites in the regulatory regions of a p53 target gene, and the ability of SMRT to localize to the 5′ and 3′ p53 binding (enhancer) sites of the p21 gene (48, 49) was therefore assessed by chromatin immunoprecipitation (ChIP) experiments (Fig. 5A). First, immunoprecipitation of chromatin with a p53-specific antibody followed by qPCR demonstrated that p53 binding to these sites was present at low levels under basal conditions, particularly for the 5′ site, and strongly stimulated upon doxorubicin treatment (Fig. 5B). Moreover, very little signal was obtained with the IgG negative control, confirming the specific binding of p53 to these sites. Similar to p53, a doxorubicin-induced enrichment of precipitated DNA for both the 5′ and 3′ sites was detected following ChIP with the SMRT-specific antibody. In contrast, neither p53 nor SMRT binding to an upstream nonspecific site of the p21 gene (Fig. 5A) was substantially increased over that for the IgG negative control (Fig. 5B, inset), and together these data demonstrate that p53 and SMRT are recruited to the p21 enhancer regions in a doxorubicin-dependent manner, consistent with a role for SMRT as a p53 coactivator.
FIG 5.

SMRT associates with p53 binding sites on the p21 gene. (A) Schematic representation of the upstream region of the p21 gene, and the positions of the primers used in qPCR to target the 5′ and 3′ p53 binding and nonspecific sites are indicated by arrows. TSS, transcription start site. (B) Chromatin immunoprecipitation assays showing doxorubicin-induced recruitment of SMRT to p53 binding sites of the p21 gene promoter, as well as the upstream nonspecific (NS; p53 response element-deficient) site (inset). MCF-7 cells were treated with vehicle (Veh; PBS) or doxorubicin (Dox; 1 μM) for 6 h, followed by chromatin isolation and immunoprecipitation using antibodies for SMRT or p53 in parallel with the appropriate IgG negative control. Immunoprecipitated DNA was quantitated by qPCR using the indicated primers. Data represent the averages ± SEMs of three independent experiments.
SMRT interacts with p53.
To further define SMRT promotion of p53-mediated gene transcription, SMRT interaction with the p53 protein was assessed. First, the ability of SMRT and p53 to coexist in the same complex was tested by coimmunoprecipitation assay in MCF-7 cells transfected with a Flag epitope-tagged SMRT expression plasmid. With a p53 antibody, Flag-tagged SMRT was immunoprecipitated in doxorubicin-treated cells, while this could not be achieved with the IgG negative control, indicating the presence of a specific SMRT-p53 complex (Fig. 6A, top). Western blot assays indicated the levels of SMRT and p53 in these cells (Fig. 6A, middle and bottom, respectively). Second, the presence of a SMRT and p53 complex was evaluated by coimmunoprecipitation assay in cells of the HCT116 cell line, a colon carcinoma cell line with abundant wild-type p53 expression. Endogenous SMRT protein was coimmunoprecipitated along with p53 protein by an anti-p53 antibody from lysates obtained from cells treated with doxorubicin but not by the IgG negative control antiserum (Fig. 6B, middle), and reciprocally, the endogenous p53 protein was coimmunoprecipitated along with SMRT protein by an anti-SMRT antibody (Fig. 6B, right). As expected, doxorubicin treatment strongly increased p53 expression (Fig. 6B, input). Similar results were also observed in ER-negative but p53-positive C4-12 breast cancer cells after immunoprecipitation with anti-p53 antibody (data not shown). Collectively, these results demonstrate that endogenous SMRT and p53 proteins can be detected in complexes within cells of different origins using anti-p53 and anti-SMRT antibodies, respectively.
FIG 6.

SMRT binds to p53 both in vivo and in vitro. (A) MCF-7 cells were transfected with an expression vector for Flag epitope-tagged SMRT and treated with increasing concentrations of doxorubicin. Cell lysates were immunoprecipitated (IP) with p53 antibody or IgG, and immunoprecipitates were assessed by Western blotting with Flag antibody (top). Levels of SMRT (middle) and p53 (bottom) for input cell lysates were assessed by Western blotting with their respective antibodies. (B) HCT116 cells were treated with increasing doses of doxorubicin for 16 h, and total cell lysates were used for immunoprecipitation using antibodies for p53 or SMRT in parallel with the appropriate IgG negative control. Immunoprecipitated protein complexes were resolved by SDS-PAGE and analyzed by Western blotting using the indicated antibodies. (C) GST pulldown assays showing the interaction of p53 with full-length SMRTτ protein or its truncated mutants. Shown at the left are schematic representations of full-length SMRT and its truncated mutants. The bacterially expressed and purified GST-tagged p53 protein was incubated with in vitro-translated 35S-labeled SMRTτ or its truncated mutants, and the p53-bound SMRT or deletion mutants were detected by autoradiography. Input represents 20% of the corresponding amount of in vitro-translated 35S-labeled lysates. (D) Densitometric quantification of protein bands from panel C normalized to the amounts in the corresponding input lanes and represented relative to the amount of SMRTτ. Rel., relative.
Finally, to determine if SMRT interacts with p53 in vitro, GST pulldown assays employing 35S-labeled full-length SMRTτ protein produced in an in vitro translation system and bacterially expressed and purified recombinant GST-p53 protein were performed. The pulldown using glutathione-agarose beads revealed the association of SMRT with GST-p53 but not with the control GST alone, indicating an interaction between SMRT and p53 proteins (Fig. 6C, top). Thereafter, to define the region of SMRT that interacts with p53, C-terminal and N-terminal fragments of SMRT (SMRT-CT and SMRT-NT, respectively) were tested for their ability to bind to p53. Pulldown studies demonstrated that GST-p53, but not the GST control, was able to interact strongly with SMRT-NT, whereas the interaction between the SMRT-CT deletion mutant and GST-p53 was very weak (Fig. 6C, middle and bottom). The observation that GST-p53 binds preferentially to the SMRT N terminus rather than the C-terminal fragment (Fig. 6D) suggests that the former is the primary interaction domain of SMRT for p53 binding.
