

Conspectus
Our cellular genome is continuously exposed to a wide spectrum of exogenous and endogenous DNA damaging agents. These agents can lead to formation of an extensive array of DNA lesions including single- and double-stranded breaks, inter- and intrastrand cross-links, abasic sites, and modification of DNA nucleobases. Persistence of these DNA lesions can be both mutagenic and cytotoxic, and can cause altered gene expression and cellular apoptosis leading to aging, cancer, and various neurological disorders. To combat the deleterious effects of DNA lesions, cells have a variety of DNA repair pathways responsible for restoring damaged DNA to its canonical form. Here we examine one of those repair pathways, the base excision repair (BER) pathway, a highly regulated network of enzymes responsible for repair of modified nucleobase and abasic site lesions.
The enzymes required to reconstitute BER in vitro have been identified, and the repair event can be considered to occur in two parts: (1) excision of the modified nucleobase by a DNA glycosylase, and (2) filling the resulting “hole” with an undamaged nucleobase by a series of downstream enzymes. DNA glycosylases, which initiate a BER event, recognize and remove specific modified nucleobases and yield an abasic site as the product. The abasic site, a highly reactive BER intermediate, is further processed by AP endonuclease 1 (APE1), which cleaves the DNA backbone 5′ to the abasic site, generating a nick in the DNA backbone. After action of APE1, BER can follow one of two subpathways, the short-patch (SP) or long-patch (LP) version, which differ based on the number of nucleotides a polymerase incorporates at the nick site. DNA ligase is responsible for sealing the nick in the backbone and regenerating undamaged duplex.
Not surprisingly, and consistent with the idea that BER maintains genetic stability, deficiency and/or inactivity of BER enzymes can be detrimental and result in cancer. Intriguingly, this DNA repair pathway has also been implicated in causing genetic instability by contributing to the trinucleotide repeat expansion associated with several neurological disorders.
Within this Account, we outline the chemistry of the human BER pathway with a mechanistic focus on the DNA glycosylases that initiate the repair event. Furthermore, we describe kinetic studies of many BER enzymes as a means to understand the complex coordination that occurs during this highly regulated event. Finally, we examine the pitfalls associated with deficiency in BER activity, as well as instances when BER goes awry.
1. Introduction
DNA nucleobases are chemically reactive; this reactivity leads to a wide variety of nucleobase modifications that can occur by oxidation, alkylation, deamination, or hydrolysis. Indeed, more than 70 modified nucleobases have been identified in vitro, of which ∼15 have been found in cellular DNA.1,2 Additionally, hydrolysis of the carbon–nitrogen bond that adjoins the nucleobase to the deoxyribose sugar, the glycosidic bond, results in loss of the nucleobase and formation of an abasic site. Examples of several modified nucleobases are shown in Figure 1.
Figure 1.

Examples of DNA lesions formed by oxidation, alkylation, deamination, and hydrolysis of canonical nucleobases. 5hU is a product of deamination and oxidation. Shown are 8-oxoG (8-oxo-7,8-dihydroguanine), Tg (thymine glycol), 5,6DHU (5,6-dihydrouracil), hmU (hydroxymethyluracil), 5hC (5-hydroxycytosine), FapyG (4,6-diamino-5-formamidopyrimidine G), FapyA (4,6-diamino-5-formamidopyrimidine A), Gh (guanidinohydantoin), Sp (spiroiminodihydantoin), 3meA (3-methyladenine), 7meG (7-methylguanine), 7meA (7-methyladenine), 3meG (3-methylguanine), eA (1,N6-ethenoadenine), U (uracil), X (xanthine), Hx (hypoxanthine), 5hU (5-hydroxyuracil), and an abasic site.
Many of these DNA lesions are highly mutagenic when formed in cellular DNA, meaning they are mispaired by a DNA polymerase during replication. For example, 8-oxo-7,8-dihydroguanine (8oxoG), which should be paired with C during replication, as it is a lesion derived from G, can also be paired with A to form a Hoogsteen base pair.3 In addition, DNA lesions can be cytotoxic meaning that they cause a polymerase to stall and halt DNA replication, leading to cellular apoptosis.
Due to the extensive range of lesions formed, and the deleterious effects they can have, the ability to repair the damaged DNA is integral to genomic stability and cell viability. The BER pathway, comprising several enzymes including a DNA glycosylase, APE1, DNA polymerase, and DNA ligase, along with several accessory proteins, is responsible for recognizing and repairing these modified nucleobases and abasic sites. The proceeding sections describe the role(s) of each of the BER enzymes. In this Account, we focus on the BER pathway in humans, with homologous repair enzymes present in bacteria and yeast.
2. DNA Glycosylase
A DNA glycosylase initiates the BER pathway, and is responsible for recognizing and binding specific nucleobase lesions, and flipping the targeted nucleobase into the active site to catalyze cleavage of the glycosidic bond (Figure 2, STEP 1). There are at least 11 known human DNA glycosylases; some have activity on a variety of nucleobase lesions, others are specific for just one or two DNA lesions. Preferred substrate lesion(s) for each glycosylase are listed in Table 1.
Figure 2.
