

Abstract
Interaction of Notch receptors with Delta- and Serrate-type ligands is an evolutionarily conserved mechanism that mediates direct communication between adjacent cells and thereby regulates multiple developmental processes. Posttranslational modifications of both receptors and ligands are pivotal for normal Notch pathway function. We have identified by mass spectrometric analysis two serine and one threonine phosphorylation sites in the intracellular domain of the mouse Notch ligand DLL1. Phosphorylation requires cell membrane association of DLL1 and occurs sequentially at the two serine residues. Phosphorylation of one serine residue most likely by protein kinase B primes phosphorylation of the other serine. A DLL1 variant, in which all three identified phosphorylated serine/threonine residues are mutated to alanine and valine, was more stable than wild-type DLL1 but had reduced relative levels on the cell surface and was more effectively cleaved in the extracellular domain. In addition, the mutant variant activated Notch1 significantly less efficient than wild-type DLL1 in a coculture assay in vitro. Mice, however, whose endogenous DLL1 was replaced with the phosphorylation-deficient triple mutant developed normally, suggesting compensatory mechanisms under physiological conditions in vivo.
INTRODUCTION
The evolutionarily conserved Notch signaling pathway mediates local interactions between adjacent cells, which are of central importance for the regulation of developmental processes in a wide variety of different tissues and species, and mutations in its components cause human diseases (reviewed in references 1 to 7). The Notch gene of Drosophila as well as its vertebrate homologues encode large receptors that, at the surface of a cell, interact with products of the Delta and Serrate genes acting as ligands. Notch, Delta, and Serrate (called Jagged in vertebrates) encode transmembrane proteins with specific numbers of epidermal growth factor (EGF)-like repeats in their extracellular domains (8–10). The Notch protein is proteolytically processed in the Golgi network and present as a noncovalently linked heterodimeric receptor at the cell surface (11, 12). Ligand binding induces two subsequent proteolytic cleavages by ADAM proteases and γ-secretase, releasing the intracellular domain of Notch (NICD). NICD translocates to the nucleus and, by complexing with the transcriptional regulator suppressor of hairless [su(h)], activates transcription of bHLH genes of the enhancer of split [e(spl)] family (13–19). Their gene products in turn regulate the transcription of other downstream effector genes. Similar to the Notch receptors, ligands can be cleaved by ADAM proteases releasing the ectodomain (“ectodomain shedding”), followed by γ-secretase-mediated generation of the intracellular domains (20–24), the significance of which for Notch signaling is unclear.
Posttranslational modifications such as glycosylation, ubiquitination or phosphorylation of receptor and ligands are critical for normal Notch pathway function. For example, modification of NOTCH by O-fucosylation of specific S or T residues in certain EGF motifs (25, 26), followed by further modification of O-fucose residues by Fringe (FNG) proteins (26, 27) modulates the NOTCH response to ligands in a context-dependent manner (28–31). Ubiquitination of Notch receptors by E3 ligases antagonistically modulates the amount of receptor that is available for ligand binding at the cell surface by regulating trafficking to distinct internalization pathways, but the physiologically important ubiquitination sites and the consequences of their alternative usage are not well understood on the molecular level (reviewed in references 32 to 36). Phosphorylation at S/T residues has been observed in the intracellular domains (NICDs) of Drosophila and vertebrate Notch receptors (37–41). NICD phosphorylation has been associated with nuclear translocation (17, 42) and with both positive and negative modulation of Notch activity: phosphorylation of Notch1 NICD by glycogen synthase kinase 3β (GSK3β) inhibited proteasomal degradation (43) and led to enhanced Notch activity, a finding consistent with the role of shaggy, the Drosophila homologue of GSK3β, as a positive modulator of Notch signaling (44). In contrast, GSK3β-dependent phosphorylation of NOTCH2 appears to negatively regulate NOTCH2 activity (41). Phosphorylation of NOTCH1-ICD by Nemo-like kinase (NLK) suppresses NOTCH1 activity by interfering with the formation of an active transcriptional complex, whereas NLK phosphorylation of NOTCH3 enhanced NOTCH3-ICD activity (40).
Also, Notch ligands are posttranslationally modified, and their activity is subject to complex regulation. Like Notch, ligands are modified by O-fucosylation (45). However, Drosophila Delta is functional without O-fucosylation (46), and the significance of such a modification for ligand function in vertebrates is unknown. Similar to Notch, modification of the ligands by ubiquitination regulates their activity. Ubiquitination is essential for endocytosis of ligands, which has been shown to be critical for their ability to activate Notch. Endocytosis has been suggested to convert by an as-yet-ill-defined process initially inactive to active ligands that are recycled back to the cell surface or to direct ligands to specialized membrane microdomains. Alternatively, endocytosis of ligand bound to the extracellular domain of the Notch receptor was suggested to generate a pulling force that exposes the S2 cleavage site to ADAM protease. These models are not mutually exclusive, and both endocytic events might be required for productive Notch signaling (reviewed in references 32 to 36, 47, and 48). In addition to endocytosis and recycling, ligand activity is regulated by proteolysis. Similar to Notch, ligands are subject to consecutive proteolytic cleavages by ADAM proteases and γ-secretase, leading to ectodomain shedding and the subsequent release of the intracellular domains (ICDs) into the cytoplasm. Both contribution to downregulation of Notch signaling or to relief of cis-inhibition have been suggested as functions for ectodomain shedding. However, its physiological role has to be demonstrated. The release of the ICDs of Notch ligands, their detection in the nucleus, and the effects of overexpressed ICDs in cultured cells led to the idea that these ICDs have signaling functions in ligand-expressing cells (48; reviewed in reference 49). However, overexpression of different forms of mouse DLL1ICD in mice did not affect embryonic development (50), arguing against such a role. Collectively, these findings indicate that the activity of Notch ligands is regulated on multiple levels.
Here, we report on the phosphorylation of the mouse Notch ligand DLL1 in cultured cells and in vivo. We found that DLL1 is phosphorylated at three S/T residues in the intracellular domain and that the phosphorylation of two sites occurs sequentially, and phosphorylation most likely by protein kinase B (PKB) primes for the phosphorylation of another residue. Phosphorylation of all three sites requires association with the plasma membrane. In vitro, mutation of the phosphorylation sites increased the half-life of the protein but reduced relative cell surface levels, enhanced ectodomain shedding, and significantly attenuated DLL1 ligand activity. However, mutation of all three phosphosites in vivo had no obvious effects on processes regulated by DLL1 such as somitogenesis, myogenesis, neurogenesis, and marginal zone B cell development, suggesting that in vivo compensatory mechanisms are in place to maintain physiological DLL1 activity.
MATERIALS AND METHODS
Cell culture and transfection.
Chinese hamster ovary (CHO), HEK293 (Human embryonic kidney), HeLa, and L-cells were cultured in Dulbecco modified Eagle medium (DMEM)–F-12 (Invitrogen) cell culture medium supplemented with 10% fetal calf serum (FCS; Invitrogen), 1% penicillin-streptomycin (Gibco), and 1% GlutaMAX (Gibco). Cells were transfected with jetPEI (Polyplus-Transfection) or Perfectin (Genlantis) according to the manufacturers' instructions. CHO cells containing an attP-site were generated by transfection of the pHZ-attP (51) plasmid and selection with 500 μg of hygromycin B/ml. Transgenes were cloned into the attB pNC-attB vector (51) after removal of enhanced green fluorescent protein (eGFP), and recombination was mediated by transient expression of phiC31 integrase. Cells were selected for correct integration with 250 μg of zeocin/ml.
Antibodies.
The primary antibodies used in Western blots were as follows: phospho-specific rabbit anti-pT638, -pS693, and -pS696 antibodies (generated by Biogenes GmbH, Berlin, Germany), using peptides C-KVRYP-pT-VDYNL (pT638), C-RKRPE-pS-VYSTS (pS693), and C-PESVY-pS-TSKDT (pS696), respectively; anti-DLL1 (ab85346; Abcam; directed against amino acids 148 to 162 of human DLL1 for the detection of mouse, chicken, and Xenopus DLL1); rat anti-DLL1 1F9 (52) affinity purified with a synthetic peptide (Genecust; CPGPMVVDLSERHMESQG) coupled to SulfoLink resin (Thermo Scientific) according to the manufacturer's instructions; anti-Flag M2 POD (Sigma); antihemagglutinin (anti-HA) 3F10 POD (Roche); and anti-His–horseradish peroxidase (Miltenyi Biotec). β-Actin detected with mouse anti-β-actin antibodies (clone C4; MP Biomedicals) or anti-sodium potassium ATPase antibody (ab353; Abcam) was used as a loading control. Horseradish peroxidase-conjugated secondary anti-rat, -mouse, -rabbit, -chicken, or -goat antibodies were obtained from GE Healthcare or Dianova. The antibodies used in immunoprecipitations were anti-Flag M2 (Sigma), anti-DLL1-c20 antibody (used for immunoprecipitation of endogenous DLL1 [Santa Cruz]), and the rabbit non-phospho-specific antibodies anti-T638, anti-S693, and anti-S696 (generated by Biogenes GmbH). The primary antibodies used for the immunofluorescence staining of CHO cells included DLL1 2A5 (50), anti-Rab11 clone 47/Rab11 BD (Transduction Laboratories), anti-Rab5 clone Rab5-65 (Sigma), and anti-Rab6a clone 3G3 (Sigma). Secondary antibodies were conjugated to Alexa Fluor 488, 555, or 633 (Life Technologies).
Immunoprecipitation.
CHO, HEK293, or L-cell cell lines stably or transiently expressing mouse, Xenopus, or chicken DLL1 were harvested at 90% confluence at 24 h posttransfection in ice-cold cell lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 [Applichem]) supplemented with protease inhibitor cocktail tablets (Roche) and treated with PhosSTOP (Roche) or left untreated. The cells were lysed on ice for 30 min, sonicated, and centrifuged at 13,000 × g at 4°C for 10 min. The supernatant was collected and incubated with the appropriate antibody for 2 h or overnight at 4°C on an end-to-end rotator, followed by incubation with Sepharose G-beads (GE Healthcare) overnight at 4°C on an end-to-end rotator. The beads were washed three times in ice-cold cell lysis buffer, mixed with sample buffer, boiled for 5 min at 99°C, and either loaded onto sodium dodecyl sulfate (SDS) gels or stored at −20°C. If not otherwise indicated, immunoprecipitations of mDLL1-tagged proteins were performed by incubating the lysates with directly coupled anti-Flag M2 affinity gel (Sigma) or EZview red anti-HA affinity gel (Sigma). XDLL1 or cDLL1 were precipitated with anti-Flag M2 antibody (XDLL1) or anti-non-pS693 antibody (cDLL1).
32P metabolic labeling of HEK293 cells.
Labeling of cells was performed overnight with 0.1 mCi of 32P-labeled phosphoric acid/ml in cell culture medium supplemented with 10% dialyzed FCS (Invitrogen). The cells were then lysed in (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 1% Triton X-100, 1% deoxycholate [DOC], 0.1% SDS) and subjected to immunoprecipitation as described above with the following differences. DNA was sheered by passing the lysate through a 19-gauge needle attached to a 1-ml syringe. Before the lysate was mixed with the appropriate antibodies, it was precleared by centrifugation at 3,000 × g and incubation of the supernatant for 1 h at 4°C on an end-to-end rotator. SDS gels were dried and used for autoradiography (GE Healthcare) for 24 or 72 h.
Pulse-chase analysis.
