

Abstract
Drug release from liposomal formulations is governed by a complex interplay of kinetic (i.e., drug permeability) and thermodynamic factors (i.e., drug partitioning to the bilayer surface). Release studies under sink conditions that attempt to mimic physiological conditions are insufficient to decipher these separate contributions. The present study explores release studies performed under nonsink conditions coupled with appropriate mathematical models to describe both the release kinetics and the conditions in which equilibrium is established. Liposomal release profiles for a model anticancer agent, topotecan, under nonsink conditions provided values for both the first-order rate constant for drug release and the bilayer/water partition coefficient. These findings were validated by conducting release studies under sink conditions via dynamic dialysis at the same temperature and buffer pH. A nearly identical rate constant for drug release could be obtained from dynamic dialysis data when appropriate volume corrections were applied and a mechanism-based mathematical model was employed to account for lipid bilayer binding and dialysis membrane transport. The usefulness of the nonsink method combined with mathematical modeling was further explored by demonstrating the effects of topotecan dimerization and bilayer surface charge potential on the bilayer/water partition coefficient at varying suspension concentrations of lipid and drug.
Keywords: liposomes, nanoparticles, lipid bilayers, membrane partitioning, ultrafiltration, dynamic dialysis, drug release
Introduction
Mathematical models for assessing drug permeability and predicting in vivo drug release from nanoparticle formulations would be useful both in the design phase and during preclinical testing where avoiding the extensive use of animals would be highly desirable. Such models would facilitate the design of formulations with adjustable and predictable drug release rates for patient-specific treatment regimens. Mechanism-based models applicable to liposomal systems would need to account for three main factors affecting drug release: (1) the escaping tendency or effective concentration of the entrapped (permeable) drug species which serves as the driving force for liposomal release; (2) drug speciation and species permeability–area products for lipid bilayer transport;1−7 and (3) the environmental conditions in which drug release occurs both during the in vitro release characterization and in vivo.3,8 The intraliposomal driving force for transport likely depends on such factors as pH-dependent drug speciation, self-association, complexation, precipitate formation, membrane binding, and drug degradation/interconversion kinetics. The driving force for liposomal release and the membrane permeability–area product are closely linked and dependent on which drug species account for the release.3,9−11 The environmental conditions (e.g., temperature, pH, sink conditions or lack thereof, presence of permeable buffer species, lipid-bilayer perturbing components, etc.) also impact both the driving forces and permeability coefficients. Thus, robust mechanism-based models for predicting liposomal drug release may be quite complex. Translation of release parameters generated in vitro to the prediction of drug release in vivo may be particularly challenging. The necessary corrections will likely vary depending on the in vitro method employed to study drug release.
A number of methods currently exist to monitor in vitro drug release from nanoparticles,12−14 but extrapolation to predict in vivo release often requires an adjustment for the absence of sink conditions in the in vitro experiments as well as other possible environmental differences. For example, one popular method to monitor in vitro drug release from nanoparticles is dynamic dialysis. Dynamic dialysis uses a large reservoir in an attempt to provide the sink conditions necessary to drive drug release to completion. Meanwhile, the nanoparticles remain concentrated within the small volume compartment and separated from the reservoir by a semipermeable membrane.4,8,9,15−18 Unfortunately, a large reservoir volume does not ensure sink conditions within the dialysis chamber itself. Depending on the nanoparticle release kinetics and the extent of drug binding to the nanoparticle, transport across the dialysis membrane may become rate-limiting.3,8 Corrections for drug binding to the nanoparticles and the barrier properties of the dialysis membrane are therefore crucial when employing dynamic dialysis for predictive modeling.3,4,8,19 In some cases, incomplete release has been observed even though approximate sink conditions (based on overall drug concentration gradients) were maintained due to factors such as pH differences or drug binding phenomena. Such factors reduce the thermodynamic activity gradient for the permeable species, resulting in the achievement of equilibrium and subsequently incomplete release.20−23 Finally, even if the above concerns relating to sink conditions are properly taken into account, a separate set of experiments in addition to dynamic dialysis would be needed. These additional experiments would be required to quantify the species-dependent membrane binding of the drug and its influence on observed release kinetics for the construction of a mechanism-based release model.4,17
A method to evaluate drug release kinetics under well-defined nonsink conditions when combined with the appropriate mechanistic release model would allow simultaneous determination of the kinetic and thermodynamic parameters governing release kinetics. This method would also provide a more robust assessment of nanoparticle formulations. This study demonstrates the utility of a novel ultrafiltration method to analyze drug release from nanoliposomal formulations under nonsink conditions using the model anticancer agent topotecan (TPT). With the appropriate mathematical models, the liposomal drug release parameters generated under nonsink conditions were shown to be comparable with those obtained from dynamic dialysis. This nonsink method was also used to simultaneously characterize membrane binding of the drug and its dependence on both drug and lipid concentrations in suspension.
Experimental Section
Materials
Powders of 1,2-distearoyl-sn-glycero-3-phosphatidylcholine (DSPC, >99% purity) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (m-PEG DSPE, MW = 2806, >99% purity) were purchased from Avanti Polar Lipids (Alabaster, AL). Topotecan hydrochloride was purchased from AK Scientific (Union City, CA). Float-A-Lyzer G2 dialysis tubes (100,000 MWCO) were purchased from Spectrum Laboratories (Rancho Dominguez, CA). Millipore semimicro ultrafiltration centrifugation devices (regenerated cellulose, NMWL: 30,000), 100 nm pore size Nuclepore polycarbonate membranes, solvents, and buffer salts were purchased from Fisher Scientific (Florence, KY). All solvents were of HPLC grade.
Preparation and Characterization of DSPC/m-PEG DSPE Liposomes
Large unilamellar vesicles were formed using a film hydration and extrusion process as reported previously with slight modifications.3,24 Briefly, DSPC and m-PEG DSPE (95:5 mol/mol) lipids were weighed and dissolved in chloroform, and aliquots of the resulting solutions were distributed into separate vials. Chloroform was subsequently evaporated under a stream of nitrogen gas, and the residue was vacuum-dried at 40 °C for 6 h. For release studies, TPT was passively loaded into liposomes by hydrating the dried lipid film with TPT solutions (0.25 mM in pH 4.0, 50 mM formate buffer adjusted to an ionic strength of 0.3 with NaCl) to achieve 40 or 90 mg lipid/mL suspensions. These suspensions were extruded 10 times through two stacked 100 nm pore size Nuclepore polycarbonate membranes using a Liposofast extrusion device at 60 °C to obtain unilamellar vesicles with TPT in the intravesicular solution. Blank liposome suspensions (40 mg lipid/mL) used in spiking experiments for dynamic dialysis and ultrafiltration validation were made under the same conditions as passively loaded liposomes without TPT present in the hydrating solution.
Liposome characterization included particle size measurements by dynamic light scattering (DLS) and lipid content analyses using HPLC with evaporative light scattering detection (ELSD) as previously reported.11 Particle size data were used to monitor liposome stability and in combination with information on the number of vesicles in suspension (based on lipid content) and bilayer surface density data from the literature to calculate liposomal volumes necessary for the mathematical models.3,25−27
Release of TPT from DSPC/m-PEG DSPE Liposomes
All release studies were conducted in a water-jacketed incubator maintained at 37 °C.
Sephadex Column Removal of Unencapsulated Drug from Passively Loaded Liposome Suspensions
To compare release studies using dynamic dialysis (sink conditions) and ultrafiltration (nonsink conditions), 0.7 mL of 40 mg lipid/mL suspensions was passed through a Sephadex PD-10 column to separate liposomes from the unencapsulated drug. The first 4.75 mL was collected and diluted to 15 mL of suspension using the same buffer used for lipid hydration (without drug). Next, 4.5 mL of this suspension was either transferred to dialysis tubes or 7 mL glass vials with a rubber stopper. Release studies under either sink or nonsink conditions were performed in triplicate. Additional studies of the concentration dependence of binding to the DSPC bilayer utilized 90 mg lipid/mL suspensions and 0.25 or 0.7 mL aliquots passed through a Sephadex column. In these instances, the first 1.5 mL of eluent was discarded, and the next 3.25 mL containing the liposome suspension was collected and transferred to 7 mL glass vials with a rubber stopper.