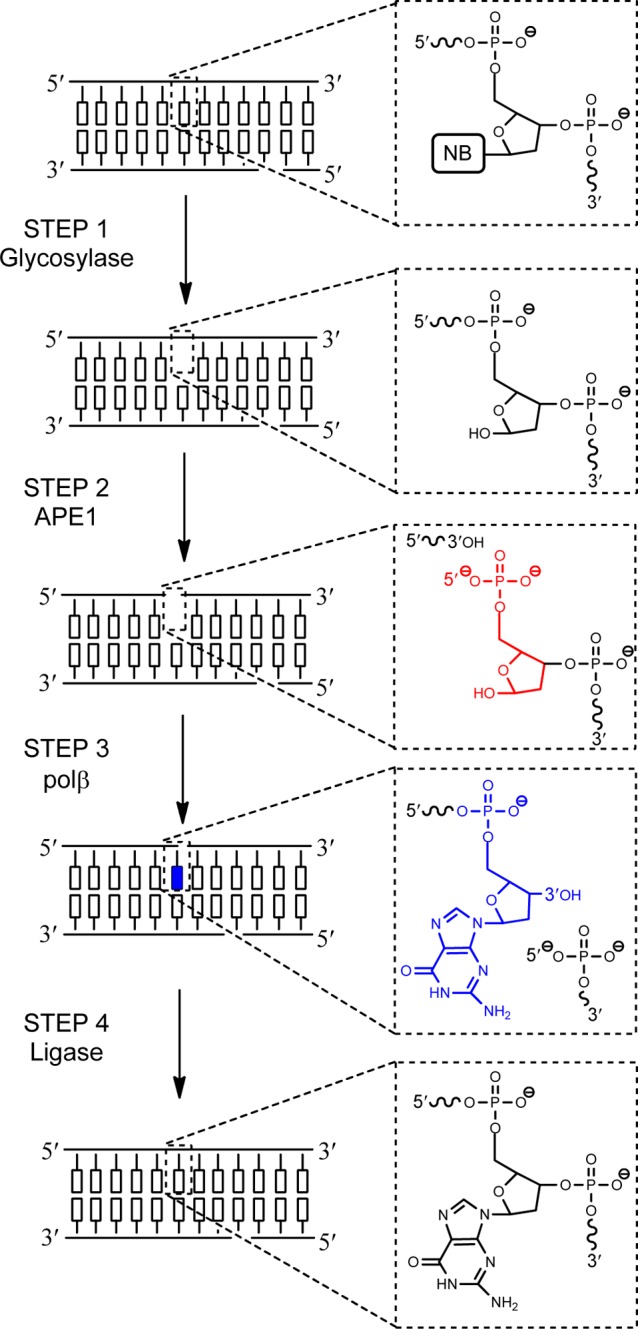
A modified DNA nucleobase lesion (NB) is recognized and removed by a glycosylase creating an abasic site (STEP 1). APE1 cleaves the DNA backbone 5′ to the abasic site, creating a nick with 3′-OH and 5′-dRP (red) termini (STEP 2). Pol β removes the 5′-dRP and inserts an unmodified nucleotide at the 3′-OH (blue) (STEP 3). Finally, a ligase seals the nick between the 3′-OH of the newly incorporated nucleotide and 5′-phosphate in the backbone (STEP 4).
Table 1. Human DNA Glycosylases and Their Preferred Lesion Substrates (E. coli homologue provided in parentheses).
| monofunctional DNA glycosylases | substrate(s) |
|---|---|
| UNG1/2 (UDG)ad | U |
| MBD4b | T:G; U:G; hmU:Gc |
| TDGb | T:G; U:G; T:C; T:Tc |
| SMUG1a (MUG) | U; hmU |
| MUTYHb (MutY) | A:8oxoGb |
| AAGb (AlkA) | 3meA; 7meG; eA; Hx; X |
| bifunctional DNA glycosylases | substrate(s) |
|---|---|
| OGG1b (Fpg) | 8oxoG; FapyG |
| NTH1b (Nth) | FapyG; Tg; 5,6DHU, 5hC; 5hU |
| NEIL1a (Nei) | Sp; Gh; FapyG; FapyA; 5,6DHT, 5,6DHU |
| NEIL2d (Nei) | Sp; Gh; 5hU; 5,6DHT, 5,6DHU; 5hC |
| NEIL3d (Nei) | Sp; Gh; FapyG; FapyA |
Activity on single-stranded and double-stranded DNA.
Activity on double-stranded DNA.
N:N represents a mispair where nucleobase removed is in bold.
Prefers lesions in single-stranded DNA.
The ability of a DNA glycosylase to find its substrate among the excess of unmodified nucleobases present in the genome has been likened to finding a needle in a haystack. Much research has been dedicated to understanding this process. Most models include short-range sliding along DNA, with the glycosylase probing and extruding individual nucleobases; in doing so, the glycosylase can identify its substrate(s) and catalzye cleavage of the glycosidic bond.4 Subsequent BER enzymes are also thought to employ a sliding mechanism, which allows for enzyme processivitiy; indeed, several DNA glycosylases have been shown to remove multiple DNA lesions during a single binding event.5−7
Some glycosylases work on lesions in both single- and double-stranded DNA, and others only work on double-stranded DNA.8−10 Interestingly, some glycosylases have a preference for lesions in single-stranded bulged or bubble structures.11,12 It is also noteworthy that not all DNA glycosylases remove modified nucleobases. For example, MUTYH removes the A from a 8oxoG:A mispair.13 This activity prevents the point mutation that would result if 8oxoG were removed instead. Furthermore, there are glycosylases that work on canonical DNA nucleobases that are mispaired, for example, TDG, which removes T from T:G mispairs.14
DNA glycosylases can be divided into two categories: monofunctional and bifunctional (Table 1). Monofunctional DNA glycosylases use an activated water molecule to hydrolyze the glycosidic bond, affording an abasic site product. Bifunctional DNA glycosylases utilize an amino group of the enzyme for nucleophilic attack and in addition to glycosidic bond cleavage, catalyze β-elimination of the DNA backbone 3′ to the abasic site via formation of a Schiff base, creating a single-stranded break with 3′-α,β-unsaturated aldehyde and 5′-phosphate termini. Some bifunctional DNA glycosylases can also perform δ-elimination to yield a 3′-phosphate. Notably, it has been proposed that for some bifunctional glycosylases, the β-elimination strand cleavage may be bypassed in vivo, with the subsequent BER enzyme, APE1, acting directly on the abasic site.15,16
Figure 3 shows a proposed SN1 (DN*AN) mechanism for DNA glycosylases in which nucleobase removal progresses through two oxocarbenium-ion-like transition states (TS) (Figure 3A,C) and a distinct oxocarbenium intermediate (Figure 3B). TS analysis of the E. coli homologues of UNG and MUTYH, UDG and MutY, respectively, has been performed using kinetic isotope effect (KIE) measurements. Examination of primary 13C and 15N KIEs, along with secondary deuterium and 15N KIEs, suggests a strongly dissociative TS with extensive oxocarbenium character for both UDG and MutY.17,18 KIE measurements for other glycosylases remain to be performed.
Figure 3.

