

ABSTRACT
Ikaros is a zinc finger DNA-binding protein that regulates chromatin remodeling and the expression of genes involved in the cell cycle, apoptosis, and Notch signaling. It is a master regulator of lymphocyte differentiation and functions as a tumor suppressor in acute lymphoblastic leukemia. Nevertheless, no previous reports described effects of Ikaros on the life cycle of any human lymphotropic virus. Here, we demonstrate that full-length Ikaros (IK-1) functions as a major factor in the maintenance of viral latency in Epstein-Barr virus (EBV)-positive Burkitt's lymphoma Sal and MutuI cell lines. Either silencing of Ikaros expression by small hairpin RNA (shRNA) knockdown or ectopic expression of a non-DNA-binding isoform induced lytic gene expression. These effects synergized with other lytic inducers of EBV, including transforming growth factor β (TGF-β) and the hypoxia mimic desferrioxamine. Data from chromatin immunoprecipitation (ChIP)-quantitative PCR (qPCR) and ChIP-sequencing (ChIP-seq) analyses indicated that Ikaros did not bind to either of the EBV immediate early genes BZLF1 and BRLF1. Rather, Ikaros affected the expression of Oct-2 and Bcl-6, other transcription factors that directly inhibit EBV reactivation and plasma cell differentiation, respectively. IK-1 also complexed with the EBV immediate early R protein in coimmunoprecipitation assays and partially colocalized with R within cells. The presence of R alleviated IK-1-mediated transcriptional repression, with IK-1 then cooperating with Z and R to enhance lytic gene expression. Thus, we conclude that Ikaros plays distinct roles at different stages of EBV's life cycle: it contributes to maintaining latency via indirect mechanisms, and it may also synergize with Z and R to enhance lytic replication through direct association with R and/or R-induced alterations in Ikaros' functional activities via cellular signaling pathways.
IMPORTANCE This is the first report showing that the cellular protein Ikaros, a known master regulator of hematopoiesis and critical tumor suppressor in acute lymphoblastic leukemia, also plays important roles in the life cycle of Epstein-Barr virus in B cells.
INTRODUCTION
Epstein-Barr virus (EBV) is a ubiquitous human gamma herpesvirus frequently associated with Burkitt's lymphoma (BL), Hodgkin's lymphoma, posttransplant lymphoproliferative disease (PTLD), nasopharyngeal carcinoma (NPC), and occasionally, T-cell lymphoma and gastric cancer (1). Primary infection can cause mononucleosis, after which EBV establishes latency in memory B cells, occasionally reactivating into lytic replication, especially during plasma cell differentiation (1, 2).
The switch from latency to lytic replication is regulated by the expression of the BZLF1 and BRLF1 viral immediate early (IE) genes and their encoded proteins, Z and R, respectively. During latency, cellular factors strongly repress transcription from their promoters, Zp and Rp (3–5). Reactivation into lytic replication involves the loss of these repressors together with the addition of activators of these promoters (1, 6–8). Z and R then activate each other's promoters to amplify their lytic-inducing effects and to cooperatively turn on the expression of early (E) genes involved in viral genome lytic replication (1, 9) and, subsequently, the expression of late genes that encode virion structural proteins (1). Z can induce reactivation in most epithelial and B-cell lines, while R can do likewise in some epithelial cell lines (1). Factors known to activate transcription from Zp and Rp include transforming growth factor β (TGF-β), B-cell receptor cross-linking, phorbol esters, butyrate, ionophores, and hypoxia (8, 10, 11).
Z is a bZIP transcription factor. It binds AP-1-like sites called Z-responsive elements (ZREs), preferentially activating transcription from the methylated forms of its target promoters, including the methylated EBV genomes present in latently infected B cells (12, 13). The cellular transcription factors Oct-2, Pax-5, p65 subunit of NF-κB, and c-Myc promote EBV latency in part by interacting with Z, inhibiting its functional activities (14–17).
R is a 605-amino acid protein (see Fig. 7A below). Its amino-terminal region contains overlapping dimerization and DNA-binding domains (DBDs), while its carboxy-terminal region contains acidic and accessory activation domains (AD) (18, 19). All gamma herpesviruses encode an R-like protein, with their DBDs exhibiting high homology. R directly activates many EBV genes, including BMRF1 (encoding early antigen diffuse [EAD]), BMLF1 (encoding SM), and BALF2, by binding GC-rich motifs known as R-responsive elements (RREs) (20). R also indirectly activates many genes, including c-Myc, by interacting with cellular transcription factors like Sp1, MCAF1, and Oct-1 or by altering cellular signaling pathways (21–25). In addition, two EBV-encoded early proteins affect R's activities: BRRF1 activates phosphorylation of c-Jun, which then synergizes with R to activate Zp (26, 27), and LF2 binds R, redistributing it to the cytoplasm (28).
FIG 7.

Conserved hydrophobic amino acid residues 249, 250, 254, and 255 of R are critical for its interaction with Ikaros. (A) Schematic showing R's DNA-binding, dimerization, nuclear localization (NLS), and accessory and acidic activation domains (AD). Numbers indicate amino acid residues. Deletion mutants analyzed in coimmunoprecipitation assays are shown; kinks denote internally deleted regions. (B) Immunoblot showing coimmunoprecipitation of R mutant variants with IK-1. 293T cells in 6-well plates were cotransfected as follows: lanes 1 and 8, 0.28 μg pcDNA3-HA-IK-1; lanes 2 and 9, 0.25 μg pcDNA3-R; lanes 3 and 10, 0.45 μg pcDNA3-R-M1; lanes 4 and 11, 0.30 μg pcDNA3-R-M2; lanes 5 and 12, 0.31 μg pcDNA3-HA-IK-1 plus 0.25 μg pcDNA3-R; lanes 6 and 13, 0.25 μg pcDNA3-HA-IK-1 plus 0.45 μg pcDNA3-R-M1; and lanes 7 and 14, 0.28 μg pcDNA3-HA-IK-1 plus 0.30 μg pcDNA3-R-M2; total DNA was brought up to 0.70 μg per well with pcDNA3.1 where needed. Whole-cell extracts were prepared 48 h later, and complexes were coimmunoprecipitated with anti-HA tag antibody. (C) Alignment of amino acid residues 248 to 256 of EBV R with similar residues from the R-like proteins of some other gamma herpesviruses. Conserved hydrophobic residues are emphasized by boxes. The substitution mutations present in quadruple mutant R-QM are shown. (D) Immunoblot showing reduced coimmunoprecipitation of mutant R-QM with IK-1. 293T cells in 6-well plates were cotransfected as follows: lanes 1 and 6, 0.20 μg pcDNA3-HA-IK-1; lanes 2 and 7, 0.20 μg pcDNA3-R; lanes 3 and 8, 0.20 μg pcDNA3-R-QM; lanes 4 and 9, 0.36 μg pcDNA3-HA-IK-1 plus 0.20 μg pcDNA3-R; and lanes 5 and 10, 0.36 μg pcDNA3-HA-IK-1 plus 0.20 μg pcDNA3-R-QM; total DNA was brought up to 0.56 μg per well with pcDNA3.1 where needed. Whole-cell extracts were prepared and processed as described in the legend for panel B. (E) Immunoblot showing failure of mutant R-QM to disrupt EBV latency. 293T-EBV cells in a 12-well plate were transfected with the indicated amounts of pcDNA3-R or pcDNA3-R-QM plus pcDNA3.1 to bring total DNA to 0.3 μg per well and were harvested 48 h later. (F) Luciferase reporter assays showing failure of mutant R-QM to activate the EBV SM (BMLF1) promoter. BJAB cells were coelectroporated with 1.7 μg pCpGL-SMp reporter plasmid, 0.4 μg pcDNA3-eGFP, and the indicated amounts of pcDNA3-R or pcDNA3-R-QM (plus vector pcDNA3.1 to bring total DNA to 2.7 μg per sample). Luciferase activities were determined 44 h later. Data were normalized internally to the amount of protein in each lysate and externally to basal activity observed in the absence of R. Immunoblot analysis was also performed to determine WT and mutant R protein levels. WB, Western blot.
Ikaros, encoded by the cellular Ikzf1 gene, is a member of the Kruppel zinc finger family of transcription factors. It is predominantly expressed in hematopoietic cells (29) but can also be detected in the brain and pituitary gland (30). Ikaros is a key regulator of lymphopoiesis, contributing to B lineage specification, commitment, and maturation (31). It functions as a tumor suppressor in B-cell acute lymphoblastic leukemia (B-ALL), with somatic mutations of Ikzf1 present in a large percentage of B-ALLs (32).
Full-length Ikaros, IK-1, contains four amino-terminal zinc fingers that mediate DNA binding to motifs resembling 5′-GGGAA-3′ and two carboxy-terminal zinc fingers required for dimerization with itself and other members of this family (see Fig. 8A below) (33). Thirteen isoforms have been identified that result from alternatively spliced transcripts or mutation of the Ikzf1 gene (34, 35). The most abundant Ikaros isoforms in human lymphoid cells are IK-1 and IK-H. IK-H, containing 20 more amino acids than IK-1, preferentially associates with the regulatory regions of genes activated by Ikaros (36). Among the numerous smaller Ikaros isoforms are IK-2, which lacks the first amino-terminal zinc finger, and IK-6, which lacks all four amino-terminal zinc fingers and has a dominant-negative function, inhibiting IK-1's activities (37–39).
FIG 8.

Ikaros domains involved in its interaction with R. (A) Schematic diagrams showing structures of IK-1, IK-H, IK-6, and deletion variants studied here. Numbers indicate amino acid residues. F1 to F6 denote zinc fingers. +/−, +, and ++ denote interaction with R that was less than, similar to, or greater than that observed with IK-1, respectively. (B, C, and D) Immunoblots showing coimmunoprecipitation of R with Ikaros deletion variants. (B) 293T cells in 6-well plates were cotransfected as follows: lanes 1 and 6, 0.1 μg pcDNA3-R; lanes 2 and 7, 0.1 μg pcDNA3-R plus 0.2 μg pcDNA3-HA-IK-1; lanes 3 and 8, 0.1 μg pcDNA3-R plus 0.9 μg pcDNA3-HA-IKΔ1-310; lanes 4 and 9, 0.1 μg pcDNA3-R plus 0.9 μg pcDNA3-HA-IKΔ311-415; and lanes 5 and 10, 0.1 μg pcDNA3-R plus 0.9 μg pcDNA3-HA-IKΔ416-460; total DNA was brought up to 1.0 μg per well with pcDNA3.1 where needed. Whole-cell extracts were prepared 48 h later, and protein was immunoprecipitated with anti-HA tag antibody. (C) 293T cells in 6-well plates were cotransfected as follows: lanes 1 and 6, 0.2 μg pcDNA3-R; lanes 2 and 7, 0.2 μg pcDNA3-HA-IK-1; lanes 3 and 8, 0.2 μg pcDNA3-HA-IKΔZF5; lanes 4 and 9, 0.2 μg pcDNA3-R plus 0.36 μg pcDNA3-HA-IK-1; and lanes 5 and 10, 0.2 μg pcDNA3-R plus 0.36 μg pcDNA3-HA-IKΔZF5; total DNA was brought up to 0.56 μg per well with pcDNA3.1 where needed. Whole-cell extracts were processed as described above. (D) 293T cells were cotransfected and processed as described for the experiment whose results are shown in panel C, except with pcDNA3-HA-IKΔZF6 in place of pcDNA3-HA-IKΔZF5. (E) Immunoblot showing coimmunoprecipitation of R with eGFP-fused IK416-519. 293T cells in 6-well plates were cotransfected with 0.1 μg pcDNA3-R and 0.9 μg pcDNA3-HA-eGFP-2XNLS or 0.2 μg pcDNA3-HA-eGFP-2XNLS-IK416-519 plus 0.7 μg pcDNA3.1. Whole-cell extracts were processed as described above, except that blots were probed with anti-GFP antibody.
Ikaros can either activate or repress the transcription of its target genes, doing so via direct binding, inducing chromatin remodeling (29, 40–42), or recruiting to pericentromeric heterochromatin (43–45). Ikaros represses in association with the nucleosome remodeling and deacetylase (NuRD) complex, Mi-2β, Sin3A, and Sin3B, in a histone deacetylase (HDAC)-dependent manner or with CtBP and CtIP in an HDAC-independent manner (46–48). It activates in association with Brg-1, a catalytic subunit of the SWI/SNF chromatin remodeling complex (49, 50). Ikaros is involved in regulating genes involved in B-cell lineage, DNA repair, cell cycle, apoptosis, JAK-STAT, and Notch signaling (46, 51). Its activities are regulated by posttranslational modifications, including phosphorylation and sumoylation (52–54).