Nonsink Release Studies Measured by Ultrafiltration
Glass vials containing the liposome suspensions were placed on a Thermo Cimerac iPoly 15 multipoint stirrer insulated with 1.5 in. of Styrofoam to minimize heating from the stir plate and subsequently maintained at a suspension temperature of 37.4 ± 0.6 °C. Liposome suspensions were stirred at 200 rpm over the time course of the release study (∼96 h) using 10 × 5 mm Teflon stir bars. The encapsulated drug was monitored by ultrafiltration of 100 μL samples taken throughout the duration of the release studies.
Ultrafiltration was chosen as it has been used in previous studies with liposomes as a method in which the encapsulated drug may be separated from released drug.11,28 Each sample was diluted with chilled (4 °C) buffer to 450 μL to quench drug release and ultrafiltered using an Amicon Ultra 0.5 mL centrifugal filter device with a 30,000 MWCO Ultracel membrane. Samples were centrifuged in these cartridges at 14,000 rpm for 10 min in an Eppendorf 5417R maintained at 4 °C. During centrifugation, liposome integrity was maintained as suspensions were concentrated but not dried completely due to the conical geometry of the ultrafiltration membrane. Concentrated suspensions (26 ± 2 μL) were recovered by inverting and centrifuging the cartridge at 2000 rpm for another 2 min. After recovery of the concentrate, 400 μL of chilled buffer was added, and the process was repeated to ensure complete removal of membrane-bound extravesicular drug. The final concentrate from this second cycle was analyzed by HPLC after dilution into the calibration range of TPT standards. Chilled methanol (−20 °C) was used to disrupt the vesicles and minimize solvent evaporation during sample dilution. Samples that had not been ultrafiltered (20–100 μL) were also taken and immediately diluted in chilled methanol to determine the total amount of TPT and any extravesicular drug present at the beginning of the release study.
Dynamic Dialysis under Sink Conditions
Dialysis tubes (Float-A-Lyzer G2, 100,000 MWCO) containing 4.5 mL of liposome suspension were placed in 900 mL reservoirs containing pH 4.0 formate buffer pre-equilibrated at 37 °C. Aliquots (20 uL) were removed from the dialysis tube over a 48 h period and immediately diluted in chilled methanol for TPT analysis by HPLC.
Dialysis Tube Swelling Studies
Changes in the suspension volume within the dialysis tube during release studies may produce errors in the observed loss of drug during dynamic dialysis. To correct for this, the rate of swelling as measured by the volume of sample within the dialysis tubes at equilibrium must be determined. Fresh dialysis tubes of the same make as those used in dynamic dialysis studies were filled with 4 mL of the same buffer as that in the reservoir. These tubes were then allowed to sit in reservoirs at the same conditions used in dynamic dialysis studies. The volume in these tubes was monitored over time using a 10 mL graduated cylinder.
TPT Dimerization
Several reports have indicated that TPT self-associates to form dimers,29−31 the tendency of which may be pH dependent.31 Self-association of TPT may result in liposomal membrane binding coefficients that are concentration dependent if only the monomeric form is involved in binding. Since previous characterization of TPT self-association has been in the neutral pH range,29−31 studies were conducted to assess TPT dimerization at the conditions in which release studies were performed. Apparent extinction coefficients were calculated for varying concentrations of TPT (1–250 μM) dissolved in the same buffer employed for release studies. Absorbance was measured at wavelengths of 360, 376, 378, 380, 382, 384, 386, and 388 nm using a Varian Cary 50 UV–vis spectrophotometer. NSG quartz cuvettes (NSG Precision Cells, Farmingdale, NY) with 2 and 10 mm path lengths were used to stay within the analytical range of the instrument.
HPLC Analyses
Samples from release and validation studies were analyzed for TPT and lipid concentration by HPLC as reported previously.3,11 TPT samples were analyzed with a previously developed HPLC method utilizing fluorescence detection.11 TPT lactone standards were prepared in chilled, acidified methanol over a concentration range of 20–200 nM. Samples were diluted to within this concentration range using chilled methanol. Samples were either immediately injected or stored at −20 °C for no more than 48 h before analysis.
Lipid analysis was performed using an HPLC coupled to an ELSD (Sedere, Inc., Lawrenceville, NJ) as previously reported.3,11 DSPC standards and samples were dissolved in 80% chloroform/19.5% methanol/0.5%(v/v) of 30% (vol) NH4OH solution. Standards spanned the concentration range of 0.05–0.3 mg DSPC/mL. Lipid samples from release studies (50–150 μL) were dried at room temperature under N2. Once dried, samples were redissolved in the above-mentioned solvent mixture to be within the calibration range of DSPC standards.
Model Development and Data Analysis
Mechanistic models for liposomal release have been previously developed to account for the additional resistance contributed by the dialysis membrane in dynamic dialysis studies.3,8,9 The general concepts applicable to liposomal systems are depicted in Scheme 1. By developing appropriate models, the rate of drug release applicable to sink conditions can be extracted from a variety of release methods. Such a case is illustrated here by using mathematical models to analyze and compare the kinetics of liposomal release of TPT under sink and nonsink conditions. All fitting of release kinetics and dimerization data was performed using Micromath Scientist nonlinear regression software utilizing a weighting factor of 2.
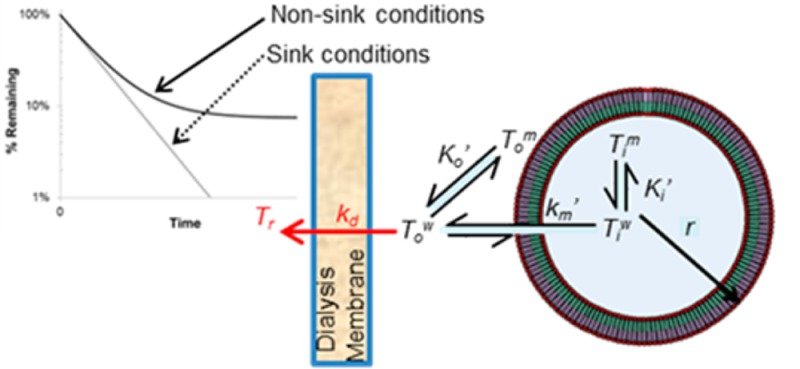
Scheme 1. Illustration of the Relevant Kinetic and Equilibrium Processes Applicable in Developing a Mathematical Model for Liposomal Drug Release As Determined by Dynamic Dialysis.
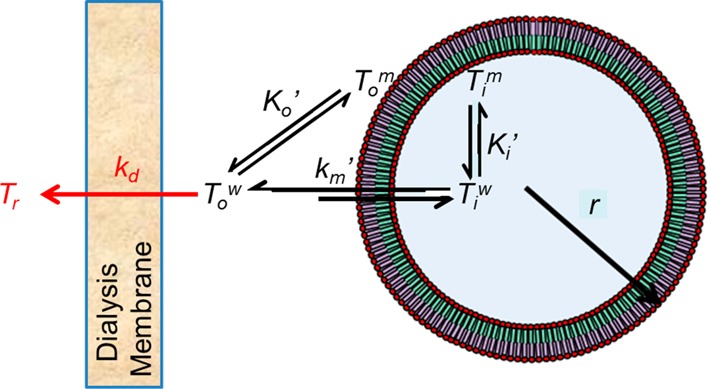
The volume compartments of a liposome with radius, r, are highlighted along with the kinetic and binding components governing drug release. The blue core is the inner aqueous volume, Viw, while the green and purple sections refer to the inner, Vi, and outer, Vow, membrane volumes, respectively. The rate of liposomal drug release depends on the rate constant, km′, and the difference in the unbound inner and outer aqueous drug concentrations, Tiw and Tow, respectively, while the apparent intravesicular, Ki′, and extravesicular, Ko′, binding coefficients govern the equilibrium between drug bound to the inner or outer lipid membrane, Tim and Tom, respectively, and the corresponding unbound drug in the intravesicular or extravesicular compartments, respectively. The rate constant kd reflects the diffusion of drug across the dialysis membrane driven by the concentration gradient Tow – Tr. All notations in red refer to aspects unique to dynamic dialysis conditions.