Proposed SN1 (DN*AN) mechanism for DNA glycosylases which proceeds through two oxocarbenium-ion-like transition states (A, C) and a distinct oxocarbenium intermediate (B). Monofunctional DNA glycosylases use H2O as the nucleophile (Nu) yielding an abasic site product (D), while bifunctional DNA glycosylases use an active site amine with formation of a Schiff base (E) prior to β-elimination and hydrolysis to yield a nick in the DNA backbone with 3′-α,β-unsaturated aldehyde and 5′-phosphate termini (F). The corresponding transition state and intermediate pyrrolidine analogues are shown in G, H, and I. Chu et al reports use of both H and I as analogues of the transition state C.
In addition to TS analysis by KIE measurements, electrophoretic mobility shift assays (EMSA) also support a DN*AN mechanism. Using several E. coli monofunctional (AlkA, MutY) and bifunctional (Fpg, Nth) and human monofunctional (AAG, TDG) glycosylases, tight binding between a pyrrolidine abasic site analogue, which mimics the oxocarbenium intermediate, (Figure 3H) and the glycosylase was revealed.19,20 Interestingly, this binding event is strong enough to inhibit activity of many of the glycosylases on their prototypic substrate lesion(s). A more recent EMSA study using pyrrolidine abasic site analogues that mimic the two oxocarbenium TS (Figure 3G,I), demonstrated binding of bifunctional glycosylases Fpg, Nei, OGG1, and NEIL1 to mimics of both TS.21 Interestingly, OGG1 and NEIL1 displayed different binding preferences for the two TS mimics, suggesting alternate modes of recognition and catalysis for these bifunctional glycosylases.
3. AP Endonuclease
The enzyme following a DNA glycosylase in the BER pathway is AP endonuclease 1 (APE1). APE1, a Mg2+-dependent enzyme, is responsible for incising the DNA backbone at abasic sites, creating a nick with 3′-OH and 5′-deoxyribose phosphate (dRP) termini (Figure 2, STEP 2). Abasic sites are highly mutagenic and cytotoxic, and can also form protein–DNA and DNA–DNA cross-links.22 Therefore, repair of abasic sites by APE1 is critical in maintaining genomic integrity. For APE1, an activated water molecule has been implicated as the nucleophile for strand incision. A Mg2+ ion is also required; the divalent metal ion coordinates an oxygen of the 5′-phosphate, increasing its electrophilicty and also orienting the DNA backbone within the APE1 active site.23,24
4. Polymerase β
Polymerase β (pol β) follows APE1 in the BER pathway and has two catalytic functions: (1) it converts the 5′-dRP to a 5′-phosphate using its dRP lyase activity and (2) in a Mg2+-dependent reaction catalyzes incorporation of a single nucleotide to the 3′-OH of the nick (Figure 2, STEP 3).25 Nucleotide incorporation and dRP chemistry of pol β occur at separate active sites, although evidence suggests that both catalytic events occur during a single pol β/DNA binding event. Notably, the rate of dRP removal by pol β is 20-fold faster than incorporation, and therefore, it is postulated that dRP removal occurs prior to nucleotide incorporation.26
5. DNA Ligase
The final step of the BER pathway is sealing of the nick in the backbone by a DNA ligase (Figure 2, STEP 4). Both DNA ligase I (Lig1) and DNA ligase III (Lig3) have been implicated in nick sealing by catalyzing formation of a phosphodiester bond between the 3′-OH of the newly incorporated nucleotide and the 5′-phosphate of its neighbor. Human ligases require ATP and Mg2+ for activity, and their mechanism involves three distinct steps: (1) enzyme adenylation at an active site lysine, (2) adenylyl transfer to the 5′-phosphate of the nick, and (3) nucleophilic attack of the 3′-OH to seal the nick and release AMP.27
It is known that for activity in vivo, Lig3 requires the presence of X-ray repair cross-complementing protein 1 (XRCC1); this protein, described later in the Account, has no known catalytic function, but rather acts as a scaffold.28 While it has traditionally been thought that Lig3 is the major ligase in short-patch BER (vide infra) in the nucleus, it was recently reported that Lig1 is the major ligase in nuclear short-patch BER while Lig3 is essential for mitochondrial short-patch BER.29,30
6. Long-Patch BER
The pathway described above is typically referred to as short-patch BER (SP-BER), in which pol β removes the 5′-dRP group at the gap site and inserts a single nucleotide. Under conditions where the 5′-dRP group is modified such that the dRP lyase activity of pol β is blocked, an alternate pathway, long-patch BER (LP-BER), is utilized (Figure 4).31,32 In LP-BER, multiple nucleotides are incorporated at the gap site by polymerase β, δ, or ε (Figure 4, STEP 3). The polymerase incorporates, on average, 2–6 nucleotides at the gap site but this number can increase depending on the lesion as well as the sequence context.33 The incorporation of multiple nucleotides at the gap site generates a displaced single-stranded flap of DNA, another key feature of LP-BER. This flap must be removed by flap endonuclease 1 (FEN1) (Figure 4, STEP 4) so that DNA ligase, Lig1 in LP-BER, can seal the nick (Figure 4, STEP 5). Importantly, an accessory protein, proliferating cell nuclear antigen (PCNA), has been implicated in binding and coordinating the activity of many LP-BER enzymes.34
Figure 4.

Long-patch BER. STEP 1 and 2 are the same as in Figure 2. An oxidized abasic site which is known to require LP-BER, 2-deoxyribonolactone, is shown. Pol β, δ, or ε inserts multiple nucleotides at the 3′-OH nick site generated by APE1 (STEP 3; we show insertion of three G’s (blue)). FEN1 removes the 5′-flap (STEP 4), and Lig1 seals the nick in the backbone (STEP 5).