A role for Ikaros in the life cycle of a virus has only been reported for the mink cell focus-inducing virus MCF247, a nonacute murine leukemia virus (55). In this case, Ikaros enhances transcription from the viral promoter via sequence-specific binding in the U3 region; virus mutated in this site replicates less efficiently in thymocytes and induces T-cell lymphomas with a delayed onset in newborn mice.
Despite its critical roles in lymphocyte development and tumor suppression, no previous studies have examined the effects of Ikaros on the life cycle of any human lymphotropic virus, including EBV, which harnesses the B-cell differentiation program to regulate its latent-lytic switch. Here, we show that knockdown of Ikaros by small hairpin RNAs (shRNAs) induces reactivation in EBV-positive (EBV+) B-cell lines, an effect that synergizes with other lytic inducers of EBV. It does so by affecting the expression of some cellular factors known to inhibit EBV reactivation and plasma cell differentiation. Ikaros also complexes with R; the presence of R alleviates Ikaros-mediated repression. Ikaros may then synergize with R and Z to enhance reactivation. Thus, we conclude that Ikaros plays important roles in regulating EBV's latent-lytic switch in B cells.
MATERIALS AND METHODS
Cells.
Sal (gift from Alan Rickinson) is a W promoter (Wp)-restricted BL cell line coinfected with wild-type (WT) and EBNA2-deleted EBV genomes (56, 57). Akata, MutuI, and KemI (gifts from Kenzo Takada, Alan Rickinson, and Jeff Sample, respectively) are EBV+ BL cell lines in type I latency, expressing only EBNA1 (58). MutuIII and KemIII are cell lines derived from the same tumors as MutuI and KemI, but they maintain a type III latency program (59, 60). EBV-negative (EBV−) Mutu (gift from John Sixbey) was derived from MutuI (61). BJAB is another EBV− BL cell line (gift from Bill Sugden). BJAB-EBV was derived from BJAB by infection with the EBV strain B95.8 BAC, p2089 (62). The lymphoblastoid cell lines (LCLs) D4 (63) and WT3333 in type III latency were derived from in vitro infection of primary B cells with EBV. Simian virus 40 (SV40)-infected human embryonic kidney 293T cells were purchased from ATCC. 293T-EBV cells were generated by transfection of 293T cells with p2089 (R. J. Kraus, X. Yu, S. Sathiamoorthi, N. Ruegsegger, D. M. Nawandar, S. C. Kenney, and J. E. Mertz, unpublished data). All of the B-cell lines and 293T were maintained in RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals or HyClone/Thermo Scientific) and 100 units/ml penicillin plus 100 μg/ml streptomycin (Pen&Strep) or 100 μg/ml of the antimicrobial Primocin (InvivoGen). The 293T-EBV cells were grown in RPMI supplemented with 10% FBS, 100 μg/ml hygromycin B, and Pen&Strep or 100 μg/ml Primocin. All cells were maintained at 37°C in a 5% CO2 incubator.
Plasmids.
The expression plasmids pcDNA3-HA-IK-H and pcDNA3-HA-IK-1 encode hemagglutinin (HA)-tagged human IK-H and IK-1, respectively (36). The firefly luciferase reporter pGL4.15-c-Mycp (gift from Chunhua Song) contains nucleotides (nt) −1,936 to +525 of the c-Myc promoter cloned into pGL4.15 (Promega). The renilla luciferase reporter pRom-Hes1p contains nt −860 to +200 of the cellular Hes1 promoter (Switchgear Genomics). The firefly luciferase reporters pCpGL-SMp and pCpGL-BALF2p contain the EBV BMLF1 (EBV nt 84,311 to 84,922) and BALF2 (EBV nt 164,776 to 165,375) promoters, respectively, cloned into pCpGL-Basic (12). The mammalian expression plasmids p3xFLAG-Z (gift from Paul Lieberman) and pSG5-Z (gift from Diane Hayward) contain EBV Z cDNA and genomic DNA cloned into p3xFLAG-myc-CMV24 (Sigma) and pSG5 (Agilent Technologies), respectively. The expression plasmids pcDNA3-R and pcDNA3-R-V5 encode wild-type and V5-tagged EBV R, respectively (28). Plasmid pCVL-EμB29-MCS-T2A-GFP (number 525) (gift from David Rawlings) is a B-cell lentivirus expression vector (64). The expression vector pCDH-EF1-MSC-EF1-GFP+Puro (abbreviated pCDH-EF1; gift from Stacy Hagemeier) was constructed by substituting the EF1 promoter for the murine stem cell virus (MSCV) promoter in pCDH-MSCV-MCS-EF1-GFP+Puro (CD713B-1; System Biosciences). Plasmid DNAs were purified using plasmid plus midi kits (Qiagen).
Cloning.
Plasmids expressing mutant variants of EBV R were constructed as follows. Plasmid pcDNA3-RΔ416-605-V5 was generated by PCR amplification with an oligonucleotide (5′-GGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACG-3′) containing the V5 epitope tag immediately downstream from amino acid residues 1 to 415 of the R open reading frame (ORF). Plasmids pcDNA3-RΔ350-408, pcDNA3-RΔ280-360, pcDNA3-RΔ233-280 (encoding R deletion variant R-M1), and pcDNA3-RΔ249-256 (encoding R-M2) were generated by overlap extension PCR with appropriate primer pairs to contain the deletion mutations indicated below. The quadruple mutant pcDNA3-R-QM was constructed from pcDNA3-R by overlap extension PCR using oligonucleotides that encode the four amino acid substitution mutations V249R, L250A, L254R, and L255A in R. All PCR products were then cloned into the NotI/XbaI sites of pcDNA3.1.
The Ikaros expression plasmids pcDNA3-HA-IK-6 (containing Δ54-283), pcDNA3-HA-IKΔ1-310, pcDNA3-HA-IKΔ311-415, pcDNA3-HA-IKΔ416-460, pcDNA3-HA-IKΔ462-484 (IKΔZF5), and pcDNA3-HA-IKΔ485-519 (IKΔZF6) were constructed likewise, using a forward primer encoding an HA epitope tag (5′-TACCCATACGATGTTCCAGATTACGCT-3′) located immediately amino terminal of the Ikaros ORF. Lentiviruses expressing the nontagged Ikaros isoforms 525-IK-H, 525-IK-1, and 525-IK-6 were generated by PCR amplification of the Ikaros ORFs from pcDNA3-HA-IK-H, pcDNA3-HA-IK-1, and pcDNA3-HA-IK-6, respectively, followed by cloning into the NotI/PacI sites of vector 525. The expression plasmids pCDH-EF1-HA-IK-1 and pCDH-EF1-HA-IK-6 were generated by PCR amplification of the sequences from the corresponding HA-tagged Ikaros isoforms followed by cloning into the NheI/BamHI sites of pCDH-EF1. The expression plasmid pcDNA3-HA-eGFP-2XNLS-IK-416-519 encodes HA-tagged enhanced green fluorescent protein (eGFP) linked to two copies of the SV40 nuclear localization signal (NLS) fused with amino acid residues 416 to 519 of Ikaros; it was generated by PCR amplification of the eGFP-encoding sequences from vector 525 and amino acid residues 416 to 519 of Ikaros from pcDNA3-HA-IK-1 using the following primer pairs: F1 (5′-ACTGGCGGCCGCACCATGTACCCATACGATGTTCCAGATTACGCTAATGGGGCCGCAATGGTGAGCAAGGGCGAGG-3′) and R1 (5′-TACCTTTCTCTTCTTTTTTGGATCAACTTTCCTCTTTTTCTTAGGGTCAGATCTGAGTCCGGACTTGTACAGCTCGTCCATGCC-3′) and F2 (5′-GACCCTAAGAAAAAGAGGAAAGTTGATCCAAAAAAGAAGAGAAAGGTAGATACGGCCGCAAACCACATCGCCCCGCAC-3′) and R2 (5′-AGCCTCTAGATTACTAGCTCATGTGGAAGCGGTGC-3′). The first-round PCR products were then mixed together and amplified with primers F1 and R2, followed by cloning into the NotI/XbaI sites of pcDNA3.1. The expression plasmid pcDNA3-HA-eGFP-2XNLS was constructed from pcDNA3-HA-eGFP-2XNLS-IK-416-519 by using the primer pair F1 and R3 (5′-AGCCTCTAGATTACTATGCGGCCGTATCTACCTTTC-3′) and cloning into the NotI/XbaI sites of pcDNA3.1. All plasmid constructs were verified by sequencing.
Transient transfections.
293T and 293T-EBV cells were transfected with Lipofectamine 2000 (Invitrogen) or TransIT-LT1 (Mirus). BJAB and BJAB-EBV cells were electroporated by nucleofection (Lonza). In brief, 2.7 × 106 cells per sample were pelleted, resuspended in 100 μl of buffer V, combined with 2.5 to 2.8 μg DNA, transferred into Ingenio cuvettes (Mirus), and electroporated with a Nucleofactor II device using the G-016 program.
Infection of B cells with lentivirus.
For knockdown of protein expression, pLKO.1 lentiviral vectors expressing the nontargeting shRNA control #1 (number 1864; Addgene) or control #2 (SHC002; Sigma) or five shRNAs targeting Ikaros (RHS4533-EG10320; Thermo Scientific) were used to produce lentivirus, following the protocol of Open Biosystems. In brief, 293T cells in 10-cm dishes were cotransfected with 4 μg lentiviral vector(s), 1.4 μg packaging plasmid pCMV-dR8.2 dvpr (number 8455; Addgene), and 0.6 μg of a plasmid encoding vesicular stomatitis virus G (VSVG) (gift from Bill Sugden). Medium containing the lentivirus(es) was harvested 72 h later, filtered through an 0.8-μm pore-size filter, and added to the cells. Infected cells were selected 72 h later by incubation with 1 μg/ml puromycin for 4 days and then incubated for 24 h with 100 pM TGF-β1 (R&D Systems) where indicated below before harvesting.
For protein overexpression, 293T cells in 10-cm dishes were cotransfected with 4 μg of the indicated lentivirus expression vector (525-IK-H, 525-IK-1, 525-IK-6, or 525), 0.7 μg psPAX2 (number 12260; Addgene), 0.7 μg pCMV-dR8.2 dvpr, and 0.6 μg VSVG-encoding plasmid. The medium was collected after 72 h, processed, and used to infect cells as described above.
Immunoblot analysis.
Proteins were processed as previously described, with the following modifications (65). In brief, whole-cell extracts were prepared by lysis in SUMO lysis buffer containing inhibitor cocktail (1× protease inhibitor cocktail set III [EMD Millipore], 1× Halt protease and phosphatase inhibitor cocktail [Thermo Scientific], 1 mM phenylmethylsulfonyl fluoride [PMSF]). Following sonication, the protein concentration was determined using Pierce 660-nm protein assay reagent (Thermo Scientific) in the presence of ionic detergent compatibility reagent (Thermo Scientific). The proteins were resolved by electrophoresis in SDS 10% TGX polyacrylamide gels (Bio-Rad) and transferred to nitrocellulose membranes (Whatman). The membranes were blocked with phosphate-buffered saline (PBS) containing 5% skim milk, 0.05% Tween 20 and incubated overnight with the primary antibody at 4°C. The following antibodies were used: anti-Z (1:500, sc-53904; Santa Cruz Biotechnology), anti-R (1:2,000, 11–008; Argene), anti-EAD (1:500, VP-E608; Vector Laboratories), anti-Bcl-6 (1:200, sc-70414; Santa Cruz Biotechnology), and anti-Oct-2 (1:6,000; sc-233; Santa Cruz Biotechnology) antibodies. Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-horseradish peroxidase (HRP) antibody (1:4,000, A00192; Genscript) served as a loading control.
For detection of Ikaros, an amount of 0.7 to 1.5 μg of whole-cell extract was separated by electrophoresis in 4 to 12% Bis-Tris polyacrylamide gels (Invitrogen) in morpholinepropanesulfonic acid (MOPS) SDS running buffer and transferred to nitrocellulose membranes. Anti-Ikaros antibodies were used to detect the IK-H, IK-1, and IK-2/3 isoforms (1:5,000, number 200503; Intrepid Biosciences) and the IK-6 isoform (1:1,000, AF4984 [R&D Systems] or 1:1,000, number 9034 [Cell Signaling]). The secondary antibody was HRP-conjugated anti-mouse (1:2,500 to 6,000, number 31430 [Thermo Scientific] or 1:2,500 to 6,000, A00160 [Genscript]) or anti-rabbit (1:2,500, NA9340V; GE Healthcare) antibody. The blots were developed with Luminata Crescendo Western HRP substrate (Millipore). The band intensities were quantified using ImageJ software and internally normalized to GAPDH.
Coimmunoprecipitation assays.