DLL1 or phosphomutant DLL1 expressing CHO-attP-cells were cultured on 60-mm cell culture dishes to 100% confluence. The cells were washed three times with prewarmed phosphate-buffered saline (PBS) and cultured with cysteine-methionine-free RPMI 1640 cell culture medium (Sigma) for 2 h under normal culture conditions. The cells were washed once with prewarmed PBS and pulse-labeled in RPMI 160 medium supplemented with 250 μCi of 35S-labeled methionine for 1 h. For the chase, cells were washed with PBS and medium was replaced with RPMI medium (Sigma). The cells were lysed at different time points in cell lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, supplemented with protease inhibitor cocktail tablets [Roche]) for 30 min on ice. DNA was sheered by passing the lysate through a 19-gauge needle attached to a 1-ml syringe. Lysates were centrifuged for 10 min at 10,000 × g. For preclearing, the supernatant was incubated with Sepharose G-beads (GE) for 60 min on an end-to-end rotator at 4°C. For immunoprecipitation of radiolabeled DLL1-protein lysates were centrifuged for 1 min at 3,000 × g. The supernatant was incubated with anti-Flag M2 affinity gel (Sigma) for 5 h or overnight. Proteins were eluted from the beads by boiling in sample buffer and separated by SDS-PAGE. Dried gels were exposed to autoradiography film. The intensity of the bands were measured from scans using ImageJ. Half-life times were calculated using Prism software (GraphPad) based on a one-phase decay fit.
Notch activation assay.
To determine Notch activation by wild-type (wt) and phosphomutant DLL1 HeLa cells stably expressing Notch1 (HeLaN1 [15]) were transfected with pGa981-6, a Notch1-firefly luciferase reporter construct (53), and for normalization to the transfection efficiency with a plasmid (Promega) driving constitutive expression of Renilla luciferase. At 18 to 20 h posttransfection 5 × 105 HeLa-Notch1 were cocultured with 5 × 105 wt) CHO cells or CHO cells expressing wt or phosphomutant DLL1, respectively, for 18 to 20 h. After coculture, the cells were lysed in lysis buffer (Promega), and the luciferase activity was measured using a dual-luciferase reporter assay (Promega). The relative transactivation potential was calculated relative to the activation obtained by wt CHO cells.
Ubiquitination pulldown assay.
HEK93 cells were transiently transfected with an His-tagged ubiquitin expression vector and cotransfected with wt DLL1 or phosphomutant DLL1 expression vectors in a 100-mm cell culture dish. At 24 to 48 h posttransfection, the cells were treated with 25 μM proteasome inhibitor MG132 (Sigma) for 3 h. Cells were washed in cold PBS, lysed in 1 ml of urea buffer (8 M urea, 10 mM imidazole, 0.1 M phosphate buffer [pH 8]) and sonified on ice. Subsequently, lysates were centrifuged for 10 min at 10,000 × g, and the supernatants were incubated with Talon- or nickel-NTA Superflow beads (Clontech/Qiagen) for 4 h on an end-to-end rotator at room temperature. The beads were washed three times with urea buffer and resuspended with sample buffer containing 200 mM imidazole.
Mass spectrometry.
DLL1-Flag or DLL1TMICD-HA were immunoprecipitated as described above. For elution of the proteins, the resin was prepared according to the manufacturer's instructions and loaded onto a disposable plastic column (Thermo Scientific). The resin was then incubated with 0.3 M glycine (pH 3.5) for 5 min on ice. The proteins were eluted by gravity flow in vials containing 0.5 M Tris-HCl (pH 8.0) and 1.5 M NaCl. After acetone precipitation, the dried pellet was mixed with sample buffer and separated by SDS-PAGE. The respective bands were excised and in-gel digested using trypsin as described previously (54). The resulting peptide mixtures were purified by using Stage-Tips (55) and subjected to liquid chromatography-tandem mass spectrometry (MS/MS) analysis on an Easy-NanoLC liquid chromatographer (Proxeon, Odense, Denmark) coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Peptides were loaded at a flow rate of 500 nl/min onto an in-house-made C18 nano-HPLC column (bead diameter, 3 μm; column size, 15 cm by 75 μm) using high-pressure liquid chromatography (HPLC) solvent A (0.5% acetic acid in water) and eluted at a flow rate of 200 nl/min using a segmented gradient of 5 to 35% of HPLC solvent B (0.5% acetic acid in 80% acetonitrile) over 100 min. The LTQ-Orbitrap XL was operated in the positive-ion mode. The full scan mass spectra were performed in the Orbitrap mass analyzer at a resolution of 60,000, whereas the MS/MS fragmentation five most intense ions (the “top-5 method”) was performed in the linear ion trap. Multistage activation of frequencies corresponding to neutral loss of phosphoric acid (−98, −49, and −32.6 thomsons relative to the precursor ion) was performed; dynamic exclusion was set to 90 s. The resulting mass spectra were processed by using the MaxQuant software suite (v1.0.13.13) and subjected to a database search against the ipi.MOUSE.v3.64 protein database (56,751 entries) using the Mascot search engine. The database search parameters were set as follows: the precursor mass tolerance was 7 ppm; the fragment mass tolerance was 0.5 Da; the fixed modification was carbamidomethylation (C); and the variable modifications were oxidation (M), acetylation (protein N terminus), and phosphorylation (S/T/Y). The false discovery rate was set to 1% at the peptide and protein level, and the resulting phosphopeptide spectra were manually validated. The MS analysis led to identification of two DLL1 phosphopeptides comprising three phosphorylation events in the ECD domain: at T638, S693, and S696 (Fig. 1D; see Table S1 in the supplemental material).
FIG 1.
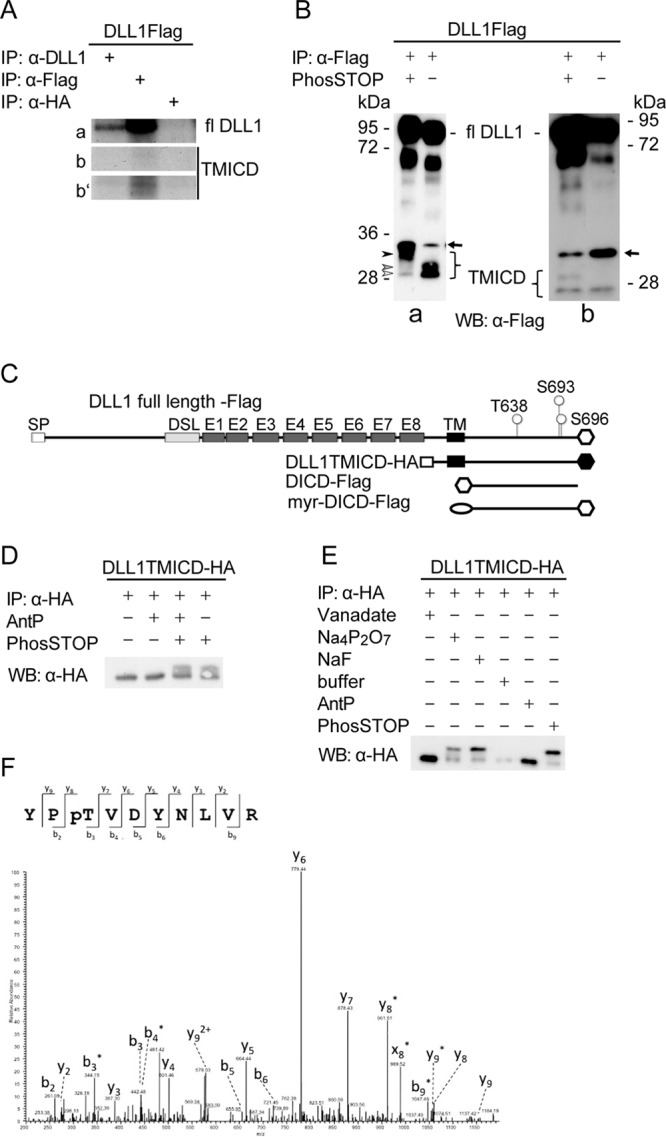
DLL1 is phosphorylated in its intracellular domain. (A) Autoradiography of immunoprecipitations of metabolically 32P-labeled Flag-tagged full-length DLL1 transiently expressed in HEK293 cells. Row a shows the region of the gel containing full-length DLL1; row b shows the region of the gel where TMICD cleavage product is expected at the same exposure time as row a. Row b′ is a longer exposure of row b. (B) Western blot (WB) analysis of immunoprecipitations from lysates of CHO cells stably expressing Flag-tagged DLL1 after SDS-PAGE in Phos-tag gels. Note that DLL1 full-length and TMICD run at lower apparent molecular sizes in the sample without phosphatase inhibitor (PhosSTOP). The absence of phosphorylated DLL1 in the lysate lacking phosphatase inhibitors indicates phosphatase activity in that lysate. (C) Schematic representation of DLL1 variants used in the in vitro experiments. The position of the phosphorylated residues is indicated in the full-length protein by hollow circles. SP, signal peptide; DSL, DSL domain; E1 to E8, EGF-like repeats 1 to 8; TM, transmembrane domain; white hexagon, Flag tag; black hexagon, HA tag; ellipsoid, myristoylation signal. (D and E) WB analyses of stably overexpressed HA-tagged DLL1TMICD in CHO cells in the presence or absence of PhosSTOP, Antarctic phosphatase (AntP), and phosphatase inhibitors as indicated. (F) Mass spectrum from the phosphopeptide YP[pT]VDYNLVR of DLL1. The fragment ions y8, x8, and b3 point to T638 as the phosphorylation site. The precursor ion was measured with mass deviation of 1.3 ppm (see Table S1 in the supplemental material for details and data on the other phosphosites). WB, Western blot; anti-DLL1, MAb 1F9.
Detection of endogenous phosphorylated DLL1.
Posterior halves of embryonic day 10.5 CD1 wild-type embryos or fetal kidneys were dissected and collected in ice-cold cell lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% Nonidet-P40 [Roche]) treated with PhosSTOP (Roche) or left untreated. Lysates were homogenized with a micropestle in Eppendorf tubes, subjected to sonification, centrifuged at 13,000 × g at 4°C for 10 min, and then immunoprecipitated with the indicated antibodies. In a second experiment, lysates were additionally incubated with anti-DLL1-c20 blocked with a double amount of the corresponding peptide to demonstrate the specificity of the immunoprecipitation for DLL1.
Phos-tag SDS-PAGE.
A 2.5 μM concentration of Phos-tag AAL107 (NARD Institute, Ltd.) was used in SDS-PAGE analyses according to the manufacturer's instructions.
Generation of DLL1 expression plasmids.
pTracer-CMV vector was used for the expression of either mDLL1-Flag wild-type or phosphomutant protein. Mutagenesis of single or double phosphomutant DLL1 variants was performed using a Stratagene QuikChange site-directed mutagenesis kit according to the manufacturer's instructions with the following primers: T638-V, GCTTTAAGGTCCGATACCCCGTTGTGGACTATAACCTCGTTC and GAACGAGGTTATAGTCCACAACGGGGTATCGGACCTTAAAGC; S693-A, CAGAAAAAGGCCAGAGGCTGTCTACTCTACTTCAAAGGACAC and GTGTCCTTTGAAGTAGAGTAGACAGCCTCTGGCCTTTTTCTG; and S696-A, GCCAGAGTCTGTCTACGCCACTTCAAAGGACACCAAGTACCA and TGGTACTTGGTGTCCTTTGAAGTGGCGTAGACAGACTCTGGC.