7. Kinetics of BER Enzymes
Much of our knowledge about the chemistry and substrate specificity of each BER enzyme has been gathered from extensive kinetic studies. The catalytic pathway that an enzyme follows in converting substrate to product is represented by a minimal kinetic scheme, such as that shown in Figure 5 for glycosylases.35 Arrows represent distinct steps along the pathway and k represents the rate associated with that step. (Figure 5 is used as an example and represents the minimal kinetic scheme for some, but not all, BER enzymes). The number of reactions an enzyme can catalyze per unit of time, where reaction is defined as encompassing the entire kinetic scheme and converting the unbound substrate to unbound product, is represented by kcat. Therefore, kcat is defined by the slow rate-determining step (RDS) of the catalytic pathway. For most BER enzymes, kcat is defined by product release (k3 in Figure 5); as we examine below, this slow rate of product release is an important feature of BER enzymes, and may serve to coordinate individual steps of the pathway. The kcat of BER enzymes range from as slow as 0.05 min–1 to as fast as 50 s–1 (Table 2). Although kcat is useful for considering an individual BER enzyme, the best way to kinetically compare enzymes is to examine their catalytic efficiency, defined as kcat/KM (where KM represents the substrate concentration at which the enzyme has reached half maximal velocity). Because catalytic efficiency reflects both the rate at which the enzyme completes the entire catalytic cycle, together with how well the enzyme binds a particular substrate, this term best represents how efficiently an enzyme works. UNG is currently the most catalytically efficient BER enzyme known with kcat/KM of 500 s–1 μM–1 (Table 2).
Figure 5.

Minimal kinetic scheme for DNA glycosylases. Three steps are depicted: binding of glycosylase enzyme (E) to the DNA substrate (DNAS) (k1/k–1), glycosidic bond cleavage (and β-elimination, when applicable) (k2), and DNA product (DNAP) release (k3).
Table 2. Kinetic Parameters of BER Enzymesab.
| enzyme | kcat | kcat/KM (s–1 μM–1) | kchemistry | ref |
|---|---|---|---|---|
| UNG | 50 s–1 | 500 | 115 s–1 | (8), 70 |
| OGG1 | 0.05 min–1 | 0.03 | 40 min–1 | (71), 72 |
| APE1 | 2 s–1 | 100 | ≥700 s–1 | (36, 73−75) |
| polβ (insertion) | 0.45 s–1 | 1.5 | 2–20 s–1c | (38), 69 |
| polβ (dRP Lyase) | 0.075 s–1 | 0.15 | 2 s–1d | 26, 76 |
| Lig1 | 0.04 s–1 | 0.4 | 12 s–1 | (27), (69) |
Rates determined at 37 °C.
Portion of table adapted from ref (69).
Rate of insertion depends on dNTP.
Determined at 15 °C. Value represents the slow phase of a biphasic time course; the fast phase was too fast to measure.
In addition to catalytic efficiency, it is important to call attention to rate of chemistry, kchemistry (k2 in Figure 5) (i.e., cleavage of the DNA backbone by APE1 or insertion of a nucleotide by pol β). For most, if not all BER enzymes, kchemistry is much faster than kcat. For example, APE1 cleaves DNA at a rate ≥700 s–1, making APE1 one of the fastest BER enzymes. Although kcat of APE1 is defined by product release and is quite slow, ∼2 s–1, this slow turnover may be overcome by its high copy number which is estimated to be 350 000–7 000 000 molecules per cell.36,37 Measuring rates of kchemistry not only provides an understanding of how fast each BER enzyme carries out its required task in BER, but also provides an understanding of substrate specificity. For instance, the rate of nucleotide insertion by pol β varies depending on the nucleotide inserted.38 Furthermore, for many BER enzymes, rates are affected by concentration of a required cofactor. A notable example is Lig1, which requires Mg2+ and ATP. At saturating concentrations of Mg2+, enzyme adenylation defines kcat, whereas at limiting concentrations of Mg2+, nick-sealing defines kcat.27
8. Coordination during SP- and LP-BER
The BER pathway is a highly coordinated process. This coordination is evident in various kinetic studies, as well as by the presence of scaffold accessory proteins. As stated above, the RDS of many BER enzymes is product release. It has been postulated that such a kinetic scheme allows for hand-off of DNA between enzymes of the BER pathway, and prevents exposure of mutagenic and cytotoxic repair intermediates. Such a hand-off, which has also been likened to “passing of a baton”,39 suggests a cascade of enzymes acting much like an assembly line. Furthermore, it is known that some BER enzymes stimulate slow product release of the enzyme that precedes it in the cascade. For example, APE1 stimulates the rate of product release of many DNA glycosylases.15,40−42 Likewise, Lig1 plays a role in regulating multinucleotide incorporation of pol δ and ε,43 while FEN1 stimulates and coordinates dRP lyase activity of pol β.44
As an alternative to the “passing of a baton” scheme, it has also been proposed that participation of several scaffolding accessory proteins suggests formation of a preassembled BER complex.45 These scaffolds have no known catalytic function and are not required to reconstitute BER in vitro, but are necessary for efficient BER in vivo. The scaffold protein XRCC1 can bind APE1, pol β, and Lig3, forming a complex at lesion sites during SP-BER.28 Furthermore, PCNA acts as a processivity clamp for pol β, δ, and ε to aid in nondissociative, accurate DNA replication.46,47 PCNA has been shown to complex with many BER enzymes, such as UNG, AAG, MUTYH, NEIL1, APE1, FEN1, and Lig1;48−51 accordingly, PCNA has been referred to as a docking station or communication point for such enzymes. Due to extensive interactions among BER enzymes, it has been proposed that a preassembled PCNA/BER enzyme complex slides along DNA searching for lesions.52 PARP1, poly [ADP-ribose] polymerase 1, has also been proposed to contribute to BER. PARP1 poly(ADP)-ribosylates several proteins, including itself, and has a defined role in sensing DNA single-strand breaks, but specific role(s) of PARP1 in BER remain unclear.53 It remains to be determined which model is followed in vivo, “passing of a baton” or a preassembled complex; it is possible that a combination of both is at work during SP- and LP-BER.
