

ABSTRACT
Hepatitis C virus (HCV) infects 180 million people worldwide and is a leading cause of liver diseases such as fibrosis, cirrhosis, and hepatocellular carcinoma. It has been shown that HCV can spread to naive cells using two distinct entry mechanisms, “cell-free” entry of infectious extracellular virions that have been released by infected cells and direct “cell-to-cell” transmission. Here, we examined host cell requirements for HCV spread and found that the cholesterol uptake receptor NPC1L1, which we recently identified as being an antiviral target involved in HCV cell-free entry/spread, is also required for the cell-to-cell spread. In contrast, the very low density lipoprotein (VLDL) pathway, which is required for the secretion of cell-free infectious virus and thus has been identified as an antiviral target for blocking cell-free virus secretion/spread, is not required for cell-to-cell spread. Noting that HCV cell-free and cell-to-cell spread share some common factors but not others, we tested the therapeutic implications of these observations and demonstrate that inhibitors that target cell factors required for both forms of HCV spread exhibit synergy when used in combination with interferon (a representative inhibitor of intracellular HCV production), while inhibitors that block only cell-free spread do not. This provides insight into the mechanistic basis of synergy between interferon and HCV entry inhibitors and highlights the broader, previously unappreciated impact blocking HCV cell-to-cell spread can have on the efficacy of HCV combination therapies.
IMPORTANCE HCV can spread to naive cells using distinct mechanisms: “cell-free” entry of extracellular virus and direct “cell-to-cell” transmission. Herein, we identify the host cell HCV entry factor NPC1L1 as also being required for HCV cell-to-cell spread, while showing that the VLDL pathway, which is required for the secretion of cell-free infectious virus, is not required for cell-to-cell spread. While both these host factors are considered viable antiviral targets, we demonstrate that only inhibitors that block factors required for both forms of HCV entry/spread (i.e., NPC1L1) exhibit synergy when used in combination with interferon, while inhibitors that block factors required only for cell-free spread (i.e., VLDL pathway components) do not. Thus, this study advances our understanding of HCV cell-to-cell spread, provides mechanistic insight into the basis of drug synergy, and highlights inhibition of HCV spread as a previously unappreciated consideration in HCV therapy design.
INTRODUCTION
Hepatitis C virus (HCV) is a leading cause of liver disease (1, 2). Following exposure and an acute, usually asymptomatic infection, only 20% of individuals clear the virus, while up to 80% develop a chronic infection. Over an extended period of decades, infected patients are at high risk of developing severe liver disease which may include steatosis, fibrosis, cirrhosis, and hepatocellular carcinoma (3). In fact, HCV is the leading cause of liver transplantation in the United States (4, 5). Until recently, interferon (IFN) and ribavirin combination therapy was the only clinically approved treatment option for HCV infection, yet it is ineffective in up to 50% of patients. Moreover, treatment with interferon can have severe side effects, including flu-like symptoms, fatigue, and psychiatric manifestations. In 2012, the first small-molecule drugs targeting the NS3/4A protease were approved by the FDA to treat HCV genotype 1 infections. However, monotherapy with these direct-acting antivirals (DAAs) leads to the rapid emergence of resistance mutants (6, 7), and therefore these virus-targeted inhibitors are currently approved only for use in combination with interferon and ribavirin, which increases the barrier to escape. While promising anti-HCV drugs are in the pipeline, the development of pan-effective, well-tolerated, low-cost, interferon-free treatment combinations remains an important goal.
Because entry into permissive cells is the first essential step in establishing productive infection, viral entry is considered a promising antiviral target. However, after genome replication and assembly of progeny virus particles in the initially infected cell, HCV infection can spread to infect additional cells by one of two different entry routes: “cell-free” entry of infectious extracellular virions that have been released by infected cells and direct “cell-to-cell” transmission. While long-range dissemination of infection is facilitated by the secretion of cell-free virus particles from infected cells, which can travel through the body and enter into host cells that are not necessarily contiguous, cell-to-cell spread has the advantage of allowing the virus to rapidly enter neighboring cells while being shielded from neutralizing host antibodies.
HCV utilizes multiple host molecules for initial cell-free entry into cells. Glycosaminoglycans (GAGs) (8, 9), liver/lymph node-specific intercellular adhesion molecule 3-grabbing integrin (L-SIGN) (10, 11), and the low-density lipoprotein receptor (LDLR) (12, 13) have been implicated in preliminary attachment followed by a cascade of additional host cell factors, including the scavenger receptor B type 1 (SR-B1) (14), CD81 (15), claudin-1 (CLDN1) (16), occludin (OLCN) (17, 18), and the Niemann-Pick C1-like 1 (NPC1L1) cholesterol receptor (19). The requirements for subsequent viral cell-to-cell spread are also under investigation, and results thus far suggest that SR-B1, CLDN1, and OCLN are important for cell-to-cell spread (20). The involvement of CD81 is less clear-cut, with reports showing data in support of, as well as against, the requirement for CD81 in cell-to-cell spread (20–22). In addition, epidermal growth factor receptor (EGFR) and EphA2 were identified to be HCV entry cofactors, required for both cell-free entry and cell-to-cell spread (23). More recently, we also demonstrated that the transferrin receptor 1 (TfR1) is involved in HCV cell-free entry but less involved in cell-to-cell spread (24). Thus, while some factors participate in both mechanisms of HCV spread, some factors appear to be unique to one process or the other.
For this reason, we examined the requirements for different known cellular antiviral targets in HCV cell-to-cell spread with a focus on how this may impact therapeutic strategies. Importantly, we show that HCV cell-to-cell spread is dependent on the newly identified HCV entry factor NPC1L1 and thus efficiently blocked by ezetimibe, the FDA-approved drug that specifically inhibits internalization of NPC1L1 (25, 26). In contrast, we show that disruption of the very low density lipoprotein (VLDL) pathway by naringenin or small interfering RNA (siRNA) inhibition has no effect on cell-to-cell spread, suggesting that intracellular virus particles are capable of spreading cell to cell prior to maturation via the VLDL pathway, a pathway which is required for infectious viral particle secretion into the extracellular milieu and thus is required for cell-free virus spread (27, 28). We tested the therapeutic implications of these findings and found that inhibitors that target cellular factors required for both forms of HCV spread exhibit synergy when used in combination with interferon, while inhibitors that block only cell-free spread do not.
MATERIALS AND METHODS
Cells.
Huh7 cells (29) (also known as Huh7/scr cells [30, 31] and Huh7-1 cells [32] in other manuscripts) were obtained from Francis Chisari (The Scripps Research Institute, La Jolla, CA). Cells were cultured in complete Dulbecco's modified Eagle's medium (cDMEM) (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (HyClone), 100 units/ml penicillin, 100 mg/ml streptomycin, and 2 mM l-glutamine (Gibco Invitrogen, Carlsbad, CA). Notably, for the majority of the spread focus assays, we utilized nondividing Huh7 cell cultures as previously described in detail (33, 34) in order to avoid issues of cell division during the assay. Briefly, Huh7 cells were plated in collagen-coated 96-well plates and allowed to reach confluence, at which point the medium was replaced with medium containing 1% dimethyl sulfoxide (DMSO). The cells were then incubated for 20 days, with medium changes every other day. During this period, the cells remain metabolically active but stop dividing and exhibit a more differentiated state, as indicated by the upregulation of hepatocyte-specific markers (34) and induction of phase I and phase II drug metabolism (35).
Viruses.
The plasmid containing the full-length HCV JFH-1 genome (pUC-JFH1) was provided by Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) (36–38). Intergenotypic chimeric virus constructs containing the region encoding the core, E1, E2, p7, and partial/complete NS2 of each genotype substituted in a JFH-1 backbone were provided by Jens Bukh (Copenhagen University Hospital, Denmark) (39). These included GT1a H77C/JFH1 (40), GT1b HC-JC4/JFH1, GT2b HC-J8/JFH1 (39), GT4a ED43/JFH1 (40), GT5a SA13/JFH1 (41), GT6a HK6a/JFH1, and GT7a QC69/JFH1 (39). Protocols for HCV RNA in vitro transcription, HCV RNA electroporation, and propagation of HCV cell culture-derived (HCVcc) viral stocks have been described in detail elsewhere (42). Briefly, in vitro transcribed HCV genomic RNA was electroporated into Huh7 cells. Medium containing the virus was collected from the cells, and virus stocks were prepared by amplification in naive Huh7 cells. Infectious virus units were quantified by limiting dilution titer assays (42, 43) followed by immunostaining of HCV-positive foci as described below.
Reagents and antibodies.