Site-directed mutagenesis was performed on a Dll1 fragment (bp 1605 to 2199 of the open reading frame) and reintroduced into pTracer-CMV-Dll1. A Dll1 fragment (bp 1605 to 2199) mutated at all three phosphosites was synthesized (Entelechon). The S693/S696 DLL1 double mutant was generated by site-directed mutagenesis with the triple mutant as a template using the following primers: V638-T, GCTTTAAGGTCCGATACCCCACTGTGGACTATAACCTCGTTC and GAACGAGGTTATAGTCCACAGTGGGGTATCGGACCTTAAAGC.
All vector constructs were generated by using standard cloning techniques and were verified by sequencing. mDll1-ICD fused to a 5′ myristoylation signal from rat PSD-Zip70 protein (56) was synthesized (Entelechon) and cloned into pIRES-puro3 (Clontech).
Generation of the targeting construct and genotyping of mice.
A DLL1-knock-in targeting vector described previously (46) was used for the generation of a phosphomutant DLL1 mouse. A NotI-SpeI fragment of this vector containing the coding sequence of the intracellular domain was cloned into pZErO-1 (Invitrogen). The wild-type Mfe1-HpaI fragment was replaced with a fragment carrying mutations in all three phospho-sites generated by gene synthesis (Geneart), and the mutated NotI-SpeI fragment was reintroduced into the targeting vector as a NotI-RsrII fragment. Mice were genotyped by using DNA prepared from tail biopsy specimens by PCR with the following primers: Dll1 wild-type allele, CTGAAGCGACCTGGCCCTGATAGCAC and CTGCGCGGGGAAGGGGCG; Dll1Dll1wtki, CTTCAACTGTGAGAAGAAGATG and CCTTTGAAGTAGAGTAGACAGA; and Dll1Dll1T638V,S693A,S696A, CTTCAACTGTGAGAAGAAGATG and CCTTTGAAGTGGCCTAGACGGC.
Surface biotinylation.
Surface biotinylation was performed as described previously (52) with the following modifications. Cells were washed three times with ice-cold PBS supplemented with 1 mM MgCl2 and 0.1 mM CaCl2 (PBS-C/M) at 80% confluence and kept on ice for 10 min before the cells were incubated with 0.25 mg of Sulfo-NHS-LC (Pierce)/ml on ice for 40 min. The cells were washed in ice-cold PBS-C/M and incubated with DMEM supplemented with 100 mmol of glycine for 15 min on ice to quench the biotin reaction. Next, the cells were washed twice in ice-cold PBS-C/M and lysed in lysis buffer (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% Triton X-100, 0.25% DOC, 0.1% SDS) supplemented with Complete protease inhibitor cocktail tablets and PhosSTOP (Roche) on ice for 30 min, subjected to sonification, and centrifuged for 10 min at 4 ° C to remove cellular debris. NeutrAvidin beads (Pierce) were washed in lysis buffer and used for the pulldown of biotinylated DLL1 species.
Endocytosis assay.
Biotinylation of cell surface proteins was performed as described above. Instead of Sulfo-NHS-LC, cleavable Sulfo-NHS-SS-Biotin (Pierce) was used. Endocytosis was then assayed as described previously (57) with the following minor modification. Before lysis, all cells were incubated with 5 mg iodoacetamide/ml in PBS for 15 min to quench the MesNa (Sigma) reaction.
Kinase inhibitor assay.
Single-copy wild-type DLL1 expressing CHO cells at 80% confluence were incubated for 24 or 48 h with the indicated concentrations of the selective InSolution Akt Inhibitor V (Triciribine) or with the selective InSolution casein kinase I (CK1) inhibitor D4476 (Calbiochem), respectively. As a control, cells were also incubated with dimethyl sulfoxide or left untreated. After 24 h, the cells were washed in ice-cold PBS and then subjected to immunoprecipitation as described above.
MMP inhibition.
For the inhibition of matrix-metallo-proteases (MMPs), equal amounts of CHO cells were treated with different concentrations of the MMP inhibitors TAPI-2 or GM6001 for 6 h, washed in cold PBS, and lysed in cell lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) supplemented with a protease inhibitor cocktail tablet (Roche) for 30 min on ice. After centrifugation at 8,000 × g, the supernatant was mixed with sample buffer. The samples were boiled for 5 min at 99°C minutes and loaded onto SDS gels.
Quantification of proteins.
Proteins detected in Western blots were quantified by using ImageJ with β-actin for normalization. All calculations are based on a minimum of three independent Western blots.
Whole-mount in situ hybridization.
Whole mount in situ hybridizations were performed according to standard procedures (58). Pictures were taken with the Leica Z6 APO macroscope with a Leica DFC420 camera and Leica Firecam software, v3.4.1.
Immunofluorescence staining.
Stable DLL1 or phosphomutant DLL1 CHO-attP cells were grown on gelatin-coated coverslips to 50 to 60% confluence. Cells were washed in cold PBS and fixed for 10 min in methanol on ice and washed again three times in PBS. Nonspecific binding was blocked by incubating the cells for 1 h in PBS supplemented with 10% goat serum. The cells were incubated for 1 h with primary antibodies in blocking solution, washed three times for 5 min each time with PBS, and incubated with specific secondary fluorescence-coupled antibodies in blocking solution for 1 h in the dark. The cells were then washed three times in PBS and incubated for 10 min in PBS containing DAPI (4′,6′-diamidino-2-phenylindole) for nuclear staining. Subsequently, the cells were washed twice in PBS. Samples were mounted on microscope slides with Pro-Long antifade gold mounting medium (Life Technologies). Pictures were taken with a Leica TCS SP2 AOBS scanhead mounted on an DM IRB inverse microscope. Pictures were processed by using Leica Application Suite advanced fluorescence software (Leica AF). For tonal value correction of whole pictures, Adobe Photoshop was used.
RESULTS
DLL1 is phosphorylated at S/T residues in its intracellular domain.
First, we analyzed phosphorylation of DLL1 by immunoprecipitation of DLL1 from lysates of transiently DLL1-Flag-expressing HEK293 cells metabolically labeled with 32P using a monoclonal antibody (MAb) directed against the extracellular domain of DLL1 or against the C-terminal Flag tag. Both immunoprecipitations pulled down full-length DLL1 that had incorporated 32P (Fig. 1A, row a). Labeled protein corresponding to the size of TMICD, a fragment containing the transmembrane and intracellular domain that is generated by ADAM proteases releasing the ectodomain (20–24), was only detected after immunoprecipitation with the anti-Flag antibody and extended exposure time (Fig. 1A, row b′), most likely reflecting that TMICD is less prevalent or stable than full-length DLL1. A similar result was obtained with metabolically labeled CHO cells (data not shown). Next, we analyzed DLL1 phosphorylation using Phos-tag {1,3-bis[bis(pyridin-2-ylmethyl)amino]propan-2-olatodizinc(II) complex} gels, which allow for the detection of phosphorylated proteins based on their lower mobility in SDS-PAGE (59). We immunoprecipitated DLL1 from lysates of CHO cells expressing DLL1-Flag in the presence or absence of phosphatase inhibitors (PhosSTOP), separated the proteins by SDS-PAGE using Phos-tag gels, and analyzed them by Western blotting with anti-Flag antibodies (Fig. 1B). In the lysate treated with PhosSTOP full-length DLL1 migrated at a higher apparent molecular size consistent with phosphorylation (Fig. 1B, blot a). In this lysate, we weakly detected a protein migrating at ∼28 Da (white arrowhead in Fig. 1B, blot a), a very weak protein band slightly above (gray arrowhead in Fig. 1B, blot a) and a strong band at ∼32 kDa (black arrowhead in Fig. 1B, blot a). In the lysate not treated with PhosSTOP, the most slowly migrating protein was not detected, and the two faster-migrating proteins were readily detected in comparable amounts (Fig. 1B, blot a). We interpret these results such that the protein migrating at ∼32 kDa represents the phosphorylated TMICD and the fragments migrating at lower molecular sizes represent partially and nonphosphorylated TMICD. Since the presumed TMICD proteins migrated at higher apparent molecular masses in Phos-tag gels than predicted, we analyzed immunoprecipitated DLL1 from lysates of CHO cells expressing DLL1-Flag in the presence or absence of PhosSTOP by Western blotting after conventional SDS-PAGE (Fig. 1B, blot b). In these blots, we observed a single band migrating at the expected size of TMICD at 28 kDa (24) in the absence of PhosSTOP, and an additional band migrating slightly higher, supporting the notion that these bands correspond to phosphorylated and nonphosphorylated TMICD fragments. The severe reduction of protein species with lower mobility in lysates without PhosSTOP also suggests that DLL1 is dephosphorylated under these conditions. The additional band below full-length DLL1 (Fig. 1B, blots a and b) represents a cleavage product of unknown significance, which we frequently observe in cultured cells. Since this fragment is detected by antibodies directed against the C-terminal flag tag, as well as by antibodies directed against the proximal extracellular domain (data not shown), most likely this fragment lacks the N-terminal portion of DLL1.
To test whether the extracellular domain of DLL1 is required for phosphorylation of the ICD, we used overexpressed hemagglutinin (HA)-tagged TMICD, which lacks the extracellular domain (except for the signal peptide and 21 N-terminal amino acids; DLL1TMICD-HA) (Fig. 1C). DLL1TMICD-HA is phosphorylated (Fig. 1D), indicating that the presence of the extracellular domain and thus interaction with Notch is not required for this modification. To address whether S/T and/or Y residues are phosphorylated, lysates of CHO cells stably expressing DLL1TMICD-HA were treated with vanadate, an inhibitor of phosphotyrosine phosphatases, or Na4P2O7 or NaF, inhibitors of phosphoserine/phosphothreonine phosphatases, or Antarctic phosphatase that completely dephosphorylates proteins (60), or with PhosSTOP, a general inhibitor of phosphatases. Samples treated with both Na4P2O7 and NaF showed a band pattern indistinguishable from PhosSTOP-treated samples, whereas vanadate-treated samples showed a single band migrating at the same apparent molecular size as the sample treated with Antarctic phosphatase or the sample kept in the absence of phosphatase inhibitors (Fig. 1E). Collectively, the results of these experiments suggested that the intracellular domain of DLL1 (DICD) is phosphorylated at S and/or T and not Y residues. To identify the residues that are phosphorylated, we purified overexpressed DLL1 and TMICD from CHO cells in the presence of PhosSTOP and subjected the purified protein to mass spectrometric analysis on an LTQ-Orbitrap mass spectrometer. Consistent with our inhibitor analysis, mass spectrometry identified phosphorylation at S and T residues (T638, S693, and S696, indicated in Fig. 1C) with a sequence coverage of the ICD of 75% (Fig. 1F; see Table S1 in the supplemental material).
DLL1 is phosphorylated in vivo.
To be able to directly detect phosphorylated DLL1, we generated polyclonal antibodies directed against peptides containing phospho-S693, phospho-S696, or phospho-T638 (BioGenes, Berlin, Germany). In Western blot analyses, these antibodies detected Flag-tagged wild-type DLL1 that was immunoprecipitated from L, HeLa, and CHO cells in the presence of phosphatase inhibitors but not a DLL1 variant that had all three phosphosites mutated (T638V, S693A, and S696A) (Fig. 2A). Similarly, DLL1 that was immunoprecipitated from CHO cells in the absence of phosphatase inhibitors was not recognized by these antibodies (Fig. 2A). These analyses indicated that the antibodies specifically recognize DLL1 that is phosphorylated at the respective S/T residues and that DLL1 is phosphorylated in cell lines from different mammalian species. To analyze whether DLL1 is phosphorylated in vivo, DLL1 was immunoprecipitated from mouse E10.5 embryo lysates in the presence or absence of phosphatase inhibitors and analyzed by Western blotting with the antiphosphopeptide antibodies or an MAb directed against the extracellular domain of DLL1. The MAb detected a band at the molecular weight corresponding to DLL1 under both conditions (Fig. 2B, blot a). In contrast, anti-pS693 antibodies detected a specific band only in immunoprecipitates from phosphatase inhibitor-treated lysates (arrow in Fig. 2B) in several independent experiments, indicating that endogenous DLL1 is phosphorylated in early embryos at S693. anti-pT638 and anti-pS696 antibodies did not detect specific bands in these experiments (data not shown). Similarly, we detected phosphorylation of DLL1 at S693 in lysates of fetal kidneys (Fig. 2B, blot b), which express DLL1 at high levels during nephron formation (61, 62), indicating that phosphorylation is not restricted to early development.