9. Deficiency in BER
The BER pathway is responsible for repair of many modified DNA nucleobase lesions, and deficiency and/or inactivity of any BER enzyme can have deleterious cellular outcomes. Deficiency or inactivity of DNA glycosylases can lead to various cancers. For instance, some mutations in the OGG1 gene, which inactivate the glycosylase, are linked to esophageal, lung squamous cell carcinomas, orolaryngeal, kidney, and gastric cancers. Similarly, mutations in MUTYH are linked to a form of colorectal cancer known as MUTYH-associated polyposis.54 This mutation leads to MUTYH variants that have decreased affinity and catalytic activity on 8oxoG:A mispairs.55 These are just two examples of several, in which inactivity of a DNA glycosylase leads to cancer. A single-nucleotide polymorphism in APE1 causes increased risk of colorectal cancer.56 Furthermore, APE1 activity is essential for cell viability.57,58 This requirement for APE1 is due to the fact that cells rely on APE1 for 95% of all endonuclease activity.59 In contrast, for DNA glycosylases, polymerases, and ligases, there can be substrate overlap and therefore another enzyme may be able to compensate for enzyme deficiency. Mice that produce ∼50% of normal levels of pol β have increased amounts of single-stranded breaks and chromosomal aberrations, and are hypersensitive to DNA damaging agents.60 Furthermore, mutations in pol β have been detected in ∼30% of tumors in humans.61 Interestingly, in conjunction with DNA damaging agents, APE1 and pol β are also targets for cancer therapy, with aim of inducing apoptosis in cancer cells by inhibiting APE1 or pol β activity.62,63
10. When BER Goes Awry
Although BER is essential for genetic integrity, there are instances when initiation of BER contributes to genetic instability; in these instances, we consider that BER has gone awry. In particular, repair of 8oxoG in a CAG/CTG repeat sequence of the huntingtin gene is linked to expansion of the sequence.64 This expansion is the molecular basis for Huntington’s disease. Thus, while BER of 8oxoG typically minimizes the point mutations cause by this modified nucleobase, when repair occurs in the CAG/CTG sequence context, genetic instability results.
Repair of 8oxoG in CAG/CTG repeat sequences has been shown to follow LP-BER, even in the absence of a modified 5′-dRP group.65 Pol β incorporates multiple nucleotides at the gap site, displacing a 5′-flap of CAG repeats. This 5′-flap can fold on itself, forming a hairpin structure that is refractory to cleavage by FEN1, but can be ligated by Lig1, leading to incorporation of excess CAG repeats. Furthermore, Gs in the incorporated CAG hairpin are a hotspot for damage, leading to formation of additional 8oxoG lesions.66 In addition to this accumulation of damage, OGG1 has reduced activity on 8oxoG within these CAG hairpins, leading to persistence of the lesion.66,67 Through DNA replication or nick-induced gap filling synthesis, the damage-containing hairpin can be reincorporated into duplex DNA, regenerating a substrate for BER. Thus, a toxic cycle is initiated in which DNA incrementally expands and is reoxidized.
11. Final Remarks
In this Account, we highlight the enzymes involved in the BER pathway. While the substrate specificity and kinetic parameters of many of these enzymes have largely been defined, several questions remain in the field. For example, much remains unknown about the energetics associated with DNA glycosylases sliding and probing the genome for nucleobase damage, or how such mechanisms occur in the context of chromatin. Indeed, understanding mechanisms in which BER enzymes carry out chemistry on DNA packaged within chromatin is also ongoing.68 Furthermore, considering that several enzymes are involved in successful completion of a BER event, how is their activity coordinated to avoid exposing mutagenic and cytotoxic intermediates? Consequently, these gaps in knowledge form the basis for much of the current research in the field and answers will broaden our understanding of this essential repair pathway and its contributions to genetic stability.
Acknowledgments
We are grateful to NIH/NIEHS for their financial support of our work on BER (ES019296). We also recognize the excellent work of our colleagues in the field, and apologize to those whose work could not be discussed given the length of the Account.
Biographies
Kelly Schermerhorn received her B.S. in Biomedical Science with a minor in Chemistry from Quinnipiac University in Hamden, CT. She is currently a doctoral candidate in the Chemistry Department at Brown University. Her current research focuses on the kinetics of BER enzymes.
Sarah Delaney is an Associate Professor of Chemistry at Brown University. She received her B.A. in Chemistry from Middlebury College, where she conducted undergraduate research with Professor Sunhee Choi. After receiving her Ph.D. from the California Institute of Technology under the guidance of Professor Jacqueline K. Barton, she conducted postdoctoral research as a Damon Runyon Postdoctoral Fellow with John M. Essigmann at the Massachusetts Institute of Technology. Professor Delaney’s research interests include the chemistry and biology of DNA damage and repair.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Cadet J.; Douki T.; Ravanat J. Oxidatively Generated Base Damage to Cellular DNA. Free Radical Biol. Med. 2010, 49, 9–21. [DOI] [PubMed] [Google Scholar]