Mathematical Model of TPT Release from Unilamellar Liposomes: Nonsink Conditions
A mechanistic, mathematical model is required to obtain both drug permeability and membrane binding from release studies. Several models describing drug loading and release have already been developed;3,6,7,9,32−34 however, only a few have been tested, and these studies have only examined release under sink conditions.3,9,34
The apparent rate constant governing drug release from a liposome is a function of the drug’s apparent permeability coefficient, Pm′, through the bilayer and the radius, r, of the particle. This is shown below in eq 1.3,25
| 1 |
While km′ may be dependent on the respective permeabilities of each species of drug present in solution, such a distinction cannot be made here as multiple conditions (e.g., pH) must be explored to determine each specie’s contribution. Therefore, the km determined here applies to the specific pH chosen for these experiments (which is satisfactory for comparing these different release methods).
Liposomal drug release is dependent on the driving force developed by the effective concentration gradient between unbound, intra-, and extra-vesicular drug concentrations (Tiw and To, respectively). This is expressed by eqs 2a and 2b.
| 2a |
| 2b |
These differential equations describe bilayer-limited Fickian diffusion at a pseudo-steady-state. The term fv symbolizes the ratio of total entrapped volume (the product of the total number of vesicles, n, and intravesicular volume of a single liposome, ViT) to total extravesicular volume, V0, thus accounting for the difference in volumes of the inner and outer compartments. Derivation of the concentrations of unbound drug in the intra- and extra-vesicular compartments in terms of total intra- and extra-vesicular drug concentrations (Ti and To, respectively) can be found in the Appendix along with the initial conditions assumed for these differential equations.
Dynamic Dialysis Model of Drug Release from Unilamellar Liposomes: Sink Conditions
Dynamic dialysis is advantageous for maintaining sink conditions as it provides a large reservoir capable of maintaining the driving force for drug release. Because nanoparticles cannot cross the dialysis membrane, significant dilution of the nanoparticle suspension during drug release is avoided, and the concentration of drug remaining in the suspension versus time can be quantified. This is depicted in Scheme 1. Mathematically, the differential equation governing transport in the vesicle is the same as eq 2a, where Tow refers to the unbound extravesicular TPT within the dialysis tube. A release rate constant for transport of liposomally released drug from the dialysis tube, kd, must be added to eq 2b to describe transport from the extravesicular compartment of the dialysis tube into the reservoir compartment. This is expressed by eq 3 with portions in bold font identifying the term unique to dynamic dialysis.
| 3 |
In these studies, the suspension concentration of TPT within the dialysis tube at any time (Td) is sampled. This concentration would naturally be composed of intra- and extra-vesicular TPT as shown by eq 4.
| 4 |
Derivation of the unbound drug concentrations in dynamic dialysis is the same as that in the nonsink condition case (see Appendix). The initial conditions for the rate equations pertaining to dynamic dialysis depend on whether the experiment involves passively loaded drug-containing liposomes or blank liposomes spiked with free drug as described in the Appendix.
Concentration Corrections for Ultrafiltration Recovery and Dialysis Compartment Volume
For nonsink release studies, the recovery of intra- and extra-vesicular drug after ultrafiltration must be accounted for to accurately assess release kinetics. In dialysis experiments under sink conditions, the volume of the nanoparticle suspension within the dialysis tubes may fluctuate. Mathematical corrections for the recovery of intra- and extra-vesicular drug after ultrafiltration and volume changes within the dialysis tube (and their subsequent effects on sample removal during release studies) are explained in detail in the Appendix.
Determination of the TPT Dimerization Constant (K2)
Self-association of TPT in solution has been previously reported30,31 and may affect observed binding due to the different binding affinities of the drug in its monomeric (T1) and dimeric (T2) forms and the effects of binding on the bilayer surface charge. The two forms of TPT in solution can be related by a dimerization constant, K2, as shown by eq 5.
| 5 |
K2 was determined under the conditions for drug release from the change in extinction coefficient as a function of concentration (see Appendix).
Results
Validation of Analytical Methods and Liposome Particle Characterization
TPT concentrations were analyzed using a previously validated HPLC method with fluorescence detection.11 A linear response for TPT lactone (4.5 min retention time) was observed between 20 and 200 nM using excitation and emission wavelengths of 380 and 550 nm, respectively. TPT concentrations in samples taken from release studies and size exclusion experiments ranging from 0.2–2 μM were determined by diluting samples with chilled methanol into the concentration range of standards.
Phospholipid content was determined using an HPLC method previously developed and validated.3,11 ELSD was employed due to the lack of a chromophore/fluorophore in the lipid molecules. A peak retention time of 7.9 min and a linear relationship between the logarithm of peak area and DSPC concentration were observed from 0.05–0.3 mg DSPC/mL, similar to that previously reported.3,11
Separation of passively loaded TPT liposomes from an unencapsulated drug was achieved with a Sephadex size exclusion column. Figure 1 compares the elution profiles of an aqueous solution of TPT in the absence of liposomes and a suspension of passively loaded TPT-containing liposomes. Both TPT and liposomes detected using HPLC and DLS, respectively, were present in the peak eluting in the 2.5–5 mL range, while the solution of TPT in the absence of liposomes did not produce a peak in this range.
Figure 1.
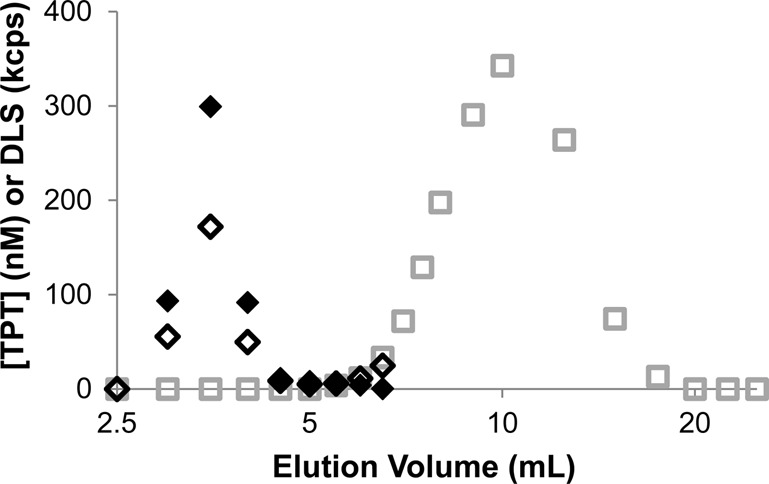
Elution profiles of free (□) or liposomal TPT (◊) analyzed by HPLC. The DLS intensity profile generated by liposomes (⧫) is also shown to indicate the separation of free from entrapped drug.
Particle size was determined by DLS for the liposomes before and after the conclusion of release studies. The average particle size in five independent release studies (with 95% confidence interval) was 98 ± 2 nm before studies began and 100 ± 3 nm after release studies were concluded. Because phospholipids undergo acid-catalyzed ester hydrolysis,35−37 the stability of the phospholipid bilayer under acidic conditions for extended periods of time could lead to lipid loss during the release study. This possibility was examined by monitoring lipid content in solution using HPLC with an ELSD. Figure 2 demonstrates that liposomal suspensions employed in release studies conducted under nonsink conditions exhibited no lipid loss during the 96 h period in which release was monitored.