DMSO stocks of naringenin (Sigma, St. Louis, MO), ezetimibe (Sequoia Research Products), the HCV protease inhibitor ITMN 5537 (Intermune), and the TfR1 inhibitor ferristatin (NSC306711; National Cancer Institute) were diluted in complete medium to the indicated concentrations. Human alpha interferon (IFN-α; PBL Biomedical Laboratories, Piscataway, NJ) was diluted in complete medium to 100 units/ml. Rabbit anti-human CLDN1 polyclonal antibody from AbCam (Cambridge, MA) and the rabbit anti-human NPC1L1 polyclonal antibodies targeting extracellular loops 1 and 2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) or Cayman Chemicals (Ann Arbor, MI), respectively. The human anti-HCV E2 glycoprotein monoclonal antibody (MAb AR3A or C1) from Dennis Burton and Mansun Law (The Scripps Research Institute, La Jolla, CA) and the mouse anti-HCV NS5A 9E10 monoclonal antibody from Charles Rice (Rockefeller University, New York, NY) have both been previously described (29, 44, 45). The horseradish peroxidase (HRP)-conjugated anti-human, anti-mouse, and anti-rabbit secondary antibodies were purchased from Pierce (Rockford, IL). Negative-control mouse and rabbit IgG antibodies were obtained from Santa Cruz Biotechnology. The cell-tracking fluorescent probe 5-chloromethylfluorescein diacetate (CMFDA) was purchased from Invitrogen and used to label live Huh7 cell (5 μM) as described by Krieger et al. (46). Hoechst 33342 was purchased from Sigma Chemicals (St. Louis, MO).
Cell-to-cell spread focus size assay.
Confluent monolayers of cells were infected with the indicated focus-forming units (FFU) of HCVcc per well. After incubation for 16 h, the inoculum was removed, cells were rinsed with phosphate-buffered saline (PBS), and medium containing 10 μg/ml anti-E2 was added to neutralize cell-free virus infection. This concentration of anti-E2 was chosen based on a previous publication showing effective neutralization in analogous spread assays (22) and our own confirmatory titration analysis (data not shown). Importantly, to ensure that all secreted cell-free virus was blocked by anti-E2 during the entire course of the assay, the medium from the experimental wells was also harvested at the end of each assay for titration on naive Huh7 cell monolayers to confirm continued neutralization (Fig. 1D). Additional indicated treatments were performed in parallel with the anti-E2 for the duration of the experiment to assess the effect of different host factors on HCV cell-to-cell spread. For antibody inhibition experiments, rabbit polyclonal anti-NPC1L1 (Santa Cruz Biotechnology) was used at 36 μg/ml, rabbit polyclonal anti-claudin-1 was used at 30 μg/ml, and control rabbit anti-IgG (Becton, Dickinson) was used at 36 μg/ml, in a total volume of 100 μl. To eliminate the effects of cell division on focus size, where possible, spread assays were performed or repeated in the nondividing cell culture system described above; however, for antibody-blocking experiments, the assay was performed only in growing Huh7 cells, as the nongrowing cultures contain considerably more cells and thus require a proportionately larger amount of antibody, resulting in toxicity due to the increased amount of sodium azide. To determine the degree of cell division during these growing cell assays, cell counts from triplicate wells at the time of infection and at 72 h postinfection (hpi), the time of fixing, were taken and found to be 12,000 and 15,000 cells/well, respectively. At the same time, the remaining wells were fixed and stained to detect HCV-positive foci. The number of HCV-positive cells per focus was used as readout of HCV cell-to-cell spread. Notably, based on the fact that the number of cells in each well increased less than a fold during the course of the focus assay, we concluded that cell division alone could account for not only 1 cell focus, but also 2 cell foci.
FIG 1.

NPC1L1 is required for HCV cell-to-cell spread. Confluent Huh7 cells were infected with 100 FFU JFH-1 HCVcc. At 16 hpi, neutralizing antibody against HCV E2 alone or together with antibodies specific for claudin-1 or NPC1L1 were added to the cultures as indicated. At 72 hpi, cells were fixed and stained for HCV E2 to visualize HCV-positive foci as well as the fluorescent dye Hoechst 33342 to visualize cell nuclei. (A) Representative HCV-positive foci formed in the presence of antibodies to HCV-E2 alone or HCV-E2 plus control IgG, anti-claudin-1, or anti-NPC1L1 were photographed using a Nikon Eclipse TE2000U with a 20× objective lens. The left panel shows HCV-positive foci; the right panel shows the nuclei of cells in the same fields. (B) Total number of foci in each well. Results are the averages of foci counted ± standard deviations (SD) in triplicate wells. (C) Quantification of cell-to-cell spread. The number of cells in each focus was counted. Foci were scored as either 1 or 2 cells in size (no spread) or ≥3 cells (exhibiting cell-to-cell spread). The percentage of foci that were 1 or 2 cells in size is graphed. Results are averaged from triplicate wells ± SD. Significant differences relative to anti-E2 controls are indicated (*, P < 0.05; **, P < 0.001 by analysis of variance [ANOVA]). Results are representative of three independent experiments. (D) Confirmation of anti-E2 neutralization. Culture medium was harvested at 72 hpi from experimental wells incubated with or without anti-HCV E2 in the cell-to-cell focus spread assay and titrated on naive Huh7 cells to screen for the presence of nonneutralized infectious virus (UT [untreated]). Results are the averages of foci counted in three wells; error bars indicate ±SD from the mean.
Immunostaining of HCV foci.
Immunostaining of HCV E2-positive foci has been previously described (42). Briefly, fixed cells were first incubated with 1× PBS containing 0.3% (vol/vol) hydrogen peroxide (Fisher, Fairlawn, NJ) to block endogenous peroxidase. Following three rinses with 1× PBS, cells were blocked for 1 h with 1× PBS containing 0.5% (vol/vol) Triton X-100 (Fisher), 3% (wt/vol) bovine serum albumin (BSA) (Sigma), and 10% (vol/vol) FBS. The primary human monoclonal HCV E2 antibody C1 was diluted to 2.3 μg/ml in 1× PBS containing 0.5% (vol/vol) Triton X-100 and 3% (wt/vol) BSA and incubated with cells for 1 h at room temperature. Bound anti-E2 was subsequently detected by 1 h of incubation with a 1:1,000 dilution of an HRP-conjugated anti-human antibody (Pierce) followed by a 30-min incubation with an AEC detection substrate (BD Biosciences). Alternatively, the NS5A protein was detected by incubation at room temperature with 1× PBS containing 0.5% (vol/vol) Triton X-100 and 3% (wt/vol) BSA and a 1:500 dilution of the monoclonal NS5A antibody 9E10, followed by 1 h of incubation with a 1:500 dilution of an HRP-conjugated anti-mouse antibody (Vector Laboratories, California) and subsequent detection with AEC substrate (BD Biosciences). Cells were washed with distilled water (dH2O), and foci were quantified and photographed using a Nikon TE2000U microscope (Nikon Instruments).
RNA interference (RNAi) knockdown experiments.
Pools of 3 small interfering RNAs (siRNAs) targeting PI4K, ApoB, ApoE, and MTP were purchased from Ambion (Austin, TX). CLDN1-specific siRNAs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and control siRNAs against enhanced green fluorescent protein (EGFP) were purchased from Qiagen (Valencia, CA). Huh7 cells were reverse transfected using Lipofectamine RNAiMax transfection reagent as per the manufacturer's instructions. Specifically, transfection mix containing 1 μl RNAiMax and 25 nM siRNA in Opti-MEM (Invitrogen) was incubated for 20 min at room temperature. A flask of Huh7 cells were trypsinized, and 10,000 cells were then reseeded with the transfection mix in 96-well BioCoat plates. At 24 h posttransfection, 200 μl of fresh cDMEM was added to each well, and at the indicated times posttransfection cultures were inoculated with 100 FFU of JFH-1 HCVcc per well. To assess RNA knockdown, total cellular RNA was harvested in 1× nucleic acid purification lysis solution (Applied Biosystems) from duplicate parallel wells at various times posttransfection for quantitative reverse transcription-PCR (qRT-PCR) analysis.
Inhibitor kinetic experiments.
Huh7 cells were infected with JFH-1 HCVcc and allowed to reach steady-state infection levels. HCV-infected cells were seeded in a 96-well plate. The next day, the cells were treated with the following alone and in combination as indicated: medium alone, 30 μM ezetimibe, 100 units/ml IFN-α, 50 μM ferristatin, and 200 μM naringenin. Medium and drugs were replaced every 24 h. At 24, 48, 72, or 96 h posttreatment, total cellular RNA was harvested in 1× nucleic acid purification lysis solution (Applied Biosystems) from triplicate wells for qRT-PCR analysis.
RNA isolation and qRT-PCR.