- Gates K. S. An Overview of Chemical Processes That Damage Cellular DNA: Spontaneous Hydrolysis, Alkylation, and Reaction with Radicals. Chem. Res. Toxicol. 2009, 22, 1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S.; Takeshita M.; Grollman A. P. Insertion of Specific Bases During DNA Synthesis Past the Oxidation-Damaged Base 8-oxodG. Nature 1991, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- Friedman J.; Stivers J. Detection of Damaged DNA Bases by DNA Glycosylase Enzymes. Biochemistry 2010, 49, 4957–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley M.; Llyod R. Processivity of Uracil DNA Glycosylase. Mutat. Res. 1993, 294, 109–116. [DOI] [PubMed] [Google Scholar]
- Francis A.; David S. S. Escherichia coli MutY and Fpg Utilize a Processive Mechanism for Target Location. Biochemistry 2003, 42, 801–810. [DOI] [PubMed] [Google Scholar]
- Hudglin M.; O’Brien P. J. Human Alkyladenine DNA Glycosylase Employs a Processive Search for DNA Damage. Biochemistry 2008, 47, 11434–11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B.; Slupphaug G.; Mol C.; Arvai A.; Peterson S.; Tainer J. A.; Krokan H. Excision of Cytosine and Thymine from DNA by Mutants of Human Uracil-DNA Glycosylase. EMBO J. 1996, 15, 3442–3447. [PMC free article] [PubMed] [Google Scholar]
- Nash H. M.; Lu R.; Lane W.; Verdine G. L. The Critical Active-Site Amine of the Human 8-Oxoguanine DNA Glycosylase, hOgg1: Direct Identification, Ablation and Chemical Reconstitution. Chem. Biol. 1997, 4, 693–702. [DOI] [PubMed] [Google Scholar]
- Hazra T. K.; Kow Y.; Hatahet Z.; Imhoff B.; Boldogh I.; Mokkapati S.; Mitra S.; Izumi T. Identification and Characterization of a Novel Human DNA Glycosylase for Repair of Cytosine-Derived Lesions. J. Biol. Chem. 2002, 277, 30417–30420. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Krishnamurthy N.; Burrows C. J.; David S. S. Mutation versus Repair: NEIL1 Removal of Hydantoin Lesions in Single-Stranded, Bulge, Bubble, and Duplex DNA Contexts. Biochemistry 2010, 49, 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Liu M.; Fleming A.; Burrows C. J.; Wallace S. S. Neil3 and NEIL1 DNA Glycosylases Remove Oxidative Damages from Quadruplex DNA and Exhibit Preferences for Lesions in the Telomeric Sequence Context. J. Biol. Chem. 2013, 288, 27263–27272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels M.; Tchou J.; Grollman A.; Miller J. A Repair System for 8-Oxo-7,8-Dihydrodeoxyguanine. Biochemistry 1992, 31, 10964–10968. [DOI] [PubMed] [Google Scholar]
- Wiebauer K.; Jiricny J. Mismatch-Specific Thymine DNA Glycosylase and DNA Polymerase Beta Mediate the Correction of GT Mispairs in Nuclear Extracts from Human Cells. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 5842–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal A. E.; Hickson I.; Boiteux S.; Radicella J. P. Mechanism of Stimulation of the DNA Glycosylase Activity of hOGG1 by the Major Human AP Endonuclease: Bypass of the AP Lyase Activity Step. Nucleic Acids Res. 2001, 29, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalhus B.; Forsbring M.; Helle I.; Vik E.; Forstrøm R.; Backe P.; Alseth I.; Bjoras M. Separation-of-Function Mutants Unravel the Dual-Reaction Mode of Human 8-Oxoguanine DNA Glycosylase. Structure 2011, 19, 117–127. [DOI] [PubMed] [Google Scholar]
- Werner R. M.; Stivers J. T. Kinetic Isotope Effect Studies of the Reaction Catalyzed by Uracil DNA Glycosylase: Evidence for an Oxocarbenium Ion-Uracil Anion Intermediate. Biochemistry 2000, 39, 14054–14064. [DOI] [PubMed] [Google Scholar]
- McCann J.; Berti P. J. Transition-State Analysis of the DNA Repair Enzyme MutY. J. Am. Chem. Soc. 2008, 130, 5789–5797. [DOI] [PubMed] [Google Scholar]
- Schärer O. D.; Ortholand J.-Y.; Ganesan A.; Ezaz-Nikpay K.; Verdine G. L. Specific Binding of the DNA Repair Enzyme AlkA to a Pyrrolidine-Based Inhibitor. J. Am. Chem. Soc. 1995, 117, 6623–6624. [Google Scholar]
- Schärer O. D.; Nash H. M.; Jiricny J.; Laval J.; Verdine G. L. Specific Binding of a Designed Pyrrolidine Abasic Site Analog to Multiple DNA Glycosylases. J. Biol. Chem. 1998, 273, 8592–8597. [DOI] [PubMed] [Google Scholar]
- Chu A. M.; Fettinger J. C.; David S. S. Profiling Base Excision Repair Glycosylases with Synthesized Transition State Analogs. Bioorg. Med. Chem. Lett. 2011, 21, 4969–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski J. T.; Wong R.; McKnight J.; Bowman G.; Greenberg M. M. Rapid DNA-Protein Cross-Linking and Strand Scission by an Abasic site in a Nucleosome Core Particle. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 22475–22480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilay G.; Mol C.; Robson C.; Walker L.; Cunningham R. P.; Tainer J. A.; Hickson I. D. Identification of Critical Active-Site Residues in the Multifunctional Human DNA Repair Enzyme HAP1. Nat. Struct. Biol. 1995, 2, 561–568. [DOI] [PubMed] [Google Scholar]
- Erzberger J. P.; Wilson D. M. The Role of Mg2+ and Specific Amino Acid Residues in the Catalytic Reaction of the Major Human Abasic Endonuclease: New Insights from EDTA-Resistant Incision of Acyclic Abasic Site Analogs and Site-Directed Mutagenesis. J. Mol. Biol. 1999, 290, 447–457. [DOI] [PubMed] [Google Scholar]
- Beard W. A.; Wilson S. H. Structure and Mechanism of DNA Polymerase β. Chem. Rev. 2006, 106, 361–382. [DOI] [PubMed] [Google Scholar]