Figure 2.

Lipid content was monitored during nonsink release studies. The line indicates the average of all measured lipid concentrations and shows that the lipid content remained constant throughout the release experiments. Error bars represent 95% confidence intervals.
Recovery from Ultrafiltration and Volume Changes in Dynamic Dialysis
Corrections were required to obtain the true release profiles from changes in drug concentration observed by both ultrafiltration and dynamic dialysis methods. Values for the percentage of TPT and lipid recovered after ultrafiltration have been reported previously under similar conditions.11 The percentage of lipid recovered was used to determine the actual amount of intravesicular drug present in samples as trace amounts of extravesicular TPT still present after separation by Sephadex8 would lead to a lower % of TPT recovered.11
Additionally, any extra-vesicular drug still present after ultrafiltration could also lead to an overestimation in the binding coefficient observed. To determine the percentage of extra-vesicular drug present in the retentate after ultrafiltration, blank liposome suspensions were spiked with TPT followed by immediate ultrafiltration. Using similar drug and lipid concentrations as those employed in release studies, the percentage of extravesicular TPT recovered during ultrafiltration was determined to be 1.5 ± 0.2%. This recovery was similar to the 1.4% that would be expected based on the 26 μL of ultrafiltrate suspension that was retained after ultrafiltration. For nonsink release studies, the initial concentration of extravesicular drug was never more than 0.2% of the drug concentration used to load the liposomes.
Dynamic dialysis studies also required corrections in drug concentration due to increases or decreases in volume within the dialysis tube. Additionally, the effect of sample removal also needed to be taken into account. For these dynamic dialysis studies, 4.5 mL of solution was initially observed to fill the dialysis tubes to the top of the dialysis membrane. However, these tubes swelled during release studies. To correct for the effect of observed volume changes on drug concentration, the rate of volume swelling was determined. This was achieved by filling a fresh set of dialysis tubes initially with 4 mL (Vo) of buffer solution, then monitoring volume changes over 72 h at the same conditions used in dynamic dialysis release studies. The rate of swelling, kv, and tube volume at equilibrium, Veq, could be determined using the equation below.
| 6 |
The resulting swelling profile of the dialysis tubes (see Supporting Information, Figure S1a) resulted in a kv of 0.13 ± 0.02 h–1, while Veq varied greatly between dialysis tubes (ranging from 4.8–5.3 mL). Using this rate constant and the Veq determined for each dialysis tube, the loss of lipid observed in dynamic dialysis studies could be accounted for using the correction factors described by eqs A9–A12c (see Appendix and Figure S2, Supporting Information). These equations were then applied to TPT concentrations obtained during dynamic dialysis studies to reflect drug loss due only to liposomal release.
Comparison of Release Studies under Nonsink and Sink Conditions
In addition to these corrections, the parameters calculated in Table 1, which describe the ratio of aqueous and membrane volumes for the intra-vesicular compartment (a and b, respectively) and the extra-vesicular compartment (c and d respectively), along with the ratio of entrapped and external volume (fv), were required for model fitting (see Appendix for a more detailed explanation) and calculated using previously reported values and equations.25,27 With this information, the kinetic parameters for drug release under nonsink and sink conditions could be compared. For simplicity and because equilibrium is nearly reached in these nonsink studies, Ki′ and Ko′ are assumed to be equivalent at the end of these studies and thus referred to from this point on as K′. Fitting of release profiles from 0.48 mg lipid/mL suspensions under nonsink conditions as shown in Figure 3A resulted in a k′m of 0.51 ± 0.05 h–1 and K′ of 73 ± 2. For dialysis studies, drug transport across the dialysis membrane may affect observed drug release.3,8 As such, release profiles from passively loaded liposome suspensions and blank liposome suspensions spiked with TPT were simultaneously fit to determine both k′m and the rate constant for TPT transport across the dialysis membrane (kd). Because K′ cannot be determined from dynamic dialysis studies, it was held constant at the value determined from the nonsink studies. Using this value and the parameters listed in Table 1, k′m and kd were simultaneously fit as shown by Figure 3B, resulting in values of 0.50 ± 0.04 h–1 and 0.79 ± 0.13 h–1, respectively. The release profile of passively loaded liposomes in Figure 3B also exhibits a lag time consistent with accumulation of released drug within the dialysis tube caused by the noninstantaneous rate of drug transport across the dialysis membrane.3,8 The values of k′m determined from both methods are nearly identical and show that nonsink studies can simultaneously provide accurate release rate constants along with drug binding information.
Table 1. Volume Parameters Used When Comparing Release Studies of Liposome Suspensions under Nonsink and Sink Conditions.
| lipid suspension concns | a | b | c | d | fv |
|---|---|---|---|---|---|
| 0.48 mg/mL (nonsink) | 0.85 | 0.15 | 0.99982 | 0.00018 | 0.00122 |
| 0.51 mg/mL (dialysis) | 0.85 | 0.15 | 0.99980 | 0.00020 | 0.00135 |
Figure 3.

Comparison of the release profiles of TPT from DSPC/mPEG-DSPE liposomes obtained from ultrafiltration (A) and dynamic dialysis (B) methods at pH 4.0, 37 °C. (A) The release profiles of TPT under nonsink conditions are shown for suspensions of 0.48 (■), 5.44 (gray circle), and 15.3 (gray triangle) mg lipid/mL along with the fits of these data to the mathematical model describing release under nonsink conditions (represented by the lines of corresponding color). The inset at the top right compares the approach to equilibrium occurring under nonsink conditions to a simulated profile of release under sink conditions (--). (B) The release profiles of TPT using dynamic dialysis. After correcting for volume swelling and sampling of the dialysis tube, TPT release from passively loaded liposomes (■) and blank liposome suspensions spiked with free drug (□) were fit simultaneously, producing their respective release profiles (— and --). Error bars indicate the standard deviation at each time point of triplicate release experiments.
Drug and Lipid Concentration Effects on Drug Partitioning Probed by the Nonsink Method
Further validation of the nonsink method to examine release kinetics was performed by varying the suspension concentration of lipid. For these studies, the same initial concentration of TPT was used to passively load the three different lipid suspensions. This was done to avoid drug self-association effects on release kinetics (i.e., to maintain the same intra-vesicular driving force between the studies). Because each suspension reaches equilibrium with a different amount of drug released due to the nonsink conditions, the t1/2 expression includes parameters for both TPT permeability (k′m) and partitioning (K′), as defined below (see Appendix for full derivation).
| 7 |
The fitted release profiles for the three suspensions at varying lipid concentration in Figure 3A resulted in similar release half-lives (see Table 2), indicating this method is useful over a wide range of lipid concentrations. Altering the suspension concentration of lipid to validate the nonsink method’s ability to determine release kinetics also allowed critical evaluation of the membrane binding coefficient determined from these release studies. The apparent binding coefficients (K′) were observed to vary depending on the lipid concentration (spanning a 30-fold range). The resulting fits of K′ were 73 ± 2, 46 ± 6, and 23 ± 3 for the 0.48, 5.44, and 15.3 mg lipid/mL suspensions, respectively.
Table 2. Values Used to Calculate the Intrinsic DSPC Bilayer/Water Partition Coefficients for TPT Species at pH 4 and 37 °Ca.
| lipid
suspensions |
|||
|---|---|---|---|
| parameters | 0.48 mg/mL | 5.44 mg/mL | 15.3 mg/mL |
| total TPT (μM) | 0.94 | 4.99 | 15.44 |
| t1/2 (h) | 17 ± 2 | 19 ± 2 | 19 ± 3 |
| K′ | 73 ± 2 | 46 ± 4 | 23 ± 3 |
| f1 | 0.99 | 0.95 | 0.83 |
| ΣiCi | 0.6 | 0.6 | 0.6 |
| δ1 | 1.3 ± 0.1 | 1.8 | 2.3 |
| δ2 | 1.6 ± 0.3 | 3.2 | 5.3 |
95% confidence intervals are shown where applicable.