Total intracellular RNA was purified using an ABI PRISM 6100 Nucleic Acid PrepStation (Applied Biosystems), as per the manufacturer's instructions. Purified RNA was used for cDNA synthesis using TaqMan reverse transcription reagents (Applied Biosystems), followed by SYBR green qRT-PCR (Roche Diagnostics, Indianapolis, IN) using Applied Biosystems 7300 real-time thermocyclers (Applied Biosystems). Thermal cycling consisted of an initial 10-min denaturation step at 95°C followed by 40 cycles of denaturation (15 s at 95°C) and annealing/extension (1 min at 60°C). HCV RNA was quantified relative to standard curves comprised of serial dilutions of the pUC-JFH-1 plasmid and normalized to cellular GAPDH, while ApoB, ApoE, MTP, or CLDN1 RNA levels were determined by the threshold cycle (ΔΔCT) method. The PCR primers used to amplify each were as follows: HCV (47), 5′-GCC TAG CCA TGG CGT TAG TA −3′ (sense) and 5′-CTC CCG GGG CACTCG CAA GC-3′ (antisense); human GAPDH (29), 5′-GAA GGT GAA GGT CGG AGT C-3′ (sense) and 5′-GAA GAT GGT GAT GGG ATT TC-3′ (antisense); human ApoB, 5′-TCA TTT CTG CCC TCC TGG TT-3′ (sense) and 5′-ACA TTG CCC TTC CTC GTC TT-3′ (antisense); human ApoE, 5′ GTT CTG TGG GCT GCG TTG −3′ (sense) and 5′-CTC TGT CTC CAC CGC TTG CT-3′ (antisense); human MTP, 5′-GGA GCT CTG GCG GTC-3′ (sense) and 5′-CTT CTT TCT TCG CAG TCC TG-3′ (antisense); human claudin-1, 5′-GTG GAG GAT TTA CTC CTA TGC CG-3′ (sense) and 5′ ATC AAG GCA CGG GTT GCT T −3′ (antisense).
RESULTS
Antibody blocking of NPC1L1 prevents HCV cell-to-cell spread.
Having recently identified the cholesterol uptake receptor NPC1L1 as a cellular factor required for cell-free HCV entry (19), we examined the dependence of HCV cell-to-cell transmission on NPC1L1. We used a previously established antibody-blocking approach (22), in which a neutralizing antibody to the HCV E2 glycoprotein was used to block cell-free virus spread while the role of various host cell receptors in cell-to-cell spread was determined using specific blocking antibodies. Specifically, confluent Huh7 cells were inoculated with 100 FFU JFH-1 HCVcc per well for 16 h to allow time for sufficient amounts of initial viral entry (48). Cells were then washed, and cell-free virus spread was blocked by the addition of anti-E2 at a concentration which neutralizes extracellular virus infection of cells (22). Under these conditions, cultures were cotreated with antibodies to NPC1L1 to determine if blocking of NPC1L1 prevents cell-to-cell spread. Additional, parallel cultures were cotreated with IgG or anti-CLDN1 as negative and positive controls for inhibition of HCV cell-to-cell spread, respectively. At 72 hpi, cell monolayers were fixed and immunostained for HCV E2-positive foci and counterstained with Hoechst 33342 to observe individual nuclei (Fig. 1A). The number of total foci observed was slightly reduced when factors involved in initial cell-free infection were blocked (Fig. 1B), likely because some HCV infection initiation can continue up to 24 h after inoculation in vitro (48). However, differences in HCV cell-to-cell spread were assessed by counting the number of cells per foci (Fig. 1C). Because the number of cells counted in parallel representative wells increased less than a fold during the course of the focus assay, cell division alone should account for only 1 or 2 cell foci. Consistent with this, the spread inhibiting anti-CLDN1 treatment resulted in more than 90% of foci being restricted to only 1 or 2 cells in size (Fig. 1C). Hence, we scored 1 or 2 cell foci as infections that had not spread cell to cell. Foci containing 3 or more cells were scored as infections that had exhibited some degree of cell-to-cell spread and are referred to as “multicellular” foci. As expected, most of the foci formed in the cultures treated with either the irrelevant IgG control or the HCV E2 antibody alone contained 3 or more HCV-positive cells (Fig. 1C). However, HCV cell-to-cell spread was inhibited to a similar extent in the presence of antibody to NPC1L1 compared to that of the anti-CLDN1-treated cultures (Fig. 1C). Finally, to confirm complete anti-E2 neutralization of extracellular virus during the course of the experiment, the titer of the culture medium was determined on naive cells (Fig. 1D).
Blocking NPC1L1 internalization with ezetimibe prevents HCV cell-to-cell spread.
We recently reported that the drug ezetimibe, which blocks NPC1L1 internalization, inhibits HCV infection initiation in cultured cells and in chimeric mice transplanted with human hepatocytes (19). To confirm the role of NPC1L1 in HCV spread and verify that this FDA-approved drug inhibits HCV cell-to-cell spread, confluent Huh7 cells were inoculated with 100 FFU JFH-1 HCVcc per well for 16 h to allow cell-free infection to initiate in single cells, before being washed and then incubated with increasing concentrations of ezetimibe. Once again, extracellular HCV entry was blocked by the addition of anti-E2 to neutralize cell-free virus in all cultures. As we had previously observed (19), the addition of ezetimibe postinoculation did not alter the initial infection of naive Huh7 cells as indicated by the equivalent number of independent infection initiation events (i.e., focus number) observed in ezetimibe-treated compared to mock-treated controls (Fig. 2A). However, the foci stained at 72 hpi exhibited a distinct difference in size and were again scored as 1- or 2-cell “no spread” foci or ≥3-cell “multicellular” foci generated by cell-to-cell spread (Fig. 2B). Consistent with the conclusion that NPC1L1 is required for HCV cell-to-cell spread, we observed a dose-dependent inhibition in the size of the E2-positive foci formed in the presence of ezetimibe, with the 30 μM dose of ezetimibe resulting in 84% of foci being restricted to 1 or 2 cells compared with only 13% of cell foci being 1 or 2 cells in size in the mock-treated sample (Fig. 2C). To further ensure that the multicellular foci were not a result of cell division over the time course of the experiment, we repeated the same experiment in nongrowing Huh7 cells (as described in Materials and Methods) and obtained similar results (data not shown).
FIG 2.
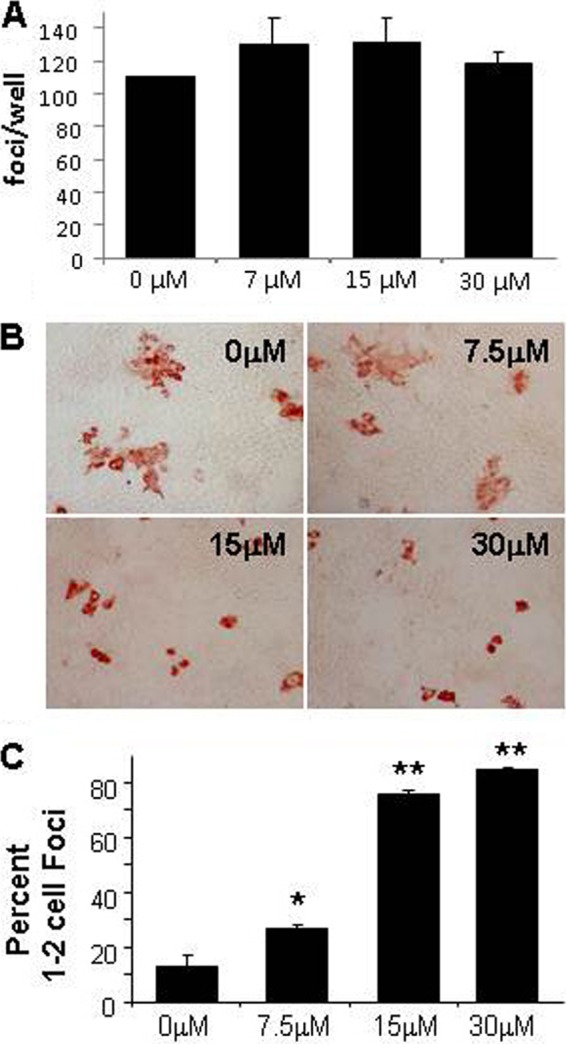
Inhibition of HCV cell-to-cell spread by ezetimibe. Confluent Huh7 cells were infected with approximately 100 FFU JFH-1 HCVcc. At 16 hpi, cells were washed and medium containing neutralizing anti-HCV E2 plus 30 μM, 15 μM, 7.5 μM, or 0 μM ezetimibe was added. Cells were fixed and stained for HCV at 72 hpi to visualize HCV-positive foci. (A) Total number of HCV-positive foci in each well. Results are graphed as the averages ± SD from triplicate wells. (B) Representative fields of HCV-positive foci photographed using a Nikon Eclipse TE2000U with a 20× objective lens. (C) Quantification of cell-to-cell spread. The percentages of the total number of foci containing only 1 or 2 cells are graphed. Results are averages from three replicate wells ± SD. Significant differences relative to mock control are indicated (**, P < 0.001; *, P < 0.05 by ANOVA). Results are representative of three experiments performed in both growing and nongrowing cells.