- Prasad R.; Shock D.; Beard W. A.; Wilson S. H. Substrate Channeling in Mammalian Base Excision Repair Pathways: Passing the Baton. J. Biol. Chem. 2010, 285, 40479–40488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. R.; Conrad J.; Wahl D.; O’Brien P. J. Kinetic Mechanism of Human DNA Ligase I Reveals Magnesium-Dependent Changes in the Rate-Limiting Step that Compromise Ligation Efficiency. J. Biol. Chem. 2011, 286, 23054–23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott K. W. XRCC1 and DNA Strand Break Repair. DNA Repair (Amsterdam) 2003, 2, 955–969. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Katyal S.; Lee Y.; Zhao J.; Rehg J.; Russell H. R. DNA ligase III is Critical for mtDNA Integrity but Not Xrcc1-Mediated Nuclear DNA Repair. Nature 2011, 471, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D.; Furda A.; Gao Y.; Artus J.; Brunet E.; Hadjantonakis A.; Van Houten B.; Shuman S.; McKinnon P.; Jasin M. Crucial role for DNA ligase III in Mitochondria but Not in Xrcc1-Dependent Repair. Nature 2011, 471, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung J.-S.; DeMott M.; Demple B. Long-Patch Base Excision DNA Repair of 2-Deoxyribonolactone Prevents the Formation of DNA-Protein Cross-links with DNA Polymerase Beta. J. Biol. Chem. 2005, 280, 39095–39103. [DOI] [PubMed] [Google Scholar]
- Horton J. K.; Prasad R.; Hou E.; Wilson S. H. Protection Against Methylation-Induced Cytotoxicity by DNA Polymerase b-Dependent Long Patch Base Excision Repair. J. Biol. Chem. 2000, 275, 2211–2218. [DOI] [PubMed] [Google Scholar]
- Singhal R. K.; Prasad R.; Wilson S. H. DNA Polymerase Beta conducts the Gap-Filling Step in Uracil-Initiated Base Excision Repair in a Bovine Testis Nuclear Extract. J. Biol. Chem. 1995, 270, 949–957. [DOI] [PubMed] [Google Scholar]
- Moldovan G.-L.; Pfander B.; Jentsch S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [DOI] [PubMed] [Google Scholar]
- Porello S. L.; Leyes A.; David S. S. Single-Turnover and Pre-Steady-State Kinetics of the Reaction of the Adenine Glycosylase MutY with Mismatch-Containing DNA Substrates. Biochemistry 1998, 37, 14756–14764. [DOI] [PubMed] [Google Scholar]
- Strauss P. R.; Beard W. A.; Patternson T. A.; Wilson S. H. Substrate Binding by Human Apurinic/Apyrimidinic Endonuclease Indicates a Briggs-Haldane Mechanism. J. Biol. Chem. 1997, 272, 1302–1307. [DOI] [PubMed] [Google Scholar]
- Demple B.; Herman T.; Chen D. S. Cloning and Expression of APE, the cDNA Encoding the Major Human Apurinic Endonuclease: Definition of a Family of DNA Repair Enzymes. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 11450–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J.; Kraynov V.; Zhong X.; Werneburg B. G.; Tsai M.-D. DNA Polymerase Beta: Effects of Gapped DNA Substrates on dNTP Specificity, Fidelity, Processivity and Conformational Changes. Biochem. J. 1998, 331, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S. H.; Kunkel T. A. Passing the Baton in Base Excision Repair. Nat. Struct. Biol. 2000, 7, 176–178. [DOI] [PubMed] [Google Scholar]
- Hill J.; Hazra T.; Izumi T.; Mitra S. Stimulation of Human 8-Oxoguanine-DNA Glycosylase by AP-Endonuclease: Potential Coordination of the Initial Steps in Base Excision Repair. Nucleic Acids Res. 2001, 29, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald M.; Drohat A. C. Coordinating the Initial Steps of Base Excision Repair: Apurinic/apyrimidinic Endonuclease 1 Actively Stimulates Thymine DNA glycosylase by Disrupting the Product Complex. J. Biol. Chem. 2008, 283, 32680–32690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin M. R.; O’Brien P. J. Human AP endonuclease 1 Stimulates Multiple-Turnover Base Excision by Alkyladenine DNA Glycosylase. Biochemistry 2009, 48, 6022–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascucci B.; Stucki M.; Jónsson Z.; Dogliotti E.; Hübscher U. Long Patch Base Excision Repair with Purified Human Proteins. DNA ligase I as Patch Size Mediator for DNA Polymerases Delta and Epsilon. J. Biol. Chem. 1999, 274, 33696–33702. [DOI] [PubMed] [Google Scholar]
- Prasad R.; Dianov G.; Bohr V.; Wilson S. H. FEN1 Stimulation of DNA Polymerase Beta Mediates an Excision Step in Mammalian Long Patch Base Excision Repair. J. Biol. Chem. 2000, 275, 4460–4466. [DOI] [PubMed] [Google Scholar]
- Dianov G. L.; Hübscher U. Mammalian Base Excision Repair: the Forgotten Archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers P. M. Saccharomyces cerevisiae replication factor C. II. Formation and activity of complexes with the proliferating cell nuclear antigen and with DNA polymerases delta and epsilon. J. Biol. Chem. 1991, 226, 22698–22706. [PubMed] [Google Scholar]
- Kedar P.; Kim S.; Robertson A.; Hou E.; Prasad R.; Horton J. K.; Wilson S. H. Direct Interaction Between Mammalian DNA Polymerase Beta and Proliferating Cell Nuclear Antigen. J. Biol. Chem. 2002, 277, 31115–31123. [DOI] [PubMed] [Google Scholar]
- Maga G.; Hubscher U. Proliferating Cell Nuclear Antigen (PCNA): A Dancer with Many Partners. J. Cell. Sci. 2003, 116, 3051–3060. [DOI] [PubMed] [Google Scholar]
- Dou H.; Theriot C.; Das A.; Hegde M.; Matsumoto Y.; Boldogh I.; Hazra T.; Bhakat K.; Mitra S. Interaction of the Human DNA Glycosylase NEIL1 with Proliferating Cell Nuclear Antigen. The Potential for Replication-Associated Repair of Oxidized Bases in Mammalian Genomes. J. Biol. Chem. 2008, 283, 3130–3140. [DOI] [PubMed] [Google Scholar]
- Xia L.; Zheng L.; Lee H.-W.; Bates S.; Federico L.; Shen B.; O’Connor T. R. Human 3-Methyladenine-DNA Glycosylase: Effect of Sequence Context on Excision, Association with PCNA, and Stimulation by AP Endonuclease. J. Mol. Biol. 2005, 346, 1259–1274. [DOI] [PubMed] [Google Scholar]
- Chang D.-Y.; Lu A.-L. Functional Interaction of MutY Homolog with Proliferating Cell Nuclear Antigen in Fission Yeast, Schizosaccharomyces pombe. J. Biol. Chem. 2002, 277, 11853–11858. [DOI] [PubMed] [Google Scholar]