Lastly, to define the region of p53 that interacts with SMRT, the 35S-labeled SMRT-NT fragment was tested for its interaction with recombinant p53 deletion mutants, with one lacking the C-terminal tetramerization and regulatory domains and the other lacking both the N-terminal transactivation and central DNA binding domains (Fig. 7A). Deletion of the C-terminal domain (CTD) resulted in the complete loss of the ability of GST-p53(1-300), in which p53 consisted of the region from aa 1 to 300, to interact with SMRT-NT and contrasted with the robust interaction of GST-p53 wild-type protein with this fragment of SMRT. However, the p53 deletion mutant GST-p53(300-393), in which the p53 sequence consisted of only the CTD from aa 300 to 393, strongly interacted with SMRT-NT; quantitative assessment of these assays revealed that the p53 CTD binds to SMRT as well as full-length p53 does, indicating that the p53 CTD is the primary location for this transcription factor's interaction with SMRT protein (Fig. 7C).
FIG 7.

The C-terminal regulatory domain of p53 is essential for SMRT-p53 interaction. (A) Schematic representations of full-length p53 and its truncated mutant derivatives used in the GST pulldown assay. TAD, transactivation domain; Pro, proline-rich domain; DBD, DNA binding domain; Tet, tetramerization; RD, regulatory domain. (B) The bacterially expressed and purified GST-tagged p53 wild-type protein or its truncated mutants were incubated with in vitro-translated 35S-labeled SMRT-NT, followed by immunoprecipitation of the protein complexes using glutathione-agarose beads and SDS-PAGE. The SMRT-NT that bound to p53 or p53 mutants was detected by autoradiography. Input represents 20% of the amount of in vitro-translated 35S-labeled SMRT-NT lysates. (C) Densitometric quantification of protein bands normalized to the amount in the corresponding input lanes and represented relative to the amount of the p53 wild type.
Regulation of p53 transcriptional activity by components of the SMRT corepressor complex.
Having found that wild-type p53 primarily binds to the SMRT N terminus, which also serves as the interaction region for multiple components of the SMRT corepressor complex, particularly HDAC3, GPS2, and TBL1 (20, 22), the roles of these proteins in SMRT-mediated, p53-dependent gene expression were investigated. Depletion of GPS2 and TBL1 significantly reduced the extent of doxorubicin-induced p21 mRNA expression in comparison to that in control siRNA-transfected MCF-7 cells (Fig. 8A and B); this is consistent with a previous report demonstrating that overexpression of GPS2, also known as AMF1, can stimulate the expression of the p53-dependent target gene, p21 (50). In contrast, RT-qPCR analyses showed a significant increase in p21 mRNA levels in cells depleted of HDAC3 and treated with doxorubicin (Fig. 8C). Increased expression of p21 mRNA in HDAC3-depleted cells was not due to altered p53 expression, since the levels of this protein were comparable for MCF-7 cells transfected with control and HDAC3 siRNAs (Fig. 8D).
FIG 8.

Effect of HDAC3, GPS2, and TBL1 depletion on p21 mRNA levels. MCF-7 cells were transfected with 20 pmol of control siRNA or siRNA specific for GPS2 (A), TBL1 (B), or HDAC3 (C). After 48 h, the cells were treated with vehicle (PBS; filled bars) or doxorubicin (0.5 μM; white bars), and p21 mRNA levels were quantitated by RT-qPCR and normalized to the signals obtained for 18S RNA. The changes in the p21 mRNA levels were represented relative to the level of control siRNA in doxorubicin-treated cells. Values represent the means ± SEMs of data from four (GPS2 and HDAC3) and three (TBL1) independent experiments. (D) Effect of HDAC3 depletion on p53 levels. MCF-7 cells were transfected with 20 pmol of control or HDAC-specific siRNA, followed by treatment with vehicle or doxorubicin for 24 h. Cells were lysed, and proteins were resolved in SDS-polyacrylamide gels and immunoblotted with p53 and actin (loading control) antibodies. (E) Chromatin immunoprecipitation assay showing doxorubicin-induced recruitment of p53 at the p21 gene promoter relative to that at enhancer sites. MCF-7 cells were treated with vehicle (PBS) or doxorubicin (1 μM) for 6 h, followed by chromatin isolation and immunoprecipitation using p53 antibody in parallel with the IgG negative control. Immunoprecipitated DNA was quantitated by qPCR using the indicated primers. Data represent averages ± SEMs of three independent experiments. The changes in the recruitment levels were represented relative to the amount of p53 recruited to the 5′ enhancer site following doxorubicin treatment. (F) Effect of NCoR depletion in MCF-7 cells on p21 mRNA levels assessed by RT-qPCR. Cells were transfected with control siRNA or NCoR siRNA, and p21 mRNA levels were quantitated as described for panels A to C. P values were determined by Student's t test. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.0001.
It has been reported previously that HDAC3 siRNA reduces the basal expression of p21 protein in colon cancer cells, as well as the activity of a p21 promoter-luciferase reporter gene (51), and that siRNA-mediated SMRT downregulation increases the activity of the p21 promoter (lacking the upstream p53 enhancer sites), which is largely dependent upon the Sp1 transcription factor (39). Therefore, in order to determine whether the effect of increased p21 mRNA expression in doxorubicin-treated, HDAC3 siRNA-transfected MCF-7 cells could be due to the loss of a p53-SMRT-HDAC3-repressive effect on the promoter, the association of p53 with the p21 promoter was analyzed by ChIP. In comparison to the established p53 enhancer binding sites, the binding of p53 to the p21 promoter was negligible in control and doxorubicin-treated cells (Fig. 8E), indicating that p53-SMRT stimulation is largely mediated by the upstream enhancer region. Interestingly, doxorubicin induction of p21 mRNA levels was significantly higher in NCoR-depleted than control cells (Fig. 8F). Knockdowns of GPS2, TBL1, HDAC3, and NCoR for these experiments were verified by RT-qPCR and/or Western blot analyses (data not shown). Taken together with the finding that HDAC3 requires activation by either SMRT or NCoR (25), these data suggest that the repressive effect of HDAC3 in doxorubicin-treated MCF-7 cells is mediated primarily via an NCoR-HDAC3 complex, while a p53-SMRT-GPS2-TBL1 complex stimulates p21 mRNA expression.
p53 binds to the SMRT deacetylase activation domain.