To confirm the role of NPC1L1 in the cell-to-cell spread of other HCV genotypes, we used intergenotypic HCVcc chimeras in which the core-to-E2 region of the JFH-1 clone had been replaced with analogous regions of various HCV genotypes (39–41). Here, nongrowing Huh7 cells were infected with the different chimeric viruses, and after 16 h, the cells were washed and 0 μM or 30 μM ezetimibe was added to parallel cultures. Since the chimeras encode unique E2 proteins and thus may have differing sensitivities to inhibition by the anti-E2 antibody, in this experiment we added methyl cellulose to all cultures as an alternative means of minimizing cell-free virus spread. At 72 hpi, the cells were fixed and stained with an antibody to HCV NS5A to detect the size of the foci formed. In all cases, the size of foci formed by each chimera was reduced in the presence of ezetimibe relative to the mock-treated control culture, confirming not only that all of these genotypes utilize the NPC1L1 receptor for cell-to-cell spread, but also that the spread of all the chimeric viruses can be inhibited by ezetimibe (Fig. 3).
FIG 3.

Ezetimibe exhibits pan-genotypic inhibition of HCV cell-to-cell spread. Nongrowing Huh7 cells were infected with approximately 50 FFU wild-type JFH-1 HCVcc or the indicated chimeric constructs containing the core-to-E2 glycoprotein region from different HCV genotypes. At 16 hpi, cells were washed and medium containing 0.25% methyl cellulose with 0 μM or 30 μM ezetimibe was added. Cells were fixed at 72 hpi and stained for HCV NS5A to detect infected cell foci. Representative foci illustrating the difference in size were photographed using a Nikon Eclipse TE2000U with a 20× objective lens. The experiment was performed with duplicate samples, and results are representative of three experiments.
Inhibition of the VLDL pathway by naringenin prevents secretion of cell-free virus but does not prevent cell-to-cell spread.
It has been demonstrated that infectious HCV particles are lipidated and released from cells via the VLDL secretory pathway, and thus this cellular pathway had emerged as an HCV antiviral target for inhibiting the secretion of infectious HCV from cells (27, 28, 49). While it has been shown that inhibiting the VLDL pathways prevents cell-free virus secretion/spread, we were interested to determine whether inhibiting the VLDL secretory pathway also blocks cell-to-cell spread. Because the flavonoid extract naringenin has been shown to inhibit microsomal transfer protein (MTP) activity, thereby decreasing ApoB secretion and consequently inhibiting HCV particle secretion (28, 50), we examined the effect of naringenin on the cell-to-cell spread of HCV by performing a focus size assay. Nongrowing Huh7 cells were infected with 100 FFU JFH-1 HCVcc per well. After 16 h, the inoculum was removed and naringenin or vehicle was added to the infected cell cultures at 200 μM, a concentration that inhibits virus particle secretion by 80 to 90% (28, 50) (Fig. 4A). As a control to ensure that the decrease in infectious HCV particle secretion detected in our titration assay was not due to effects of naringenin inhibiting virus entry into cells during the titration assay, we inoculated cells with virus in the presence of naringenin at the same concentrations used in our titer determination assay and found that there was no difference in the number of foci obtained when cells were inoculated with naringenin or the vehicle (data not shown). Importantly, when anti-E2 was added to all cultures to neutralize extracellular virus and the cells were fixed and stained at 72 hpi to detect the size of the HCV-positive cell foci, we observed that the size of the foci in the wells treated with naringenin were similar to that observed in the cultures treated with the vehicle, suggesting no detectable inhibition of cell-to-cell spread (Fig. 4B).
FIG 4.

Inhibition of the VLDL pathway by naringenin does not inhibit HCV cell-to-cell spread. (A and B) Nongrowing Huh7 cells were infected with approximately 100 FFU JFH-1 HCVcc. At 16 hpi, cultures were washed and subsequently mock treated or treated with naringenin. (A) Infectious HCVcc in the culture medium was measured at 48 h posttreatment by titration on naive Huh7 cells. The averages of triplicate wells ± SD are graphed as percentages relative to the mock-treated control. (B) To monitor cell-to-cell spread, parallel wells were incubated in the presence of neutralizing antibody against HCV E2 during treatment with 200 μM naringenin or vehicle. Cells were then fixed and stained with antibody to HCV E2 at 72 hpi. Representative foci were photographed using a Nikon Eclipse TE2000U with a 20× objective lens. Results are representative of six experiments. (C and D) Nongrowing Huh7 cells were infected with approximately 50 FFU wt JFH-1 HCVcc or the indicated chimeric viruses bearing the core through E2 glycoprotein region of different HCV genotypes. At 16 hpi, the cells were washed and medium containing 0.25% methyl cellulose with 0 μM or 200 μM naringenin was added. Cells were fixed at 5 days postinfection and stained for HCV NS5A. (C) Representative foci were photographed using a Nikon Eclipse TE2000U with a 20× objective lens. (D) Average number of cells per focus ± SD formed under each treatment is graphed. Significant differences relative to the mock-treated control are indicated (*, P < 0.05 by ANOVA). The experiment was performed with duplicate samples, and results are representative of three experiments.
To determine if naringenin affects spread of other HCV genotypes and more rigorously test the effect, nongrowing Huh7 cells were infected with the different intergenotypic chimeric viruses (50 FFU/well), and the assay was extended to 5 days to allow more time for cell-to-cell spread. After 16 h, the cells were washed and mock treated or treated with 200 μM naringenin. Methyl cellulose was added to each well to minimize cell-free virus spread. Five days postinoculation, the cells were fixed and stained to detect the number of HCV NS5A-positive cells per focus. For all chimeric viruses, except for the J8(2b)/JFH-1 chimera, the size of the focus observed in the naringenin-treated samples was similar to that observed in the control mock-treated wells, indicating that cell-to-cell spread was not inhibited by naringenin (Fig. 4C and D). Notably, the foci in the wells treated with naringenin tended to be more compact than those in the control wells overlaid with methyl cellulose alone, suggesting that perhaps methyl cellulose is not as effective at preventing local cell-free virus spread as naringenin; however, it was still clear that significant cell-to-cell spread occurred in the presence of naringenin. The one exception was the J8(2b)/JFH-1 chimera, which consistently produced smaller foci even in mock-treated cells and in the presence of naringenin exhibited even smaller foci, suggesting that unlike the other viruses screened, the spread of this particular chimeric virus is inefficient and may be affected by the VLDL pathway.
RNAi silencing of ApoB, ApoE, or MTP does not prevent HCV cell-to-cell spread.
To more specifically examine the role of different components of the VLDL pathway in cell-to-cell spread, we used siRNAs to individually silence ApoB, ApoE, or MTP. In addition, one set of cells was transfected with a positive-control siRNA targeting claudin-1, a factor known to be involved in HCV cell-to-cell spread (20), and another set of cells was transfected with a negative-control siGFP. At 24 h post-siRNA transfection, cells were infected with 100 FFU JFH-1 HCVcc, and at 16 h postinoculation, anti-E2 antibody was added to prevent cell-free virus spread. The cells were fixed and stained at 5 days postinfection to detect the number and size of the HCV-positive foci formed.
The level of silencing of the targeted genes at 24 h and 6 days after siRNA transfection was 90 to 95%, except for the CLDN1 control, which had not remained fully silenced by 6 days posttransfection (Fig. 5A). As indicated by the similar number of total foci observed in each well, silencing of ApoB, ApoE, MTP, or GFP did not inhibit initial cell-free-mediated virus infection; however, as expected, silencing of CLDN1 prior to infection resulted in markedly reduced focus formation (Fig. 5B). Likewise, the foci that were observed in CLDN1-knockdown cultures consisted mainly of 1 or 2 cells, consistent with inhibition of cell-to-cell spread (Fig. 5C). In contrast, we observed that the foci formed in ApoB-, ApoE- or MTP-silenced cultures were multicellular and comparable in size to the foci observed in the control siGFP-transfected cells (Fig. 5C). Interestingly, the foci formed in ApoE-silenced cells were perhaps slightly smaller, but this was not statistically significant (Fig. 5D). As a functional control to confirm sufficient target gene knockdown, we also assessed the effect of ApoB, ApoE, or MTP silencing on secretion of infectious HCV particles by collecting medium from the wells at 48 hpi and 72 hpi for titration on naive Huh7 cells. Consistent with a previous report (51), ApoB silencing resulted in a 40% decrease in infectious virus secreted compared to cells transfected with siGFP, while silencing of ApoE or MTP resulted in an 80 to 90% decrease in secreted infectious virus (Fig. 5E). These results were confirmed in the nongrowing cell culture system, and identical results were obtained (data not shown).
FIG 5.