FIG 2.
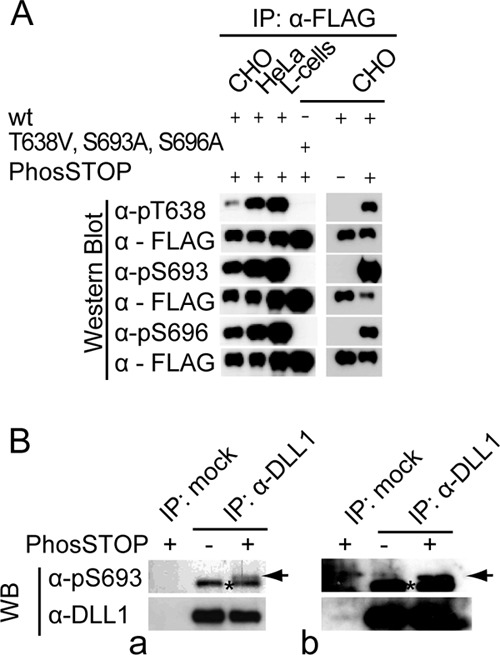
DLL1 is phosphorylated in various cell lines and in vivo. (A) Western blot (WB) analysis with phospho-specific or anti-Flag antibodies of immunoprecipitations (IP) of Flag-tagged DLL1 stably expressed in mammalian (CHO, Chinese hamster; HeLa, human; L, mouse) cell lines as indicated. wt, wild-type DLL1; T638V, S693A, S696A, DLL1 mutated at all three phosphosites. (B) WB analysis with anti-pS693 and anti-DLL1 (MAb 1F9) antibodies of DLL1 immunoprecipitated from E10.5 wt embryos (a) or from embryonic day 15.5 (E15.5) to E16.5 fetal kidneys (b) in the presence or absence of phosphatase inhibitor. Note that the anti-pS693 antibody detects a unique band only in the fraction treated with the phosphatase inhibitor (arrow; the asterisk indicates background band; mock, IP without antibody). α-DLL1 IP, c20 (Santa Cruz).
Phosphorylation of DLL1 requires membrane association.
To get first hints as to where in the cell DLL1 is phosphorylated, we compared the phosphorylation status of full-length DLL1 and DLL1TMICD-HA, which are both transmembrane proteins, with the phosphorylation of the intracellular domain of DLL1 (DICD-Flag, Fig. 1C) expressed as a cytosolic protein. Phosphorylation at S693 was readily detected in full-length DLL1, as well as in DLL1TMICD expressed in CHO cells (Fig. 3A). In contrast, phosphorylation at S693 was not detected when DICD was expressed as cytoplasmic protein (Fig. 3A). In contrast to DLL1 and DLL1TMICD, DICD is not presented at the cell surface (Fig. 3B), suggesting that membrane anchoring or association is a prerequisite for S693 phosphorylation. To test this idea, we generated a DICD variant that was artificially directed to the membrane by addition of an N-terminal myristoylation site (myr-DICD-Flag; Fig. 1C). Indeed, phosphorylation at all three sites was observed in this DICD variant (Fig. 3C), suggesting that phosphorylation of DLL1 occurs after DLL1 has reached the cell membrane.
FIG 3.
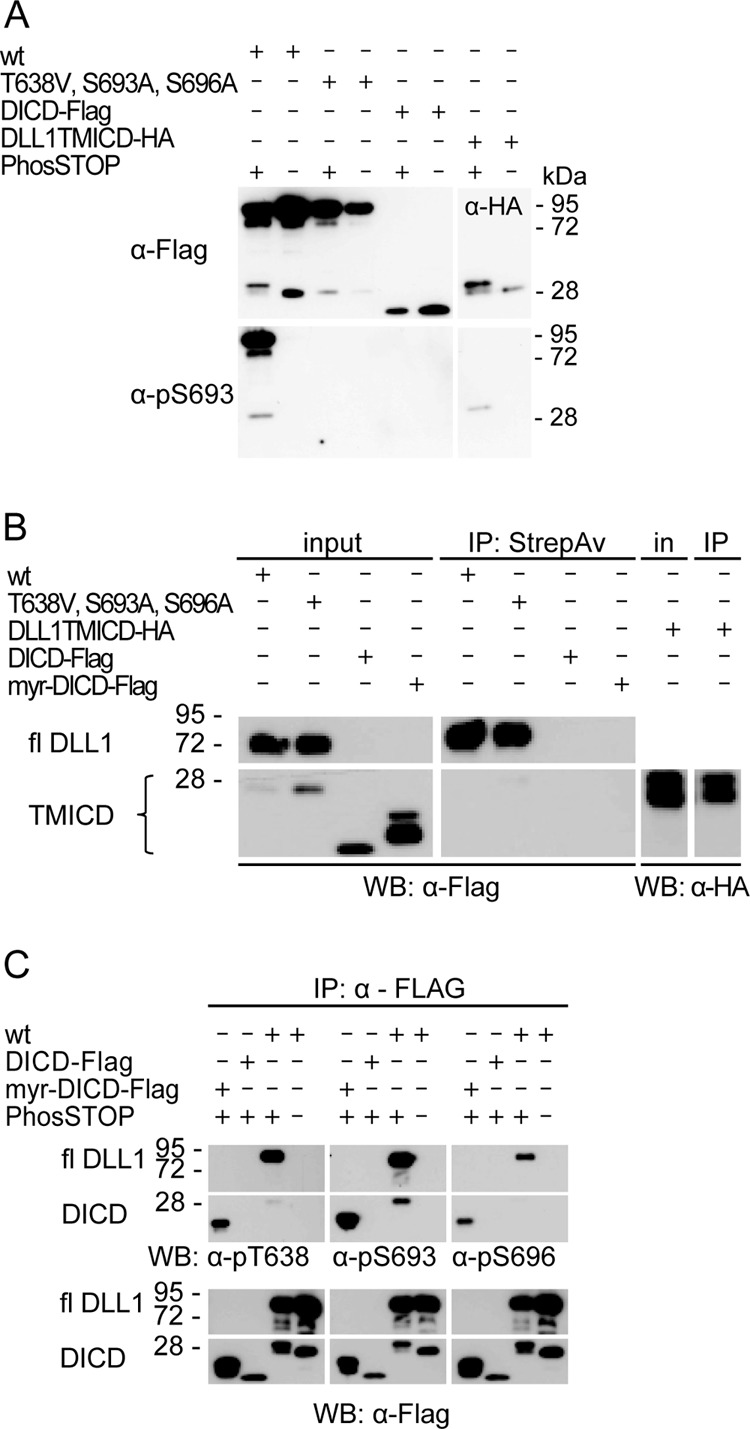
Phosphorylation of DLL1 requires membrane association. (A) Western blot (WB) analysis of immunoprecipitations (IPs) from lysates of cell lines expressing tagged DLL1 protein variants with anti-Flag or anti-pS693 antibodies. Only variants anchored to the cell membrane are phosphorylated. (B) Surface biotinylation assay with DLL1 variants immunoprecipitated with anti-Flag or anti-HA antibodies indicates that DLL1 mutated at all three phosphosites is present at the cell surface. (C) WB analysis of immunoprecipitations of DLL1 variants with phospho-specific and anti-Flag antibodies in the presence or absence of a phosphatase inhibitor indicates that myr-DICD that is not exposed extracellularly (see panel B) is phosphorylated at all three sites. wt, wild-type DLL1-Flag; T638V, S693A, S696A, DLL1-Flag mutated at all three phosphosites; DLL1TMICD-HA, DLL1 with a deletion of the extracellular domain; DICD-Flag, DLL1 intracellular domain; myr-DICD, DICD with N-terminal myristoylation signal. StrepAv, streptavidin.
Phosphorylation of S693 and S696 occurs sequentially.
To test whether phosphorylation at one of the phosphosites is a prerequisite for phosphorylation at any of the other sites, we generated mutations at individual phosphorylation sites or combinations thereof. Mutant proteins were expressed in CHO cells, and the phosphorylation status at the three sites was determined by Western blotting analyses of cell lysates using the phospho-specific antibodies. These analyses showed that phosphorylation of S696 required the presence of S693 (asterisks in Fig. 4A), whereas phosphorylation of T638 and S693 occurred irrespective of mutations in the other sites (Fig. 4A). S693 is part of a consensus sequence (RXRXXS/T) recognized by AKT/PKB (63, 64), which is recruited to and activated at the cell membrane (reviewed in reference 65). Since DLL1 phosphorylation depends on membrane association, AKT/PKB is a possible candidate for the kinase that phosphorylates S693 of DLL1. Indeed, treatment of DLL1 expressing CHO cells with the AKT/PKB inhibitor Triciribine caused a significant decrease in S693 phosphorylation, as well as phosphorylation of S696 (Fig. 4B). S693 and S696 are parts of a pSXXS consensus sequence, in which serine residue S696 can be phosphorylated by CK1 subsequent to phosphorylation of residue S693 (66, 67), raising the possibility that CK1 is the kinase phosphorylating S696. However, treatment of DLL1 expressing CHO cells with the CK1 inhibitor D4476 at different concentrations gave inconsistent results, low concentrations inhibiting and higher concentrations enhancing S696 phosphorylation (data not shown), which did not allow us to confirm CK1 as the relevant kinase.
FIG 4.

Sequential phosphorylation of mouse DLL1 and phosphorylation of chick and frog DLL1 homologues. (A) Expression and phosphomodification analyses of DLL1 phosphomutant variants stably expressed in CHO cell lines with phospho-specific and anti-Flag antibodies. Note that the anti-pS696 antibody detects no bands (missing bands indicated by asterisks) in immunoprecipitation (IP) fractions with DLL1S693A mutant proteins (“T638V, S693A, S696A” indicates mutations of the specific phosphosites). (B) Western blot (WB) analysis of DLL1 phosphorylation at S693 and S696 in the presence of the PKB inhibitor Triciribine. Inhibition of PKB significantly reduces phosphorylation at both sites in a concentration-dependent manner. (C) CLUSTAL W alignment of the mDLL1 C terminus and vertebrate homologues. Phosphorylated residues detected by mass spectrometric analysis of mDLL1 are indicated by arrowheads (dark-shaded region indicates conserved amino acids). (D) WB analysis with anti-pS693 and anti-pS696 antibodies of immunoprecipitated mouse (mDLL1), Xenopus (XDLL1), and chick (cDLL1) DLL1 in the presence or absence of a phosphatase inhibitor. wt, wild-type DLL1; combinations of T638V, S693A, and S696A, DLL1 mutated at the respective phosphosites; h, human; ZF, zebrafish.
Chicken DLL1 and Xenopus DLL1 are phosphorylated in mammalian cell lines.