For exogenously expressed proteins, 293T cells in 6-well plates were transfected with various amounts of DNAs as indicated below, using Lipofectamine 2000 or Trans-IT-LT1. After incubation for 44 to 48 h, the cells were lysed in TNE lysis buffer (50 mM Tris, pH 8.0, 0.15 M NaCl, 5 mM EDTA, 1% NP-40) containing inhibitor cocktail, followed by sonication twice for 20 s. The protein lysate (350 to 400 μg in 500-μl volume) was diluted 2-fold with TNE wash buffer (50 mM Tris, pH 8.0, 0.15 M NaCl, 5 mM EDTA, 0.5% NP-40) and incubated with rotation at 4°C for 2 h with 1 μg anti-HA tag antibody (ab9110; Abcam). Chromatin immunoprecipitation (ChIP)-grade protein G magnetic beads (Cell Signaling) were added (30 μl/reaction mixture volume), and incubation with rotation was continued at 4°C overnight. The magnetic beads were washed four times with TNE wash buffer. The proteins were eluted with SDS loading buffer and processed for immunoblot analysis.
Coimmunoprecipitation of endogenous proteins was performed as previously described, with some modifications (66). Sal cells in 10-cm plates were incubated with 100 pM TGF-β1 for 3 days to induce EBV lytic replication, pelleted, and lysed by the addition of 20 volumes of Triton-X lysis buffer 1 (25 mM Tris-Cl, pH 8.0, 10 mM KCl, 10 mM MgCl2, 2 mM EDTA, 10% glycerol, 1% Triton X-100, 10 μM ZnCl2) containing inhibitor cocktail, followed by sonication twice for 20 s. For each ml of Triton-X lysis buffer 1, 88 μl of 5 M NaCl was added, and the extract was incubated at 0°C for 1 h with occasional mixing, sonicated twice for 20 s, and cleared by centrifugation at 10,000 × g for 30 min at 4°C. The resulting supernatant was diluted 2.8-fold with lysis buffer 2 (25 mM Tris-HCl, pH 8.0, 5 mM EDTA, 10 μM ZnCl2) containing inhibitor cocktail. An amount of 400 μg of this lysate was incubated with rotation at 4°C for 2 h with a 1:1,000 dilution of a rabbit polyclonal anti-Ikaros C-terminal sequence (CTS) (67) or anti-IgG isotype control (number 2729; Cell Signaling) antibody. Protein G magnetic beads were added, and incubation was continued at 4°C overnight. The beads were washed with buffer (50 mM Tris, pH 8.0, 0.15 M NaCl, 5 mM EDTA, 0.5% NP-40, 10 μM ZnCl2). The proteins were eluted and processed as described above.
ChIP assays.
ChIP assays were performed essentially as previously described (12). Cells were cross-linked by incubation with 1% fresh paraformaldehyde at room temperature for 10 min, quenched by the addition of 125 mM glycine, and lysed by Dounce homogenization. The lysate was sonicated thrice for 30 s to yield DNA fragments of approximately 500 bp. The DNA-protein complexes were immunoprecipitated by incubation at 4°C overnight with 2 μg anti-Ikaros (sc-13039X; Santa Cruz Biotechnology), anti-HA tag (ab9110; Abcam), anti-V5 (ab15828; Abcam), or IgG control (number 2729; Cell Signaling) antibody. The immunoprecipitated DNA-protein complexes were sequentially washed at 4°C with gentle rocking for 5 min with low-salt, high-salt, lithium chloride, and Tris-EDTA buffers, respectively. The cross-linking was reversed by incubation at 65°C overnight, and the DNA was purified with a Qiagen gel extraction kit.
Ikaros ChIP-seq analysis.
Ikaros chromatin immunoprecipitation-sequencing (ChIP-seq) data from LCL GM12878 were downloaded from the ENCODE data repository (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhTfbs/). Sequence reads were mapped to the B95-8 genome (V01555.2) using the Burrows-Wheeler Aligner (BWA) (68). The position-specific read depth was calculated with a python script and displayed on a local installation of the UCSC genome browser. For positive controls, we downloaded the ENCODE data from the same ChIP-seq experiment for the cellular genes Ebf1 and CDKN1A.
qPCR.
Quantification of ChIPed DNA was performed by quantitative PCR (qPCR) using iTaq universal SYBR green supermix (Bio-Rad) or SsoAdvanced universal SYBR green supermix (Bio-Rad) and an ABI Prism 7900 real-time PCR system (Applied Biosystems). The primers were as follows: Zp, FWD (5′-GCCATGCATATTTCAACTGGGCTG-3′) and REV (5′-TGCCTGTGGCTCATGCATAGTTTC-3′); Rp, FWD (5′-CCAGCCAGATGTTCAGGAACCAAA-3′) and REV (5′-GCATGGGCGGGACAATCGCAATATAA-3′); SMp, FWD (5′-AATGTCTGCGCCATGATAGAGGGA-3′) and REV (5′-CGGTTTGCTCAAACGTGACATGGA-3′); Ebf1p, FWD (5′-GGGTTAGTGTGCCTGTGTTTAG-3′) and REV (5′-CTGCTGGATGGAGATTCTGTTT-3′); Mcl1p, FWD (5′-GCTCGCCACTTCTCACTTC-3′) and REV (5′-AGGCCAAACATTGCCAGT-3′); and CDKN1Ap, FWD (5′-TGCCGAAGTCAGTTCCTTGTGG-3′) and REV (5′-GCCGCTCTCTCACCTCCTCTG-3′).
The input samples were diluted to 5%, 1%, and 0.2% with distilled water containing 100 μg/ml sheared salmon sperm DNA (Ambion). A standard curve was calculated from the threshold cycle (CT) of the input dilution series and used to calculate the relative amount of each specific DNA present in the samples after ChIP. All assays were performed in triplicate.
Immunofluorescence assay.
Sal cells were incubated for 24 h with 200 pM TGF-β1 prior to seeding onto poly-d-lysine-coated glass coverslips (BD Biosciences), drying, fixing by incubation at room temperature for 25 min with 4% paraformaldehyde in PBS, washing with Tris-buffered saline (TBS), and permeabilizing by incubation for 10 min with 0.2% Triton X-100 in PBS. The cells were then incubated for 1 h with blocking solution (1% bovine serum albumin, 0.5% donkey serum, 0.5% goat serum in PBS) and for 1 h with rabbit anti-Ikaros CTS antibody (1:100), mouse anti-R antibody (1:80, 11-008; Argene), and 4′,6-diamidino-2-phenylindole (DAPI) (1:1,000; Invitrogen) in blocking solution. After washing with TBS, the cells were incubated for 1 h with goat anti-rabbit Alexa Fluor 488 (1:500, A11008; Molecular Probes) and goat anti-mouse Alexa Fluor 594 (1:500, A11032; Molecular Probes) antibodies in blocking solution. The coverslips were washed and mounted with ProLong gold antifade reagent (Invitrogen). Images were taken with a Nikon Eclipse Ti confocal microscope with an apochromatic 1.40 numeric aperture, ×60 oil objective lens (Nikon) plus 3× optical zoom. Z stacks were collected using 2.5- to 3.0-μm optical sections.
Reporter assays.
293T cells were transfected with the DNAs indicated below (200 ng total DNA per well in 24-well plates) using TransIT-LT1. BJAB cells were electroporated with (i) 1.7 μg pCpGL-SMp reporter plasmid, (ii) 0.4 μg eGFP, and (iii) various amounts (indicated below) of pcDNA3-R wild type, its quadruple mutant pcDNA3-R-QM, and/or pcDNA3 empty vector as described above. The cells were harvested 44 to 48 h posttransfection. To measure the promoter activities of the pCpGL-SMp, pGL4.15, and pGL4.15-c-Mycp reporters, the cells were lysed in 1× passive lysis buffer (Promega) and clarified by centrifugation, and firefly luciferase activities were determined with a VICTOR X5 multilabel plate reader (PerkinElmer) using Promega's luciferase assay reagent. To measure the promoter activities of the pRom and pRom-Hes1p reporters, the cells were lysed in 1× LightSwitch luciferase assay reagent (Switchgear Genomics), and renilla luciferase activity was quantified likewise. Protein expression was verified by immunoblot analysis. For each condition, two or more independent experiments were performed in triplicate.
RESULTS
Ikaros contributes to maintenance of EBV latency in B cells.
Given that Ikaros is both a master regulator of lymphopoiesis and a tumor suppressor in B-ALL, we hypothesized that it also plays a key role in regulating EBV's life cycle. As a first step toward testing this possibility, we determined by immunoblot analysis the relative levels of Ikaros protein present in multiple EBV− and EBV+ B-cell lines. Consistent with Ikaros being present in hematopoietic stem cells through the mature B-cell stage (69), we observed expression of Ikaros in EBV− BL, EBV+ type I latency BL, Wp-restricted BL, type III latency BL, and LCL cells (Fig. 1, lane 1, lanes 2, 4, and 5, lane 3, lanes 6 and 7, and lanes 8 and 9, respectively). The amount of Ikaros was usually higher in the EBV+ type I latency and Wp-restricted cell lines than in the type III latency ones, with little or no IK-H observed in the latter (Fig. 1, lanes 2 to 5 versus lanes 6 to 9). The non-DNA-binding Ikaros isoforms were not detected (Fig. 2C and D; also data not shown).
FIG 1.
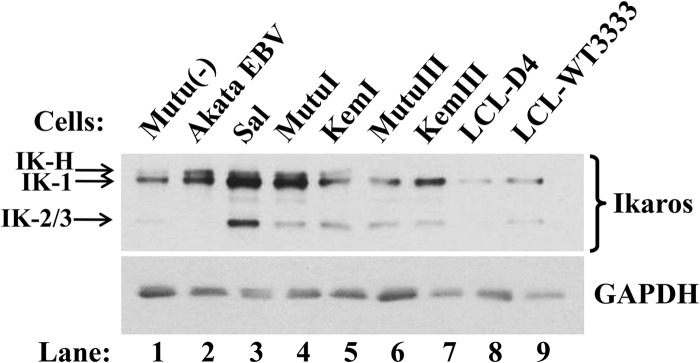
Ikaros is present in EBV+ B-cell lines. Immunoblot shows relative levels of endogenous Ikaros isoforms in a variety of EBV− and EBV+ B-lymphocytic cell lines. Whole-cell protein (0.8 μg per lane) was probed for Ikaros. GAPDH served as a loading control.
FIG 2.

Both knockdown of Ikaros and expression of a dominant-negative isoform, IK-6, enhance lytic EBV reactivation. (A) Immunoblots showing relative levels of some lytic EBV-encoded proteins following shRNA knockdown of Ikaros and incubation without (−) or with (+) TGF-β1. Sal and MutuI cells were infected for 3 days with lentivirus expressing nontargeting shRNA (Control #1) or a combination of five shRNAs targeting Ikaros, incubated for 4 days in the presence of puromycin (1 μg/ml), and then incubated for 24 h in the absence or presence of TGF-β1 (100 pM) immediately prior to preparing whole-cell extracts. (B) Immunoblots showing lytic EBV proteins following superinfection of Sal cells expressing the indicated shRNAs with lentivirus expressing IK-1 and incubation with TGF-β1. Cells were infected for 24 h with lentiviruses expressing nontargeting shRNAs (Control #1 and Control #2) or a combination of four shRNAs targeting Ikaros, superinfected for 2 days with 525 lentivirus expressing IK-1 (IK-1) or 525 as empty vector (Control), selected for 5 days with puromycin, and then incubated for 24 h with TGF-β1. (C) Immunoblots showing lytic EBV proteins following infection of Sal cells for 3 days with lentiviruses expressing the indicated isoforms of Ikaros, followed by incubation for 24 h with 0.2 mM hypoxia mimic DFO (+) or with DMSO as a control (−). (D) Immunoblots showing lytic EBV proteins following infection of MutuI cells for 3 days with lentiviruses expressing the indicated isoforms of Ikaros and incubation for 24 h with TGF-β1.
We next asked whether Ikaros might contribute to the maintenance of EBV latency in some B-cell lines that express Ikaros at high levels. To do so, we examined whether knockdown of Ikaros expression in MutuI and Sal cells induced lytic reactivation. Cells were infected with lentiviruses expressing five shRNAs targeting the coding region and 3′-untranslated region (UTR) of Ikaros mRNA or nontargeting shRNA (control #1). We achieved Ikaros knockdown of approximately 60 to 80% (Fig. 2A). Interestingly, this decrease in Ikaros levels led to significant increases in the synthesis of the lytic EBV IE Z and R and E EAD proteins compared to their synthesis in the control cells (Fig. 2A, lane 2 versus lane 1 and lane 6 versus lane 5). Similar results were obtained using four different shRNAs targeting the Ikaros coding region (Fig. 2B, lanes 1 to 3) or one targeting only the 3′-UTR of Ikaros mRNAs (data not shown). Thus, Ikaros contributes to the maintenance of EBV latency in some BL cell lines.