Because this release model accounts for the differences in aqueous and membrane volumes encountered under the various conditions studied, the apparent binding coefficients should not be different between these studies. However, the cationic charge of TPT at pH 4.0 in conjunction with the varying suspension concentrations of TPT may have an effect on observed binding coefficients. Both of these variables may be accounted for with the consideration of drug self-association and the change in bilayer surface potential due to the binding of cationic drug. To assess whether either or both effects contribute toward the variation in K′ observed experimentally, TPT dimerization in solution and the varying surface potential at the lipid membrane–solution interface were evaluated and used to determine intrinsic binding coefficients for the monomeric and dimeric forms of TPT binding to the DSPC/m-PEG DSPE bilayer.
In general, the intrinsic binding coefficient, Ki0, for any species “i” (in this case TPT) capable of binding to the lipid membrane may be expressed by eq 8.
| 8 |
Essentially, Ki0 represents the equilibrium partition coefficient at infinitely dilute concentrations within the membrane and aqueous phases (Ti→0 and Ti→0m, respectively) when the membrane surface charge is zero. These intrinsic partition coefficients can be related to the observed partition coefficient at higher TPT concentrations as illustrated by eq 9.
| 9 |
Here, f1 and f2 account for the fractions of total TPT in the monomeric and dimeric forms, respectively, as defined by eq A17. Values of f1 corresponding to the conditions at the end of each release study were calculated from the dimerization constant (K2) obtained by fitting the dependence of the TPT extinction coefficient on concentration (Figure 4A) to the dimer model described by eqs A17 and A18. The estimated value of K2 is 6700 ± 600 M–1.
Figure 4.
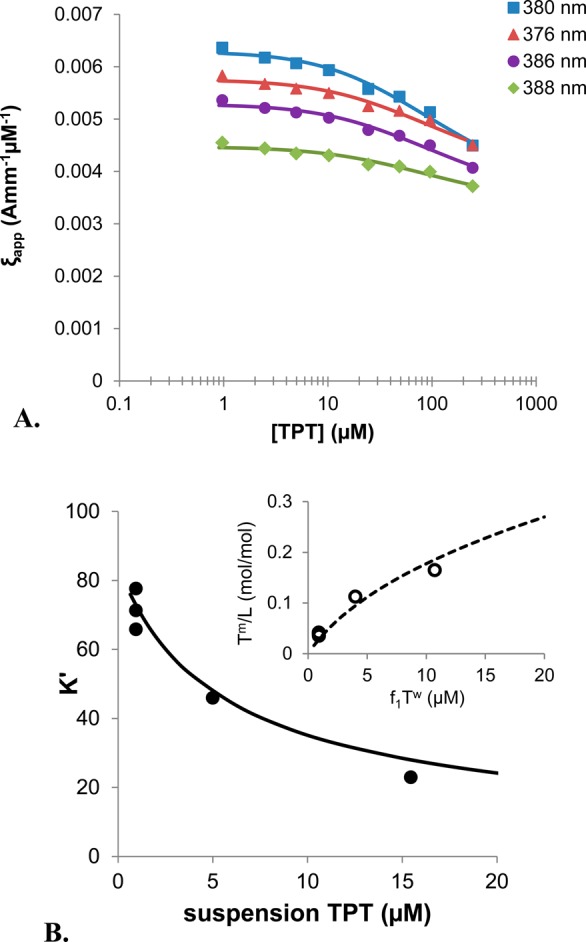
(A) Apparent extinction coefficients of TPT as a function of concentration at pH 4 were simultaneously fit to the dimer equations (A17 and A18) to determine a dimerization constant, K2. The plot shows extinction coefficients at 380 (blue), 376 (red), 386 (purple), and 388 (green) nm wavelengths along with lines of the corresponding color to represent the fit of the data to the dimerization model. Only four of the eight wavelengths used are shown above for clarity. (B) Using K2 and correcting for the changes in bilayer surface potential described by the Gouy–Chapman theory, the apparent binding coefficient, K′, observed at the three lipid concentrations used in nonsink release studies (●) was used to determine the intrinsic binding coefficient, K10 with eq 9, and the values provided in Table 2. The resulting fit of K′ to eq 9 is shown (solid line) and correlates with the reduction in binding experimentally observed with the three TPT suspension concentrations studied. The inset to the top right compares the nonlinear relationship of bound drug-to-lipid ratio, Tm/L, with increasing concentration of unbound, monomeric drug, f1Tu, predicted by the Gouy–Chapman equation (dotted line) with that determined from nonsink release studies (○).
The δ values account for the effects of changes in membrane surface potential on species binding with increasing drug concentration. Because TPT is primarily cationic at pH 4, its ability to bind to the bilayer surface will also depend on the membrane surface potential. Using the Gouy–Chapman theory as previously described by Austin and co-workers,38 this effect may be calculated for any partitioned species with charge z using the correction factor δz. This correction factor is calculated with the following equation.38
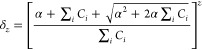 |
10 |
Here, α = σ/(2000RTε0εr), where σ is the surface charge density due to the concentration of TPT bound to the bilayer, ε0 is the permittivity of a vacuum, and εr is the relative permittivity of water. This correction is also dependent upon the bulk concentration of all electrolytes in solution, ΣiCi, and the charge of the TPT species of interest as both monomer (1+) and dimer (2+) forms are present in the concentration range studied.31
Using the values reported in Table 2 to account for dimerization and the membrane surface potential, K10 was determined to be 80 ± 20, while the partition coefficient for the dimer, K20, was found to be negligible. In Figure 4B, the profile generated by eq 9 using the fitted value of K1 along with the dimer constant, K2, correlates well with the experimentally observed apparent binding constants, K′. The inset in Figure 4B also demonstrates the nonlinearity observed in the plot of bound drug-to-lipid ratio, Tm/L, versus unbound monomeric drug concentration conforms to the Gouy–Chapman theory.
Discussion
Effect of Experimental Parameters on Extent of Drug Release under Nonsink Conditions
For the nonsink experiments, the extent of drug release is highly dependent upon two primary factors: fraction of volume encapsulated (fv) and the apparent membrane binding of drug to the liposomal bilayer (K′). The effect of these factors can be appreciated by examining the percentage of total drug released as defined by the following equation.
| 11 |
Here, Mo,∞ refers to the total mass of extra-vesicular drug at equilibrium, and MT is the total mass of the drug in the suspension.
Using the nonsink model, X can be simulated under a variety of experimental conditions (e.g., different lipid concentrations, particles sizes, drug binding coefficients etc.). Figure 5A and B illustrates two of the main experimental parameters affecting the total amount of drug released. Here, simulations were conducted to determine the expected percentage of drug released, X, for varying values of binding coefficients, K′, in Figure 5A and as a function of the ratio of entrapped volume, fv (i.e., liposome concentration), in Figure 5B. In Figure 5A, the plot shows that increasing values of K′ result in less drug released into the extra-vesicular compartment due to a higher amount bound to the membrane leaflet. For Figure 5B, the increasing values of fv result in less drug released because a larger fraction of the total volume is within the intra-vesicular compartment.
Figure 5.

Effect of experimental parameters on total drug release at equilibrium. (A) Keeping the suspension concentration constant at 0.5 mg/mL, simulations using the equations describing the nonsink model were used to determine the % of released drug, X, as a function of varying values of drug binding coefficients, K′. These simulations are plotted for several common diameters of liposomes. (B) To illustrate the effect entrapped volume, fv, has on the amount of drug released under nonsink conditions, simulations were conducted in which K′ was held constant at 90. The plot shows that increasing fv (i.e., increasing amount of liposomes) reduces the amount of drug released as the volume fraction entrapped increases (i.e., the number of liposomes in the suspension increases). The lines illustrate this trend for liposomes of different diameters indicated by the legend in the upper right corner of the plot. (C) This nomograph provides a general method for estimating the amount of released drug. The plot relates all experimental conditions affecting the amount of drug released during a nonsink release study including the drug binding coefficient, K′, and the volume compartments present in the suspension (a and b for intravesicular aqueous and membrane compartments, and c and d for extravesicular aqueous and membrane compartments) to X (as indicated by the labeled, horizontal lines). This relationship is highly dependent upon the fraction of entrapped volume, E, as the slope steepens dramatically with increasing E (and subsequently higher lipid concentration).