S693 (Fig. 4C, dark gray arrowhead) is present in DLL1 homologues in all analyzed vertebrate species and embedded in a highly similar sequence context but was not found in Drosophila Delta (results not shown). S696 (Fig. 4C, light gray arrowhead) is present in vertebrate DLL1 ligands except in zebrafish. T638 (white arrowhead in Fig. 4C) is the least well conserved phosphosite residue and is replaced by S in chicken and Xenopus and by other amino acids in human and zebrafish. Because S693 and S696 are present in chicken (cDLL1) and Xenopus (XDLL1) proteins in a conserved sequence context, we analyzed whether these DLL1 proteins are also phosphorylated when expressed in mammalian (CHO) cells. In Western blots anti-pS693 antibodies detected immunoprecipitated cDLL1 in phosphatase inhibitor-treated but not in untreated lysates (Fig. 4D), indicating phosphorylation of cDLL1 at S693. anti-pS696 antibodies gave a strong signal with cDLL1 in phosphatase inhibitor-treated lysates and a much weaker signal in lysates without inhibitor (Fig. 4D), suggesting that cDLL1 is also phosphorylated at S696 in CHO cells. No signal with anti-pS693 antibodies was detected with immunoprecipitated XDLL1 (Fig. 4D). However, anti-pS696 antibodies reacted with XDLL1 in phosphatase inhibitor-treated but not in untreated lysates, indicating phosphorylation at S696. Thus, also chicken and Xenopus DLL1 expressed in mammalian cells are phosphorylated at S residues equivalent to S693 and S696 in mouse DLL1, suggesting that the chicken and Xenopus DLL1 proteins are also substrates for kinases in these species.
Phosphorylation of DLL1 is required for full ligand activity in vitro.
To test whether phosphorylation of DLL1 affects its function, we generated CHO cells stably expressing wild-type and mutant DLL1 from single transgene copies inserted at identical genomic locations via attP site integration (51). Transcript levels of both DLL1 variants were indistinguishable by semiquantitative reverse transcription-PCR (data not shown). In contrast, protein levels of the DLL1 variant with the mutated phosphosites were ∼4-fold higher (Fig. 5Aa), suggesting increased protein stability. To analyze this possibility, we determined the protein half-life of wt and phosphomutant DLL1 protein by 35S pulse-chase labeling of CHO cells stably expressing these proteins. These analyses showed that in CHO cells wt DLL1 has a half-life of 144 min, whereas the half life of phosphomutant DLL1 protein was 249 min (Fig. 5Bb). To analyze whether phosphorylation of DLL1 affects Notch activation, we cocultured the CHO cells expressing wild-type or triple-phosphomutant DLL1 with HeLa cells stably expressing Notch1 (HeLaN1 cells) that allow for efficient detection of Notch activation (15) and determined the Notch activation using a luciferase reporter. Despite higher protein levels, the DLL1 variant with the mutated phosphosites activated Notch only about half as efficiently as wild-type DLL1 in the cocultivation assay (Fig. 5C), indicating that phosphorylation of DLL1 is required for efficient Notch activation in the in vitro coculture assay.
FIG 5.

DLL1 phosphorylation is required for full ligand activity in vitro and affects surface presentation, ectodomain shedding, and endocytosis. (A) Expression analysis (a) and quantitation (b) of full-length wt DLL1 and DLL1 T638V, S693A, S696A expressed from single-copy transgene insertions in CHO cells. Values in panel b represent the means of eight independent measurements (P < 0.001). (B) Nonlinear fit of data obtained from 35S pulse-chase experiments (wt [n = 2]; T638V, S693A, S696A [n = 3]). The calculated half-life times are 144 and 249 min for the wild-type or phosphomutant proteins, respectively (P = 0.02). (C) Notch1 transactivation assay of CHO cells stably expressing DLL1 wild-type or phosphomutant proteins from a single-copy insertion, indicating a lower transactivation potential of the phosphomutant protein compared to wt DLL1. Values represent the means of eight independent measurements (P = 0.0119). (D) Percentage of wt DLL1 and DLL1 T638V, S693A, S696A present on the cell surface. Values represent the means of four independent measurements (P = 0.013289). (E) Western blot (WB) of cell lysates of CHO cells stably expressing DLL1 wild-type or phosphomutant protein probed with an anti-Flag antibody (a) and quantification of TMICD (b). Values represent the means of 12 independent measurements (P < 0.0001). (F) WB analyses of lysates of CHO cells expressing wt and phosphomutant DLL1 in the absence or presence of the γ-secretase inhibitor DAPT, indicating that phosphorylation is not required for γ-secretase cleavage. Asterisks indicate the γ-secretase-dependent cleavage product. The double band in the presence of PhosSTOP most likely reflect the phosphorylated and unphosphorylated cleavage product. (G) Quantitation of endocytosis of wt and phosphomutant DLL1. Equally filled (DLL1 wt) and open (phospho-mutant) shapes represent the corresponding results of one experiment. In each single experiment the phosphomutant protein was endocytosed more efficiently. (H) Immunofluorescence of CHO cells stably expressing wild-type or phosphomutant DLL1. Cells were stained for DLL1 and the indicated markers for markers for vesicular trafficking. Arrows point to colocalization of DLL1 with the indicated markers. wt, wild-type DLL1; T638V, S693A, S696A, DLL1 mutated at the three phosphosites. Error bars indicate the standard errors of the mean.
DLL1 T638V, S693A, S696A is present at the cell surface (Fig. 3B) but might be less abundant there than wild-type DLL1, which could be a potential explanation for the reduced activation of Notch by DLL1 T638V, S693A, S696A. Therefore, we compared the total level of each full-length protein with its level on the cell surface. A total of 68% of wild-type DLL1 and 32% of DLL1 T638V, S693A, S696A were present on the cell surface (Fig. 5D), indicating that relative cell surface levels of the mutant protein are reduced. During these analyses, we noticed that in lysates of cells expressing wt DLL1 the C-terminal DLL1TMICD fragment appeared significantly less abundantly than in lysates of DLL1 T638V, S693A, S696A-expressing cells (Fig. 5Ea). Quantitative analysis showed that on average the TMICD cleavage fragment of DLL1 T638V, S693A, S696A made up roughly one-third of the total DLL1 protein, whereas only ca. 12% of wt DLL1 were present as TMICD (Fig. 5Eb), indicating that phosphomutant DLL1 is cleaved in the ectodomain more efficiently than wild-type DLL1. To further analyze the sensitivity of wt and phosphomutant DLL1 protein to matrix metalloproteases (MMPs), we quantitated the relative amounts of TMICD DLL1 that were obtained in the presence of increasing concentrations of GM6001 and TAPI2, inhibitors of MMPs and ADAMs (68, 69). Treatment of DLL1-expressing CHO cells with GM6001 over a range from 10 to 40 μM showed no consistent and reproducible inhibition of cleavage of either wt or of phosphomutant DLL1 protein in three independent experiments (data not shown), suggesting that the protease(s) that cleaves DLL1 in CHO cells was not effectively inhibited by GM6001. In CHO cells expressing wild-type DLL1 treated with 31.25 μM TAPI-2, the lowest effective inhibitor concentration in two independent experiments, the TMICD/total DLL1 ratio decreased 2-fold. Increased inhibitor concentrations did not change the ratio further (Fig. 5Ec). Similarly, in CHO cells expressing phosphomutant DLL1 treated with 31.25 μM TAPI-2 the ratio of TMICD/total decreased ∼1.4-fold, which was not changed by increasing the TAPI-2 concentration (Fig. 5Ec). Thus, cleavage of both DLL1 proteins is slightly inhibited, but because there was considerable protease activity that was not inhibited by TAPI2 in these experiments, we could not determine the difference in sensitivity toward these proteases. To test whether the loss of phosphorylation at T638, S693, and S696 affects DLL1 cleavage by γ-secretase, we analyzed DLL1 C-terminal fragments of wild-type DLL1 that were generated in the presence or absence of the γ-secretase inhibitor DAPT and of PhosSTOP, respectively, and of the phosphomutant variant in the presence or absence of DAPT. In the presence of DAPT, the TMICD levels of both wild-type and phosphomutant DLL1 were enhanced, and a DAPT-sensitive cleavage product of wt DLL1 was obtained both in the absence and in the presence of PhosSTOP, as well as with the phosphomutant protein (Fig. 5F), indicating that both phosphorylated and nonphosphorylated TMICDs are substrates for γ-secretase.
Ubiquitylation of DSL ligands by Neuralized and Mindbomb (Mib) E3 ligases appears to be required for ligand endocytosis, for generating fully active ligands, and to contribute to ligand degradation, but how different ubiquitylation states are regulated is unclear (reviewed in references 32 to 34, 36, and 70 to 72). Phosphorylation can regulate ubiquitylation (reviewed in reference 73). Therefore, we analyzed the ubiquitylation of wild-type and phosphomutant DLL1 in HEK293 cells, which endogenously express Mib1 (74), the critical E3 ligase required for Jagged/Delta mediated Notch signaling in mouse development (75, 76) in ubiquitin pulldown assays. We observed complex patterns of ubiquitylated DLL1 both with the wild-type and with the phosphomutant protein but could not detect consistent changes between the proteins in different experiments (data not shown), which did not allow us to unambiguously conclude that ubiquitylation of DLL1 is altered in DLL1 T638V, S693A, S696A. Altered ubiquitylation states might be revealed by effects on endocytosis. Therefore, we compared the endocytosis of wt and phosphomutant DLL1 proteins by using reversible cell surface biotinylation. In four independent experiments, we consistently observed that more phosphomutant protein was endocytosed after 20 min (Fig. 5G). However, due to the large variability between different experiments, the difference was statistically not significant (P = 0.12). To test whether the apparently different endocytosis rate might lead to different intracellular localization of wt and phosphomutant DLL1, we analyzed the distribution of both proteins in comparison to various markers for vesicular trafficking. Both wt and phosphomutant DLL1 colocalized similarly with these markers (Fig. 5H), suggesting that the endocytotic routes of both proteins do not obviously differ.
Phosphorylation of DLL1 is dispensable for DLL1 function during development and postnatally for marginal zone B cell development.
To analyze the physiological significance of DLL1 phosphorylation, we have mutated the three phosphosites in mice by integrating a triple phosphomutant Delta1 “minigene” into the Delta1 locus (Fig. 6A and B). Homozygous mice carrying a wild-type Dll1 “minigene” introduced by a similar strategy (Dll1tm2Gos, here to referred to as Dll1Dll1wtki) are viable and fertile and appear phenotypically normal (77), indicating that the minigene can functionally replace endogenous Delta1. Likewise, mice homozygous for the Dll1Dll1T638V,S693A,S696A allele were obtained at the expected Mendelian ratio, appeared phenotypically normal and were fertile.
FIG 6.