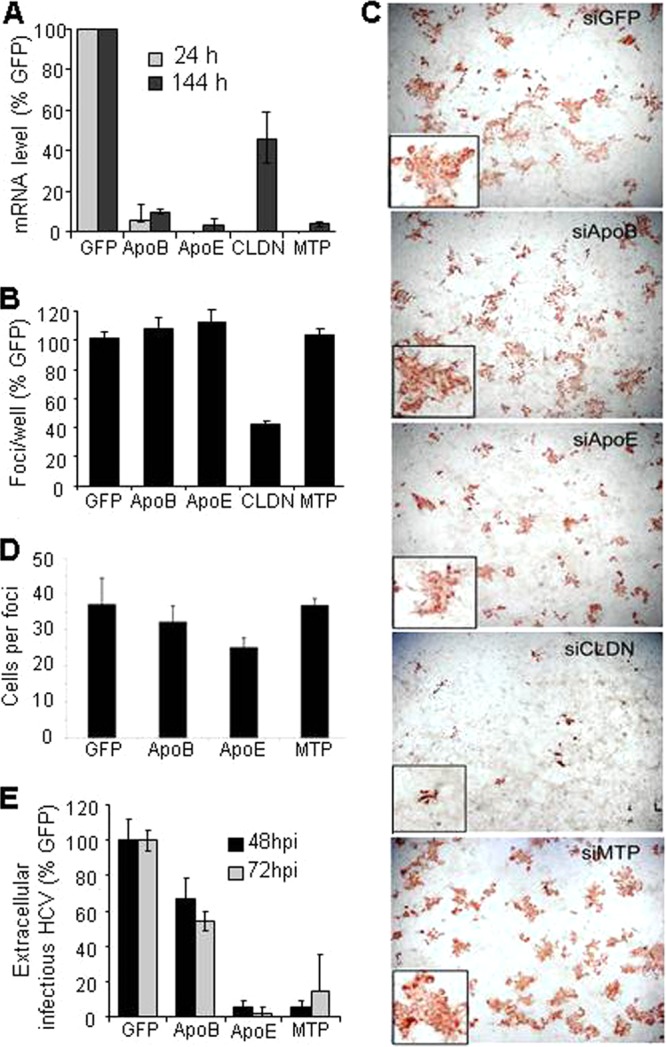
ApoB, ApoE, and MTP are not required for HCV cell-to-cell spread. Nongrowing Huh7 cells were transfected with siRNAs targeting ApoB, ApoE, MTP, claudin-1, or GFP (negative control). After 24 h, cultures were inoculated with 100 FFU JFH-1 HCVcc. At 16 hpi, cells were washed and medium containing neutralizing anti-E2 was added to limit cell-free virus spread. Cells were fixed at 5 days postinfection and stained for HCV E2 to detect infected cells. (A) Levels of target gene mRNA were quantified by qRT-PCR at days 1 and 6 posttransfection, and the averages of duplicate samples ± SD are graphed as percentages of levels in siGFP-silenced cells. (B) Number of foci observed in cultures transfected with indicated siRNA. Averages of duplicate wells ± SD are graphed as percentages of number of foci observed in cells transfected with control siGFP. (C) Morphology of foci observed in cultures transfected with the indicated siRNA. Insets show magnification of a single focus. Wells were photographed using a Nikon Eclipse TE2000U with a 4× objective lens or a 10× objective lens. (D) Average number of cells per focus ± SD. The number of cells in the foci under each condition were not significantly different from the control (as determined by ANOVA). (E) Effect of indicated siRNA-mediated knockdown on HCV secretion at 48 hpi (72 h posttransfection) and 72 hpi (96 h posttransfection). Infectious virus was quantified by titration of medium from the silenced cells on naive Huh7 cells. Average FFUs per ml ± SD are graphed as a percentage of infectious virus released from control siGFP cells. Results are representative of three experiments performed in both growing and nongrowing Huh7 cells.
Blocking HCV cell-to-cell spread determines synergy potential between entry/egress inhibitors and antiviral agents that block intracellular virus production.
The use of an entry inhibitor or an egress inhibitor alone may be of limited use in patients with chronic HCV infection, since many target cells are already infected. However, one would predict that these inhibitors might significantly enhance the efficacy of treatment when used in combination with antivirals that reduce intracellular HCV amplification (e.g., interferon, protease inhibitors, or polymerase inhibitors) because they could potentially prevent infection of new target cells, protect cells cured of HCV from reinfection, and prevent the spread of viral escape mutants, which commonly arise during treatment with direct-acting antivirals. Consistent with the prediction of enhanced efficacy, we previously showed that ezetimibe synergizes with interferon to inhibit chronic HCV infection (B. Sainz, Jr., N. Barretto, D. N. Martin, N. Hiraga, M. Imamura, X. Yu, K. Chayama, W. A. Alrefai, S. L. Uprichard, presented at the 18th International Symposium on Hepatitis C Virus and Related Viruses, Seattle, WA, 8 to 12 September 2011). However, having now discovered that ezetimibe also blocks cell-to-cell spread of HCV into secondary target cells, we were interested to determine if the synergy observed between ezetimibe and interferon was related to the ability of ezetimibe to block not only cell-free spread but also cell-to-cell spread.
To address this question, we first examined the synergy potential between an inhibitor of HCV amplification and the mainstay of anti-HCV therapy, alpha interferon (IFN-α) and the secretion inhibitor naringenin, which like ezetimibe would be expected to block cell-free HCV infection but unlike ezetimibe does not prevent cell-to-cell spread of the virus (Fig. 4). Thus, we established parallel cultures of chronically infected Huh7 cells and subsequently treated them with ezetimibe, naringenin, or IFN-α, alone or in combination (Fig. 6A). Treatments were replenished every 24 h. Triplicate cultures were harvested at 24, 48, 72, or 96 h posttreatment for quantification of intracellular HCV RNA levels by qRT-PCR. As expected, monotreatment with ezetimibe alone did not reduce HCV levels in chronically infected cultures, in which all the cells are already infected with HCV prior to treatment (19), while treatment with IFN-α alone resulted in a log decrease in HCV copies. However, combination treatment with IFN-α and ezetimibe resulted in a synergistic 1.5- to 2-log reduction of HCV copies (Fig. 6A). Interestingly, blocking cell-free spread with naringenin actually increased intracellular HCV copies by about 50%, most likely due to inhibition of secretion causing an accumulation of intracellular HCV particles. Similarly, combination treatment with IFN-α and naringenin resulted in higher intracellular HCV levels than treatment with IFN-α alone. Thus, no evidence of synergy or additive benefit was observed when IFN-α was combined with an egress inhibitor that blocks only cell-free spread. In all of the naringenin-containing samples, the secretion of extracellular virus secretion was blocked, as confirmed by determining the titer of the supernatants on naive cells (data not shown).
FIG 6.
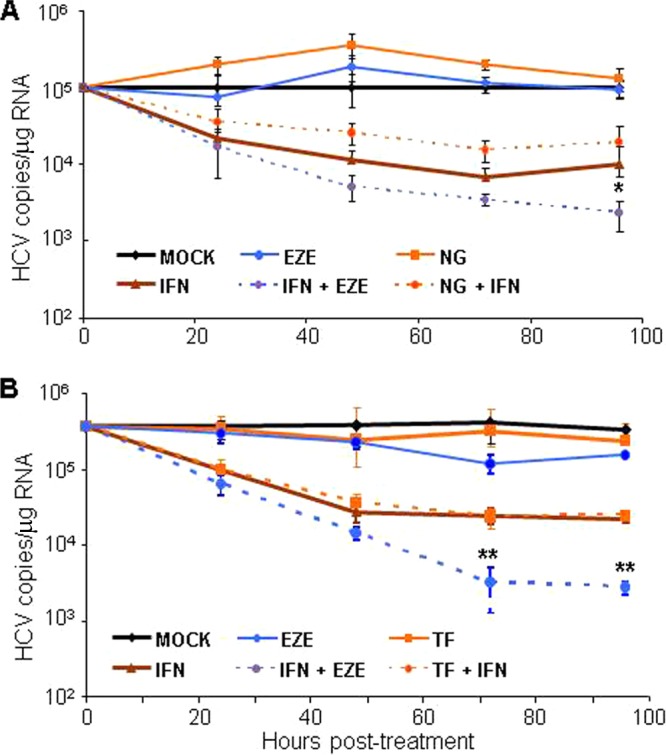
Inhibiting cell-to-cell spread affects antiviral synergy potential between HCV entry/egress inhibitors and interferon. Chronically HCV-infected Huh7 cells were seeded in 96-well plates. (A) Cultures were mock treated (black) or treated with 100 units/ml IFN-α (brown), 200 μM naringenin (NG; orange), or 30 μM ezetimibe (EZE; blue) either alone (solid lines) or in combination (dotted lines) as indicated. (B) Cultures were mock treated (black) or treated with 100 units/ml IFN-α (brown), 50 μM ferristatin (TF; orange), an inhibitor of transferrin receptor-1, or 30 μM EZE (blue) either alone (solid lines) or in combination (dotted lines) as indicated. At 24, 48, 72, or 96 h posttreatment (hpt), intracellular RNA was extracted. HCV and cellular GAPDH RNA levels were quantified by qRT-PCR. HCV copies were normalized to cellular GAPDH and are expressed as average number of HCV copies/μg RNA in triplicate samples ± SD. Significant differences between the interferon-treated samples and the IFN-EZE treatment are indicated (**, P < 0.001; *, P < 0.05 by F-test or ANOVA). Results are representative of three experiments.