It would also be convenient to generalize these relationships so that the extent of drug release from liposomes under nonsink conditions could be estimated for a wide array of experimental conditions. Such a relationship is illustrated by Figure 5C. This nomograph was constructed by noting that at equilibrium, the concentrations of unbound, aqueous drug in the intra- and extra-vesicular solutions will be equal.
| 12 |
This relationship can then be rewritten in terms of total concentration of intra- and extra-vesicular drug using the previous derived fraction of unbound intra- and extra-vesicular drug (eqs A4a and A4b) and rearranged to the following ratio.
| 13 |
Furthermore, one can specify the percent of drug released, X, in terms of the total suspension concentration of drug present in solution, T, for Ti and To as expressed by eqs 14a and 14b.
| 14a |
| 14b |
Here, the fraction of total volume entrapped, E, may be expressed in terms of the previously defined ratio of entrapped to external volume, E = fv/(1 + fv). These equations can be substituted into eq 13 and rearranged into the following equation.
| 15 |
This relationship is linear as shown in Figure 5C with E/(1 – E) providing the slope. Here, the slopes of lines are shown based on varying values of E, and the horizontal lines indicate the percent of drug which would be released at equilibrium.
The above calculations and simulations assumed that all released drug, whether membrane-bound or free in extravesicular solution, was removed during ultrafiltration due to the low binding observed in these studies. This assumption can be assessed based on the dilutions made during ultrafiltration and drug binding coefficients. On the basis of the highest binding coefficient obtained during these experiments (73), there would be less than a 2.5% change in the total amount of drug removed over the range of lipid concentrations (0.48–15.3 mg lipid/mL) used in these studies. For drugs with higher membrane binding, a similar analysis shows that a 0.5 mg lipid/mL suspension would have less than a 3% change in the amount of drug removed for a lipophilic compound having a binding coefficient of 2400.
Applicability to Drug Release Characterization for Other Drugs and/or Nanoparticle Formulations
The mathematical model described here should be adaptable to other drugs and nanoparticle formulations. For every drug–nanoparticle combination, careful consideration should be given to which components of the current model are relevant and whether additional terms are necessary. For example, an evaluation of the effect of pH on release requires consideration of drug speciation as the ionization of the drug may have an effect on observed release.3,11 Other effects such as drug precipitation, complexation, or degradation may be taken into account by including relevant equilibrium equations to solve for the fraction of total drug free to permeate the membrane or by adding relevant kinetic terms (e.g., degradation/interconversion11 or dissolution rate constant) into the rate equation. More generally, the nonsink method and model may be applicable to other agents as well as other types of nanoparticles (e.g., a current application of similar methodology underway in this laboratory involves doxorubicin-conjugated polymeric micelles).
Validation of the percent recovery and percentage of free drug removed is critical when considering the use of ultrafiltration to isolate the drug remaining within the nanoparticle. Significant binding of drug to the ultrafiltration membrane may interfere with the removal of released drug by a washing step. In such cases, other methods that can separate (e.g., size-exclusion) or distinguish (e.g., spectroscopic techniques) entrapped from the released drug may be more appropriate yet still amenable to the nonsink mathematical model used here.
Conclusions
The liposomal release kinetics and lipid bilayer partitioning of the anticancer agent TPT were simultaneously determined by ultrafiltering liposomal suspensions under nonsink conditions at various times. Dynamic dialysis was used to validate these findings by providing a nearly identical release rate constant. The nonsink method was also able to probe the concentration dependence of TPT binding to the bilayer and revealed that binding was dependent on the surface potential at the bilayer interface and TPT dimerization. The nonsink method provides a reliable way to obtain both kinetic and thermodynamic descriptors. This method may also be useful in future mechanistic studies of liposomal drug release kinetics where dynamic dialysis studies are complicated by drug binding to the dialysis membrane or the observed release is rate-limited by drug transport through the dialysis membrane. The parameter values and methodology provided may have utility in the development of models capable of providing in vitro–in vivo correlations; however, environmental in vivo factors that may alter release rates would have to be investigated and incorporated into mechanistic models to yield useful, predictive relationships for liposomal formulations.
Acknowledgments
This project was supported by Grant Number R25CA153954 from the National Cancer Institute.
Glossary
Abbreviations
- TPT
topotecan
- DSPC
1,2-distearoyl-sn-glycero-3-phosphatidylcholine
- m-PEG DSPE
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000]
Appendix
Derivation of Unbound Drug Concentration for Modeling of Release Studies at Sink and Nonsink Conditions
Binding of drug to the phospholipid membrane interface has been reported previously with other chemotherapeutics and lipophilic drugs.3−5,8,19,38 Such binding will reduce the driving force for drug transport, resulting in the need for a mathematical model that includes this effect on release kinetics. Such a model was developed based on previous models (which account for drug binding) to describe the concentration of unbound drug in terms of total intra- and extra-vesicular drug concentration and its subsequent effect on release kinetics.3,9 The relationship is the same for release studies conducted under nonsink and sink conditions and is derived below.
The total amount of drug inside (Mi,T) and outside (Mo,T) the vesicle can be expressed in terms of the contributions of aqueous and membrane bound components. Equations A1a and A1b express these mass balances.
| A1a |
| A1b |
In these equations and from this point on, the superscripts “w” and “m” represent unbound drug in the aqueous compartment and membrane-bound drug, respectively; the subscripts i and o refer to the intra- and extra-vesicular compartments. These mass balance equations can be expressed in terms of concentrations using the ratios of the aqueous to membrane volume in the inner and outer compartments defined in eqs A2a-e (see Scheme 1)
| A2a-e |
thus producing eqs A3a and A3b for total drug concentration within, Ti, and outside, To, the vesicles:
| A3a |
| A3b |
Next, the concentration gradient of aqueous, unbound drug must be solved in terms of total drug encapsulated. This is done by incorporating an apparent volume-normalized membrane binding coefficient describing the equilibrium between TPT bound at the interface of the bilayer membrane and that in solution for the intravesicular, Ki′, and extravesicular, Ko′, compartments. These binding constants may differ if there are differences in the intra- versus extra-vesicular environments (e.g. pH gradients, ionic strength differences, etc.) or prior to equilibrium when drug concentrations may differ dramatically between the inner and outer compartments. In the present study of passively loaded liposomes, the intra-vesicular and extra-vesicular compartments were at the same pH and buffer concentration throughout the experiment. At equilibrium, both compartments contained the same drug concentration. Under these conditions, we found that a single K′ could be assumed (Ki′ = Ko′) without diminishing the quality of the fit of the model to the data.
With this assumption, the model may refer to both as K′, and Tiw and Tow may be described by eqs A4a and A4b
| A4a |
| A4b |
with K′ = Tm/Tw. Using these substitutions, eqs 2a, 2b, and 3 can be rewritten in terms of total intra- and extra-vesicular drug concentrations.