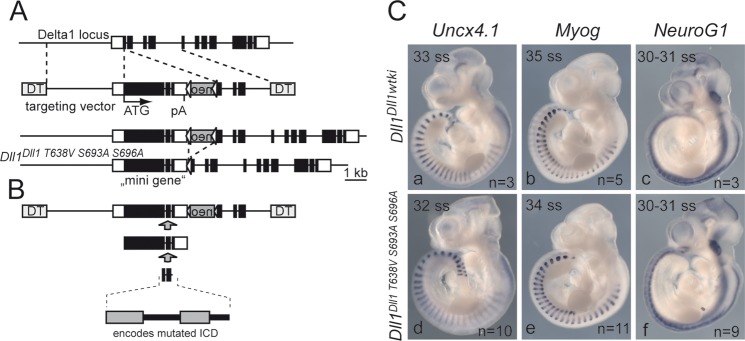
Targeting strategy and analysis of mutant embryos. (A) Schematic representation of targeting vector comprising a minigene, including 4 kb of the promoter region (providing the 5′ region of homology), exons 1 to 9 as cDNA followed by the remainder of the Delta1 gene (intron 9, exon 10, intron 10, and exon 11). After homologous recombination, the minigene replaces the coding portion of exon 1 and most of exon 2 analogously to our targeting strategy used to generate a lacZ knock-in allele of Delta1 (78), which has no apparent effect on the regulation of the Dll1 locus in vivo (61). The neo gene can be removed by Cre-mediated site-specific recombination. Coding exons are indicated by black boxes; the 5′ and 3′ untranslated regions are indicated by white boxes. Triangles flanking the neo gene indicate loxP sites. (B) A fragment encoding the intracellular domain with the three mutated phosphosites was synthesized and introduced into the targeting vector by conventional cloning. (C) Whole-mount in situ hybridization of wild-type and phosphosite mutant embryos. Markers for anterior-posterior somite patterning (Uncx4.1), myogenesis (MyoG), and neurogenesis (NeuroG1) were expressed indistinguishably in wild-type and mutant embryos. Shown are age-matched embryos at the indicated somite stage (ss). DT, diphtheria toxin A chain; pA, polyadenylation signal.
During embryonic development, DLL1 is required for anterior-posterior somite patterning, and muscle and neuronal differentiation (77–81). To detect potential effects caused by the mutations abolishing DLL1 phosphorylation during embryogenesis, we analyzed the expression of representative markers for these processes by in situ hybridization. Uncx4.1 expression marks posterior somite compartments (82) and is severely downregulated and irregular in mutants lacking DLL1 function or Notch activity (81). All analyzed mutant embryos (n = 10) at somite stages (ss) 29 to 32 had Uncx4.1 expression patterns indistinguishable from Dll1Dll1wtki embryos (Fig. 6Cd and data not shown). During muscle differentiation DLL1-mediated Notch activity is required to prevent premature and excessive muscle differentiation (77, 83). To analyze whether early muscle differentiation was affected, we analyzed expression of Myog, which marks differentiating myoblasts in somites (84). In Dll1Dll1T638V,S693A,S696A embryos (n = 4 at ss 34 to 35; n = 7 at ss 30 to 31) Myog expression was indistinguishable from expression in Dll1Dll1wtki embryos (Fig. 6Ce and data not shown). To address whether neuronal differentiation is affected, we analyzed the expression of the Notch target NeuroG1, which is expressed in sensory neuron precursors (85). Like the other markers, the expression of NeuroG1 in Dll1Dll1T638V,S693A,S696A embryos (n = 5 at ss 30 to 32; n = 4 at ss 29 to 30) was virtually identical to Dll1Dll1wtki embryos (Fig. 6Cf and data not shown). Our marker gene analysis suggests that somitogenesis, myogenesis, and neurogenesis proceed normally in phosphomutant DLL1 embryos.
In the adult animal, DLL1 is critical for the development of marginal zone (MZ) B cells (86, 87). These cells are located at the interface between red pulp and white pulp of the spleen (88). DLL1 expressed by splenic red pulp endothelial cells is required for the development and maintenance of Notch2-expressing MZ B cells (89, 90). In order to test whether phosphorylation regulates DLL1-dependent processes postnatally, we analyzed the splenic B cell compartment of homozygous Dll1Dll1T638V,S693A,S696A mice compared to homozygous Dll1Dll1wtki mice by flow cytometry. Spleens from homozygous Dll1Dll1T638V,S693A,S696A mice displayed a composition of MZ B cells and conventional follicular B cells that was indistinguishable from spleens from Dll1Dll1wtki mice (Fig. 7), suggesting that phosphorylation of DLL1 is not required for the development and/or maintenance of splenic MZ B cells.
FIG 7.
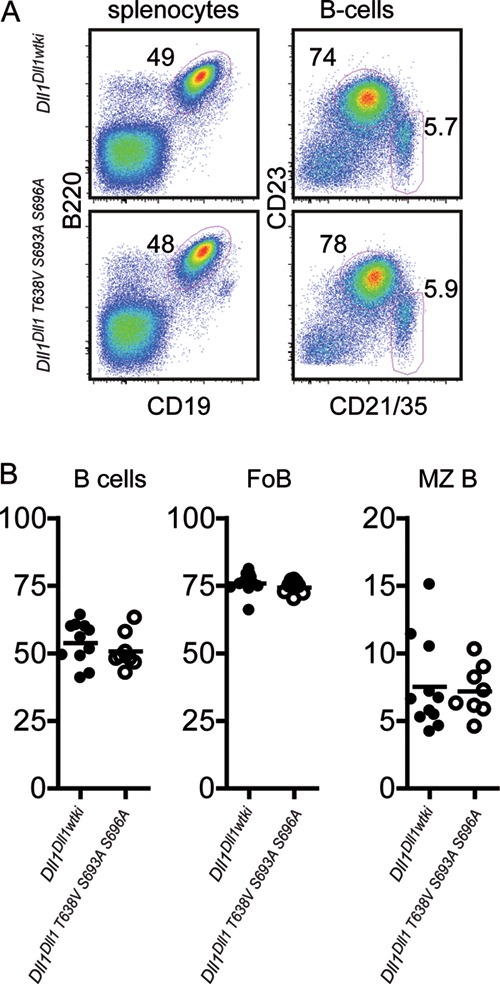
Phosphorylation of DLL1 is dispensable for the development of MZ B cells. (A) Splenocytes from Dll1Dll1wtki and Dll1Dll1,T638V,S693A,S696A mice were analyzed flow cytometrically for the expression of CD19, B220, CD23, and CD21/35. The right panels are electronically gated on CD19+ B220+ B cells. The data are representative for two independent experiments. Numbers indicate the percentages of the circled cell populations. (B) Cumulative analysis of results presented in panel A from two independent experiments. Each dot represents an individual mouse. Follicular (Fo) B cells are defined as CD23hi CD21/35lo. MZ B cells are defined as CD23lo CD21/35hi.
DISCUSSION
We have identified three S/T residues in the intracellular domain of the Notch ligand DLL1 that are phosphorylated in cell lines of various mammalian species. Antibodies specific for pS693 detected phosphorylated DLL1 in vivo, whereas anti-pT638 and anti-pS696 antibodies did not detect phosphorylated DLL1 in embryo lysates. This might indicate the absence of or a reduced phosphorylation at these residues in vivo at the analyzed stage and tissues. An alternative explanation could be that these antibodies are not sensitive enough to detect DLL1 phosphorylation at T638 and S696 in animal tissues, which is a likely possibility since anti-pT638 and anti-pS696 antibodies detected phosphorylated DLL1 less efficiently than the anti-pS693 antibodies also in overexpressing cell lines (see, for example, Fig. 3C). We thus favor the idea that DLL1 is phosphorylated at all three sites also in vivo but was not detected at all sites due to sensitivity differences of the antibodies.
Phosphorylation requires membrane association of DLL1 (Fig. 3B). Phosphorylation of myristoylated DICD also indicates that phosphorylation at the identified sites does not require interaction of DLL1 with Notch receptors in trans or other extracellular components. The efficient surface biotinylation of a DLL1 variant in which all three identified phosphosites were mutated (Fig. 3B) also indicated that phosphorylation at these sites is not essential for presentation on the cell surface, although the portion of mutant DLL1 on the cell surface was reduced. Our results suggest that phosphorylation of DICD occurs after DLL1 has reached the cell membrane. We cannot distinguish at present whether phosphorylated DLL1 at the cell surface represents newly synthesized protein that has reached the surface, DLL1 that has been endocytosed and recycled to the membrane, or DLL1 that has been endocytosed and is targeted for degradation.
Our analysis of DLL1 variants carrying mutations at individual phosphorylation sites or combinations thereof showed that phosphorylation of T638 and S693 occurred irrespective of mutations in the other sites. However, phosphorylation of S696 required the presence of S693, suggesting that phosphorylation at S693 is a prerequisite for phosphorylation at S696, and these phosphorylations occur sequentially. Consistent with S693 being part of a consensus sequence (RXRXXS/T) recognized by AKT/PKB (63, 64), inhibition of AKT/PKB caused a severe reduction of phosphorylation at S693 and at S696. This is consistent with loss of phosphorylation at S696 in the DLL1 S693A variant (Fig. 4A) and supports the notion that S693 and S696 are phosphorylated sequentially. In Xenopus, however, we detected phosphorylation only at S696 but not at S693. Thus, Xenopus DLL1 could be phosphorylated at S696 independently from S693. Alternatively, this could be a detection problem. Phosphorylated S693 in Xenopus DLL1 might not be recognized by the anti-pS693 antibodies, which appears likely due to three amino acid changes in Xenopus DLL1 compared to the peptide used for the generation of anti-pS693 of mouse (Fig. 4C). Thus, also XDLL1 might be phosphorylated sequentially at both S residues.
Mutation of the three identified phosphosites led to a longer half-life and increased overall abundance in cultured cells in vitro but attenuated the activity of DLL1 as a Notch ligand. Loss of phosphorylation caused alterations in various biochemical properties of DLL1, which might in part be interrelated and together could contribute to the reduced ability of phosphomutant DLL1 to activate Notch in the coculture assay. First, the relative amount of mutant protein on the cell surface was reduced to roughly 50% (Fig. 5D), which could be a plausible explanation for reduced ligand activity. However, because the steady-state level of full-length mutant protein was increased ∼4-fold, this is unlikely to solely account for the reduced capacity to activate Notch in the in vitro coculture assay. Second, DLL1 T638, S693, S696 was extracellularly cleaved more efficiently than wild-type DLL1. Cleavage in the extracellular domain has been associated with downregulation of Notch signaling (91, 92) and thus could contribute to the reduced activity of phosphomutant DLL1. Third, we observed that there is apparently increased endocytosis of phosphomutant DLL1 in CHO cells. Enhanced endocytosis might reduce cell surface levels and thereby reduce Notch activation. However, endocytosis of DLL1 (and other ligands) has been associated with the ability of ligands to activate Notch and not with downregulation of ligand activity (reviewed in references 32 to 36, 47, and 48). Consistent with this view, blocking DLL1 endocytosis correlated with increased DLL1 cleavage and reduced Notch activation (93). In contrast, in our experiments enhanced endocytosis correlated with increased DLL1 cleavage and reduced ligand activity, although the effects of the mutated phosphosites on endocytosis and DLL1 cleavage could be unrelated. Potentially, the loss of phosphorylation leads to increased endocytosis and abnormal recycling that alters the distribution of DLL1 on the cell surface or affects an unknown activation step of DLL1 such as posttranslational modification during endocytosis that is required for full ligand activity (reviewed in references 34 and 47). However, we cannot rule out that the altered amino acid sequence due to the exchange of the serine or threonine residues that are phosphorylated rather than the actual loss of phosphorylation underlies the observed changes in biochemical properties.
Surprisingly, despite the clearly reduced effectivity of DLL1 T638, S693, S696 to function as a Notch ligand in the in vitro assay, mice homozygous for mutations at all three phosphosites developed normally without any apparent defects in various processes that depend on DLL1 function during embryonic or B cell development. However, we cannot formally exclude that there are subtle differences or differences in specific cell populations that we did not detect and which do not have obviously recognizable consequences. Overall, our in vivo analyses indicate that phosphorylation of DLL1 is not essential for ligand function under physiological conditions. This suggests that the effects causing the observed reduced activity of phosphomutant DLL1 in vitro are less important or compensated in vivo or that the reduced activity that was found in the in vitro transactivation assay is still sufficient to activate Notch to functional levels under physiological conditions.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Redeker for the Flag-DICD expressing CHO cell line, Alain Israël for HeLaN1 cells, David Ish-Horowicz, Chris Kintner, and Domingos Henrique for X-Delta1 and chicken DLL1 constructs and cDNAs, Michele Calos for attP, attB, and integrase plasmids, Gerry Weinmaster for the tagged-Notch expression vector, and Matthias Gaestel and Michael Stauber for helpful discussions and critical comments on the manuscript.