Ikaros knockdown enhances reactivation by lytic inducers.
TGF-β1 is a physiological inducer of EBV reactivation. If Ikaros truly functions to maintain latency, knockdown of Ikaros might synergize with TGF-β1 to enhance reactivation. This is what we observed. Incubation of Sal and MutuI cells with 100 pM TGF-β1 for 24 h led to increases in the levels of Z, R, and EAD similar to those observed in cells infected with lentiviruses encoding shRNAs targeting Ikaros (Fig. 2A, lane 3 versus lane 2 and lane 7 versus lane 6, respectively); the combination of Ikaros shRNAs plus TGF-β1 synergistically enhanced the expression of Z, R, and EAD compared to the effect of either agent by itself (Fig. 2A, lane 4 versus lanes 2 and 3 and lane 8 versus lanes 6 and 7).
To exclude the possibility that the Ikaros shRNAs induced EBV lytic gene expression via indirect, nonspecific effects, we also tested whether the overexpression of IK-1 could reverse this effect. Sal cells were infected for 24 h with lentiviruses expressing Ikaros shRNAs prior to superinfection with a lentivirus expressing IK-1, followed by puromycin selection for 5 days and incubation with TGF-β1 for 24 h immediately prior to harvest. Under these conditions, IK-1 accumulated to a high level regardless of the presence of Ikaros shRNAs (Fig. 2B, lanes 4 to 6); it completely blocked the EBV reactivation normally induced by TGF-β1 (Fig. 2B, lanes 4 and 5 versus lanes 1 and 2, respectively). IK-1 overexpression even prevented the high-level synergistic reactivation observed with Ikaros shRNAs plus TGF-β1 (Fig. 2B, lane 6 versus lane 3). Thus, we conclude that the reactivation observed following treatment of B cells with shRNAs targeting Ikaros is, indeed, due to the reduction in Ikaros protein levels.
Given that the shRNAs concurrently targeted all Ikaros isoforms, we likewise investigated the roles of IK-H and IK-6 in regulating EBV latency. Ectopic expression of dominant-negative isoform IK-6 increased EBV reactivation in Sal cells, as evidenced by enhanced synthesis of R and EAD (Fig. 2C, lane 4 versus lane 1). IK-6 but not IK-H or IK-1 also enhanced TGF-β1-induced lytic gene expression in MutuI cells (Fig. 2D, lane 4 versus lanes 1 to 3).
Hypoxia induces EBV lytic replication in some EBV+ cell lines (11). Thus, we examined whether IK-6 also synergizes with the hypoxia mimic desferrioxamine (DFO) to enhance reactivation. Incubation of Sal cells for 24 h with DFO modestly enhanced EBV lytic gene expression (Fig. 2C, lane 5 versus lane 1). Ectopic expression of IK-6 together with DFO treatment dramatically induced reactivation relative to the effect of either inducer by itself (Fig. 2C, lane 8 versus lanes 4 and 5). These findings confirm that IK-1 contributes to maintenance of EBV latency in B cells, since inactivating its function by the addition of this dominant-negative isoform induces lytic replication both by itself and in synergy with the EBV lytic inducers DFO and TGF-β1.
Ikaros does not bind to Zp or Rp.
To begin to understand how Ikaros helps maintain EBV latency, we performed ChIP assays to examine whether endogenous Ikaros in latently infected B cells binds to either of the EBV IE promoters, Zp and Rp. Chromatin obtained from Sal cells was immunoprecipitated with Ikaros-specific versus isotype control antisera, followed by quantitative real-time PCR analysis with appropriate primers. Ikaros bound to the cellular Ebf1 promoter, as expected (51), but not to Zp or Rp (Fig. 3A). Similar results were observed with MutuI cells (data not shown). To exclude the possibility that Ikaros associates with Zp and/or Rp at locations considerably removed from their transcription start sites, we also analyzed ChIP-seq data for Ikaros in the EBV+ LCL GM12878 obtained from the ENCODE database. We observed excellent peaks of Ikaros bound to the cellular Ebf1 and CDKN1A promoters, as expected (51), yet we saw no enrichment above input of DNA sequences located anywhere near the BZLF1 and BRLF1 regions of the EBV genome (Fig. 3B, middle and bottom versus top, respectively). Thus, we conclude that Ikaros does not bind either Zp or Rp during latency.
FIG 3.

Endogenous Ikaros does not associate with either Zp or Rp. (A) Results of ChIP-qPCR assays for Ikaros binding. Sal cells were processed for ChIP with an Ikaros-specific or IgG control antibody. Recovered DNA was subjected to qPCR with primers spanning the EBV Z (BZLF1) and R (BRLF1) promoters and the cellular Ebf1 promoter as a positive control. Error bars show standard deviations. (B) ChIP-seq data from the EBV+ LCL GM12878, downloaded from the ENCODE consortium website, of Ikaros binding to the EBV Z and R promoters and the positive-control cellular EBf1 and CDKN1A promoters. The top one of each pair of histograms shows the Ikaros binding densities over the indicated region of the genome, while the bottom shows the input DNA across the same region as a control. Open reading frames of the Z, R, Ebf1, and CDKN1A genes are shown as lines, with arrows indicating the direction of transcription.
Ikaros affects levels of some B-cell-specific transcription factors.
EBV establishes long-term latency in B cells, undergoing reactivation when they differentiate into plasma cells (2). Some B-cell-specific factors (e.g., Oct-2 and Pax-5) promote EBV latency (14, 15), while some plasma-cell-specific factors (e.g., XBP-1s and BLIMP-1) promote EBV lytic replication (6, 7, 70, 71). To further understand how Ikaros contributes to EBV latency, we examined the effect of changing its level on the expression of some cellular factors known to play key roles in regulating EBV's latent-lytic switch or B-cell differentiation into plasma cells. Knockdown of Ikaros in EBV+ MutuI and Sal cells decreased the levels of Oct-2 by 40% to 50% (Fig. 4A; also data not shown), while overexpression of IK-1 increased it by 2-fold (Fig. 4B). Knockdown of Ikaros also decreased the level of Bcl-6 by 70%, while not decreasing the level of Pax-5 (Fig. 4A; also data not shown). Others have shown that Ikaros upregulates Ebf1 expression (which negatively regulates Blimp-1) (51, 72) and downregulates Irf4 expression (which directly activates Blimp-1 transcription) (39, 73). Thus, we conclude that IK-1 indirectly contributes to EBV latency by regulating the levels of some cellular factors known to play direct roles in the maintenance of EBV latency and/or B-cell differentiation, including Oct-2 (which inhibits Z's activities) (14) and Bcl-6 (which represses Blimp-1 and promotes the expression of Bach2, which negatively regulates Blimp-1 and downregulates Irf4 expression) (73).
FIG 4.

Ikaros regulates the levels of some key players in B-cell differentiation. (A and B) Changes in levels of the indicated cellular transcription factors following knockdown (A) or overexpression (B) of Ikaros. (A) EBV+ MutuI cells were infected for 3 days with lentivirus expressing nontargeting shRNA (Control #1) or a combination of five shRNAs targeting Ikaros (Ikaros) and then incubated for 5 days in the presence of puromycin. Whole-cell extracts were processed for immunoblot analyses. (B) MutuI cells were infected for 4 days with lentivirus 525 expressing IK-1 (IK-1) or with the empty vector (Control) prior to harvesting for immunoblot analyses. (C) Differences in mRNA levels of some key transcription factors in memory B and plasma cells. Expression levels in memory B cells and in vitro-generated plasma cells and bone marrow plasma cells were visualized with Expression Atlas (experiment E-MEXP-2360; http://www-test.ebi.ac.uk/gxa/experiment/E-MEXP-2360/ENSG00000185811/cell_type) (74). Arrows indicate significant up- and downregulation. Error bars indicate maximum and minimum values; top of light, medium, and dark regions of each bar indicates 75th, 50th, and 25th percentile, respectively.
We hypothesized that Ikaros levels might decrease during the differentiation of B cells into plasma cells, along with other factors that inhibit EBV reactivation. To examine this possibility, we analyzed expression microarray data (74) for the levels of several factors known to be critical regulators of EBV's latent-lytic switch and/or B-cell differentiation. As expected, the RNA levels of Pax-5 dropped significantly while BLIMP-1 levels increased dramatically from memory B cells to plasma cells (Fig. 4C). The levels of Oct-2, Pax-5, ZEB1, and YY1, negative regulators of Z's activities or BZLF1 expression (14, 15, 62, 75), also declined. Unexpectedly, the level of Ikaros RNA did not decline significantly. Since Ikaros activity is heavily regulated by various mechanisms at a posttranslational level (52–54, 76), we hypothesize that its function likely changes during the transition of B cells into plasma cells. However, Ikaros protein levels could also be changing, given reports of poor correlation between them and Ikaros RNA levels (e.g., see reference 77).
Ikaros interacts and colocalizes with R.
Oct-2 and Pax-5 inhibit Z's activities by interacting with it (14, 15). Thus, we asked whether Ikaros might do likewise. First, we performed coimmunoprecipitation assays by cotransfecting 293T cells with expression plasmids encoding HA-tagged IK-1 and Z or R. While Z did not immunoprecipitate with IK-1 (Fig. 5A, lane 6), R did (Fig. 5B, lane 8). The latter interaction was confirmed by coimmunoprecipitation in the opposite direction by cotransfecting 293T cells with plasmids expressing HA-tagged IK-1 and V5-tagged R; IK-1 coimmunoprecipitated with R (data not shown).
FIG 5.

Ikaros interacts with R but not Z. (A) Immunoblot showing failure of Z to coimmunoprecipitate with Ikaros. 293T cells in a 6-well plate were cotransfected with 0.06 μg p3xFLAG-Z and 0.2 μg pcDNA3-HA-IK-1 (IK-1 + Z) or either expression plasmid (Z or IK-1) plus empty vector pcDNA3.1. Whole-cell extracts were prepared 48 h later, and proteins were immunoprecipitated (IP) with an anti-HA-tag antibody. (B) Immunoblot showing coimmunoprecipitation of Ikaros isoforms and R. 293T cells in a 6-well plate were cotransfected with 0.1 μg pcDNA3-R and either 0.6 μg pCDH-EF1-HA-IK-6 (R + IK-6), 0.2 μg pCDH-EF1-HA-IK-1 plus 0.4 μg empty vector pCDH-EF1 (R + IK-1), or 0.6 μg empty vector pCDH-EF1 (R). Whole-cell extracts were prepared 48 h later and incubated for 20 min at room temperature with 800 U of Omnicleave endonuclease (Epicentre) per sample (+) or the same volume of dilution buffer (−) prior to processing as described in the legend for panel A. (C) Immunoblot showing coimmunoprecipitation of endogenous Ikaros and R. Sal cells were incubated for 72 h without (−) or with (+) TGF-β1 to induce EBV reactivation prior to preparation of whole-cell extracts and immunoprecipitation with anti-Ikaros or IgG antibody.
Since IK-1 and R are both DNA-binding proteins, we performed several controls to ensure that this observed coimmunoprecipitation was truly due to direct protein-protein interactions. First, Z is also a DNA-binding protein, yet it did not coimmunoprecipitate with IK-1. Second, incubation of the cell extract with OmniCleave (an endonuclease that degrades both single- and double-stranded DNA and RNA) prior to immunoprecipitation had little effect on the amount of R coimmunoprecipitating with IK-1 (Fig. 5B, lane 8 versus lane 11). Third, IK-6, which lacks a DBD, interacted with R as strongly as did IK-1 both in the absence and presence of OmniCleave endonuclease (Fig. 5B, lane 9 versus lane 8 and lane 12 versus lane 11). Thus, we conclude that IK-1 complexes with R within cells overexpressing these proteins.
To confirm whether this Ikaros/R interaction also occurred under physiological conditions, Sal cells were incubated with TGF-β1 to induce R synthesis prior to harvesting. Two percent of the R protein present in the cell lysate coimmunoprecipitated with the endogenous Ikaros proteins (Fig. 5C, lane 6). Thus, endogenous Ikaros associates with R within EBV+ cells induced into lytic replication.
Given that Ikaros and R form complexes, we hypothesized that they partially colocalize within cells. To examine this possibility, we performed indirect immunofluorescence assays with Sal cells following incubation with TGF-β1 to induce R synthesis. Regardless of TGF-β1 treatment, confocal fluorescence images showed the normal speckled nuclear staining pattern expected for endogenous Ikaros (Fig. 6). Thus, the presence of R did not significantly alter the localization of Ikaros. When R was present, R partially colocalized with Ikaros. Thus, we conclude that Ikaros and R partially colocalize during lytic replication in B cells.
FIG 6.