Initial Conditions for Release Studies Performed under Nonsink and Sink Conditions
Initial conditions must be provided to solve the differential equations for either condition. For nonsink release studies, both the initial concentrations of intra- and extra-vesicular drugs were determined by analyzing the total suspension concentration of the drug, T, and drug concentration after ultrafiltration of suspension when the release study began, Ti,0. This is shown by the equations below:
| A5a |
| A5b |
For sink conditions using the dynamic dialysis method, the initial conditions are dependent on the loading condition of the liposome suspension. For passively-loaded liposomes, the initial conditions are as follows:
| A6a |
| A6b |
where S is the initial suspension concentration of TPT within the dialysis tube. To accurately discern the rate of transport of drug through the dialysis membrane, a suspension of blank liposomes spiked with free TPT was used. While the rate equations are the same as those for the passively-loaded drug, the initial conditions are not and are expressed below:
| A7a |
| A7b |
Corrections for Ultrafiltration Recovery and Dialysis Bag Swelling during Sampling
The concentration of TPT determined by HPLC analysis of ultrafiltered samples, while mostly composed of intravesicular TPT, may require corrections due to the ultrafiltration process. The observed concentration obtained from ultrafiltration, Tu, must be interpreted correctly to accurately model drug release. This can be accomplished by expressing K′ = Tm/Tw with eq A8.
| A8 |
Here, the percentage of intra-vesicular (ω) and extra-vesicular drug (φ) recovered in the ultrafiltrate were determined with validation studies.
Ideally, the concentration in samples from dynamic dialysis studies at any sample time, n, would be dependent upon only diffusive transport process. This suspension concentration, Td,n, can be determined from the observed concentration within the dialysis tube, Td,n′, by accounting for volume changes due to sample removal and dialysis bag shrinking/swelling. These effects are expressed by eq A9.
| A9 |
The factors xv,n and xs,n correct for volume swelling in the dialysis tube and the mass removed due to sample collection since the previous time point, respectively.
The correction factor for volume change in the dialysis tube, xv,n, is:
| A10 |
where Vn is the volume present in the dialysis tube at sample time, n, and Vn–1 is the volume present after removing the sample for analysis at the previous time point (n – 1). The following equation describes the volume change occurring between time points:
| A11 |
where kv is the rate constant, and Veq is the volume in the dialysis tube at hydrostatic equilibrium. Vn–1 is the volume at the previous sampling time, and tΔn is the time interval between the samples.
In addition to swelling, the mass removed with each sample, while small, could cumulatively result in a substantial amount of lipid removal and subsequently encapsulated drug removed from the dialysis tube. Because of volume swelling, the amount of mass taken from the previous sampling must be accounted for at each sampling. The correction factors for the first, second, and any later sample (xs,1, xs,2, and xs,n, respectively) are:
| A12a |
| A12b |
| A12c |
Here, L0 is the lipid concentration in the initial suspension in the dialysis tube, Ls and Vs are the lipid concentration and volume of sample taken, respectively (at the denoted sample number), and V0 is the initial volume of suspension added to the dialysis chamber.
Half-Life (t1/2) Calculation for Nonsink Release Studies
Because equilibrium is achieved with a different extent of the drug released due to changes in membrane binding of the drug, the effects of membrane binding on release may be observed by calculating the half-life to equilibrium (t1/2) from these nonsink release studies. This calculation starts by solving for t1/2 with the rearrangement of eq 2a and substituting for To with the mass balance To = fv(Ti,0 – Ti).
| A13 |
Next, the term km′fofvTi,0 may be solved for by assuming equilibrium where (dTi)/(dt) = 0 and Ti = Tieq and A13 becomes eq A14.
| A14 |
Substituting A14 back into A13 and rearranging provides eq A15.
| A15 |
Equation A15 takes on the general form of a first-order reaction. Upon integration and substituting eqs A4a and A4b for fiwand fo, respectively, eq 7 is produced by solving for t1/2 as the time at which the amount of drug encapsulated is halfway to equilibrium (Ti – Tieq = 0.5(Ti,0 – Ti)).
Determination of TPT Dimerization Constant (K2)
Self-association of TPT in solution may influence binding due to different binding affinities of the monomeric (T1) and dimeric (T2) forms. The relationship between these forms was expressed previously by eq 5 via the dimerization constant, K2. The two forms may also be related by mass balance in which the total concentration of TPT in solution, T, may be written as the sum of these species as shown in eq A16.
| A16 |
Using these equations, the fraction of monomer present in solution, f1, can be solved as expressed by eq A17.
| A17 |
In solution, both monomeric and dimeric forms of TPT have their own unique extinction coefficients (ε1,i and ε2,i, respectively) at any wavelength, i, which contribute to the apparent extinction coefficient, εapp. This is shown by eq A18.
| A18 |
Using eqs A17 and A18, the concentration dependence of εapp,i was fit at multiple wavelengths simultaneously to determine K2, ε1,i, and ε2,i.
Supporting Information Available
Rate of dialysis tube swelling. This material is available free of charge via the Internet at http://pubs.acs.org.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Xiang T.-X.; Anderson B. D. Influence of Chain Ordering on the Selectivity of Dipalmitoylphosphatidylcholine Bilayer Membranes for Permeant Size and Shape. Biophys. J. 1998, 75, 2658–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang T.-X.; Anderson B. D. Liposomal Drug Transport: A molecular Perspective From Molecular Dynamics Simulations in Lipid Bilayers. Adv. Drug Delivery Rev. 2006, 58, 1357–1378. [DOI] [PubMed] [Google Scholar]
- Joguparthi V.; Xiang T.-X.; Anderson B. D. Liposome Transport of Hydrophobic Drugs: Gel Phase Lipid Bilayer Permeability and Partitioning of the Lactone Form of a Hydrophobic Camptothecin, DB-67. J. Pharm. Sci. 2008, 97, 400–420. [DOI] [PubMed] [Google Scholar]
- Joguparthi V.; Feng S.; Anderson B. D. Determination of Intraliposomal pH and its Effect on Membrane Partitioning and Passive Loading of a Hydrophobic Camptothecin, DB-67. Int. J. Pharm. 2008, 352, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi S.; Anderson B. Bilayer Composition, Temperature, Speciation Effects and the Role of Bilayer Chain Ordering on Partitioning of Dexamethasone and Its 21-Phosphate. Pharm. Res. 2013, 1–16. [DOI] [PubMed] [Google Scholar]
- Čeh B.; Lasic D. D. A Rigorous Theory of Remote Loading of Drugs Into Liposomes. Langmuir 1995, 11, 3356–3368. [DOI] [PubMed] [Google Scholar]
- Čeh B.; Lasic D. D. A Rigorous Theory of Remote Loading of Drugs into Liposomes: Transmembrane Potential and Induced pH-Gradient Loading and Leakage of Liposomes. J. Colloid Interface Sci. 1997, 185, 9–18. [DOI] [PubMed] [Google Scholar]