Given that the SMRT-interacting proteins GPS2 and TBL1, but not HDAC3, can stimulate p53-dependent gene expression and that GPS2 and TBL1 also bind directly to the N terminus of SMRT, the necessity of the amino acids within SMRT that bind to these proteins, aa 167 to 207 and aa 227 to 297, respectively (22), for binding to p53 was evaluated by GST pulldown assays. A panel of SMRT-NT deletion mutants was constructed beginning with SMRT-NTΔ36-254, which contains the same deletion of aa 36 to 254 found in the N terminus of the SMRTβ splice variant (12), and extending to a SMRT-NTΔ36-480 deletion mutant, which lacks SMRT's RD1 as well as the HDAC3-binding DAD (Fig. 9A, right). The three mutants (SMRT-NTΔ36-254, SMRT-NTΔ36-312, and SMRT-NTΔ36-388), which retain the DAD, could effectively bind to p53 protein, indicating that neither the TBL1 nor the GPS2 binding site is required for SMRT interaction with this transcription factor (Fig. 9A, left). In contrast, a complete loss of p53 binding was observed for the SMRT-NTΔ36-480 mutant, which lacks the RD1 and DAD regions, indicating that SMRT amino acids 389 to 481 are required for SMRT-NT binding to p53. The ability of the DAD-containing region to bind to p53 was confirmed in a GST pulldown experiment in which a small fragment of SMRT consisting of amino acids 255 to 480 and lacking both RD1 and RD2 was able to effectively interact with the p53 protein.
FIG 9.

The SMRT DAD binds to p53. (A) Bacterially expressed GST-tagged p53 protein was incubated with the in vitro-translated, 35S-labeled N terminus of SMRT (SMRT-NTΔ36-254), its deletion mutants, or the SMRT DAD fragment (residues 255 to 480) alone. Shown at the right are schematic representations of the SMRT deletion mutants and the DAD fragment. The protein complexes were immunoprecipitated by glutathione-agarose beads and resolved by SDS-PAGE, and the bound SMRT mutants or the DAD fragment was detected by autoradiography. Input represents 20% of the corresponding amount of the in vitro-translated, 35S-labeled lysates. (B) Competition between p53 and HDAC3 for binding to the SMRT DAD. Gal4-SMRT (DAD) was expressed in HCT116 cells and immobilized onto anti-Gal4 agarose beads to serve as bait. To this, in vitro-translated 35S-labeled p53 was added with or without in vitro-translated, 35S-labeled HDAC3. The protein complexes bound to agarose beads were resolved by SDS-PAGE, and the bound HDAC3 and p53 proteins were detected by autoradiography. Input represents 1 or 0.1% of the amount of in vitro-translated 35S-labeled lysates for HDAC3 and p53, respectively. (C) HCT116 cells were transfected with 1 μg expression vectors for control (pCR3.1) or Flag epitope-tagged SMRTτ or SMRT DADm and treated for 16 h with vehicle (−) or 0.5 μM doxorubicin (+). Cell lysates were subjected to immunoprecipitation using antibodies for Flag (lanes F) or an IgG negative control (lanes I). Immunoprecipitated protein complexes were resolved by SDS-PAGE and analyzed by Western blotting (WB) using p53 antibody (top). Relative input levels of p53 (middle) and actin (bottom) in the cell lysates were assessed by Western blot analyses. (D) HCT116 cells were transfected with 250 ng expression vectors for SMRTτ, SMRTΔ36-254, and SMRT DADm or an empty-vector control along with 1 μg of vector containing the p21-Luc reporter gene. Cells were harvested at 48 h after transfection, and luciferase activity was measured. The activity for the SMRT mutants is shown relative to that of SMRTτ. Values represent the averages ± SEMs (n = 2).
As p53 effectively binds to SMRT through a region encompassing the DAD, which is also essential for the recruitment and activation of HDAC3 enzymatic activity (19), it was hypothesized that p53 and HDAC3 could compete for interaction with the SMRT DAD, ultimately yielding either a functional SMRT-HDAC3 corepressor complex or a form of SMRT able to coactivate p53 transcriptional activity. In order to test this possibility, a competition assay was performed to evaluate the binding of p53 and HDAC3 proteins to the DAD-containing fragment of SMRT from aa 255 to 480. This portion of SMRT was transiently overexpressed in HCT116 cells as a Gal4-DBD fusion protein. Cell lysates from these cells were incubated with anti-Gal4 agarose beads to generate SMRT(255-480)-bound agarose beads that served as the bait. HDAC3 and p53 were expressed as 35S-labeled proteins in an in vitro translation system and used to compete for binding to the SMRT-DAD. Addition of HDAC3 to the binding reaction mixture attenuated but did not completely block p53 binding to the SMRT-DAD fragment, indicating competition between p53 and HDAC3 for binding to the region of SMRT from aa 255 to 480 (Fig. 9B) and suggesting that HDAC3 is highly unlikely to be required for optimal binding of p53 to SMRT. This was furthered tested by assessing the ability of wild-type and DAD mutant SMRT to coimmunoprecipitate p53 from HCT116 (p53-positive) cells transfected with expression vectors for either wild-type SMRT or a DAD mutant (DADm) of SMRT (Y470A) which cannot bind to HDAC3 or activate HDAC3 activity (19). As shown in Fig. 9C, mutation of the DAD does not inhibit SMRT interaction with p53 in coimmunoprecipitation assays. Finally, to test whether SMRT binding and activation of HDAC3 were required for the ability of SMRT to coactivate p53 transcriptional activity, transactivation experiments with wild-type SMRT, SMRTΔ36-254 (which corresponds to SMRTβ), and the SMRT DADm were conducted with HCT116 cells. Neither the loss of binding sites for GPS2 and TBL1 (as shown by the SMRTβ construct) nor the inability to interact with HDAC3 via the DAD significantly impacted the ability of SMRT to coactivate p53-dependent gene expression (Fig. 9D). Taken together, these data demonstrate that SMRT promotion of p53 transcriptional activity is HDAC3 independent.
DISCUSSION
Maintenance of the genome is pivotal for protecting organisms against the harmful consequences of DNA damage, which, if not properly dealt with, can lead to genomic instability, cancer, and cell death (52, 53). The tumor suppressor protein p53 plays a critical role in preserving genomic integrity by mediating cellular responses to genotoxic insults through transcriptional regulation of genes involved in cell cycle arrest, DNA repair, apoptosis, cellular senescence, and ageing (1, 2). Consistent with this, the p53 gene is mutated in ∼50% of all cancers, making it the single most frequently inactivated gene in human cancers (54, 55). Like other transcription factors, the function of p53 is affected by transcriptional coregulators, and given the importance of this regulator to genomic integrity, it is important to fully understand the molecular mechanisms by which p53 activity is modulated. Our study reveals that the nuclear receptor coregulator SMRT, which is best known for its ability to inhibit the transcriptional activity of many members of the nuclear receptor superfamily of transcription factors through recruitment of HDAC3, directly binds to p53 and serves as a coactivator of its transcriptional activity. Cells depleted of SMRT expression exhibit abrogated p53 transcriptional activity, decreased doxorubicin induction of endogenous p53 target gene expression without alteration of p53 protein levels, and a compromised DNA damage response. Moreover, this is an HDAC3-independent effect of SMRT. Collectively, these data indicate a biological role for SMRT in mediating DNA damage responses through promotion of p53 transcriptional activity and suggest that SMRT, via p53, protects the genome from genotoxic insults that may contribute to carcinogenesis.