As an alternative and perhaps more equivalent method of determining the therapeutic importance of inhibiting HCV cell-to-cell spread, we used a TfR1 inhibitor, as TfR1 is another recently identified host cell factor reported to be involved in cell-free entry of HCV into cells but not required for cell-to-cell-mediated HCV spread (24). Although this inhibitor (NSC306711) has been named ferristatin, it is not related to the statin class of cholesterol-reducing drugs but instead acts by binding to TfR1 and causing it to internalize and degrade in the absence of cargo (52). Hence, we established chronically infected Huh7 cell cultures and treated them with ezetimibe, ferristatin, or IFN-α, alone or in combination with treatments being replenished every 24 h (Fig. 6B). Intracellular HCV copies were quantified from triplicate cultures by qRT-PCR at 24, 48, 72, or 96 h posttreatment. As expected of entry inhibitors, neither ezetimibe nor ferristatin alone reduced intracellular HCV load in chronically infected cultures, while cells treated with IFN-α resulted in a log decrease in HCV levels. Although cells treated with both IFN-α and ezetimibe showed a 1.5- to 2-log decrease in intracellular HCV load, combination treatment with IFN-α and ferristatin showed no enhancement of HCV inhibition, with intracellular HCV RNA levels similar to those in cells treated with IFN-α alone. Therefore, analogous to what we observed with naringenin, combination treatment with IFN-α plus an entry inhibitor that blocks only cell-free virus entry exhibited no synergistic reduction of HCV in combination with IFN.
DISCUSSION
HCV can spread to naive cells via cell-free virion entry and cell-to-cell transmission. Cell-to-cell transmission has recently been the subject of many studies, because this appears to be a major route of HCV dissemination, which likely enables the virus to escape host neutralizing antibody responses and presumably facilitates the establishment of chronic infection. In this report, we show that HCV cell-to-cell spread is dependent on the newly described HCV entry factor NPC1L1 (Fig. 1) and can be prevented by treatment with the NPC1L1 inhibitor ezetimibe (Fig. 2 and 3). Although the host VLDL pathway has been shown to be involved in the secretion of infectious HCV particles into the extracellular environment (27, 28) and is thus required for cell-free HCV spread, we demonstrate that the VLDL pathway is dispensable for infectious HCV cell-to-cell transmission (Fig. 4 and 5). With the cellular VLDL pathway and other cellular factors required for HCV cell-free spread being a focus of HCV antiviral drug development research, we explored the combinatorial potential of inhibitors to determine the extent to which blocking cell-to-cell spread impacts synergy with the HCV amplification inhibitor, IFN-α. We tested three entry/egress inhibitors (i.e., ezetimibe, naringenin, and ferristatin), which have all been shown to effectively block HCV cell-free entry or egress (19, 24, 27) and thereby cell-free spread. However, only ezetimibe, which additionally blocks cell-to-cell HCV spread (Fig. 2 and 3), exhibited synergistic HCV inhibition when used in combination with IFN. Thus, we have identified differences and similarities in the cellular factors involved in HCV cell-free spread versus cell-to-cell spread and illustrate how these distinctions are clinically relevant for the design of effective anti-HCV combination therapies.
Cellular factors involved in HCV cell-to-cell spread.
Prior reports have shown that HCV cell-to-cell spread requires the cellular factors SR-B1, claudin-1, and occludin (20); however, the requirement for CD81 has been under debate. While one group used a panel of anti-CD81 antibodies and observed inhibition of cell-to-cell spread (20), another group showed that HCV cell-to-cell spread could still occur in a CD81-deficient cell line (21). Recently, we identified TfR1 as an HCV entry factor involved in HCV cell-free entry but not required cell-to-cell spread. Blocking of NPC1L1 with antibody or ezetimibe prevented HCV cell-to-cell spread as efficiently as blocking of claudin-1, indicating that NPC1L1 plays a critical role in HCV cell-to-cell spread, and this was found to be true for all the HCV genotypes tested. In polarized hepatocytes, NPC1L1 is thought to be present primarily on the apical canalicular surface (53, 54). This surface is in proximity to the intercellular tight junctions where the claudin-1 and occludin proteins, known to be involved in cell-to-cell spread of HCV, reside; thus, it is tempting to speculate that NPC1L1 may interact with these molecules during HCV spread. However, whether NPC1L1 (or the other HCV entry factors) function similarly in cell-free entry and cell-to-cell spread of HCV remains to be determined, and the development of fully polarized hepatocyte cell culture models would potentially offer additional insights as efforts continue in this area.
While many of the factors that have been investigated for their role in HCV cell-to-cell spread are those known to be involved in HCV cell-free entry, we were additionally interested to determine whether factors required for infectious HCV cell-free egress might also be involved in cell-to-cell spread. Specifically, current models propose that once assembled, HCV particles utilize the cellular VLDL pathway to acquire host cell lipoproteins that are necessary for secretion of infectious virus. This is based on literature demonstrating that secretion of infectious HCV particles depends on a functional VLDL pathway, notably MTP (27, 51), ApoB (27, 28), and ApoE (51, 55). While we confirmed that these factors are indeed involved in secretion of extracellular infectious virus to various extents, we observed efficient cell-to-cell spread in the presence of naringenin (Fig. 4) and after knockdown of ApoB, ApoE, or MTP (Fig. 5). Although the foci formed in the presence of naringenin and after ApoE knockdown had numbers of cells comparable to those of the controls, morphologically they were notably more compact. Because it is likely that the anti-E2 or methylcellulose added to all cultures to limit cell-free spread during these experiments may not have been 100% effective at eliminating extremely localized cell-free spread, this more compact phenotype could reflect a more enhanced inhibition of cell-free spread in these cultures; however, other explanations have not been ruled out.
Notably, in contrast to all the other HCV genotypes tested in which cell-to-cell spread was unaffected by inhibition of the VLDL pathway by naringenin, cell-to-cell spread (or perhaps very localized cell-free spread) of the J8(2b)/JFH-1 clone appeared to be reduced by naringenin. The reason for this difference is unknown but could be related to the fact that this chimera inherently did not appear to spread as efficiently as the other clones even in the mock-treated wells (in which all mechanisms of spread would be occurring), suggesting that the chimeric virus generated from this clone may have inherent deficiencies that make it less physiologically relevant for this particular analysis. Somewhat analogously, during the preparation of this report, Hueging et al. published a study in which they used a transcomplementation system in 293T cells to show that ApoB and MTP are not required for the formation of infectious virion particles (56, 57). Based on the results observed in this transcomplementation system, the authors also concluded that ApoE is required for HCV cell-to-cell spread. However, they did note that the HCV particles produced in these 293T cells have a specific infectivity 5-fold lower than that of particles produced in Huh7 cells. As such, we speculate that the disparate results observed in that study regarding the involvement of ApoE in HCV cell-to-cell spread may be related to the nature of the HCV particles produced in transcomplemented cells; perhaps like J8(2b)/JFH-1 chimeric virus, the 293T-generated particles are inherently less competent for spread and thus not representative of authentic HCV particles.
The fact that HCV cell-to-cell spread of all but one chimeric virus tested occurs in Huh7 cells when the VLDL pathway is inhibited suggests that typically HCV is capable of spreading cell to cell prior to maturation via the VLDL pathway and that virions destined to spread cell to cell may be trafficked via a distinct intracellular pathway(s). This notion of alternate routes of viral particle trafficking is in line with that of Mothes et al. in their recent articles regarding viral cell-to-cell spread (58–60) and raises questions regarding the nature of the viral particle that is transferred during cell-to-cell spread. Notably, earlier reports that the majority of infectious HCV particles within infected cells have a higher density than that of extracellular infectious particles (30) may indeed be consistent with intracellular infectious particles being generated in the absence of trafficking through the entire VLDL pathway. As such, future efforts will involve investigating other host cell proteins/pathways which have been implicated in HCV morphogenesis and secretion to determine whether these factors are required for cell-to-cell spread. Factors involved in initial virion assembly will likely be necessary for both cell-free and cell-to-cell spread, while those involved in trafficking or late-stage maturation may have differing importance in these two processes and may indirectly provide insight into the nature of the viral particle involved in cell-to-cell spread. In this regard, the exosome pathway is of interest, as several reports have documented that HCV RNA is secreted from infected cells in exosomes, which has been proposed to transfer the infection when taken up by other cells (61–64). While by nature this would appear to fall within the category of extracellular (i.e., cell-free) spread, a role for exosome pathway components in cell-to-cell spread cannot be ruled out.