Confocal immunofluorescence microscopy showing that Ikaros partially colocalizes with R within cells. EBV+ Sal cells were incubated for 24 h without (−) or with (+) TGF-β1 (200 pM) to induce EBV reactivation prior to fixation and processing for staining with anti-Ikaros and anti-R antibodies and DAPI. Nuclear DNA appears as blue, Ikaros as green, R as red, and Ikaros-R colocalization as yellow.
Conserved amino acids within R's DBD are important for binding Ikaros.
To begin to understand the biological significance of the Ikaros-R interaction, we mapped the domain of R required for its interaction with Ikaros. Coimmunoprecipitation assays were performed in 293T cells cotransfected with plasmids expressing HA-tagged-IK-1 and wild-type or deletion variants of R, all of which retained the NLS (Fig. 7). Initial experiments involving the R variants RΔ416-605, RΔ350-408, and RΔ280-360 indicated that the dimerization/DBD region was sufficient for interaction with IK-1 (data not shown).
To determine likely regions of R necessary for interaction with Ikaros, we performed an in silico analysis using ANCHOR (http://anchor.enzim.hu/) to predict disordered regions of R based upon the principal that disordered regions of proteins cannot form favorable intrachain interactions to fold on their own and, thus, frequently gain stabilizing energy by interacting with partners. We found that amino acid residues 249 to 256 of R came up as one of the candidate regions. Coimmunoprecipitation assays performed with HA-tagged-IK-1 plus wild-type (WT) R, RΔ233-280 (R-M1), or RΔ249-256 (R-M2) indicated that IK-1 did not interact with either R-M1 or R-M2 (Fig. 7B). Thus, one or more of the residues within the sequence from 249 to 256 is necessary for R's interaction with IK-1.
A multialignment analysis with the corresponding residues of R-like proteins encoded by other gamma herpesviruses indicated that the hydrophobic residues 249, 250, 254, and 255 are highly conserved (Fig. 7C). To determine whether these conserved residues are involved in interaction with IK-1, we constructed R-QM, an R variant containing substitution mutations in these four hydrophobic residues. This mutant exhibited a 75 to 80% reduction in its binding affinity for IK-1 compared to that of WT R (Fig. 7D), while an R variant containing alanine substitution mutations in residues 251 to 253 bound IK-1 as well as WT R (data not shown). Therefore, R residues 249, 250, 254, and/or 255 are important for the formation of IK-1/R complexes.
We next looked at R-QM's functional activities. To test for an ability to disrupt latency, we transfected R expression plasmids into 293T-EBV cells. While WT R readily induced EAD synthesis, R-QM failed to do so (Fig. 7E). We also examined the transcriptional activity of R-QM in a B-cell environment by performing luciferase reporter assays in EBV− BJAB cells. As expected, WT R strongly activated transcription from EBV's early lytic SM promoter; however, R-QM failed to do so even though it accumulated in cells to levels similar to the levels of WT R (Fig. 7F). Therefore, we conclude that R's residues 249, 250, 254, and/or 255 are critical for transcriptional activity, as well as for associating with Ikaros.
Ikaros binds R via its C-terminal domain.
To begin to understand how R modulates Ikaros' functions, we likewise mapped the domains of Ikaros involved in binding R. Coimmunoprecipitation assays were performed in 293T cells cotransfected with plasmids expressing WT R and HA-tagged-Ikaros isoforms or deletion variants (Fig. 8). Given that the naturally occurring isoforms, IK-H, IK-1, and IK-6 all interacted with R (Fig. 5B; also data not shown), we knew that (i) the extra 20 amino acids present in IK-H do not affect R binding and (ii) residues 54 to 283, including the entire DBD of Ikaros, are not necessary for this interaction. The deletion variants IKΔ311-415 and IKΔ416-460 also fully retained their ability to bind R (Fig. 8B, lanes 9 and 10 versus lane 7). The deletion of residues 1 to 310 decreased the interaction with R by approximately 70% (Fig. 8B, lane 8 versus lane 7), suggesting that a subset of these N-terminal amino acids contributes directly or indirectly to R binding.
The C-terminal zinc fingers of Ikaros (ZF5 and ZF6) are required for protein dimerization, high-affinity DNA binding, and transcriptional activity (78). Thus, we examined likewise whether they affect R binding. Variant IKΔZF5 interacted with R significantly better than did full-length IK-1 (Fig. 8C, lane 10 versus lane 9). Variant IKΔZF6 also bound R significantly better than did full-length IK-1, given that it accumulated to a much lower level than IK-1 and yet coimmunoprecipitated only 2-fold less R (Fig. 8D, lane 10 versus lane 9). Thus, dimerization of Ikaros is not required for its interaction with R; rather, IK-1 preferentially binds R as a monomer.
Previous reports showed that the association of Ikaros with Sin3, Mi-2, and HDAC2 involves both its N- and C-terminal domains (47). To examine this possibility for R binding, we constructed plasmids that express HA-tagged eGFP fused to SV40's NLS without (eGFP) or with IK-1 amino acid residues 416 to 519 (eGFP-IK416-519), respectively. Fusion with eGFP improved protein stability, and the SV40 NLS ensured it was delivered to the nucleus. eGFP-IK416-519 but not eGFP bound R in our coimmunoprecipitation assay (Fig. 8E, lane 4 versus lane 3). Thus, we conclude that both the N- and C-terminal domains of Ikaros contribute to its forming complexes with R, with its C-terminal residues 416 to 519 being sufficient.
Lack of significant effects of Ikaros and R on each other's chromatin occupancy.
Since Ikaros binding to R may involve some critical residues within R's DBD, we hypothesized that the presence of Ikaros might interfere with sequence-specific DNA binding by R. To test this possibility, we examined by quantitative ChIP assays R's ability to bind a well-known target promoter in the absence versus presence of Ikaros. For this experiment, we chose 293T-EBV cells because (i) they lack endogenous Ikaros, (ii) they contain EBV DNA, allowing for detection of R binding to the EBV SM promoter, and (iii) IK-1 ectopically expressed in 293T cells has a phosphorylation pattern similar to the one observed with endogenous Ikaros in B cells (79). The cells were cotransfected with plasmids expressing V5-tagged R plus various amounts of HA-tagged-IK-1 and processed 2 days later for both immunoblot analysis to verify the expression of R and IK-1 and ChIP with anti-V5 tag or isotype control antibody. As expected, R bound the SM promoter strongly; the presence of IK-1 had little effect on or slightly increased R binding (Fig. 9A). Thus, the presence of Ikaros does not inhibit sequence-specific DNA binding by R, at least for the SM promoter.
FIG 9.

Ikaros and R do not affect each other's chromatin occupancy. (A) ChIP-qPCR assays for R binding to the EBV SM promoter. 293T-EBV cells in 10-cm plates were cotransfected with the indicated amounts of pcDNA3-HA-IK-1 (IK-1) and/or pcDNA3-R-V5 (R) along with pcDNA3.1 DNA to bring the total DNA up to 9.0 μg per plate. ChIP assays were performed 44 h posttransfection with anti-V5 tag or IgG antibody, followed by qPCR with primers spanning the SM promoter. (B, C, and D) ChIP-qPCR assays for Ikaros binding to the cellular Ebf1, Mcl1, and CDKN1A promoters, as indicated. EBV− BJAB cells were coelectroporated with 1.5 μg pcDNA3-R-V5 (R) or pcDNA3.1 (−) plus 0.3 μg pcDNA3-eGFP and 1.0 μg pCpGL-SMp. The cells were processed 48 h later for ChIP with an Ikaros-specific or IgG antibody followed by qPCR with primers spanning the indicated promoters. All assays were performed in triplicate, with normalization to input DNA levels. Error bars show standard deviations.
We likewise investigated whether R affects sequence-specific DNA binding by endogenous Ikaros. EBV− BJAB cells were coelectroporated with a plasmid expressing V5-tagged R or the empty vector together with an SMp-luciferase reporter as an internal control to verify the synthesis of R. ChIP was performed 2 days later with polyclonal anti-Ikaros or isotype control antibody. The presence of R did not significantly affect the binding of endogenous Ikaros to Ebf1, Mcl1, and CDKN1A, known cellular targets of Ikaros (Fig. 9B to D). Therefore, the presence of R does not inhibit the binding of Ikaros to some of its targets. However, we cannot exclude the possibility that R affects Ikaros binding to other promoters not tested here or vice versa or that the amount of R synthesized in this experiment was insufficient to bind most of the endogenous Ikaros even though it activated 346-fold transcription from the cotransfected SMp-luciferase reporter.
Effects of Ikaros and R on each other's transcriptional activities.
Regardless of whether Ikaros affects R's DNA-binding activity or vice versa, they could well affect each other's transcriptional activities through direct and/or indirect mechanisms. To test this possibility, we first examined whether R affected Ikaros-mediated repression of c-Myc and Hes1, two of its well-known targets (40, 80). 293T cells were cotransfected with reporters expressed from these promoters together with various amounts of plasmids expressing V5-tagged R and HA-tagged IK-1 and harvested 2 days later for luciferase assays and immunoblot analyses to verify the expression of R and IK-1. Ectopic expression of IK-1 repressed basal transcription from the c-Myc and Hes1 promoters by up to 50% and 75%, respectively; the addition of R fully reversed this repression (Fig. 10A and B). On the other hand, IK-1 in reporter assays in EBV− NPC HONE-1 cells failed to inhibit R-mediated activation of transcription from the EBV SM and BHLF1 promoters, two of R's direct targets (data not shown). We also performed reporter assays in BJAB-EBV cells, which contain endogenous Ikaros and are not reactivated by the addition of R. As expected, the ectopic expression of R led to high-level activation of transcription from the EBV BALF2 promoter; however, coexpression of IK-1 slightly enhanced this activation rather than inhibiting it (Fig. 10C). Thus, the presence of R alleviates Ikaros-mediated repression, but IK-1 does not inhibit R-mediated activation.
FIG 10.

Effects of Ikaros and R on each other's transcriptional activity. (A and B) Luciferase assays showing that R alleviates repression by Ikaros. 293T cells in 24-well plates were cotransfected with 70 ng reporter pGL4.15-c-Mycp (A) or pROM-Hes1p (B) and the indicated amounts of pcDNA3-HA-IK-1 (IK-1) and/or pcDNA3-R-V5 (R) plus pcDNA3.1 for total DNA of 200 ng per well. Luciferase activities were measured 44 h later, with assays performed in triplicate. Data were normalized externally to the basal activity observed for each reporter in the absence of R and IK-1. Immunoblots at the bottom of each panel show the relative levels of R and IK-1 present in these extracts. (C) Luciferase assays showing that IK-1 enhances, not inhibits, activation by R. EBV+ BJAB cells were infected for 2 days with lentivirus expressing IK-1 (IK-1) or the empty vector (Control). Subsequently, the cells were coelectroporated with 1.6 μg pCpGL-BALF2p and the indicated amounts of pcDNA3-R-V5 plus pcDNA3.1 for total DNA of 2.5 μg per 2.7 × 106 cells. Luciferase activities were measured 48 h later, with assays performed in triplicate. Data were normalized internally to the amount of protein in each lysate and externally to the basal activity observed under each condition in the absence of R. Error bars show standard deviations. (D and E) Immunoblots showing that IK-1 synergizes with R and Z to induce high-level lytic gene expression. (D) 293T-EBV cells in 6-well plates were cotransfected with the indicated amounts of pcDNA3-HA-IK-1 and pcDNA3-R plus pcDNA3.1 for total DNA of 0.24 μg per well and harvested 48 h later. (E) BJAB-EBV cells were infected for 3 days with 525 lentivirus expressing IK-1 (IK-1) or 525 as empty vector (Control). Subsequently, the cells were coelectroporated with 0.8 μg pSG5-Z and/or pcDNA3-R-V5 plus pSG5 and pcDNA3.1 for total DNA of 2.5 μg per 2.7 × 106 cells and were harvested 48 h later.
We also investigated the effect of Ikaros on R's ability to disrupt latency. As expected, ectopic expression of R but not of IK-1 induced some lytic gene expression in 293T-EBV cells (Fig. 10D, lane 2 versus lane 3). Interestingly, cotransfection with both plasmids led to much higher-level synthesis of EAD than was observed with R by itself (Fig. 10D, lane 4 versus lane 2). We confirmed this unexpected synergistic effect of IK-1 on reactivation using more physiologically relevant BJAB-EBV cells, in which Z is the initial inducer of lytic replication. The ectopic expression of R, IK-1, and R plus IK-1 all failed to induce EAD synthesis (Fig. 10E, lanes 2, 5, and 6, respectively). Z induced low-level EAD synthesis, which may have been slightly enhanced when coexpressed with IK-1 (Fig. 10E, lane 3 versus lane 7). The addition of IK-1 together with Z and R strongly enhanced lytic gene expression (Fig. 10E, lane 8 versus lane 4), indicating that IK-1 synergized with R plus Z to reactivate EBV. Thus, we conclude that Ikaros may switch from a negative to a positive factor in helping to induce EBV lytic gene expression once Z and R are present.