As a regulator of genomic integrity, a number of pathways exist to promote p53-dependent gene expression (3, 56). Some of these are involved in modulating p53 posttranslational modifications (e.g., acetylation) that ultimately stabilize the protein, while other cellular factors (e.g., coactivators) interact with p53 at the regulatory regions of target genes and potentiate their expression. The identification of SMRT as a positive regulator of p53 transcriptional activity expands the cohort of transcription factors that can be coactivated by this coregulator. Although SMRT is widely recognized as a corepressor of type II nuclear receptors, such as for thyroid hormone and retinoic acid (11), previous work from our laboratory has established SMRT to be a cell type-specific coactivator of ERα (15), an estrogen-regulated member of the nuclear receptor superfamily. Not all ERα target genes are positively regulated by SMRT, but for those coactivated by this regulator (e.g., cyclin D1, progesterone receptor), E2 treatment induces the recruitment of SMRT to gene regulatory regions and stimulates ERα transcriptional activity. At present, our data suggest that SMRT regulation of p53 is limited to coactivation, and thus, while it may be considered a dual coactivator/corepressor of ERα (15), the available information indicates only a coactivator role relative to p53 and, thus, adds SMRT to an expanding group of p53 coactivators including CBP/p300, pCAF, JMY, ADA3, and MAML1 (4, 5, 57–59). Interestingly, several proteins previously shown to stimulate p53 transcriptional activity, namely, the TATA box binding protein (TBP)-associated factors (TAFs), TAFII-32/70, and AMF1 (also known as GPS2), can also bind to SMRT (50, 60, 61). TAFII-32/70, which binds to the N-terminal transactivation domain of p53, promotes p53-mediated transcription (62–64), while GPS2 associates with p53 in vivo and in vitro and augments p53-dependent expression of genes such as the gene for p21, CDKN1A (50). By virtue of being able to bind to both p53 and SMRT, any of these proteins could potentially play a role in promoting SMRT-p53 interactions and stimulating p53 target gene expression.
In addition to activating gene expression, p53 can repress transcription directed from various viral and cellular promoters (2, 43), with proteins such as mSin3a tethering p53 to a repressor complex containing HDAC1 (7). Other HDACs (e.g., HDACs 2, 3, and 4) can also modulate p53 function through its deacetylation and downregulation of p53-dependent gene activation (9). In contrast, our results demonstrate that silencing of HDAC3 expression increased the levels of doxorubicin-induced p21 mRNA, without altering p53 expression, indicating that a loss of gene repression rather than an increase in p53 protein was mediating the increase in p21 mRNA levels. The deacetylase activity of HDAC3 is dependent upon the activation achieved via association of this enzyme with either SMRT or NCoR (19, 25), and based upon the depletion of NCoR but not SMRT to stimulate doxorubicin-induced expression of p21 mRNA, we propose a model in which NCoR and HDAC3 limit p53 transcriptional activity on target genes, such as CDKN1A. Thus, even though NCoR and SMRT are closely related paralogs that can repress the activity of many different transcription factors (11, 20), these studies add p53 to the growing list of instances in which SMRT and NCoR perform distinct physiological functions (13–16, 65–68).
The core SMRT corepressor complex encompasses a tetramer of the exchange factor TBL1 and/or TBLR1 and two molecules each of GPS2 and SMRT (19, 21, 22, 69, 70). Structural characterization experiments demonstrate that the N termini of GPS2 (aa 1 to 53) and TBL1 (aa 1 to 71) bind to the RD1 domain of SMRT in the region from aa 167 to 297 (22). Conversely, a distinct region of GPS2 (aa 103 to 250) mediates binding of this protein to the central DNA binding region of p53 (aa 161 to 333) (50), while the DAD of SMRT binds to the C terminus of p53. Collectively, the ability of SMRT (reported herein) and GPS2 (50) to bind to and coactivate p53 transcriptional activity, as well as the established ability of SMRT and GPS2 to bind to one another (22), all through nonoverlapping sites, suggests that SMRT and GPS2 can work together to stimulate p53 transcriptional activity. Indeed, our data demonstrating that silencing of SMRT, GPS2, or TBL1 leads to a significant decrease in doxorubicin-induced p53 transcriptional activity, as measured by the p21-Luc assay, are consistent with the hypothesis that these proteins function as a SMRT-GPS2-TBL1 complex to coactivate p53 transcriptional activity. However, SMRT is subject to extensive alternative splicing, leading to the generation of multiple forms of this coregulator (71). One such splice variant, SMRTβ, lacks aa 34 to 254 due to exclusion of exons 2 to 6 and therefore the GPS2 binding site and a portion of the TBL1 interaction domain (12, 72). As SMRTβ retains the ability to coactivate p53 in p21-Luc transactivation assays, it appears that GPS2 is not required for SMRT stimulation of p53 function. However, this does not preclude the possibility that by promotion of SMRT-GPS2-p53 interactions, as well as through its apparent interactions with the p300 coregulator protein (50), GPS2 enhances SMRT coactivation of p53-dependent gene expression.