The therapeutic potential of inhibiting cell-to-cell spread.
Regardless of the molecular details involved, we examined the therapeutic implications of the finding that some host cell factors that prevent cell-free HCV spread may not affect HCV cell-to-cell spread. Because monotherapy with inhibitors of HCV spread have no effect on infection levels in chronically infected cultures in which all the cells are already infected, we focused on the synergy potential of these inhibitors when used in combination with the clinically used HCV inhibitor, interferon. As expected, when comparing inhibitors that block only cell-free entry/spread (i.e., naringenin or ferristatin) to an inhibitor that blocks both cell-free and cell-to-cell spread (i.e., ezetimibe), none of the treatments were sufficient on their own to reduce HCV levels. However, ezetimibe synergized with IFN, increasing intracellular HCV RNA decline (Fig. 6), while the agents that block only cell-free virus spread, either by inhibiting secretion of infectious virus (i.e., naringenin) or cell-free entry (ferristatin), did not. Thus, our data suggest that entry/egress inhibitors that block both cell-free and cell-to-cell HCV spread might be more clinically useful for enhancing HCV clearance when used in combination with antivirals that reduce HCV intracellular production (e.g., IFN, protease inhibitors, polymerase inhibitors). Figure 7 provides a simplistic illustration of the different effects that blocking NPC1L1 and blocking the VLDL pathway have on cell-free and cell-to-cell spread.
FIG 7.
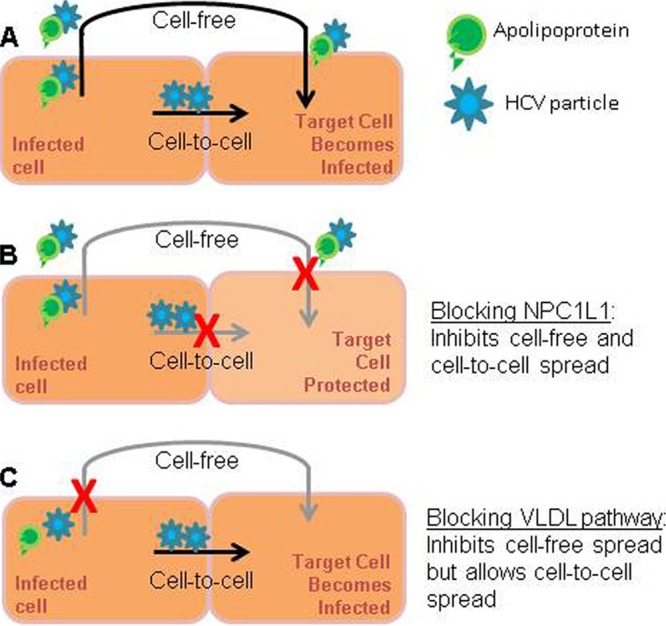
Blocking different modes of HCV spread. Cells may be infected by cell-free infection or cell-to-cell spread. (A) Infected cells produce lipidated, infectious cell-free virions, as well as particles which may not be lipidated but may still be able to be transmitted from cell to cell; (B) blocking the HCV entry factor NPC1L1 inhibits both modes of infection and the target cell is protected; (C) blocking the cellular VLDL pathway results in inhibition of secretion of infectious lipidated cell-free virion particles; however, in the absence of cell-free secretion, infectious particles are still able to be transmitted from cell to cell, and the target cell is infected.
These results also suggest that while inhibitors targeting VLDL secretion may result in lower serum HCV levels in patients, targeting the VLDL pathway may have limited therapeutic potential. Perhaps more concerning is that inhibiting HCV secretion with naringenin resulted in the intracellular accumulation of HCV RNA in treated cells (Fig. 6A), which may cause an increase in virus-induced cell stress and ultimately be harmful to the infected hepatocytes rather than cure them. Relevant to this, we also observed an increase in intracellular HCV when naringenin was used in combination with protease inhibitors (data not shown).
In terms of entry inhibitors, our data stress the important distinction between entry inhibitors that are able to inhibit cell-to-cell spread or not. As such, entry inhibitors identified in drug screens using HCVpp should be subjected to a secondary screening in a system which includes effects on cell-to-cell spread. Analogous considerations would be relevant to ongoing efforts to create HCV inhibitors and neutralizing antibodies to prevent infection of the liver graft after liver transplantation, where inhibiting both methods of virus transmission would be beneficial.
The fact that synergy was observed only when interferon was combined with an inhibitor that blocks both means of HCV spread also provides insight into the molecular basis of the synergy itself. In particular, it suggests that the synergy potential between entry/egress inhibitors and inhibitors that block intracellular HCV amplification is directly related to the ability of the entry/egress inhibitors to block HCV spread (i.e., not cell-free entry or egress as previously assumed). If synergy is dependent on blocking viral spread, this then implies that the mechanism of the synergy may be related to the ability of the spread inhibitor to protect cured cells from reinfection (Fig. 7). In the context of IFN-free cocktails of DAA, HCV spread inhibitors should also enhance treatment response by preventing the spread and subsequent amplification of viral escape variants that may arise to the DAAs that may be present in the cocktail.
In summary, in addition to identifying the involvement of NPC1L1 in cell-to-cell spread, our data also highlight that there are clinically relevant differences between HCV cell-free spread and cell-to-cell spread, as we have shown that host VLDL pathway factors are involved in cell-free secretion but not cell-to-cell spread. These data reveal the previously unrealized need to examine inhibitors of egress and entry for their ability to prevent both routes of HCV spread and provide proof-of-concept that this consideration is important in a therapeutic context.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01-AI078881 and R21-AI097809.
We thank Jens Bukh for kindly providing us with the HCV chimeric constructs. We also acknowledge helpful discussions with members of the Uprichard lab.
Footnotes
Published ahead of print 19 February 2014
REFERENCES
- 1.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115. 10.1111/j.1469-0691.2010.03432.x [DOI] [PubMed] [Google Scholar]
- 2.Chak E, Talal AH, Sherman KE, Schiff ER, Saab S. 2011. Hepatitis C virus infection in U.S.A.: an estimate of true prevalence. Liver Int. 31:1090–1101. 10.1111/j.1478-3231.2011.02494.x [DOI] [PubMed] [Google Scholar]
- 3.Seeff LB. 2002. Natural history of chronic hepatitis C. Hepatology 36:S35–S46. 10.1053/jhep.2002.36806 [DOI] [PubMed] [Google Scholar]
- 4.El-Serag HB, Rudolph KL. 2007. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132:2557–2576. 10.1053/j.gastro.2007.04.061 [DOI] [PubMed] [Google Scholar]
- 5.O'Leary JG, Lepe R, Davis GL. 2008. Indications for liver transplantation. Gastroenterology 134:1764–1776. 10.1053/j.gastro.2008.02.028 [DOI] [PubMed] [Google Scholar]
- 6.Lange CM, Sarrazin C, Zeuzem S. 2010. Review article: specifically targeted anti-viral therapy for hepatitis C—a new era in therapy. Aliment Pharmacol. Ther. 32:14–28. 10.1111/j.1365-2036.2010.04317.x [DOI] [PubMed] [Google Scholar]
- 7.Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462. 10.1053/j.gastro.2009.11.055 [DOI] [PubMed] [Google Scholar]
- 8.Barth H, Schafer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, Kinoshita-Toyoda A, Toida T, Van Kuppevelt TH, Depla E, Von Weizsacker F, Blum HE, Baumert TF. 2003. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 278:41003–41012. 10.1074/jbc.M302267200 [DOI] [PubMed] [Google Scholar]
- 9.Germi R, Crance JM, Garin D, Guimet J, Lortat-Jacob H, Ruigrok RW, Zarski JP, Drouet E. 2002. Cellular glycosaminoglycans and low density lipoprotein receptor are involved in hepatitis C virus adsorption. J. Med. Virol. 68:206–215. 10.1002/jmv.10196 [DOI] [PubMed] [Google Scholar]
- 10.Gardner JP, Durso RJ, Arrigale RR, Donovan GP, Maddon PJ, Dragic T, Olson WC. 2003. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 100:4498–4503. 10.1073/pnas.0831128100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lozach PY, Lortat-Jacob H, de Lacroix de Lavalette A, Staropoli I, Foung S, Amara A, Houles C, Fieschi F, Schwartz O, Virelizier JL, Arenzana-Seisdedos F, Altmeyer R. 2003. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 278:20358–20366. 10.1074/jbc.M301284200 [DOI] [PubMed] [Google Scholar]
- 12.Monazahian M, Bohme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R. 1999. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J. Med. Virol. 57:223–229. [DOI] [PubMed] [Google Scholar]
- 13.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. 1999. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. U. S. A. 96:12766–12771. 10.1073/pnas.96.22.12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017–5025. 10.1093/emboj/cdf529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. 10.1126/science.282.5390.938 [DOI] [PubMed] [Google Scholar]
- 16.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805 [DOI] [PubMed] [Google Scholar]
- 17.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. 10.1038/nature07684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T. 2009. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 83:2011–2014. 10.1128/JVI.01888-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sainz B, Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18:281–285. 10.1038/nm.2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brimacombe CL, Grove J, Meredith LW, Hu K, Syder AJ, Flores MV, Timpe JM, Krieger SE, Baumert TF, Tellinghuisen TL, Wong-Staal F, Balfe P, McKeating JA. 2011. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J. Virol. 85:596–605. 10.1128/JVI.01592-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witteveldt J, Evans MJ, Bitzegeio J, Koutsoudakis G, Owsianka AM, Angus AG, Keck ZY, Foung SK, Pietschmann T, Rice CM, Patel AH. 2009. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 90:48–58. 10.1099/vir.0.006700-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, McKeating JA. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24. 10.1002/hep.21959 [DOI] [PubMed] [Google Scholar]