This study was supported by Deutsche Forschungsgemeinschaft grant GO449/10 to A.G. and by funding from the Cluster of Excellence (From Regenerative Biology to Reconstructive Therapy).
Footnotes
Published ahead of print 21 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00965-13.
REFERENCES
- 1.Louvi A, Artavanis-Tsakonas S. 2012. Notch and disease: a growing field. Semin. Cell Dev. Biol. 23:473–480. 10.1016/j.semcdb.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch U, Radtke F. 2011. Mechanisms of T cell development and transformation. Annu. Rev. Cell Biol. 27:539–562. 10.1146/annurev-cellbio-092910-154008 [DOI] [PubMed] [Google Scholar]
- 3.Bolos V, Grego-Bessa J, de la Pompa JL. 2007. Notch signaling in development and cancer. Endocrine Rev. 28:339–363. 10.1210/er.2006-0046 [DOI] [PubMed] [Google Scholar]
- 4.Gridley T. 2007. Notch signaling in vascular development and physiology. Development 134:2709–2718. 10.1242/dev.004184 [DOI] [PubMed] [Google Scholar]
- 5.Yoon K, Gaiano N. 2005. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat. Neurosci. 8:709–715. 10.1038/nn1475 [DOI] [PubMed] [Google Scholar]
- 6.Artavanis-Tsakonas S, Rand MD, Lake RJ. 1999. Notch signaling: cell fate control and signal integration in development. Science 284:770–776. 10.1126/science.284.5415.770 [DOI] [PubMed] [Google Scholar]
- 7.Radtke F, Fasnacht N, Macdonald HR. 2010. Notch signaling in the immune system. Immunity 32:14–27. 10.1016/j.immuni.2010.01.004 [DOI] [PubMed] [Google Scholar]
- 8.Wharton KA, Johansen KM, Xu T, Artavanis-Tsakonas S. 1985. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 43:567–581. 10.1016/0092-8674(85)90229-6 [DOI] [PubMed] [Google Scholar]
- 9.Vässin H, Bremer KA, Knust E, Campos-Ortega JA. 1987. The neurogenic gene Delta of Drosophila melanogaster is expressed in neurogenic territories and encodes a putative transmembrane protein with EGF-like repeats. EMBO J. 6:3431–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas U, Speicher SA, Knust E. 1991. The Drosophila gene Serrate encodes an EGF-like transmembrane protein with a complex expression pattern in embryos and wing discs. Development 111:749–761 [DOI] [PubMed] [Google Scholar]
- 11.Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S. 1997. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell 90:281–291. 10.1016/S0092-8674(00)80336-0 [DOI] [PubMed] [Google Scholar]
- 12.Pan D, Rubin GM. 1997. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during drosophila and vertebrate neurogenesis. Cell 90:271–280. 10.1016/S0092-8674(00)80335-9 [DOI] [PubMed] [Google Scholar]
- 13.Fortini ME, Artavanis-Tsakonas S. 1994. The suppressor of hairless protein participates in notch receptor signaling. Cell 79:273–282. 10.1016/0092-8674(94)90196-1 [DOI] [PubMed] [Google Scholar]
- 14.Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israël A. 1995. Signaling downstream of activated mammalian Notch. Nature 377:355–358. 10.1038/377355a0 [DOI] [PubMed] [Google Scholar]
- 15.Jarriault S, Le Bail O, Hirsinger E, Pourquié O, Logeat F, Strong C, Brou C, Seidah N, Isra A. 1998. Delta-1 activation of notch-1 signaling results in HES-1 transactivation. Mol. Cell. Biol. 18:7423–7431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kopan R, Schroeter EH, Weintraub H, Nye JS. 1996. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc. Natl. Acad. Sci. U. S. A. 93:1683–1688. 10.1073/pnas.93.4.1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kidd S, Lieber T, Young MW. 1998. Ligand-induced cleavage and regulation of nuclear entry of Notch in Drosophila melanogaster embryos. Genes Dev. 12:3728–3740. 10.1101/gad.12.23.3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schroeter E, Kisslinger J, Kopan R. 1998. Notch-1 signaling requires ligand-induced proteolytic release of intracellular domain. Nature 393:382–386. 10.1038/30756 [DOI] [PubMed] [Google Scholar]
- 19.Struhl G, Adachi A. 1998. Nuclear access and action of notch in vivo. Cell 93:649–660. 10.1016/S0092-8674(00)81193-9 [DOI] [PubMed] [Google Scholar]
- 20.Six E, Ndiaye D, Laabi Y, Brou C, Gupta-Rossi N, Israel A, Logeat F. 2003. The Notch ligand Delta1 is sequentially cleaved by an ADAM protease and gamma-secretase. Proc. Natl. Acad. Sci. U. S. A. 100:7638–7643. 10.1073/pnas.1230693100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeuchi T, Sisodia S. 2003. The Notch ligands, Delta1 and Jagged2, are substrates for presenilin-dependent “gamma-secretase” cleavage. J. Biol. Chem. 278:7751–7754. 10.1074/jbc.C200711200 [DOI] [PubMed] [Google Scholar]
- 22.Bland CE, Kimberly P, Rand MD. 2003. Notch-induced proteolysis and nuclear localization of the Delta ligand. J. Biol. Chem. 278:13607–13610. 10.1074/jbc.C300016200 [DOI] [PubMed] [Google Scholar]
- 23.LaVoie MJ, Selkoe DJ. 2003. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J. Biol. Chem. 278:34427–34437. 10.1074/jbc.M302659200 [DOI] [PubMed] [Google Scholar]
- 24.Dyczynska E, Sun D, Yi H, Sehara-Fujisawa A, Blobel CP, Zolkiewska A. 2007. Proteolytic processing of delta-like 1 by ADAM proteases. J. Biol. Chem. 282:436–444. 10.1074/jbc.M605451200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moloney DJ, Shair LH, Lu FM, Xia J, Locke R, Matta KL, Haltiwanger RS. 2000. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J. Biol. Chem. 275:9604–9611. 10.1074/jbc.275.13.9604 [DOI] [PubMed] [Google Scholar]
- 26.Shao L, Moloney D, Haltiwanger RS. 2003. Fringe modifies O-fucose on mouse Notch1 at epidermal growth factor-like repeats within the ligand-binding site and the Abruptex region. J. Biol. Chem. 278:7775–7782. 10.1074/jbc.M212221200 [DOI] [PubMed] [Google Scholar]
- 27.Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, Vogt TF. 2000. Fringe is a glycosyltransferase that modifies Notch. Nature 406:369–375. 10.1038/35019000 [DOI] [PubMed] [Google Scholar]
- 28.Panin V, Papayannopoulos V, Wilson R, Irvine KD. 1997. Fringe modulates Notch-ligand interactions. Nature 387:908–912. 10.1038/43191 [DOI] [PubMed] [Google Scholar]
- 29.Hicks C, Johnston SH, diSibio G, Collazo A, Vogt TF, Weinmaster G. 2000. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat. Cell Biol. 2:515–520. 10.1038/35019553 [DOI] [PubMed] [Google Scholar]
- 30.Dale JK, Maroto M, Dequeant ML, Malapert P, McGrew M, Pourquie O. 2003. Periodic notch inhibition by lunatic fringe underlies the chick segmentation clock. Nature 421:275–278. 10.1038/nature01244 [DOI] [PubMed] [Google Scholar]
- 31.Morimoto M, Takahashi Y, Endo M, Saga Y. 2005. The Mesp2 transcription factor establishes segmental borders by suppressing Notch activity. Nature 435:354–359. 10.1038/nature03591 [DOI] [PubMed] [Google Scholar]
- 32.Bray S. 2006. Notch signaling: a simple pathway becomes complex. Nat. Rev. Mol. Cell. Biol. 7:678–689. 10.1038/nrm2009 [DOI] [PubMed] [Google Scholar]
- 33.Chitnis A. 2006. Why is delta endocytosis required for effective activation of notch? Dev. Dyn. 235:886–894. 10.1002/dvdy.20683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Borgne R. 2006. Regulation of Notch signalling by endocytosis and endosomal sorting. Curr. Opin. Cell Biol. 18:213–222. 10.1016/j.ceb.2006.02.011 [DOI] [PubMed] [Google Scholar]
- 35.Fortini ME, Bilder D. 2009. Endocytic regulation of Notch signaling. Curr. Opin. Genet. Dev. 19:323–328. 10.1016/j.gde.2009.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopan R, Ilagan MXG. 2009. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137:216–233. 10.1016/j.cell.2009.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kidd S, Baylies MK, Gasic GP, Young MW. 1989. Structure and distribution of the Notch protein in developing Drosophila. Genes Dev. 3:1113–1129. 10.1101/gad.3.8.1113 [DOI] [PubMed] [Google Scholar]
- 38.Redmond L, Oh SR, Hicks C, Weinmaster G, Ghosh A. 2000. Nuclear Notch1 signaling and the regulation of dendritic development. Nat. Neurosci. 3:30–40. 10.1038/71104 [DOI] [PubMed] [Google Scholar]
- 39.Shimizu K, Chiba S, Hosoya N, Kumano K, Saito T, Kurokawa M, Kanda Y, Hamada Y, Hirai H. 2000. Binding of Delta1, Jagged1, and Jagged2 to Notch2 rapidly induces cleavage, nuclear translocation, and hyperphosphorylation of Notch2. Mol. Cell. Biol. 20:6913–6922. 10.1128/MCB.20.18.6913-6922.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishitani T, Hirao T, Suzuki M, Isoda M, Ishitani S, Harigaya K, Kitagawa M, Matsumoto K, Itoh M. 2010. Nemo-like kinase suppresses Notch signaling by interfering with formation of the Notch active transcriptional complex. Nat. Cell Biol. 12:278–285. 10.1038/ncb2028 [DOI] [PubMed] [Google Scholar]
- 41.Espinosa L, Ingles-Esteve J, Aguilera C, Bigas A. 2003. Phosphorylation by glycogen synthase kinase-3 beta downregulates Notch activity, a link for Notch and Wnt pathways. J. Biol. Chem. 278:32227–32235. 10.1074/jbc.M304001200 [DOI] [PubMed] [Google Scholar]
- 42.Han X, Ju J-H, Shin I. 2012. Glycogen synthase kinase 3-β phosphorylates novel S/T-P-S/T domains in Notch1 intracellular domain and induces its nuclear localization. Biochem. Biophys. Res. Commun. 423:282–288. 10.1016/j.bbrc.2012.05.111 [DOI] [PubMed] [Google Scholar]
- 43.Foltz DR, Santiago MC, Berechid BE, Nye JS. 2002. Glycogen synthase kinase-3β modulates notch signaling and stability. Curr. Biol. 12:1006–1011. 10.1016/S0960-9822(02)00888-6 [DOI] [PubMed] [Google Scholar]
- 44.Ruel L, Bourouis M, Heitzler P, Pantesco V, Simpson P. 1993. Drosophila shaggy kinase and rat glycogen synthase kinase-3 have conserved activities and act downstream of Notch. Nature 362:557–560. 10.1038/362557a0 [DOI] [PubMed] [Google Scholar]
- 45.Panin V, Shao L, Lei L, Moloney D, Irvine KD, Haltiwanger RS. 2002. Notch ligands are substrates for protein O-fucosyltransferase-1 and Fringe. J. Biol. Chem. 277:29945–29952. 10.1074/jbc.M204445200 [DOI] [PubMed] [Google Scholar]
- 46.Okajima T, Irvine KD. 2002. Regulation of notch signaling by o-linked fucose. Cell 111:893–904. 10.1016/S0092-8674(02)01114-5 [DOI] [PubMed] [Google Scholar]
- 47.Le Bras S, Loyer N, Le Borgne R. 2011. The multiple facets of ubiquitination in the regulation of notch signaling pathway. Traffic 12:149–161. 10.1111/j.1600-0854.2010.01126.x [DOI] [PubMed] [Google Scholar]