The essential role of the SMRT DAD region for recruitment and activation of HDAC3 activity was demonstrated in previous studies showing that mutations in the DAD or deletion of this domain affected HDAC3 interaction with SMRT and thus eliminated corepressor stimulation of HDAC3 activity (19). In addition, mice with point mutations in the DADs of SMRT and NCoR had no detectable HDAC3 activity, despite having normal levels of this deacetylase (25). Through a combination of coimmunoprecipitation and GST pulldown assays, the region of SMRT from aa 389 to 480 encompassing the DAD was found to be essential for p53 interaction with the N terminus of SMRT, although comparisons of the interactions between p53 and SMRT-NTΔ5-312 and SMRT-NTΔ5-254 suggested that the region of SMRT from aa 250 to 312, which is N terminal to the DAD and binds to TBL1, may be required to mediate the maximal interaction of p53 with SMRT. The former, necessary region (aa 389 to 480) aligns very closely to the region of SMRT from aa 395 to 489 shown previously to mediate the corepressor's interaction with HDAC3 (19), suggesting that the same region of SMRT is employed for binding to HDAC3 and p53, with SMRT serving as a corepressor with the former and as a coactivator for the latter. This suggests that HDAC3 and p53 can physically compete for binding to SMRT, or alternatively, there may be distinct pools of SMRT, one in which the corepressor is bound to HDAC3 and can repress gene expression and another in which SMRT is available to bind to p53 and ultimately contributes to activation of gene expression. Most importantly, our collective data indicate that SMRT coactivation of p53 is independent of HDAC3 activation by the DAD, and we therefore propose a model (Fig. 10) in which DNA damage increases p53 expression, whereupon the C terminus of this transcription factor binds to the DAD of SMRT, and in conjunction with GPS2, TBL1, and CBP/p300, this HDAC3-independent SMRT complex can stimulate p53 transcriptional activity.
FIG 10.
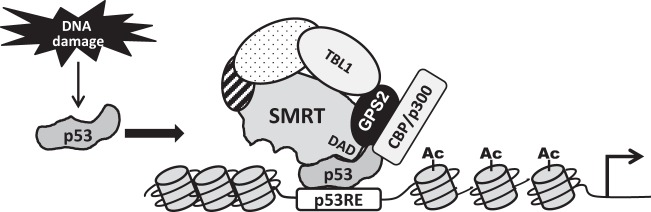
Proposed model for SMRT coactivation of p53 transcriptional activity. Model for SMRT coactivation of p53 transcriptional activity. In response to DNA damage, p53 expression is stabilized and p53 binds to the regulatory regions of its target genes. The C terminus of p53 binds to the DAD region of SMRT, effectively limiting the ability of HDAC3 to bind to p53-bound SMRT and repress p53 target genes. Instead, a SMRT complex containing GPS2, TBL1, and, potentially, CBP/p300 is able to achieve an open chromatin structure with acetylated (Ac) histones that promotes expression of p53 target genes.
The ability of p53 and HDAC3 to bind to the same domain of SMRT highlights the importance of understanding cellular factors that can influence SMRT interactions with these two transcriptional regulators. Recent crystallographic determinations of the SMRT DAD-HDAC3 structure reveal that the interaction is dependent upon IP4 binding to the SMRT DAD (24). Thus, cellular IP4 availability may be one factor regulating SMRT corepressor versus coactivator potential. One of the enzymes that controls IP4 synthesis is inositol polyphosphate multikinase (73), which binds to p53 (74), thereby placing it in an appropriate location for influencing SMRT-HDAC3 interactions. Reduced expression levels of phosphatidylinositol phosphatases, such as PTEN or INPP4B, in cancers (75–77) could also be expected to influence the relative affinity of SMRT for HDAC3 and, consequently, the availability of SMRT for coactivation of p53. At this time, it is not clear what, if any, signaling pathways directly regulate SMRT-p53 interactions. However, the C-terminal domain of p53, required for binding to SMRT, is subject to extensive posttranslational modifications which have the potential to impact protein-protein interactions (78).
In its coactivation role, SMRT must be able to interact with proteins that can promote gene expression, quite possibly acting as a type of scaffold protein. A significant portion of the SMRT structure is highly disordered, suggesting that it serves as a hub protein for transient yet highly specific binding to other transcriptional regulatory proteins that promote p53-dependent gene expression (20, 79, 80). Conceivably, this could occur through SMRT-mediated recruitment and stabilization of complexes of positive regulators of transcription, and/or it may be due to SMRT facilitating a positively acting p53 posttranslational modification by another protein with enzymatic activity. For instance, SMRT has been shown to bind to members of the p160 family of steroid receptor coactivators and stimulate their intrinsic transcriptional activity (30). Members of this family, SRC-1 and, potentially, SRC-3, can coactivate p53 transcriptional activity (81), and SMRT and SRC coactivators are therefore likely candidates for jointly promoting p53-dependent gene expression. Moreover, the SRC coactivators can bind to CBP/p300 (82), which are known positive regulators of p53 (4, 5, 8), and this implicates the possibility of a SMRT-SRC-CBP/p300 complex as a potential stimulator of p53-dependent gene expression. It should be noted that SMRT binds to the C-terminal region of p53, which, as noted above, is heavily modified by posttranslational marks that can lead to p53 stabilization or stimulation of its transcriptional activity (78). As a scaffold, SMRT may recruit or stabilize the binding of other proteins, such as acetyltransferases (e.g., SRC-3), that can modify p53 and ultimately increase the expression of its target genes.
Our microarray results revealed that SMRT depletion decreases the expression of a group of genes involved in the DNA damage response and provide insight into the importance of SMRT's coactivator role in mediating DNA damage responses. Indeed, decreased expression of DNA damage response genes was initially noted in cells naive to genotoxic stress, indicating that SMRT regulates the basal transcription of these genes and may provide to cells protection against the intrinsic DNA damage (e.g., replication errors) that arises during cell division, thus extending the transcriptional regulatory role of SMRT to protection of the genome.
With respect to the DNA damage resulting from genotoxic stresses, other investigators have demonstrated that a reduction of SMRT but not NCoR enhanced the sensitivity of HeLa cells to ionizing radiation or DNA damage-inducing drugs, presumably through loss of the ability of SMRT to promote the inherent repressive activity of the Ku70 subunit of the DNA-dependent protein kinase (DNA-PK) complex that is required for double-strand break repair via the nonhomologous end-joining (NHEJ) pathway (68). More recently, it was demonstrated that knockdown of SMRT affected the expression of a number of genes that are regulated upon cellular exposure to the DNA-damaging agent cisplatin by an unknown mechanism (67). In particular, SMRT but not NCoR inhibited apoptosis associated with DNA damage, at least in part through inhibition of the expression of the Wip1 phosphatase that negatively regulates proapoptotic proteins, such as Chk2 (67). Taken together, the ability of SMRT to contribute to cellular responses to DNA damage via transcriptional regulation of the expression of genes involved in apoptosis (i.e., WIP1) and the repair of DNA damage via homologous recombination (i.e., RAD51, BRCA1, and CHEK1) and SMRT's interaction with the Ku70 subunit of the DNA-PK required for NHEJ repair pathways place SMRT in a position to coordinately effect the necessary alterations in chromatin architecture and the activity of the DNA repair pathways so that cells can effectively mount responses to DNA damage incurred either through normal cell division processes or as a result of external stresses to the cell.