DISCUSSION
Here, we tested the hypothesis that Ikaros contributes to the regulation of EBV's life cycle. First, we demonstrated that both knockdown of Ikaros expression and inhibition of Ikaros function by a dominant-negative isoform induce lytic gene expression in EBV+ B-cell lines (Fig. 2). The mechanism by which Ikaros promotes EBV latency does not involve direct binding to EBV's IE BZLF1 or BRLF1 genes (Fig. 3); rather, Ikaros does so indirectly, in part by influencing the levels of cellular factors that directly inhibit Z's activities or B-cell differentiation into plasma cells (Fig. 4). When R is present, Ikaros can form complexes with it and partially colocalize within cells (Fig. 5 and 6). The amino acid residues important for this IK/R interaction primarily lie within a highly conserved DBD of R (Fig. 7) and the C-terminal domain of Ikaros (Fig. 8). The presence of R alleviates Ikaros-mediated transcriptional repression while not significantly affecting its DNA-binding activity (Fig. 9 and 10). Ikaros may also synergize with R and Z to induce high-level reactivation (Fig. 10). Thus, we conclude that Ikaros plays important roles in EBV's life cycle: it contributes to the maintenance of latency via indirect mechanisms, and it may also synergize with Z and R to enhance lytic replication through direct association with R and/or R-induced alterations in Ikaros' functional activities via cellular signaling pathways.
Downregulation of Ikaros by EBV in type III latency.
Ikaros is expressed throughout hematopoiesis from stem cells to mature B cells (81). It continues to be expressed even in plasma cells (Fig. 4C) (74). We found that Ikaros is usually expressed at lower levels in EBV+ B cells in type III latency than in type I latency and Wp restriction (Fig. 1). Proper splicing and synthesis of Ikaros requires FoxO1, which is negatively regulated by phosphatidylinositol 3-kinase (PI3K) signaling (82). EBV-encoded LMP1 and LMP2A downregulate FoxO1 expression via PI3K-mediated nuclear export (83). The EBV latency III program also induces the expression of cellular microRNA-27a (miR-27a), which targets Ikaros mRNA (84, 85). Thus, EBV likely utilizes LMP1, LMP2A, and miR-27a to downregulate Ikaros expression in type III latency. It may do so because Ikaros can suppress cell cycle progression, induce apoptosis (86), and inhibit Notch signaling (87), thereby likely interfering with some EBNA2 and LMP2A functions (88, 89). Interestingly, HIV-1 also downregulates Ikaros, doing so via its TAR microRNAs (90).
Effects of Ikaros isoforms on EBV latency.
EBV+ B cells in type I latency contain several isoforms of Ikaros (Fig. 1). Knockdown of all of them with shRNAs induced EBV lytic gene expression (Fig. 2A), while overexpression of IK-1 inhibited the reactivation induced by TGF-β (Fig. 2B). IK-H is functionally distinct from IK-1. It potentiates binding by IK-1 to DNA with two Ikaros-binding sites, while inhibiting binding to DNA with only one site; it also binds to genes upregulated by Ikaros but not to genes repressed by Ikaros (36, 37). Nevertheless, as opposed to IK-6, whose expression reactivated EBV, IK-H did not significantly affect lytic gene expression in our assays (Fig. 2C and D).
Ikaros promotes EBV latency by indirect mechanisms.
We failed to find by ChIP-qPCR assays Ikaros associated near the transcription initiation sites of either Zp or Rp in Sal and MutuI cells (Fig. 3A). We also failed to observe effects of IK-1 on transcription from Zp and Rp in reporter assays performed in EBV− NPC HONE-1 cells (data not shown). ChIP-seq data from LCLs showed lack of binding of Ikaros anywhere near Zp or Rp (Fig. 3B). However, given that LCLs express all latent EBV proteins and usually contain lower levels of Ikaros, ChIP-seq profiles of Ikaros might be different in LCLs than in type I and Wp-restricted B cells. Thus, although we cannot yet definitively rule out the possibility that Ikaros may regulate BZLF1 and/or BRLF1 gene expression in type I latency by binding to regions somewhat removed from their transcription initiation sites, our findings suggest that Ikaros's contribution to the maintenance of EBV latency likely is not primarily via direct repression of IE gene expression.
We found that Ikaros induced the expression of the B-cell-specific factor Oct-2 (Fig. 4A and B), which inhibits Z's functions, preventing lytic reactivation (14). Ikaros also positively regulated the expression of Bcl-6, which maintains the germinal center B-cell phenotype and inhibits plasma cell differentiation (73). Thus, Ikaros indirectly promotes EBV latency at least in part by sustaining the expression of Oct-2 and Bcl-6. Nevertheless, while the expression levels of both Bcl-6 and Oct-2 decrease during plasma cell differentiation (91, 92), the RNA levels of Ikaros were not significantly different (Fig. 4C). It is likely that changes in the posttranslational modifications of Ikaros alter its activities to enable B-cell differentiation and EBV lytic replication.
Ikaros forms complexes with R.
The cellular factors Oct-2, Pax-5, p65 subunit of NF-κB, and c-Myc promote EBV latency by interacting with Z (14–17). Here, we showed that Ikaros interacts with R, partially colocalizing with it within the nuclei of cells (Fig. 5 and 6). Unfortunately, we could not definitively demonstrate that this protein-protein interaction is important for Ikaros' roles in EBV's life cycle because the region of R necessary for this interaction mapped to residues that are also critical for R's transcriptional activities (Fig. 7). We also cannot exclude the possibility that these residues of R do not directly interact with Ikaros, given that the substitution mutations we introduced might lead to improper folding of R, thereby inhibiting its ability to bind Ikaros directly or indirectly as a component of multiprotein complexes. Given their highly conserved nature (Fig. 7C), Ikaros may also interact with the R-like proteins of some other gamma herpesviruses.
Unlike that of EBV, Rta of Kaposi's sarcoma-associated herpesvirus (KSHV) binds RBP-Jκ, utilizing the Notch pathway for lytic reactivation (93). The region of KSHV Rta necessary for this binding likely involves its leucine-rich repeat region (i.e., residues 246 to 270) (93), which overlaps the corresponding residues of EBV R critical for Ikaros binding. Interestingly, Ikaros can bind the same DNA sequences as RPB-Jκ; it represses the Notch target gene Hes1 by competing with RPB-Jκ for binding to Hes1p (87). The fact that EBV R interacts with the Notch signaling suppressor Ikaros while EBNA2 and -3 interact with the Notch signaling mediator RPB-Jκ supports the notion that EBV exploits Notch signaling during latency, while KSHV exploits it during reactivation.
Both the N- and C-terminal regions of Ikaros contributed to its binding to R, with residues 416 to 519 being sufficient for this interaction (Fig. 8). Ikaros variants lacking either zinc finger 5 or 6 interacted considerably more strongly with R than did full-length IK-1. The latter finding suggests that Ikaros may preferentially complex with R as a monomer, with the resulting protein complex exhibiting distinct biological functions that favor lytic reactivation, as compared to Ikaros homodimers that promote latency.
R alters Ikaros' transcriptional activities.
While the presence of R did not significantly alter Ikaros DNA binding (Fig. 9B to D), it did eliminate Ikaros-mediated transcriptional repression of some known target genes (Fig. 10A and B). The simplest explanation for this finding is that Ikaros/R complexes preferentially contain coactivators rather than corepressors, while continuing to bind many, if not all of Ikaros' usual targets. Alternatively, R activates cellular signaling pathways that indirectly lead to alterations in Ikaros' posttranslational modifications (e.g., phosphorylations and sumoylations), thereby modulating its transcriptional activities and/or the coregulators with which it complexes. Unfortunately, we could not distinguish between these two nonmutually exclusive possibilities because we lacked an R mutant that was defective in its interaction with Ikaros but retained its transcriptional activities.
The presence of R frequently also led to decreased levels of endogenous Ikaros in B cells (Fig. 10C, for example). This effect was also observed in 293T cells cotransfected with 0.1 to 0.5 μg of R and IK-1 expression plasmids per well of a 6-well plate; the addition of the proteasome inhibitor MG-132 partially reversed this effect (data not shown). Thus, by analogy to KSHV Rta-induced degradation of cellular silencers (94), R-induced partial degradation of Ikaros might serve as a third mechanism for alleviating Ikaros-promoted EBV latency. Probably, all three mechanisms contribute to R's effects on Ikaros.
Ikaros may also synergize with R and Z to induce reactivation.
Unlike Pax-5 and Oct-2, which inhibit Z's function directly, the presence of Ikaros did not inhibit R's activities. For example, Ikaros did not inhibit R's DNA binding to the SM promoter (Fig. 9A). IK-1 also failed in reporter assays to inhibit R-mediated activation of the EBV SM and BHLF1 promoters in EBV− HONE cells (data not shown), and it even slightly enhanced R-mediated activation of the BALF2 promoter in B cells (Fig. 10C). Rather, coexpression of IK-1 and R synergistically enhanced the expression of the viral DNA polymerase processivity factor, EAD, in 293T-EBV cells (Fig. 10D). Given that the expression of R induces Z synthesis in 293T-EBV cells and that R and Z form complexes with MCAF1 (9), we hypothesize that Ikaros may enhance EBV lytic gene expression in part as one of multiple components of R/MCAF1/Z complexes. Consistent with this possibility, we found that overexpression of IK-1 together with Z and R synergistically induced EAD synthesis in BJAB-EBV cells 8-fold or more above the levels observed with two or one of these three factors (Fig. 10E). Taking all of our findings together, we conclude that Ikaros plays important roles in EBV's life cycle: it contributes to the maintenance of EBV latency via indirect mechanisms, and it may also promote lytic replication in cooperation with R and Z through direct association with R and/or R-induced alterations in Ikaros' functional activities via cellular signaling pathways.
Synergistic reactivation was not observed when IK-1 was overexpressed in the presence of lytic inducers (Fig. 2). However, lytic inducers typically only induce reactivation in a small subset of the cells, i.e., 2% of MutuI cells incubated with TGF-β1 for 24 h (8), while we infected most of the cells with the IK-1-expressing lentivirus. In addition, our transfection and electroporation methods used for the experiments whose results are shown in Fig. 10 delivered high levels of the R and Z expression plasmids to a fairly high percentage of the cells. Therefore, both the percentage of the cells coexpressing R and IK-1 and the molar ratio of R to IK-1 were much lower in the experiments whose results are shown in Fig. 2 than in those whose results are shown in Fig. 10. However, we do not exclude the possibility that the observed difference was a consequence of the use of different cell lines.
Model for Ikaros regulation of EBV.
We propose a working model for Ikaros-mediated regulation of EBV's life cycle (Fig. 11). Ikaros recruits coactivators via interaction with Brg-1, a subunit of the SWI/SNF remodeling complex (49, 50), activating the expression of negative regulators of EBV lytic infection (e.g., Oct-2) (Fig. 11A). Ikaros also associates with the NuRD complex, CtBP, or CtIP corepressors (46–48), inhibiting the expression of cellular genes that promote lytic EBV replication (Fig. 11B). In addition, it regulates the expression of several B-cell-specific factors to maintain B-cell identity and to inhibit differentiation into plasma cells (e.g., EBF1 and Bcl-6) (51). In the presence of lytic-inducing stimuli, R and Z are synthesized and some cellular repressors (e.g., Pax-5 and Oct-2) are lost (14, 15). The presence of R may directly and/or through cellular signaling pathways indirectly downregulate the level of Ikaros by a proteasome-dependent mechanism, alleviating Ikaros-mediated repression (Fig. 11C). R also alters the transcriptional activities of Ikaros, leading to loss of some cellular repressors and corepressors (Fig. 11D) and gain of some cellular activators and coactivators (Fig. 11E) of EBV lytic gene expression. Concomitantly, Ikaros enhances R's ability to activate its target genes through R-responsive elements (RRE), either directly by protein-protein interaction or indirectly through cellular signaling pathways (Fig. 11F), and to activate EBV lytic gene expression via Z-responsive elements (ZRE) as a component of R/MCAF1/Z complexes (Fig. 11G) (9).
FIG 11.

Working model for regulation of EBV's life cycle by Ikaros. See Discussion for details.
In summary, Ikaros may act as both a brake and a driver of lytic replication, depending upon the stage of EBV's life cycle. It promotes EBV latency in B cells by indirect mechanisms, such as sustaining the expression of Oct-2 and inhibiting B-cell differentiation into plasma cells. Once R is synthesized during reactivation, R either directly or indirectly affects the levels and functional activities of Ikaros, attenuating repression by Ikaros and favoring lytic reactivation over latency; Ikaros may also synergize with Z and R to enhance lytic gene expression.