- Balakrishnan L.; Brandt P.; Lindsey-Boltz L.; Sancar A.; Bambara R. A. Long Patch Base Excision Repair Proceeds via Coordinated Stimulation of the Multienzyme DNA Repair Complex. J. Biol. Chem. 2009, 284, 15158–15172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher A.; Hochegger H.; Takeda S.; Caldecott K. W. Poly(ADP-Ribose) Polymerase 1 Accelerates Single-Strand Break Repair in Concert with Poly(ADP-Ribose) Glycohydrolase. Mol. Cell. Biol. 2007, 27, 5597–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Tassan N.; Chmiel N.; Maynard J.; Fleming N.; Livingston A.; Williams G.; Hodges A.; Davies D.; David S. S.; Sampson J. R.; Cheadle J. P. Inherited Variants of MYH Associated with Somatic G:C→T:A Mutations in Colorectal Tumors. Nat. Genet. 2002, 30, 227–232. [DOI] [PubMed] [Google Scholar]
- Pope M. A.; Chmiel N. H.; David S. S. Insight into the Functional Consequences of hMYH Variants Associated with Colorectal Cancer: Distinct Differences in the Adenine Glycosylase Activity and the Response to AP Endonucleases of Y150C and G365D Murine MYH. DNA Repair (Amsterdam) 2005, 4, 315–325. [DOI] [PubMed] [Google Scholar]
- Karahalil B.; Bohr V.; Wilson D. Impact of DNA Polymorphisms in Key DNA Base Excision Repair Proteins on Cancer Risk. Hum. Exp. Toxicol. 2012, 31, 981–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S.; Smeyne R.; Wallace J.; Curran T. The Redox/DNA Repair Protein, Ref-1, is Essential for Early Embryonic Development in Mice. Proc. Natl. Acad. Sci.U.S.A. 1996, 93, 8919–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung H.; Demple B. A Vital Role for Ape1/Ref1 Protein in Repairing Spontaneous DNA Damage in Human Cells. Mol. Cell 2005, 17, 463–470. [DOI] [PubMed] [Google Scholar]
- Doetsch P.; Cunningham R. The Enzymology of Apurinic/Apyrimidinic Endonucleases. Mutat. Res. 1990, 236, 173–201. [DOI] [PubMed] [Google Scholar]
- Cabelof D. C.; Guo Z.; Raffoul J. J.; Sobol R. W.; Wilson S. H.; Richardson A.; Heydari A. R. Base Excision Repair Deficiency caused by Polymerase Beta Haploinsufficiency: Accelerated DNA Damage and Increased Mutational Response to Carcinogens. Cancer Res. 2003, 63, 5799–5807. [PubMed] [Google Scholar]
- Starcevic D.; Dalal S.; Sweasy J. B. Is There a Link Between DNA Polymerase Beta and Cancer?. Cell Cycle 2004, 3, 996–999. [PubMed] [Google Scholar]
- Fishel M. L.; Kelley M. R. The DNA Base Excision Repair Protein Ape1/Ref-1 as a Therapeutic and Chemopreventive Target. Mol. Aspects Med. 2007, 28, 375–395. [DOI] [PubMed] [Google Scholar]
- Yang J.; Parsons J.; Nicolay N. H.; Caporali S.; Harrington C. F.; Singh R.; Finch D.; D’Atri S.; Farmer P. B.; Johnston P. G.; McKenna W. G.; Dianov G.; Sharma R. A. Cells Deficient in the Base Excision Repair Protein DNA Polymerase Beta are Hypersensitive to Oxaliplatin Chemotherapy. Oncogene 2010, 29, 463–468. [DOI] [PubMed] [Google Scholar]
- Kovtun I. V.; Liu Y.; Bjoras M.; Klungland A.; Wilson S. H.; McMurray C. T. OGG1 Initiates Age-dependent CAG Trinucleotide Expansion in Somatic Cells. Nature 2007, 447, 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Prasad R.; Beard W. A.; Hou E. W.; Horton J. K.; McMurray C. T.; Wilson S. H. Coordination Between Polymerase Beta and FEN1 can Modulate CAG Repeat Expansion. J. Biol. Chem. 2009, 284, 28352–28366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarem D. A.; Wilson N. R.; Delaney S. Structure-dependent DNA Damage and Repair in a Trinucleotide Repeat Sequence. Biochemistry 2009, 48, 6655–6663. [DOI] [PubMed] [Google Scholar]
- Jarem D. A.; Wilson N. R.; Schermerhorn K. M.; Delaney S. Incidence and Persistence of 8-oxo-7,8-dihydroguanine within a Hairpin Intermediate Exacerbates a Toxic Oxidation Cycle Associated with Trinucleotide Repeat Expansion. DNA Repair (Amsterdam) 2011, 10, 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odell I.; Wallace S.; Pederson D. Rules of Engagement for Base Excision Repair in Chromatin. J. Cell. Physiol. 2013, 228, 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava D.; Berg B. J.; Prasad R.; Molina J. T.; Beard W. A.; Tomkinson A. E.; Wilson S. H. Mammalian Abasic Site Base Excision Repair. Identification of the Reaction Sequence and Rate-Determining Steps. J. Biol. Chem. 1998, 273, 21203–21209. [DOI] [PubMed] [Google Scholar]
- Drohat A.; Jagashree J.; Ferguson E.; Stivers J. T. Role of Electrophilic and General Base Catalysis in the Mechanism of Escherichia coli Uracil DNA Glycosylase. Biochemistry 1999, 38, 11866–11875. [DOI] [PubMed] [Google Scholar]
- Asagoshi K.; Yamada T.; Terato H.; Ohyama Y.; Monden Y.; Arai T.; Nishimura S.; Aburatani H.; Lindahl T.; Ide H. Distinct Repair Activities of Human 7,8-Dihydro-8-oxoguanine DNA Glycosylase and Formamidopyrimidine DNA Glycosylase for Formamidopyrimidine and 7,8-Dihydro-8-oxoguanine. J. Biol. Chem. 2000, 275, 4956–4964. [DOI] [PubMed] [Google Scholar]
- Leipold M.; Workman H.; Muller J.; Burrows C. J.; David S. S. Recognition and Removal of Oxidized Guanines in Duplex DNA by the Base Excision Repair Enzymes hOGG1, yOGG1, and yOGG2. Biochemistry 2003, 42, 11373–11381. [DOI] [PubMed] [Google Scholar]
- Xu Y.-J.; DeMott M. S.; Hwang J. T.; Greenberg M. M.; Demple B. Action of Human Apurinic Endonuclease (Ape1) on C1′-oxidized Deoxyribose Damage in DNA. DNA Repair (Amsterdam) 2003, 2, 175–185. [DOI] [PubMed] [Google Scholar]
- Maher R.; Bloom L. B. Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J. Biol. Chem. 2007, 282, 30577–30585. [DOI] [PubMed] [Google Scholar]
- Schermerhorn K.; Delaney S. Transient-State Kinetics of Apurinic/Apyrimidinic (AP) Endonuclease 1 Acting on an Authentic AP Site and Commonly Used Substrate Analogs: The Effect of Diverse Metal Ions and Base Mismatches. Biochemistry 2013, 52, 7669–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R.; Beard W. A.; Strauss P. R.; Wilson S. H. Human DNA Polymerase Beta Deoxyribose Phosphate Lyase. Substrate Specificity and Catalytic Mechanism. J. Biol. Chem. 1998, 273, 15263–15270. [DOI] [PubMed] [Google Scholar]