ACKNOWLEDGMENTS
We thank Lawrence Donehower, Mitchell Lazar, and Mengtao Li for providing plasmids and gratefully acknowledge the technical support of Cheryl Parker.
S.K. was supported by a postdoctoral fellowship award (PDF 0707868) from the Susan G. Komen for the Cure Foundation. This work was supported, in part, by the Genomic and RNA Profiling Core at the Baylor College of Medicine with funding from an NIH NCI grant (P30CA125123) and the expert assistance of Lisa D. White and by Public Health Service grant DK53002 to C.L.S. from the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Published ahead of print 21 January 2014
REFERENCES
- 1.Laptenko O, Prives C. 2006. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Diff. 13:951–961. 10.1038/sj.cdd.4401916 [DOI] [PubMed] [Google Scholar]
- 2.Rinn JL, Huarte M. 2011. To repress or not to repress: this is the guardian's question. Trends Cell Biol. 21:344–353. 10.1016/j.tcb.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 3.Dai C, Gu W. 2010. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 16:528–536. 10.1016/j.molmed.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu W, Shi X-L, Roeder RG. 1997. Synergistic activation of transcription by CBP and p53. Nature 387:819–823. 10.1038/42972 [DOI] [PubMed] [Google Scholar]
- 5.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. 1997. Binding and modulation of p53 by p300/CBP coactivators. Nature 387:823–827. 10.1038/42981 [DOI] [PubMed] [Google Scholar]
- 6.Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, Berger SL. 1999. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19:1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy M, Ahn J, Walker KK, Hoffman WH, Evans RM, Levine AJ, George DL. 1999. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 13:2490–2501. 10.1101/gad.13.19.2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL. 2001. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 8:1243–1254. 10.1016/S1097-2765(01)00414-2 [DOI] [PubMed] [Google Scholar]
- 9.Juan L-J, Shia W-J, Chen M-H, Yang W-M, Seto E, Lin Y-S, Wu C-W. 2000. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 275:20436–20443. 10.1074/jbc.M000202200 [DOI] [PubMed] [Google Scholar]
- 10.Perissi V, Jepsen K, Glass CK, Rosenfeld MG. 2010. Deconstructing repression: evolving models of co-repressor action. Nat. Rev. Genet. 11:109–123. 10.1038/nrg2736 [DOI] [PubMed] [Google Scholar]
- 11.Privalsky ML. 2004. The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annu. Rev. Physiol. 66:315–360. 10.1146/annurev.physiol.66.032802.155556 [DOI] [PubMed] [Google Scholar]
- 12.Ordentlich P, Downes M, Xie W, Genin A, Spinner NB, Evans RM. 1999. Unique forms of human and mouse nuclear receptor corepressor SMRT. Proc. Natl. Acad. Sci. U. S. A. 96:2639–2644. 10.1073/pnas.96.6.2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hermanson O, Jepsen K, Rosenfeld MG. 2002. N-CoR controls differentiation of neural stem cells into astrocytes. Nature 419:934–939. 10.1038/nature01156 [DOI] [PubMed] [Google Scholar]
- 14.Jepsen K, Hermanson O, Onami TM, Gleiberman AS, Lunyak V, McEvilly RJ, Kurokawa R, Kumar V, Liu F, Seto E, Hedrick SM, Mandel G, Glass CK, Rose DW, Rosenfeld MG. 2000. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 102:753–763. 10.1016/S0092-8674(00)00064-7 [DOI] [PubMed] [Google Scholar]
- 15.Peterson TJ, Karmakar S, Pace MC, Gao T, Smith CL. 2007. The silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) corepressor is required for full estrogen receptor-α transcriptional activity. Mol. Cell. Biol. 27:5933–5948. 10.1128/MCB.00237-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonas BA, Privalsky ML. 2004. SMRT and N-CoR corepressors are regulated by distinct kinase signaling pathways. J. Biol. Chem. 279:54676–54686. 10.1074/jbc.M410128200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jepsen K, Rosenfeld MG. 2002. Biological roles and mechanistic actions of co-repressor complexes. J. Cell Sci. 115:689–698 [DOI] [PubMed] [Google Scholar]
- 18.Goodson M, Jonas BA, Privalsky MA. 2005. Corepressors: custom tailoring and alterations while you wait. Nucl. Recept. Signal. 3:e003. 10.1621/nrs.03003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guenther MG, Barak O, Lazar MA. 2001. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 21:6091–6101. 10.1128/MCB.21.18.6091-6101.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watson PJ, Fairall L, Schwabe JWR. 2012. Nuclear hormone receptor co-repressors: structure and function. Mol. Cell. Endocrinol. 348:440–449. 10.1016/j.mce.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. 2000. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 19:4342–4350. 10.1093/emboj/19.16.4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oberoi J, Fairall L, Watson PJ, Yang J-C, Czimmerer Z, Kampmann T, Goult BT, Greenwood JA, Gooch JT, Kallenberger BC, Nagy L, Neuhaus D, Schwabe JWR. 2011. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 18:177–184. 10.1038/nsmb.1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Codina A, Love JD, Li Y, Lazar MA, Neuhaus D, Schwabe JWR. 2005. Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc. Natl. Acad. Sci. U. S. A. 102:6009–6014. 10.1073/pnas.0500299102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watson PJ, Fairall L, Santos GM, Schwabe JWR. 2012. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 481:335–340. 10.1038/nature10728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.You S-H, Lim H-W, Sun Z, Broache M, Won K-J, Lazar MA. 2013. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 20:182–187. 10.1038/nsmb.2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, Liu XS, Lazar MA. 2011. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331:1315–1319. 10.1126/science.1198125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green AR, Burney C, Granger CJ, Paish EC, El-Sheikh S, Rakha EA, Powe DG, Macmillin RD, Ellis IO, Stylianou E. 2008. The prognostic significance of steroid receptor co-regulators in breast cancer: co-repressor NCOR2/SMRT is an independent indicator of poor outcome. Breast Cancer Res. Treat. 110:427–437. 10.1007/s10549-007-9737-y [DOI] [PubMed] [Google Scholar]