ACKNOWLEDGMENTS
We thank Alan Rickinson, Jeff Sample, Bill Sugden, Kenzo Takada, and Xianming Yu for cell lines, Stacy Hagemeier, Diane Hayward, Paul Lieberman, David Rawlings, Chunhua Song, and Bill Sugden for plasmids, and members of the Johannsen, Kenney, and Mertz laboratories for suggestions and discussions.
This work was supported by U.S. Department of Health and Human Services NIH grants AI07034, CA22443, and CA14520 to J.E.M. and S.C.K. and HL095120 to S.D. T.I. is a Royal Thai Government Scholar with funding from the National Science and Technology Development Agency of Thailand.
Footnotes
Published ahead of print 12 February 2014
REFERENCES
- 1.Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2700 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79:1296–1307. 10.1128/JVI.79.2.1296-1307.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis AL, Wang Z, Yu X, Mertz JE. 2010. Either ZEB1 or ZEB2/SIP1 can play a central role in regulating the Epstein-Barr virus latent-lytic switch in a cell-type-specific manner. J. Virol. 84:6139–6152. 10.1128/JVI.02706-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu X, McCarthy PJ, Lim HJ, Iempridee T, Kraus RJ, Gorlen DA, Mertz JE. 2011. The ZIIR element of the Epstein-Barr virus BZLF1 promoter plays a central role in establishment and maintenance of viral latency. J. Virol. 85:5081–5090. 10.1128/JVI.02615-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gruffat H, Manet E, Sergeant A. 2002. MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 3:141–146. 10.1093/embo-reports/kvf031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun CC, Thorley-Lawson DA. 2007. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J. Virol. 81:13566–13577. 10.1128/JVI.01055-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhende PM, Dickerson SJ, Sun X, Feng WH, Kenney SC. 2007. X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J. Virol. 81:7363–7370. 10.1128/JVI.00154-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iempridee T, Das S, Xu I, Mertz JE. 2011. Transforming growth factor beta-induced reactivation of Epstein-Barr virus involves multiple Smad-binding elements cooperatively activating expression of the latent-lytic switch BZLF1 gene. J. Virol. 85:7836–7848. 10.1128/JVI.01197-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang LK, Chuang JY, Nakao M, Liu ST. 2010. MCAF1 and synergistic activation of the transcription of Epstein-Barr virus lytic genes by Rta and Zta. Nucleic Acids Res. 38:4687–4700. 10.1093/nar/gkq243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kenney SC. 2007. Reactivation and lytic replication of EBV. Chapter 25 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 11.Jiang JH, Wang N, Li A, Liao WT, Pan ZG, Mai SJ, Li DJ, Zeng MS, Wen JM, Zeng YX. 2006. Hypoxia can contribute to the induction of the Epstein-Barr virus (EBV) lytic cycle. J. Clin. Virol. 37:98–103. 10.1016/j.jcv.2006.06.013 [DOI] [PubMed] [Google Scholar]
- 12.Wille CK, Nawandar DM, Panfil AR, Ko MM, Hagemeier SR, Kenney SC. 2013. Viral genome methylation differentially affects the ability of BZLF1 versus BRLF1 to activate Epstein-Barr virus lytic gene expression and viral replication. J. Virol. 87:935–950. 10.1128/JVI.01790-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woellmer A, Arteaga-Salas JM, Hammerschmidt W. 2012. BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog. 8:e1002902. 10.1371/journal.ppat.1002902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson AR, Kwek SS, Kenney SC. 2012. The B-cell specific transcription factor, Oct-2, promotes Epstein-Barr virus latency by inhibiting the viral immediate-early protein, BZLF1. PLoS Pathog. 8:e1002516. 10.1371/journal.ppat.1002516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raver RM, Panfil AR, Hagemeier SR, Kenney SC. 2013. The B-cell-specific transcription factor and master regulator Pax5 promotes Epstein-Barr virus latency by negatively regulating the viral immediate early protein BZLF1. J. Virol. 87:8053–8063. 10.1128/JVI.00546-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutsch DE, Holley-Guthrie EA, Zhang Q, Stein B, Blanar MA, Baldwin AS, Kenney SC. 1994. The bZIP transactivator of Epstein-Barr virus, BZLF1, functionally and physically interacts with the p65 subunit of NF-kappa B. Mol. Cell. Biol. 14:1939–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez A, Jung EJ, Yin Q, Cayrol C, Flemington EK. 2001. Role of c-myc regulation in Zta-mediated induction of the cyclin-dependent kinase inhibitors p21 and p27 and cell growth arrest. Virology 284:159–169. 10.1006/viro.2001.0923 [DOI] [PubMed] [Google Scholar]
- 18.Hardwick JM, Tse L, Applegren N, Nicholas J, Veliuona MA. 1992. The Epstein-Barr virus R transactivator (Rta) contains a complex, potent activation domain with properties different from those of VP16. J. Virol. 66:5500–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manet E, Rigolet A, Gruffat H, Giot JF, Sergeant A. 1991. Domains of the Epstein-Barr virus (EBV) transcription factor R required for dimerization, DNA binding and activation. Nucleic Acids Res. 19:2661–2667. 10.1093/nar/19.10.2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heilmann AM, Calderwood MA, Portal D, Lu Y, Johannsen E. 2012. Genome-wide analysis of Epstein-Barr virus Rta DNA binding. J. Virol. 86:5151–5164. 10.1128/JVI.06760-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamson AL, Darr D, Holley-Guthrie E, Johnson RA, Mauser A, Swenson J, Kenney S. 2000. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J. Virol. 74:1224–1233. 10.1128/JVI.74.3.1224-1233.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee YH, Chiu YF, Wang WH, Chang LK, Liu ST. 2008. Activation of the ERK signal transduction pathway by Epstein-Barr virus immediate-early protein Rta. J. Gen. Virol. 89:2437–2446. 10.1099/vir.0.2008/003897-0 [DOI] [PubMed] [Google Scholar]
- 23.Ragoczy T, Miller G. 2001. Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. J. Virol. 75:5240–5251. 10.1128/JVI.75.11.5240-5251.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Speck SH, Chatila T, Flemington E. 1997. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol. 5:399–405. 10.1016/S0966-842X(97)01129-3 [DOI] [PubMed] [Google Scholar]
- 25.Robinson AR, Kwek SS, Hagemeier SR, Wille CK, Kenney SC. 2011. Cellular transcription factor Oct-1 interacts with the Epstein-Barr virus BRLF1 protein to promote disruption of viral latency. J. Virol. 85:8940–8953. 10.1128/JVI.00569-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong GK, Delecluse HJ, Gruffat H, Morrison TE, Feng WH, Sergeant A, Kenney SC. 2004. The BRRF1 early gene of Epstein-Barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. J. Virol. 78:4983–4992. 10.1128/JVI.78.10.4983-4992.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagemeier SR, Barlow EA, Kleman AA, Kenney SC. 2011. The Epstein-Barr virus BRRF1 protein, Na, induces lytic infection in a TRAF2- and p53-dependent manner. J. Virol. 85:4318–4329. 10.1128/JVI.01856-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heilmann AM, Calderwood MA, Johannsen E. 2010. Epstein-Barr virus LF2 protein regulates viral replication by altering Rta subcellular localization. J. Virol. 84:9920–9931. 10.1128/JVI.00573-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merkenschlager M. 2010. Ikaros in immune receptor signaling, lymphocyte differentiation, and function. FEBS Lett. 584:4910–4914. 10.1016/j.febslet.2010.09.042 [DOI] [PubMed] [Google Scholar]
- 30.Asa SL, Ezzat S. 2009. The pathogenesis of pituitary tumors. Annu. Rev. Pathol. 4:97–126. 10.1146/annurev.pathol.4.110807.092259 [DOI] [PubMed] [Google Scholar]
- 31.Sellars M, Kastner P, Chan S. 2011. Ikaros in B cell development and function. World J. Biol. Chem. 2:132–139. 10.4331/wjbc.v2.i6.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kastner P, Dupuis A, Gaub MP, Herbrecht R, Lutz P, Chan S. 2013. Function of Ikaros as a tumor suppressor in B cell acute lymphoblastic leukemia. Am. J. Blood Res. 3:1–13 [PMC free article] [PubMed] [Google Scholar]
- 33.Molnar A, Georgopoulos K. 1994. The Ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol. Cell. Biol. 14:8292–8303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tijchon E, Havinga J, van Leeuwen FN, Scheijen B. 2013. B-lineage transcription factors and cooperating gene lesions required for leukemia development. Leukemia 27:541–552. 10.1038/leu.2012.293 [DOI] [PubMed] [Google Scholar]
- 35.Iacobucci I, Storlazzi CT, Cilloni D, Lonetti A, Ottaviani E, Soverini S, Astolfi A, Chiaretti S, Vitale A, Messa F, Impera L, Baldazzi C, D'Addabbo P, Papayannidis C, Lonoce A, Colarossi S, Vignetti M, Piccaluga PP, Paolini S, Russo D, Pane F, Saglio G, Baccarani M, Foa R, Martinelli G. 2009. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell'Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood 114:2159–2167. 10.1182/blood-2008-08-173963 [DOI] [PubMed] [Google Scholar]
- 36.Ronni T, Payne KJ, Ho S, Bradley MN, Dorsam G, Dovat S. 2007. Human Ikaros function in activated T cells is regulated by coordinated expression of its largest isoforms. J. Biol. Chem. 282:2538–2547. 10.1074/jbc.M605627200 [DOI] [PubMed] [Google Scholar]
- 37.Li Z, Perez-Casellas LA, Savic A, Song C, Dovat S. 2011. Ikaros isoforms: the saga continues. World J. Biol. Chem. 2:140–145. 10.4331/wjbc.v2.i6.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, Sharpe A. 1994. The Ikaros gene is required for the development of all lymphoid lineages. Cell 79:143–156. 10.1016/0092-8674(94)90407-3 [DOI] [PubMed] [Google Scholar]
- 39.Schjerven H, McLaughlin J, Arenzana TL, Frietze S, Cheng D, Wadsworth SE, Lawson GW, Bensinger SJ, Farnham PJ, Witte ON, Smale ST. 2013. Selective regulation of lymphopoiesis and leukemogenesis by individual zinc fingers of Ikaros. Nat. Immunol. 14:1073–1083. 10.1038/ni.2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma S, Pathak S, Mandal M, Trinh L, Clark MR, Lu R. 2010. Ikaros and Aiolos inhibit pre-B-cell proliferation by directly suppressing c-Myc expression. Mol. Cell. Biol. 30:4149–4158. 10.1128/MCB.00224-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshida T, Ng SY, Georgopoulos K. 2010. Awakening lineage potential by Ikaros-mediated transcriptional priming. Curr. Opin. Immunol. 22:154–160. 10.1016/j.coi.2010.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bottardi S, Zmiri FA, Bourgoin V, Ross J, Mavoungou L, Milot E. 2011. Ikaros interacts with P-TEFb and cooperates with GATA-1 to enhance transcription elongation. Nucleic Acids Res. 39:3505–3519. 10.1093/nar/gkq1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JH, Ebersole T, Kouprina N, Noskov VN, Ohzeki J, Masumoto H, Mravinac B, Sullivan BA, Pavlicek A, Dovat S, Pack SD, Kwon YW, Flanagan PT, Loukinov D, Lobanenkov V, Larionov V. 2009. Human gamma-satellite DNA maintains open chromatin structure and protects a transgene from epigenetic silencing. Genome Res. 19:533–544. 10.1101/gr.086496.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG. 1997. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 91:845–854. 10.1016/S0092-8674(00)80472-9 [DOI] [PubMed] [Google Scholar]
- 45.Koipally J, Heller EJ, Seavitt JR, Georgopoulos K. 2002. Unconventional potentiation of gene expression by Ikaros. J. Biol. Chem. 277:13007–13015. 10.1074/jbc.M111371200 [DOI] [PubMed] [Google Scholar]
- 46.Payne KJ, Dovat S. 2011. Ikaros and tumor suppression in acute lymphoblastic leukemia. Crit. Rev. Oncog. 16:3–12. 10.1615/CritRevOncog.v16.i1-2.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koipally J, Georgopoulos K. 2002. A molecular dissection of the repression circuitry of Ikaros. J. Biol. Chem. 277:27697–27705. 10.1074/jbc.M201694200 [DOI] [PubMed] [Google Scholar]
- 48.Koipally J, Georgopoulos K. 2002. Ikaros-CtIP interactions do not require C-terminal binding protein and participate in a deacetylase-independent mode of repression. J. Biol. Chem. 277:23143–23149. 10.1074/jbc.M202079200 [DOI] [PubMed] [Google Scholar]