- 48.D'Souza B, Miyamoto A, Weinmaster G. 2008. The many facets of Notch ligands. Oncogene 27:5148–5167. 10.1038/onc.2008.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zolkiewska A. 2008. ADAM proteases: ligand processing and modulation of the Notch pathway. Cell. Mol. Life Sci. 65:2056–2068. 10.1007/s00018-008-7586-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Redeker C, Schuster-Gossler K, Kremmer E, Gossler A. 2013. Normal development in mice overexpressing the intracellular domain of DLL1 argues against reverse signaling by DLL1 in vivo. PLoS One 8:e79050. 10.1371/journal.pone.0079050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keravala A, Calos MP. 2008. Site-specific chromosomal integration mediated by phiC31 integrase. Methods Mol. Biol. 435:165–173. 10.1007/978-1-59745-232-8_12 [DOI] [PubMed] [Google Scholar]
- 52.Geffers I, Serth K, Chapman G, Jaekel R, Schuster-Gossler K, Cordes R, Sparrow DB, Kremmer E, Dunwoodie SL, Klein T, Gossler A. 2007. Divergent functions and distinct localization of the Notch ligands DLL1 and DLL3 in vivo. J. Cell Biol. 178:465–476. 10.1083/jcb.200702009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minoguchi S, Taniguchi Y, Kato H, Okazaki T, Strobl LJ, Zimber-Strobl U, Bornkamm GW, Honjo T. 1997. RBP-L, a transcription factor related to RBP-Jκ. Mol. Cell. Biol. 17:2679–2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1:2856–2860. 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- 55.Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75:663–670. 10.1021/ac026117i [DOI] [PubMed] [Google Scholar]
- 56.Konno D, Ko J-A, Usui S, Hori K, Maruoka H, Inui M, Fujikado T, Tano Y, Suzuki T, Tohyama K, Sobue K. 2002. The postsynaptic density and dendritic raft localization of PSD-Zip70, which contains an N-myristoylation sequence and leucine-zipper motifs. J. Cell Sci. 115:4695–4706. 10.1242/jcs.00127 [DOI] [PubMed] [Google Scholar]
- 57.Heuss SF, Ndiaye-Lobry D, Six EM, Israel A, Logeat F. 2008. The intracellular region of Notch ligands Dll1 and Dll3 regulates their trafficking and signaling activity. Proc. Natl. Acad. Sci. U. S. A. 105:11212–11217. 10.1073/pnas.0800695105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilkinson DG. 1992. In situ hybridization: a practical approach, p 75–83 Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 59.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5:749–757. 10.1074/mcp.T500024-MCP200 [DOI] [PubMed] [Google Scholar]
- 60.Jonas BA, Privalsky ML. 2004. SMRT and N-CoR corepressors are regulated by distinct kinase signaling pathways. J. Biol. Chem. 279:54676–54686. 10.1074/jbc.M410128200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beckers J, Clark A, Wünsch K, Hrabe De Angelis M, Gossler A. 1999. Expression of the mouse Delta1 gene during organogenesis and fetal development. Mech. Dev. 84:165–168. 10.1016/S0925-4773(99)00065-9 [DOI] [PubMed] [Google Scholar]
- 62.Cheng H-T, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, Kopan R. 2007. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 134:801–811. 10.1242/dev.02773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. 1996. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 399:333–338. 10.1016/S0014-5793(96)01370-1 [DOI] [PubMed] [Google Scholar]
- 64.Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129:1261–1274. 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fayard E, Tintignac LA, Baudry A, Hemmings BA. 2005. Protein kinase B/Akt at a glance. J. Cell Sci. 118:5675–5678. 10.1242/jcs.02724 [DOI] [PubMed] [Google Scholar]
- 66.Flotow H, Roach PJ. 1991. Role of acidic residues as substrate determinants for casein kinase I. J. Biol. Chem. 266:3724–3727 [PubMed] [Google Scholar]
- 67.Rena G, Woods YL, Prescott AR, Peggie M, Unterman TG, Williams MR, Cohen P. 2002. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 21:2263–2271. 10.1093/emboj/21.9.2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Levy DE, Lapierre F, Liang W, Ye W, Lange CW, Li X, Grobelny D, Casabonne M, Tyrrell D, Holme K, Nadzan A, Galardy RE. 1998. Matrix metalloproteinase inhibitors: a structure-activity study. J. Med. Chem. 41:199–223. 10.1021/jm970494j [DOI] [PubMed] [Google Scholar]
- 69.Mohler KM, Sleath PR, Fitzner JN, Cerretti DP, Alderson M, Kerwar SS, Torrance DS, Otten-Evans C, Greenstreet T, Weerawarna K. 1994. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature 370:218–220. 10.1038/370218a0 [DOI] [PubMed] [Google Scholar]
- 70.Le Borgne R, Bardin A, Schweisguth F. 2005. The roles of receptor and ligand endocytosis in regulating Notch signaling. Development 132:1751–1762. 10.1242/dev.01789 [DOI] [PubMed] [Google Scholar]
- 71.Nichols JT, Miyamoto A, Olsen SL, D'Souza B, Yao C, Weinmaster G. 2007. DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J. Cell Biol. 176:445–458. 10.1083/jcb.200609014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weinmaster G, Fischer JA. 2011. Notch ligand ubiquitylation: what is it good for? Dev. Cell 21:134–144. 10.1016/j.devcel.2011.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hunter T. 2007. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28:730–738. 10.1016/j.molcel.2007.11.019 [DOI] [PubMed] [Google Scholar]
- 74.Zhang L, Gallagher PJ. 2009. Mind bomb 1 regulation of cFLIP interactions. Am. J. Physiol. Cell Physiol. 297:C1275–C1283. 10.1152/ajpcell.00214.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koo B-K, Yoon M-J, Yoon K-J, Im S-K, Kim Y-Y, Kim C-H, Suh P-G, Jan YN, Kong Y-Y. 2007. An obligatory role of mind bomb-1 in notch signaling of mammalian development. PLoS One 2:e1221. 10.1371/journal.pone.0001221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koo B-K, Lim H-S, Song R, Yoon M-J, Yoon K-J, Moon J-S, Kim Y-W, Kwon M-C, Yoo K-W, Kong M-P, Lee J, Chitnis AB, Kim C-H, Kong Y-Y. 2005. Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development 132:3459–3470. 10.1242/dev.01922 [DOI] [PubMed] [Google Scholar]
- 77.Schuster-Gossler K, Cordes R, Gossler A. 2007. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc. Natl. Acad. Sci. U. S. A. 104:537–542. 10.1073/pnas.0608281104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hrabe De Angelis M, McIntyre J, Gossler A. 1997. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature 386:717–721. 10.1038/386717a0 [DOI] [PubMed] [Google Scholar]
- 79.Rocha SF, Lopes SS, Gossler A, Henrique D. 2009. Dll1 and Dll4 function sequentially in the retina and pV2 domain of the spinal cord to regulate neurogenesis and create cell diversity. Dev. Biol. 328:54–65. 10.1016/j.ydbio.2009.01.011 [DOI] [PubMed] [Google Scholar]
- 80.Ramos C, Rocha S, Gaspar C, Henrique D. 2010. Two Notch ligands, Dll1 and Jag1, are differently restricted in their range of action to control neurogenesis in the mammalian spinal cord. PLoS One 5:e15515. 10.1371/journal.pone.0015515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feller J, Schneider A, Schuster-Gossler K, Gossler A. 2008. Noncyclic Notch activity in the presomitic mesoderm demonstrates uncoupling of somite compartmentalization and boundary formation. Genes Dev. 22:2166–2171. 10.1101/gad.480408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Neidhardt LM, Kispert A, Herrmann BG. 1997. A mouse gene of the paired-related homeobox class expressed in the caudal somite compartment and in the developing vertebral column, kidney and nervous system. Dev. Genes Evol. 207:330–339. 10.1007/s004270050120 [DOI] [PubMed] [Google Scholar]
- 83.Vasyutina E, Lenhard DC, Wende H, Erdmann B, Epstein JA, Birchmeier C. 2007. RBP-J (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc. Natl. Acad. Sci. U. S. A. 104:4443–4448. 10.1073/pnas.0610647104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arnold HH, Braun T. 2000. Genetics of muscle determination and development. Curr. Top. Dev. Biol. 48:129–164 [DOI] [PubMed] [Google Scholar]
- 85.Ma Q, Chen Z, del Barco Barrantes I, de la Pompa JL, Anderson DJ. 1998. neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron 20:469–482. 10.1016/S0896-6273(00)80988-5 [DOI] [PubMed] [Google Scholar]
- 86.Hozumi K, Negishi N, Suzuki D, Abe N, Sotomaru Y, Tamaoki N, Mailhos C, Ish-Horowicz D, Habu S, Owen MJ. 2004. Delta-like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nat. Immunol. 5:638–644. 10.1038/ni1075 [DOI] [PubMed] [Google Scholar]
- 87.Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, Yamaguchi T, Yamamoto G, Seo S, Kumano K, Nakagami-Yamaguchi E, Hamada Y, Aizawa S, Hirai H. 2003. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity 18:675–685. 10.1016/S1074-7613(03)00111-0 [DOI] [PubMed] [Google Scholar]
- 88.Pillai S, Cariappa A, Moran ST. 2005. Marginal zone B cells. Annu. Rev. Immunol. 23:161–196. 10.1146/annurev.immunol.23.021704.115728 [DOI] [PubMed] [Google Scholar]
- 89.Moriyama Y, Sekine C, Koyanagi A, Koyama N, Ogata H, Chiba S, Hirose S, Okumura K, Yagita H. 2008. Delta-like 1 is essential for the maintenance of marginal zone B cells in normal mice but not in autoimmune mice. Int. Immunol. 20:763–773. 10.1093/intimm/dxn034 [DOI] [PubMed] [Google Scholar]
- 90.Tan JB, Xu K, Cretegny K, Visan I, Yuan JS, Egan SE, Guidos CJ. 2009. Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches. Immunity 30:254–263. 10.1016/j.immuni.2008.12.016 [DOI] [PubMed] [Google Scholar]
- 91.Sun D, Li H, Zolkiewska A. 2008. The role of Delta-like 1 shedding in muscle cell self-renewal and differentiation. J. Cell Sci. 121:3815–3823. 10.1242/jcs.035493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hicks C, Ladi E, Lindsell C, Hsieh JJ-D, Hayward SD, Collazo A, Weinmaster G. 2002. A secreted Delta1-Fc fusion protein functions both as an activator and inhibitor of Notch1 signaling. J. Neurosci. Res. 68:655–667. 10.1002/jnr.10263 [DOI] [PubMed] [Google Scholar]
- 93.Heuss SF, Tarantino N, Fantini J, Ndiaye-Lobry D, Moretti J, Israel A, Logeat F. 2013. A glycosphingolipid binding domain controls trafficking and activity of the mammalian notch ligand Delta-like 1. PLoS One 8:e74392. 10.1371/journal.pone.0074392 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.