- 28.Smith CL, Migliaccio I, Chaubal V, Wu MF, Pace MC, Hartmaier R, Jiang S, Edwards DP, Gutiérrez MC, Hilsenbeck SG, Oesterreich S. 2012. Elevated nuclear expression of the SMRT corepressor in breast cancer is associated with earlier tumor recurrence. Breast Cancer Res. Treat. 136:253–265. 10.1007/s10549-012-2262-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karmakar S, Foster EA, Smith CL. 2009. Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-α transcriptional activity. Endocrinology 150:1588–1596. 10.1210/en.2008-1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karmakar S, Gao T, Pace MC, Oesterreich S, Smith CL. 2010. Cooperative activation of cyclin D1 and progesterone receptor gene expression by the SRC-3 coactivator and SMRT corepressor. Mol. Endocrinol. 24:1187–1202. 10.1210/me.2009-0480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Wu RC, O'Malley BW. 2009. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 9:615–630. 10.1038/nrc2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hermeking H, Lengauer C, Polyak K, He T-C, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 1997. 14-3-3σ is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1:3–11. 10.1016/S1097-2765(00)80002-7 [DOI] [PubMed] [Google Scholar]
- 33.Chang P-C, Li M. 2008. Kaposi's sarcoma-associated herpesvirus K-cyclin interacts with Cdk9 and stimulates Cdk9-mediated phosphorylation of p53 tumor suppressor. J. Virol. 82:278–290. 10.1128/JVI.01552-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon H-G, Chan DW, Reynolds AB, Qin J, Wong J. 2003. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein kaiso. Mol. Cell 12:723–734. 10.1016/j.molcel.2003.08.008 [DOI] [PubMed] [Google Scholar]
- 35.Yoon H-G, Choi Y, Cole PA, Wong J. 2005. Reading and function of a histone code involved in targeting corepressor complexes for repression. Mol. Cell. Biol. 25:324–335. 10.1128/MCB.25.1.324-335.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264. 10.1093/biostatistics/4.2.249 [DOI] [PubMed] [Google Scholar]
- 37.Tusher VG, Tibshirani R, Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 98:5116–5121. 10.1073/pnas.091062498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackson JG, Pereira-Smith OM. 2006. p53 is preferentially recruited to the promoters of growth arrest genes p21 and GADD45 during replicative senescence of normal human fibroblasts. Cancer Res. 66:8356–8360. 10.1158/0008-5472.CAN-06-1752 [DOI] [PubMed] [Google Scholar]
- 39.Wilson AJ, Byun D-S, Nasser S, Murray LB, Ayyanar K, Arango D, Figueroa M, Melnick A, Kao GD, Augenlicht LH, Mariadason JM. 2008. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol. Biol. Cell 19:4062–4075. 10.1091/mbc.E08-02-0139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu W, Konduri SD, Bansal S, Nayak BK, Rajasekaran SA, Karuppayil SM, Rajasekaran AK, Das GM. 2006. Estrogen receptor-α binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 281:9837–9840. 10.1074/jbc.C600001200 [DOI] [PubMed] [Google Scholar]
- 41.Kho PS, Wang Z, Zhuang L, Li Y, Chew J-L, Ng H-H, Liu ET, Yu Q. 2004. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J. Biol. Chem. 279:21183–21192. 10.1074/jbc.M311912200 [DOI] [PubMed] [Google Scholar]
- 42.Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, Kovioja T, Ignatiev I, Kel A, Taipale J, Selivanova G. 2012. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Diff. 19:1992–2002. 10.1038/cdd.2012.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang B, Xiao Z, Ko HL, Ren EC. 2010. The p53 response element and transcriptional repression. Cell Cycle 9:870–879. 10.4161/cc.9.5.10825 [DOI] [PubMed] [Google Scholar]
- 44.Mah LJ, El-Osta A, Karagiannis TC. 2010. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24:679–686. 10.1038/leu.2010.6 [DOI] [PubMed] [Google Scholar]
- 45.Kurz EU, Douglas P, Lees-Miller SP. 2004. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J. Biol. Chem. 279:53272–53281. 10.1074/jbc.M406879200 [DOI] [PubMed] [Google Scholar]
- 46.Soldani C, Scovassi AI. 2002. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis 7:321–328. 10.1023/A:1016119328968 [DOI] [PubMed] [Google Scholar]
- 47.Kolb RH, Greer PM, Cao PT, Cowan KH, Yan Y. 2012. ERK1/2 signaling plays an important role in topoisomerase II poison-induced G2/M checkpoint activation. PLoS One 7:e50281. 10.1371/journal.pone.0050281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu W, Ip MM, Podgorsak MB, Das GM. 2009. Disruption of estrogen receptor α-p53 interaction in breast tumors: a novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res. Treat. 115:43–50. 10.1007/s10549-008-0044-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Resnick-Silverman L, St Clair S, Maurer M, Zhao K, Manfredi J. 1998. Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev. 12:2102–2107. 10.1101/gad.12.14.2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng YC, Kuo F, Breiding DE, Wang YF, Mansur CP, Androphy EJ. 2001. AMF1 (GPS2) modulates p53 transactivation. Mol. Cell. Biol. 21:5913–5924. 10.1128/MCB.21.17.5913-5924.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. 2006. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 281:13548–13558. 10.1074/jbc.M510023200 [DOI] [PubMed] [Google Scholar]
- 52.Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461:1071–1078. 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Negrini S, Gorgoulis VG, Halazonetis TD. 2010. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11:220–228. 10.1038/nrm2858 [DOI] [PubMed] [Google Scholar]
- 54.Beckerman R, Prives C. 2010. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2:a000935. 10.1101/cshperspect.a000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. 2007. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 26:2157–2165. 10.1038/sj.onc.1210302 [DOI] [PubMed] [Google Scholar]
- 56.Riley T, Sontag E, Chen P, Levine A. 2008. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9:402–412. 10.1038/nrm2395 [DOI] [PubMed] [Google Scholar]