- 49.Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E, Winandy S, Viel A, Sawyer A, Ikeda T, Kingston R, Georgopoulos K. 1999. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 10:345–355. 10.1016/S1074-7613(00)80034-5 [DOI] [PubMed] [Google Scholar]
- 50.O'Neill DW, Schoetz SS, Lopez RA, Castle M, Rabinowitz L, Shor E, Krawchuk D, Goll MG, Renz M, Seelig HP, Han S, Seong RH, Park SD, Agalioti T, Munshi N, Thanos D, Erdjument-Bromage H, Tempst P, Bank A. 2000. An ikaros-containing chromatin-remodeling complex in adult-type erythroid cells. Mol. Cell. Biol. 20:7572–7582. 10.1128/MCB.20.20.7572-7582.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iacobucci I, Iraci N, Messina M, Lonetti A, Chiaretti S, Valli E, Ferrari A, Papayannidis C, Paoloni F, Vitale A, Storlazzi CT, Ottaviani E, Guadagnuolo V, Durante S, Vignetti M, Soverini S, Pane F, Foa R, Baccarani M, Muschen M, Perini G, Martinelli G. 2012. IKAROS deletions dictate a unique gene expression signature in patients with adult B-cell acute lymphoblastic leukemia. PLoS One 7:e40934. 10.1371/journal.pone.0040934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dovat S, Song C, Payne KJ, Li Z. 2011. Ikaros, CK2 kinase, and the road to leukemia. Mol. Cell. Biochem. 356:201–207. 10.1007/s11010-011-0964-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uckun FM, Ma H, Zhang J, Ozer Z, Dovat S, Mao C, Ishkhanian R, Goodman P, Qazi S. 2012. Serine phosphorylation by SYK is critical for nuclear localization and transcription factor function of Ikaros. Proc. Natl. Acad. Sci. U. S. A. 109:18072–18077. 10.1073/pnas.1209828109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomez-del Arco P, Koipally J, Georgopoulos K. 2005. Ikaros SUMOylation: switching out of repression. Mol. Cell. Biol. 25:2688–2697. 10.1128/MCB.25.7.2688-2697.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DiFronzo NL, Leung CT, Mammel MK, Georgopoulos K, Taylor BJ, Pham QN. 2002. Ikaros, a lymphoid-cell-specific transcription factor, contributes to the leukemogenic phenotype of a mink cell focus-inducing murine leukemia virus. J. Virol. 76:78–87. 10.1128/JVI.76.1.78-87.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly G, Bell A, Rickinson A. 2002. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat. Med. 8:1098–1104. 10.1038/nm758 [DOI] [PubMed] [Google Scholar]
- 57.Kelly GL, Stylianou J, Rasaiyaah J, Wei W, Thomas W, Croom-Carter D, Kohler C, Spang R, Woodman C, Kellam P, Rickinson AB, Bell AI. 2013. Different patterns of Epstein-Barr virus latency in endemic Burkitt lymphoma (BL) lead to distinct variants within the BL-associated gene expression signature. J. Virol. 87:2882–2894. 10.1128/JVI.03003-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Habeshaw G, Yao QY, Bell AI, Morton D, Rickinson AB. 1999. Epstein-barr virus nuclear antigen 1 sequences in endemic and sporadic Burkitt's lymphoma reflect virus strains prevalent in different geographic areas. J. Virol. 73:965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gregory CD, Rowe M, Rickinson AB. 1990. Different Epstein-Barr virus-B cell interactions in phenotypically distinct clones of a Burkitt's lymphoma cell line. J. Gen. Virol. 71(Pt 7):1481–1495. 10.1099/0022-1317-71-7-1481 [DOI] [PubMed] [Google Scholar]
- 60.Rowe M, Khanna R, Jacob CA, Argaet V, Kelly A, Powis S, Belich M, Croom-Carter D, Lee S, Burrows SR, Trowsdale J, Moss DJ, Rickinson AB. 1995. Restoration of endogenous antigen processing in Burkitt's lymphoma cells by Epstein-Barr virus latent membrane protein-1: coordinate up-regulation of peptide transporters and HLA-class I antigen expression. Eur. J. Immunol. 25:1374–1384. 10.1002/eji.1830250536 [DOI] [PubMed] [Google Scholar]
- 61.Chodosh J, Holder VP, Gan YJ, Belgaumi A, Sample J, Sixbey JW. 1998. Eradication of latent Epstein-Barr virus by hydroxyurea alters the growth-transformed cell phenotype. J. Infect. Dis. 177:1194–1201. 10.1086/515290 [DOI] [PubMed] [Google Scholar]
- 62.Yu X, Wang Z, Mertz JE. 2007. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog. 3:e194. 10.1371/journal.ppat.0030194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hong GK, Kumar P, Wang L, Damania B, Gulley ML, Delecluse HJ, Polverini PJ, Kenney SC. 2005. Epstein-Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. J. Virol. 79:13984–13992. 10.1128/JVI.79.22.13984-13992.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sather BD, Ryu BY, Stirling BV, Garibov M, Kerns HM, Humblet-Baron S, Astrakhan A, Rawlings DJ. 2011. Development of B-lineage predominant lentiviral vectors for use in genetic therapies for B cell disorders. Mol. Ther. 19:515–525. 10.1038/mt.2010.259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adamson AL, Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 75:2388–2399. 10.1128/JVI.75.5.2388-2399.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klenova E, Chernukhin I, Inoue T, Shamsuddin S, Norton J. 2002. Immunoprecipitation techniques for the analysis of transcription factor complexes. Methods 26:254–259. 10.1016/S1046-2023(02)00029-4 [DOI] [PubMed] [Google Scholar]
- 67.Hahm K, Cobb BS, McCarty AS, Brown KE, Klug CA, Lee R, Akashi K, Weissman IL, Fisher AG, Smale ST. 1998. Helios, a T cell-restricted Ikaros family member that quantitatively associates with Ikaros at centromeric heterochromatin. Genes Dev. 12:782–796. 10.1101/gad.12.6.782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wojcik H, Griffiths E, Staggs S, Hagman J, Winandy S. 2007. Expression of a non-DNA-binding Ikaros isoform exclusively in B cells leads to autoimmunity but not leukemogenesis. Eur. J. Immunol. 37:1022–1032. 10.1002/eji.200637026 [DOI] [PubMed] [Google Scholar]
- 70.Buettner M, Lang A, Tudor CS, Meyer B, Cruchley A, Barros MH, Farrell PJ, Jack HM, Schuh W, Niedobitek G. 2012. Lytic Epstein-Barr virus infection in epithelial cells but not in B-lymphocytes is dependent on Blimp1. J. Gen. Virol. 93:1059–1064. 10.1099/vir.0.038661-0 [DOI] [PubMed] [Google Scholar]
- 71.Vrzalikova K, Vockerodt M, Leonard S, Bell A, Wei W, Schrader A, Wright KL, Kube D, Rowe M, Woodman CB, Murray PG. 2011. Down-regulation of BLIMP1alpha by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 117:5907–5917. 10.1182/blood-2010-09-307710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kikuchi H, Nakayama M, Takami Y, Kuribayashi F, Nakayama T. 2012. EBF1 acts as a powerful repressor of Blimp-1 gene expression in immature B cells. Biochem. Biophys. Res. Commun. 422:780–785. 10.1016/j.bbrc.2012.05.099 [DOI] [PubMed] [Google Scholar]
- 73.Alinikula J, Nera KP, Junttila S, Lassila O. 2011. Alternate pathways for Bcl6-mediated regulation of B cell to plasma cell differentiation. Eur. J. Immunol. 41:2404–2413. 10.1002/eji.201141553 [DOI] [PubMed] [Google Scholar]
- 74.Jourdan M, Caraux A, De Vos J, Fiol G, Larroque M, Cognot C, Bret C, Duperray C, Hose D, Klein B. 2009. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood 114:5173–5181. 10.1182/blood-2009-07-235960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Montalvo EA, Cottam M, Hill S, Wang YJ. 1995. YY1 binds to and regulates cis-acting negative elements in the Epstein-Barr virus BZLF1 promoter. J. Virol. 69:4158–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma H, Qazi S, Ozer Z, Zhang J, Ishkhanian R, Uckun FM. 2013. Regulatory phosphorylation of Ikaros by Bruton's tyrosine kinase. PLoS One 8:e71302. 10.1371/journal.pone.0071302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Payne KJ, Huang G, Sahakian E, Zhu JY, Barteneva NS, Barsky LW, Payne MA, Crooks GM. 2003. Ikaros isoform x is selectively expressed in myeloid differentiation. J. Immunol. 170:3091–3098 [DOI] [PubMed] [Google Scholar]
- 78.Sun L, Liu A, Georgopoulos K. 1996. Zinc finger-mediated protein interactions modulate Ikaros activity, a molecular control of lymphocyte development. EMBO J. 15:5358–5369 [PMC free article] [PubMed] [Google Scholar]
- 79.Gurel Z, Ronni T, Ho S, Kuchar J, Payne KJ, Turk CW, Dovat S. 2008. Recruitment of ikaros to pericentromeric heterochromatin is regulated by phosphorylation. J. Biol. Chem. 283:8291–8300. 10.1074/jbc.M707906200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kathrein KL, Chari S, Winandy S. 2008. Ikaros directly represses the notch target gene Hes1 in a leukemia T cell line: implications for CD4 regulation. J. Biol. Chem. 283:10476–10484. 10.1074/jbc.M709643200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Papathanasiou P, Attema JL, Karsunky H, Hosen N, Sontani Y, Hoyne GF, Tunningley R, Smale ST, Weissman IL. 2009. Self-renewal of the long-term reconstituting subset of hematopoietic stem cells is regulated by Ikaros. Stem Cells 27:3082–3092. 10.1002/stem.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alkhatib A, Werner M, Hug E, Herzog S, Eschbach C, Faraidun H, Kohler F, Wossning T, Jumaa H. 2012. FoxO1 induces Ikaros splicing to promote immunoglobulin gene recombination. J. Exp. Med. 209:395–406. 10.1084/jem.20110216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shore AM, White PC, Hui RC, Essafi A, Lam EW, Rowe M, Brennan P. 2006. Epstein-Barr virus represses the FoxO1 transcription factor through latent membrane protein 1 and latent membrane protein 2A. J. Virol. 80:11191–11199. 10.1128/JVI.00983-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mavrakis KJ, Van Der Meulen J, Wolfe AL, Liu X, Mets E, Taghon T, Khan AA, Setty M, Rondou P, Vandenberghe P, Delabesse E, Benoit Y, Socci NB, Leslie CS, Van Vlierberghe P, Speleman F, Wendel HG. 2011. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat. Genet. 43:673–678. 10.1038/ng.858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cameron JE, Fewell C, Yin Q, McBride J, Wang X, Lin Z, Flemington EK. 2008. Epstein-Barr virus growth/latency III program alters cellular microRNA expression. Virology 382:257–266. 10.1016/j.virol.2008.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He LC, Xu HZ, Gu ZM, Liu CX, Chen GQ, Wang YF, Wen DH, Wu YL. 2011. Ikaros is degraded by proteasome-dependent mechanism in the early phase of apoptosis induction. Biochem. Biophys. Res. Commun. 406:430–434. 10.1016/j.bbrc.2011.02.062 [DOI] [PubMed] [Google Scholar]
- 87.Kleinmann E, Geimer Le Lay AS, Sellars M, Kastner P, Chan S. 2008. Ikaros represses the transcriptional response to Notch signaling in T-cell development. Mol. Cell. Biol. 28:7465–7475. 10.1128/MCB.00715-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anderson LJ, Longnecker R. 2009. Epstein-Barr virus latent membrane protein 2A exploits Notch1 to alter B-cell identity in vivo. Blood 113:108–116. 10.1182/blood-2008-06-160937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kohlhof H, Hampel F, Hoffmann R, Burtscher H, Weidle UH, Holzel M, Eick D, Zimber-Strobl U, Strobl LJ. 2009. Notch1, Notch2, and Epstein-Barr virus-encoded nuclear antigen 2 signaling differentially affects proliferation and survival of Epstein-Barr virus-infected B cells. Blood 113:5506–5515. 10.1182/blood-2008-11-190090 [DOI] [PubMed] [Google Scholar]
- 90.Ouellet DL, Vigneault-Edwards J, Letourneau K, Gobeil LA, Plante I, Burnett JC, Rossi JJ, Provost P. 2013. Regulation of host gene expression by HIV-1 TAR microRNAs. Retrovirology 10:86. 10.1186/1742-4690-10-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. 2004. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J. Immunol. 173:1158–1165 [DOI] [PubMed] [Google Scholar]
- 92.Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. 2002. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17:51–62. 10.1016/S1074-7613(02)00335-7 [DOI] [PubMed] [Google Scholar]
- 93.Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 16:1977–1989. 10.1101/gad.996502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang Z, Yan Z, Wood C. 2008. Kaposi's sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 82:3590–3603. 10.1128/JVI.02229-07 [DOI] [PMC free article] [PubMed] [Google Scholar]