- 23.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17:589–595. 10.1038/nm.2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. U. S. A. 110:10777–10782. 10.1073/pnas.1301764110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinglass AB, Kohler M, Schulte U, Liu J, Nketiah EO, Thomas A, Schmalhofer W, Williams B, Bildl W, McMasters DR, Dai K, Beers L, McCann ME, Kaczorowski GJ, Garcia ML. 2008. Extracellular loop C of NPC1L1 is important for binding to ezetimibe. Proc. Natl. Acad. Sci. U. S. A. 105:11140–11145. 10.1073/pnas.0800936105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang TY, Chang C. 2008. Ezetimibe blocks internalization of the NPC1L1/cholesterol complex. Cell Metab. 7:469–471. 10.1016/j.cmet.2008.05.001 [DOI] [PubMed] [Google Scholar]
- 27.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82:2120–2129. 10.1128/JVI.02053-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nahmias Y, Goldwasser J, Casali M, van Poll D, Wakita T, Chung RT, Yarmush ML. 2008. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 47:1437–1445. 10.1002/hep.22197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299. 10.1073/pnas.0503596102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gastaminza P, Kapadia SB, Chisari FV. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 80:11074–11081. 10.1128/JVI.01150-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 80:11082–11093. 10.1128/JVI.01307-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sainz B, Jr, Barretto N, Uprichard SL. 2009. Hepatitis C virus infection in phenotypically distinct Huh7 cell lines. PLoS One 4:e6561. 10.1371/journal.pone.0006561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu X, Sainz B, Jr, Uprichard SL. 2009. Development of a cell-based hepatitis C virus infection fluorescent resonance energy transfer assay for high-throughput antiviral compound screening. Antimicrob. Agents Chemother. 53:4311–4319. 10.1128/AAC.00495-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sainz B, Jr, Chisari FV. 2006. Production of infectious hepatitis C virus by well-differentiated, growth-arrested human hepatoma-derived cells. J. Virol. 80:10253–10257. 10.1128/JVI.01059-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi S, Sainz B, Jr, Corcoran P, Uprichard S, Jeong H. 2009. Characterization of increased drug metabolism activity in dimethyl sulfoxide (DMSO)-treated Huh7 hepatoma cells. Xenobiotica 39:205–217. 10.1080/00498250802613620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. 10.1053/j.gastro.2003.09.023 [DOI] [PubMed] [Google Scholar]
- 37.Kato T, Furusaka A, Miyamoto M, Date T, Yasui K, Hiramoto J, Nagayama K, Tanaka T, Wakita T. 2001. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J. Med. Virol. 64:334–339. 10.1002/jmv.1055 [DOI] [PubMed] [Google Scholar]
- 38.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796. 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. 2009. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377. 10.1002/hep.22673 [DOI] [PubMed] [Google Scholar]
- 40.Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, Alter HJ, Eugen-Olsen J, Bukh J. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. U. S. A. 105:997–1002. 10.1073/pnas.0711044105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen TB, Gottwein JM, Scheel TK, Hoegh AM, Eugen-Olsen J, Bukh J. 2008. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J. Infect. Dis. 198:1756–1765. 10.1086/593021 [DOI] [PubMed] [Google Scholar]
- 42.Yu X, Uprichard SL. 2010. Cell-based hepatitis C virus infection fluorescence resonance energy transfer (FRET) assay for antiviral compound screening. Curr. Protoc. Microbiol. Chapter 17:Unit 17.5. 10.1002/9780471729259.mc1705s18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knipe D, Howley PM. 2007. Principles of virology, 5th ed, vol 1, p 30–43 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 44.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27. 10.1038/nm1698 [DOI] [PubMed] [Google Scholar]
- 45.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- 46.Krieger SE, Zeisel MB, Davis C, Thumann C, Harris HJ, Schnober EK, Mee C, Soulier E, Royer C, Lambotin M, Grunert F, Dao Thi VL, Dreux M, Cosset FL, McKeating JA, Schuster C, Baumert TF. 2009. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology 51:1144–1157. 10.1002/hep.23445 [DOI] [PubMed] [Google Scholar]
- 47.Komurian-Pradel F, Perret M, Deiman B, Sodoyer M, Lotteau V, Paranhos-Baccala G, Andre P. 2004. Strand specific quantitative real-time PCR to study replication of hepatitis C virus genome. J. Virol. Methods 116:103–106. 10.1016/j.jviromet.2003.10.004 [DOI] [PubMed] [Google Scholar]
- 48.Sabahi A, Marsh KA, Dahari H, Corcoran P, Lamora JM, Yu X, Garry RF, Uprichard SL. 2010. The rate of hepatitis C virus infection initiation in vitro is directly related to particle density. Virology 407:110–119. 10.1016/j.virol.2010.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr, Ye J. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 104:5848–5853. 10.1073/pnas.0700760104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldwasser J, Cohen PY, Lin W, Kitsberg D, Balaguer P, Polyak SJ, Chung RT, Yarmush ML, Nahmias Y. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J. Hepatol 55:963–971. 10.1016/j.jhep.2011.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang J, Luo G. 2009. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 83:12680–12691. 10.1128/JVI.01476-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Horonchik L, Wessling-Resnick M. 2008. The small-molecule iron transport inhibitor ferristatin/NSC306711 promotes degradation of the transferrin receptor. Chem. Biol. 15:647–653. 10.1016/j.chembiol.2008.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, Ioannou YA, Davies JP, Nilsson LM, Yu L. 2007. Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J. Clin. Invest. 117:1968–1978. 10.1172/JCI30060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu L, Bharadwaj S, Brown JM, Ma Y, Du W, Davis MA, Michaely P, Liu P, Willingham MC, Rudel LL. 2006. Cholesterol-regulated translocation of NPC1L1 to the cell surface facilitates free cholesterol uptake. J. Biol. Chem. 281:6616–6624. 10.1074/jbc.M511123200 [DOI] [PubMed] [Google Scholar]
- 55.Benga WJ, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, Schuster C. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51:43–53 [DOI] [PubMed] [Google Scholar]
- 56.Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A, Doerrbecker J, Gumz F, Haid S, Wolk B, Kaderali L, Pietschmann T. 2014. Apolipoprotein E co-determines tissue-tropism of hepatitis C virus and it is crucial for viral cell-to-cell transmission by contributing to a post-envelopment step of assembly. J. Virol. 88:1433–1446. 10.1128/JVI.01815-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. 2008. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J. Virol. 82:7034–7046. 10.1128/JVI.00118-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J. Virol. 84:8360–8368. 10.1128/JVI.00443-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sherer NM, Jin J, Mothes W. Directional spread of surface-associated retroviruses regulated by differential virus-cell interactions. J. Virol. 84:3248–3258. 10.1128/JVI.02155-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. 2007. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 9:310–315. 10.1038/ncb1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JA, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus HW, Haagmans BL, Baumert TF, van der Laan LJ. 2013. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. U. S. A. 110:13109–13113. 10.1073/pnas.1221899110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. 2012. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12:558–570. 10.1016/j.chom.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan Q, Ramakrishnaiah V, Henry S, Fouraschen S, de Ruiter PE, Kwekkeboom J, Tilanus HW, Janssen HL, van der Laan LJ. 2012. Hepatic cell-to-cell transmission of small silencing RNA can extend the therapeutic reach of RNA interference (RNAi). Gut 61:1330–1339. 10.1136/gutjnl-2011-300449 [DOI] [PubMed] [Google Scholar]
- 64.Tamai K, Shiina M, Tanaka N, Nakano T, Yamamoto A, Kondo Y, Kakazu E, Inoue J, Fukushima K, Sano K, Ueno Y, Shimosegawa T, Sugamura K. 2012. Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology 422:377–385. 10.1016/j.virol.2011.11.009 [DOI] [PubMed] [Google Scholar]