- 57.Wang T, Kobayashi T, Takimoto R, Denes AE, Snyder EL, el-Deiry WS, Brachmann RK. 2001. hADA3 is required for p53 activity. EMBO J. 20:6404–6413. 10.1093/emboj/20.22.6404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao Y, Katzman RB, Delmolino LM, Bhat I, Zhang Y, Gurumurthy CB, Germaniuk-Kurowska A, Reddi HV, Solomon A, Zeng MS, Kung A, Ma H, Gao Q, Dimri G, Stanculescu A, Miele L, Wu L, Griffin JD, Wazer DE, Band H, Band V. 2007. The notch regulator MAML1 interacts with p53 and functions as a coactivator. J. Biol. Chem. 282:11969–11981. 10.1074/jbc.M608974200 [DOI] [PubMed] [Google Scholar]
- 59.Zuchero JB, Coutts AS, Quinlan ME, Thanque NB, Mullins RD. 2009. p53-cofactor JMY is a multifunctional actin nucleation factor. Nat. Cell Biol. 11:451–459. 10.1038/ncb1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farmer G, Colgan J, Nakatani Y, Manley JL, Prives C. 1996. Functional interaction between p53, the TATA-binding protein (TBP), and TBP-associated factors in vivo. Mol. Cell. Biol. 16:4295–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muscat GE, Burke LJ, Downes M. 1998. The corepressor N-CoR and its variants RIP13a and RIP13Δ1 directly interact with the basal transcription factors TFIIB, TAFII32 and TAFII70. Nucleic Acids Res. 26:2899–2907. 10.1093/nar/26.12.2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buschmann T, Lin Y, Aithmitti N, Fuchs SY, Lu H, Resnick-Silverman L, Manfredi JJ, Ronai Z, Wu X. 2001. Stabilization and activation of p53 by the coactivator protein TAFII31. J. Biol. Chem. 276:13852–13857. 10.1074/jbc.M007955200 [DOI] [PubMed] [Google Scholar]
- 63.Thut CJ, Chen JL, Klemm R, Tjian R. 1995. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science 267:100–104. 10.1126/science.7809597 [DOI] [PubMed] [Google Scholar]
- 64.Wang W, Nahta R, Huper G, Marks JR. 2004. TAFII70 isoform-specific growth suppression correlates with its ability to complex with the GADD45a protein. Mol. Cancer Res. 2:442–452 [PubMed] [Google Scholar]
- 65.Hoberg JE, Yeung F, Mayo MW. 2004. SMRT derepression by the IκB kinase α: a prerequisite to NF-κB transcription and survival. Mol. Cell 16:245–255. 10.1016/j.molcel.2004.10.010 [DOI] [PubMed] [Google Scholar]
- 66.Jepsen K, Solum D, Zhou T, McEvilly RJ, Kim H-J, Glass CK, Hermanson O, Rosenfeld MG. 2007. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 450:415–419. 10.1038/nature06270 [DOI] [PubMed] [Google Scholar]
- 67.Scafoglio C, Smolka M, Zhou H, Perissi V, Rosenfeld MG. 2013. The co-repressor SMRT delays DNA damage-induced caspase activation by repressing pro-apoptotic genes and modulating the dynamics of checkpoint kinase 2 activation. PLoS One 8:e59986. 10.1371/journal.pone.0059986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu J, Palmer C, Alenghat T, Li Y, Kao G, Lazar MA. 2006. The corepressor silencing mediator for retinoid and thyroid hormone receptor facilitates cellular recovery from DNA double-strand breaks. Cancer Res. 66:9316–9322. 10.1158/0008-5472.CAN-06-1902 [DOI] [PubMed] [Google Scholar]
- 69.Guenther MG, Lane WS, Fischle W, Verdin E, Lazar MA, Shiekhattar R. 2000. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 14:1048–1057. 10.1101/gad.14.9.1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, Kalkum M, Chait BT, Roeder RG. 2002. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 9:611–623. 10.1016/S1097-2765(02)00468-9 [DOI] [PubMed] [Google Scholar]
- 71.Goodson ML, Jonas BA, Privalsky ML. 2005. Alternative mRNA splicing of SMRT creates functional diversity by generating corepressor isoforms with different affinities for different nuclear receptors. J. Biol. Chem. 280:7493–7503. 10.1074/jbc.M411514200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cote S, McNamara S, Brambilla D, Bianchini A, Rizzo G, Victoria del Rincon S, Grignani F, Nervi C, Miller WH. 2004. Expression of SMRTβ promotes ligand-induced activation of mutated and wild-type retinoid receptors. Blood 104:4226–4335. 10.1182/blood-2003-10-3583 [DOI] [PubMed] [Google Scholar]
- 73.Chakraborty A, Kim S, Snyder SH. 2011. Inositol pyrophosphates as mammalian cell signals. Sci. Signal. 4:re1. 10.1126/scisignal.2001958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu R, Sen N, Paul BD, Snowman AM, Rao F, Vandiver MS, Xu J, Snyder SH. 2013. Inositol polyphosphate multikinase is a coactivator of p53-mediated transcription and cell death. Sci. Signal. 6:ra22. 10.1126/scisignal.2003405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM. 2011. INPP4B: the new kid on the PI3K block. Oncotarget 2:321–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caffrey JJ, Darden T, Wenk MR, Shears SB. 2001. Expanding coincident signaling by PTEN through its inositol 1,3,4,5,6-pentakisphosphate 3-phosphatase activity. FEBS Lett. 499:6–10. 10.1016/S0014-5793(01)02500-5 [DOI] [PubMed] [Google Scholar]
- 77.Di Cristofano A, Pandolfi PP. 2000. The multiple roles of PTEN in tumor suppression. Cell 100:387–390. 10.1016/S0092-8674(00)80674-1 [DOI] [PubMed] [Google Scholar]
- 78.Bode AM, Dong Z. 2004. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4:793–805. 10.1038/nrc1455 [DOI] [PubMed] [Google Scholar]
- 79.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. 2006. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2:e100. 10.1371/journal.pcbi.0020100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singh GP, Ganapathi M, Dash D. 2007. Role of intrinsic disorder in transient interactions of hub proteins. Proteins 66:761–765. 10.1002/prot.21281 [DOI] [PubMed] [Google Scholar]
- 81.Lee S-K, Kim H-J, Kim JW, Lee JW. 1999. Steroid receptor coactivator-1 and its family members differentially regulate transactivation by the tumor suppressor protein p53. Mol. Endocrinol. 13:1924–1933. 10.1210/me.13.11.1924 [DOI] [PubMed] [Google Scholar]
- 82.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S-C, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. 1996. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85:403–414. 10.1016/S0092-8674(00)81118-6 [DOI] [PubMed] [Google Scholar]