- Modi S.; Anderson B. D. Determination of Drug Release Kinetics from Nanoparticles: Overcoming Pitfalls of the Dynamic Dialysis Method. Mol. Pharmacol. 2013, 10, 3076–3089. [DOI] [PubMed] [Google Scholar]
- Joguparthi V.; Anderson B. D. Liposomal Delivery of Hydrophobic Weak Acids: Enhancement of Drug Retention Using a High Intraliposomal pH. J. Pharm. Sci. 2008, 97, 433–454. [DOI] [PubMed] [Google Scholar]
- Tejwani R. W.; Stouch T. R.; Anderson B. D. Substituent Effects on the Ionization and Partitioning of p-(Aminoethyl)Phenols and Structurally Related Compounds: Electrostatic Effects Dependent on Conformation. J. Pharm. Sci. 2009, 98, 4534–4544. [DOI] [PubMed] [Google Scholar]
- Fugit K. D.; Anderson B. D. The Role of pH and Ring-Opening Hydrolysis Kinetics on Liposomal Release of Topotecan. J. Controlled Release 2014, 174, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzen U.; Østergaard J. Physico-Chemical Characterization of Liposomes and Drug Substance–Lliposome Interactions in Pharmaceutics Using Capillary Electrophoresis and Electrokinetic Chromatography. J. Chromatogr. A 2012, 1267, 32–44. [DOI] [PubMed] [Google Scholar]
- Domingo C.; Saurina J. An Overview of the Analytical Characterization of Nanostructured Drug Delivery Systems: Towards Green and Sustainable Pharmaceuticals: A Review. Anal. Chim. Acta 2012, 744, 8–22. [DOI] [PubMed] [Google Scholar]
- Cho E. J.; Holback H.; Liu K. C.; Abouelmagd S. A.; Park J.; Yeo Y. Nanoparticle Characterization: State of the Art, Challenges, and Emerging Technologies. Mol. Pharmacol. 2013, 10, 2093–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham S. A.; Edwards K.; Karlsson G.; Hudon N.; Mayer L. D.; Bally M. B. An Evaluation of Transmembrane Ion Gradient-Mediated Encapsulation of Topotecan Within Liposomes. J. Controlled Release 2004, 96, 449–461. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Shi Y.; Chen Y.; Ye J.; Sha X.; Fang X. Multifunctional Pluronic P123/F127 Mixed Polymeric Micelles Loaded With Paclitaxel for the Treatment of Multidrug Resistant Tumors. Biomaterials 2011, 32, 2894–2906. [DOI] [PubMed] [Google Scholar]
- Gajbhiye V.; Ganesh N.; Barve J.; Jain N. K. Synthesis, Characterization and Targeting Potential of Zidovudine Loaded Sialic Acid Conjugated-Mannosylated poly(Propyleneimine) Dendrimers. Eur. J. Pharm. Sci. 2013, 48, 668–679. [DOI] [PubMed] [Google Scholar]
- Liko F.; Erdoğan S.; Özer Y. A.; Vural I. In vitro Studies on 5-Florouracil-Loaded DTPA-PE Containing Nanosized Pegylated Liposomes for Diagnosis and Treatment of Tumor. J. Lipos. Res. 2013, 23, 61–69. [DOI] [PubMed] [Google Scholar]
- Maurer-Spurej E.; Wong K. F.; Maurer N.; Fenske D. B.; Cullis P. R. Factors Influencing Uptake and Retention of Amino-Containing Drugs in Large Unilamellar Vesicles Exhibiting Transmembrane pH Gradients. Biochim. Biophys. Acta, Biomembr. 1999, 1416, 1–10. [DOI] [PubMed] [Google Scholar]
- Park J.; Cho Y.; Son Y.; Kim K.; Chung H.; Jeong S.; Choi K.; Park C.; Park R.-W.; Kim I.-S.; Kwon I. Preparation and Characterization of Self-Assembled Nanoparticles Based on Glycol Chitosan Bearing Adriamycin. Colloid Polym. Sci. 2006, 284, 763–770. [Google Scholar]
- Ferraretto A.; Sonnino S.; Soria M. R.; Masserini M. Characterization of Biotinylated Liposomes Sensitive to Temperature and pH: New Tools for Anti-Cancer Drug Delivery. Chem. Phys. Lipids 1996, 82, 133–139. [DOI] [PubMed] [Google Scholar]
- Lu D.-X.; Wen X.-T.; Liang J.; Zhang X.-D.; Gu Z.-W.; Fan Y.-J. Novel pH-Sensitive Drug Delivery System Based on Natural Polysaccharide for Doxorubicin Release. Chin. J. Polym. Sci. 2008, 26, 369–374. [Google Scholar]
- Atyabi F.; Farkhondehfai A.; Esmaeili F.; Dinarvand R. Preparation of Pegylated Nano-Liposomal Formulation Containing SN-38: In Vitro Characterization and in Vivo Biodistribution in Mice. Acta. Pharm. 2009, 59, 133. [DOI] [PubMed] [Google Scholar]
- Liu J.-J.; Hong R.-L.; Cheng W.-F.; Hong K.; Chang F.-H.; Tseng Y.-L. Simple and Efficient Liposomal Encapsulation of Topotecan by Ammonium Sulfate Gradient: Stability, Pharmacokinetic and Therapeutic Evaluation. Anticancer. Drugs 2002, 13, 709–717. [DOI] [PubMed] [Google Scholar]
- Xiang T.-X.; Anderson B. D. Permeability of Acetic Acid Across Gel and Liquid-Crystalline Lipid Bilayers Conforms to Free-Surface-Area theory. Biophys. J. 1997, 72, 223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig A.; Seelig J. Dynamic Structure of Fatty Acyl Chains in a Phospholipid Bilayer Measured by Deuterium Magnetic Resonance. Biochemistry (Moscow) 1974, 13, 4839–4845. [DOI] [PubMed] [Google Scholar]
- Huang C. H. A 13C and 2H Nuclear Magnetic Resonance Study of Phosphatidylcholine/Cholesterol Interactions: Characterization of Liquid-Gel Phases. Biochemistry (Moscow) 1993, 32, 11. [DOI] [PubMed] [Google Scholar]
- Tejwani R. W.; Anderson B. D. Influence of Intravesicular pH Drift and Membrane Binding on the Liposomal Release of a Model Amine-Containing Permeant. J. Pharm. Sci. 2008, 97, 381–399. [DOI] [PubMed] [Google Scholar]
- Bocian W.; Kawecki R.; Bednarek E.; Sitkowski J.; Pietrzyk A.; Williamson M. P.; Hansen P. E.; Kozerski L. Multiple Binding Modes of the Camptothecin Family to DNA Oligomers. Chem.—Eur. J. 2004, 10, 5776–5787. [DOI] [PubMed] [Google Scholar]
- Strel’tsov S.; Oleinikov V.; Ermishov M.; Mochalov K.; Sukhanova A.; Nechipurenko Y.; Grokhovsky S.; Zhuze A.; Pluot M.; Nabiev I. Interaction of Clinically Important Human DNA Topoisomerase I Poison, Topotecan, With Double-Stranded DNA. Biopolymers 2003, 72, 442–454. [DOI] [PubMed] [Google Scholar]
- Strel’tsov S. A.; Grokhovskii S. L.; Kudelina I. A.; Oleinikov V. A.; Zhuze A. L. Interaction of Topotecan, DNA Topoisomerase I Inhibitor, with Double-Stranded Polydeoxyribonucleotides. 1. Topotecan Dimerization in Solution. Mol. Biol. 2001, 35, 365–373. [PubMed] [Google Scholar]
- Clerc S.; Barenholz Y. A Quantitative Model for Using Acridine Orange as a Transmembrane pH Gradient Probe. Anal. Biochem. 1998, 259, 104–111. [DOI] [PubMed] [Google Scholar]
- Zucker D.; Marcus D.; Barenholz Y.; Goldblum A. Liposome Drugs’ Loading Efficiency: A Working Model Based on Loading Conditions and Drug’s Physicochemical Properties. J. Controlled Release 2009, 139, 73–80. [DOI] [PubMed] [Google Scholar]
- Modi S.; Xiang T.-X.; Anderson B. D. Enhanced Active Liposomal Loading of a Poorly Soluble Ionizable Drug Using Supersaturated Drug Solutions. J. Controlled Release 2012, 162, 330–339. [DOI] [PubMed] [Google Scholar]
- Grit M.; Crommelin D. J. A. Chemical Stability of Liposomes: Implications for Their Physical Stability. Chem. Phys. Lipids 1993, 64, 3–18. [DOI] [PubMed] [Google Scholar]
- Grit M.; Crommelin D. J. A. The Effect of Aging on the Physical Stability of Liposome Dispersions. Chem. Phys. Lipids 1992, 62, 113–122. [DOI] [PubMed] [Google Scholar]
- Grit M.; de Smidt J. H.; Struijke A.; Crommelin D. J. A. Hydrolysis of Phosphatidylcholine in Aqueous Liposome Dispersions. Int. J. Pharm. 1989, 50, 1–6. [Google Scholar]
- Austin R. P.; Barton P.; Davis A. M.; Manners C. N.; Stansfield M. C. The Effect of Ionic Strength on Liposome–Buffer and 1-Octanol–Buffer Distribution Coefficients. J. Pharm. Sci. 1998, 87, 599–607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.